 Satoshi Watanabe
Satoshi Watanabe Tomohiro Koshiyama
Tomohiro Koshiyama- Department of Chemical Engineering, Kyoto University, Kyoto, Japan
Metal nanoparticles (NPs) are key materials used in a broad range of industries. Among the various synthetic routes of NPs, liquid-phase chemical reactions are promising because of their versatility in reaction conditions as well as their potential productivity. However, because the synthesis of NPs involves not only chemical reactions but also nucleation and growth processes, which are typically higher-order reactions in terms of the concentration, a small degree of nonuniformity in the concentration during mixing of reaction solutions can easily result in a wide size distribution of the resultant particles. A typical solution to this problem is to slow the rate of reactions compared with that of mixing; however, as a result, the synthetic processes often require long reaction periods and complex procedures. In this study, we applied a microreactor with excellent mixing performance to NP synthesis to simplify and intensify the processes. We synthesized nickel and platinum-cobalt alloy NPs as model materials. For the Ni NP synthesis, we demonstrated that the quick mixing provided by the microreactor enabled the precise control of the residence time, and consequently, monodispersed Ni NPs with an average size of 3.8 nm were synthesized. For the Pt-Co bimetallic system, the microreactor successfully produced Pt-Co alloy NPs, while batch-type synthesis with weaker mixing intensity resulted in a bimodal mixture of larger Pt NPs and smaller Co NPs. For both Ni and Pt-Co, monodispersed NPs were synthesized by simply mixing the reaction solutions in the microreactor at room temperature. These results demonstrate that the mixing process plays a key role in NP synthesis, and application of a microreactor enables the establishment of a facile and robust synthetic process.
Introduction
Metal particles exhibit excellent chemical and physical properties, and thus can be incorporated in various applications, including catalysts, electronics, photonics, sensing, and imaging (Xia et al., 2009). Downsizing metal particles to the nanoscale is a promising approach to fully exploit their potential not only by enlarging specific surface areas, but also by drawing properties that are not available in bulk crystals. To optimize their functions suitable for desired applications, it is crucial to control the average size of nanoparticles (NPs) with a sharp size distribution because the particle size dominantly governs the resultant properties. Among the various synthetic routes of NPs, liquid-phase techniques are attractive because of their ease of handling and versatility in reaction conditions. A typical synthesis method is the polyol process (Fiévet et al., 2018), in which polyols such as ether glycols and 1,2-diols serve both as a solvent and a reducing agent. Such polyols have been widely used because of their ability to control the particle size and applicability to various metals such as Fe, Ni, Pd, Ag, Pt, Au, and Pb. However, there is still room for improvement regarding synthesis times, reaction temperatures, and solvents; accordingly, process intensification is strongly required to achieve more energy-saving and greener synthesis.
In this study, we focused on Ni as the target material. Several methods have been reported for the liquid-phase synthesis of Ni NPs as well as the gas-phase synthesis using sputtering (Bussamara et al., 2013) and microplasma techniques (Lin et al., 2018). A typical synthesis method is a water-in-oil microemulsion technique, in which microemulsions formed in an oil phase serve not only as reactors to provide Ni ions with confined reaction fields for reduction reactions, but also as stabilizers against aggregation after NP formation (Chen and Wu, 2000; Wu et al., 2012). Another technique is an organometallic approach, which utilizes the spontaneous decomposition of organometallic precursors such as Ni(C8H12)2 (Ely et al., 1999; Domínguez-Crespo et al., 2009). The polyol process has also been modified to use an additional reducing agent (Wu and Chen, 2003; Couto et al., 2007; Eluri and Paul, 2012). Using these methods, Ni NPs with average diameters of 3–10 nm were successfully synthesized. However, these methods use organic solvents and require heating during the synthesis. Although aqueous syntheses were attempted, the resultant particle sizes were larger (>10 nm) than those obtained using organic solvents, and the size distributions were relatively wide (Chen and Hsieh, 2002; Wu and Chen, 2004; Park et al., 2006; Jiang et al., 2013), indicating that the aqueous synthesis of monodisperse Ni NPs is challenging.
A general strategy for the formation of NPs is to quickly produce many small nuclei by bringing the reaction solution to a high supersaturation condition. In fact, the above aqueous syntheses use a strong reducing agent, hydrazine, following the general strategy. However, the reduction reaction of Ni ions by hydrazine involves multiple steps (Park et al., 2006) and accordingly, simple batch reactors, as applied in the above aqueous syntheses, would not be effective because the reaction sequence is difficult to precisely control with their intrinsically weak mixing intensity. This leads to the idea of introducing an alternative reaction device with high mixing performance, which would be a possible solution to the problem of synthesizing monodisperse Ni NPs in an aqueous system. In our previous studies, we applied a tailor-made microreactor to the synthesis of core-shell-type particles including SiO2@Au (Watanabe et al., 2015), SiO2@Ag (Maw et al., 2019), and metal-organic framework (MOF)@MOF (Fujiwara et al., 2021) and succeeded in process intensification by taking advantage of the excellent mixing performance of the microreactor. One of the key achievements is the establishment of a sequential flow process involving two separate reaction steps for the synthesis of SiO2@Au particles. This result indicates that the microreactor is a suitable reaction device for controlling the multiple reaction steps involved in Ni NP synthesis.
Similar to the case of monometallic NPs, the synthesis of bimetallic alloy NPs also requires process intensification. Although a variety of alloy NPs have been synthesized, their syntheses mostly rely on the polyol process (Kobayashi et al., 2015), and the development of a room-temperature aqueous synthetic method will further accelerate the industrial applications of alloy NPs. Ideally, a completely mixed solution in which constituent metal atoms are uniformly distributed would result in the spontaneous formation of alloy NPs through nucleation and subsequent growth if the solid solution state is thermodynamically more stable than the phase-separated state in which monometallic NPs of two metal components are separately formed. Our strategy to realize a reaction solution close to the ideal situation is to quickly reduce metal ions using a strong reducing agent. For this purpose, the microreactor is a promising reaction tool because it offers quick and homogeneous mixing of metal ions and reductant molecules. We selected Pt-Co bimetallic NPs as a model system because they are promising materials for electrocatalytic applications (Huang et al., 2020; Shi et al., 2021).
In this study, we employed the microreactor for the liquid-phase synthesis of Ni and Pt-Co alloy NPs. For the Ni NP synthesis, we constructed a sequential flow process by connecting two microreactors and investigated the effects of the residence time between the two microreactors on the resultant particle size and distribution. We also determined the roles of the microreactors in Ni NP synthesis through a comparison with a Y-shaped mixer with weaker mixing intensity and proposed a possible mechanism for the formation of Ni NPs. For the Pt-Co alloy NP synthesis, we used a single microreactor in which a mixed solution of Pt and Co ions was mixed with a reducing agent, and investigated the effects of mixing devices and precursors of Pt and Co on the size and structure of the resultant particles.
Experimental Section
Materials
Cetyltrimethylammonium bromide (CTAB, >98%), cobalt (II) chloride hexahydrate (CoCl2.6H2O, 98%), cobalt (II) nitrate hexahydrate (Co(NO3)2·6H2O, 98%), hexachloroplatinic (IV) acid (H2PtCl6, 8 wt% aqueous solution), nickel (II) chloride (NiCl2, 98%), polyvinylpyrrolidone (PVP, molecular weight Mw ∼ 1,300,000), potassium hexachloroplatinate (IV) (K2PtCl6, 99.99%), potassium tetrachloroplatinate (II) (K2PtCl4, 99.99%), and sodium borohydride (NaBH4, 99%) were purchased from Sigma-Aldrich Co. LLC. (United States). Anhydrous hydrazine (N2H4, >98.0%) and sodium hydroxide (NaOH, 0.1 M) were purchased from Tokyo Chemical Industry Co., Ltd. (Japan) and Kishida Chemical Co. Ltd. (Japan), respectively. All chemicals were used as received. Solutions with the desired concentrations were prepared by dissolving the original chemicals in ultrapure water (>18 MΩ cm, Arium mini, Sartorius, Germany).
Microreactor
The microreactor used in this study is a central-collision-type microreactor, the disassembly drawing of which is shown in Figure 1. In the microreactor, each of the two inlet solutions is branched into seven streams and the branched fourteen streams in total intensively collide with each other at the mixing zone into a single outlet stream so that the collision of the streams breaks the fluids into micron-sized segments to shorten the diffusion distance (Nagasawa et al., 2005). The microreactor has a cylindrical shape made of stainless steel with the diameter of 4 cm and height of 2.4 cm; the channel width of the branched streams is 100 µm and that of the outlet is 360 µm. In our previous study, we evaluated the mixing performance of the microreactor through a chemical test reaction and demonstrated that its characteristic mixing time is shorter than 1 ms, which is one to three orders of magnitude faster than those of a batch reactor and a millimeter-sized Y-shaped mixer (Watanabe et al., 2017).
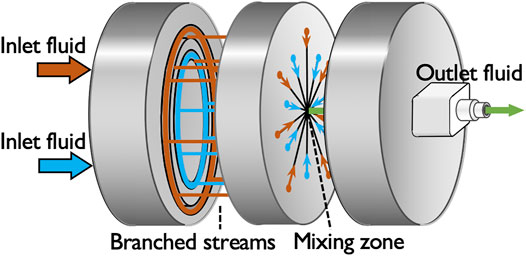
FIGURE 1. Disassembly drawing of the central-collision type microreactor.
Synthesis and Characterization of Ni NPs
Figure 2 shows a schematic of the sequential flow process comprising two microreactors for Ni NP synthesis. A premixed aqueous solution of NiCl2 and CTAB was first mixed with an N2H4 aqueous solution in microreactor A, and the mixed solution from the outlet was then directly injected into microreactor B to be mixed with a NaOH solution. The reaction solution from the outlet of microreactor B was collected in a glass vial and stirred for 20 h using a magnetic stirrer. Syringe pumps were used to flow the solutions into the microreactors at the flow rate of 10 ml/min for each syringe.
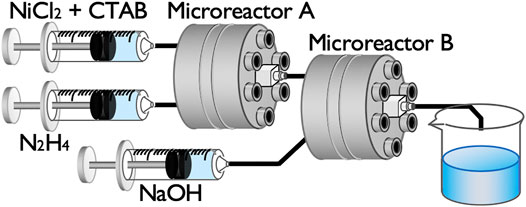
FIGURE 2. Schematic of the sequential flow process for the synthesis of Ni NPs.
In this sequential flow process, we assumed the following reaction scheme (Park et al., 2006). The reaction in microreactor A produces nickel–hydrazine complexes.
Mixing with NaOH in microreactor B induces a ligand exchange reaction to produce nickel (II) hydroxide and hydrazine.
Ni(OH)2 (Eq. 2) was reduced to zero-valent Ni by N2H4 to produce Ni NPs.
A typical concentration set after mixing in the two microreactors was [NiCl2] = 25 mM [CTAB] = 25 mM [N2H4] = 500 mM, and [NaOH] = 20 mM. We varied the residence time τ between microreactors A and B by changing the tube length connecting the microreactors. The tube used was made of silicone rubber with the inner diameter of 1.5 mm. Tube lengths of 1, 3, 20, and 40 cm corresponded to residence times of 0.053, 0.16, 1.1, and 2.1 s, respectively, based on a simple calculation, τ = πd2L/4v, where d is the inner diameter, L is the tube length, and v is the flow rate. We also synthesized Ni NPs using a Y-shaped mixer (VFY106, polypropylene mini fitting with an inner diameter of 1.5 mm, ISIS Co., Ltd., Japan) in place of either microreactor A or microreactor B (i.e. mixing in a Y-shaped mixer followed by microreactor B or mixing in microreactor A followed by a Y-shaped mixer in sequential flow processes) to investigate the effect of the mixing intensity. All the experiments were conducted at room temperature.
Transmission electron microscopy (TEM) images of the synthesized particles were obtained using a JEM-1010 (JEOL Ltd., Japan) TEM instrument. The average particle size and size distribution were calculated by measuring the size of at least 200 particles from the TEM images. The elemental compositions of the particles were measured using X-ray photoelectron spectroscopy (XPS; ESCA-3400, Shimadzu Corp., Japan). The crystal structures of the particles were analyzed via powder X-ray diffraction (XRD) using Cu Kα radiation (MultiFlex, Rigaku Corp., Japan). The absorption spectra of the reaction solution of nickel–hydrazine complexes were measured using a Mutispec-1500 (Shimadzu Corp., Japan) UV-visible spectrophotometer.
Synthesis and Characterization of Pt-Co Alloy NPs
The same microreactor used in the Ni NP synthesis was used as the reaction device. A premixed aqueous solution of Pt and Co ions and PVP was mixed with an aqueous solution of a strong reducing agent, NaBH4. Both solutions were injected into the microreactor using a syringe pump at the flow rate of 10 ml/min for each syringe. The mixed reaction solution from the outlet was collected in a glass vial. We also used a batch reactor instead of a microreactor, in which NaBH4 solution was added to a premixed solution of Pt and Co ions and PVP in a glass vial under stirring at 1,500 rpm on a magnetic stirrer.
K2PtCl4, H2PtCl6, or K2PtCl6 were used as metallic precursors for Pt and CoCl2 or Co(NO3)2 for Co. The molar ratios of [NaBH4]/[Mn+total] and [PVPmonomer]/[Mn+total] were set to 1.4 and 1.1, respectively, where Mn+total is the sum of the Pt and Co ions and PVPmonomer is the number of moles of PVP monomer units, and [Mn+total] was fixed as 0.20 mM. A typical concentration set after the mixing was [Pt ion] = 0.10 mM [Co ion] = 0.10 mM [NaBH4] = 0.28 mM, and [PVPmonomer] = 0.22 mM. Experiments were conducted at room temperature, unless otherwise stated.
Scanning TEM–energy dispersive X-ray (STEM-EDX) analyses were conducted to obtain STEM and elemental distribution mapping images of the resultant particles using a JEM-2100F (JEOL Ltd., Japan) field emission transmission electron microscope equipped with a JED-2300 (JEOL Ltd., Japan) analysis station. TEM images of the particles were obtained using a JEM-2100F microscope (JEOL Ltd., Japan). The average particle size and size distribution were calculated by measuring the size of at least 200 particles from the TEM images. The XRD patterns of the particles were obtained using an UltimaIV/285/DX (Rigaku Corp., Japan) X-ray diffractometer. The residual amounts of Pt and Co ions in the resultant suspension were measured using an inductively coupled plasma (ICP) instrument (ISPS-7500, Shimadzu Corp., Japan). ICP measurements were conducted for supernatants obtained by centrifuging suspensions at 15,000 rpm for 10 min twice.
Results and Discussion
Synthesis of Monodispersed Ni NPs
As a representative result, Figure 3 presents the resultant particles synthesized at the residence time τ of 0.053 s between microreactors A and B. As shown in Figures 3A,B, nearly spherical NPs formed with an average diameter of 3.8 ± 0.6 nm. The XPS result of the as-synthesized particles exhibited no specific peaks of binding energies of Ni metal, as shown in Figure 3C, which could be due to the CTAB molecules surrounding the particle surfaces. After Ar ion etching of the particles for 30 s, two broad peaks appeared around 870 and 853 eV in the XPS spectrum, and further etching for 60 and 90 s intensified and sharpened these two peaks. Because the peaks at 870 and 853 eV were attributed to Ni 2p1/2 and Ni 2p3/2, respectively, these results suggest that the resultant particles were comprised of Ni metal. A slight peak was observed for the spectra before etching and after 30 s of etching at around 857 eV. This could be attributed to the formation of thin nickel oxide layers on the Ni NP surfaces. The oxidation of Ni surfaces was assumed to proceed during drying in air because it has been reported that Ni particles in suspensions are not oxidized by dissolved oxygen (Chen and Hsieh, 2002). Figure 3D displays the XRD patterns of the as-synthesized particles. The three peaks at 2θ = 44.5, 51.8, and 76.4° were due to the diffractions of the {111}, {200}, and {220} faces of the fcc crystal of Ni, demonstrating the formation of Ni NPs with fcc crystal structures. The synthesized Ni NPs were comparable to those obtained from methods using organic solvents in terms of the average size and size distribution; therefore, our synthetic process using the microreactor was demonstrated to successfully achieve the aqueous and room-temperature synthesis of monodisperse Ni NPs.
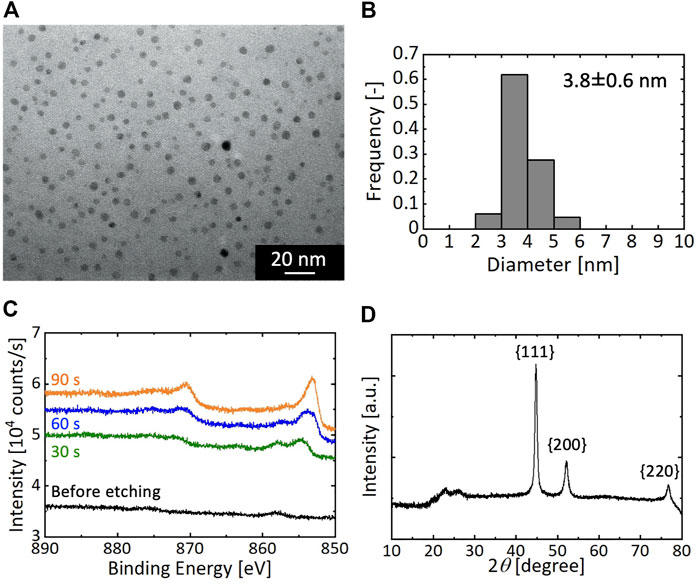
FIGURE 3. Synthetic result of Ni NPs with the residence time τ of 0.053 s (A) TEM image (B) Particle size distribution (C) XPS result; and (D) XRD pattern.
Figure 4 shows TEM images and corresponding size distributions of Ni NPs synthesized for different τ values of 2.1, 1.1, and 0.16 s. The synthetic condition of τ = 2.1 s yielded particles with large size variation of 2–10 nm, and particularly the formation of particles larger than 5 nm is noticeable in the TEM result (Figure 4A). In contrast, syntheses with τ < 2.1 s produced particles with much sharper size distributions (Figures 4B,C). To further investigate the effect of τ, we conducted an experiment with a longer τ on the order of minutes. Because the tube length on the order of 10 m was required to realize the long residence time (e.g. 56 m for 5-min residence time), which was impractical in terms of the power of the syringe pump we used, we divided the sequential flow process into two separate mixing processes. In the experiment, we first mixed a pre-mixed solution of NiCl2 and CTAB with a N2H4 solution in the microreactor and collected the reaction solution in a glass vial. After 5 min, we mixed the reaction solution with a NaOH solution in the microreactor. This synthetic process resulted in the formation of Ni particles with a considerably wide size range, as shown in Figure 5, in which particles even larger than 10 nm were observed. These results demonstrate that the residence time shorter than ca. 1 s was required for the formation of monodispersed Ni NPs.
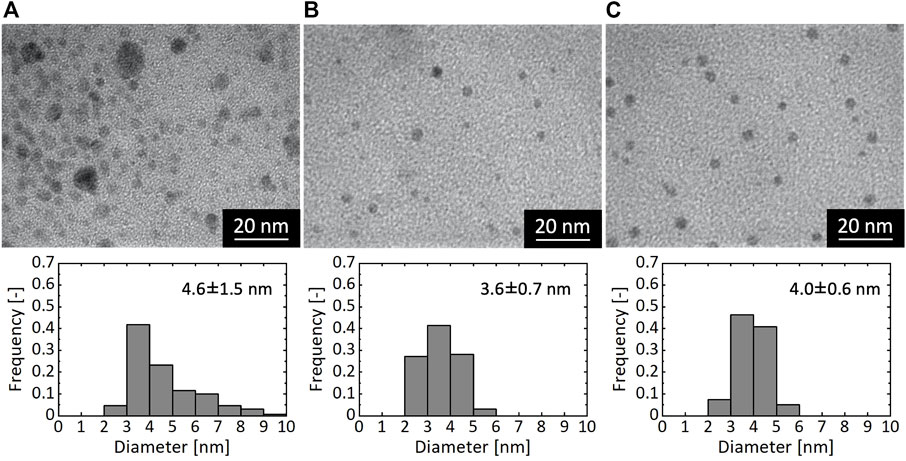
FIGURE 4. TEM images and particle size distributions of resultant Ni NPs synthesized with different residence times of (A) 2.1 s (B) 1.1 s, and (C) 0.16 s.

FIGURE 5. TEM image of resultant Ni NPs synthesized with the prolonged residence time.
The residence time was assumed to change the reaction progress of the nickel–hydrazine complex formation. The formation process of the complexes was traced by measuring the absorbance at 589 nm (Figure 6). In this experiment, a mixed solution of NiCl2 and N2H4 from the microreactor was collected in a cuvette, which was set in the UV-visible spectrophotometer, and the time course of absorbance was measured. As shown in Figure 6, the intensity increased with time and reached a constant value in 10 s, suggesting that the reaction was completed in 10 s to form stable complexes.
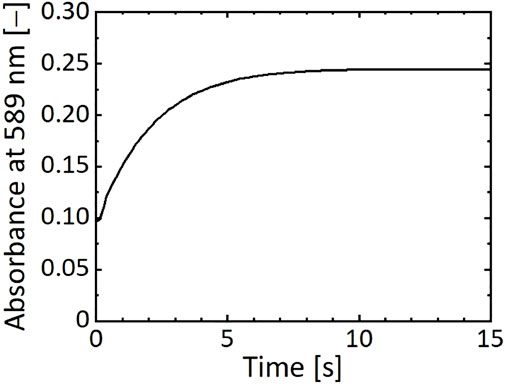
FIGURE 6. Time course of absorbance at 589 nm of a reaction solution of NiCl2 and N2H4 mixed in microreactor.
Based on these results, we propose a possible mechanism. It has been reported that an excess amount of hydrazine, as used in this study, leads to the formation of stable three-coordinate complex structures of Ni ions with hydrazine molecules [Ni(N2H4)3]2+, through intermediate species with one- and two-coordinate structures [Ni(N2H4)]2+ and [Ni(N2H4)2]2+ (Gilbert and Evans, 1951; Li et al., 1999). Under a long residence time condition, the dominant species would be [Ni(N2H4)3]2+ at the moment of mixing with a NaOH solution in microreactor B, while the intermediate species [Ni(N2H4)]2+ and [Ni(N2H4)2]2+ reacted with NaOH molecules under short residence time conditions. Because the intermediates are assumed to be less stable than the three-coordination complex, the ligand exchange reaction (Eq. 2) would readily and quickly release N2H4 molecules, thereby leading to the subsequent reduction of Ni ions. In contrast, the ligand exchange reaction of [Ni(N2H4)3]2+ would be slower, possibly because of the higher activation energy required to replace N2H4 molecules with OH−ions from the stable complexes with three coordination, which consequently leads to a slower reduction reaction and results in the formation of particles with a wider size distribution, even though quick and homogeneous mixing is provided by the microreactor. In this manner, the key requirement for the synthesis of monodisperse Ni NPs is trapping the intermediate complex species to react with NaOH molecules before the formation of stable three-coordination complexes. To satisfy this requirement, the precise control of the residence time on the order of milliseconds is critical, which is enabled by the excellent mixing performance of the microreactor.
To investigate the effects of the mixing intensity on the Ni NP synthesis, we used a Y-shaped mixer instead of the microreactors in the sequential flow process with the residence time fixed at 0.16 s. Under the present flow rate condition, the characteristic mixing times of the microreactor and Y-shaped mixer were determined as 0.3 ms and 0.2 s, respectively (Watanabe et al., 2017). As shown in Figure 7A, only a few particles were observed in the case of the Y-shaped mixer used in place of microreactor A, which is in contrast to Figure 4C. This is because the two inflow solutions of NiCl2/CTAB and N2H4 were not well mixed due to weak mixing in the Y-shaped mixer, and the complex formation reaction did not proceed, resulting in a lack of nickel–hydrazine complexes at the moment of mixing with the NaOH solution. Figure 7B shows a TEM image of the particles synthesized using the Y-shaped mixer instead of microreactor B. Although microreactor A was used for complex formation combined with a short residence time (0.16 s), the resultant particles showed a wide size variation, which is contrary to Figure 4C. This could be due to insufficient mixing in the Y-shaped mixer, by which the ligand exchange and subsequent reduction reactions did not proceed uniformly, resulting in a prolonged nucleation period to yield particles of varied sizes.

FIGURE 7. TEM images of resultant particles synthesized using a Y-shaped mixer in place of (A) microreactor A and (B) microreactor B.
These results demonstrate that both microreactors A and B play crucial roles in the synthesis of monodispersed Ni NPs. The quick and homogeneous mixing in microreactor A enables the formation of a sufficient amount of intermediate complexes during the residence time, and that in microreactor B allows instantaneous nucleation by bringing a mixed solution into a highly supersaturated condition to produce many small nuclei. Here, we emphasize that setting a short residence time on the order of milliseconds only works if a mixing device with a characteristic mixing time shorter than the desired residence time is used. In that sense, the microreactor is a favorable reaction tool, and we successfully achieved aqueous and room-temperature synthesis of monodisperse Ni NPs by taking advantage of the excellent mixing performance provided by the microreactor.
Synthesis of Pt-Co Alloy NPs
Figure 8 shows TEM images, particle size distributions, and STEM and EDX mapping images of resultant particles synthesized using the microreactor (Figure 8A) and a batch reactor (Figure 8B), in which K2PtCl4 and CoCl2 were used as precursors and [Mn+total] was 0.20 mM. In the case of the microreactor, resultant particles exhibited a monomodal distribution with the average diameter of 10.4 nm. In contrast, the batch reactor resulted in a rather bimodal distribution with particles around 8 nm and those larger than 15 nm. As shown in the STEM and EDX mapping images, the microreactor produced solid solution particles, in which Pt and Co atoms were uniformly distributed with a Pt:Co atomic ratio of 0.66:0.34 based on an EDX compositional analysis. In the case of the batch reactor, NPs of separate components were observed with smaller domains for Co and larger ones for Pt, suggesting that the bimodal distribution from the batch reactor was attributed to the formation of smaller and larger particles with the main components of Co and Pt, respectively. Figure 9 presents the XRD patterns of resultant particles from the microreactor and the batch reactor compared with those of Pt and Co metals as references (Davey, 1925; Owen and Jones, 1954). For both reactors, the main peaks appeared in between the peaks of the {111} faces of Pt and Co references and the second ones in between those of the {200} faces, indicating the formation of Pt-Co alloys (Huang et al., 2020). However, the peaks for the batch reactor were flat-topped, which was particularly clear in the second peak from 46.4° to 47.1°, demonstrating a larger variation in the composition of resultant particles, which agrees with the EDX analysis result (Figure 8B).
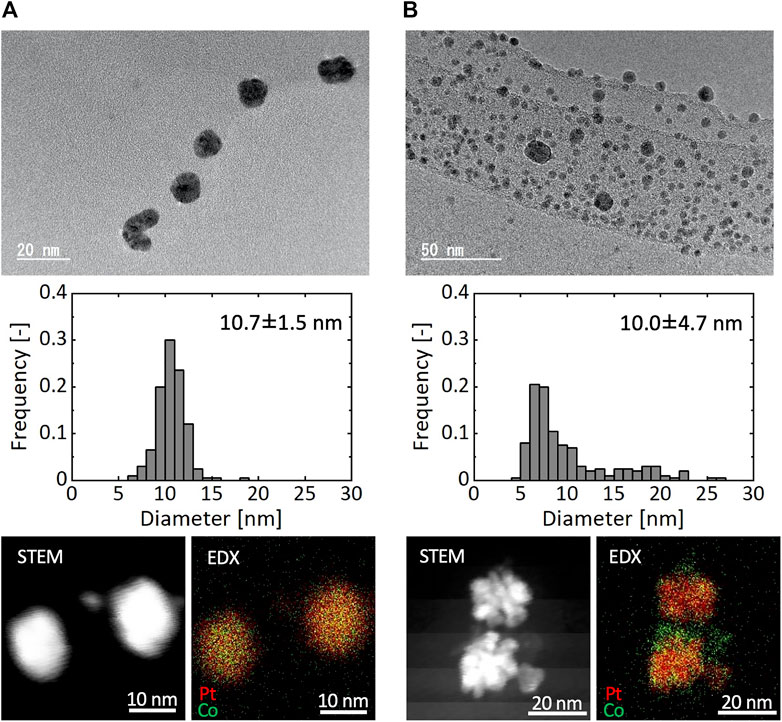
FIGURE 8. TEM image, particle size distribution, and STEM and EDX mapping images of Pt and Co bimetallic particles synthesized by (A) microreactor and (B) batch reactor.
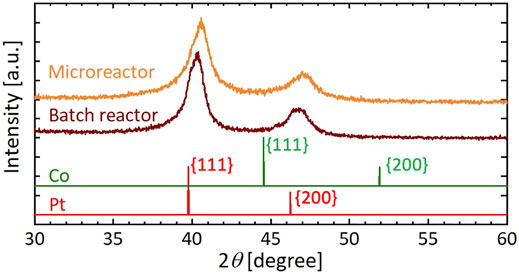
FIGURE 9. XRD patterns of Pt and Co bimetallic particles synthesized using microreactor and batch reactor compared with those of Pt and Co metals as references.
Because the mixing device is the only difference in the synthetic processes, these results are attributable to the difference in the mixing intensity of the batch reactor and the microreactor. The possible mechanism is as follows: In the case of the batch reactor, a reaction solution is segregated immediately after mixing to have a concentration distribution due to the weaker mixing, which leads to local deficiency of reducing agents relative to metal ions. In this situation, Pt ions would be readily reduced due to their lower ionization tendency than that of Co ions to induce preferred nucleation and subsequent growth of Pt, which would produce larger Pt NPs. Consequently, the resultant particles showed a bimodal distribution comprising larger Pt and smaller Co NPs. In contrast, the reaction solution in the microreactor is more uniform, and accordingly, the reduction reaction of both Pt and Co ions would simultaneously proceed to yield Pt-Co alloy NPs.
The proposed mechanism suggests that a larger amount of Co ions is consumed in the reaction in the microreactor. Accordingly, we measured the reaction ratios of Pt and Co ions, xPt and xCo, respectively, based on the ICP analyses, in which Pt and Co ions remaining in the supernatants of the reaction suspensions were quantified. The measured reaction ratios were xPt = 0.91 and xCo = 0.59 for the microreactor and xPt = 0.93 and xCo = 0.40 for the batch reactor; the higher reaction ratio of Co ions for the microreactor demonstrates an enhanced reduction reaction in the microreactor, which also supports the proposed mechanism. It should be noted that the measured composition ratio of the alloy NP, Pt:Co = 0.66:0.34, reflects the reaction ratio, xPt:xCo = 0.91:0.59, rather than the ratio in the feed solutions, Pt:Co = 1:1. In this manner, the mixing process influences the microscopic structures of the resultant alloy NPs, and simply intensifying the mixing ability enables the synthesis of Pt-Co alloy NPs from an aqueous solution at room temperature.
Figure 10 displays EDX mapping images of resultant particles synthesized with different combinations of precursors. The use of K2PtCl4 produced Pt-Co alloy NPs for both CoCl2 and Co(NO3)2 (Figures 10A,B). Meanwhile, the use of H2PtCl6 resulted in a bimodal mixture of larger Pt-Co alloy NPs and smaller NPs with a major component of Pt (Figures 10C,D), even with the strong mixing of the microreactor. We confirmed that K2PtCl6 yielded a similar result to the case of H2PtCl6. These results demonstrate that the valence number of Pt ions plays a more crucial role in the synthesis of monodisperse Pt-Co alloy NPs than the type of Co precursors, and PtCl42– ions are more favorable than PtCl62– ions. Because PtCl62– ions are reduced to zero-valent Pt through two-step sequential reactions by way of PtCl42– ions with similar standard reduction potentials (PtCl62–/PtCl42–: +0.68 V vs standard hydrogen electrode (SHE) and PtCl42–/Pt: +0.76 V vs SHE (Jiang et al., 2015)), the reduction reaction of PtCl62– ions would be slowed and the reduction rate difference would arise between Co ions. Due to this rate difference, Pt ions that are later reduced could result in the formation of smaller Pt NPs. Therefore, a mismatch in the reduction rate of constituent metal ions results in a large variation in size and composition of NPs, and an appropriate choice of precursors with a similar reduction rate is another critical factor for the synthesis of Pt-Co alloy NPs.
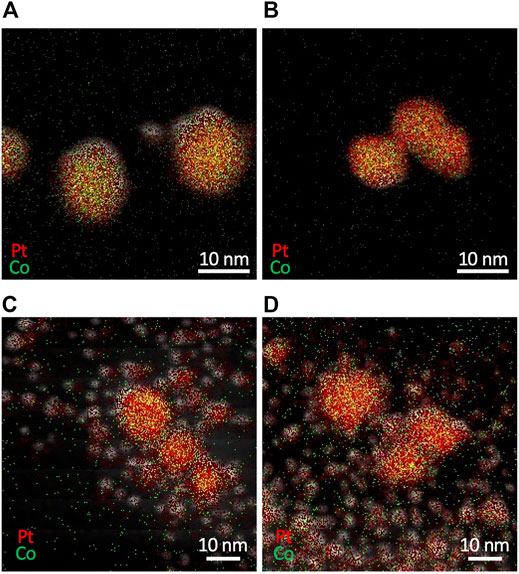
FIGURE 10. EDX mapping images of Pt and Co bimetallic particles synthesized from different combinations of precursors of (A) K2PtCl4 and CoCl2 (B) K2PtCl4 and Co(NO3)2 (C) H2PtCl6 and CoCl2, and (D) H2PtCl6 and Co(NO3)2.
The particle size can be increased by the increase in the temperature. As shown in Figure 11A, a synthesis at a higher temperature, in which the microreactor and vial were immersed in a water bath at a temperature of 95°C, produced alloy NPs with an average diameter of 17.9 ± 4.1 nm, which were larger than those synthesized at room temperature (Figure 8A). This would be because of a lower degree of supersaturation at a higher temperature, leading to the formation of a smaller number of larger nuclei. The composition ratio of alloy NPs changed under a different Pt:Co ratio in feed solutions. Figure 11B presents typical resultant particles synthesized with a concentration set of [K2PtCl4] = 0.15 mM and [CoCl2] = 0.05 mM. The Pt:Co atomic ratio in the resultant particles was measured to be 0.74:0.26 based on an EDX compositional analysis, which reflected the Pt:Co ratio of three in the feed solutions. Meanwhile, setting a higher Co ratio to Pt in feed solutions ([K2PtCl4] = 0.05 mM and [CoCl2] = 0.15 mM) resulted in the formation of larger Co and smaller Pt particles as shown in Figure 11C, although we observed some alloy NPs with a Pt:Co atomic ratio of 0.58:0.42 (Figure 11D), which would be because of a mismatch in the reduction rate caused by the concentration change in feed solutions. Although further optimization in the experimental condition will be needed, these results demonstrate a great potential of our technique to control both the particle size and composition ratio of alloy NPs.
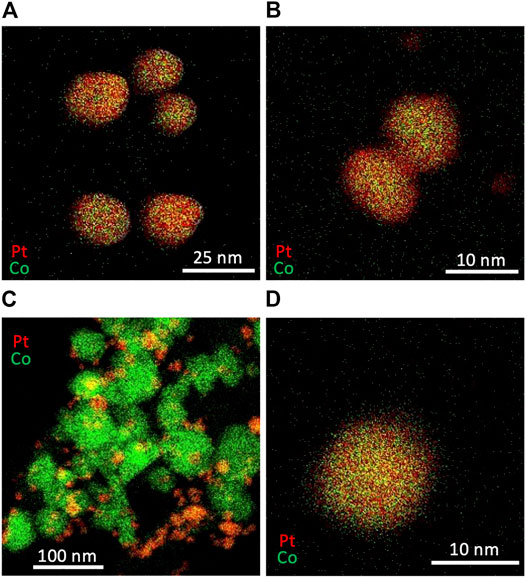
FIGURE 11. EDX mapping images of Pt and Co bimetallic particles synthesized with different concentrations and temperature (A) [K2PtCl4] = 0.10 mM [CoCl2] = 0.10 mM, 95°C (B) [K2PtCl4] = 0.15 mM [CoCl2] = 0.05 mM, 95°C, and (C) and (D) [K2PtCl4] = 0.05 mM [CoCl2] = 0.15 mM, 95°C.
Conclusion
We applied the microreactor to the liquid-phase synthesis of monometallic Ni and bimetallic Pt-Co alloy NPs. In the Ni NP synthesis, we constructed a sequential flow process by connecting two microreactors to manipulate the multiple steps involved in the reaction. This flow process successfully achieved the aqueous and room-temperature synthesis of monodisperse Ni NPs. Our investigation demonstrated that a residence time shorter than ca. 1 s between the microreactors was the key requirement to realize a sharp size distribution so that intermediate nickel–hydrazine complex species were trapped to react with NaOH molecules in the second microreactor, which was enabled by the excellent mixing ability of the microreactor. In the synthesis of bimetallic Pt-Co alloy NPs, we mixed a premixed solution of Pt and Co precursors with a reducing agent in the microreactor. Simultaneous reduction of both Pt and Co ions is crucial for obtaining uniform NPs in a solid solution state. The microreactor successfully produced Pt-Co alloy NPs because of the uniform concentration field in the reaction solution provided by the strong mixing of the microreactor, while a batch-type reaction resulted in a large variation in particle size and composition, possibly because of a local concentration distribution due to its weak mixing intensity. In this manner, “mixing” plays critical roles in NP synthesis, and the process intensification was successfully achieved by applying the microreactor to the NP synthesis to develop aqueous and room-temperature synthetic processes of Ni and Pt-Co alloy NPs. Here, we emphasize that the mixing intensity required for a successful synthesis is naturally determined by the rate of a target reaction; microreactors will not be necessary for slow reactions on the order of minutes or hours, but normal flow reactors will suffice. Therefore, a key to successful process intensification is to strategically select a reactor with a desired mixing intensity based on a deep understanding of the target reactions.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author Contributions
SW designed the research project of nanoparticle synthesis using the microreactor, acquired funding, supervised the project, and wrote the manuscript. KT and TW conducted experiments on the synthesis of Ni NPs and Pt-Co alloy NPs, respectively. MM gave much advice and many instructions as one of the supervisors and acquired funding.
Funding
This work was partly supported by JSPS KAKENHI (20H02504 and 23656490), Hosokawa Powder Technology Foundation, and The Information Center of Particle Technology, Japan.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bussamara, R., Eberhardt, D., Feil, A. F., Migowski, P., Wender, H., de Moraes, D. P., et al. (2013). Sputtering Deposition of Magnetic Ni Nanoparticles Directly onto an Enzyme Surface: A Novel Method to Obtain a Magnetic Biocatalyst. Chem. Commun. 49, 1273–1275. doi:10.1039/c2cc38737a
Chen, D.-H., and Hsieh, C.-H. (2002). Synthesis of Nickel Nanoparticles in Aqueous Cationic Surfactant Solutions. J. Mater. Chem. 12, 2412–2415. doi:10.1039/b200603k
Chen, D.-H., and Wu, S.-H. (2000). Synthesis of Nickel Nanoparticles in Water-In-Oil Microemulsions. Chem. Mater. 12, 1354–1360. doi:10.1021/cm991167y
Couto, G. G., Klein, J. J., Schreiner, W. H., Mosca, D. H., de Oliveira, A. J. A., and Zarbin, A. J. G. (2007). Nickel Nanoparticles Obtained by a Modified Polyol Process: Synthesis, Characterization, and Magnetic Properties. J. Colloid Interf. Sci. 311, 461–468. doi:10.1016/j.jcis.2007.03.045
Davey, W. P. (1925). Precision Measurements of the Lattice Constants of Twelve Common Metals. Phys. Rev. 25, 753–761. doi:10.1103/PhysRev.25.753
Domínguez-Crespo, M. A., Ramírez-Meneses, E., Montiel-Palma, V., Torres Huerta, A. M., and Dorantes Rosales, H. (2009). Synthesis and Electrochemical Characterization of Stabilized Nickel Nanoparticles. Int. J. Hydrogen Energ. 34, 1664–1676. doi:10.1016/j.ijhydene.2008.12.012
Eluri, R., and Paul, B. (2012). Synthesis of Nickel Nanoparticles by Hydrazine Reduction: Mechanistic Study and Continuous Flow Synthesis. J. Nanopart Res. 14, 800. doi:10.1007/s11051-012-0800-1
Ely, T. O., Amiens, C., Chaudret, B., Snoeck, E., Verelst, M., Respaud, M., et al. (1999). Synthesis of Nickel Nanoparticles. Influence of Aggregation Induced by Modification of Poly(Vinylpyrrolidone) Chain Length on Their Magnetic Properties. Chem. Mater. 11, 526–529. doi:10.1021/cm980675p
Fiévet, F., Ammar-Merah, S., Brayner, R., Chau, F., Giraud, M., Mammeri, F., et al. (2018). The Polyol Process: A Unique Method for Easy Access to Metal Nanoparticles with Tailored Sizes, Shapes and Compositions. Chem. Soc. Rev. 47, 5187–5233. doi:10.1039/c7cs00777a
Fujiwara, A., Watanabe, S., and Miyahara, M. T. (2021). Flow Microreactor Synthesis of Zeolitic Imidazolate Framework (ZIF)@ZIF Core-Shell Metal-Organic Framework Particles and Their Adsorption Properties. Langmuir 37, 3858–3867. doi:10.1021/acs.langmuir.0c03378
Gilbert, E. C., and Evans, W. H. (1951). Complex Formation between Nickel Ion and Hydrazine in Solution. J. Am. Chem. Soc. 73, 3516–3518. doi:10.1021/ja01151a518
Huang, L., Zheng, C. Y., Shen, B., and Mirkin, C. A. (2020). High‐Index‐Facet Metal‐Alloy Nanoparticles as Fuel Cell Electrocatalysts. Adv. Mater. 32, 2002849. doi:10.1002/adma.202002849
Jiang, B., Li, C., Imura, M., Tang, J., and Yamauchi, Y. (2015). Multimetallic Mesoporous Spheres through Surfactant-Directed Synthesis. Adv. Sci. 2, 1500112. doi:10.1002/advs.201500112
Jiang, Z., Xie, J., Jiang, D., Wei, X., and Chen, M. (2013). Modifiers-Assisted Formation of Nickel Nanoparticles and Their Catalytic Application to P-Nitrophenol Reduction. CrystEngComm 15, 560–569. doi:10.1039/c2ce26398j
Kobayashi, H., Kusada, K., and Kitagawa, H. (2015). Creation of Novel Solid-Solution Alloy Nanoparticles on the Basis of Density-Of-States Engineering by Interelement Fusion. Acc. Chem. Res. 48, 1551–1559. doi:10.1021/ar500413e
Li, Y. D., Li, L. Q., Liao, H. W., and Wang, H. R. (1999). Preparation of Pure Nickel, Cobalt, Nickel-Cobalt and Nickel-Copper Alloys by Hydrothermal Reduction. J. Mater. Chem. 9, 2675–2677. doi:10.1039/a904686k
Lin, L., Li, S., Hessel, V., Starostin, S. A., Lavrijsen, R., and Zhang, W. (2018). Synthesis of Ni Nanoparticles with Controllable Magnetic Properties by Atmospheric Pressure Microplasma Assisted Process. Aiche J. 64, 1540–1549. doi:10.1002/aic.16054
Maw, S. S., Watanabe, S., and Miyahara, M. T. (2019). Flow Synthesis of Silver Nanoshells Using a Microreactor. Chem. Eng. J. 374, 674–683. doi:10.1016/j.cej.2019.05.210
Nagasawa, H., Aoki, N., and Mae, K. (2005). Design of a New Micromixer for Instant Mixing Based on the Collision of Micro Segments. Chem. Eng. Technol. 28, 324–330. doi:10.1002/ceat.200407118
Owen, E. A., and Jones, D. M. (1954). Effect of Grain Size on the Crystal Structure of Cobalt. Proc. Phys. Soc. B 67, 456–466. doi:10.1088/0370-1301/67/6/302
Park, J. W., Chae, E. H., Kim, S. H., Lee, J. H., Kim, J. W., Yoon, S. M., et al. (2006). Preparation of Fine Ni Powders from Nickel Hydrazine Complex. Mater. Chem. Phys. 97, 371–378. doi:10.1016/j.matchemphys.2005.08.028
Shi, Y., Lyu, Z., Zhao, M., Chen, R., Nguyen, Q. N., and Xia, Y. (2021). Noble-metal Nanocrystals with Controlled Shapes for Catalytic and Electrocatalytic Applications. Chem. Rev. 121, 649–735. doi:10.1021/acs.chemrev.0c00454
Watanabe, S., Hiratsuka, T., Asahi, Y., Tanaka, A., Mae, K., and Miyahara, M. T. (2015). Flow Synthesis of Plasmonic Gold Nanoshells via a Microreactor. Part. Part. Syst. Charact. 32, 234–242. doi:10.1002/ppsc.201400126
Watanabe, S., Ohsaki, S., Fukuta, A., Hanafusa, T., Takada, K., Tanaka, H., et al. (2017). Characterization of Mixing Performance in a Microreactor and its Application to the Synthesis of Porous Coordination Polymer Particles. Adv. Powder Techn. 28, 3104–3110. doi:10.1016/j.apt.2017.09.005
Wu, S.-H., and Chen, D.-H. (2003). Synthesis and Characterization of Nickel Nanoparticles by Hydrazine Reduction in Ethylene Glycol. J. Colloid Interf. Sci. 259, 282–286. doi:10.1016/s0021-9797(02)00135-2
Wu, S.-H., and Chen, D.-H. (2004). Synthesis and Stabilization of Ni Nanoparticles in a Pure Aqueous CTAB Solution. Chem. Lett. 33, 406–407. doi:10.1246/cl.2004.406
Wu, X., Xing, W., Zhang, L., Zhuo, S., Zhou, J., Wang, G., et al. (2012). Nickel Nanoparticles Prepared by Hydrazine Hydrate Reduction and Their Application in Supercapacitor. Powder Techn. 224, 162–167. doi:10.1016/j.powtec.2012.02.048
Keywords: microreactor, nanoparticle, mixing, nucleation, residence time, nickel, platinum-cobalt alloy
Citation: Watanabe S, Koshiyama T, Watanabe T and Miyahara MT (2021) Room-Temperature Synthesis of Ni and Pt-Co Alloy Nanoparticles Using a Microreactor. Front. Chem. Eng. 3:780384. doi: 10.3389/fceng.2021.780384
Received: 21 September 2021; Accepted: 01 December 2021;
Published: 15 December 2021.
Edited by:
Jun Yue, University of Groningen, NetherlandsReviewed by:
Liangliang Lin, Jiangnan University, ChinaJean-Marc COMMENGE, Université de Lorraine, France
Copyright © 2021 Watanabe, Koshiyama, Watanabe and Miyahara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satoshi Watanabe, bmFiZUBjaGVtZS5reW90by11LmFjLmpw