
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. Biol. , 17 June 2024
Sec. Structure, Spectroscopy & Imaging
Volume 3 - 2024 | https://doi.org/10.3389/fchbi.2024.1423430
This article is part of the Research Topic Bioluminescence and Fluorescence Molecular Imaging in Chemical Biology View all 6 articles
 Cristina Quiroga1†
Cristina Quiroga1† Marcelo Incerti2Diego Benitez1
Marcelo Incerti2Diego Benitez1 Eduardo Manta2
Eduardo Manta2 Andrea Medeiros1,3*
Andrea Medeiros1,3* Marcelo A. Comini1*
Marcelo A. Comini1*Trypanosoma cruzi is the etiologic agent of Chagas’ disease, a debilitating and mortal zoonotic illness that threatens the lives of several millions of people in the American continent. The acute phase is underdiagnosed and there is no curative treatment for the chronic stage. This unicellular pathogen colonizes cells from different tissues wherein it replicates and remains hidden from immune recognition. There is an urgent need to develop new chemotherapy, which requires robust screening bioassays against the amastigote, a proliferative and drug-resilient intracellular stage of the parasite. With this aim, here we present the generation and characterization of a bioluminescent reporter cell line of the highly infective strain Dm28c from T. cruzi. Constitutive and stable expression of the reporter gene (a red-shifted luciferase from Photinus pyralis) was achieved in the different developmental stages of the pathogen. The transgene did not affect parasite growth and differentiation. The bioluminescence signal displayed a linear correlation with the parasite number. A 96-well screening assay against the amastigote stage has been established and validated with a small compound library. The bioassay proved sensitive, robust, and amenable for high throughput applications. The reporter cell line may prove highly valuable to address different goals in the early phase of the drug discovery process against American trypanosomiasis.
Chagas disease (CD) is a zoonosis caused by the protozoan parasite Trypanosoma cruzi, which is transmitted by hematophagous hemipteran of the subfamily Triatominae. The World Health Organization classifies this disease as “neglected” and a silent ailment. It is endemic in 21 countries across Latin America, with an estimated 6 to 8 million people suffering from the disease, about 50.000 annual deaths, and 75 million people being at risk of infection. Alarmingly, only 30% of CD cases are diagnosed (DNDI, 2024).
The pathogen presents a complex life cycle involving four major developmental stages: epimastigotes (non-infective), metacyclic trypomastigotes (infective and non-replicative), amastigotes (intracellular and replicative), and trypomastigotes (extracellular and non-replicative), and several intermediate forms. The first two stages occur in the gut of the insect vector while the other are present in the mammalian host. For almost 50 years, Chagas’s disease has been treated with two old-fashioned nitroheterocyclic pro-drugs, nifurtimox (NFX), and benzinidazole (BNZ), that are effective only against the acute stage of the infection (Chao et al., 2020). In the chronic stage of the disease, the parasite remains resilient to these and other drugs (i.e., azoles). To some extent, this is because T. cruzi displays a lower replication/metabolic rate (Sanchez-Valdez et al., 2019; Ward et al., 2020) and/or the drugs do not reach or stay enough time to exert their anti-parasitic action in the body niches colonized by the pathogen.
Despite the World Health Organization has raised attention towards CD by including it in the list of neglected tropical diseases (NTD), private investment in drug discovery remains scarce, leading to a notable lack of chemotherapeutic options (Weng et al., 2018). Several academic groups and, in the last decade, public-private partnership initiatives (i.e., DNDi) have contributed to nominating novel molecular targets or compounds as drug candidates against CD (Bhattacharya et al., 2020; Gabaldón-Figueira et al., 2023). It is well known that the drug discovery process is a long path with multiple bottlenecks that, at the hit discovery stage, include: assay development (for compound screening) and hit-to-lead optimization. For a phenotypic-based screening, is of utmost importance that the cell-based assay resembles the physiological conditions (medium, cell model, pathogen stage and strain, etc.) under which the drug is expected to act at the organismal level (e.g., drug crossing host cell membrane). In this regard, a diversity of screening systems has been implemented in the search for chemicals active against T. cruzi (Chatelain and Ioset, 2018). They used axenic epimastigotes (Scarim et al., 2021) or trypomastigotes (Lara et al., 2018; Gulin et al., 2021; Scarim et al., 2021), and intracellular amastigotes (Bettiol et al., 2009; Engel et al., 2010; De Rycker et al., 2016; Yang et al., 2017; Lara et al., 2018; Scarim et al., 2021), and relied in non-automatized microscopic counting (Engel et al., 2010; Alonso-Padilla et al., 2015), colorimetric (Buckner et al., 1996; Bettiol et al., 2009; Yang et al., 2017), luminescent (Bot et al., 2010; MacLean et al., 2018), fluorescent (Canavaci et al., 2010; Henriques et al., 2014), or high-content microscopy assays (Sykes and Avery, 2013; Yang et al., 2017; Gonçalves et al., 2018; Benítez et al., 2022) to measure the trypanocidal effect of compounds. The metabolic needs and proteomic repertoire of epimastigotes and axenic amastigotes are different from the clinically relevant infective stages (trypomastigotes and intracellular amastigotes; (Canavaci et al., 2010; Villalta and Rachakonda, 2019), therefore, assays based on the last have a higher predictive value for the identification of drug candidates (Gabaldón-Figueira et al., 2023). Concerning the assay read-out methods, the microscopy techniques are laborious (i.e., when non-automatized), provide limited information (e.g., only a minor population of the sample is analyzed, hence, leading to high intra-assay variability), require expensive equipment for data acquisition or analysis (i.e., fluorescence microscope with automatized microplate screening platform) and their throughput capacity is low to medium (Alonso-Padilla and Rodríguez, 2014; Yang et al., 2017). Although the colorimetric methods based on reagents metabolized by endogenous or reporter enzymes (e.g., β-galactosidase) improve the readout rate and cell population coverage, they suffer from low sensitivity and specificity (e.g., host cell and compounds may interfere with color development (Young et al., 1993; Buckner et al., 1996; Sharma et al., 2021). Comparatively, bioluminescence-based assays offer the highest signal-to-noise ratio and sensitivity. This class of assays relies on luciferase activity that emits light upon the ATP-mediated decarboxylation of its substrate luciferin. Luciferase (LUC) can be genetically encoded (reporter) or added exogenously and the bioluminescent signal is proportional to the cellular content of the reporter gene or ATP, respectively. Bot et al. (2010) were the first to report the generation of a transgenic T. cruzi (strain CL-Brener) expressing a firefly LUC that has been used for compound screening. The reporter gene had no noticeable effects on trypanosome growth, differentiation, and infectivity, and proved very sensitive (e.g., it detected 10 parasites in a mammalian cell infection). The assay was standardized in a 96-well plate format (Bot et al., 2010) and, so far, has not been applied to drug screening. Several years later, and devoid of details about their generation and characterization, LUC-reporter cell lines of T. cruzi (strain Dm28c and Tulahuen) were used to evaluate the activity of naphthoquinones (Lewis et al., 2015; Lara et al., 2018; Silberstein et al., 2018) and extracts or compounds from natural sources (Tayama et al., 2023) in tissue-culture derived trypomastigotes and intracellular amastigotes. Also, the conventional ATP bioluminescence assay (Cell Titer-Glo cell viability kit from Promega) has been used to assess the viability of infective T. cruzi (from different strains) exposed to compounds (Lara et al., 2018) and clinical drugs (MacLean et al., 2018).
Recently, novel LUC variants, namely, a red-shifted and a nanoluciferase, with increased luminescence emission have been expressed in T. cruzi (Lewis et al., 2014; Taylor et al., 2019; Ward et al., 2020). Worth noting, these and other LUC-reporter cell lines have attracted more interest for investigating host-parasite interaction and drug response (e.g., relapse to treatment and pre-clinical assessment of hits) by in vivo imaging (Hyland et al., 2008; Andriani et al., 2011; Henriques et al., 2014; Lewis et al., 2014; 2015; Calvet et al., 2017; Silva-Dos-Santos et al., 2017; Sánchez-Valdéz and Padilla, 2019; Taylor et al., 2019; Mann et al., 2020) rather than for in vitro screening of compounds (Bot et al., 2010; Lara et al., 2018; Tayama et al., 2023).
The development of phenotypic screening assays against the intracellular form of T. cruzi has been challenging and demands standardized conditions to assess properly drug susceptibility (Yang et al., 2017) and mitigate experimental artifacts contributing to the high attrition rate or laboratory-dependent variability (Chatelain and Ioset, 2018). With this goal in mind, here we report the development of a high-throughput bioluminescence assay for evaluating the activity of chemical compounds against the amastigote stage of T. cruzi strain Dm28c, clone TcI (a highly virulent strain associated with oral outbreaks and fully assemble genome sequence; Berná et al., 2018). The reporter gene, a red-shifted LUC from Photinus pyralis (Branchini et al., 2010), is expressed in a stable and constitutive fashion in all developmental stages of the pathogen. Standardized conditions for cell infection and (single-step) assay readout are described. The robustness of the assay was validated by screening a small compound library.
Unless otherwise stated all chemical reagents were of analytical grade and purchased from SIGMA-ALDRICH (owned by Merck KGaA). The chemical compounds subjected to screening were available in our laboratory via collaborations quoted and acknowledged in Section 3.2 and Acknowledgements. The synthesis and preparation of the benzoisotiazolone derivatives and the rotenoid compound will be published elsewhere.
The epimastigote stage of the wild-type T. cruzi Dm28c (Dm28c-WT) was cultured in vitro in liver infusion tryptose (LIT) medium (Camargo, 1963; Rodríguez Durán et al., 2021) supplemented with 10% v/v fetal bovine serum (FBS, Gibco®), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco®) and hemin 0.02 g/L. The cultures (5 mL in 25 cm2 plug seal-cap culture-flask from Corning®) were incubated at 28°C and maintained in logarithmic growth (log-phase) by seeding parasites in fresh culture medium at a density of 5 × 104 cells/mL every 5 days. Cells were counted under a light microscope (Neubauer chamber). This procedure was applied to synchronize the growth of cultures from the wild-type and transgenic cell line.
In a total volume of 160 μL, 27 µg of the plasmid pTRIX2-RE9h (Lewis et al., 2014)) was linearized with AatII (0.75 U/µL) and AscI (0.6 U/µL) in Cut Smart Buffer (reagents from New England Biolabs) by overnight incubation at 37°C. The digested DNA was precipitated with sodium acetate 0.3 M (PanReac AppliChem) and isopropanol (0.7 volumes) and, after centrifugation (10,000 g at 4°C for 10 min), resuspended in 5 µL of UltraPureTM DNase/RNase-Free Distilled Water (Invitrogen). Parasites (∼65 million) in the log-phase, were washed with Phosphate-Buffered Saline (PBS) and then with HEPES-buffered saline (50 mM HEPES, 140 mM NaCl, 1.5 mM Na2HPO4.2H2O pH 7.4) after centrifugation at 10,000 g at 4°C for 10 min. The cell pellet was resuspended in 100 µL of Basic Parasite NucleofectorTM Kit1 solution, mixed with linearized DNA and electroporated with an AmaxaTM NucleofectorTM II (Lonza), program U-033. Transfected parasites were transferred to a 25 cm2 plug seal-cap culture-flask containing 2 mL of culture medium without selection antibiotic and incubated overnight at 28°C. Next, and within 1 week, the concentration of the selection antibiotic (G418 disulfate salt) was incremented stepwise from 125 μg/mL up to 500 μg/mL. To confirm the efficacy of the selective pressure, epimastigotes electroporated in the absence of DNA vector and subjected to identical G418-selection were included as a control. From the second week post-transfection onwards, the parasite cultures were maintained in a medium containing 250 μg/mL G418. The transgenic cell line was called Dm28c-luc.
Highly infective metacyclic trypomastigotes (MT) from both cell lines (WT and Dm28c-luc) were obtained by starvation (Daghero et al., 2023). Briefly, epimastigotes in the log-phase were seeded to 5 × 104 cells/mL in 5 mL of LIT medium (25 cm2 plug seal-cap culture-flask) and incubated for 14 days at 28°C and then used to infect Vero cells as described next (Scheme 1, Step 1).
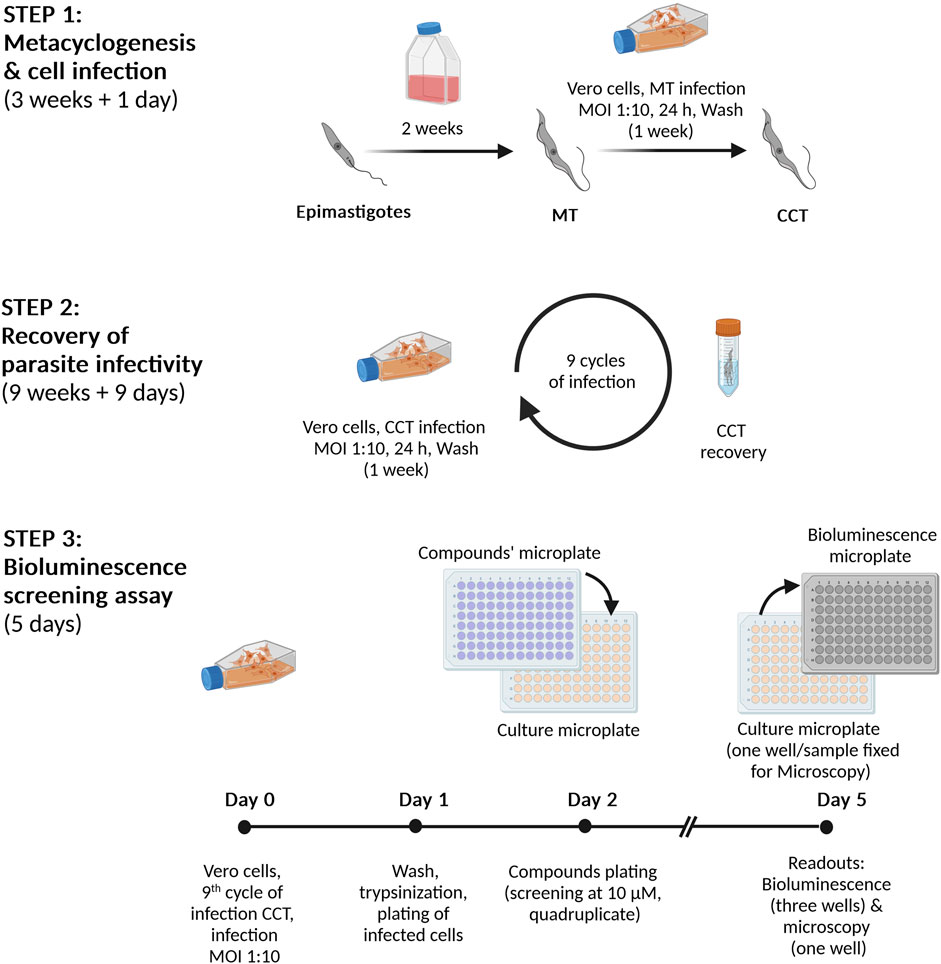
Scheme 1. Infection and bioluminescent assay procedures. Step 1. Metacyclogenesis and cell infection. Epimastigotes (reporter cell line Dm28c-luc) are incubated for 2 weeks in culture medium until nutritional starvation, which induces the differentiation into metacyclic trypomastigotes (MT). Then, Vero cells are infected with MT at a multiplicity of infection (MOI) of 1:10, cells:parasites. Seven days post-infection, cell culture trypomastigotes (CCT) are harvested from the culture supernatant. Step 2. Preparation of highly infective trypomastigotes. Vero cells are infected (MOI 1:10) with CCT obtained in Step 1 and the emerging CCT are collected and further used in repeated infection cycles (a total of nine). Step 3. Bioluminescent assay. On Day 0, Vero cells are infected with CCT (infection cycle 9) and, after 24 h (Day 1), the cell monolayer is detached and the cells are transferred to a 96-well microplate. After 24 h incubation at 37°C and 5% CO2 (Day 2), the compounds/drugs are added in quadruplicate replica at the corresponding concentrations (10 µM for primary screening or serial dilutions for IC50 determinations). The microplate is incubated for 72 h at 37°C with 5% CO2 and, at Day 5, the content from three replicate wells is transferred to a 96-well black microplate, treated with the Bright-GloTM Luciferase Assay System (PROMEGA) and bioluminescence signal read in a luminometer. The cells from the fourth well are fixed and analyzed by microscopy.
Vero cells (ATCC CCL-81) were cultivated at 40% confluency (about 1.2 × 106 cells) in a 25 cm2 plug vented-cap culture-flask (Corning®) in Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen) supplemented with 10% v/v FBS, 100 U/mL of penicillin, and 100 μg/mL streptomycin at 37°C and 5% CO2. MT were counted in a Neubauer chamber, centrifuged at 1,250 g for 10 min at 20°C, resuspended in culture medium (DMEM +10% v/v FBS) and co-incubated with Vero cells at a multiplicity of infection (MOI) of 1:10, cells:parasites. After incubation for 24 h at 37°C and 5% CO2, the cell monolayer was washed three times with pre-warmed (37°C) PBS before adding 5 mL of maintenance medium (DMEM with 2% v/v FBS). Five days post-infection, the culture supernatant was replaced with fresh maintenance medium to avoid culture acidification (Scheme 1, Step 1 and 2). The presence of cell culture trypomastigotes (CCT) emerging from lysed host cells was monitored daily by microscopy. When the content of CCT in the culture supernatant was about 50–60% of the total parasite burden (i.e., one week post-infection), the extracellular parasites were transferred to a 15 mL conical tube and harvested by centrifugation (1,250 g for 10 min at 20°C). Four mL of growth medium were carefully added to the pellet and the sample was incubated for 4 h (37°C, 5% CO2) to allow trypomastigotes to move outside the pellet of cells and debris (Eufrásio and Cordeiro, 2020). The CCT present in the supernatant were counted in a Neubauer chamber and used to infect Vero cells exactly as described above. This procedure was repeated nine times (Scheme 1, Step 2), which yielded a consistent infection rate (expressed as the percentage of the number of Vero cells infected with T. cruzi versus the total number of Vero cells) of 80–100%. The CCT isolated at cycle #9 were cryopreserved (1-2 million parasites in LIT complete medium supplemented with 10% v/v glycerol) at −80°C until use.
Amastigotes-like forms (Supplementary Figure S1) were obtained upon incubation of 106 CCT/mL in Brain Heart Infusion (BHI) medium at 37°C and 5% CO2 for 72–96 h.
LUC activity was measured in samples from different developmental stages of the reporter Dm28c-luc or Dm28c-WT (negative bioluminescence control) cell lines subjected to different growth conditions using the Bright-GloTM Luciferase Assay System reagent (BG-LAS, E2650 Promega).
Parasites at defined or unknown cell densities, depending on the experiment, were washed and, after centrifugation (1,250 g for 10 min at 20°C), resuspended in 100 µL of PBS 1% w/v Glucose. The cells were transferred to a 96-well black microplate (Greiner Bio-One) and 100 µL of BG-LAS was added per well. The luminescence signal was measured in a LUMIstar OPTIMA Microplate luminometer (BMG LABTECH) using the following settings (time expressed in seconds: s): 5 s shaking, 5 s/well acquisition, 0.2 s measurement delay, maximum gain value, and 25°C. All samples were analyzed in triplicates.
The bioluminescent signal (BL) is expressed as a percentage relative to the signal obtained for 1 × 106 parasites (i.e., epimastigotes in early/mid log-phase or amastigotes), and calculated as follows:
For statistical comparisons, the One-Way ANOVA test was applied.
On Day 0 (Scheme 1, Step 3), 1.2 × 106 Vero cells in log-phase are resuspended in 5 mL of culture medium (DMEM +10% FBS +100 U/mL of penicillin, and 100 μg/mL streptomycin), transferred to a 25 cm2 vented cap culture-flask (Corning®) and incubated for 4 h at 37°C and 5% CO2 to allow cellular adherence. Next, the cells are incubated with 1.2 × 107 CCT from cycle 9th of infection (see Section 2.3) at 37°C and 5% CO2 for 4 h or 24 h to achieve an infection rate of ∼30% or ≥50% (see Section 2.5.3 for determination of infection rate), respectively. Thereafter (Day 1), the cell monolayer is washed with PBS, and then incubated with 1 mL Trypsin-EDTA 0.25% w/v (Thermo Fisher Scientific) for 5 min at 37°C and 5% CO2 to detach all cells. The cells are resuspended in culture medium (1.25 × 105 cells/mL) and transferred to a 96-well culture microplate (120 µL/well or 15,000 cells/well, which covers 90–100% of the well surface) and incubated for 24 h at 37°C and 5% CO2.
Working solutions of NFX (8 mM) and BNZ (10 mM), both prepared in dimethyl sulfoxide (DMSO) 100% v/v from 25 mM stocks, were used to prepare serial 1:2 dilutions in culture medium. The range of concentrations tested for nifurtimox and benznidazole was from 40 to 0.1 μM and from 100 to 0.1 μM, respectively.
On Day 2, the culture supernatant from the microplate is replaced with 100 µL/well containing different concentrations of nifurtimox or benznidazole, and DMSO 0.5% v/v (negative control) prepared in maintenance medium. A blank condition consisted of wells lacking cells but containing 100 µL of maintenance medium supplemented with 0.5% v/v DMSO. The microplate was then incubated for 72 h at 37°C with 5% CO2. Three replicates were assayed for each drug concentration and the negative control.
On Day 5, the microplate was washed twice with 150 µL/well PBS 1X and then the cells were fixed with 4% v/v paraformaldehyde (40 µL/well) for 20 min at room temperature. After washing with 100 µL PBS, the cellular DNA was stained with methyl green (100 µL/well of a 0.0004% w/v solution prepared in PBS) for 10 min at room temperature. Upon washing with PBS, images (TIFF format 16 bit) from multiple fields of each well were taken in an inverted epifluorescence microscope (Olympus IX-81) using a 40× objective lens. A minimum of 250 host cells were analyzed per sample (well) using the “Trainable Weka Segmentation” plugin (Arganda-Carreras et al., 2017) from Fiji´s software (Schindelin et al., 2012). The percentage of infected cells (or infection rate) for each condition tested was estimated as follows:
For calculating the EC50, the % infection was further normalized to the values corresponding to non-infected cells (0% infection) or infected and non-treated cells (100% infection) and calculated by fitting the concentration-response curves to a four-parameter sigmoid equation using GraphPad Prism software (version 8.0).
Vero cells were infected with Dm28c-luc CCT exactly as described in Section 2.5.1 (Scheme 1 Day 0 and 1), except that the incubation with CCT was performed only for 24 h, which yielded an average infection rate of 65% (n = 4 assays).
Stock (25 mM) and working (2 mM) solutions of the chemical compounds to be screened were prepared in DMSO 100% v/v. To obtain a 10 µM assay solution, 6 μL from each working solution was diluted in 1,194 μL of maintenance medium, mixed thoroughly, and solubility was checked by nacked eye and microscope examination.
On Day 2, the culture supernatant from the microplate is replaced with 100 µL of maintenance medium containing the compounds of interest (10 µM), DMSO 0.5% v/v (negative control) or nifurtimox 5 µM (EC50; positive control). The blank condition consisted of wells lacking cells but containing 100 µL of maintenance medium supplemented with 0.5% v/v DMSO. The microplate was then incubated for 72 h at 37°C with 5% CO2. Supplementary Figure S2 shows the microplate layout used for the screening assay.
On Day 5, randomly selected wells from each sample were promptly inspected with an inverted microscope (40× objective lens) to verify the degree of cellular infection. Next, 100 µL/well of BG-LAS was added to the samples using an 8-channel micropipette, and the mixture was homogenized by carefully pipetting up and down to facilitate cell lysis. Immediately thereafter, the entire volume from each well (approximately 200 µL) was transferred to a 96-well black microplate (bioluminescent microplate) for subsequent reading in the luminometer. The bioluminescent signal was measured using the instrument and settings described in Section 2.4.
Parasite viability was calculated using the formula:
where BL refers to the mean of the bioluminescent signal corresponding to the tested compound (cpd), the blank (blank, maintenance medium containing 0.5% v/v DMSO), or the negative control (neg, infected cells treated with maintenance medium containing 0.5% v/v DMSO). The Z′ factor (Zhang et al., 1999) was determined according to the following formula:
where SD and BL refer to the standard deviation and mean bioluminescence value of the corresponding blank and negative samples.
EC50 values were calculated by fitting concentration-response curves to a four-parameter sigmoid equation using GraphPad Prism software (version 8.0).
Epimastigotes from the strain Dm28c were transfected with a solution containing or lacking (mock control) the pTRIX2-RE9h vector coding for a red-shifted LUC from P. pyralis. About 1 week after selection, parasites resistant to G418 were observed in samples transfected with the reporter vector (Dm28c-luc parasites) but not in the mock control. First, LUC activity was tested in samples containing different amounts of Dm28c-luc epimastigotes obtained from synchronized (log-phase) cultures using two different commercial kits from Promega (Figure 1A): the Luciferase Assay System (LAS) and the BG-LAS. LAS is a non-homogeneous assay that requires the removal of culture supernatant and cell wash before lysis, which is then followed by the addition of the Luciferase Assay Reagent (LAR). Although sensitive, the light generated by this assay has a half-life of 10 min. In contrast, the BG-LAS kit does not require sample pre-processing (medium removal and cell wash) and has a longer half-life (>3-fold compared to LAS). Although both assays were able to reveal LUC activity in Dm28c-luc epimastigotes, the performance of the BG-LAS was far superior to the LAS (Figure 1A). For the BG-LAS, the bioluminescence signal displayed an excellent correlation with parasite number (linear regression slope = 0.57 and R2= 0.9997) embracing four orders of magnitude dynamic range (signal from 103 to 107 parasites). For our reporter cell model, the LAS displayed a dynamic range of three orders of magnitude and a narrower linear range of signal quantification (e.g., signal emitted by 105 to 107 parasites). Furthermore, the bioluminescence signal generated with the BG-LAS was on average 7.3-fold higher than that produced with the LAS. Based on these results, all further bioluminescence assays were performed using the BG-LAS kit.
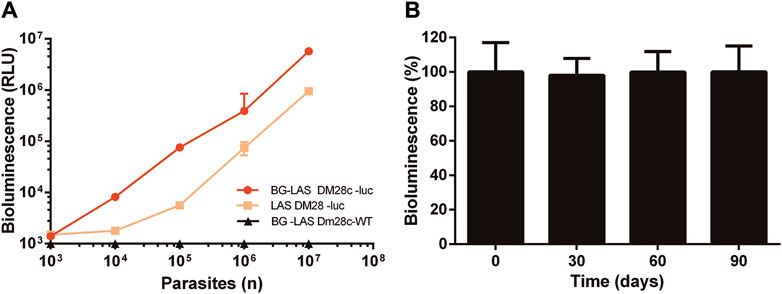
Figure 1. Bioluminescence correlates with parasite number and is stable in long-term cultures of Dm28c-luc epimastigotes. LUC activity was determined in Dm28c-luc epimastigotes grown to log-phase and in the absence of selection antibiotic for (A) 5 days using the LAS or BG-LAS kits (Dm28c-WT epimastigotes were used as a control for the BG-LAS) or (B) for 90 days using the BG-LAS kit. The cells were harvested, cell number adjusted to 1 × 106 epimastigotes (if needed) and analyzed for LUC activity (details in Section 2.4). Statistical analysis revealed no significant differences in signal (One-Way ANOVA) compared to day 0. For all plots, the errors are expressed as one standard deviation (SD) of the mean value (n=3).
In order to confirm the stable integration of the reporter gene, the luciferase activity was monitored at different time points in log-phase Dm28c-luc epimastigotes grown for long term in the absence of selection antibiotic. During this experiment, the parasites were maintained in log-phase by repeated subcultures every 5 days (see details in Section 2.2). As shown in Figure 1B, the bioluminescence signal remained high and stable for at least 3 months of continued and synchronized culture.
Next, different experiments were performed to characterize the growth phenotype (Figure 2A), differentiation, and infective capacity (Figure 2B; Supplementary Figure S3) of the reporter cell line. Based on the stable expression of LUC (Figure 1B) and to avoid a bias due to G418 effect on cell growth, all the assays pointed out above were performed in the absence of selective pressure.
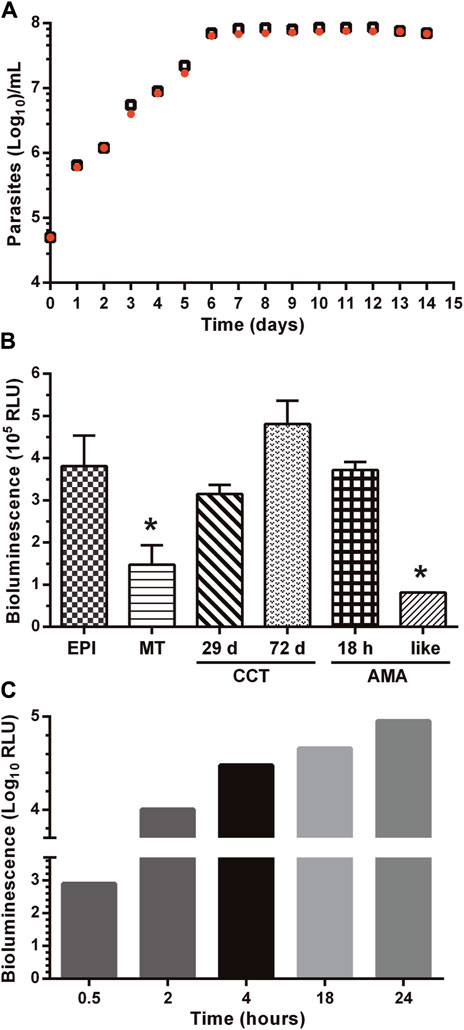
Figure 2. LUC expression does not affect the growth phenotype and differentiation capacity of the reporter parasites. (A) Synchronized epimastigotes from the parental (Dm28c-WT, black empty squares) and transgenic (Dm28c-luc; orange filled circles) cell lines cultured in the absence of the selection antibiotic were seeded at 5
As shown in Figure 2A, there were no differences in the proliferative capacity, maximum cell density, and growth curve profile of epimastigotes from the Dm28c-luc cell line compared to the wild-type strain.
The capacity of the Dm28c-luc cell line to undergo differentiation through the major life-cycle stages was evaluated along with LUC activity. In axenic culture conditions, replicative epimastigotes were forced to differentiate to metacyclic trypomastigotes (MT) by nutrient starvation. At variance with epimastigotes, MT are highly infective and non-replicative. There were no differences in the time of appearance of MT (∼14 days) in starved cultures of Dm28c-luc and Dm28c-WT. However, upon transition from the epimastigote to the MT stage, LUC activity dropped significantly (i.e. 2.5-fold) (Figure 2B). This result agrees with the nutritional stress to which parasites were subjected to differentiate to MT and with the transcriptional downregulation of energy metabolism reported for this non-replicative stage (Cruz-Saavedra et al., 2020). In several biological replicates conducted under identical experimental conditions (e.g., MOI 1:10), Dm28c-luc MT proved infective to VERO cells although at a lower ratio (10–20% infected cells) than those from the parental strain (80% infected cells; Supplementary Figure S4). Despite this observation, inside the mammalian cells, the Dm28c-luc MT were capable of differentiating into amastigotes that showed an steady increase in LUC activity (Figure 2C). The transgenic amastigotes replicated and transformed into trypomastigotes with similar kinetics to parasites from the WT strain. Similar to the WT strain, mechanical cell lysis by intracellular Dm28c-luc trypomastigotes was observed at 72–96 h post-infection. Importantly, during amastigogenesis (i.e., differentiation process from MT to amastigotes inside the host cell) and within 24 h, LUC activity increased by two orders of magnitude, in line with the metabolic and proliferative de-repression occurring in this stage.
To determine whether the lower infectivity displayed by Dm28c-luc MT is a permanent or transient phenotypic feature, we performed 10th successive cycles of host cell re-infection with trypomastigotes isolated from Vero cells (the so-called, cell culture trypomastigotes: CCT). By measuring LUC activity in CCT obtained at different infection cycles, we were able to confirm that this procedure did not affect the expression of the reporter gene, which produced a bioluminescence signal similar (re-infection cycle 4th and 9th corresponding to 29 and 72 days in culture, respectively) than that achieved by epimastigotes in log-phase (Figure 2B).
Applying this experimental approach, we observed that upon the 7th re-infection cycle, the Dm28c-luc cell line recovered an infectivity of Vero cells similar to WT parasites (Supplementary Figure S4). Importantly, intracellular amastigotes derived from an infection experiment that lasted 77 days (10 re-infection cycles) displayed a high bioluminescence signal (Figure 2B). Furthermore, Dm28c-luc CCT were incubated in BHI medium for 96 h at 37°C and 5% CO2, which led to the appearance of morphologically spherical, amastigote-like forms (Supplementary Figure S1). Under this growth condition, LUC activity was the lowest observed when compared to that detected in other developmental forms of T. cruzi, and suggests a transcriptional or metabolic downregulation in the amastigote-like parasites obtained in vitro.
Finally, we performed a comparative evaluation of the sensitivity of the reporter cell line towards the two clinical drugs used to treat Chagas’ disease, NFX and BNZ. Vero cells were infected at different rates (30% and 50%) with the Dm28c-luc or Dm28c-WT cell lines and the number of infected cells upon treatment was determined microscopically (further technical details in Section 2.5). Independently of the infection rate, the LUC-reporter cell line displayed a similar degree of sensitivity against NFX (EC50 = 1.8–5.3 µM) and BNZ (EC50 = 9.9–10.4 µM) than the parental strain (EC50 for NFX = 2.5–3.4 µM and BNZ = 11.4–11.6 µM; Table 1; Supplementary Figure S5). Statistical analysis (unpaired t-test) revealed no significant differences (p > 0.05) between EC50 values for each drug and cell line. Worth noting, NFX was 3- to 5-fold more potent than BNZ against Dm28c-luc and WT. The EC50 values obtained for these drugs under our assay conditions and the observed lower efficiency of BNZ to inhibit amastigote growth/survival agree well with data recently reported in a drug sensitivity study performed on 21 T. cruzi strains (Revollo et al., 2019).
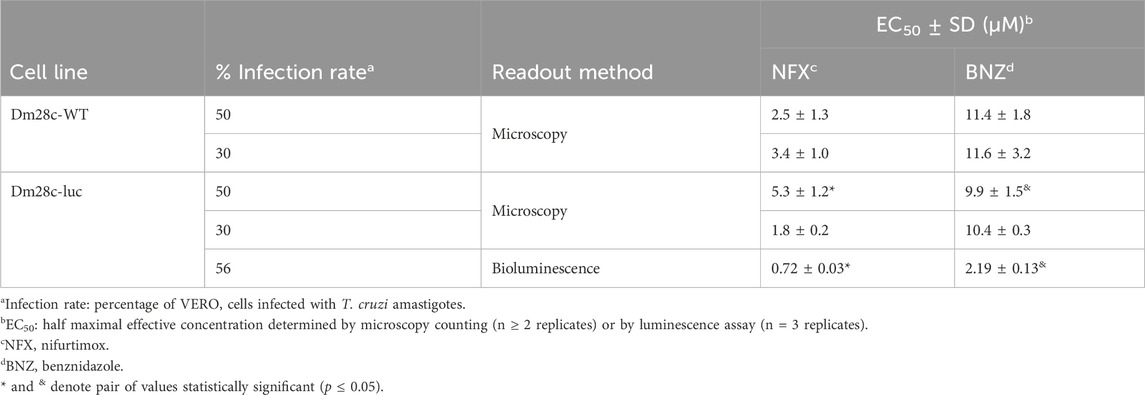
Table 1. Drug sensitivity of amastigotes from the bioluminescent and parental T. cruzi (strain Dm28c) cell lines.
Some important parameters of a bioassay relate to signal stability, sensitivity, and linearity. The stability of the bioluminescence signal produced by amastigotes infecting VERO cells was determined at different time points. Upon addition of the BG-LAS reagent, bioluminescence reached a maximum and stable level within 10–45 min and began to decrease thereafter (Figure 3A). This readout´s time window is suitable for low-to high-throughput screening applications since it provides sufficient time for handling several microplates and reduces the possibility of inter-assay variations in signal measurements.
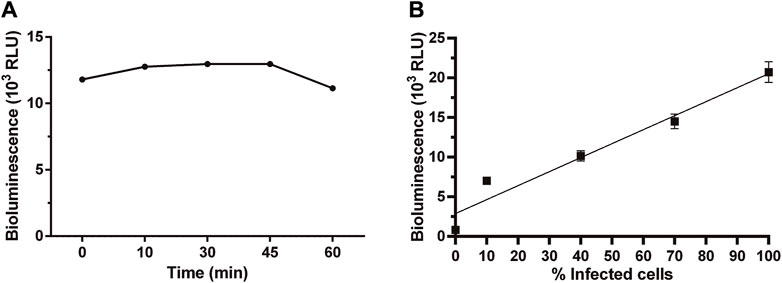
Figure 3. Stability and linearity of bioluminescent signal in Dm28c-luc amastigotes. Bioluminescence signal measured in samples of amastigote-infected cells at 72 h post-infection (A) at different time points upon addition of the BG-LAS reagent, and (B) with different infection ratios (i.e., percentage of infected cells), obtained by serial dilution of a culture sample where ∼100% of the VERO cells were infected (the 0 value correspond to non-infected cells). For technical details on the procedure, see section 2.3 and 2.4.
The bioluminescence signal emitted by intracellular amastigotes also showed a good linear correlation with the percentage of infected cells or, in other words, the amount of intracellular amastigotes (Figure 3B, R2 = 0.93 for linear regression plot). According to microscopical counting (29 ± 8 amastigotes/VERO cell) and the number of Vero cells/well, the signal detected at the lowest infection rate analyzed (10%) corresponds to about 4 × 104 amastigotes. Though lower infection rates were not experimentally tested, considering that: a) the signal/cell for amastigotes (1.7 RLU) is similar to that calculated for epimastigotes (1.5 RLU), b) a signal-to-background ratio of 288 ± 16, and c) the linear correlation between signal and parasite number (Figure 3B; Figure 1C), we estimated that our bioluminescence assay is able to detect ∼200 viable amastigotes/well. A similar bioluminescence assay for intracellular amastigotes reported a detection limit of 10 amastigotes/well using the LAS kit, which includes removal of the growth medium before lysis and substrate addition (Bot et al., 2010). The comparatively 10-fold lower sensitivity is likely consequence of the higher signal-to-background ratio of our assay.
Next, a concentration-response experiment was performed to estimate the potency of NFX and BNZ by the bioluminescence assay. The EC50 determined for intracellular amastigotes (infection ratio of 56%) was 0.7 µM for NFX and 3-fold higher for BNZ, 2.2 µM (Table 1; Supplementary Figure S5). The EC50 value obtained for NFX with our bioluminescence assay (0.72 μM) is higher but within the same order of magnitude to that determined for amastigotes from the CL-Brener strain (0.24 μM) using also a bioluminescence assay (Bot et al., 2010). The difference in NFX potency between both assays can be attributed to the 10-fold lower number of infected cells (1,500 cells/well) used in the assay of (Bot et al., 2010). Despite differences in bioluminescence assay setup (e.g., T. cruzi strains, MOI ∼1, in-house reagents for assay), the EC50 of BNZ determined for the Dm28c-luc (2.2 µM) was within the range reported for bioluminescent amastigotes from the strains Y (1.1 μM; (Lara et al., 2018) and Tulahuen 2 (2.8–4.9 µM; (Tayama et al., 2023).
Interestingly, compared to the microscopy counting method, the EC50 values for NFX and BNZ (at a 50% infection rate) determined the bioluminescence assay are 8- to 5-fold lower, respectively (Table 1). This difference is statistically significant (p ≤ 0.05) and can be ascribed to the fact that the microscopy analysis is based on counting the number of infected cells, which does not allow to discriminate the proportion of intracellular amastigotes alive or death. Therefore, this approach is prone to underestimate drug potency. In contrast, our LUC reporter system provides a more accurate measurement of parasite viability since starved amastigotes (which morphologically may look alive-like) produce a significant lower bioluminescence signal than those metabolically active (Figures 2B,C).
The performance of the bioluminescence assay for compounds’ screening was validated by testing a set of diverse chemical molecules (n = 51). The mini-library (Supplementary Table S1) included 35 compounds with unreported (34 benzoisothiazolones and 1 rotenoid) or previously reported activity against trypanosomatids: 4 quinones (Ballesteros-Casallas et al., 2023), 7 sesquiterpene lactones (Schmidt et al., 2002; 2014; Sülsen et al., 2016; Wulsten et al., 2017; Elso et al., 2020), 2 steroids (Ortíz et al., 2021), 2 paullones (Sousa et al., 2014; Medeiros et al., 2020), and 1 platinum complex (Rivas et al., 2019). Briefly, the 96-well screening assay consisted in incubating the compounds (assay concentration of 10 µM) for 72 h with VERO cells infected with amastigotes (infection ratio 65 ± 6%, n = 4 assays) and, thereafter, measuring LUC activity with the BG-LAS kit. Four independent screenings were performed, which yielded the following assay quality parameters: Z’ factor = 0.77 ± 0.06, coefficient of variation (CV) = 6.28 ± 0.47%. These values largely exceeded the cut-off points (Z′ factor >0.5 and CV < 20%) set for rating a HTS bioassay as satisfactory (Iversen et al., 2012).
The screening identified 20 hits (defined as compounds that tested at 10 μM reduce cell viability ≥50%; Figure 4; Supplementary Table S1). Thirteen of them corresponded to compounds not yet tested against T. cruzi (12 benzoisothiazolones and 1 rotenoid, which is one of the singletons), whereas the remaining ones corresponded to previously characterized molecules (2 quinones, 4 sesquiterpene lactones and 1 platinum complex). Restricting the analysis to the last group, the two quinones identified as hits were recently shown to have sub-µM EC50 against T. cruzi amastigotes (compounds 9 and 14 in (Ballesteros-Casallas et al., 2023). All sesquiterpene lactones assayed displayed anti-T. cruzi activity (cell viability ≤61%) and the two most potent: helenalin acetate (2% viability) and budlein A (26% viability), have already been reported to be one-digit μM inhibitors of T. cruzi amastigotes (EC50 = 6.93 μM and 1.75 μM, respectively; Schmidt et al., 2002; 2014). The metal-based compound that reduced amastigote viability to 2% is a heterobiplatinum 1,1′-bis(diphenylphosphino)ferrocene derivative with sub-μM potency against bloodstream T. brucei (EC50 = 170 nM, Pt-dppf-L4 in (Rivas et al., 2019). In addition, both paullones tested against T. cruzi amastigotes presented hit-like activities (53% and 63% viability) and were previously reported to be active against Leishmania infantum promastigotes (FS-554 in Sousa et al., 2014) and amastigotes (compound 20 in (Medeiros et al., 2020) with EC50 of 112 µM and 0.9 µM, respectively.
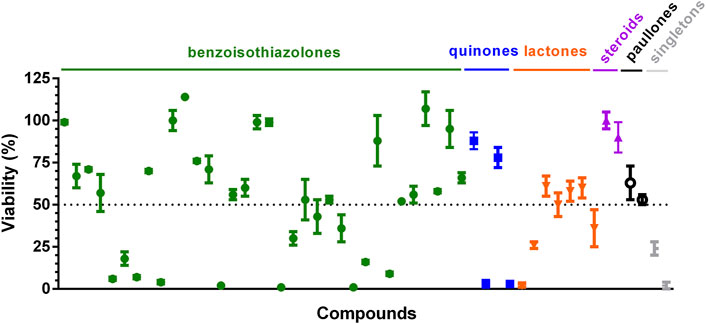
Figure 4. Validation of bioluminescent assay for compound screening. Compounds were assayed at 10 μM against VERO cells infected with T. cruzi Dm28-luc amastigotes. After 72 h incubation with compounds, bioluminescence signal was measured with the BG-LAS kit to determine cell viability. The dotted line indicates the cut-off value for hit compounds (viability ≤50%). Mean and error values (n = 3) are shown. The compounds labeled as singletons are (from left to right): one rotenoid and one platinum complex.
As stated in the introductory section, our study was preceded by others evaluating different expression systems (i.e., episomal or integrative, constitutive or inducible (Taylor et al., 2011); and LUC variants (P. pyralis LUC and nanoLUC) in T. cruzi. Among them, and based on early evidence (Lewis et al., 2015), we opted for selecting an expression vector (pTRIX2) that integrates into the non-transcribed RRNA spacer and harbors a ribosomal DNA (rDNA) promoter for constitutive and homogenous transgene expression (Costa et al., 2018). Regarding the reporter gene, and bearing in mind future in vivo applications of the cell line, we preferred to use a red-shifted variant of the P. pyralis LUC (PpyRE9H or RE9HLUC (Branchini et al., 2010); because it proved suitable for deep-tissue imaging in animal infection models (Lewis et al., 2015) and its activity, at variance with nanoLUC (England et al., 2016), is ATP-dependent, which offers the possibility to correlate light signal with the cells’ metabolic status (Benítez et al., 2020). Despite being monophyletic, T. cruzi can be classified into lineages or discrete typing units (DTUs) that present different clinical manifestations or ecological distributions (Brenière et al., 2016). The strain selected in our study (Dm28c) belongs to DTU I, has been associated with human infections in domicile cycles transmission in endemic areas (Nielebock et al., 2020) and its genome is known (Berná et al., 2018), which is essential for experimental approaches requiring the genetic manipulation of the parasite.
The LUC reporter cell line generated in our study showed a stable genomic integration (at least for 77 and 90 days in the absence of antibiotic selective pressure for amastigotes and epimastigotes, respectively) and constitutive expression of the transgene across the different developmental stages of the parasite. Importantly, expression of the LUC did not alter the growth phenotype, capability to differentiate, and drug sensitivity of the pathogen. Nonetheless, upon transformation to MT, we observed a reduced capability of the reporter cell line to infect mammalian cells. By performing at least seven successive cell infection cycles, the infectivity of the reporter cell line was restored to a level comparable to that of the parental wild-type strain. The reason for this transient deficiency is unknown (i.e., impaired host-pathogen recognition or parasite internalization) and further investigation of this issue may shed light on the host-pathogen factors modulating infectivity. However, this in vitro procedure, which mimics the in vivo infection cycle, is regularly used by other researchers to increase the in vitro infectivity of different T. cruzi strains (Hyland et al., 2008; Canavaci et al., 2010; Henriques et al., 2012; 2014; Lewis et al., 2014).
As anticipated for a constitutively expressed protein, whose function relies on the cellular content of ATP, luciferase activity achieved maximal levels in the proliferative extracellular (epimastigote) and intracellular (amastigote) stages of T. cruzi. In addition, LUC activity correlated linearly with parasite number and their metabolic status (i.e., starved MT and amastigote-like parasites presented low LUC activity), rendering this reporter system suitable to assess pathogen viability. In this regard, the bioluminescence assay proved more sensitive, accurate, and throughput than microscopy counting for determining the load of viable amastigotes infecting mammalian cells. The microscopy method is efficient when the infection rates are lower than 30%, allowing for the use of automated counting software tools such as Fiji (ImageJ). However, for a screening method that requires infection rates above 50% to ensure a more homogeneous infection and reliable assessment of compound bioactivity, cell counting (manual or with trained software) is prone to errors and laborious since it fails to accurately discriminate non-infected from infected cells as well as in determining the precise number of intracellular amastigotes, which, according to our experience, can range from 30 to 216 parasites/cell. Furthermore, only a minor fraction of the cell population is analyzed by microscopy imaging. In contrast, the bioluminescence method provides information from the whole cell population present in the assay well, discriminating between viable and non-viable states.
A protocol for compound screening was developed and validated with a small chemolibrary. The screening assay lasts a total of 120 h, is performed in 96-well microplates, and includes the plating of Vero cells (day 1), the infection with T. cruzi Dm28c-luc (infection rate of ≥50%, day 2) followed by treatment with compounds for 72 h (triplicate samples and controls) and bioluminescence reading (≤45 min). This assay scheme has been established considering the guidelines of a thorough study that identified cell seeding sequence and compound incubation time as the major factors responsible for variation between assays for evaluating the viability of intracellular T. cruzi (Yang et al., 2017). Notably, the use of the BG-LAS kit introduced three major technical advantages to this assay: i) removal of culture medium or washing before substrate addition is not required, eliminating a risky handling step not only because of the highly infective material manipulated but also because they may lead to cell de-attachment and the consequent overestimation of compound potency, ii) a single solution contains the cell lysis reagent and the LUC substrate, thus, reducing sample handling; iii) due to stabilization of reactants, luminescence has a longer half-life (>60 min in our assay), thereby minimizing pipetting errors or incomplete mixing and providing enough time for high-throughput multiwell plates reading.
The screening assay allowed verifying the anti-T. cruzi activity of compounds previously reported to be active against trypanosomatids (quinones, sesquiterpene lactones, Pt-organic complex, and paullones) and to identify new derivatives (benzoisotiazolones) with higher potency than its congener (ebsulfur (Lu et al., 2013); as well as a new hit (a rotenoid). As shown here, across the different life- and growth stages of the parasite, LUC has an expression pattern similar to a housekeeping gene, and its activity level is proportional to the parasite’s number and energetic status. These features of our reporter expression system may help to discover novel anti-Chagasic compounds with diverse mechanisms of action (cytotoxic or cytostatic), including some acting on host molecules relevant to intracellular survival, and also to investigate environmental or molecular factors affecting LUC expression or activity. Compared to other screening methods based on colorimetry (β-galactosidase) or fluorescence (resarzurin, or GFP-like reporter proteins), requiring or not microscopy as readout, the bioluminescent assay here developed appears simpler (handling and timing) and robust (low CV% and high Z′ factor). Furthermore, its sensitivity and high-throughput-like features make it a suitable tool for large drug discovery campaigns. Besides its use for phenotyping the clinically relevant stages of the parasite (trypomastigotes and amastigotes), the reporter cell line may result in valuable in vivo applications with high translational potential, such as real-time monitoring of parasite behavior and drug efficacy profiling.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
CQ: Formal Analysis, Investigation, Methodology, Validation, Writing–original draft. MI: Writing–review and editing, Resources. DB: Investigation, Methodology, Data curation, Writing–review and editing. EM: Resources, Writing–review and editing. AM: Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing–original draft, Writing–review and editing. MC: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Resources, Supervision, Validation, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. AM and MC thank the financial support of the Comisión Sectorial de Investigación Científica, Universidad de la República, Uruguay (CSIC I + D 2019 project number 388). MC also acknowledges the support of FOCEM (MERCOSUR Structural Convergence Fund, COF 03/11). CQ received fellowships from CSIC, ANII (Agencia Nacional de Investigación e Innovación), PEDECIBA and Institut Pasteur de Montevideo. DB, AM, and MC are members of the SNI (Sistema Nacional de Investigadores, ANII, Uruguay) and PEDECIBA.
Dr. John M. Kelly (London School of Hygiene and Tropical Medicine, United Kingdom) and Dr. Carlos Robello (Institut Pasteur de Montevideo, Uruguay) are acknowledged for the gift of the pTRIX2-RE9h plasmid and the wild-type strain Dm28c from T. cruzi, respectively. Laboratorio de Química Fina, Instituto de Polo Tecnológico de Pando, Facultad de Química, Universidad de la República, Montevideo, Uruguay, for providing Benznidazole. Dr. Hugo Cerecetto and Dr. Elena Aguilera (Instituto de Química Biológica, Facultad de Ciencias, Universidad de la República, Montevideo, Uruguay) are acknowledged for providing purified nifurtimox. Dr. Valeria Sulsen (Universidad de Buenos Aires, Argentina), Dr. Thomas Schmidt (University of Münster, Germany), Dr. Cristian Salas and Dr. R. Tapia (Pontificia Universidad Católica de Chile, Chile), Dr. Nial Hamilton (Manchester University, United Kingdom), Dr. Dinorah Gambino (Universidad de la República, Uruguay) and Dr. Conrad Kunick (Technical University of Braunschweig, Germany) are gratefully acknowledged for providing compounds for testing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchbi.2024.1423430/full#supplementary-material
AMA, Amastigotes; BNZ, Benznidazole; Bioluminescent cell line of Trypanosoma cruzi, strain Dm28c (Dm28c-luc); CCT, Cell culture trypomastigotes; CD, Chagas disease; DMSO, dimethyl sulfoxide; DMEM, Dulbecco’s Modified Eagle’s Medium; EPI, Epimastigotes; FBS, Fetal Bovine Serum; LIT, Liver infusion tryptose; MT, Metacyclic trypomastigotes; NTDs, Neglected Tropical Diseases; DNDi, Neglected Diseases Initiative; NFX, Nifurtimox; T. cruzi, Trypanosoma cruzi; WT, Trypanosoma cruzi, strain Dm28c wild-type; EC50, Half Maximal Effective Concentration.
Alonso-Padilla, J., Cotillo, I., Presa, J. L., Cantizani, J., Peña, I., Bardera, A. I., et al. (2015). Automated high-content assay for compounds selectively toxic to Trypanosoma cruzi in a myoblastic cell line. PLoS Negl. Trop. Dis. 9, e0003493. doi:10.1371/journal.pntd.0003493
Alonso-Padilla, J., and Rodríguez, A. (2014). High throughput screening for anti–trypanosoma cruzi drug discovery. PLoS Negl. Trop. Dis. 8, 32599–e3266. doi:10.1371/journal.pntd.0003259
Andriani, G., Chessler, A.-D. C., Courtemanche, G., Burleigh, B. A., and Rodriguez, A. (2011). Activity in vivo of anti-trypanosoma cruzi compounds selected from a high throughput screening. PLoS Negl. Trop. Dis. 5, 12988–e1306. doi:10.1371/journal.pntd.0001298
Arganda-Carreras, I., Kaynig, V., Rueden, C., Eliceiri, K. W., Schindelin, J., Cardona, A., et al. (2017). Trainable Weka Segmentation: a machine learning tool for microscopy pixel classification. Bioinformatics 33, 2424–2426. doi:10.1093/bioinformatics/btx180
Ballesteros-Casallas, A., Quiroga, C., Ortiz, C., Benítez, D., Denis, P. A., Figueroa, D., et al. (2023). Mode of action of p-quinone derivatives with trypanocidal activity studied by experimental and in silico models. Eur. J. Med. Chem. 246, 114926. doi:10.1016/j.ejmech.2022.114926
Benítez, D., Dibello, E., Bonilla, M., and Comini, M. A. (2020). A simple, robust, and affordable bioluminescent assay for drug discovery against infective African trypanosomes. Drug Dev. Res. 83, 253–263. doi:10.1002/ddr.21634
Benítez, D., Franco, J., Sardi, F., Leyva, A., Durán, R., Choi, G., et al. (2022). Drug-like molecules with anti-trypanothione synthetase activity identified by high throughput screening. J. Enzyme Inhibition Med. Chem. 37, 912–929. doi:10.1080/14756366.2022.2045590
Berná, L., Rodriguez, M., Chiribao, M. L., Parodi-Talice, A., Pita, S., Rijo, G., et al. (2018). Expanding an expanded genome: long-read sequencing of Trypanosoma cruzi. Microb. Genomics 4, e000177. doi:10.1099/mgen.0.000177
Bettiol, E., Samanovic, M., Murkin, A. S., Raper, J., Buckner, F., and Rodriguez, A. (2009). Identification of three classes of heteroaromatic compounds with activity against intracellular trypanosoma cruzi by chemical library screening. PLoS Negl. Trop. Dis. 3, 3844–e412. doi:10.1371/journal.pntd.0000384
Bhattacharya, A., Corbeil, A., Do Monte-Neto, R. L., and Fernandez-Prada, C. (2020). Of drugs and trypanosomatids: new tools and knowledge to reduce bottlenecks in drug discovery. Genes (Basel) 11, 1–24. doi:10.3390/genes11070722
Bot, C., Hall, B. S., Bashir, N., Taylor, M. C., Helsby, N. A., and Wilkinson, S. R. (2010). Trypanocidal activity of aziridinyl nitrobenzamide prodrugs. Antimicrob. Agents Chemother. 54, 4246–4252. doi:10.1128/AAC.00800-10
Branchini, B. R., Ablamsky, D. M., Davis, A. L., Southworth, T. L., Butler, B., Fan, F., et al. (2010). Red-emitting luciferases for bioluminescence reporter and imaging applications. Anal. Biochem. 396, 290–297. doi:10.1016/j.ab.2009.09.009
Brenière, S. F., Waleckx, E., and Barnabé, C. (2016). Over six thousand trypanosoma cruzi strains classified into discrete typing units (DTUs): attempt at an inventory. PLoS Negl. Trop. Dis. 10, e0004792. doi:10.1371/journal.pntd.0004792
Buckner, F. S., Verlinde, C. L., La Flamme, A. C., and Van Voorhis, W. C. (1996). Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing beta-galactosidase. Antimicrob. Agents Chemother. 40, 2592–2597. doi:10.1128/AAC.40.11.2592
Calvet, C. M., Choi, J. Y., Thomas, D., Suzuki, B., Hirata, K., Lostracco-Johnson, S., et al. (2017). 4-aminopyridyl-based lead compounds targeting CYP51 prevent spontaneous parasite relapse in a chronic model and improve cardiac pathology in an acute model of Trypanosoma cruzi infection. PLoS Negl. Trop. Dis. 11, 00061322–e6224. doi:10.1371/journal.pntd.0006132
Camargo, E. (1964). Growth and differentiation in trypanosoma cruzi. I. Origin of metacyclic trypanosomes in liquid media. Rev. Inst. Med. Trop. Sao Paulo 6, 93–100.
Canavaci, A. M. C., Bustamante, J. M., Padilla, A. M., Perez Brandan, C. M., Simpson, L. J., Xu, D., et al. (2010). In vitro and in vivo high-throughput assays for the testing of anti-trypanosoma cruzi compounds. PLoS Negl. Trop. Dis. 4, 7400–e813. doi:10.1371/journal.pntd.0000740
Chao, C., Leone, J. L., and Vigliano, C. A. (2020). Chagas disease: historic perspective. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1866, 165689. doi:10.1016/j.bbadis.2020.165689
Chatelain, E., and Ioset, J. R. (2018). Phenotypic screening approaches for Chagas disease drug discovery. Expert Opin. Drug Discov. 13, 141–153. doi:10.1080/17460441.2018.1417380
Costa, F. C., Francisco, A. F., Jayawardhana, S., Calderano, S. G., Lewis, M. D., Olmo, F., et al. (2018). Expanding the toolbox for Trypanosoma cruzi: a parasite line incorporating a bioluminescence-fluorescence dual reporter and streamlined CRISPR/Cas9 functionality for rapid in vivo localisation and phenotyping. PLoS Negl. Trop. Dis. 12, e0006388. doi:10.1371/journal.pntd.0006388
Cruz-Saavedra, L., Vallejo, G. A., Guhl, F., Messenger, L. A., and Ramírez, J. D. (2020). Transcriptional remodeling during metacyclogenesis in Trypanosoma cruzi I. Virulence 11, 968–979. doi:10.1080/21505594.2020.1797274
Daghero, H., Pagotto, R., Quiroga, C., Medeiros, A., Comini, M. A., and Bollati-Fogolín, M. (2023). Murine colon organoids as a novel model to study Trypanosoma cruzi infection and interactions with the intestinal epithelium. Front. Cell. Infect. Microbiol. 13, 1082524. doi:10.3389/fcimb.2023.1082524
De Rycker, M., Thomas, J., Riley, J., Brough, S. J., Miles, T. J., and Gray, D. W. (2016). Identification of trypanocidal activity for known clinical compounds using a new trypanosoma cruzi hit-discovery screening cascade. PLoS Negl. Trop. Dis. 10, e0004584. doi:10.1371/journal.pntd.0004584
DNDI (2024). DNDi. Available at: https://dndi.org/diseases/chagas/.
Elso, O. G., Bivona, A. E., Sanchez Alberti, A., Cerny, N., Fabian, L., Morales, C., et al. (2020). Trypanocidal activity of four sesquiterpene lactones isolated from asteraceae species. Molecules 25, 2014. doi:10.3390/molecules25092014
Engel, J. C., Ang, K. K. H., Chen, S., Arkin, M. R., McKerrow, J. H., and Doyle, P. S. (2010). Image-based high-throughput drug screening targeting the intracellular stage of Trypanosoma cruzi, the agent of Chagas’ disease. Antimicrob. Agents Chemother. 54, 3326–3334. doi:10.1128/AAC.01777-09
England, C. G., Ehlerding, E. B., and Cai, W. (2016). NanoLuc: a small luciferase is brightening up the field of bioluminescence. Bioconjugate Chem. 27, 1175–1187. doi:10.1021/acs.bioconjchem.6b00112
Eufrásio, A. G., and Cordeiro, A. T. (2020). “In vitro image-based assay for trypanosoma cruzi intracellular forms,” in Trypanosomatids: methods and protocols. Editors P. A. M. Michels, M. L. Ginger, and D. Zilberstein (New York, NY: Springer US), 773–780. doi:10.1007/978-1-0716-0294-2_45
Francisco, A. F., Lewis, M. D., Jayawardhana, S., Taylor, M. C., Chatelain, E., and Kelly, J. M. (2015). Limited ability of posaconazole to cure both acute and chronic trypanosoma cruzi infections revealed by highly sensitive in vivo imaging. Antimicrob. Agents Chemother. 59, 4653–4661. doi:10.1128/aac.00520-15
Gabaldón-Figueira, J. C., Martinez-Peinado, N., Escabia, E., Ros-Lucas, A., Chatelain, E., Scandale, I., et al. (2023). State-of-the-Art in the drug discovery pathway for Chagas disease: a framework for drug development and target validation. Res. Rep. Trop. Med. 14, 1–19. doi:10.2147/RRTM.S415273
Gonçalves, C. S., Ávila, A. R., de Souza, W., Motta, M. C. M., and Cavalcanti, D. P. (2018). Revisiting the Trypanosoma cruzi metacyclogenesis: morphological and ultrastructural analyses during cell differentiation. Parasites Vectors 11, 83. doi:10.1186/s13071-018-2664-4
Gulin, J. E. N., Rocco, D. M., Alonso, V., Cribb, P., Altcheh, J., and García-Bournissen, F. (2021). Optimization and biological validation of an in vitro assay using the transfected Dm28c/pLacZ Trypanosoma cruzi strain. Biol. methods & Protoc. 6, bpab004. doi:10.1093/biomethods/bpab004
Henriques, C., Castro, D. P., Gomes, L. H. F., Garcia, E. S., and de Souza, W. (2012). Bioluminescent imaging of Trypanosoma cruzi infection in Rhodnius prolixus. Parasites Vectors 5, 214. doi:10.1186/1756-3305-5-214
Henriques, C., Henriques-Pons, A., Meuser-Batista, M., Ribeiro, A. S., and de Souza, W. (2014). In vivo imaging of mice infected with bioluminescent Trypanosoma cruzi unveils novel sites of infection. Parasites Vectors 7, 89. doi:10.1186/1756-3305-7-89
Hyland, K. V., Asfaw, S. H., Olson, C. L., Daniels, M. D., and Engman, D. M. (2008). Bioluminescent imaging of Trypanosoma cruzi infection. Int. J. Parasitol. 38, 1391–1400. doi:10.1016/j.ijpara.2008.04.002
Iversen, P. W., Beck, B., Chen, Y.-F., DereMS, W., Devanarayan, V., EastwoodPhD, B. J., et al. (2012). HTS assay validation. Assay. Guid. Man. 26. Available at: https://www.ncbi.nlm.nih.gov/books/NBK83783/.
Lara, L. S., Moreira, C. S., Calvet, C. M., Lechuga, G. C., Souza, R. S., Bourguignon, S. C., et al. (2018). Efficacy of 2-hydroxy-3-phenylsulfanylmethyl-[1,4]-naphthoquinone derivatives against different Trypanosoma cruzi discrete type units: identification of a promising hit compound. Eur. J. Med. Chem. 144, 572–581. doi:10.1016/j.ejmech.2017.12.052
Lewis, M. D., Fortes Francisco, A., Taylor, M. C., Burrell-Saward, H., Mclatchie, A. P., Miles, M. A., et al. (2014). Bioluminescence imaging of chronic T rypanosoma cruzi infections reveals tissue-specific parasite dynamics and heart disease in the absence of locally persistent infection. Cell. Microbiol. 16, 1285–1300. doi:10.1111/cmi.12297
Lewis, M. D., Francisco, A. F., Taylor, M. C., and Kelly, J. M. (2015). A new experimental model for assessing drug efficacy against Trypanosoma cruzi infection based on highly sensitive in vivo imaging. SLAS Discov. 20, 36–43. doi:10.1177/1087057114552623
Lu, J., Vodnala, S. K., Gustavsson, A. L., Gustafsson, T. N., Sjöberg, B., Johansson, H. A., et al. (2013). Ebsulfur is a benzisothiazolone cytocidal inhibitor targeting the trypanothione reductase of Trypanosoma brucei. J. Biol. Chem. 288, 27456–27468. doi:10.1074/jbc.M113.495101
MacLean, L. M., Thomas, J., Lewis, M. D., Cotillo, I., Gray, D. W., and De Rycker, M. (2018). Development of Trypanosoma cruzi in vitro assays to identify compounds suitable for progression in Chagas’ disease drug discovery. PLoS Negl. Trop. Dis. 12, e0006612. doi:10.1371/journal.pntd.0006612
Mann, G. S., Francisco, A. F., Jayawardhana, S., Taylor, M. C., Lewis, M. D., Olmo, F., et al. (2020). Drug-cured experimental Trypanosoma cruzi infections confer long-lasting and cross-strain protection. PLoS Negl. Trop. Dis. 14, e0007717. doi:10.1371/journal.pntd.0007717
Medeiros, A., Benítez, D., Korn, R. S., Ferreira, V. C., Barrera, E., Carrión, F., et al. (2020). Mechanistic and biological characterisation of novel N5-substituted paullones targeting the biosynthesis of trypanothione in Leishmania. J. Enzyme Inhibition Med. Chem. 35, 1345–1358. doi:10.1080/14756366.2020.1780227
Nielebock, M. A. P., Moreira, O. C., Xavier, S. C. d. C., Miranda, L. d. F. C., Lima, A. C. B. de, Pereira, T. O., et al. (2020). Association between Trypanosoma cruzi DTU TcII and chronic Chagas disease clinical presentation and outcome in an urban cohort in Brazil. PLoS One 15, e0243008. doi:10.1371/journal.pone.0243008
Ortíz, C., Moraca, F., Laverriere, M., Jordan, A., Hamilton, N., and Comini, M. A. (2021). Glucose 6-phosphate dehydrogenase from trypanosomes: selectivity for steroids and chemical validation in bloodstream trypanosoma brucei. Molecules 26, 358. doi:10.3390/molecules26020358
Revollo, S., Oury, B., Vela, A., Tibayrenc, M., and Sereno, D. (2019). In vitro benznidazole and nifurtimox susceptibility profile of trypanosoma cruzi strains belonging to discrete typing units TcI, TcII, and TcV. Pathogens 8, 197. doi:10.3390/pathogens8040197
Rivas, F., Medeiros, A., Comini, M., Suescun, L., Rodríguez Arce, E., Martins, M., et al. (2019). Pt-Fe ferrocenyl compounds with hydroxyquinoline ligands show selective cytotoxicity on highly proliferative cells. J. Inorg. Biochem. 199, 110779. doi:10.1016/j.jinorgbio.2019.110779
Rodríguez Durán, J., Muñoz-Calderón, A., Gómez, K. A., and Potenza, M. (2021). In vitro differentiation of Trypanosoma cruzi epimastigotes into metacyclic trypomastigotes using a biphasic medium. Star. Protoc. 2, 100703. doi:10.1016/j.xpro.2021.100703
Sánchez-Valdéz, F., and Padilla, A. (2019). In situ detection of dormant trypanosoma cruzi amastigotes using bioluminescent-fluorescent reporters. Methods Mol. Biol. 1955, 179–186. doi:10.1007/978-1-4939-9148-8_13
Scarim, C. B., Olmo, F., Ferreira, E. I., Chin, C. M., Kelly, J. M., and Francisco, A. (2021). Image-based in vitro screening reveals the trypanostatic activity of hydroxymethylnitrofurazone against trypanosoma cruzi. Int. J. Mol. Sci. 22, 6930. doi:10.3390/ijms22136930
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi:10.1038/nmeth.2019
Schmidt, T. J., Brun, R., Willuhn, G., and Khalid, S. A. (2002). Anti-trypanosomal activity of helenalin and some structurally related sesquiterpene lactones. Planta Med. 68, 750–751. doi:10.1055/s-2002-33799
Schmidt, T. J., Da Costa, F. B., Lopes, N. P., Kaiser, M., and Brun, R. (2014). In silico prediction and experimental evaluation of furanoheliangolide sesquiterpene lactones as potent agents against Trypanosoma brucei rhodesiense. Antimicrob. Agents Chemother. 58, 325–332. doi:10.1128/AAC.01263-13
Sharma, S. K., Poudel Sharma, S., and Leblanc, R. M. (2021). Methods of detection of β-galactosidase enzyme in living cells. Enzyme Microb. Technol. 150, 109885. doi:10.1016/j.enzmictec.2021.109885
Silberstein, E., Serna, C., Fragoso, S. P., Nagarkatti, R., and Debrabant, A. (2018). A novel nanoluciferase-based system to monitor Trypanosoma cruzi infection in mice by bioluminescence imaging. PLoS One 13, e0195879. doi:10.1371/journal.pone.0195879
Silva-Dos-Santos, D., Barreto-de-Albuquerque, J., Guerra, B., Moreira, O. C., Berbert, L. R., Ramos, M. T., et al. (2017). Unraveling Chagas disease transmission through the oral route: gateways to Trypanosoma cruzi infection and target tissues. PLoS Negl. Trop. Dis. 11, e0005507. doi:10.1371/journal.pntd.0005507
Sousa, A. F., Gomes-Alves, A. G., Benítez, D., Comini, M. A., Flohé, L., Jaeger, T., et al. (2014). Genetic and chemical analyses reveal that trypanothione synthetase but not glutathionylspermidine synthetase is essential for Leishmania infantum. Free Radic. Biol. Med. 73, 229–238. doi:10.1016/j.freeradbiomed.2014.05.007
Sülsen, V. P., Puente, V., Papademetrio, D., Batlle, A., Martino, V. S., Frank, F. M., et al. (2016). Mode of action of the sesquiterpene lactones psilostachyin and psilostachyin C on trypanosoma cruzi. PLoS One 11, e0150526. doi:10.1371/journal.pone.0150526
Sykes, M. L., and Avery, V. M. (2013). Approaches to Protozoan drug discovery: phenotypic screening. J. Med. Chem. 56, 7727–7740. doi:10.1021/jm4004279
Tayama, Y., Mizukami, S., Toume, K., Komatsu, K., Yanagi, T., Nara, T., et al. (2023). Anti-Trypanosoma cruzi activity of Coptis rhizome extract and its constituents. Trop. Med. Health 51, 12. doi:10.1186/s41182-023-00502-2
Taylor, M. C., Francisco, A. F., Jayawardhana, S., Mann, G. S., Ward, A. I., Olmo, F., et al. (2019). Exploiting genetically modified dual-reporter strains to monitor experimental trypanosoma cruzi infections and host-parasite interactions. Methods Mol. Biol. 1955, 147–163. doi:10.1007/978-1-4939-9148-8_11
Taylor, M. C., Huang, H., and Kelly, J. M. (2011). Genetic techniques in Trypanosoma cruzi. Adv. Parasitol. 75, 231–250. doi:10.1016/B978-0-12-385863-4.00011-3
Villalta, F., and Rachakonda, G. (2019). Advances in preclinical approaches to Chagas disease drug discovery. Expert Opin. Drug Discov. 14, 1161–1174. doi:10.1080/17460441.2019.1652593
Ward, A. I., Olmo, F., Atherton, R. L., Taylor, M. C., and Kelly, J. M. (2020). Trypanosoma cruzi amastigotes that persist in the colon during chronic stage murine infections have a reduced replication rate. Open Biol. 10, 200261. doi:10.1098/rsob.200261
Weng, H.-B., Chen, H.-X., and Wang, M.-W. (2018). Innovation in neglected tropical disease drug discovery and development. Infect. Dis. poverty 7, 67. doi:10.1186/s40249-018-0444-1
Wulsten, I. F., Costa-Silva, T. A., Mesquita, J. T., Lima, M. L., Galuppo, M. K., Taniwaki, N. N., et al. (2017). Investigation of the anti-leishmania (Leishmania) infantum activity of some natural sesquiterpene lactones. Molecules 22, 685. doi:10.3390/molecules22050685
Yang, G., Lee, N., Ioset, J. R., and No, J. H. (2017). Evaluation of parameters impacting drug susceptibility in intracellular Trypanosoma cruzi assay protocols. SLAS Discov. 22, 125–134. doi:10.1177/1087057116673796
Young, D. C., Kingsley, S. D., Ryan, K. A., and Dutko, F. J. (1993). Selective inactivation of eukaryotic β-galactosidase in assays for inhibitors of HIV-1 TAT using bacterial β-galactosidase as a reporter enzyme. Anal. Biochem. 215, 24–30. doi:10.1006/abio.1993.1549
Keywords: Chagas’ disease, intracellular amastigotes, strain Dm28c, red-shifted luciferase, high-throughput assay, drug discovery
Citation: Quiroga C, Incerti M, Benitez D, Manta E, Medeiros A and Comini MA (2024) Development of bioluminescent reporter Trypanosoma cruzi and bioassay for compound screening. Front. Chem. Biol 3:1423430. doi: 10.3389/fchbi.2024.1423430
Received: 25 April 2024; Accepted: 27 May 2024;
Published: 17 June 2024.
Edited by:
Giuseppe Sancataldo, University of Palermo, ItalyReviewed by:
Jyothi Sistla, Stony Brook University, United StatesCopyright © 2024 Quiroga, Incerti, Benitez, Manta, Medeiros and Comini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Medeiros, YW1lZGVpcm9AZm1lZC5lZHUudXk=; Marcelo A. Comini, bWNvbWluaUBwYXN0ZXVyLmVkdS51eQ==
†Present address: Cristina Quiroga, Facultad de Ciencias de la Vida, Universidad Regional Amazónica Ikiam, Tena, Ecuador.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.