 Laura Slappendel
Laura Slappendel Xiaodan Liu2†‡
Xiaodan Liu2†‡ Michael R. Macarthur
Michael R. Macarthur
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. Biol. , 10 September 2024
Sec. Molecular Sciences
Volume 3 - 2024 | https://doi.org/10.3389/fchbi.2024.1462351
This article is part of the Research Topic New Developments in Bioinorganic and Bioorganic Chemistry View all 5 articles
Maintaining cellular redox homeostasis is critical for cell viability and growth, with disruptions implicated in cellular responses to chemicals and drugs. This study investigates the interactions between acylfulvenes (AFs), a class of DNA alkylating drugs, and thioredoxin (Trx), a key redox regulating enzyme. AFs are semi-synthetic derivatives of the natural product illudin S. While their cytotoxic properties are widely attributed to DNA alkylation, they also react with cellular thiols, such as Trx, and the implications of these interactions remain poorly understood. Through biochemical assays with isolated E. Coli Trx, and cellular experiments in a human cell line (HeLa), we elucidate AFs’ impact on Trx activity and cellular levels. AFs, particularly hydroxymethylacylfulvene (HMAF), inhibited Trx activity by covalently modifying its active site cysteines. Drug exposure also altered cellular Trx levels and nuclear accumulation. In contrast, illudin S, which has a less selective toxicity profile for cancer cells, minimally inhibited isolated Trx. These data underscore Trx as a potential target contributing to the chemotherapeutic potential of AFs and provide insights into molecular interactions governing their impact on cancer cells.
Maintaining redox homeostasis is crucial for regulating cellular function and viability, and disruptions in the redox state of cells are characteristic of various pathologies (Trachootham et al., 2008). Cellular redox control is mediated by networks of enzymes and small molecules, like thioredoxin (Trx) and glutathione (GSH), such that cysteine (Cys) residues undergo reversible thiol/disulfide exchange reactions and act as switches to activate or induce conformational changes in proteins. Consequently, chemical reactivity of electrophilic drugs towards GSH and enzymes containing active site thiols can contribute to toxicity (Zanotto-Filho et al., 2016). DNA alkylating agents are examples of such electrophilic drugs, where their cytotoxicity is often attributed solely to DNA alkylation. However, their reactivity towards thiols could also contribute to their cytotoxicity by influencing cellular redox levels.
Acylfulvenes (AFs) are a class of semi-synthetic DNA alkylating drugs derived from illudin S, a metabolite found in the mushroom Omphalotus illudens. The narrow therapeutic window of illudin S resulted in the family of AFs with optimized characteristics (Kim et al., 2000). Hydroxymethylacylfulvene (HMAF) has a favorable selectivity profile in pre-clinical and tumor xenograft studies and advanced to clinical trials phase III. It was withdrawn however, due to lack of selectivity in cancer patients (Baekelandt, 2002). More recently, the activity of AFs is being re-evaluated by integrating genetic alterations in tumors to selectively target tumors with AF, for example, tumors with a deficiency in nucleotide excision repair (Jiang and Greenberg, 2021; Topka et al., 2021).
Following metabolic activation by reduction of the α,β unsaturated enone by alkenal/one reductase (PTGR1) (Yu et al., 2012; Neels et al., 2007), nucleophilic attack by the purine bases occurs on the cyclopropyl ring leading to aromatic ring formation by eliminating the tertiary alcohol (Figure 1). So, as a result, the overall toxicity of acylfulvenes (AFs) is believed to stem from their ability to alkylate DNA. However, due to the highly electrophilic nature of the reactive intermediate after activation/reduction, interactions with other cellular nucleophiles may influence AF activity and selectivity profiles (Le et al., 2019). Cancer cells often display mis-regulation of the Trx system which presents an attractive target for cancer therapy given the critical role of reactive oxygen species (ROS) in cell growth, proliferation and survival (Cai et al., 2012; Zhang et al., 2017; Ghareeb and Metanis, 2020). The possibility that acylfulvenes might also target Trx further enhances their potential as promising cancer therapeutics.
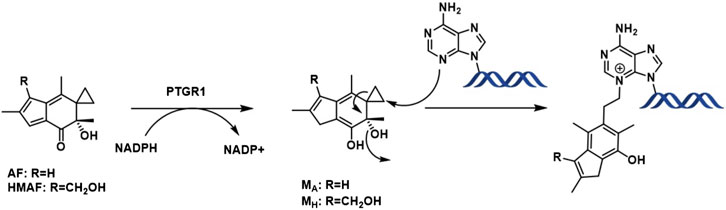
Figure 1. AF enzymatic activation and DNA alkylation.
Despite an evident role of DNA alkylation in the AF cytotoxic response (Otto et al., 2017; Pietsch et al., 2013), there is some evidence that thiol modification of certain selenoenzymes can also contribute to AF cytotoxicity (Liu et al., 2011a). Surprisingly, AF reacts faster with the redox regulating enzyme glutathione reductase (GR) than illudin S (McMorris, 1999a; McMorris et al., 1997a; Weichsel et al., 1996). Based on observations that the less cytotoxic AFs are less reactive towards small thiols (McMorris et al., 1996),and that cellular sensitivity to illudin S is modulated by cellular levels of glutathione (McMorris et al., 1990), it has been hypothesized that the differential cytotoxicity may also result from differences in the inhibitory potency of thiol-containing enzymes in cells (McMorris et al., 2000a; McMorris et al., 1997b). In previous studies, we showed that AF and HMAF covalently react with thioredoxin reductase (TrxR) active site Cys residues and inhibit TrxR activity, with a similar selectivity profile to GR (Liu et al., 2011a; Liu and Sturla, 2009a; Woynarowska and Woynarowski, 2002). For Trx, transient enzyme up-regulation and consequent depletion were observed in cancer cells but not normal cells following HMAF exposure. HMAF only formed covalent adducts that arise from reactions of thiols with the allylic alcohol position (Liu and Sturla, 2009a). Trx, the cellular substrate of TrxR, is reduced via disulfide/dithiol exchange, regulating redox-sensitive signaling proteins and transcription factors. These results confirm Trx modification by HMAF, yet it remains unknown whether illudin S interacts with Trx, how HMAF’s modification affects enzyme function, and how these properties relate to cellular enzyme actions.
In this study, we focus on the E. coli Trx, which shares a high degree of overall structure and active site similarity with human Trx (Weichsel et al., 1996; Lu and Holmgren, 2014). The dithiol/disulfide exchange activity of Trx regulates a broad spectrum of cellular processes including DNA synthesis, apoptosis, and antioxidative defense. Trx is a 12 kDa protein with a highly conserved active site (WCGPCK) among species from archaea to humans (Holmgren, 1995; Zhong et al., 2000). In mammalian cells, Trx is localized to the cytosol and Trx2 to the mitochondria. In different types of cancers, Trx levels vary and cancer cell lines with high Trx levels are highly sensitive to HMAF (Poindessous et al., 2003a). However, it is not known whether HMAF, nor AF or illudin S, inhibit Trx or how their reactivities compare (Poindessous et al., 2003a; Kelner et al., 1998).
In this study, we characterized the reactivity of illudin S, AF and HMAF toward Trx. We analyzed the inhibition susceptibility of Trx by AFs. Different inhibition potencies were observed for binding Trx by illudin S, AF and HMAF. Inhibition on isolated Trx was tested as well as nuclear localization of Trx in cells. This study evaluates Trx as a potential cellular target, especially for HMAF, and explores how controlling Trx system interactions impact chemotherapeutic outcomes as well as insight on the improved therapeutic indices of HMAF compared to illudin S and AF.
Illudin S was provided by MGI Pharma (Bloomington, MN). Acylfulvene and HMAF were synthesized according to a published procedure with illudin S as starting material (McMorris et al., 1996). Purified E.coli Trx1 was purchased from Promega Corporation (Madison, WI). Insulin from bovine pancreas, Tris base, and EDTA were obtained from Sigma Chemical (Milwaukee, WI). Dithiothreitol (DTT) was obtained from Fisher Scientific Inc. (Hanover Park, IL). Recombinant rPTGR1 was expressed and purified as published previously, and generously provided by Dr. Xiang Yu and Professor Thomas Kensler. N-biotinoyl-N-(iodoacetyl)ethylene diamine (BIAM) was purchased from Molecular Probes, Inc. (Eugene, OR). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Mediatech (Herndon, VA). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Lawrenceville, GA). Phosphate-buffered saline (PBS), 0.25% trypsin-EDTA, penicillin-streptomycin were obtained from Invitrogen (Carlsbad, CA). Tris-buffered saline was purchased from Biorad (Hercules, CA).
HPLC analysis was carried out on an Agilent 1100 series instrument with diode array detector and autosampler. Analytes were eluted with a solvent gradient involving water (A) and acetonitrile (B), at a flow rate of 1 mL/min: initial conditions, 10:90 B:A, were held for 5 min followed by linear increase to 50:50 B:A over a course of 20 min, and held 5 min. For AF analysis, analytes were eluted with a solvent gradient involving water (A) and acetonitrile (B), at a flow rate of 1 mL/min: initial conditions, 10:90 B:A, were held for 5 min followed by a linear increase to 50:50 B:A over a course of 20 min, and then further increase to 80:20 B:A in 20 min and held 5 min. A Luna 5 μm C18 (2) 100 Å 250 mm × 4.60 mm column was used (Phenomenex, Torrance, CA). Trx and DTT stock solutions were prepared in Tris-HCl buffer (50 mM, pH 7.2) containing EDTA (1 mM), i.e., TE buffer. All assays were carried out in TE buffer. Stock solutions of test compounds were prepared in DMSO, and aliquots of these solutions were added to the reaction mixture to yield a final DMSO concentration of 2% (v/v). Trx activity was determined by measuring turbidity formed from reduction of insulin by monitoring UV absorbance at 650 nm with a 96-well plate reader (Biotek, Winooski, VT) (Holmgren, 1979).
Fluorescence measurements were carried out in a 4 × 10 mm rectangular cuvette and on a spectrofluorometer (Varian Cary) at 90° in relation to the excitation source. Fluorescence was measured with excitation at 295 nm (bandpasses of 5 and 10 nm for excitation and emission respectively) and emission from 310 to 450 nm. HPLC analysis was carried out with an HPLC equipped with diode array detector and autosampler, and analytes were eluted on a 250 mm × 4.60 mm C18 column with a water-acetonitrile solvent gradient (Complete details can be found in the SI). Liquid chromatography-mass spectrometry (LC/MS) analyses were carried out with a capillary HPLC interfaced with an ion trap mass spectrometer operated in positive ion mode and equipped with an autosampler. Analytes were eluted on a Zorbax 300 SB-C3 5 μm 150 mm × 0.5 mm column with a solvent gradient involving 0.05% TFA in water and acetonitrile.
To determine whether test compounds were Trx substrates, each compound (AF and HMAF, 400 μM; Illudin S, 1 mM; reference blank, 2% DMSO) was combined individually with non-reduced Trx (4 μM) in the presence of DTT (2 mM), or pre-reduced Trx (4 μM) in TE buffer with a final volume of 200 μL and allowed to react at 25°C for 2 h. The resulting solution was extracted with ethyl acetate (EtOAc, 200 μL) and centrifuged for 5 min (6,000 g). The supernatant was collected and EtOAc was evaporated on a centrifugal vacuum concentrator (Savant, Waltham, MA). The dried material was reconstituted in 100 μL DMSO, and 50 μL was injected and analyzed by HPLC. As a negative control, the same procedure was carried out with compound only, or compound with DTT.
Reduced Trx was prepared from oxidized Trx by incubating 60 μL Trx (450 μM) with 20 μL DTT (100 mM) at 25°C for 20 min. DTT was subsequently removed with a micro bio-spin P6 column (NMWL 6000, Bio-rad, Hercules, CA) pre-equilibrated with N2-purged TE buffer according to the manufacturer’s protocols. The absence of DTT in the resulting protein solution was verified by testing with Ellman’s reagent (Ellman, 1959). The resulting reduced Trx was diluted to a final concentration of 4 μM with N2-purged TE buffer. Test samples (5 μL) of varying concentrations were added to 195 μL of pre-reduced Trx solution and allowed to react in a 96-well plate at 25°C for 2 h. Trx activities were measured by ability to reduce insulin, with DTT as the electron donor, by a published method (Holmgren, 1979). Briefly, after incubation, 50 μL DTT (10 mM) and 60 μL insulin (10 mg/mL) were added; enzyme activity was measured at 25°C by monitoring the increase of A650 due to the precipitation of reduced insulin. Samples designated as blank contained all reagents except Trx.
The degree of modification of Trx by AFs at Cys was determined with the use of the thiol-reactive biotinylation reagent N-(biotinoyl)-N`-(iodoacetyl)ethylenediamine (BIAM) (Kim et al., 2000). DMSO or varying concentrations of acyfulvene were added to prereduced Trx (0.9 μM), which was then allowed to react at 25°C for 2 h. After reaction, 1 μL of the resulting solution was removed and added to a new 1.5 mL microcentrifuge tube containing 19 μL of 100 μM BIAM in TE buffer. The resulting mixture was allowed to react at 25°C for another 30 min to alkylate the remaining free enzyme thiol groups. A sample containing 10 μL of BIAM-modified enzyme was analyzed on a 7.5% SDS-PAGE gel, and the separated proteins were transferred to a PVDF membrane. Proteins labeled with BIAM were detected by horseradish peroxidase (HRP)-conjugated streptavidin (Invitrogen, Carlsbad, CA) and enhanced chemiluminescence (ECL) detection (Pierce, Rockford, IL).
A total of 104 Hela cells were plated out on six-well tissue culture plates. Cells were allowed to grow for 3 days in DMEM with 10% fetal bovine serum, and then exposed to drugs or vehicle control (0.1% DMSO) for 12 h. After treatment, cells were washed twice with PBS. Cells were harvested and kept frozen at −80°C until use. Cell pellets were re-suspended in 0.2 mL of lysis buffer (50 mM Tris-HCl, pH 7.5; 1 mM EDTA; 0.1% Triton X-100; 1 mM phenylmethanesulfonyl fluoride; 1 mM benzamidine; 1.4 μM pepstatin A; and 2.0 μM leupeptin) and sonicated at 4°C (5 s bursts). The resulting cell lysate was centrifuged for 10 min (8,000 g, 4°C), and the supernatant was withdrawn for analysis. Supernatants were assayed for protein content with a bicinchoninic acid (BCA) kit (Pierce, Rockford, IL), gel electrophoresed, and transferred onto a PVDF membrane. The membrane was cut into two pieces according to the molecular weight marker. Blots were probed with rabbit anti-human Trx1 antibody (Proteintech 14999-1-AP) or rabbit anti-human vinculin (CST E1E9V) diluted 1:1000 followed by horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody diluted 1:10000. Bands were analyzed with an enhanced chemiluminescence protocol (ECL reagents, Pierce, Rockford, IL), and visualized on a Licor Odyssey XF. Western blots were performed in duplicates.
Hela cells were seeded at 105 cells per well in 6-well plates that contained 22 cm glass coverslips coated with collagen (10 μg/mL) and incubated for 48 h at 37°C. Cells were incubated either with test compounds, cisplatin, or DMSO (0.2%) for 8 h. Afterward, the cells were washed twice with phosphate buffer saline (PBS, pH 7.4), and then fixed with 3.7% formaldehyde in PBS for 20 min. After two washes with PBS, the cells were permeabilized with 0.1% saponin in PBS and blocked with 3% (w/v) BSA in PBS for 1 h at 25°C. The cells were then incubated for 2 h at 25°C with the anti-Trx1 monoclonal antibody (BD Transduction Laboratories) in a solution containing 0.1% saponin and 3% BSA in PBS (1:500). The cells were washed three times with PBS and incubated for 1 h at 25°C in blocking buffer containing anti-mouse IgG fluorescein isothiocyanate (FITC)-conjugate (1:1000), and 4,6-diamidino-2-phenylindole (2.5 ng/mL in PBS) to stain the cellular nuclei (1:2000). The signals were observed under a fluorescence microscope (Zeiss Axioscop 2) equipped with a digital camera (Zeiss Axiocam R2), and × 100 objective. Images were captured using Zeiss Axiovision software release 3.1 (Carl Zeiss, Inc., Thornwood, NY). Images from three independent experiments were obtained and fluorescence intensities of each image were analyzed with NIH software ImageJ (version 1.44p). Fluorescence intensities of the nuclei and entire cells in each image were measured and the percentage of nuclear fluorescence intensity relative to overall cell fluorescence intensity was calculated. The data were expressed as means ± SD, and significant differences were evaluated with Student’s t-test (GraphPad software).
Intrinsic tryptophan fluorescence is an optical indicator of the redox status of the Trx active site (Holmgren, 1972). Active site disulfide reduction results in a large increase in fluorescence, indicating a conformational change in the environment of both Trx tryptophan residues, Trp 28 and Trp 31, which are close to the active site disulfide (Cys 32 and Cys 35) (Holmgren, 1972). Thus, to evaluate drug binding to the Trx active site, we acquired fluorescence emission spectra of Trx (in the presence of DTT to ensure Trx reduction) at an excitation wavelength of 295 nm with addition of increasing concentrations of drugs. Illudin S and HMAF have absorption peaks at 280 and 330 nm (Supplementary Figure S1), respectively, which partially overlap with Trp absorption peaks. To account for this, the highest concentration of illudin S or HMAF tested was 400 μM and 200 μM, respectively, to keep the absorbance at the emission and excitation wavelengths below 0.8 AU. The final emission spectra were corrected to eliminate inner filter effects.
The cellular reductase PTGR1, together with cofactor NADPH, are known to reduce AFs to their reactive intermediate, e.g., MA and MH, which alkylates DNA (Figure 1) 8 15 36. To verify that Trx, due to its broad substrate range and versatile functions, does not reduce AFs, we combined illudin S or AFs with Trx either in the presence or absence of DTT, as well as with pre-reduced Trx (Figure 2). The resulting mixtures of three individual experiments were analyzed by HPLC. For AF, only a single peak corresponding to AF was observed to elute (26 min). There was no evidence of metabolites (such as MA Figure 1, MA) (Figure 2), but we observed the formation of a single significant product (Figure 2, 22 min) in reactions of HMAF with dithiothreitol (DTT) with or without Trx (Figure 2, HMAF, B and C). Illudin S also reacts with DTT, but to a lesser extent. This HMAF-DTT adduct was isolated by HPLC purification. Mass spectrometric analysis indicated single alkylation of HMAF by DTT (m/z 383.2, Figure 3). The mass fragmentation suggests that the product is a mixture of isomers resulting from displacement of the primary hydroxyl group of HMAF by the hydroxyl (fragment m/z 244.9) or thiol of DTT (fragment m/z 261.0).
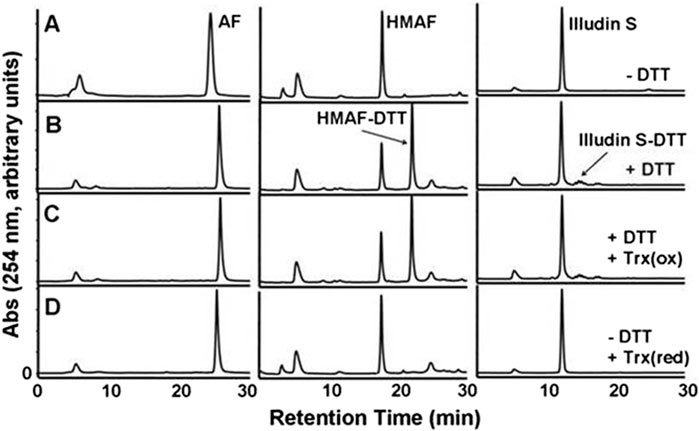
Figure 2. HPLC-UV analysis of the product of the reaction of Trx with illudin S or AFs. Test compounds (AF, 400 μM; HMAF, 400 μM; illudin S, 1 mM) were allowed to react with Trx (2 μM) in the presence of DTT 2 mM, row (C) or in the absence of DTT, but with pre-reduced Trx 2 μM, row (D) in TE buffer (500 μL) for 2 h at 25°C, followed by EtOAC extraction and HPLC analysis. Control samples were incubated in buffer in the absence row (A) or presence of DTT 20 mM, row (B) and extracted in the same manner. The peak at 5 min observed in each sample is DMSO. The retention times of AF, HMAF and illudin S are 26 min, 17 min, and 12 min respectively.
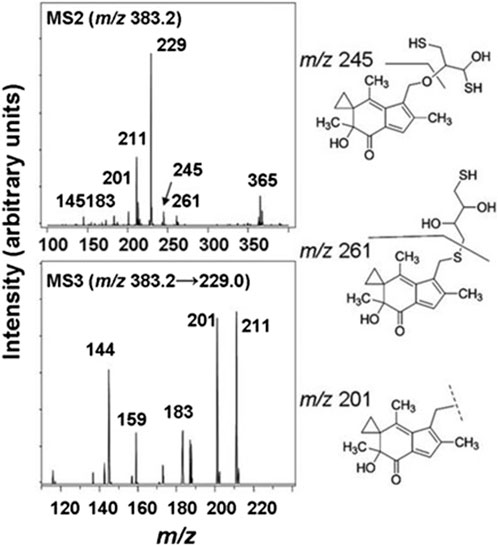
Figure 3. MS/MS spectra of the HMAF-DTT adduct, MS2 spectra of HMAF-DTT (top) and MS3 spectra of fragment 229 from MS2 (bottom).
While we showed that acylfulvenes can influence the fluorescence quenching of Trx, direct evidence of inhibiting Trx cellular function, namely, oxidizing TrxR and maintaining redox homeostasis, can be obtained from the insulin reducing activity assay. The TrxR-coupled insulin reduction assay for Trx activity has been reported previously (Holmgren, 1984). In the assay, Trx is reduced by TrxR with NADPH as the electron donor; herein solution turbidity, associated with insulin reduction, is monitored. While preliminary studies indicated that illudin S and AFs demonstrated inhibitory activity in this assay, results were confounded by interactions with TrxR (Liu et al., 2011b; Herzig et al., 1999), therefore we used an assay in which Trx catalyzes the reduction of insulin disulfides with DTT (Holmgren, 1979). The presence of DTT protects Trx from being inhibited by illudin S or AFs, due to faster reaction with DTT. Thus, no Trx inhibition was observed when test compounds were added to the reaction mixture without removing DTT (data not shown). Therefore, the experimental approach was modified such that excess DTT was removed from the reduced Trx prior to reactions with drugs. In this way, AFs blocked Trx-mediated reduction of insulin in a dose-dependent manner (Supplementary Figure S2). Trx insulin-reducing activity was inhibited by AF or HMAF with IC50 137 μM and 32.5 μM, respectively (Table 1). In contrast, an IC50 value for illudin S was not reached at 2,500 μM (Table 1). The assay was performed in triplicate and again, as negative controls, metabolites were examined to confirm a lack of significant inhibitory activity: 1000 μM MA had no influence (data not shown), 2,500 μM MH inhibited Trx activity by about 50% (Supplementary Figure S3). To also account for the influence of PTGR1 in the cellular environment, we performed an experiment where illudin S, AF and HMAF were pre-incubated with PTGR1 and NADPH before adding pre-reduced Trx to visualize inhibition. No significant changes were observed for this reaction compared to directly adding the activated metabolite (Supplementary Figure S4).
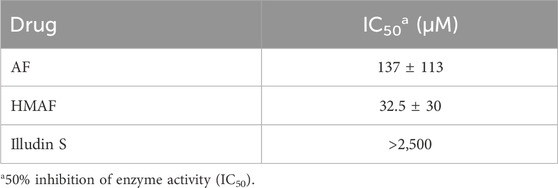
Table 1. Drug concentrations for Trx fluorescence quenching and inhibition of enzyme activity.
The inhibition of Trx-reducing activity together with the known reactivity of AFs towards thiols suggested that the Trx active site, containing only the 2 E coli Trx Cys residues (Cys (Holmgren, 1979) and Cys (Holmgren, 1972)), are modified by the drugs. N-biotinoyl-N-(iodoacetyl)ethylene diamine (BIAM), an alkylating agent used as an indicator of free thiols (Kim et al., 2000), was allowed to react with Trx that was first combined with varying concentrations of AFs. The resulting levels of protein modification were analyzed by blotting; the biotin tag was recognized by streptavidin-horseradish peroxidase and detected with enhanced chemiluminescence. With increasing drug concentration, the intensity of the BIAM-labeled protein band diminished (Figure 4). For high AF concentrations (250 and 1250 μM), the band intensity was reduced to half that of control, suggesting mono-alkylation of the protein by AF. The band almost disappeared completely at high HMAF concentration (250 μM), suggesting drug alkylation on both active site Cys residues. Furthermore, we tested whether the AF bioactivating enzyme NADPH-dependent PTGR1 could potentiate the inhibition of Trx by AFs or change the pattern of adducts formed between AFs and Trx, but it had no influence (Supplementary Figure S5).
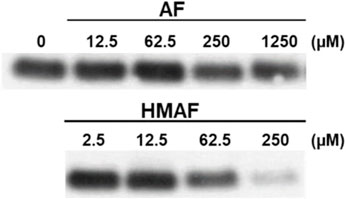
Figure 4. Modification of Cys residues in Trx by AF or HMAF. Increasing concentrations of AFs were added to Trx (0.9 μM, pre-reduced with DTT) and incubated at 25°C for 2 h. Image is representative of two independent experiments.
To evaluate whether AFs influence cellular Trx, protein levels in cells were measured after drug exposure. HeLa cells were exposed to illudin S, AF, HMAF or vehicle control (0.1% DMSO) for 12 h, cell lysates were prepared, and Trx levels were analyzed by Western blotting. The resulting data (Figure 5) suggests a dose dependent reduction in Trx following treatment with up to 100 µM of either AF or HMAF, but not illudin S.
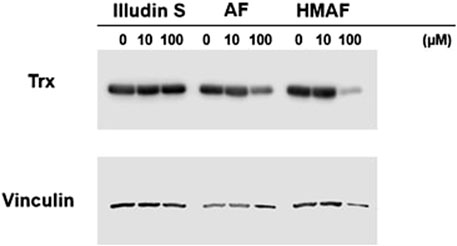
Figure 5. Inhibition of Cellular Trx by AFs. HeLa cells exposed to various concentrations of drugs for 12 h were lysed and proteins (20 μg) were separated on SDS-PAGE under reducing conditions. Vinculin was visualized as control.
Since cellular Trx levels decreased upon treatment with AFs, and isolated Trx was covalently modified by AFs, we tested whether the interaction between Trx and AFs alters the cellular localization of Trx. As a positive control, HeLa cells were treated with cisplatin, and nuclear accumulation (63%) was consistent with previous reported observations (Figure 6). Cells were treated with illudin S, AF, or HMAF, as well as DMSO, or the AF metabolites MH and MA. Trx intracellular localization was visualized by indirect immunofluorescence staining with an anti-Trx monoclonal antibody. In the absence of the drug, 44% of Trx was localized in the nucleus. In cells exposed to illudin S (0.5 μM), AF (2 μM), or HMAF (2 μM) for 10 h, the percentages of Trx signal localized to the nucleus were 57%, 58%, and 73%, respectively. This capacity to promote cellular localization was not observed for AF metabolites (10 μM, MA, 51% and MH, 37%), which were considered negative controls that share some chemical properties of metabolites AFs, but do not appear to bind to Trx or exhibit toxicity.
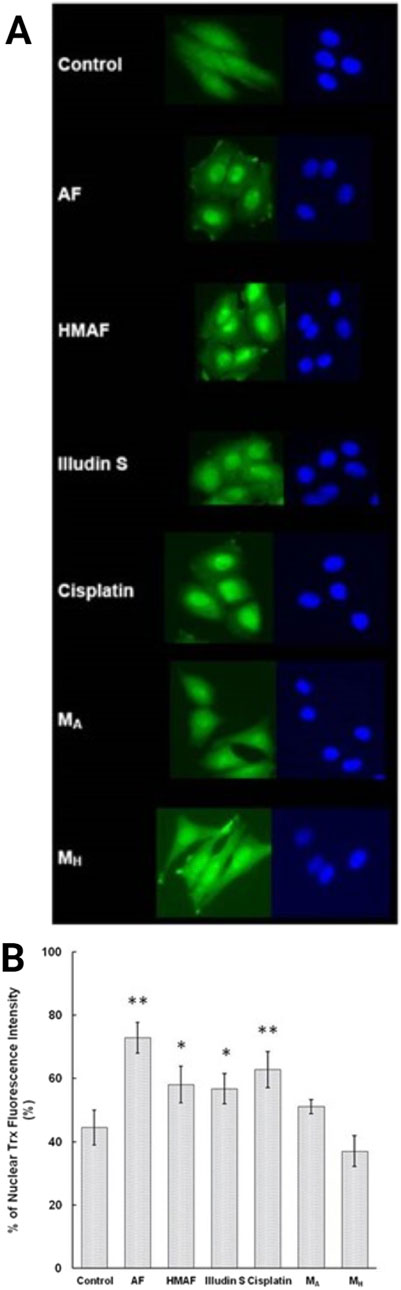
Figure 6. Indirect immunofluorescence analysis to determine Trx sub-cellular location in HeLa cells. Cells were treated with different compounds (AF, 2 μM, HMAF, 2 μM, illudin S 0.2 μM, cisplatin, 10 μM, MA, 10 μM, and MH, 10 μM) for 10 h. (A). Cellular Trx was visualized as described in the methods section. Images shown represent data from three independent experiments. (B). Quantification of the fluorescent intensity localized in nucleus versus the total cell expressed as mean ± SD. Significant differences were evaluated with Student’s t-test (GraphPad software), * p < 0.05, ** p< 0.01.
Conventional chemotherapeutics often exhibit multiple off-target effects. AFs are DNA alkylating agents, but their enone and cyclopropyl groups make them susceptible to interaction with multiple cellular targets. In the current study we aimed to compare the reactivity of illudin S and AFs towards Trx, a critical thiol-based antioxidant enzyme. We found AFs inhibit Trx to a greater extent than illudin S, with HMAF exhibiting the lowest IC50 at 32.5 ± 30 µM. The inhibition of Trx arises from monoalkylation and bis-alkylation to the active site cysteine residues by AF and HMAF, respectively. In a cellular context, levels of Trx were reduced with increasing concentration of HMAF and AF and caused nuclear localization of Trx, while illudin S did not produce these effects.
The capacity of HMAF to covalently bind to free thiols of Trx and the potential for this interaction to inhibit cellular Trx activity may be explained by the susceptibility of the allylic alcohol to nucleophilic addition. This is evident from the wide variety of thiol derivatives synthesized via direct nucleophilic substitution on the allylic alcohol 39 18 40. Illudin S lacks this allylic alcohol and it does not appear to inhibit Trx. Consistently we found no evidence by mass spectrometry analysis for covalent modification by illudin S, whereas HMAF and AF formed bis-and mono-adducts, respectively (data not shown). Furthermore, while the isolated enzyme experiments performed herein were with E. Coli Trx as a model, the human variant of the enzyme has three additional cysteine residues (positions 62, 69, and 72). While the active site is comprised of Cys 32 and Cys 35, there is evidence for regulation of Trx function after post-translational modification at the allosteric sites. (Haendeler, 2006). In further biochemical studies, it would be important to evaluate the particular sites of Trx modification for the isolated human enzyme. Finally, regarding the chemical basis of the potential interactionfor both HMAF and AF, the enone functionality is likely to be more reactive towards nucleophilic addition by thiols than illudin S. In this study, HMAF (250 uM) almost completely prevented BIAM alkylation of the E. Coli Trx active site Cys residues. Meanwhile, AF resulted in an approximately 50% reduction at 1250 μM, consistent with mono-adduct formation and preliminary MS analysis (data not shown).
Cellular levels of Trx were reduced when cells were exposed to AF and HMAF (100 µM), but not illudin S. The exposures likely reduced cell viability, as reflected by slightly lower intensity in the reference vinculin band, which may be due to an indirect decrease in Trx levels rather than direct Trx reduction, consistent with the relatively high drug concentrations relative to their IC50 values (Poindessous et al., 2003b). Nonetheless, at levels below the IC50 values, both AF and HMAF drove nuclear localization of Trx as observed by nuclear translocation assay. Consistently, the effects of illudin S on Trx nuclear localization were weaker.
As a multifunctional protein, Trx exists in the extracellular milieu, cytoplasm, and nucleus, and has a different role in each location. In an ovarian cancer study, high nuclear Trx and low cytoplasmic Trx was found to be significantly positively correlated with progression-free survival (Woolston et al., 2010). Like cisplatin, AFs and illudin S stimulated nuclear accumulation of Trx and thereby suggest induction of a general stress response (Chen et al., 2007). Surprisingly, the associated metabolites MH and MA did not cause significant nuclear translocation, despite being the active compounds alkylating DNA. These observations suggest that the reactivity of the activated metabolite, MA, substantially favors alkylating DNA rather than reacting with cellular thiols after reduction by PTGR1.
To conclude, we have characterized modes of Trx interaction and, in some cases, inactivation by the natural product illudin S and its semi-synthetic analogues AF and HMAF. The data suggests that enzyme conformational changes and inhibition are induced by HMAF and AFs, but not by illudin S. As summarized in Figure 7, in the complex environment of the cell, AFs covalently react with Trx. We also found that Trx accumulates in the nucleusafter AF exposure and leads to a decrease in Trx protein levels in a dose-dependent manner. Taken together, these observations suggest that the covalent modification of the enzyme by the drug may lead to the protein degradation and/or translocation. The chemical reactivities of illudin S and AF analogues toward Trx are consistent with reactivity trends for the enzyme GR, but inconsistent with previously reported chemical reactivity toward certain thiols45 19 46. Considering the high reactivity of HMAF towards cellular thiols, it would be interesting to investigate additional protein interactions between AFs and Cys-containing proteins using global unbiased methods.
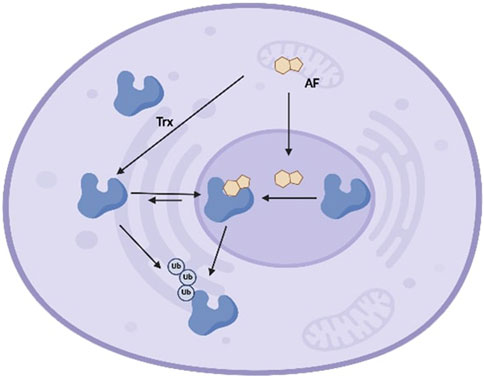
Figure 7. Schematic model of interaction between Trx and AFs in cells. Trx exists in the extracellular milieu, cytoplasm, and nucleus, and shuttles between the cytoplasm and nucleus at equilibrium. AFs enter cells and localize in either cytoplasm or nucleus, where AFs covalently react with Trx at active site cysteines. Upon alkylation, the cytoplasm-nucleus equilibrium shifts and Trx accumulates in nucleus. Finally, AFs-mediated Trx alkylation triggers enzyme degradation, leading to reduced cellular Trx.
The results of this study are a chemical basis for Trx alkylation by illudin S and its AF derivatives as a potential basis driving their relative cytotoxic profiles. We characterized a consistent association between structure-Trx Cys reactivity relationships of illudin S and its AF derivatives with cellular responses including protein degradation and nuclear translocation, as well as known cytotoxicity profiles of the drugs. These associations suggest that an additional contributing process to the mechanism of action for Afs, but not for illudin S, may involve Trx inactivation. Finally, this work provides new probes and experimental strategies for addressing the role of Trx in different cellular compartments and suggests cytosolic depletion of this enzyme as a potential strategy for overcoming drug resistance to alkylating agents.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
LS: Data curation, Formal Analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing–original draft, Writing–review and editing. XL: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing–original draft, Writing–review and editing. Michael Robert MM: Supervision, Writing–original draft, Writing–review and editing, Data curation. CS: Data curation, Investigation, Methodology, Writing–original draft, Writing–review and editing. SS: Conceptualization, Funding acquisition, Supervision, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. We acknowledge the National Cancer Institute (CA123007) and the Swiss National Science Foundation (186332) for funding, of separate research projects. Open access funding by ETH Zurich.
We thank Dr. Jiachang Gong for synthesizing AF and HMAF.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchbi.2024.1462351/full#supplementary-material
AFs, acylfulvenes; AF, acylfulvene; HMAF, hydroxymethylacylfulvene; Trx, thioredoxin; GSH, glutathione; PTGR1, alkenal/one reductase; ROS, reactive oxygen species; GR, glutathione reductase; DTT, dithiothreitol; Cys, Cysteine.
Cai, W., Zhang, L., Song, Y., Wang, B., Zhang, B., Cui, X., et al. (2012). Small molecule inhibitors of mammalian thioredoxin reductase. Free Radic. Biol. Med. 52 (2), 257–265. doi:10.1016/j.freeradbiomed.2011.10.447
Chen, X.-P., Liu, S., Tang, W.-X., and Chen, Z.-W. (2007). Nuclear thioredoxin-1 is required to suppress cisplatin-mediated apoptosis of MCF-7 cells. Biochem. Biophysical Res. Commun. 361 (2), 362–366. doi:10.1016/j.bbrc.2007.07.033
Ellman, G. L. (1959). Tissue sulfhydryl groups. Archives Biochem. Biophysics 82 (1), 70–77. doi:10.1016/0003-9861(59)90090-6
Ghareeb, H., and Metanis, N. (2020). The thioredoxin system: a promising target for cancer drug development. Chem. – A Eur. J. 26 (45), 10175–10184. doi:10.1002/chem.201905792
Gong, J., Vaidyanathan, V. G., Yu, X., Kensler, T. W., Peterson, L. A., and Sturla, S. J. (2007). Depurinating Acylfulvene−DNA adducts: characterizing cellular chemical reactions of a selective antitumor agent. J. Am. Chem. Soc. 129 (7), 2101–2111. doi:10.1021/ja0665951
Haendeler, J. (2006). Thioredoxin-1 and posttranslational modifications. Antioxidants & Redox Signal. 8 (9-10), 1723–1728. doi:10.1089/ars.2006.8.1723
Herzig, M. C., Arnett, B., MacDonald, J. R., and Woynarowski, J. M. (1999). Drug uptake and cellular targets of hydroxymethylacylfulvene (HMAF). Biochem. Pharmacol. 58 (2), 217–225. doi:10.1016/s0006-2952(99)00085-4
Holmgren, A. (1972). Tryptophan fluorescence study of conformational transitions of the oxidized and reduced form of thioredoxin. J. Biol. Chem. 247 (7), 1992–1998. doi:10.1016/S0021-9258(19)45481-1
Holmgren, A. (1979). Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J. Biol. Chem. 254 (19), 9627–9632. From NLM. doi:10.1016/s0021-9258(19)83562-7
Holmgren, A. (1984). Enzymatic reduction-oxidation of protein disulfides by thioredoxin. Methods Enzym. 107, 295–300. doi:10.1016/0076-6879(84)07019-1
Holmgren, A. (1995). Thioredoxin structure and mechanism: conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure 3 (3), 239–243. doi:10.1016/s0969-2126(01)00153-8
Jiang, H., and Greenberg, R. A. (2021). Morning for irofulven, what could be fiNER? Clin. Cancer Res. 27 (7), 1833–1835. doi:10.1158/1078-0432.Ccr-20-4708
Kelner, M. J., McMorris, T. C., Beck, W. T., Zamora, J. M., and Taetle, R. (1987). Preclinical evaluation of illudins as anticancer agents. Cancer Res. 47 (12), 3186–3189.
Kelner, M. J., McMorris, T. C., Estes, L., Samson, K. M., Bagnell, R. D., and Taetle, R. (1998). Efficacy of MGI 114 (6-hydroxymethylacylfulvene, HMAF) against the mdr1/gp170 metastatic MV522 lung carcinoma xenograft. Eur. J. Cancer 34 (6), 908–913. doi:10.1016/S0959-8049(98)00033-1
Kim, J.-R., Yoon, H. W., Kwon, K.-S., Lee, S.-R., and Rhee, S. G. (2000). Identification of proteins containing cysteine residues that are sensitive to oxidation by hydrogen peroxide at neutral pH. Anal. Biochem. 283 (2), 214–221. doi:10.1006/abio.2000.4623
Le, P., Nodwell, M. B., Eirich, J., and Sieber, S. A. (2019). A chemical proteomic analysis of illudin-interacting proteins. Chem. – A Eur. J. 25 (54), 12644–12651. doi:10.1002/chem.201902919
Liu, X., Pietsch, K. E., and Sturla, S. J. (2011a). Susceptibility of the antioxidant selenoenyzmes thioredoxin reductase and glutathione peroxidase to alkylation-mediated inhibition by anticancer acylfulvenes. Chem. Res. Toxicol. 24 (5), 726–736. doi:10.1021/tx2000152
Liu, X., Pietsch, K. E., and Sturla, S. J. (2011b). Susceptibility of the antioxidant selenoenyzmes thioredoxin reductase and glutathione peroxidase to alkylation-mediated inhibition by anticancer acylfulvenes. Chem. Res. Toxicol. 24 (5), 726–736. doi:10.1021/tx2000152
Liu, X., and Sturla, S. J. (2009a). Profiling patterns of glutathione reductase inhibition by the natural product illudin S and its acylfulvene analogues. Mol. Biosyst. 5 (9), 1013–1024. doi:10.1039/b904720d
Liu, X., and Sturla, S. J. Profiling patterns of glutathione reductase inhibition by the natural productilludin S and its acylfulvene analogues. Mol. Biosyst. 2009b, 5 (9), 1013–1024. doi:10.1039/B904720D
Lu, J., and Holmgren, A. (2014). The thioredoxin antioxidant system. Free Radic. Biol. Med. 66, 75–87. doi:10.1016/j.freeradbiomed.2013.07.036
McMorris, T. C. (1999a). Discovery and development of sesquiterpenoid derived hydroxymethylacylfulvene: a new anticancer drug. Bioorg. & Med. Chem. 7 (5), 881–886. doi:10.1016/s0968-0896(99)00016-4
McMorris, T. C. (1999b). Discovery and development of sesquiterpenoid derived hydroxymethylacylfulvene: a new anticancer drug. Bioorg. & Med. Chem. 7 (5), 881–886. doi:10.1016/S0968-0896(99)00016-4
McMorris, T., C., Hu, Y., Yu, J., Kelner, J., and Total, M. Synthesis of hydroxymethylacylfulvene, an antitumour derivative of illudin S. Chem. Commun. 1997b, (3), 315–316. doi:10.1039/A607077I
McMorris, T. C., Kelner, M. J., Wang, W., Diaz, M. A., Estes, L. A., and Taetle, R. (1996). Acylfulvenes, a new class of potent antitumor agents. Experientia 52 (1), 75–80. doi:10.1007/bf01922420
McMorris, T. C., Kelner, M. J., Wang, W., Moon, S., and Taetle, R. (1990). On the mechanism of toxicity of illudins: the role of glutathione. Chem. Res. Toxicol. 3 (6), 574–579. doi:10.1021/tx00018a013
McMorris, T. C., Lira, R., Gantzel, P. K., Kelner, M. J., and Dawe, R. (2000a). Sesquiterpenes from the basidiomycete Omphalotus illudens. J. Nat. Prod. 63 (11), 1557–1559. doi:10.1021/np9904760
McMorris, T. C., Yu, J., Estes, L. A., and Kelner, M. J. (1997a). Reaction of antitumor hydroxymethylacylfulvene (HMAF) with thiols. Tetrahedron 53 (43), 14579–14590. doi:10.1016/S0040-4020(97)01064-8
McMorris, T. C., Yu, J., Lira, R., Dawe, R., MacDonald, J. R., Waters, S. J., et al. (2001). Structure−Activity studies of antitumor agent irofulven (hydroxymethylacylfulvene) and analogues. J. Org. Chem. 66 (18), 6158–6163. doi:10.1021/jo010458z
McMorris, T. C., Yu, J., Ngo, H. T., Wang, H., and Kelner, M. J. (2000b). Preparation and biological activity of amino acid and peptide conjugates of antitumor hydroxymethylacylfulvene. J. Med. Chem. 43 (19), 3577–3580. doi:10.1021/jm0000315
Neels, J. F., Gong, J., Yu, X., and Sturla, S. J. (2007). Quantitative correlation of drug bioactivation and deoxyadenosine alkylation by acylfulvene. Chem. Res. Toxicol. 20 (10), 1513–1519. doi:10.1021/tx7001756
Otto, C., Spivak, G., Aloisi, C. M. N., Menigatti, M., Naegeli, H., Hanawalt, P. C., et al. (2017). Modulation of cytotoxicity by transcription-coupled nucleotide excision repair is independent of the requirement for bioactivation of acylfulvene. Chem. Res. Toxicol. 30 (3), 769–776. doi:10.1021/acs.chemrestox.6b00240
Pietsch, K. E., van Midwoud, P. M., Villalta, P. W., and Sturla, S. J. (2013). Quantification of acylfulvene– and illudin S–DNA adducts in cells with variable bioactivation capacities. Chem. Res. Toxicol. 26 (1), 146–155. doi:10.1021/tx300430r
Poindessous, V., Koeppel, F., Raymond, E., Comisso, M., Waters, S. J., and Larsen, A. K. (2003a). Marked activity of irofulven toward human carcinoma cells: comparison with cisplatin and ecteinascidin. Clin. Cancer Res. 9 (7), 2817–2825. From NLM.
Poindessous, V., Koeppel, F., Raymond, E., Comisso, M., Waters, S. J., and Larsen, A. K. (2003b). Marked activity of irofulven toward human carcinoma cells: comparison with cisplatin and ecteinascidin. Clin. cancer Res. official J. Am. Assoc. Cancer Res. 9 (7), 2817–2825. (acccessed 7/4/2024).
Tanasova, M., and Sturla, S. J. (2012). Chemistry and biology of acylfulvenes: sesquiterpene-derived antitumor agents. Chem. Rev. 112 (6), 3578–3610. doi:10.1021/cr2001367
Topka, S., Steinsnyder, Z., Ravichandran, V., Tkachuk, K., Kemel, Y., Bandlamudi, C., et al. (2021). Targeting germline- and tumor-associated nucleotide excision repair defects in cancer. Clin. Cancer Res. 27 (7), 1997–2010. doi:10.1158/1078-0432.Ccr-20-3322
Trachootham, D., Lu, W., Ogasawara, M. A., Valle, N. R. D., and Huang, P. (2008). Redox regulation of cell survival. Antioxidants & Redox Signal. 10 (8), 1343–1374. doi:10.1089/ars.2007.1957
Weichsel, A., Gasdaska, J. R., Powis, G., and Montfort, W. R. (1996). Crystal structures of reduced, oxidized, and mutated human thioredoxins: evidence for a regulatory homodimer. Structure 4 (6), 735–751. doi:10.1016/s0969-2126(96)00079-2
Woolston, C. M., Deen, S., Al-Attar, A., Shehata, M., Chan, S. Y., and Martin, S. G. (2010). Redox protein expression predicts progression-free and overall survival in ovarian cancer patients treated with platinum-based chemotherapy. Free Radic. Biol. Med. 49 (8), 1263–1272. doi:10.1016/j.freeradbiomed.2010.07.008
Woynarowska, B. A., and Woynarowski, J. M. (2002). Preferential targeting of apoptosis in tumor versus normal cells. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1587 (2), 309–317. doi:10.1016/S0925-4439(02)00094-7
Yu, X., Erzinger, M. M., Pietsch, K. E., Cervoni-Curet, F. N., Whang, J., Niederhuber, J., et al. (2012). Up-regulation of human prostaglandin reductase 1 improves the efficacy of hydroxymethylacylfulvene, an antitumor chemotherapeutic agent. J. Pharmacol. Exp. Ther. 343 (2), 426–433. doi:10.1124/jpet.112.195768
Zanotto-Filho, A., Masamsetti, V. P., Loranc, E., Tonapi, S. S., Gorthi, A., Bernard, X., et al. (2016). Alkylating agent-induced NRF2 blocks endoplasmic reticulum stress-mediated apoptosis via control of glutathione pools and protein thiol homeostasis. Mol. Cancer Ther. 15 (12), 3000–3014. doi:10.1158/1535-7163.Mct-16-0271
Zhang, J., Li, X., Han, X., Liu, R., and Fang, J. (2017). Targeting the thioredoxin system for cancer therapy. Trends Pharmacol. Sci. 38 (9), 794–808. doi:10.1016/j.tips.2017.06.001
Zhong, L., Arnér, E. S. J., and Holmgren, A. (2000). Structure and mechanism of mammalian thioredoxin reductase: the active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Natl. Acad. Sci. 97 (11), 5854–5859. doi:10.1073/pnas.100114897
Keywords: drug metabolism, thioredoxin, acylfulvene, illudin S, protein alkylation, anticancer drug, protein degradation
Citation: Slappendel L, Liu X, Macarthur MR, Sharpless CM and Sturla SJ (2024) Acylfulvenes covalently interact with thioredoxin as an additional cancer target. Front. Chem. Biol 3:1462351. doi: 10.3389/fchbi.2024.1462351
Received: 10 July 2024; Accepted: 26 August 2024;
Published: 10 September 2024.
Edited by:
Efstratios Stratikos, National and Kapodistrian University of Athens, GreeceReviewed by:
Norman Metanis, Hebrew University of Jerusalem, IsraelCopyright © 2024 Slappendel, Liu, Macarthur, Sharpless and Sturla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shana J. Sturla, c3R1cmxhc0BldGh6LmNo
†Present addresses: Xiaodan Liu, Max Planck Florida Institute for Neuroscience, Jupiter, Florida, United States
‡These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.