 Eduardo Sánchez-Lara
Eduardo Sánchez-Lara Roberto Favela
Roberto Favela Ivan Castillo
Ivan Castillo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Chem. Biol. , 11 March 2024
Sec. Bioinorganic Chemistry
Volume 3 - 2024 | https://doi.org/10.3389/fchbi.2024.1373647
This article is part of the Research Topic Advancing Polyoxometalates in Bioinorganic Chemistry: Applications and Biological Interactions View all articles
The design of molecular systems with capabilities to carry out the water oxidation reaction and thereby overcome the bottleneck of artificial photosynthesis is one of the scientific fields of most significant interest and urgency due to its potential to address energy demand and climate change. Nevertheless, the search for efficient and robust catalysts has been limited by the degradation of carbon-based ligands under oxidative conditions, leading to the search for fully inorganic catalysts. Polyoxometalates (POMs), an emerging class of carbon-free ligands with oxygen-enriched surfaces, offer a unique alternative as inorganic scaffolds to self-assemble and stabilize transition-metal clusters with unique redox properties. Under catalytic working conditions, POMs can undergo electron transfer reactions coupled to O2 formation without modifying their parental structure. As a result, these materials have recently entered the scene as catalytic players in designing new artificial photosynthetic platforms for water oxidation. We focus on the methods used to create these compounds, their unique structural characteristics, and how effectively they function as catalysts. We also explore the proposed mechanisms behind their ability to produce O2 and their potential use in designing photosynthetic devices.
There is a general consensus among climate scientists that human activities have contributed significantly to shifts in weather patterns (Lynas et al., 2021; Lloyd and Winsberg, 2018; Oreskes, 2004; Goudie, 2019). According to recent estimates and climate model simulations, these activities are closely related to a gradual increase in global temperature on Earth since the Industrial Revolution (
The main protagonist of these environmental changes is carbon dioxide (CO2) released into the atmosphere by burning fossil fuels (coal, oil, natural gas), used as non-renewable primary energy sources to drive and maintain modern societies’ development level. Chart 1 shows how these CO2 emissions have changed from the start of the Industrial Revolution to the present day (Ritchie et al., 2023; Yoro and Daramola, 2020; Soeder, 2021; Letcher, 2019).

CHART 1. Annual carbon dioxide (CO2) emission from fossil fuels and industry. It is interesting to note that during 2019–2020, there was a decrease in CO2 emissions worldwide of approximately 1.82 billion tons in response to policy restrictions imposed by world governments to counter the effects of the COVID-19 pandemic. Source: Our World in Data based on the Global Carbon Project (2022) (Ritchie et al., 2023).
Before the COVID-19 pandemic and according to the last IPCC information, atmospheric CO2 concentrations reached levels not seen in the previous 2 million years, and concentrations of short-lived climate forcers such as methane and nitrous oxide were higher than at any time in at least 80,000 years. These are the three greenhouse gases that have unequivocally contributed to the increase in global average temperature compared to levels observed before the development of industrial societies (Masson-Delmotte et al., 2021). If this scenario continues, we will reach a temperature increase of 4.4 °C by the end of this century, which will be irreversible for centuries and even millennia.
Faced with this challenging environment, finding renewable energy sources and technologies to exploit them to decrease our dependence on fossil fuels and reduce the effects of climate change has become one of the most critical challenges facing the world’s political and scientific agenda in recent years (Goudie, 2019).
Nuclear energy has been considered an important alternative to responding to future energy demand among the various renewable and exploitable energy resources (Fawzy et al., 2020; Prăvălie and Bandoc, 2018). It has begun to be seen as a potential short-term solution to reduce the effects of climate change. However, behind nuclear energy, some controversies remain regarding nuclear accidents and the fact that it has not been able to solve the radioactive waste problem. In addition, its use is linked to military activities. Therefore, efforts must be directed toward promoting and using eco-friendly renewable energy sources (Panwar et al., 2011).
Solar energy is another alternative source that will play a key role in decarbonizing global power systems in the coming decades (Panwar et al., 2011; Kannan and Vakeesan, 2016; Creutzig et al., 2017; Rabaia et al., 2021). Solar energy is plentiful, and as pointed out by the biochemist James Barber: Our Sun is the world’s most powerful energy source, providing the planet with more energy in a single hour than is currently derived from fossil fuels, nuclear power, and all renewable energy sources put together. Its energy supply is inexhaustible in human terms, and its use is harmless to our environment and climate (Barber, 2009; Barber and Tran, 2013).
Besides, the Sun can generate energy using conversion technologies that provide light, heat, electricity, and fuels (Crabtree and Lewis, 2007; Inganas and Sundstrom, 2015). Among these, solar fuels have the potential to be stored for future use and could be a substitute for fossil fuels (Nocera, 2017; Lewis and Nocera, 2006).
Photosynthesis is the example par excellence of absorbing, transferring, and converting solar energy into fuels as energy-rich molecules. Terrestrial plants, green algae, and cyanobacteria have performed this process for over 2 billion years, giving rise to the evolution of aerobic metabolism and complex life (Figure 1) (El-Khouly et al., 2017; Blankenship, 2022; Stirbet et al., 2020; Fischer et al., 2016). Despite oxygenic photosynthesis being the result of a long and challenging process of natural selection, it can be broken down into two fundamental chemical reactions: the oxidation of water to dioxygen (O2) and the reduction of carbon dioxide into organic matter (see Eqs (1), (2), respectively) (Blankenship, 2022; Krewald et al., 2015a). The first reaction occurs in the heart of photosystem II (PSII) (Brudvig, 2007), an enzyme complex consisting of several protein subunits located in the thylakoid membrane of chloroplasts of green plants and the inner membranes of cyanobacteria.
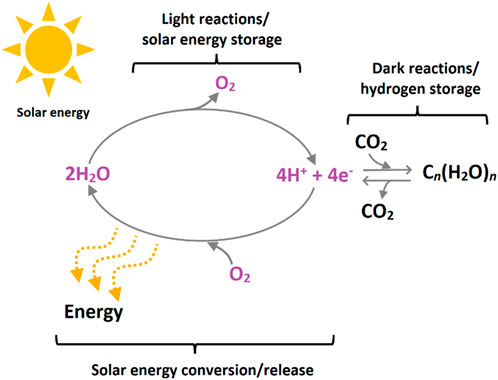
FIGURE 1. Energy flow in biology: photosynthesis transforms sunlight into chemical energy through light and dark reactions by splitting water into protons and electrons to release molecular oxygen and fix CO2 into sugar and other carbohydrates. By burning these energy-containing molecules through respiration or by combustion of carbon-based fuels, the opposite process of photosynthesis is set in motion, i.e., CO2 is released, and stored hydrogen combines with O2 to form water. Figure adapted with permission from (Barber, 2009)—copyright 2009 Royal Society of Chemistry.
The X-ray crystallographic resolution of the atomic arrangement of PSII has revealed that the active site responsible for the catalytic transformation of water into oxygen, protons, and electrons is a cluster of manganese, calcium, and oxygen atoms linked by μ-oxo bridges, named because of its activity as the oxygen-evolution center (OEC) (Ferreira, 2004; Umena et al., 2011; Young et al., 2016; Suga et al., 2015). In this way, whereas the oxygen released from the oxidation of water is available for metabolic processes in living systems and to support combustion reactions to sustain our daily lives, the electrons and protons can combine to form H2 or can eventually be used to chemically convert raw materials such as CO2 into energy-dense carbohydrates (equations (2) and (3) (Barber and Tran, 2013). The latter processes occur naturally in photosystem I (Blankenship, 2022; Nelson and Ben-Shem, 2005).
For CO2 fixation, nature has also developed biochemical processes such as the Benson-Bassham-Calvin cycle to reduce carbon dioxide into carbohydrates. Photosynthetic organisms transform about 1017 g (100 Gt) of CO2 into biomaterials and organic matter powered by the Sun (Brinkert, 2018).
The oxygen-evolving center is located at the membrane-lumen interface of the PSII and features four coordinated water molecules and several amino acid residues from the protein side chains (Ferreira, 2004; Umena et al., 2011). The water molecules surrounding the OEC and the amino acids that constitute a saturated ligand environment play a significant role in the stabilization and activity of the catalytic site toward the water oxidation process, which is driven by a photo-induced cycle based on five different oxidation states (Lubitz et al., 2019; Dau and Haumann, 2008). Due to the extraordinary catalytic ability of this small cluster to transform clean and abundant sources such as water, CO2, and solar energy into fuels, the OEC has inspired many bioinorganic chemists to rationally synthesize molecules that emulate not only its structural features but its catalytic properties towards water oxidation (Chen et al., 2021; García-Álvarez et al., 2021; Zhang et al., 2015; Sun, 2015; Chen et al., 2019; Chen et al., 2015; Yagi and Kaneko, 2000; Li et al., 2020; Mullins and Pecoraro, 2008; Yao et al., 2021; Zhang et al., 2021; Chen et al., 2022; Wiechen et al., 2014). Over time, this progress has allowed the integration of artificial photosynthetic models inspired by the chemical principles of nature.
Artificial materials with the potential to catalyze the conversion of water into O2 and reducing equivalents by mimicking the process of photosynthesis are known as water oxidation catalysts (WOCs). They can be broadly separated into metal oxides and molecular complexes (Yagi and Kaneko, 2000; Smith et al., 2013). Metal oxides were the first materials reported with the electrocatalytic ability for OER and can be traced back to 1903 with the early work of Cohen and Gläser about cobalt oxides (Matheu et al., 2019; Coehn and Gläser, 1902). By contrast, molecular complexes appeared on the scene until 1985, with the famous binuclear Ru-complex or blue-dimer with formula cis-[(H2O)Ru(bpy)2(μ-O)Ru(bpy)2(H2O)]+4, a compound featuring two [Ru(bipy)]3- units linked by an oxo bridge; where each ruthenium(III) ion is coordinated by two solvent molecules involved in the water oxidation process. Electrochemical insights of this complex showed a catalytic process similar to that of the natural OEC (Gilbert et al., 1985). However, two fundamental problems exist in many molecular WOCs: organic systems that interfere with the decrease in catalytic activity due to their oxidative degradation under typical experimental conditions and precious metals that are prohibitively expensive for large-scale artificial photosynthetic systems (Liu et al., 2018; Garrido-Barros et al., 2017; Limburg et al., 2012). Thus, one of the main motivations in the WOC community is the search for fully inorganic catalysts based on earth-abundant metals.
This review article focuses on a fascinating class of molecules that lie at the frontier between metal oxides and molecular complexes called polyoxometalates (POMs) and their role as water oxidation catalysts. In the broadest sense, POMs are discrete anionic clusters of transition metal ions self-assembled readily under one-pot reaction conditions (Long et al., 2007; 2010; Goura et al., 2021; Gumerova and Rompel, 2023). These molecules fall into two categories according to their structural composition: homo- and hetero-POMs, represented by the general formula {MmOy}n- and {XxMmOy}n-, respectively, where M are the addenda atoms such as V, Mo or W in their highest oxidation states, and X represents the heteroatoms, which are intentionally introduced into the framework during the self-assembly to modulate the degree of nuclearity of the polyoxometalate and its properties. The heteroatoms can be almost any element, but commonly found are atoms of Si, P, Al, As, etc.
Hetero-POMs are grouped into six to ten basic structures, from which they derive their huge structural diversity. The Keggin, Wells-Dawson, Anderson, Waugh, Silverton, and Lindqvist-type clusters are among the most representative structures (Wang et al., 2019; Ye et al., 2016; Li and Xu, 2011). Materials derived from Keggin and Wells-Dawson-type POMs have been used as multifunctional catalytic systems due to their high Brønsted acidity (Li et al., 2007; García-López et al., 2019; Mürtz et al., 2024). In general, polyoxometalate-based materials, and especially those incorporating redox-active transition metal ions, possess an enormous potential as water oxidation catalysts due to the following features (Bonchio et al., 2006; Proust et al., 2012; Zhang et al., 2024): (
The application of POMs as water oxidation catalysts became a significant issue for designing a purely inorganic catalyst based on a Ru-substituted polyoxotungstate, researched by two pioneering groups in 2008 (Geletii et al., 2008; Sartorel et al., 2008). Since then, a remarkable number of articles have emerged, and it is necessary to organize the most relevant information to provide the reader with a road map for further field development. This review presents a general overview of the water oxidation process performed in natural photosynthesis to analyze the structural and electronic features of the best WOC optimized over billions of years of biological evolution. Furthermore, a comprehensive section on the light-driven water oxidation processes catalyzed by TMSPs places readers on the timeline of these materials, discussing the synthetic strategies, structural features, and catalytic activity. Subsequently, these materials’ likely mechanisms of action are outlined, and finally, we report those artificial photosynthetic devices incorporating POMs.
Photosystem II is a multi-subunit enzyme in photosynthetic plants, algae, and cyanobacteria. It harnesses solar energy to drive a series of electron transfer processes that split water into its primary components: molecular oxygen, protons, and electrons (Figure 2). Although several medium-resolution 3D structures have been obtained, the catalytic site remained hidden for a long time due to structural distortion of the protein during X-ray data acquisition (Umena et al., 2011; Zouni et al., 2001; Kamiya and Shen, 2002). This problem has been gradually resolved by a series of high-resolution structures which, together with theoretical analyses, have provided new insights into the organization of the enzyme and the mechanistic pathway of light-driven water splitting preceding the formation of the O-O bond on the catalytic site.
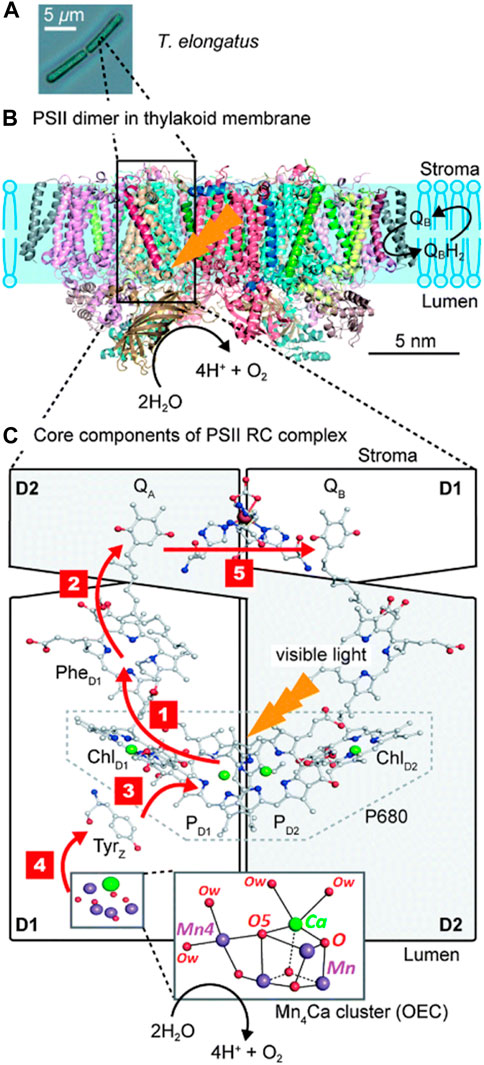
FIGURE 2. (A) The structural resolution of the photosystem II from the bacterium Thermosynechococcus elongatus has afforded a complete 3D picture of the enzyme (B). At the same time, the electronic steps involved in water oxidation have been clarified: the expanded view of the photosynthetic reaction center, highlighted with red arrows, shows the consecutive steps in which water oxidation occurs at the oxygen-evolving center (C). Step 1: charge separation prompted by electron transfer from a chlorophyll molecule (ChlD1) of the P680 system to a pheophytin (PheD1), resulting in the ion-paired system [P680•+
The X-ray diffraction of isolated photosystem II from the bacterium Thermosynechococcus elongatus at 3.5 Å resolution showed a 650.75 kDa transmembrane protein dimer. The structure comprises two nearly identical monomers divided by a double symmetry axis perpendicular to the lipid layer (Figure 2B) (Ferreira, 2004). According to the structural determination and the complete architecture of PSII, each monomer is composed of 19 protein subunits containing a significant amount of pigment molecules such as α-chlorophylls and beta-carotenes, also including molecules responsible for electron transfer processes such as heme groups, plastoquinones, pheophytin, a non-heme iron, and molecules incorporated during the crystallization process such as carbonate groups. The dimer also comprises two manganese-calcium-based clusters (Ferreira, 2004; Kato et al., 2014). The extrinsic subunits of the inner antenna, or CP43, are associated with the OEC proteins on the luminal side.
The photosynthetic process begins capturing sunlight through the antenna system (the light-harvesting complex or LHC), which is based on several hundred chromophores coupled electronically and integrated into protein molecules, including chlorophylls, carotenoids, and phycobilin molecules (Blankenship, 2022; Ferreira, 2004; Su et al., 2017). The light captured by these systems is funneled to the inner antenna located at the central core of PSII, built by a smaller number of chlorophylls and carotenoids related to the denominated CP43 and CP47 intrinsic proteins (Cardona et al., 2012; Mirkovic et al., 2017). Electronic excitation energy from the peripheral membrane antenna complex absorption is transferred and trapped in a photochemical reaction center, which is converted into electrochemical potential energy. The photosynthetic reaction centers are membrane-bound protein-pigment complexes in which the redox cofactors, i.e., chlorophyll, pheophytin, and quinone, involved in electron transfer are located perpendicular to a two-fold axis (Barber, 2003; Cox et al., 2013; Barber and Archer, 2001). When the excitation reaches a particular Chl a in the reaction center, instead of cascading the energy to a neighboring pigment molecule, an electron is transferred to a pheophytin molecule (PheD1), resulting in the first charge separation.
Chlorophyll a is part of a set of four molecules (labeled as PD1, PD2, ChlD1, and ChlD2 in Figure 2) grouped as the P680, or primary electron donor system, associated with the D1 and D2 reaction center proteins. The electron transfer pathways leading to water oxidation begin between the chlorophyll molecule (ChlD1) and pheophytin (PheD1), generating an unstable radical species or [P680•+
The oxygen-evolving center enters the scene by oxidizing a water molecule bound to the biological catalyst and the electron transferring it to a redox-active amino acid (TyzZ) of the D1 protein subunit, donating it to the P680 system, and leaving it stable and ready to absorb another photon (Figure 2C) (Vinyard et al., 2013; Barber, 2002; Lubitz et al., 2008; Nelson and Ben-Shem, 2005; Shen, 2015). Four light-driven electron transfer cycles lead to the oxidation of two water molecules with the simultaneous release of O2 to the atmosphere for the utility of biological systems and the formation of reducing equivalents in the form of energized electrons and protons for synthesizing energized molecules.
The crystal structure of PSII from the cyanobacterium T. elongatus at 3.5 Å resolution resolved the OEC as a distorted cubane-type [Mn3CaO4] cluster bonded to an external Mn (Mn4) through a single mono-μ-oxo bridge. In the X-ray crystal structure proposed by Ferreira and co-workers, the Mn─Mn and Mn─Ca distances are 2.7 Å and 3.4 Å, respectively, indicating that the elongated Mn … Ca interaction is weak, and this feature seems to play a structural role in the catalytic cycle. Concerning the protein residues, the [Mn4CaO4] cubane is wrapped by amino acids from the D1 and CP43 proteins that fulfill the function of biological ligands by binding directly to the cluster metals via the carboxylate groups of the amino acids or stabilizing it through hydrogen bonds or other intermolecular interactions (Barber and Tran, 2013; Crabtree and Lewis, 2007). The residual electron density around the cubane shows a bicarbonate molecule acting as a bridge between the Ca and Mn atoms, temporally replacing the water molecules that bind to the OEC in the active catalytic state.
Later, Umena and co-workers confirmed the distorted cubic geometry of the [Mn3CaO4] cluster at a resolution of 1.9 Å in PSII isolated from the thermophilic cyanobacterium Thermosynechococcus vulcanus but adding an oxo group that bridges an Mn atom of the cubane and the external Mn4, giving rise to a [Mn4CaO5] system organized in the shape of a distorted chair, where the cubane serves as the base of the seat and the external Mn4 and O4 atoms as the back of this rare asymmetric chair (Figure 2C) (Umena et al., 2011). This crystallographic study confirmed through electron density maps the hypothesis of Ferreira et al. that the Ca and Mn4 atoms are bonded to two water molecules each, suggesting that these solvent molecules may serve as substrates for the formation of the O─O bond on the catalyst. The environment surrounding the [Mn4CaO5] cluster does not differ much from that found in previous Ferreira’s structure. Again, some amino acids (glutamic and aspartic acids, for example,) orient their carboxylate groups towards the cubane to add stability, while others are in the second coordination sphere.
Two essential features explaining the unusual activity of the oxygen evolution center, particularly the inorganic [Mn4CaO5] cluster towards the water oxidation reaction, are its distorted polyhedral structure and unique redox chemistry. The differences between the bond distances within the cluster define its geometrical distortion. For example, in the structure proposed by Umena and co-workers, the distances around O5 are much longer compared to the other bond distances. This structural detail provides flexibility to the cluster during the catalytic process, and it has even been argued—considering the proximity of this atom to the water molecules on Ca and Mn4—that this O5 can protonate and form part of one of the substrates forming the O-O bond. From an electronic point of view, manganese has a wide range of oxidation states (Zhang and Sun, 2018; Krewald et al., 2015b; Kok et al., 1970; Armstrong, 2008). This feature is one of the reasons why nature chose this metal as the protagonist of photosynthesis since (as will be seen below) Mn rapidly changes its oxidation states during the catalytic cycle.
The steps through which oxygen evolution occurs are known as the Kok cycle or S-state cycle. It comprises four metastable intermediate states (S0, S1, S2, and S3) and a transition state, S4, through which O2 is released, and the catalyst is restored to its resting state (Ferreira, 2004; Armstrong, 2008; Cox et al., 2020; Kern et al., 2018; Pantazis, 2018). Although the PSII community has accepted the Kok cycle, the exact mechanism by which the coupling between the two water molecules in the S4 state occurs remains a mystery, leading to several proposals for the formation of the O-O bond, summarized in the oxo-oxyl coupling and the water nucleophilic attack or WNA (Shamsipur and Pashabadi, 2018; Najafpour et al., 2017; Suga et al., 2019; Vinyard et al., 2015).
In one scenario, a MnIV–oxyl radical reacts with an Mn–bridging oxo to generate the O—O bond. Some studies suggest that one of the substrate water molecules binds to the OEC during the S2 → S3 transition and is then oxidized to an oxyl radical in the S4 state to carry out the coupling and formation of the O―O bond. The second scenario aligns more with bioinspired WOCs, where a water molecule (possibly coordinated with a Ca2+ ion) attacks a terminal oxo (or oxyl) group in the OEC (Vinyard et al., 2015). While obtaining structural information on the S4 state has not been forthcoming, understanding the structural motions and reactivity of the oxygen-evolving center during the metastable states has shed light on the pathways leading to the formation and release of molecular oxygen (Pushkar et al., 2018; Capone et al., 2021). Recently, two independent research groups have used femtosecond X-ray crystallography to visualize the metastable states of the Kok cycle (Kern et al., 2018; Suga et al., 2019; Kupitz et al., 2014).
These structural studies appeal for the binding of an oxygen atom during the S2→S3 transition state from a nearby water molecule close to the OEC and agree that this oxygen can either act as one of the substrates for the formation of the O-O bond or replace the O5 position of the [Mn4CaO5] cluster during the release of molecular oxygen. In the latter case, the O-O bond formation may occur between the O5 and one of the available water molecules of the Mn4 or Ca atoms. These findings indirectly rule out the mechanism of water nucleophilic attack or the formation of a peroxo intermediate, supporting the possibility that the mechanism occurs via oxo-oxyl coupling (Kern et al., 2018). Finally, when O2 is released from the [Mn4CaO5] system, the S4 state decays to S0 with a new water molecule binding, rearranging its structure.
A question that arises from all the above is: is there a potential role for POMs in artificial photosynthesis? To address this issue, it is important to note that the term artificial photosynthesis has been used in several contexts. It is often used when an attempt is made to mimic any single stage of the natural photosynthetic process (Barber, 2009). In the broadest sense, natural photosynthesis is a process in which an organism captures and stores light energy, and the stored energy is used to drive cellular processes (Blankenship, 2022). Artificial photosynthesis, conversely, is based on the design of schemes to harness solar energy for technological purposes. Because this definition encompasses different scientific fields, “artificial photosynthesis” has been extended to any simple or complex method involving converting sunlight into useable energy. Most of these methods may or may not use the natural steps followed by photosynthesis, leading to the term being interpreted in different ways. With these definitions, we will attempt to answer the initial question.
The knowledge generated over the years about natural photosynthesis has provided a comprehensive picture of the process leading to water oxidation and CO2 reduction. However, the key to understanding the photosynthetic model lies in separating this complex photosynthetic puzzle into individual pieces and analyzing them separately (Barber, 2009; Gust et al., 2012; El-Khouly et al., 2017; Symes et al., 2013; Alstrum-Acevedo et al., 2005; Gust et al., 2001; Meyer, 1989; Kalyanasundaram and Graetzel, 2010; Concepcion et al., 2012). This strategy dramatically reduces the complex natural mechanism to its primary functional units and allows faster progress in the design of integrated artificial photosynthetic systems (Brinkert, 2018; Meyer, 1989). A typical artificial photosynthetic design includes antenna systems to harvest sunlight and generate an electrochemical potential, reaction centers for electron transfer and charge separation, and two catalytic systems for multi-electron redox processes (Barber, 2009; Blankenship, 2022; Meyer, 1989; Berardi et al., 2014; McConnell et al., 2010; Keijer et al., 2021). Individually, one catalytic unit is responsible for mimicking the function of the Mn-cluster, and the second catalytic unit is responsible for the fuel production via H+/CO2-reduction (Scheme 1) (Barber, 2009; Berardi et al., 2014). Therefore, when we mention artificial photosynthesis, we refer to a model that includes all the above characteristics.
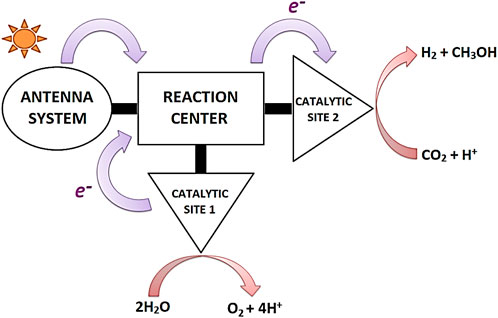
SCHEME 1. Integrated artificial device to produce clean fuels. Figure adapted with permission from (Symes et al., 2013). Copyright 2013 The Royal Society.
One of the critical issues in achieving the design of an integrated artificial device is developing modular and configurable routes to join the various components. Although there have been remarkable advances in this sense (Gust et al., 2012; Gust et al., 2001; Meyer, 1989; Keijer et al., 2021; Youngblood et al., 2009; Brimblecombe et al., 2010; Dogutan and Nocera, 2019; Imahori et al., 2003), one of the problems regarding catalysts is that they are based on expensive and rare metals such as platinum, iridium, or ruthenium, which, although having proved to operate at low overpotentials, are too costly and rare to be used in artificial photosynthetic systems for large-scale multi-electron redox processes (Gust et al., 2012; Berardi et al., 2014; Youngblood et al., 2009). Moreover, thermodynamically speaking, water oxidation is the main obstacle in the field of artificial photosynthesis, as it involves the removal of four protons and four electrons from two water molecules, together with the formation of an O─O bond on the catalyst (Du and Eisenberg, 2012). Therefore, the catalytic unit must be based on abundant and inexpensive elements and meet the molecular requirements to efficiently carry out the water oxidation mechanism.
Some POMs inspired by the cubane [Mn4CaO4] are used to overcome these obstacles. Due to their characteristics as heterogeneous catalysts, POM-based materials can be easily integrated into electrochemical devices capable of coupling water oxidation and proton reduction, achieving complete water splitting (Dashtian et al., 2024). This is the proposed role of POMs in the search for integrated photosynthetic systems. Considering that these molecules are revolutionizing the field of water oxidation catalysts, we describe the most recent advances in the following sections.
Adding to the brief introduction to POMs, it is important to note that photo-driven water oxidation is more efficient in a very attractive group of POMs denominated transition-metal-substituted polyoxometalates (abbreviated from here as TMSPs) (Han and Ding, 2018; Lauinger et al., 2017; Lv et al., 2012; Geletii et al., 2011). These compounds are obtained by combining lacunary POMs with transition-metal ions.
In the center of Scheme 2, we show the saturated structure of the Keggin type-anion, commonly referred to as α-[XM12O40]n-8; structurally, it consists of a central tetrahedral ion surrounded by eleven octahedra {MO6}. Depending on the degree of rotation (60°) of the four {M3O13} triads (each consisting of three edge-shared MO6 octahedra), the anion can be designated by the labels β-, γ-, δ-, and ε-. Often, these isomers can be surface decorated with metal coordination units (Weinstock et al., 1999; Cao et al., 2019). However, our interest is focused on lacunary species, where one or several polyhedral units of the addenda atoms (V, Mo, W, etc.) have been separated from their primitive structures.

SCHEME 2. Different lacunary POMs are derived from transformations of the saturated Keggin structure α-[XM12O40]n-8, where X=heteroatom and M=addenda atoms.
This removal results in cavity systems that can react as multidentate O-donor ligands toward various transition metal ions, replacing these empty sites. Most of the POMs discussed here are based on lacunary species of the Keggin-type anion (Scheme 2). The surprising range of TMSPs lies in the number of vacant sites available in the POM backbone, the type of transition metal that occupies these sites, and how these metals are incorporated into the native structure [Clemente-Juan and Coronado, 1999; Clemente-Juan et al., 2012; Das et al., 2020; Liu et al., 2016].
The intrinsic features of the TMSPs, derived from the combination of their components, allow them to acquire properties applicable in highly relevant research areas such as water splitting, catalysis, environmental science, magnetism, electronic materials, biomedicine, and electrochemistry, among others (Mizuno et al., 2005; Zheng and Yang, 2012; Gao et al., 2014; Wang and Yang, 2015; Bijelic et al., 2019; Horn et al., 2021). As water oxidation catalysts, TMSPs have played a significant role since they meet structural and chemical requirements that a WOC must possess, e.g.,: (
At the end of the 20th century, POMs were only used in organic catalysis despite featuring structural motifs in common with some Ru-based WOCs. It was not until 2004 that the POM community addressed the electrocatalytic generation of oxygen with a di-Ru-substituted polyoxometalate, which opened an attractive and multidisciplinary field of research (Neumann and Khenkin, 1995; Xinrong et al., 2000; Neumann and Dahan, 1997; Hill and Prosser-McCartha, 1995; Rüttinger and Dismukes, 1997). Electrochemical oxygen generation with this compound was monitored using a Clark electrode by differential pulse voltammetry in a phosphate buffer solution at pH 8. The synthesis strategy of the complex consisted of reacting Ru[CH2)2SO]4Cl2 with the polyoxometalate precursor Na12[WZnZn2(H2O)2(ZnW9O34)2], allowing the exchange of Zn(II) for Ru(III) ions and obtaining the system Na14[Ru2Zn2(H2O)2(ZnW9O34)2].42H2O (Ru2-ZnPOM) (Howells et al., 2004).
The X-ray diffraction technique revealed a system with a sandwich-type structure in which two [ZnW9O34]12- polyanions are assembled through a four-transition metal belt. This belt is formed by two Zn atoms connected to the two [ZnW9O34]12- polyanions through five oxygen atoms. At the same time, a peripherally coordinating water molecule accessible for catalytic processes occupied the sixth position (Figure 3A). On the other hand, the Ru(III) ions are placed at the center of this belt in a saturated octahedral geometry by the O atoms of the POMs scaffolds. In this scheme, the Ru-Ru distance is approximately 3.18 Å.
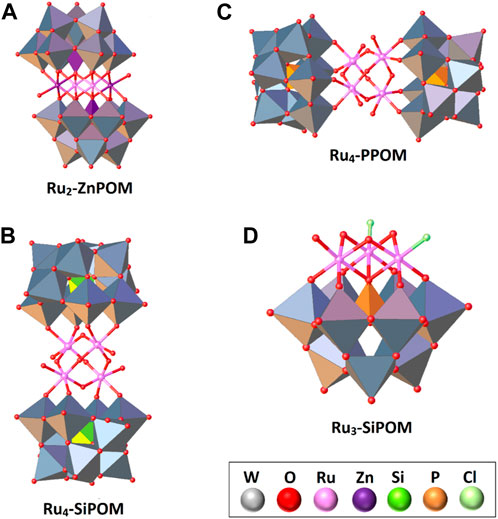
FIGURE 3. Schematic representation of selected Ru-substituted POMs. The POM-based scaffolds are shown in a polyhedral scheme, while the catalytic centers are in a ball-and-stick one. The inset shows the color code. For more information, see the text.
The authors focused on the structure-activity relationship showing that Ru-free or monosubstituted POMs with a Keggin-type topology [RuIII(H2O)PW9O39]4- did not exhibit the same activity toward O2 generation under similar experimental conditions, indicating that the sandwich-type structure allowing the incorporation of redox-active transition metal centers are fundamental to observe efficient catalytic processes (Table 1, entry 1) (Gao et al., 2020; Howells et al., 2004). Although the authors did not present experimentally supported mechanistic proposals, they pointed out that a possible route may involve redox processes during the catalytic cycle and the presence of oxygen species (OH− or H2O) as substrates for O2 generation.

TABLE 1. Water oxidation catalyzed by selected Ru-substituted POMs.
Considering the effect of Ru ions on the POM backbone, two research groups published 2008 a tetra-ruthenium POM with promising WOC activity. The complex with structural formula [RuIV4O4(OH)2(H2O)4(
The Hill group characterized the redox processes of this polyanion by analytical and electrochemical techniques. Cyclic voltammetry at acidic pH (0.1 M HCl) showed oxidation and reduction weaves corresponding to the {Ru4O6} cluster. In contrast, at pH 7 in a phosphate buffer and 0.6 mM Ru4-SiPOM, there was a significant increase in current at
On the other hand, the Bonchio group characterized the protonation states of the Ru4-SiPOM system by UV-Vis and acid-base spectrophotometric titration, confirming the observations of Hill’s group that the Ru(IV) centers in the central core undergo reversible protonation equilibria, which, added to the redox properties of ruthenium, explains the catalytic efficiency of this tetra-ruthenium POM. Water oxidation was evaluated by employing an excess of Ce(IV) salt as an oxidizing agent and Ru4-SiPOM at a concentration of 4.3 µmol in water at pH 6 (Table 1, entry 3). The O2 generation was monitored by gas chromatography, obtaining a yield of about 90%. IR and Raman spectroscopy confirmed the catalyst’s stability after the process.
In subsequent work, Geletti et al. demonstrated that the Ru4-SiPOM system integrated into an artificial photosynthetic scheme at physiological pH catalyzes the reaction 4) efficiently concerning the calculated quantum yield (Geletii et al., 2009). This system consisted of four primary components, i.e., a typical oxidizing agent ([Ru(bpy)3]3+), an electron acceptor (S2O82-), the ruthenium(IV) POM-based catalyst, and a Xe lamp as a visible light source (Table 1, entry 4). According to the authors, the quantum yield of the system (defined as O2 produced/quantum absorbed by the photosensitizer) was relatively high (almost 10%) compared to other photocatalytic systems based on molecular catalysts.
In this photosynthetic system, the photosensitizer [Ru(bpy)3]2+ acts as a visible light harvester, generating the [Ru(bpy)3]2+/[Ru(bpy)3]2+* system, which produces the oxidized [Ru(bpy)3]3+ species through photooxidation employing S2O82- as the oxidizing agent. The [Ru(bpy)3]3+ species oxidize the redox core of the polyoxometalate in a four-electron process, preparing it to oxidize two water molecules to generate O2 and reestablish the [Ru(bpy)3]2+ species (Geletii et al., 2009; Süss-Fink, 2008). Eq. 5 summarizes this process.
Bonchio and others performed kinetic studies on this type of complex photocatalytic process using time-resolved techniques (such as flash photolysis) to help clarify two physicochemical problems related to this light-driven scheme: (
With the results obtained with the system Ru4-SiPOM and interested in exploring potential structure-activity correlations in water oxidation, Besson and co-workers prepared the compound Cs9[RuIV4O5(OH)(H2O)4(γ-PW10O36)2]·17H2O (Ru4-PPOM), changing only the nature of the heteroatom (from Si to P) in the POM framework (Besson et al., 2010). The complex was obtained by reacting a cesium polyoxometalate salt, Cs7[γ-PW10O36]·H2O, with ruthenium trichloride in an acidic aqueous solution (pH 0.6) under ambient conditions. The X-ray crystal analysis showed comparable structural features with Ru4-SiPOM, i.e., a tetrahedral tetra-ruthenium(IV) oxo core sandwiched by two [γ-PW10O36]7- staggered Keggin-type anions (Figure 3C). However, an essential structure difference in charge and redox properties is that in Ru4-PPOM, the central [RuIV4O6] core was found to be monoprotonated (In Ru4-SiPOM, the central core is diprotonated). Nevertheless, the redox potentials of Ru4-PPOM and its analogous Si-POM were very similar.
The results of the photocatalytic water oxidation are shown in Table 1, entry 6. An interesting fact from this work was that the oxygen yield calculated from the sacrificial electron acceptor (S2O82-) consumption was 25% lower than that calculated for the Ru4-SiPOM compound under similar experimental conditions. Considering that the heteroatom is the only structural difference between the two catalysts, the authors addressed these contrasting results by analyzing the equilibrium constants of both systems in the limiting step leading to O2 evolution, i.e., in the final reaction RuV4 + 2H2O → RuIV4 + O2 + 4H+. This entire process is determined by four consecutive series of oxidations, including the gradual oxidation of the [RuIV4O6] system to its oxidized form [RuV4O6] by the photogenerated sensitizer [Ru(bpy)3]3+ (Han and Ding, 2018; Besson et al., 2010). For the silicon-centered system (pH 7),
In addition to tungsten-based polynuclear catalysts, some polyoxomolybdates have been electrostatically stabilized by Ru-based photosensitizers, allowing their catalytic activity for water oxidation to be studied. Incorporating a coordination complex in the POM scaffolds is a suitable strategy to obtain hybrid molecular systems in which the coordination units are grafted on the surface of POMs or act as counterions to stabilize the charge of the anionic POMs. In these systems, the organic ligands generally serve as neutral donors (Sánchez-Lara et al., 2021; Sánchez-Lara et al., 2024). A study by Gao and co-workers 2013 explored the correlation between oxygen evolution and the degree of nuclearity of three POM-ruthenium photosensitizer hybrids (Gao et al., 2013). The compounds were prepared by solvothermal conditions at 110 °C using (TBA)2[Mo6O19] and
The catalytic performance was investigated using the classical system, which employs a sacrificial agent, which, in this case, was S2O82-. An external source was not used as a photosensitizer because the compounds already include ruthenium-based II) light harvester systems in their crystal structures. The results concerning O2 generation appeared to depend on the size of the POM cluster, i.e., the catalytic efficiency obeyed the following order: α-[Mo8O26]4-
The authors propose that the mechanism of action is related to a radical cation process that takes place through the transfer of an electron from the peripheral bonds Mo=O to the [Ru(phen)3]2+ system, generating radicals on the surface of polyoxometalate Mo-O+• and oxidized species [Ru(phen)3]3+ (Gao et al., 2013). The oxidizing potential of Mo-O+• is high enough to produce O2 from water. This hypothesis was supported using free radical scavengers such as hydroquinone.
In 2012, Patzke and others conducted a complex comparative analysis between three complexes in which the redox-active core is exposed or intercalated by robust Keggin-type ligands (Car et al., 2012). The system analyzed as a possible WOC was a trivacant triruthenium-substituted POM [{Ru3O3(H2O)Cl2}(SiW9O34)] (Ru3-SiPOM) and its catalytic activity was compared with two cobalt and nickel sandwiched POMs with formulae [Co4(H2O)2(SiW9O34)2] (Co4-SiW9) and [Ni4(H2O)2(SiW9O34)2] (Ni4-SiW9), respectively. Ru3-SiPOM was obtained from an acidic aqueous solution (pH 2.5) of RuCl3·nH2O with the sodium salt α-Na10[SiW9O34]·15H2O. Precipitation of Ru3-SiPOM as a black solid was favored upon adding an excess of KCl to the solution. Due to the low tendency that presents the open systems to crystallize, the structure was proposed based on computational methods as a small [{Ru3O3(H2O)Cl2}(SiW9O34)]7- anion where the three Ru atoms are arranged in a Ru3O3Cl cluster and fused to the trivacant [SiW9O34]10- scaffold. As seen in Figure 3D, the catalytically active Ru ions are exposed on the surface of the POM, replacing some tungsten atoms of the native Keggin structure.
As in the previous cases, the light-driven catalytic activity of varying amounts of the three compounds was studied with the system containing [Ru(bpy)3]2+ and S2O82- as photosensitizer and sacrificial electron acceptors at pH 5.8 using Na2SiF6 instead of phosphate buffer to avoid decomposition of the sensitizer. Some general data, along with TON and TOF values, are summarized in Table 1 (entry 7). With this photocatalytic system, the best results were observed at a concentration of 50 µM catalyst (Ru3-SiPOM), reaching a TON value of 23 after a 2-h exposure. The authors noted that this value is lower than the tetra-ruthenium(IV)-oxo system. Interestingly, the polyoxometalate Co4-SiW9 with a sandwich-type structure showed similar activity since the TON and TOF values were very close to those observed for the Ru3-SiPOM system; the isostructural Ni4-SiW9 compound showed no activity under the same experimental conditions, indicating that the open structure and the nature of the transition metal favors catalysis.
The authors performed reference experiments by modifying the catalytic conditions to investigate whether the Ru3-SiPOM system is present as the species responsible for water oxidation throughout various photocatalytic processes. Reference experiments in buffer solutions (Na2SiF6 pH 5.8) supported by DLS and TEM confirmed the integrity of Ru3-SiPOM, as no heterogeneous particles or by-products were identified under the experimental conditions. Furthermore, additional experiments with the RuCl3·nH2O salt as a potential decomposition material of POM did not lead to O2-evolving under the experimental conditions under which the catalysis was carried out.
On the other hand, a brown precipitate was identified under dark conditions while purging the headspace vial after sensitizer addition. According to other works (Natali et al., 2012a), this pointed to the formation of an adduct between the anionic Ru3-SiPOM complex and the cationic photosensitizer (PS), Ru3-SiPOM/PS. The formation of Ru3-SiPOM/PS was corroborated by adding a solution of Ru3-SiPOM to a [Ru(bpy)3]Cl2 buffered solution (Car et al., 2012). The catalytic studies indicated that the Ru3-SiPOM/PS adduct was efficient under illumination and dark conditions, carrying out a redox process via four consecutive cycles that lead to the formation of the {RuV3SiPOM-PS} oxidizing species, which in turn oxidizes water to restore the POM/PS adduct.
Over time, research on these materials, particularly the Ru4-SiPOM system, has focused on their immobilization on the electrodes or carbon nanotube surfaces to develop modified electrochemical systems capable of performing heterogeneous catalytic processes, including water splitting (Toma et al., 2011; Anwar et al., 2014).
In the following sections of this review, we will address the cases where Earth-abundant transition metals (Co, Ni, Mn, Cu, and Fe) have been used as redox metal clusters self-assembled by robust POM ligands. Considering that Co-based systems have been appearing significantly in the specialized literature, we have decided to start with this metal ion and continue with the other transition metals according to their status in the research on this attractive field.
A good start to addressing the field of Earth-abundant transition-metal substituted POMs with catalytic capabilities for water oxidation is the famous cobalt system Na10[Co4(H2O)2(PW9O34)2] (Co4-PPOM) synthesized and structurally characterized for the first time in 1973 (Weakley et al., 1973), and re-investigated by Hill and others in 2010 as a promising WOC (Yin et al., 2010). Before this work, some Co-substituted POMs with catalytic activity for organic oxidations had already been reported, while in the water oxidation field, some cobalt oxides CoOx had shown good activity towards water oxidation under heterogeneous conditions (Kholdeeva, 2004; Tang et al., 2010; Kanan and Nocera, 2008; Jiao and Frei, 2009). Therefore, the Co4-PPOM system filled a gap in homogeneous catalysis and was a breakthrough in synthesizing schemes based on 3 days elements. Co4-PPOM was synthesized under mild conditions in a one-pot process by mixing the corresponding precursor salts based on W, P, and Co in water. The resulting suspension was heated to reflux before adjusting the pH to 7. The solution was then saturated with sodium chloride to shift the equilibrium toward product formation.
As shown in Figure 4A, the main feature of the [Co4(H2O)2(PW9O34)2]10- polyanion is a flat central cluster featuring four cobalt atoms in an octahedral environment, resembling a fragment of a composite layer in Co(OH)2, separating two moieties of the anionic {PW9O34}9- scaffolds. The reader may notice that the POM anion in the empirical formula is the same as that used in Ru4-PPOM, as described in the previous section. An essential structural characteristic for catalytic water oxidation processes is that the sixth coordination position on the two external Co atoms of the central cluster is completed by a water molecule shielded by the POM anion that, as will be seen later, plays a pivotal role in the water oxidation mechanism. In the crystal structure, ten Na+ ions placed in available positions in the crystal lattice stabilize the high anionic charge of Co4-PPOM.
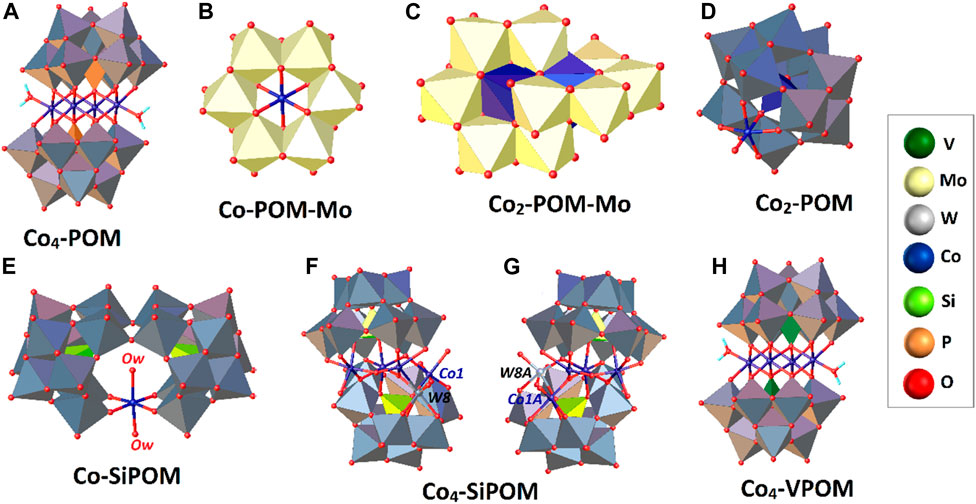
FIGURE 4. Schematic representation of selected Co-substituted POMs. The POM-based scaffolds are shown in a polyhedral scheme, while the catalytic centers are in a ball-and-stick one. The inset shows the color code. For more information, see the text.
To gain insight into the O2 generation, the authors compared the catalytic behavior of the Co4-PPOM complex with seven different Co-substituted POMs, featuring a variation of the Co core structure and the type of POM framework. In the cyclic voltammogram (CV), the complex showed a significant increase in the catalytic current at low overpotential (1.2 V vs Ag/AgCl) in phosphate buffer (pH 8) at 1 mM Co4-PPOM. This current was attributed to water oxidation. Interestingly, this catalytic phenomenon was not observed for the other cobalt POMs because they were not stable under the experimental conditions for water oxidation. Under specific requirements given in Table 2 (entry 1), a high TON value was reached (>1,000 in ca. 3 min), which is one of the highest values reported for a molecular WOC.

TABLE 2. Water oxidation catalyzed by different Co-substituted POMs.
The stability of Co4-PPOM was confirmed through several spectroscopic and electrochemical techniques such as UV-Vis, 13P NMR, IR spectroscopies, and CV before and after catalysis and over time. Poisoning experiments were also carried out using bipyridine as a chelating agent of potential free Co2+ released if catalyst decomposition occurs. Since no significant changes in the catalytic activity of water oxidation were observed, it was postulated that polyoxotungstate ligands prevent the formation of Co particles. Some theoretical calculations supported the thesis that POM is almost inert during catalysis and stabilizes the central Co core. This last argument justifies using polyhedral structures based on POMs to stabilize active sites where water oxidation occurs.
An intriguing experimental debate that began by questioning the role of Co4-PPOM as a true molecular catalyst highlighted the importance of distinguishing the catalytic activity of the parent compound and its potential decomposition products formed under the strongly oxidizing conditions required, especially when these products are active species with water oxidation capabilities. In this sense, Stracke and Finke proposed, through an electrochemical approach supported by several spectroscopic and microscopic tools such as UV-Vis, SEM, and EDX, that the catalytic activity observed for the Co4-PPOM compound was due to the in situ deposition of cobalt particles (oxide or hydroxide solids) on the working electrode surface under oxidizing conditions (
In response to these concerns, Hill, Geletti, et al. proposed the following experimental approaches:
A set of studies reported between 2012 and 2014 confirmed that including a polynuclear cobalt, core is not essential to observe water oxidation catalysis by showing that POMs containing mono- and di-cobalt cores can exhibit water oxidation. First, Sakai and co-workers evaluated in 2012 the WOC activity of two Co-containing Anderson- and Evans-Showell-type anions, i.e., [CoMo6O24H6]3- (Co-POM-Mo) and [Co2Mo10O38H4]6- (Co2-POM-Mo), respectively (Tanaka et al., 2012). Both anions are used extensively as starting materials to produce a variety of salts and functionalized metal complexes (An et al., 2017; Sang et al., 2018). In the [CoMo6O24H6]3- anion, six assembled {MoO6} octahedral units are arranged around a central CoO6 system to form the Anderson-type polyoxometalate in Figure 4B, while the [Co2Mo10O38H4]6- anion can be seen as a dimer of two monolacunary Anderson [CoMo5H2O19]3- clusters, assembled via two hexacoordinate CoO6 units with shared edges, producing an acute angle of nearly 45° between them (Figure 4C) (An et al., 2017).
Catalytic studies showed that these small Mo-based POMs have capabilities for O2 generation at basic pH under the typical visible light-driven system based in the [Ru(bpy)2]2+/Na2S2O8 pair. The control experiments carried out with Co-free polyanions, [PMo12O40]3- and [Mo7O24]6-, did not show catalytic activity, indicating that the mononuclear cobalt sites are responsible for O2 formation (Tanaka et al., 2012). The highest TON values for these two polyoxometalate systems at
Subsequently, a dinuclear Keggin-type POM doped with two mixed-valence cobalt atoms (CoIII/CoII) was reported in 2013 as an efficient molecular WOC using a typical photocatalytic setup (Song et al., 2013). As noted in Figure 4D, the complex is compatible with the structural formula K7[CoIIICoII(H2O)W11O39]·15H2O (Co2POM), where one CoIII ion was incorporated within the POM backbone acting as a central heteroatom, and the CoII ion replaces one of the addenda W atoms in a peripheral position. The compound was synthesized in several steps using a methodology previously reported by Baker and McCutcheon (Baker and McCutcheon, 1956).
The catalytic activity of Co2-POM was investigated and compared under the same conditions (Table 2, entry 11) with four Co-substituted Keggin anions, two of which exhibited the formula [Xn+Co2+W11O39](12-n)- (XW11; Xn+ = Si4+, and P5+), and the other two were analogs of Co2-POM, i.e., [Co2+W12O40] and [Co3+W12O40]. Of all these complexes, only the disubstituted Co2-POM system showed catalytic activity (Table 2, entry 11), suggesting that the two Co atoms’ redox properties in the POM network favored the water oxidation processes. However, considering the other POMs established as control groups did not exhibit any catalytic activity, the experimental conditions set for a particular compound may not be suitable for other cobalt/POM systems (Song et al., 2013). Five stability studies using various analytical techniques (e.g., DLS, cyclic voltammetry, catalyst recycling studies, etc.) confirmed that the Co2-POM compound is responsible for the activity observed under the typical photocatalytic scheme.
Finally, the complex with formula K10[Co-(H2O)2(γ-SiW10O35)2]23·H2O (Co-SiPOM) reported in 2006 by Ulrich Kortz’s group, who studied it from a structural and electrochemical point of view (Bassil et al., 2006), was re-investigated by Ding’s group in 2014 as a potential WOC (Xiang et al., 2014). The X-ray crystal structure of this polyanion is shown in Figure 4E; an accessible octahedral CoII ion was incorporated into the framework of the lacunary polyoxometalate [Si2W20O70]12- through four W-O-Co μ2-oxo bridges. Interestingly, the inner coordination sphere of this central core is saturated with two possibly active water molecules located axial to each other. The water oxidation catalyzed by this compound was characterized by CV in borate buffer (pH 8), observing a catalytic current at
The O2 generation was performed in the ordinary photocatalytic system using a visible light source, the Ru(bpy)3]2+ complex, and the electron acceptor Na2S2O8. The activity was compared with two structural analogs based on Ni and Mn and six mono, di, or tetra cobalt-substituted POMs, including the Co4-PPOM. In summary, the Co-SiPOM catalyst was active toward O2 evolution with results comparable to the Co4-PPOM compound (Table 2, entry 12). At the same time, the Ni and Mn analogs and the other systems evaluated did not show any catalytic activity under these experimental conditions. As in the previous cases, the stability of the catalyst against hydrolysis was studied in detail using various practical approaches.
The water oxidation activity of these four POMs featuring mono and dicobalt cores was influenced by the CoII/III system’s intrinsic redox properties and the coordination environments imposed by the polyoxometalate-based ligands. In Co-SiPOM, the Si heteroatom does not appear to be essential for water oxidation; however, in Co-POM-Mo, Co2-POM, and Co-SiPOM, where external cobalt ions are accessible to water molecules, the catalytic activity seems to be higher. On the other hand, the water oxidation activity of Co2-POM-Mo and Co2-POM seemed to improve through an electron transfer between the two Co ions incorporated in the POM scaffold.
In parallel with these efforts, some works were conducted to study the effect of replacing the PO43- group in Co4-PPOM on its electronic and catalytic properties. Hill and co-workers first attempted such a direction by keeping the same tetranuclear cobalt core but using the Si-centered POM used in Ru4-SiPOM. However, the authors failed to obtain crystals of the desired isostructural complex ([Co4(H2O)2[
The activity of Co4-SiPOM was examined for light-driven water oxidation at pH seven to nine using the typical photocatalytic platform. From the kinetics of O2 formation, its yield increases at higher pH, with a maximum TON value of
Subsequently, a POM isostructural to the previously discussed Co4-PPOM was reported, which showed remarkable water oxidation activity (Table 2, entry 14) (Lv et al., 2014). The structure of this compound retains the same central {Co4} core as its analog phosphate-centered Co4-PPOM. However, the phosphorus heteroatom was replaced by the tetrahedral vanadate(V) ion, resulting in a different structural formula Na10[Co4(H2O)2(VW9O34)2]·35H2O (Co4-VPOM; Figure 4H).
Although both compounds exhibit the same structural arrangement, they display different electronic structures due to the redox properties of vanadium, influencing the catalytic performance. DFT calculations on the crystal structure of the Co4-VPOM system supported this hypothesis, which showed that the HOMO level consists mainly of d orbitals from Co2+ ions, with some mixing of the oxygen orbitals of the inorganic tungsten fragments [VW9O34]. In contrast, the LUMO orbital consists mainly of the 3 d orbitals of vanadium with some mixing of the cobalt central core. The fact that vanadium works with a d0 electronic configuration allows for electronic transfers and redox processes that were not observed for the Co4-PPOM system. Magnetic studies established the differences in the electronic features, revealing the ferromagnetic nature of Co4-VPOM. A more complex analysis of the charge transfer phenomena in this compound was studied using two X-ray spectroscopic techniques and published elsewhere (Liu et al., 2018).
Although we focus this review on light-driven schemes, it is important to note that heterogeneous water oxidation catalysis, a system where the POMs appear stable and robust, has been reported. This methodology is helpful in poorly soluble systems or where it is necessary to avoid the formation of metal particles (e.g., CoOx) due to the inherent decomposition of the catalyst under oxidizing conditions. Carbon pastes modified with cobalt-substituted POM have shown a strong electrochemical oxidation wave, which is absent in pure carbon paste electrodes under the same catalytic conditions, suggesting the participation of catalytic processes promoted by the Co-POM components (Haider et al., 2019; Arens et al., 2020; Soriano-López et al., 2023). This versatility of exploring catalytic water oxidation using various methodologies favors exploring artificial photosynthetic systems, a topic we study below.
One of the most significant advances in designing abundant transition metal-substituted POMs is the self-assembly and stabilization of clusters with bio-inspired cubic topologies akin to the manganese catalytic center in PSII. In this context, Han and others described in 2014 four cobalt molecular photocatalysts, i.e, [(Co4(OH)3(PO4)}(SiW9O34)4]32- (CoPi-SiPOM), [{Co4(OH)3(PO4)}(GeW9O34)4]32- (CoPi-GePOM), [{Co4(OH)3(PO4)}4(PW9O34)4]28- (CoPi-PPOM) and (Co4(OH)3(PO4)4}(AsW9O34)4]28- (CoPi-AsPOM), which have in common a central core with a cubic structure reminiscent of the natural photosynthetic center [Mn3CaO4] of PSII (Han et al., 2014). The compounds were synthesized at basic pH using a one-pot approach using abundant and economically accessible salts (e.g., CoCl2.6H2O and Na3PO4.12H2O). As can be seen from the chemical formulae, the same POM framework (XW9O34)n- was used, but the heteroatom was different in each complex (X = Si, Ge, P, and As).
The main structure of these complexes consisted of a tetranuclear cobalt/phosphate {Co16(PO4)(OH)12} center, enclosing a {Co4O4} cubane-type cluster; at its vertices defined by O atoms, four tetrahedral phosphates are linked. Additionally, the cubane is surrounded by four {Co3} cobalt units, as shown in Figure 5A. The central {Co16(PO4)(OH)12} unit is intercalated by four trivacant [XW9O34]n- anions (X = Si, Ge, P, and As) that confer stability and form a structural scheme with a symmetry that is close to Td (Figures 5A,B). The Co2+ ions have an irregular octahedral geometry with short Co-O bonds in the range 2.064(9)-2.332(10) Å.
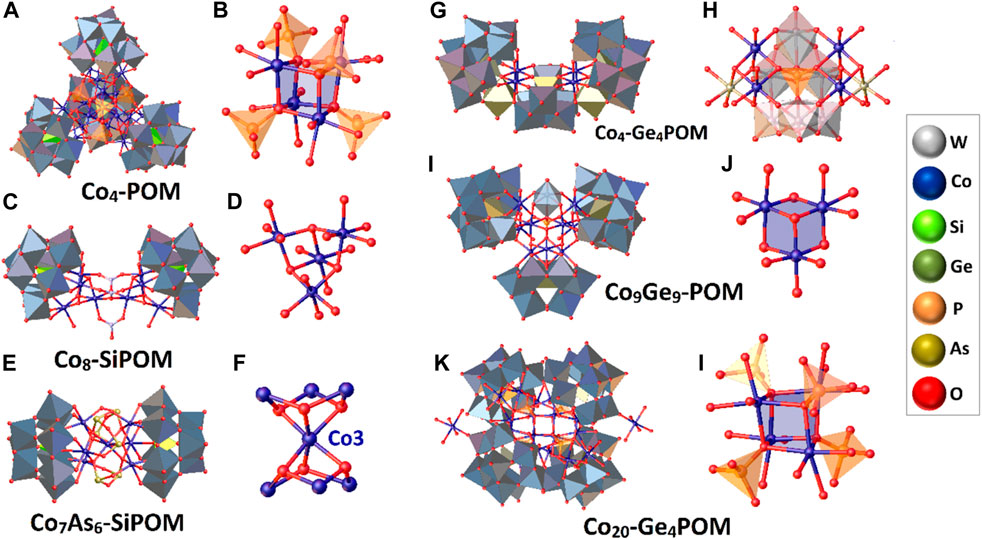
FIGURE 5. Schematic representation of selected Co-substituted POMs. The POM-based scaffolds are shown as polyhedra, while the cobalt active sites are represented as ball-and-stick. The inset shows the color code.For more information, see the text.
The authors used the photocatalytic system based on [Ru(bpy)3]2+/S2O82- in buffer solutions in a pH range between seven to nine and at different catalyst concentrations to study photocatalytic water oxidation. The trend in catalytic performance under visible light was CoPi-PPOM ≤ CoPi-SiPOM < CoPi-AsPOM < CoPi-GePOM, with the highest TON value of 38.7 (Table 2, entries 15–18) (Han et al., 2014). These results highlight the non-innocent role of the heteroatom in the redox processes leading to water oxidation. To confirm the protection conferred by inorganic ligands, the authors performed a series of experiments to detect species released by the structural decomposition of these complexes. Experiments using DLS ruled out the formation of Co nanoparticles after the catalytic process. At the same time, 31P NMR showed no shifts, suggesting decomposition processes of the polyoxometalate or the central tetrameric core stabilized by the phosphate groups. On the other hand, isolating the POMs from the solutions using bulky organic cations allowed the determination of the concentration of remaining cobalt in solution below 1 μM, which does not account for the observed water oxidation processes.
Qiao and co-workers reported an isostructural complex in which the PO43- groups were replaced by the tetrahedral VO43- anion, resulting in the high-nuclear mixed metal–oxo cluster-containing POM, [{Co4(OH)3(VO4)}4(SiW9O34)4]32- (Qiao et al., 2019). This molecular catalyst showed efficient visible-light-driven CO2 reduction, which suggests using POMs in artificial photosynthetic platforms to support the multielectronic catalytic processes required for water oxidation and CO2 reduction reactions.
Inspired by the OEC in photosystem II, Ding and co-workers achieved a milestone in the field of POM-WOCs by reporting a cobalt-substituted POM, [(A-α-SiW9O34)2Co8(OH)6(H2O)2(CO3)3]16- (Co8-SiPOM), with high hydrolytic stability and a very high TON value (Figure 5C) (Wei et al., 2015). The complex crystallized at pH 9 from a sodium acetate solution containing Na10[A-α-SiW9O34], CoSO4, and K2CO3. A single-crystal was analyzed through X-ray crystallography, revealing two symmetry-related central {Co4O3} cores bridged by two carbonate CO32- ligands and stabilized by two trivacant lacunary [A-α-SiW9O34] systems; each cobalt {Co4O3} core incorporated one aqua ligand (Figure 5D). The Co
Experiments to determine the catalytic activity were carried out at pH eight to nine in a borate buffer solution using the above system (1 mM of the photosensitizer, 5 mM of sodium persulphate, and 2 μM of the catalyst). From the O2 production kinetics, it was observed that at pH 9, higher catalytic activity was obtained (43.6% O2 yield and a TON value of 545). In contrast, at pH 8, the O2 generation decreased by 50% (with a TON of 256), indicating that a slight pH variation can considerably influence water oxidation activity. Experiments varying the catalyst concentration determined that at 0.5 μM of Co8-SiPOM, the highest kinetic values recorded among this type of molecular catalysts (Table 2, entry 19) are achieved compared to other systems enclosing cubane-type metal clusters. However, comparisons can only be considered indirect since the catalysts have been studied under different experimental conditions. Stability studies show Co8-SiPOM was oxidatively and hydrolytically stable (Han and Ding, 2018; Wei et al., 2015).
Following a similar approach to that described above, a cobalt-arsenic core with a double quasi-cubane arrangement stabilized by two POM ligands [{Co7As6O9}(A-α-SiW9O34)2]12- (Co7As6-SiPOM) was synthesized in 2016 (Figure 5E) (Chen et al., 2016). Light-violet crystals were obtained at room temperature following a one-pot synthetic strategy: addition of sodium arsenite NaAsO2 to a basic solution containing CoCl6.6H2O and Na10[A-α-SiW9O34].18H2O, allowing the incorporation of As3+ ions into the central core. The crystal structure consists of a rare asymmetric {Co6As6O9(OH)6} fragment, which encloses a seventh CoII atom (Co3) with a distorted octahedral geometry. Furthermore, the three edge-shared {CoO6} units that complete the {Co3SiW9} Keggin-type structures form two quasi-cubane fragments related through the central Co3 atom (Figure 5F), generating a unique structural motif intercalated by two
The water oxidation activity was tested under photochemical conditions by irradiating with visible light a basic solution (borate buffer, pH 8) containing the cobalt-arsenic catalyst (1 µM), the photosensitizer (1 mM), and sodium persulphate (50 mM). This photocatalytic system, which we will analyze later, favors oxidation processes leading to water splitting by the POM system. Under the experimental conditions shown in Table 2, entry 20, a TON value of 115.2 and an initial TOF of 0.14 s-1 were achieved. The maximum values for O2 yield (38.4%) and O2 evolution amount (19.2 μmol) were obtained for 20 μM of Co7As6-SiPOM after 60 min illumination. Above 30 µM of the catalyst, an insoluble solid appears in the solution caused by the ion pairing between the anionic polyoxometalate and the photosensitizer, decreasing water oxidation activity. As will be seen in other cases, this phenomenon seems to be expected when using the oxidizing agent [Ru(bpy)]2+ and high catalyst concentrations.
The catalytic activity under the same experimental conditions was comparable to that of Co8-SiPOM, possibly because both compounds contain similar cubane-type clusters, i.e., a double-quasi-cubane core stabilized by two robust POM ligands. However, Co7As6-SiPOM exhibited better activity than other cobalt-based catalysts featuring cubane or quasi-cubane cores, including CoPi-SiPOM that features a {Co4O4} moiety (Chen et al., 2016). Additionally, the photocatalytic activity of Co7As6-SiPOM appears to be twice that of redox-active Co4-VPOM under similar experimental conditions. Multiple stability analyses on Co7As6-SiPOM concluded that this compound is the dominant catalyst.
Bonchio group reported in 2017 a series of cobalt complexes stabilized with heteroatoms derived from phosphate and silicon incorporated into polyoxotungstate networks (Natali et al., 2017). Cobalt central cores were differentiated by the degree of nuclearity and the number of coordination-active water molecules, namely, Co9 =Co9(µ-OH)3(H2O)6(HPO4)2; Co15 = {Co9(µ-OH)3(H2O)9Cl2} and six Co(H2O)5 units at peripheral sites of the POM surfaces; Co16 = {Co16(µ-OH)12(PO2)4}. The reported systems were structurally stable in water and examined as WOCs in phosphate buffer at pH 8 in the typical sacrificial cycle, Ru(bpy)32+/S2O82-. The authors studied in detail the formation of Ru(bpy)32+/POM ionic pairs, observing that the charge of the anion is involved in this association, decreasing the emission of the photosensitizer, which was resolved by modifying the ionic strength of the solvent. The best results regarding oxygen evolution (dependent on concentration) were observed for Co15, which contains the most significant amount of coordination water molecules concerning Co9 and Co16. Flash photolysis studies confirmed this trend.
Three POM-WOCs with non-, quasi-, or complete {Co4O4} units structurally similar to the oxygen-evolution center were recently prepared (Al-Sayed et al., 2021). The complexes with empirical formulae [Ge4PCo4(H2O)2W24O94]·38H2O (Co4-Ge4POM), [Ge3PCo9(OH)5(H2O)4W30O115]·45H2O (Co9-Ge3POM) and, [Ge4P4Co20(OH)14(H2O)18W36O150]·61H2O (Co20-Ge4POM) were obtained at neutral pH combining the POM precursor salts, GeO2/Na2WO4.2H2O, K8[γ-GeW10] and K8Na2[α-GeW9]·25H2O with CoCl2 and Na3PO4 in water. The solutions were heated at 80 °C/10 min to activate the PO43- groups. Single crystals of these complexes were obtained after filtering and cooling the reaction mixtures to 20°C.
The crystal structure of Co4-Ge4POM can be considered a dimeric double-sandwich structure, in which two [GeW9O34]9- anions and a central [PW6O26]11- bridge are separated by two {Co2GeO13} units (Figures 5G, H). In Co6-PPOM, two {Co2GeW9} polyanions and one {Co3GeW9} fragment are bridged via three O atoms from a phosphate group. The fourth O atom of this ligand is linked to two octahedral {CoO5(H2O)} units and a single [WO6] site, building a {Co3O4(H2O)2} partial cubane structure, with two exchangeable water molecules as potential substrates for oxidation (Figures 5I, J) (Al-Sayed et al., 2021). Finally, the structure of Co20-Ge4POM is a tetrameric unit composed of a {Co4O4} cubane-type core stabilized by four PO43- groups and sandwiched by four robust
The O2 evolution kinetics for the three POM systems were evaluated in borate buffer solutions under the conditions indicated in Table 2, entries 21–23. In this way, Co9-GePOM and Co20-GePOM exhibited TONs of 21.6 and 20.4, while Co4-GePOM reached a TON of 11.30, comparable to the non-cubane Co4-PPOM catalyst (TON 10.90 and TOF 0.015 s-1) under the same conditions. The TOF values for Co4-GePOM, Co9-GePOM, and Co20-GePOM were 0.047, 0.069 and 0.060 s-1, respectively. Studies at different catalyst concentrations (2
On the other hand, pH-dependent studies (pH = 7.5–9) established that at pH 8.5, the WOC activity had a better catalytic performance, with a decrease in activity at pH 9.0 due to the decomposition of the [Ru(bpy)]2+ system. The pH effect on catalytic activity generally followed a trend according to the protonation state and nuclearity of the central units stabilized by POMs: Co4-GePOM
Hill’s group made the first report on Ni-substituted POMs in 2012. The complex was synthesized by reacting the POM precursor salt Na10[SiW9O34] with NiCl2-6H2O and KCl in an aqueous medium at pH 6.8; again, the addition of the alkaline salt to favors the stability of the anionic polyoxometalate and its subsequent crystallization from the mother solution (Zhu et al., 2012a). Green needle crystals obtained by slow evaporation under ambient conditions were studied by X-ray diffraction, revealing a Wells-Dawson-type open structure [Si2W18O66]16- composed by the assembly of two [A-β-SiW9O34]10- systems communicated through a pair of W-O-W bridging bonds. The resulting open structure allowed the incorporation of a central pentanuclear core {Ni5(OH)6(OH2)3}4+, which is of particular interest for water oxidation processes because the metal ions have water molecules in their coordination sphere, which are accessible for catalytic processes.
During catalytic experiments, the precipitation of a poorly soluble adduct formed between Ni5-SiPOM and the photosensitizer was observed in dark and light-driven reactions. The Ni5-SiPOM–[Ru(bpy)3]n+ ion-pair was identified as the catalytically active species since the filtered solutions of these systems before the photo-illumination showed no O2 evolution. The kinetic conditions of catalytic O2 evolution in the presence of 2 μM of Ni5-SiPOM are presented in Table 3 (entry 24) (Zhu et al., 2012b). Although Ni5-SiPOM appears to be an effective catalyst, no detailed studies were conducted to interrogate the electronic or photophysical characteristics of the ion pair generated during the catalytic cycle. The association of the photosensitizer with the POM anion positively affected the stability of Ni5-SiPOM since studies showed no nickel hydroxide/oxide particle formation under the conditions employed.
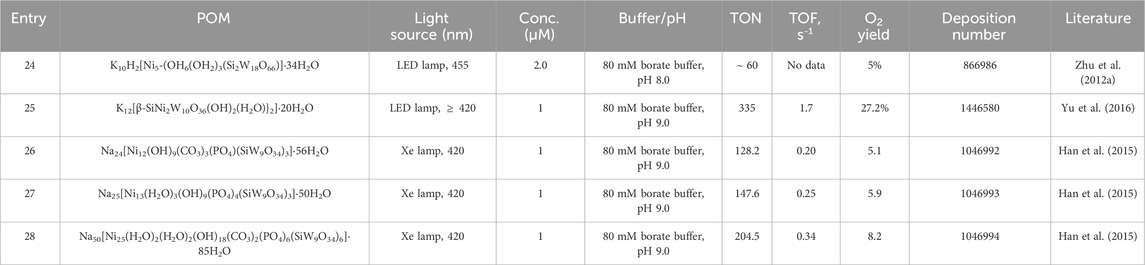
TABLE 3. Water oxidation catalyzed by different Ni-substituted POMs.
On the other hand, Ding and others designed a dimeric POM enclosing the Ni cluster [{β-SiNi2W10O36(OH)2(H2O)}2]12- (Ni2-SiPOM) that efficiently catalyzes water oxidation under visible light (Yu et al., 2016). Ni2-SiPOM was prepared in a one-pot scheme by combining the salt K8[β-SiW11O39] with NiSO4.6H2O in a potassium acetate solution under gentle warming and was separated in the solid-state as the hydrate potassium salt, K12[β-SiNi2W10O36(OH)2(H2O)}2]·20H2O (Figure 6C). The centrosymmetric polyanion Ni2-SiPOM is compatible with two divacant lacunary [β-SiW10O36]10- anions, which complete their Keggin-type structures incorporating two independent octahedral NiII ions. The corresponding asymmetric unit [{β-SiNi2W10O36(OH)2(H2O)}2]12- grows and is linked with another symmetry-related fragment through two μ3-oxygen atoms (O1 and O1’), resulting in a perfect parallelogram with Ni‒O bond distances in the range of 2.0 Å as shown in Figure 6D.
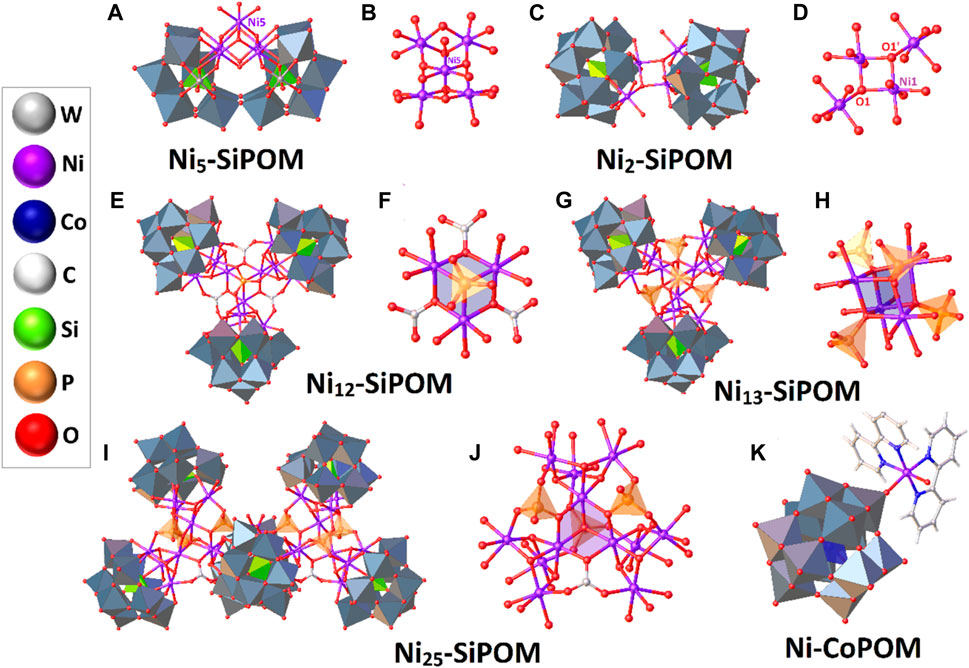
FIGURE 6. Schematic representation of selected Ni-substituted POMs. The POM-based scaffolds are shown in a polyhedral scheme, while the nickel active sites are in a ball-and-stick one. The inset shows the color code. For more information, see the text.
The catalytic activity was evaluated using the typical water oxidation system. The optimal conditions shown in Table 3, entry 25, were established by modifying different experimental conditions, such as the pH value of the buffer solutions and the concentrations of the catalyst, photosensitizer, and oxidizing agent. At concentrations above 15 μM, the catalytic activity decreases due to the appearance of heterogeneous species formed between the POM and the ruthenium-based photosensitizer (Yu et al., 2016). Several experiments confirmed that Ni2-SiPOM is the dominant catalyst, ruling out the formation of soluble ions or nickel particles during water oxidation.
Following the structural rules of the [Mn4CaO5] cluster in plants, three POMs ligands stabilizing nickel centers with a cubane-type assembly, Na24[Ni12(OH)9(CO3)3(PO4)(SiW9O34)3]·56H2O (Ni12-SiPOM), Na25[Ni13(H2O)3(OH)9(PO4)4(SiW9O34)3]·50H2O (N13-SiPOM), and Na50[Ni25(H2O)2(H2O)2(OH)18(CO3)2(PO4)6(SiW9O34)6]·85H2O (Ni25-SiPOM) were prepared, characterized by single crystal X-ray diffraction, and studied as molecular water oxidation catalysts (Figures 6E, G, I) (Han et al., 2015). The systems were prepared in water at basic pH using N10[A-α-SiW9O34].18H2O, NiCl2.6H2O, and Na3PO4.12H2O. Furthermore, to obtain Ni12-SiPOM and Ni25-SiPOM as crystalline materials, it was necessary to add Na2CO3 and heat the reaction medium below 90 °C to activate the PO43- groups.
Ni12-SiPOM and N25-SiPOM contain the partial cubane {Ni3O4} cluster encapsulated by PO43- and CO32- ligands (Figures 6F–J), with central units formulated as {Ni3(CO3)3(PO4)} and {Ni3(CO3)2(PO4)}, respectively. In Ni12-SiPOM, the system {Ni3(CO3)3(PO4)} is covered by the carbon-free [A-α-SiW9O34]-(NiOH)3] scaffolds. However, in N25-SiPOM, two central {Ni3(CO3)2(PO4)} cores are connected through a {Ni(H2O)2} bridge, resulting in a complex entity {Ni25(H2O)2(OH)18(CO3)2(PO4)6}, stabilized by several [A-α-SiW9O34]-(NiOH)3] units. On the other hand, in Ni13-SiPOM the {Ni4(H2O)3(PO4)4} cubane is wrapped by three {[A-α-SiW9O34]-(NiOH)3} fragments. It is noteworthy that the fully inorganic {Ni4(H2O)3(PO4)4)} moiety shown in Figure 6H encloses a central {Ni4O4} cubane, which structurally mimics the assembly present in the biological catalyst responsible for water oxidation in photosynthetic organisms.
Water oxidation was performed under the photocatalytic system outlined above. The catalysts showed the following order of efficiency Ni12SiPOM < Ni13-SiPOM < Ni25-SiPOM with TONmax = 204.5 (see Table 3, entries 26–28), demonstrating that the nuclearity of the internal clusters influenced the water oxidation processes. The authors tested the activity of these compounds with various polyoxotungstates stabilizing different arrangements of Ni ions, which did not show any photocatalytic activity under controlled experimental conditions (pH 9) (Han et al., 2015). The ability of the Ni12SiPOM, Ni13-SiPOM, and Ni25-SiPOM to efficiently sustain the water oxidation processes was explained by the ability of the photosensitizer to oxidize the POMs systems, by the nuclearity of the Ni centers affecting the number of possible active sites for the water molecules, and finally, by the cube-like arrangement of these nickel clusters. Stability studies of the catalysts ruled out the formation of nickel hydroxides, oxides, or other species during the catalytic process and the structural decomposition of the POMs as a function of time under oxidizing conditions.
In 2018, Das and others studied the electrocatalytic water oxidation process of a POM-supporting nickel(II) complex with interesting structural features, which was resolved by X-ray crystallography and other techniques as [Ni(bpy)3]3][{Ni(bpy)2(H2O)}{HCoW12O40}]·3H2O (Ni-CoPOM) (Singh et al., 2018). The complex was obtained through solvothermal synthesis at 160 °C from a suspension formed by small quantities of K6[CoW12O40]·6H2O, Ni(OAc)2·4H2O, and 2,2′-bipyridine in an acidic solution. The hydrothermal reaction conditions allowed the covalent grafting of the [HCoW12O40]5- anion by {NiII(bpy)2(H2O)} (Figure 6K). Additionally, the asymmetric unit contains 1.5 discrete [Ni(bpy)3]2+ complexes stabilizing the POM anionic charge and is complemented by three lattice water molecules in general positions.
Several electrochemical experiments were conducted to determine the water oxidation activity of Ni-CoPOM and identify the true catalyst since several metal ions in the crystal structure can support WOC activity. The experiments showed that the [Ni(bpy)2(H2O)]2+ unit linked to the POM structure is the catalytically active species and that its activity is related to both the redox nature of the Ni2+ ions and its coordination environment. However, the Co2+ ion enclosed in [HCoW12O40] may also have an active role in the global catalytic performance. On the other hand, the nickel ions within [Ni(bpy)3]2+ were discarded as potential WOC centers since they are coordinatively saturated.
[Ni(bpy)2(H2O)}{HCoW12O40] catalyzed the oxygen evolution reaction at neutral pH in phosphate buffer with an overpotential of 476 mV and a high TOF of 18.5 s-1 O2 evolved per mol of Ni ion at a current density of 1 mA cm-2 (Singh et al., 2018). The same research group recently reported a similar system with a copper(II)-bipyridine complex attached to the [HCoW12O40]5- anion; the active unit [{Cu(bpy)(H2O)}2[CoW12O40]] is an efficient electrocatalyst for the hydrogen evolving reaction in a near-neutral medium (Singh et al., 2021). These results introduce a new way of developing POM-based electrocatalysts for water splitting.
Mimicking the photosynthetic oxygen-evolving center with artificial systems to overcome the demanding water oxidation processes has been one of the most challenging issues bioinorganic chemists have faced over the past 2 decades. Nevertheless, after several efforts, a significant breakthrough in this direction was achieved when Zhang and co-workers synthesized 2015 the first artificial [Mn4CaO5] cluster through a two-step procedure using low-cost commercial chemicals (Zhang et al., 2015; Chen et al., 2017; Li et al., 2020). The artificial complex contains an asymmetric Mn3CaO4 unit attached to the fourth external manganese and is stabilized by eight (CH3)3CCO2− pendant units, forming a complex with the structural formula [Mn4CaO4(ButCO2)8(ButCO2H)2(py)] (But = tert-butyl; py = pyridine) (Zhang et al., 2015). In the molecule, the synthetic Mn4CaO4 cluster is similar to the catalytic structure in photosystem II (Figure 7) (Umena et al., 2011). Despite the remarkable structural similarity, the synthetic OEC was not stable under the established experimental conditions, and the catalytic performance could not be determined due to the rapid decomposition of cubane in the presence of water in the solution studied, which is essential for determining the WOC activity.
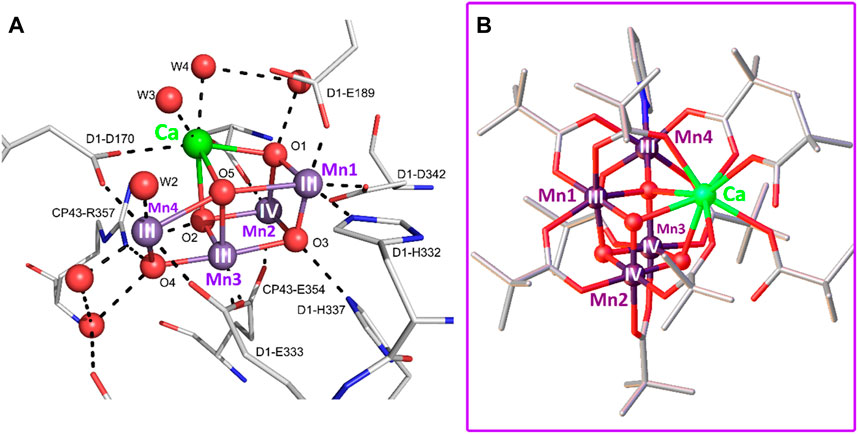
FIGURE 7. (A) Part of the molecular structure of the {Mn4O5Ca} cubane from the oxygen-evolving center of photosystem II at a resolution of 1.9 Å (Figure modified from https://en.wikipedia.org/wiki/Oxygen-evolving_complex) and (B) crystal structure of the artificial Mn4CaO4 system design by Zhang and coworkers in 2005. In both cases, the oxidation states are highlighted on the Mn ions (Umena et al., 2011; Zhang et al., 2015).
Using POMs as multidentate ligands to direct the assembly of cubane-type clusters may be a good design strategy to overcome the structural issues observed in artificial Mn4CaO4 models; this will lead to a molecule without the exact structural features of the biological catalyst (such as its organic environment). However, the incorporation of polynuclear Mn clusters into POM-based frameworks can not only solve the stability problem but also improve catalytic activity through charge transfer, redox processes, and the rapid removal of reducing equivalents generated by the water-splitting reaction, thus providing modern platforms for photosynthetic water oxidation.
An example of the above is the compound reported by Cronin’s group in 2011 formulated as K18[Mn2Mn4(μ3-O2)-(H2O)(β-SiW8O31)(β-SiW9O34)(γ-SiW10O36)]·40H2O (Mn5-SiPOM) (Mitchell et al., 2011). According to the authors, Mn5-SiPOM represented the first model of an Mn-cubane wrapped by POM scaffolds (Figure 8A). The synthetic strategy consisted of adequate monitoring of both the temperature and pH, allowing the isomerization of the starting material {γ-SiW10O36} into the {β-SiW8O31} and {β-SiW9O34} polyanions. During the self-assembly process, the three lacunary species encapsulated a mixed-valence {MnIII2MnII4O4}2+ cubane, which closely resembles that found in the structure of the PSII (Figure 8B). This Mn cluster may be described as a {Mn5O4} cubane-type cluster linked to an external MnII atom via one of the μ3–O vertices of the cube.
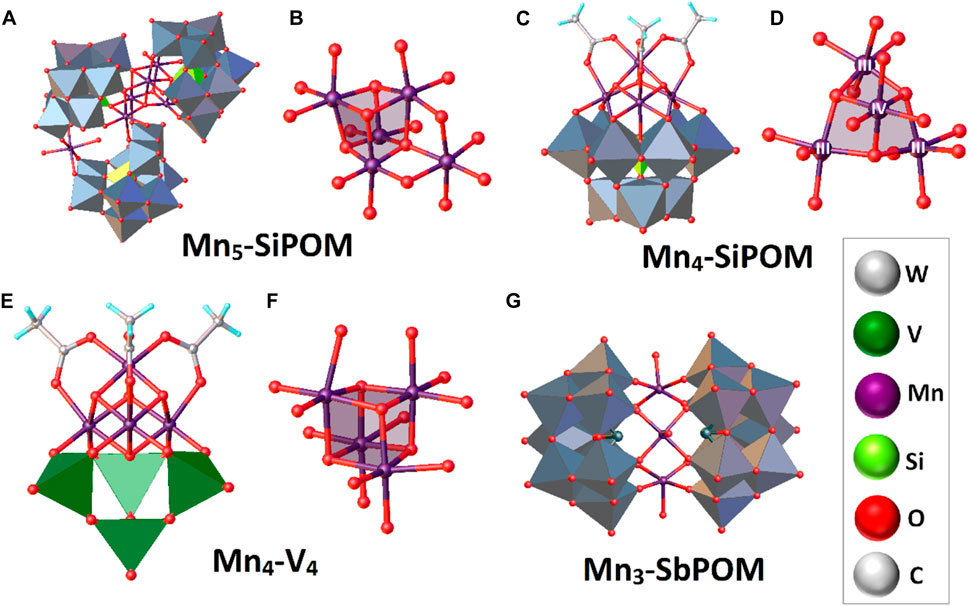
FIGURE 8. Schematic representation of selected Mn-substituted POMs. The POM-based scaffolds are shown in a polyhedral scheme, while the manganese-active sites are in a ball-and-stick one. The inset shows the color code. For more information, see the text.
From an electrochemical point of view, Mn5-SiPOM exhibited an interesting and similar redox activity both in solution and solid state, showing that the compound is stable under electrochemical conditions. Its stability was confirmed by ESI-MS experiments, revealing that Mn5-SiPOM is exceptionally stable in H2O/CH3CN mixture (Mitchell et al., 2011). Moreover, the complex was also able to form microtubular architectures through morphogenesis when a crystal was exposed to an aqueous solution containing bulky cations. Although the WOC activity of the Mn5-SiPOM compound has not been studied in detail, the design of materials inspired by this approach may open a window for the development of electronically interesting POM-based catalytic devices.
Scancola, Kortz Bonchio, and coworkers prepared the first Mn-substituted POM with WOC activity in 2014 (Al-Oweini et al., 2014). The complex shown in Figure 8C with formula Na3.5K2.5[MnIII3MnIVO3(CH3COO)3(A-α-SiW9O34].20H2O was synthesized under mild conditions by reacting the polyoxometalate sodium salt [α-SiW9O34]10-, prepared previously with a mixed-valence manganese complex [MnIII8MnIV4O12(CH3COOH)16(H2O)4]·2CH3COOH.4H2O; the reaction was carried out at pH 6 using a sodium acetate buffer solution; KCl was also used to stabilize the anionic charge of the resulting complex. The system crystallized in the centrosymmetric space group
The experiment to corroborate water oxidation activity was conducted electrochemically in a Na2SiF6/NaHCO3 buffer solution (pH 5.2) using a 0.5 µM concentration of Mn4-SiPOM. In the cyclic voltammogram, a strong catalytic current was observed at an onset potential of 1.25 V (vs. Ag/AgCl), which was attributed to water oxidation; this behavior was not observed in the buffer solution or the unpolished electrode, suggesting that the catalytic process was homogeneous.
Once the electrochemical conditions under which the catalyst oxidizes water were established, oxygen production in the photo-induced system was tested using [Ru(bpy)3]2+ and S2O82-, and a wide range of catalyst Mn4-SiPOM concentrations (from 6.3 to 50 µM) at pH 5.2 using the buffer noted above (Table 4, entry 29). Under illumination, O2 evolved immediately (although with low yields), reaching a plateau after 30 min of exposure. The oxygen formation depended on catalyst concentration and persulfate consumption, while the low O2 yield between 1.2 and 3.7% was attributed to the gradual decomposition of the photosensitizer under illumination. Control experiments with manganese(II) ions introduced as MnSO4.H2O led to insignificant O2 production even at high concentrations, indicating that the action mechanism leading to water oxidation involves the four Mn ions of the redox active {MnIII3MnIVO3} core.
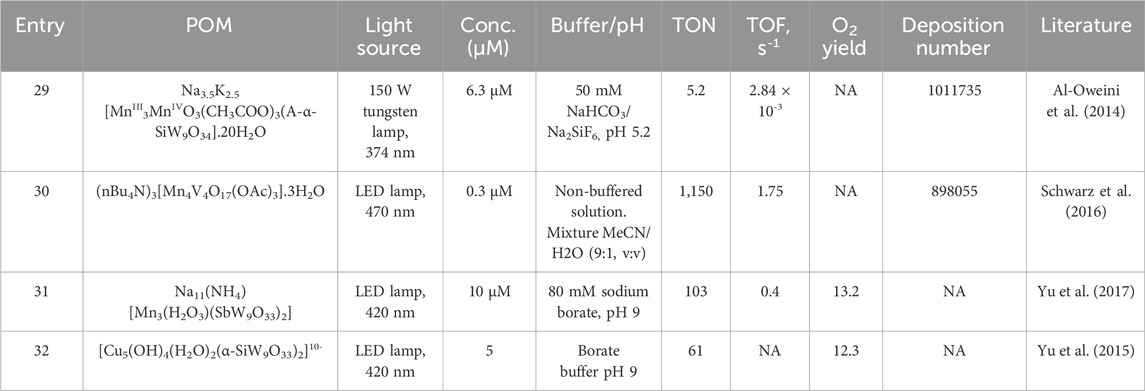
TABLE 4. Water oxidation catalyzed by different Cu-and-Mn-substituted POMs.
On the same basis, Streb’s group synthesized 2016 a molecular vanadium-manganese compound using one-pot self-assembly from readily available precursors, formulated as (n-Bu4N)3[Mn4V4O17(OAc)3]·3H2O (Mn4V4) (Schwarz et al., 2016). Crystallization of Mn4V4 was achieved by solvent diffusion of diethyl ether into an acetonitrile solution containing (n-Bu4N)4[V4O14]4H2O, Mn(OAc)3, and (n-Bu4N)MnO4. X-ray studies showed a central {Mn4O4} oxo core stabilized by two different kinds of ligands, i.e., a vanadate species [V4O13]6- and three acetate groups (Figure 8E). While [V4O13]6- coordinated through O atoms from two tetrahedral VO43- units, the acetate ions were coordinated through carboxylate groups, resulting in the anionic entity [Mn4V4O17(OAc)3]3- with idealized C3v symmetry. Three (n-Bu4N)+ cations stabilize the anion in the crystal structure. The authors indicate that the Mn‒O distances and Mn···Mn interactions in this artificial cubane are similar to those in the cubic [Mn4CaO4] assembly from the oxygen evolution center. In addition, the oxidation states of the manganese ions, calculated using bond valence sum (BVS) from the crystal structure, are compatible with a central {MnIII2MnIV2O4}6+ unit (Figure 8F), analogous to the metastable S1 state of the natural catalytic cycle.
Electrochemical analyses in anhydrous MeCN revealed that Mn4V4 undergoes successive one-electron oxidation processes leading to the metastable electronic states S2 (MIIIMnIV) and S3 (MnIV) of the water oxidation cycle; these redox events were confirmed by EPR through comparison of the spectrum of the native compound {MnIII2MnIV2O4}6+ with the one- and two-electron oxidized systems obtained by bulk electrolysis, i.e., {MIIIMnIV3O4}7+ and {MnIV4O4}8+. The water oxidation activity of Mn4V4 in the standard system [Ru(bpy)3]2+/S2O82- was studied in a non-buffered MeCN/H2O solution. The catalytic results support the electrochemical findings by showing that Mn4V4 is a very active WOC with a maximum TON of 1,150 and TOF of 1.75 s-1, using a concentration of 0.3 μM catalyst (Table 4, entry 30) (Schwarz et al., 2016). The TON values decreased considerably at concentrations above 0.6 μM, possibly due to the formation of adducts between the photosensitizer and the polyanion. Finally, deactivation of the catalytic activity was observed in recovering/recycling experiments, indicating that the experimental conditions could limit WOC performance.
Subsequently, Ding and others studied the O2 production by a manganese antimoniotungstate, [Mn3(H2O)3(SbW9O33)2]12- (Mn3-SbPOM) and compared its catalytic activity with that of two structural analogs with different numbers of manganese ions and other heteroatoms in their structure: [Mn(H2O)5(PW9O34)2]9- and [Mn3(H2O)3(AsW9O33)2]12-(Yu et al., 2017). The abbreviated compound Mn3-SbPOM was synthesized following a process previously described by Bernt Krebs in 1998, which consisted of reacting the Na9[SbW9O33] salt with MnCl2.4H2O in an aqueous medium (pH 6–7) (Bösing et al., 1998). After the addition of NH4NO3 solution, the complex crystallized by slow evaporation as a mixed ammonium-sodium salt Na11(NH4)[Mn3(H2O)3(SbW9O33)2]. The resulting crystal structure corresponds to two kegging-type [SbW9O33]9- networks associated with three Mn2+ ions (Figure 8G). Each manganese atom has a distorted square pyramidal geometry with the apical position filled by a water molecule. Under a photochemical setup, the catalytic performance of Mn3-SbPOM afforded TON and TOF values of 103 and 0.4 s-1 (Table 4, entry 31). In contrast, its analogs showed low O2 photocatalytic activity. No soluble ions or Mn oxides were detected during the water oxidation process.
In 2019, the compound [Mn(H2O)3)2(H2W12O42)]6-, which consists of a polymeric network extending in 2D through manganese and tungsten centers, was prepared and characterized. The basic unit can be formulated as [Mn(H2O)3)2(H2W12O42)]n6n-, which is stabilized by hydrated alkali metal ions such as sodium and potassium. An essential feature of this system is that the manganese centers are solvated by three water molecules, which can be used for catalytic processes. This complex undergoes electron transfer processes leading to the formation of manganese species in high oxidation states that promote water oxidation (Teillout et al., 2019). On the other hand, six Mn-substituted POMs selected from literature with different structural features were recently studied as electrocatalytic WOCs in solution and composite films. The controlled experiments indicated that the Mn ions’ content, oxidation state, and central core affected the electrocatalytic water oxidation activity (Wu et al., 2022).
It has been exemplified that an effective strategy in the search for new POM-WOCs is to test previously reported compounds with interesting structural features whose water oxidation activity has yet to be studied. This is the case of the first copper-substituted polyoxometalate studied as WOC, [Cu5(OH)4(H2O)2(α-SiW9O33)2]10- (Cu5-SiPOM). This compound had already been studied in 2005 as a material with interesting magnetic properties but was not explored as a catalyst for water oxidation until 2015 by Ding and others (Figure 9A) (Nellutla et al., 2005; Yu et al., 2015). Cu5-SiPOM was prepared by reacting the potassium salt K10[α-SiW9O34] with CuCl2.2H2O in a hot acidic aqueous solution (pH 4.8). Crystals of the corresponding hydrated potassium salt, K10[Cu5(OH)4(H2O)2(α-SiW9O33)2].18H2O (Figure 9B) were isolated by slow evaporation. The structure was described as a dimeric polyanion formed of two [α-SiW9O34]10- frameworks fused through two bridging W–O bonds, which enclosed a central [Cu5(OH)4(H2O)2]6+ core, in which the Cu2+ ions have a Jahn-Teller distorted octahedral geometry. The Cu—O distances in the copper cluster are in the range of 1.907–2.387 Å.
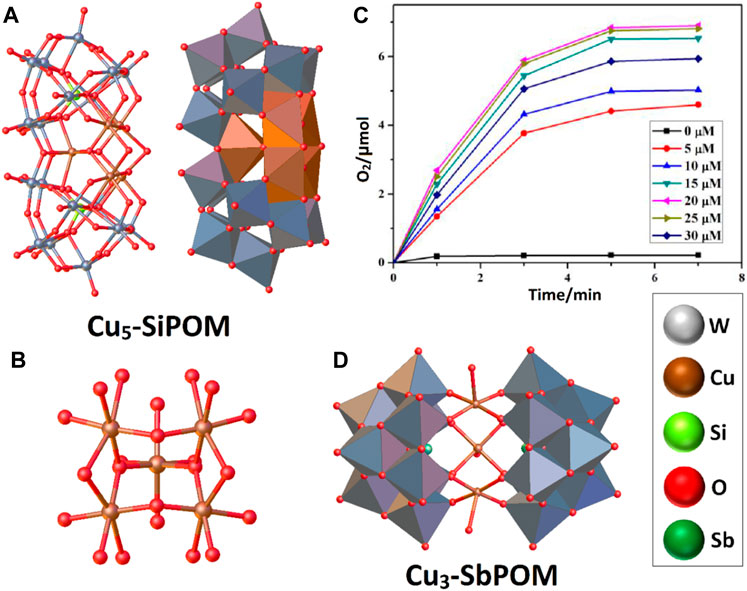
FIGURE 9. (A, B) Ball-and-stick and polyhedral representation of Cu5-SiPOM and it is central {Cu(OH)4(H2O)2}6+ fragment (C) Kinetics of O2 formation system using different concentrations of Cu5-SiPOM. The figure was reproduced with permission from Yu et al., 2015. Copyright 2015 Royal Society of Chemistry. (D) Ball-and-stick and polyhedral scheme of Cu3-SbPOM. The inset shows the color code. For more information, see the text.
The light-driven water oxidation of the Cu5-SiPOM system was compared under the same conditions with that of four copper POMs, which differ in the number of copper centers and polyoxometalate networks: [{Cu(H2O)SiW8O31}2]12-, [Cu4(H2O)2(OH)4(Si2W16O58]8-, [Cu6Cl(SbW9O33)2]7-, and [Cu20Cl(OH)24(H2O)12(P8W48O184)]25-. Of these, only Cu5-SiPOM showed oxygen evolution, indicating that the number of copper centers is unrelated to catalytic activity. A possible answer to the activity of this complex can be found in the arrangement of the [Cu5(OH)4(H2O)2]6+ cluster, in its protonation state, and in its stabilization with the robust tungsten-based ligands [SbW9O33]9- (Yu et al., 2015). In the best experimental conditions shown in Table 4, entry 32, and in a concentration range of 5–30 μM, the copper system showed O2 evolution under illumination in the typical photocatalytic system, reaching a plateau after 5 min, as seen in Figure 9C. Above 20 μM, the catalytic activity decreased due to interactions between the polyoxometalate and the photosensitizer. Several experiments confirmed that Cu5-SiPOM is the active and dominant catalyst.
Subsequently, one Cu(II)-substituted POM was reported to exhibit water oxidation capabilities at neutral pH, mimicking the conditions in which the OEC performs water oxidation. The complex was reported by Ding’s group in 2018 as a catalyst for O2 formation through homogeneous electrocatalytic water oxidation (Yu et al., 2018). The antimony-containing polyoxometalate [Cu3(H2O)3[(SbW9O33)2]12- (Cu3-SbPOM) was prepared following a synthetic strategy reported by Kortz, which consisted of reacting CuCl2.2H2O with Na9[SbW9O33]19.5H2O in an aqueous solution at pH 6.2. Single crystals of the complex Na12[(SbW9O33)2Cu3(H2O)3] were obtained by slow evaporation. The crystal structure features two [α-SbW9O33]9- polyanions related by three equivalent Cu2+ ions, resulting in a sandwich-type design (Figure 9D). The compound shows structural features similar to the Mn3-SbPOM manganese system described in the previous section. Different approaches confirmed electrocatalytic water oxidation and the system’s stability under electrochemical conditions.
Due to its abundance on Earth, iron is a very cheap metal and less toxic than other transition metals; therefore, iron compounds can play a leading role as WOCs (Liu et al., 2019). Nevertheless, water oxidation catalysis of Fe-substituted POMs began to be reported only recently. In 2005, Ding and co-workers informed a Fe-POM complex formulated through X-ray crystallography as Na27[FeIII11(H2O)14-(OH)2(W2O10)2(α-SbW9O33)6]·103H2O (Fe11-SbPOM) obtained via a facile one-pot synthesis by combining FeCl3 and Na9[α-SbW9O33] in an acidic aqueous medium (Du et al., 2015a). Compound Fe11-SbPOM was crystallized by slow evaporation at room temperature. The solid-state structure consisted of a large entity with a nanoscale size of 1.75
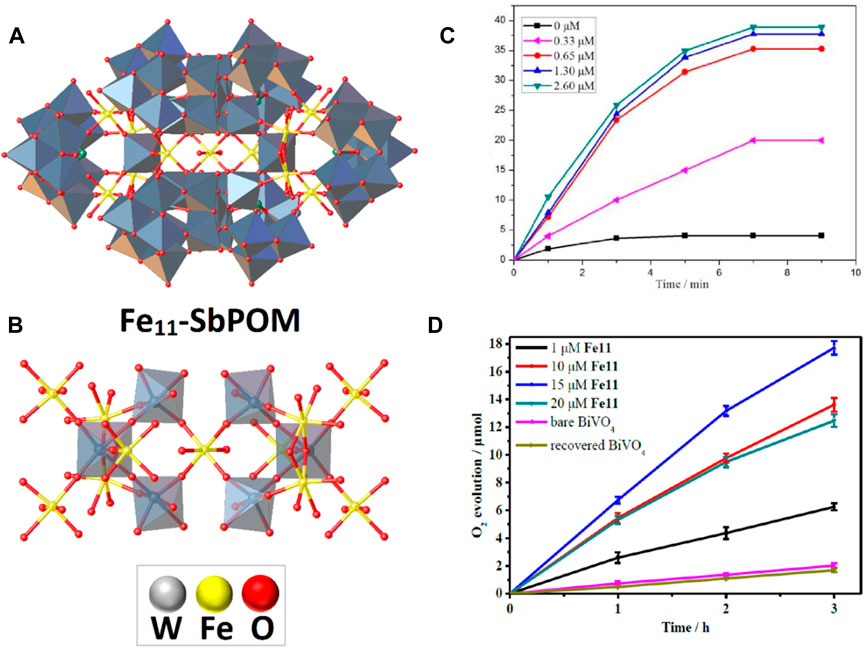
FIGURE 10. (A, B) Ball-and-stick and polyhedral representation of Fe11-SbPOM and it is central [Fe11(H2O)14(OH)2(W2O10)2]27+ unit (C) Kinetics of O2 formation in the photocatalytic system using different concentrations of Fe11-SbPOM. The figure was reproduced with permission from Du et al. (2015a). Copyright 2015 Royal Society of Chemistry (D) Kinetics of O2 evolution in the photocatalytic system using different concentrations of Fe11-SbPOM. The figure was reproduced with permission from Hu et al. (2019). Copyright 2019 Royal Society of Chemistry. For more information, see the text.
Subsequently, the same research group described a water oxidation system using bismuth vanadate BiVO4 instead of the typical ruthenium photosensitizer, NaIO3 as the sacrificial agent, and the molecular Fe11-SbPOM complex as the catalyst (Hu et al., 2019). Under optimal conditions, the system evolved O2 in a yield of 19.1%, sustaining an initial TOF of 173 h-1 in an acetate buffer at pH 3, under which Fe11-SbPOM is stable (Figure 10 d). Another spherical giant POM of formula, Mo72Fe30O252(CH3COO)12-[Mo2O7(H2O)]2[H2Mo2O8(H2O)](H2O)91]·nH2O, was studied as water oxidation catalysts in the [Ru(bpy)3]2+/persulfate system at pH 9, showing a TON of 13.99 with a TOF of 3 min-1 (Kaushik et al., 2020).
From the previous sections, it is evident that the mechanism through which different transition-metal-substituted POMs generate O2 is discussed mainly concerning the artificial photosynthetic system involving the [Ru(bpy)3]2+/S2O82- pair, which is active only under visible light and in the presence of the catalyst (Geletii et al., 2008; Han and Ding, 2018; Geletii et al., 2009; Natali et al., 2012a; Kaledin et al., 2010; Huang et al., 2012). This system is widely used to study the ability of a molecular system to oxidize water. In the case of TMSPs, it is one of the most valuable ways to generate O2 and reducing equivalents (Figure 11). Therefore, it seems appropriate to describe the functioning of this system in more detail.
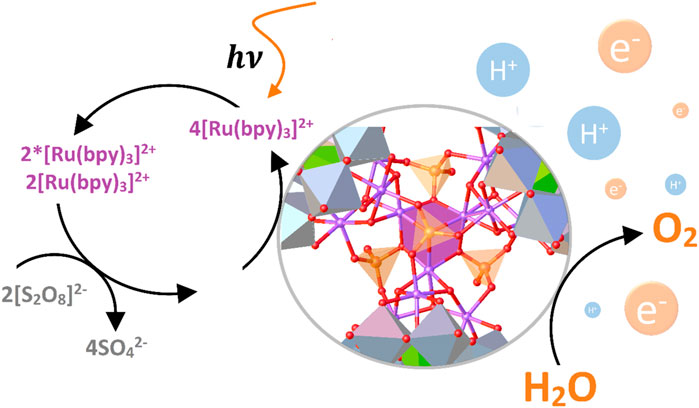
FIGURE 11. Schematic representation of the light-driven water oxidation system catalyzed by TMSPs.
Similar to natural photosynthesis, the process starts with the absorption of visible light by four photosensitizer molecules to generate the pair 2[Ru(bpy)3]2+/2[Ru(bpy)3]2+•. Of these two systems formed, the excited species 2[Ru(bpy)3]2+• can either relax naturally or be quenched by the sacrificial electron acceptor (S2O82-) via biomolecular or unimolecular electron transfer processes that we will not discuss here. This light-driven multi-electron process forms two oxidized [Ru(bpy)3]3+ molecules and two SO4−•sulfate species, which have the role of oxidizing the remaining ruthenium complexes to the corresponding oxidized [Ru(bpy)3]3+ forms (Bensaid et al., 2017; Piccinin and Fabris, 2011; Piccinin et al., 2013). The active site of the TMSPs transfers electrons to the [Ru(bpy)3]3+ systems to re-establish the light-driven cycle, leaving the positively charged polyoxometalate [TMSP]n+ (Figure 11). Regardless of the number of metal ions incorporated into the POM structure, the active site must transfer four electrons to the reduced ruthenium(II) complexes to form a highly oxidized [TMSPs]4+ species capable of oxidizing water.
However, this light-driven water oxidation system does not provide molecular information to understand the steps leading to the evolution of O2, such as the deprotonation of the water molecules linked to the active site or the intra- or intermolecular pathways leading to the formation of the O‒O bond and ultimately to the release of O2 from the catalyst (Taira et al., 2023). Most of the reports that have addressed these questions consist of theoretical studies based on DFT calculations supported by experimental data (Soriano-López et al., 2018; Lang et al., 2013). Regardless of the central active core’s structural arrangement, the proposed mechanisms are governed by chemical reactions involving proton-coupled electron transfer (PCET) processes.
In this context, theoretical studies focus on studying in detail the evolution of the active center during the catalytic cycle that ends with the generation of O2 and the re-establishment of the catalyst. Experimentally, water oxidation is carried out by applying a potential that leads to the polyoxometalate’s oxidation that forces the catalytic process. With the help of computational analysis, it is feasible to elucidate two fundamental features: i) the oxidative nature of the catalytic site, i.e., the ability of the central core to lose electrons to reach high oxidation states, and ii) the rate of the non-electrochemical or molecular steps, i.e., the nucleophilic water attack (WNA) mechanism that is facilitated by the oxidized state of TMSPs and further by proton transfer events governed by its acid/base capabilities (Soriano-López et al., 2017). The steps in the catalytic cycle leading to water oxidation are summarized in Figure 12 and will be discussed in more detail below.
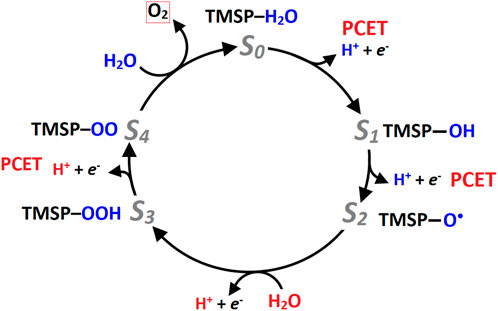
FIGURE 12. O2-evolution mechanism for TMSPs considered as a single-site catalyst. The figure was adapted with permission from Azmani et al., 2021—copyright 2021 Royal Society of Chemistry.
The first step is to characterize the molecular properties of the catalyst in the resting state by DFT, which we will call S0. Some studies have shown that in the resting structure, the water molecules are coordinated to the catalytic center through weak TMSP···OH2 bonds and in a scheme that favors the intermolecular interactions between the H-atoms of these water molecules and the neighboring O atoms from the POM scaffolds. Subsequently, the computational characterization is extended to all possible structural intermediates S1‒S4 of the catalytic process following a Kok-type scheme. According to this approach, the first step of the catalytic process starts from the resting state S0 and consists of the loss of a proton from the H2O molecule linked to the active central core (Figure 12). This event leads to the deprotonated species TMSPox‒OH (S1) with an oxidized metal center and a deprotonated aquo group forming a hydroxo group; according to the reaction, TMSP‒H2O → TMSPox‒OH + H+ + e-.
The step to the S2 state involves the elimination of the remaining H-atom, forming a metal‒oxyl group via the process TMSPox‒OH → TMSP‒O• + H+ + e-. For the particular case of cobalt sandwich-type POMs that incorporate two water molecules in their central belt (see Figure 4A), the second removal of the H-atom occurs through a chemical reaction involving a concerted PCET process at a potential of 1.48 V, in which the electron and proton are transferred in a single step from the hydroxyl group [S1(TMSP-CoIII‒OH) → S2(TMSP-CoIII‒O•)] (Soriano-López et al., 2017). Thermodynamically speaking, the second oxidation is much more demanding than the loss of the first electron, so the applied potential for the two-electron process is determined by this step, leading to the active state S2. Theoretical studies have determined that these processes lead to a shortening of the Co‒O bond of the catalytic center, confirming that the solvated sites in POMs featuring a sandwich-type design are activated for the formation of the O‒O bond. Similar electronic and structural changes have been calculated for the structural analog of Fe (Azmani et al., 2021).
In photosystem II, the coupling of the O-O bond on the manganese-calcium cluster has been debated for several years. The most accepted mechanisms are: (
Returning to the mechanism of TMSPs, the nucleophilic attack of a solvent molecule on the active TMSP‒O• site leads to the formation of an intermediate species (in some works referred to as S3’) with a hydroperoxo group TMSP‒OOH (Figure 12). During the transition state, the proton of the -OOH fragment can be transferred from the external water molecule to a nearby basic O atom of the polyoxometalate, which can eventually be released into solution via a PCET process leading to the S3 state (Soriano-López et al., 2017; Piccinin et al., 2013). While the last PCET event requires relatively low energy and leads to the formation of the TMSP‒OO species (S4), the final step of the O‒O coupling mechanism involves the release of O2 from the S4 state following the coordination of another water molecule to the catalyst, regenerating the S0 state of the polyoxometalate (TMSP-OH2). This mechanistic route is energetically more favorable than any intramolecular reaction mechanism involving coupling between two reactive species (e.g., two oxyl groups) on the same POM due to the large distances between the active sites coordinating water molecules.
Nevertheless, it has been observed that for some Mn-substituted POMs with a peripheral water molecule, nucleophilic water attack and oxo-oxo coupling are thermodynamically competitive towards O‒O bond formation. In the latter case, two oxyl radicals from two discrete POM molecules react to form the peroxo group. The process of O2 evolution via the oxo-oxo coupling mechanism is relatively straightforward. An O2 molecule dissociates from an intermediate species, and the catalyst returns to the ground state after coordinating a new water molecule (Su et al., 2019).
The number of reports on artificial photosynthesis designs incorporating POMs is scarce compared to the number of articles analyzing the efficiency of these materials as WOCs (Paul et al., 2022). This is due primarily to the enormous complexity involved in imitating the molecular assembly of the natural process. Although this complexity is clearly beyond the scope of synthetic chemists due to the living nature of this process, i.e., it is evolving nature, the understanding of natural photosynthesis has gradually allowed us to move toward the design of artificial systems (Bozal-Ginesta and Durrant, 2019; Balzani et al., 2008; Wang et al., 2022; Collings and Critchley, 2007). As noted above, this progress is based on the specific design of individual photosynthetic devices and on employing molecular engineering to assemble these systems homogeneously and functionally into an integrated device by exploiting covalent and non-covalent interactions (Alstrum-Acevedo et al., 2005; Keijer et al., 2021; Bozal-Ginesta and Durrant, 2019). Although significant progress has been made in this direction, it has yet to be possible to unite the various components into an integrated functional system; this is undoubtedly one of the current challenges of modern science.
The photosynthesis study shows that the different biological components that act together to oxidize water and reduce carbon dioxide are separated by complex cell membranes with a well-defined space between them (Nelson and Ben-Shem, 2004; Chabi et al., 2017; Cooper et al., 2011). This approach must be met in artificial devices to avoid degradation of the different components used under water oxidation, mainly when organic oxidants such as bipyridine-derived agents [Ru(bpy)3]2+ show instability under electrochemical conditions (Liu et al., 2017). To avoid these problems, Cronin and others have developed micrometer-scale microtubule architectures and synthetic inorganic cells called iCHELLs, formed in situ when certain crystalline polyoxometalate-based materials are brought into contact with solutions containing partially soluble organic cations (Copper et al., 2011; Cooper and Cronin, 2009). These iCHELLs can be individual compartments to separate processes with specific environments (Figures 13A, B). Regarding the topic at hand, a scheme based on tubular architectures or iCHELLs using POMs will allow the isolation of components used for water oxidation, such as the photosensitizer or the electronic sacrificial agent, avoiding oxidative degradation of the [Ru(bpy)3]2+ agent or ionic pairing between this species and POM ions when the latter are used at high concentrations.
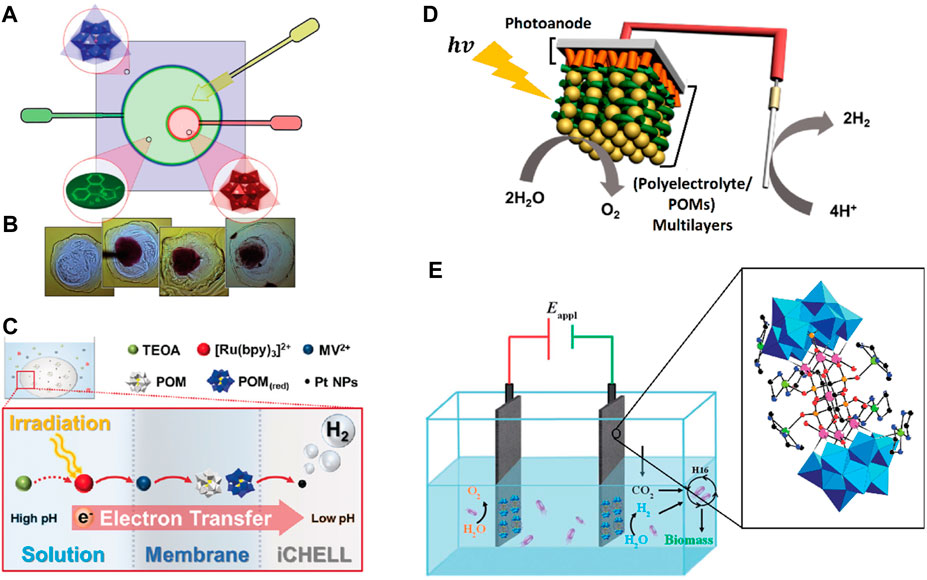
FIGURE 13. Integrated systems for artificial photosynthetic reactions. (A) A scheme that shows the formation of different confined reaction spaces or iCHELLs. An iCHELL (green area) is formed on an acidic solution containing Keggin-type POMs (blue area) by injecting a bulky organic cations solution. Another solution of a different POM (red area) is injected into the first inorganic cell to form a second iCHELL. A reagent can be injected inside these separate systems to initiate a chemical reaction with specific conditions (yellow arrow). (B) Formation of an iCHELL using H3[Mo12PO40], followed by the formation of another iCHELL inside the first one formed with H₃[W₁₂PO₄₀], the second cell is made with KMnO4 and by adding H2O2 the interior of this space is colored. The figure was reproduced with permission from (Symes et al., 2013). Copyright 2013 The Royal Society. (C) The schematic represents the system for H2 evolution applying confined space formation based on the construction of iCHELLs. The figure was reproduced with permission from Nakanishi et al., 2018. Copyright 2018 Wiley-VCH. (D) A system constructed using the layer-by-layer approach employing photoelectrodes modified with cationic electrolytes and sandwich-type tetracobalt(II) POMs for water oxidation. The figure was reproduced with permission from Jeon et al. (2017). Copyright 2017 American Chemical Society. (E) An electrochemical cell was designed with POM-based electrodes and carbon cloth for microbial CO2 conversion. Figure reproduced with permissions from (Wang et al., 2018). Copyright 2018 Royal Society of Chemistry.
Using iCHELLs to carry out photosynthetic processes has been implemented for the hydrogen evolution reaction (HER), which is essential in directing it to the water-splitting schemes. The system was carried out by forming three confined spaces with different controlled environments (such as pH) through which hydrogen evolution occurs (Nakanishi et al., 2018). First, the membranes were constructed using the [PW12O40]3- and [SiW12O40]4- Keggin-type anions on a solution of methyl viologen (MV), which serves as an electronic mediator and contains both the photosensitizer [Ru(bpy)3]2+ and the triethanolamine-based sacrificial electron donor (TEOA). The inside of the iCHELL has Pt nanoparticles that catalyze hydrogen evolution. As can be seen in Figure 13C, the electron transfer process starts under visible light irradiation to the solution containing the photosensitizer and sacrificial electron donor; the electron transfer migrates across the membrane formed by the POMs and is facilitated by both these systems (through oxidation processes) and the electron mediator (MV). Finally, these electrons are transferred to the interior of the iCHELL, which contains a significant concentration of protons from the dissociation of H3[PW12O40] polyacids. The platinum-based catalyst in this confined space uses these electrons to reduce protons and generate molecular hydrogen.
This system based on iCHELLs or microtubule architectures can be used for many chemical reactions that require independent spaces with special characteristics, leading to efficient catalytic processes and avoiding degradation of the components used. Considering that one of the challenges of artificial photosynthesis is the integration of the various elements into a single functional scheme, this method can be an essential step towards achieving global artificial photosynthetic systems that include both water oxidation reaction and CO2 reduction for the development of raw chemicals.
Another practical example of integrating a polyoxometalate molecule with proven WOC activity (i.e., Co4-PPOM) into an artificial photosynthetic model was reported by Ryu and co-workers (Jeon et al., 2017). The authors designed a functional device using the layer-by-layer technique, assembling several units to produce an integrated platform. In this case, the separate systems consisted of photoelectrodes (based on BiVO4, Fe2O3, TiO2, or gold), different kinds of cationic polyelectrolytes, and the cobalt-substituted POM. The assembly process was based on coating the photoelectrode substrates with the positively charged polyelectrolytes, forming a cationic surface layer covered by the anionic POMs (Jeon et al., 2017). This process is repeated several times until a photoanode with the anchored catalyst for water oxidation is constructed (Figure 13D). The activity of the photoanodes using this technique was significantly improved after modification in terms of stability and catalytic properties, regardless of the type of substrate and polyelectrolyte used, indicating that the tailored electrodes performed better activity toward water oxidation reaction compared to the photoelectrodes that were not coated with the anionic POMs.
The cases we have reviewed in this section refer to processes in which the role of POMs is limited to the water oxidation reaction or the formation of functional membranes that can separate electron transfer processes. Although POMs are integrated into artificial photosynthetic devices in both cases, few examples in the literature refer to systems in which the water oxidation reaction and the H2 evolution or CO2 reduction are carried out using modified polyoxometalate-based electrodes. An interesting example in this regard was reported in 2018 by Wang et al., 2018. The authors synthesized a water-insoluble compound of complex formula, Na2[Cu(en)2]6[{PW9O34)Co3(OH)-(H2O)2(O3PC(O))C3H6NH3)PO3)}2Co]45·H2O (Cu6Co7-POM). The structure comprises a sandwich design based on the [PW9O34]9- anions, which intercalate rare central heptanuclear cobalt core whose external O atoms are linked to copper/ethylenediamine (en) complexes, {Cu(en)2}. Two of these copper(II) complexes are also functionalized to the O atoms of the two robust [PW9O34]9- ligands.
The Cu6Co7-POM complex was integrated into carbon cloth through a simple process, and SEM characterized the surface modification of the electrodes. At the same time, other studies identified the presence of polyoxometalate building blocks. Subsequently, the authors assessed the ability of these modified electrodes to perform both the O2 evolution reaction and the H2 evolution reaction under neutral conditions using phosphate buffer. According to linear sweep voltammetry (LSV), the system could perform both reactions at an overpotential below 450 mV, much lower than those reported by other systems using cobalt-phosphate particles (Co-Pi) reported by Nocera (Surendranath et al., 2009).
On the other hand, these same improved electrodes were used for electro-microbial CO2 conversion using the bacterium Ralstonia eutropha. As can be seen in Figure 13E, the system consisted of a single electrochemical cell in which the bacteria were grown at a constant potential (between 1.8 and 2.2 V) using these electrodes as anode and cathode. In contrast, the electrolyte was a biological medium (pH 7) saturated with gaseous CO2. In this configuration, the bacteria consumed most of the hydrogen produced at the cathode to live and grow, generating biomass. According to the authors, this process was carried out with an efficiency ten times higher than the natural photosynthetic process.
We have presented in this review how an artificial photosynthetic device is a complex unit with functions that, although designed independently, must be efficiently coupled to achieve one of the most important and thermodynamically demanding reactions that support life on Earth: the oxidation of water. This reaction takes place by plants with an efficiency of only 3%. However, the photosynthetic machinery is the best biological model for harnessing the Sun’s energy to generate molecules considered solar fuels. Undoubtedly, designing a module with similar features would be a considerable step to mitigate the effects of climate change by reducing the consumption of fossil fuels. However, the water oxidation reaction is the bottleneck in artificial photosynthesis since it involves not only the removal of four electrons and four protons from two solvent molecules but also a specific molecular pathway for the formation of O‒O bonds. Nature solved this problem using the oxygen-evolving center in PSII, and scientists have found inspiration in this model to design an efficient catalyst based on cheap and abundant metals.
We have also seen how POMs feature some essential characteristics required to design a biomimetic OEC that can be functionally integrated into a photosynthetic device. However, current research has focused mainly on the catalytic evaluation against water oxidation that different TMSPs present. Despite this, we can draw valuable conclusions from the systems published since 2008 with Ru4-SiPOM and point out some characteristics that can be considered for the synthetic design and catalytic evaluation of new TMSPs.
First, systems based on transition metals as the active site of the molecule are essential to achieve the incorporation of water molecules and sustain the multi-electronic events required for the water oxidation reaction. Second, the nature of this active site (i.e., structure, nuclearity, type of metal ion, oxidation states) must strongly influence the catalytic performance. Although catalytic centers based on Co, Ni, and Mn with cubic structures analogous to the natural OEC stabilized with POMs have been reported, in most examples, these active sites are saturated by the inorganic scaffolds, making incorporating water molecules difficult. On the other hand, some sandwich-type structures or open systems present active centers that contain water molecules per se, favoring the oxidation reaction through nucleophilic mechanisms. Third, the POMs that support the active center can be involved in the catalytic process when active redox heteroatoms are included in their structure, as in the case of the vanadate anion. In this sense, substituting heteroatoms based on P or Si by V can modify the electronic behavior of the system through charge transfers, affecting the catalytic performance.
The points made above must be addressed by rational experimental work. The synthetic strategies of most of the compounds discussed consist of conventional one-pot self-assembly reactions. However, the structural features of the TMSPs obtained by this method dramatically differ from those obtained by hydrothermal strategies, as evidenced in tungsten-based lacunary species. Thus, hydrothermal synthesis may increase the number of accessible WOC candidates. On the other hand, an impressive number of TMSPs in the crystallographic literature have yet to be studied as catalysts, so a statistical analysis supported by the theoretical studies performed on specific POM structures can serve as a basis for selecting ideal complexes for oxygen evolution.
A few examples of artificial photosynthetic schemes include materials based on POMs since this field is still in its infancy. However, considering the characteristics of these molecules, it is not an exaggeration to assume that in the future, these materials will be integrated into photosynthetic platforms as possible WOCs or to carry out the hydrogen evolution reaction. Furthermore, they could also be used to form membranes that separate the different components of photosynthetic units and solve the oxidation problem of Ru-based organometallic dyes used as light-harvesting centers. Finally, advances in the coming years on the natural photosynthetic process, specifically on PSII and the OEC, will directly impact the development of a new generation of artificial systems that can be integrated into a functional device for generating solar fuels. After all, natural photosynthesis is more than two billion years ahead of us.
ES-L: Investigation, Writing–original draft, Writing–review and editing. RF: Investigation, Writing–original draft. IC: Funding acquisition, Investigation, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. IC thanks Conahcyt (A1-S-8682) and DGAPA-PAPIIT (IN217020) for financial support.
ES-L gratefully acknowledges DGAPA-UNAM for the postdoctoral scholarship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Chl a, Chlorophyll a; CV, Cyclic voltammetry; DFT, Density functional theory; DLS, Dynamic light scattering; EDX, Energy-dispersive X-ray analysis; HER, Hydrogen evolution reaction; iCHELLs, Synthetic inorganic cells; IPCC, International Panel on Climate Change; LBL, Layer-by-layer technique; OEC, Oxygen-evolving center; OER, Oxygen evolution reaction; PCET, Proton-coupled electron transfer processes; POMs, Polyoxometalates; POM-WOCs, Polyoxometalate water oxidation catalysts; PS, Photosensitizer; PSI, Photosystem I; PSII, Photosystem II; SEM, Scanning electron microscope; TBA, Tetrabutylammonium; TEM, Transmission electron microscopy; THpANO3, tetra-n-heptylammonium nitrate (THpANO3) toluene extraction; TMSPs, Transition-metal-substituted polyoxometalates; TOF, Turnover frequency; TON, Turnover number; WNA, Water nucleophilic attack; WOCs, Water oxidation catalysts; XPS, X-ray photoelectron spectroscopy.
Al-Ghussain, L. (2018). Global warming: review on driving forces and mitigation. Environ. Prog. Sustain. Energy 38 (1), 13–21. doi:10.1002/ep.13041
Al-Oweini, R., Sartorel, A., Bassil, B. S., Natali, M., Berardi, S., Scandola, F., et al. (2014). Photocatalytic water oxidation by a mixed-valent MnIII3MnIVO3Manganese oxo core that mimics the natural oxygen-evolving center. Angew. Chem. 126 (42), 11364–11367. doi:10.1002/ange.201404664
Al-Sayed, E., Nandan, S. P., Tanuhadi, E., Giester, G., Arrigoni, M., Madsen, G. K. H., et al. (2021). Phosphate-templated encapsulation of a {CoII4O4} cubane in germanotungstates as carbon-free homogeneous water oxidation photocatalysts. ChemSusChem 14 (12), 2529–2536. doi:10.1002/cssc.202100506
Alstrum-Acevedo, J. H., Brennaman, M. K., and Meyer, T. J. (2005). Chemical approaches to artificial photosynthesis. 2. Inorg. Chem. 44 (20), 6802–6827. doi:10.1021/ic050904r
An, H., Hou, Y., Wang, L., Zhang, Y., Yang, W., and Chang, S. (2017). Evans–showell-type polyoxometalates constructing high-dimensional inorganic–organic hybrid compounds with copper–organic coordination complexes: synthesis and oxidation catalysis. Inorg. Chem. 56 (19), 11619–11632. doi:10.1021/acs.inorgchem.7b01564
Anwar, N., Sartorel, A., Yaqub, M., Wearen, K., Laffir, F., Armstrong, G., et al. (2014). Surface immobilization of a tetra-ruthenium substituted polyoxometalate water oxidation catalyst through the employment of conducting polypyrrole and the layer-by-layer (LBL) technique. ACS Appl. Mat. Interfaces 6 (11), 8022–8031. doi:10.1021/am405295c
Arens, J. T., Blasco-Ahicart, M., Azmani, K., Soriano-López, J., García-Eguizábal, A., Poblet, J. M., et al. (2020). Water oxidation electrocatalysis in acidic media with Co-containing polyoxometalates. J. Catal. 389, 345–351. doi:10.1016/j.jcat.2020.06.006
Armstrong, F. A. (2008). Why did Nature choose manganese to make oxygen? Philosophical Trans. R. Soc. B Biol. Sci. 363 (1494), 1263–1270. doi:10.1098/rstb.2007.2223
Azmani, K., Besora, M., Soriano-López, J., Landolsi, M., Teillout, A.-L., de Oliveira, P., et al. (2021). Understanding polyoxometalates as water oxidation catalysts through iron vs cobalt reactivity. Chem. Sci. 12 (25), 8755–8766. doi:10.1039/D1SC01016F
Baker, L. C., and McCutcheon, T. P. (1956). Heteropoly salts containing cobalt and hexavalent tungsten in the Anion1. J. Am. Chem. Soc. 78, 4503–4510. doi:10.1021/ja01599a001
Balzani, V., Credi, A., and Venturi, M. (2008). Photochemical conversion of solar energy. ChemSusChem 1 (1-2), 26–58. doi:10.1002/cssc.200700087
Barber, J. (2002). P680: what is it and where is it? Bioelectrochemistry 55 (1-2), 135–138. doi:10.1016/s1567-5394(01)00141-4
Barber, J. (2003). Photosystem II: the engine of life. Q. Rev. Biophysics 36 (1), 71–89. doi:10.1017/S0033583502003839
Barber, J. (2009). Photosynthetic energy conversion: natural and artificial. Chem. Soc. Rev. 38 (1), 185–196. doi:10.1039/b802262n
Barber, J., and Archer, M. D. (2001). P680, the primary electron donor of photosystem II. J. Photochem. Photobiol. A Chem. 142 (2-3), 97–106. doi:10.1016/s1010-6030(01)00503-2
Barber, J., and Tran, P. D. (2013). From natural to artificial photosynthesis. J. R. Soc. Interface 10 (81), 20120984. doi:10.1098/rsif.2012.0984
Bassil, B. S., Dickman, M. H., Reicke, M., Kortz, U., Keita, B., and Nadjo, L. (2006). Transition metal containing decatungstosilicate dimer [M(H2O)2(γ-SiW10O35)2]10−(M = Mn2+, Co2+, Ni2+). Dalton Trans. 35, 4253–4259. doi:10.1039/b606911h
Bensaid, S., Ottone Melis, C., Hernández, S., Armandi, M., Esposito, S., Saracco, G., et al. (2017). A simple model for a complex system: kinetics of water oxidation with the [Ru(bpy)3]2+/S2O82− photosystem as catalyzed by Mn2O3 under different illumination conditions. Chem. Eng. J. 311, 143–152. doi:10.1016/j.cej.2016.11.087
Berardi, S., Drouet, S., Francàs, L., Gimbert-Suriñach, C., Guttentag, M., Richmond, C., et al. (2014). Molecular artificial photosynthesis. Chem. Soc. Rev. 43 (22), 7501–7519. doi:10.1039/c3cs60405e
Besson, C., Huang, Z., Geletii, Y. V., Lense, S., Hardcastle, K. I., Musaev, D. G., et al. (2010). Cs9[(γ-PW10O36)2Ru4O5(OH)(H2O)4], a new all-inorganic, soluble catalyst for the efficient visible-light-driven oxidation of water. Chem. Commun. 46 (16), 2784. doi:10.1039/b926064a
Bijelic, A., Aureliano, M., and Rompel, A. (2019). Polyoxometalates as potential next-generation metallodrugs in the combat against cancer. Angew. Chem. Int. Ed. 58 (10), 2980–2999. doi:10.1002/anie.201803868
Bonchio, M., Carraro, M., Sartorel, A., Scorrano, G., and Kortz, U. (2006). Bio-inspired oxidations with polyoxometalate catalysts. J. Mol. Catal. A Chem. 251 (1-2), 93–99. doi:10.1016/j.molcata.2006.02.034
Bösing, M., Nöh, A., Loose, I., and Krebs, B. (1998). Highly efficient catalysts in directed oxygen-transfer processes: synthesis, structures of novel manganese-containing heteropolyanions, and applications in regioselective epoxidation of dienes with hydrogen peroxide. J. Am. Chem. Soc. 120 (29), 7252–7259. doi:10.1021/ja974281v
Bozal-Ginesta, C., and Durrant, J. R. (2019). Artificial photosynthesis – concluding remarks. Faraday Discuss. 215 (0), 439–451. doi:10.1039/C9FD00076C
Brimblecombe, R., Koo, A., Dismukes, G. C., Swiegers, G. F., and Spiccia, L. (2010). Solar driven water oxidation by a bioinspired manganese molecular catalyst. J. Am. Chem. Soc. 132 (9), 2892–2894. doi:10.1021/ja910055a
Brinkert, K. (2018). “Energy conversion in natural and artificial photosynthesis,” in Springer series in chemical physics (Cham: Springer). doi:10.1007/978-3-319-77980-5
Brudvig, G. W. (2007). Water oxidation chemistry of photosystem II. Philosophical Trans. R. Soc. B Biol. Sci. 363 (1494), 1211–1219. doi:10.1098/rstb.2007.2217
Cao, Y., Chen, Q., Shen, C., and He, L. (2019). Polyoxometalate-based catalysts for CO2 conversion. Molecules 24 (11), 2069. doi:10.3390/molecules24112069
Capone, M., Narzi, D., and Guidoni, L. (2021). Mechanism of oxygen evolution and Mn4CaO5 cluster restoration in the natural water-oxidizing catalyst. Biochemistry 60 (30), 2341–2348. doi:10.1021/acs.biochem.1c00226
Car, P.-E., Guttentag, M., Baldridge, K. K., Alberto, R., and Patzke, G. R. (2012). Synthesis and characterization of open and sandwich-type polyoxometalates reveals visible-light-driven water oxidation via POM-photosensitizer complexes. Green Chem. 14 (6), 1680. doi:10.1039/c2gc16646a
Cardona, T., Sedoud, A., Cox, N., and Rutherford, W. (2012). Charge separation in Photosystem II: a comparative and evolutionary overview. Biochimica Biophysica Acta (BBA) - Bioenergetics 1817 (1), 26–43. doi:10.1016/j.bbabio.2011.07.012
Chabi, S., Papadantonakis, K. M., Lewis, N. S., and Freund, M. S. (2017). Membranes for artificial photosynthesis. Energy Environ. Sci. 10 (6), 1320–1338. doi:10.1039/c7ee00294g
Chen, C., Chen, Y., Yao, R., Li, Y., and Zhang, C. (2019). Artificial Mn 4 Ca clusters with exchangeable solvent molecules mimicking the oxygen-evolving center in photosynthesis. Angew. Chem. 131 (12), 3979–3982. doi:10.1002/ange.201814440
Chen, C., Li, Y., Zhao, G., Yao, R., and Zhang, C. (2017). Natural and artificial Mn4Ca cluster for the water splitting reaction. ChemSusChem 10 (22), 4403–4408. doi:10.1002/cssc.201701371
Chen, C., Xu, B., Yao, R., Chen, Y., and Zhang, C. (2022). Synthesizing mechanism of the Mn4Ca cluster mimicking the oxygen-evolving center in photosynthesis. ChemSusChem 15 (6), e202102661. doi:10.1002/cssc.202102661
Chen, C., Zhang, C., Dong, H., and Zhao, J. (2015). Artificial synthetic MnIVCa–oxido complexes mimic the oxygen-evolving complex in photosystem II. Dalton Trans. 44 (10), 4431–4435. doi:10.1039/c4dt03459g
Chen, Q., Guo, Y., Yu, Y.-H., and Zhang, M. (2021). Bioinspired molecular clusters for water oxidation. Coord. Chem. Rev. 448, 214164. doi:10.1016/j.ccr.2021.214164
Chen, W.-C., Wang, X.-L., Qin, C., Shao, K.-Z., Su, Z.-M., and Wang, E.-B. (2016). A carbon-free polyoxometalate molecular catalyst with a cobalt–arsenic core for visible light-driven water oxidation. Chem. Commun. 52 (61), 9514–9517. doi:10.1039/c6cc03763a
Clemente-Juan, J. M., and Coronado, E. (1999). Magnetic clusters from polyoxometalate complexes. Coord. Chem. Rev. 193-195, 361–394. doi:10.1016/s0010-8545(99)00170-8
Clemente-Juan, J. M., Coronado, E., and Gaita-Ariño, A. (2012). Magnetic polyoxometalates: from molecular magnetism to molecular spintronics and quantum computing. Chem. Soc. Rev. 41 (22), 7464. doi:10.1039/c2cs35205b
Coehn, A., and Gläser, M. (1902). Studien über die Bildung von Metalloxyden I. Über das anodische Verhalten von Kobalt-und Nickel-Lösungen. Z. für Anorg. Chem. 33 (1), 9–24. doi:10.1002/zaac.19020330105
Concepcion, J. J., House, R. L., Papanikolas, J. M., and Meyer, T. J. (2012). Chemical approaches to artificial photosynthesis. Proc. Natl. Acad. Sci. U.S.A. 109 (39), 15560–15564. doi:10.1073/pnas.1212254109
Concepcion, J. J., Jurss, J. W., Norris, M. R., Chen, Z., Templeton, J. L., and Meyer, T. J. (2010). Catalytic water oxidation by single-site ruthenium catalysts. Inorg. Chem. 49 (4), 1277–1279. doi:10.1021/ic901437e
Cooper, G. J. T., and Cronin, L. (2009). Real-time direction control of self fabricating polyoxometalate-based microtubes. J. Am. Chem. Soc. 131 (24), 8368–8369. doi:10.1021/ja902684b
Cooper, G. J. T., Kitson, P. J., Winter, R., Zagnoni, M., Long, D.-L., and Cronin, L. (2011). Modular redox-active inorganic chemical cells: iCHELLs. Angew. Chem. 123 (44), 0557–10560. doi:10.1002/ange.201105068
Cox, N., Pantazis, D. A., and Lubitz, W. (2020). Current understanding of the mechanism of water oxidation in photosystem II and its relation to XFEL data. Annu. Rev. Biochem. 89 (1), 795–820. doi:10.1146/annurev-biochem-011520-104801
Cox, N., Pantazis, D. A., Neese, F., and Lubitz, W. (2013). Biological water oxidation. Acc. Chem. Res. 46 (7), 1588–1596. doi:10.1021/ar3003249
Crabtree, G. W., and Lewis, N. S. (2007). Solar energy conversion. Phys. Today 60 (3), 37–42. doi:10.1063/1.2718755
Creutzig, F., Agoston, P., Goldschmidt, J. C., Luderer, G., Nemet, G., and Pietzcker, R. C. (2017). The underestimated potential of solar energy to mitigate climate change. Nat. Energy 2 (9), 17140. doi:10.1038/nenergy.2017.140
Das, V., Kaushik, R., and Hussain, F. (2020). Heterometallic 3d-4f polyoxometalates: an emerging field with structural diversity to multiple applications. Coord. Chem. Rev. 413, 213271. doi:10.1016/j.ccr.2020.213271
Dashtian, K., Shahsavarifar, S., Usman, M., Joseph, Y., Ganjali, M. R., Yin, Z., et al. (2024). A comprehensive review on advances in polyoxometalate based materials for electrochemical water splitting. Coord. Chem. Rev. 504, 215644. doi:10.1016/j.ccr.2023.215644
Dau, H., and Haumann, M. (2008). The manganese complex of photosystem II in its reaction cycle—basic framework and possible realization at the atomic level. Coord. Chem. Rev. 252 (3-4), 273–295. doi:10.1016/j.ccr.2007.09.001
Dogutan, D. K., and Nocera, D. G. (2019). Artificial photosynthesis at efficiencies greatly exceeding that of natural photosynthesis. Acc. Chem. Res. 52 (11), 3143–3148. doi:10.1021/acs.accounts.9b00380
Du, P., and Eisenberg, R. (2012). Catalysts made of earth-abundant elements (Co, Ni, Fe) for water splitting: recent progress and future challenges. Energy Environ. Sci. 5 (3), 6012. doi:10.1039/c2ee03250c
Du, X., Ding, Y., Song, F., Ma, B., Zhao, J., and Song, J. (2015a). Efficient photocatalytic water oxidation catalyzed by polyoxometalate [Fe11(H2O)14(OH)2(W3O10)2(α-SbW9O33)6]27− based on abundant metals. Chem. Commun. 51 (73), 13925–13928. doi:10.1039/C5CC04551G
Du, X., Zhao, J., Mi, J., Ding, Y., Zhou, P., Ma, B., et al. (2015b). Efficient photocatalytic H2 evolution catalyzed by an unprecedented robust molecular semiconductor {Fe11} nanocluster without cocatalysts at neutral conditions. Nano Energy 16, 247–255. doi:10.1016/j.nanoen.2015.06.025
El-Khouly, M. E., El-Mohsnawy, E., and Fukuzumi, S. (2017). Solar energy conversion: from natural to artificial photosynthesis. J. Photochem. Photobiol. C Photochem. Rev. 31, 36–83. doi:10.1016/j.jphotochemrev.2017.02.001
Fawzy, S., Osman, A. I., Doran, J., and Rooney, D. W. (2020). Strategies for mitigation of climate change: a review. Environ. Chem. Lett. 18 (18), 2069–2094. doi:10.1007/s10311-020-01059-w
Ferreira, K. N., Iverson, T. M., Maghlaoui, K., Barber, J., and Iwata, S. (2004). Architecture of the photosynthetic oxygen-evolving center. Science 303 (5665), 1831–1838. doi:10.1126/science.1093087
Fischer, W. W., Hemp, J., and Johnson, J. E. (2016). Evolution of oxygenic photosynthesis. Annu. Rev. Earth Planet. Sci. 44 (1), 647–683. doi:10.1146/annurev-earth-060313-054810
Gao, D., Trentin, I., Schwiedrzik, L., González, L., and Streb, C. (2019). The reactivity and stability of polyoxometalate water oxidation electrocatalysts. Molecules 25 (1), 157. doi:10.3390/molecules25010157
Gao, J., Cao, S., Tay, Q., Liu, Y., Yu, L., Ye, K., et al. (2013). Molecule-based water-oxidation catalysts (WOCs): cluster-size-dependent dye-sensitized polyoxometalates for visible-light-driven O2 evolution. Sci. Rep. 3 (1), 1853. doi:10.1038/srep01853
Gao, N., Sun, H., Dong, K., Ren, J., Duan, T., Xu, C., et al. (2014). Transition-metal-substituted polyoxometalate derivatives as functional anti-amyloid agents for Alzheimer’s disease. Nat. Commun. 5 (1), 3422. doi:10.1038/ncomms4422
García-Álvarez, A. C., Gamboa-Ramírez, S., Martínez-Otero, D., Orio, M., and Castillo, I. (2021). Self-assembled nickel cubanes as oxygen evolution catalysts. Chem. Commun. 57 (69), 8608–8611. doi:10.1039/d1cc03227e
García-López, E. I., Marcì, G., Krivtsov, I., Casado Espina, J., Liotta, L. F., and Serrano, A. (2019). Local structure of supported Keggin and wells–dawson heteropolyacids and its influence on the catalytic activity. J. Phys. Chem. C 123 (32), 19513–19527. doi:10.1021/acs.jpcc.9b03659
Garrido-Barros, P., Gimbert-Suriñach, C., Matheu, R., Sala, X., and Llobet, A. (2017). How to make an efficient and robust molecular catalyst for water oxidation. Chem. Soc. Rev. 46 (20), 6088–6098. doi:10.1039/C7CS00248C
Geletii, Y. V., Botar, B., Kögerler, P., Hillesheim, D. A., Musaev, D. G., and Hill, C. L. (2008). An all-inorganic, stable, and highly active tetraruthenium homogeneous catalyst for water oxidation. Angew. Chem. 120 (21), 3960–3963. doi:10.1002/ange.200705652
Geletii, Y. V., Huang, Z., Hou, Y., Musaev, D. G., Lian, T., and Hill, C. L. (2009). Homogeneous light-driven water oxidation catalyzed by a tetraruthenium complex with all inorganic ligands. J. Am. Chem. Soc. 131 (22), 7522–7523. doi:10.1021/ja901373m
Geletii, Y. V., Yin, Q., Hou, Y., Huang, Z., Ma, H., Song, J., et al. (2011). Polyoxometalates in the design of effective and tunable water oxidation catalysts. Israel J. Chem. 51 (2), 238–246. doi:10.1002/ijch.201100021
Gilbert, J., Eggleston, D. S., Murphy, W., Geselowitz, D. A., Gersten, S. W., Hodgson, D. J., et al. (1985). Structure and redox properties of the water-oxidation catalyst [(bpy)2(OH2)RuORu(OH2)(bpy)2]4+. J. Am. Chem. Soc. 107 (13), 3855–3864. doi:10.1021/ja00299a017
Goudie, A. (2019). Human impact on the natural environment: past, present and future. Hoboken, NJ: Wiley Blackwell.
Goura, J., Bassil, B. S., Ma, X., Rajan, A., Moreno-Pineda, E., Schnack, J., et al. (2021). NiII36-Containing 54-tungsto-6-silicate: synthesis, structure, magnetic and electrochemical studies. Chem. – A Eur. J. 27 (61), 15081–15085. doi:10.1002/chem.202102973
Gumerova, N. I., and Rompel, A. (2023). Speciation atlas of polyoxometalates in aqueous solutions. Sci. Adv. 9 (25), eadi0814. doi:10.1126/sciadv.adi0814
Gust, D., Moore, T. A., and Moore, A. L. (2001). Mimicking photosynthetic solar energy transduction. Acc. Chem. Res. 34 (1), 40–48. doi:10.1021/ar9801301
Gust, D., Moore, T. A., and Moore, A. L. (2012). Realizing artificial photosynthesis. Faraday Discuss. 155, 9–26. doi:10.1039/c1fd00110h
Haider, A., Bassil, B. S., Soriano-López, J., Qasim, H. M., Sáenz de Pipaón, C., Ibrahim, M., et al. (2019). 9-Cobalt(II)-Containing 27-Tungsto-3-germanate(IV): synthesis, structure, computational modeling, and heterogeneous water oxidation catalysis. Inorg. Chem. 58 (17), 11308–11316. doi:10.1021/acs.inorgchem.9b01495
Han, Q., and Ding, Y. (2018). Recent advances in the field of light-driven water oxidation catalyzed by transition-metal substituted polyoxometalates. Dalton Trans. 47 (25), 8180–8188. doi:10.1039/C8DT01291A
Han, X.-B., Li, Y.-G., Zhang, Z.-M., Tan, H.-Q., Lu, Y., and Wang, E.-B. (2015). Polyoxometalate-based nickel clusters as visible light-driven water oxidation catalysts. J. Am. Chem. Soc. 137 (16), 5486–5493. doi:10.1021/jacs.5b01329
Han, X.-B., Zhang, Z.-M., Zhang, T., Li, Y.-G., Lin, W., You, W., et al. (2014). Polyoxometalate-based cobalt–phosphate molecular catalysts for visible light-driven water oxidation. J. Am. Chem. Soc. 136 (14), 5359–5366. doi:10.1021/ja412886e
Hansen, J., Ruedy, R., Sato, M., and Lo, K. (2010). Global surface temperature change. Rev. Geophys. 48 (4). doi:10.1029/2010rg000345
Hill, C. L., and Prosser-McCartha, C. M. (1995). Homogeneous catalysis by transition metal oxygen anion clusters. Coord. Chem. Rev. 143, 407–455. doi:10.1016/0010-8545(95)01141-b
Horn, M. R., Singh, A., Alomari, S., Goberna-Ferrón, S., Benages-Vilau, R., Chodankar, N., et al. (2021). Polyoxometalates (POMs): from electroactive clusters to energy materials. Energy Environ. Sci. 14 (4), 1652–1700. doi:10.1039/d0ee03407j
Howells, A. R., Sankarraj, A., and Shannon, C. (2004). A diruthenium-substituted polyoxometalate as an electrocatalyst for oxygen generation. J. Am. Chem. Soc. 126 (39), 12258–12259. doi:10.1021/ja0495821
Hu, Q., Meng, X., Dong, Y., Han, Q., Wang, Y., and Ding, Y. (2019). A stable iron-containing polyoxometalate coupled with semiconductor for efficient photocatalytic water oxidation under acidic condition. Chem. Commun. 55 (78), 11778–11781. doi:10.1039/c9cc05726a
Huang, Z., Geletii, Y. V., Musaev, D. G., Hill, C. L., and Lian, T. (2012). Spectroscopic studies of light-driven water oxidation catalyzed by polyoxometalates. Ind. Eng. Chem. Res. 51 (37), 11850–11859. doi:10.1021/ie202950h
Imahori, H., Mori, Y., and Matano, Y. (2003). Nanostructured artificial photosynthesis. J. Photochem. Photobiol. C Photochem. Rev. 4 (1), 51–83. doi:10.1016/S1389-5567(03)00004-2
Inganas, O., and Sundstrom, V. (2015). Solar energy for electricity and fuels. Ambio 45 (S1), 15–23. doi:10.1007/s13280-015-0729-6
IPCC (2021). Climate change 2021: the physical science basis. Available at: https://www.ipcc.ch/report/ar6/wg1/.
Jeon, D., Kim, H., Lee, C., Han, Y.-J., Gu, M., Kim, B. S., et al. (2017). Layer-by-Layer assembly of polyoxometalates for photoelectrochemical (PEC) water splitting: toward modular PEC devices. ACS Appl. Mat. Interfaces 9 (46), 40151–40161. doi:10.1021/acsami.7b09416
Jiao, F., and Frei, H. (2009). Nanostructured cobalt oxide clusters in mesoporous silica as efficient oxygen-evolving catalysts. Angew. Chem. 121 (10), 1873–1876. doi:10.1002/ange.200805534
Kaledin, A. L., Huang, Z., Geletii, Y. V., Lian, T., Hill, C. L., and Musaev, D. G. (2010). Insights into photoinduced electron transfer between [Ru(bpy)3]2+ and [S2O8]2− in water: computational and experimental studies. J. Phys. Chem. A 114 (1), 73–80. doi:10.1021/jp908409n
Kalyanasundaram, K., and Graetzel, M. (2010). Artificial photosynthesis: biomimetic approaches to solar energy conversion and storage. Curr. Opin. Biotechnol. 21 (3), 298–310. doi:10.1016/j.copbio.2010.03.021
Kamdar, J., and Grotjahn, D. (2019). An overview of significant achievements in ruthenium-based molecular water oxidation catalysis. Molecules 24 (3), 494. doi:10.3390/molecules24030494
Kamiya, N., and Shen, J.-R. (2002). Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7-Å resolution. Proc. Natl. Acad. Sci. U.S.A. 100 (1), 98–103. doi:10.1073/pnas.0135651100
Kanan, M. W., and Nocera, D. G. (2008). In situ Formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+. Science 321 (5892), 1072–1075. doi:10.1126/science.1162018
Kannan, N., and Vakeesan, D. (2016). Solar energy for future world: - a review. Renew. Sustain. Energy Rev. 62 (1364-0321), 1092–1105. doi:10.1016/j.rser.2016.05.022
Kato, M., Zhang, J. Z., Paul, N., and Reisner, E. (2014). Protein film photoelectrochemistry of the water oxidation enzyme photosystem II. Chem. Soc. Rev. 43 (18), 6485–6497. doi:10.1039/c4cs00031e
Kaushik, R., Sakla, R., Amilan Jose, D., and Ghosh, A. (2020). Giant iron polyoxometalate that works as a catalyst for water oxidation. New J. Chem. 44 (9), 3764–3770. doi:10.1039/C9NJ05690D
Keijer, T., Bouwens, T., Hessels, J., and Reek, J. H. (2021). Supramolecular strategies in artificial photosynthesis. Chem. Sci. 12 (1), 50–70. doi:10.1039/D0SC03715J
Kern, J., Chatterjee, R., Young, I. D., Fuller, F. D., Lassalle, L., Ibrahim, M., et al. (2018). Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 563 (7731), 421–425. doi:10.1038/s41586-018-0681-2
Khandekar, M. L., Murty, T. S., and Chittibabu, P. (2005). The global warming debate: a review of the state of science. Pure Appl. Geophys. 162 (8-9), 1557–1586. doi:10.1007/s00024-005-2683-x
Kholdeeva, O. (2004). Co-containing polyoxometalate-based heterogeneous catalysts for the selective aerobic oxidation of aldehydes under ambient conditions. J. Catal. 226 (2), 363–371. doi:10.1016/j.jcat.2004.05.032
Kok, B., Forbush, B., and McGloin, M. (1970). COOPERATION OF CHARGES IN PHOTOSYNTHETIC O2 EVOLUTION–I. A LINEAR FOUR STEP MECHANISM. Photochem. Photobiol. 11 (6), 457–475. doi:10.1111/j.1751-1097.1970.tb06017.x
Krewald, V., Retegan, M., Cox, N., Messinger, J., Lubitz, W., DeBeer, S., et al. (2015b). Metal oxidation states in biological water splitting. Chem. Sci. 6 (3), 1676–1695. doi:10.1039/c4sc03720k
Krewald, V., Retegan, M., and Pantazis, D. A. (2016). Principles of natural photosynthesis. Top. Curr. Chem. 371, 23–48. doi:10.1007/128_2015_645
Kupitz, C., Basu, S., Grotjohann, I., Fromme, R., Zatsepin, N. A., Rendek, K. N., et al. (2014). Serial time-resolved crystallography of photosystem II using a femtosecond X-ray laser. Nature 513 (7517), 261–265. doi:10.1038/nature13453
Lang, Z.-L., Yang, G.-C., Ma, N.-N., Wen, S.-Z., Yan, L.-K., Guan, W., et al. (2013). DFT characterization on the mechanism of water splitting catalyzed by single-Ru-substituted polyoxometalates. Dalton Trans. 42 (29), 10617–10625. doi:10.1039/C3DT50666E
Lauinger, S. M., Yin, Q. R., Geletii, Y. V., and Hill, C. (2017). Polyoxometalate multielectron catalysts in solar fuel production. Adv. Inorg. Chem., 117–154. doi:10.1016/bs.adioch.2016.12.002
Letcher, T. M. (2019). Managing global warming: an interface of technology and human issues. London, United Kingdom; San Diego, Ca, United States: Academic Press, An Imprint of Elsevier.
Lewis, N. S., and Nocera, D. G. (2006). Powering the planet: chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. 103 (43), 15729–15735. doi:10.1073/pnas.0603395103
Li, F., and Xu, L. (2011). Coordination assemblies of polyoxomolybdate cluster framework: from labile building blocks to stable functional materials. Dalton Trans. 40 (16), 4024. doi:10.1039/c0dt00691b
Li, G., Ding, Y., Wang, J., Wang, X., and Suo, J. (2007). New progress of Keggin and Wells–Dawson type polyoxometalates catalyze acid and oxidative reactions. J. Mol. Catal. A Chem. 262 (1-2), 67–76. doi:10.1016/j.molcata.2006.08.067
Li, Y., Yao, R., Chen, Y., Xu, B., Chen, C., and Zhang, C. (2020). Mimicking the catalytic center for the water-splitting reaction in photosystem II. Catalysts 10 (2), 185. doi:10.3390/catal10020185
Limburg, B., Bouwman, E., and Bonnet, S. (2012). Molecular water oxidation catalysts based on transition metals and their decomposition pathways. Coord. Chem. Rev. 256 (15-16), 1451–1467. doi:10.1016/j.ccr.2012.02.021
Liu, B., Glass, E. N., Wang, R., Cui, Y., Harada, Y., Huang, D., et al. (2018). Cobalt-to-vanadium charge transfer in polyoxometalate water oxidation catalysts revealed by 2p3d resonant inelastic X-ray scattering. Phys. Chem. Chem. Phys. 20 (6), 4554–4562. doi:10.1039/c7cp06786k
Liu, J., Han, Q., Chen, L., and Zhao, J. (2016). A brief review of the crucial progress on heterometallic polyoxotungstates in the past decade. CrystEngComm 18 (6), 842–862. doi:10.1039/c5ce02378e
Liu, T., Zhang, B., and Sun, L. (2019). Iron-based molecular water oxidation catalysts: abundant, cheap, and promising. Chem. – Asian J. 14 (1), 31–43. doi:10.1002/asia.201801253
Liu, Y., Chen, G., Yiu, S.-M., Wong, C.-Y., and Lau, T.-C. (2017). Intermediates in the oxidative degradation of a ruthenium-bound 2,2′-bipyridyl-phenoxy ligand during catalytic water oxidation. ChemCatChem 10 (3), 501–504. doi:10.1002/cctc.201701319
Lloyd, E. A., and Winsberg, E. (2018). Climate modelling: philosophical and conceptual issues. Cham: Palgrave Macmillan.
Long, D.-L., Burkholder, E., and Cronin, L. (2007). Polyoxometalate clusters, nanostructures and materials: from self assembly to designer materials and devices. Chem. Soc. Rev. 36 (1), 105–121. doi:10.1039/b502666k
Long, D.-L., Tsunashima, R., and Cronin, L. (2010). Polyoxometalates: building blocks for functional nanoscale systems. Angew. Chem. Int. Ed. 49 (10), 1736–1758. doi:10.1002/anie.200902483
Lubitz, W., Chrysina, M., and Cox, N. (2019). Water oxidation in photosystem II. Photosynth. Res. 142 (1), 105–125. doi:10.1007/s11120-019-00648-3
Lubitz, W., Reijerse, E. J., and Messinger, J. (2008). Solar water-splitting into H2 and O2: design principles of photosystem II and hydrogenases. Energy Environ. Sci. 1 (1), 15. doi:10.1039/b808792j
Lv, H., Geletii, Y. V., Zhao, C., Vickers, J. W., Zhu, G., Luo, Z., et al. (2012). Polyoxometalate water oxidation catalysts and the production of green fuel. Chem. Soc. Rev. 41 (22), 7572. doi:10.1039/c2cs35292c
Lv, H., Song, J., Geletii, Y. V., Vickers, J. W., Sumliner, J. M., Musaev, D. G., et al. (2014). An exceptionally fast homogeneous carbon-free cobalt-based water oxidation catalyst. J. Am. Chem. Soc. 136 (26), 9268–9271. doi:10.1021/ja5045488
Lynas, M., Houlton, B. Z., and Perry, S. (2021). Greater than 99% consensus on human caused climate change in the peer-reviewed scientific literature. Environ. Res. Lett. 16 (11), 114005. doi:10.1088/1748-9326/ac2966
Matheu, R., Garrido-Barros, P., Gil-Sepulcre, M., Ertem, M. Z., Sala, X., Gimbert-Suriñach, C., et al. (2019). The development of molecular water oxidation catalysts. Nat. Rev. Chem. 3 (5), 331–341. doi:10.1038/s41570-019-0096-0
Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S. L., Péan, C., Berger, S., et al. (2021). Climate change 2021 the physical science basis: assessment working group I contribution to the IPCC sixth assessment report.
McConnell, I., Li, G., and Brudvig, G. W. (2010). Energy conversion in natural and artificial photosynthesis. Chem. Biol. 17 (5), 434–447. doi:10.1016/j.chembiol.2010.05.005
Meyer, T. J. (1989). Chemical approaches to artificial photosynthesis. Acc. Chem. Res. 22 (5), 163–170. doi:10.1021/ar00161a001
Mirkovic, T., Ostroumov, E. E., Anna, J. M., van Grondelle, R., Govindjee, N., and Scholes, G. D. (2017). Light absorption and energy transfer in the antenna complexes of photosynthetic organisms. Chem. Rev. 117 (2), 249–293. doi:10.1021/acs.chemrev.6b00002
Mitchell, S. G., Molina, P., Khanra, S., Miras, H. N., Prescimone, A., Cooper, G. J. T., et al. (2011). A mixed-valence manganese cubane trapped by inequivalent trilacunary polyoxometalate ligands. Angew. Chem. 123 (39), 9320–9323. doi:10.1002/ange.201102727
Mizuno, N., Yamaguchi, K., and Kamata, K. (2005). Epoxidation of olefins with hydrogen peroxide catalyzed by polyoxometalates. Coord. Chem. Rev. 249 (17-18), 1944–1956. doi:10.1016/j.ccr.2004.11.019
Mullins, C. S., and Pecoraro, V. L. (2008). Reflections on small molecule manganese models that seek to mimic photosynthetic water oxidation chemistry. Coord. Chem. Rev. 252 (3), 416–443. doi:10.1016/j.ccr.2007.07.021
Mürtz, S. D., Raabe, J.-C., Poller, M. J., Palkovits, R., Albert, J., and Kurig, N. (2024). Transition-metal substituted polyoxometalates as soluble redox mediators in electrocatalytic biomass conversion. Chem. Cat. Chem. doi:10.1002/cctc.202301632
Najafpour, M. M., Heidari, S., Balaghi, S. E., Hołyńska, M., Sadr, M. H., Soltani, B., et al. (2017). Proposed mechanisms for water oxidation by Photosystem II and nanosized manganese oxides. Biochimica Biophysica Acta (BBA) - Bioenergetics 1858 (2), 156–174. doi:10.1016/j.bbabio.2016.11.007
Nakanishi, K., Cooper, G. J. T., Points, L. J., Bloor, L. G., Ohba, M., and Cronin, L. (2018). Development of a minimal photosystem for hydrogen production in inorganic chemical cells. Angew. Chem. 130 (40), 13250–13254. doi:10.1002/ange.201805584
Natali, M., Bazzan, I., Goberna-Ferrón, S., Al-Oweini, R., Ibrahim, M., Bassil, B. S., et al. (2017). Photo-assisted water oxidation by high-nuclearity cobalt-oxo cores: tracing the catalyst fate during oxygen evolution turnover. Green Chem. 19 (10), 2416–2426. doi:10.1039/c7gc00052a
Natali, M., Berardi, S., Sartorel, A., Bonchio, M., Campagna, S., and Scandola, F. (2012a). Is [Co4(H2O)2(α-PW9O34)2]10− a genuine molecular catalyst in photochemical water oxidation? Answers from time-resolved hole scavenging experiments. Chem. Commun. 48 (70), 8808–8810. doi:10.1039/C2CC34804G
Natali, M., Orlandi, M., Berardi, S., Campagna, S., Bonchio, M., Sartorel, A., et al. (2012b). Photoinduced water oxidation by a tetraruthenium polyoxometalate catalyst: ion-pairing and primary processes with Ru(bpy)32+ photosensitizer. Inorg. Chem. 51 (13), 7324–7331. doi:10.1021/ic300703f
Nellutla, S., van Tol, J., Dalal, N. S., Bi, L.-H., Kortz, U., Keita, B., et al. (2005). Magnetism, electron paramagnetic resonance, electrochemistry, and mass spectrometry of the pentacopper(II)-Substituted tungstosilicate [Cu5(OH)4(H2O)2(A-α-SiW9O33)2]10-, A model five-spin frustrated cluster. Inorg. Chem. 44 (26), 9795–9806. doi:10.1021/ic0512633
Nelson, N., and Ben-Shem, A. (2004). The complex architecture of oxygenic photosynthesis. Nat. Rev. Mol. Cell Biol. 5 (12), 971–982. doi:10.1038/nrm1525
Nelson, N., and Ben-Shem, A. (2005). The structure of photosystem I and evolution of photosynthesis. BioEssays 27 (9), 914–922. doi:10.1002/bies.20278
Neumann, R., and Dahan, M. (1997). A ruthenium-substituted polyoxometalate as an inorganic dioxygenase for activation of molecular oxygen. Nature 388 (6640), 353–355. doi:10.1038/41039
Neumann, R., and Khenkin, A. M. (1995). Noble metal (RuIII, PdII, PtII) substituted ‘sandwich’ type polyoxometalates: preparation, characterization, and catalytic activity in oxidations of alkanes and alkenes by peroxides. Inorg. Chem. 34 (23), 5753–5760. doi:10.1021/ic00127a012
Nocera, D. G. (2017). Solar fuels and solar chemicals industry. Acc. Chem. Res. 50 (3), 616–619. doi:10.1021/acs.accounts.6b00615
Oreskes, N. (2004). The scientific consensus on climate change. Science 306 (5702), 1686. doi:10.1126/science.1103618
Pantazis, D. A. (2018). Missing pieces in the puzzle of biological water oxidation. ACS Catal. 8 (10), 9477–9507. doi:10.1021/acscatal.8b01928
Panwar, N. L., Kaushik, S. C., and Kothari, S. (2011). Role of renewable energy sources in environmental protection: a review. Renew. Sustain. Energy Rev. 15 (3), 1513–1524. doi:10.1016/j.rser.2010.11.037
Paul, A., Adhikary, S. D., Kapurwan, S., and Konar, S. (2022). En route to artificial photosynthesis: the role of polyoxometalate based photocatalysts. J. Mat. Chem. A 10 (25), 13152–13169. doi:10.1039/d2ta02243e
Piccinin, S., and Fabris, S. (2011). A first principles study of water oxidation catalyzed by a tetraruthenium-oxo core embedded in polyoxometalate ligands. Phys. Chem. Chem. Phys. 13 (17), 7666–7674. doi:10.1039/c0cp01915a
Piccinin, S., Sartorel, A., Aquilanti, G., Goldoni, A., Bonchio, M., and Fabris, S. (2013). Water oxidation surface mechanisms replicated by a totally inorganic tetraruthenium–oxo molecular complex. Proc. Natl. Acad. Sci. 110 (13), 4917–4922. doi:10.1073/pnas.1213486110
Prăvălie, R., and Bandoc, G. (2018). Nuclear energy: between global electricity demand, worldwide decarbonisation imperativeness, and planetary environmental implications. J. Environ. Manag. 209, 81–92. doi:10.1016/j.jenvman.2017.12.043
Proust, A., Matt, B., Villanneau, R., Guillemot, G., Gouzerh, P., and Izzet, G. (2012). Functionalization and post-functionalization: a step towards polyoxometalate-based materials. Chem. Soc. Rev. 41 (22), 7605. doi:10.1039/c2cs35119f
Pushkar, Y., Davis, K. M., and Palenik, M. C. (2018). Model of the oxygen evolving complex which is highly predisposed to O–O bond formation. J. Phys. Chem. Lett. 9 (12), 3525–3531. doi:10.1021/acs.jpclett.8b00800
Qiao, L., Song, M., Geng, A., and Yao, S. (2019). Polyoxometalate-based high-nuclear cobalt–vanadium–oxo cluster as efficient catalyst for visible light-driven CO2 reduction. Chin. Chem. Lett. 30 (6), 1273–1276. doi:10.1016/j.cclet.2019.01.024
Rabaia, M. K. H., Abdelkareem, M. A., Sayed, E. T., Elsaid, K., Chae, K.-J., Wilberforce, T., et al. (2021). Environmental impacts of solar energy systems: a review. Sci. Total Environ. 754, 141989. doi:10.1016/j.scitotenv.2020.141989
Ritchie, H., Roser, M., and Rosado, P. (2023). CO2 and greenhouse gas emissions. Our World in data. Available at: https://ourworldindata.org/co2-and-greenhouse-gas-emissions.
Rüttinger, W., and Dismukes, G. C. (1997). Synthetic water-oxidation catalysts for artificial photosynthetic water oxidation. Chem. Rev. 97 (1), 1–24. doi:10.1021/cr950201z
Sánchez-Lara, E., Favela, R., Tzian, K., Monroy-Torres, B., Romo-Pérez, A., Ramírez-Apan, M. T., et al. (2024). Effects of the tetravanadate [V4O12]4− anion on the structural, magnetic, and biological properties of copper/phenanthroline complexes. J. Biol. Inorg. Chem. doi:10.1007/s00775-023-02035-9
Sánchez-Lara, E., García-García, A., González-Vergara, E., Cepeda, J., and Rodríguez-Diéguez, A. (2021). Magneto-structural correlations of cyclo-tetravanadates functionalized with mixed-ligand copper(ii) complexes. New J. Chem. 45 (11), 5081–5092. doi:10.1039/d0nj06004f
Sang, X., Feng, S., Lü, Y., Zhang, Y. X., Su, F., Zhang, L., et al. (2018). A new hexamolybdate-based copper–2,2′-biimidazole coordination polymer serving as an acid catalyst and support for enzyme immobilization. Acta Crystallogr. Sect. C Struct. Chem. 74 (11), 1362–1369. doi:10.1107/s2053229618013037
Sartorel, A., Carraro, M., Scorrano, G., Zorzi, R. D., Geremia, S., McDaniel, N. D., et al. (2008). Polyoxometalate embedding of a tetraruthenium(IV)-oxo-core by template-directed metalation of [γ-SiW10O36]8−: a totally inorganic oxygen-evolving catalyst. J. Am. Chem. Soc. 130 (15), 5006–5007. doi:10.1021/ja077837f
Schwarz, B., Förster, J., Goetz, M. K., Yücel, D., Berger, C., Jacob, T., et al. (2016). Visible-light-driven water oxidation by a molecular manganese vanadium oxide cluster. Angew. Chem. Int. Ed. 55 (21), 6329–6333. doi:10.1002/anie.201601799
Shamsipur, M., and Pashabadi, A. (2018). Latest advances in PSII features and mechanism of water oxidation. Coord. Chem. Rev. 374, 153–172. doi:10.1016/j.ccr.2018.07.006
Shen, J. R. (2015). The structure of photosystem II and the mechanism of water oxidation in photosynthesis. Annu. Rev. Plant Biol. 66 (1), 23–48. doi:10.1146/annurev-arplant-050312-120129
Singh, C., Haldar, A., Basu, O., and Das, S. K. (2021). Devising a polyoxometalate-based functional material as an efficient electrocatalyst for the hydrogen evolution reaction. Inorg. Chem. 60 (14), 10302–10314. doi:10.1021/acs.inorgchem.1c00734
Singh, C., Mukhopadhyay, S., and Das, S. K. (2018). Polyoxometalate-supported bis(2,2′-bipyridine)mono(aqua)nickel(II) coordination complex: an efficient electrocatalyst for water oxidation. Inorg. Chem. 57 (11), 6479–6490. doi:10.1021/acs.inorgchem.8b00541
Smith, R. D. L., Prévot, M. S., Fagan, R. D., Zhang, Z., Sedach, P. A., Siu, M. K. J., et al. (2013). Photochemical route for accessing amorphous metal oxide materials for water oxidation catalysis. Science 340 (6128), 60–63. doi:10.1126/science.1233638
Soeder, D. J. (2021). Fracking and the environment: a scientific assessment of the environmental risks from hydraulic fracturing and fossil fuels. Springer Nature.
Solomon, S. (2007). Climate change 2007: contribution of working group I to the fourth assessment report of the intergovernmental panel on climate change. Cambridge: Cambridge University Press.
Song, F., Ding, Y., Ma, B., Wang, C., Wang, Q., Du, X., et al. (2013). K7[CoIIICoII(H2O)W11O39]: a molecular mixed-valence Keggin polyoxometalate catalyst of high stability and efficiency for visible light-driven water oxidation. Energy Environ. Sci. 6 (4), 1170. doi:10.1039/c3ee24433d
Soriano-López, J., Goberna-Ferrón, S., Vigara, L., Carbó, J. J., Poblet, J. M., and Galán-Mascarós, J. R. (2013). Cobalt polyoxometalates as heterogeneous water oxidation catalysts. Inorg. Chem. 52 (9), 4753–4755. doi:10.1021/ic4001945
Soriano-López, J., Musaev, D. G., Hill, C. L., Galán-Mascarós, J. R., Carbó, J. J., and Poblet, J. M. (2017). Tetracobalt-polyoxometalate catalysts for water oxidation: key mechanistic details. J. Catal. 350, 56–63. doi:10.1016/j.jcat.2017.03.018
Soriano-López, J., Schmitt, W., and García-Melchor, M. (2018). Computational modelling of water oxidation catalysts. Curr. Opin. Electrochem. 7, 22–30. doi:10.1016/j.coelec.2017.10.001
Soriano-López, J., Steuber, W., Mulahmetović, M., Besora, M., Clemente-Juan, J. M., O’Doherty, M., et al. (2023). Accelerating water oxidation – a mixed Co/Fe polyoxometalate with improved turnover characteristics. Chem. Sci. 14 (47), 13722–13733. doi:10.1039/D3SC04002J
Stirbet, A., Lazár, D., Guo, Y., and Govindjee, G. (2020). Photosynthesis: basics, history and modelling. Ann. Bot. 126 (4), 511–537. doi:10.1093/aob/mcz171
Stracke, J. J., and Finke, R. G. (2011). Electrocatalytic water oxidation beginning with the cobalt polyoxometalate [Co4(H2O)2(PW9O34)2]10–: identification of heterogeneous CoOx as the dominant catalyst. J. Am. Chem. Soc. 133 (38), 14872–14875. doi:10.1021/ja205569j
Su, X., Ma, J., Wei, X., Cao, P., Zhu, D., Chang, W., et al. (2017). Structure and assembly mechanism of plant C 2 S 2 M 2 -type PSII-LHCII supercomplex. Science 357 (6353), 815–820. doi:10.1126/science.aan0327
Su, X., Yan, L., and Su, Z. (2019). Theoretical insight into the performance of MnII/III-monosubstituted heteropolytungstates as water oxidation catalysts. Inorg. Chem. 58 (23), 15751–15757. doi:10.1021/acs.inorgchem.9b01806
Suga, M., Akita, F., Hirata, K., Ueno, G., Murakami, H., Nakajima, Y., et al. (2015). Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses. Nature 517 (7532), 99–103. doi:10.1038/nature13991
Suga, M., Akita, F., Yamashita, K., Nakajima, Y., Ueno, G., Li, H., et al. (2019). An oxyl/oxo mechanism for oxygen-oxygen coupling in PSII revealed by an x-ray free-electron laser. Science 366 (6463), 334–338. doi:10.1126/science.aax6998
Sun, L. (2015). A closer mimic of the oxygen evolution complex of photosystem II. Science 348 (6235), 635–636. doi:10.1126/science.aaa9094
Surendranath, Y., Dincǎ, M., and Nocera, D. G. (2009). Electrolyte-dependent electrosynthesis and activity of cobalt-based water oxidation catalysts. J. Am. Chem. Soc. 131 (7), 2615–2620. doi:10.1021/ja807769r
Süss-Fink, G. (2008). Water oxidation: a robust all-inorganic catalyst. Angew. Chem. Int. Ed. 47 (32), 5888–5890. doi:10.1002/anie.200801121
Symes, M. D., Cogdell, R. J., and Cronin, L. (2013). Designing artificial photosynthetic devices using hybrid organic–inorganic modules based on polyoxometalates. Philosophical Trans. R. Soc. A Math. Phys. Eng. Sci. 371, 20110411. doi:10.1098/rsta.2011.0411
Taira, N., Yamauchi, K., and Sakai, K. (2023). Intracluster O–O coupling pathway evidenced for an anderson-type single-cobalt polymolybdate water oxidation catalyst. ACS Catal. 13 (5), 3211–3223. doi:10.1021/acscatal.2c05925
Tanaka, S., Annaka, M., and Sakai, K. (2012). Visible light-induced water oxidation catalyzed by molybdenum-based polyoxometalates with mono- and dicobalt(III) cores as oxygen-evolving centers. Chem. Commun. 48 (11), 1653–1655. doi:10.1039/C2CC16821A
Tang, J., Yang, X., Zhang, X. W., Wang, M., and Wu, C. D. (2010). A functionalized polyoxometalate solid for selective oxidation of styrene to benzaldehyde. Dalton Trans. 39 (14), 3396. doi:10.1039/b924041a
Teillout, A. L., de Oliveira, P., Marrot, J., Howell, R. C., Vilà, N., Walcarius, A., et al. (2019). Synthesis, crystal structure, electrochemistry and electro-catalytic properties of the manganese-containing polyoxotungstate, [(Mn(H2O)3)2(H2W12O42)]6−. Inorganics 7 (2), 15. doi:10.3390/inorganics7020015
Toma, F. M., Sartorel, A., Carraro, M., Bonchio, M., and Prato, M. (2011). Dendron-functionalized multiwalled carbon nanotubes incorporating polyoxometalates for water-splitting catalysis. Pure Appl. Chem. 83 (8), 1529–1542. doi:10.1351/pac-con-10-11-12
Umena, Y., Kawakami, K., Shen, J.-R., and Kamiya, N. (2011). Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473 (7345), 55–60. doi:10.1038/nature09913
Vickers, J. W., Lv, H., Sumliner, J. M., Zhu, G., Luo, Z., Musaev, D. G., et al. (2013). Differentiating homogeneous and heterogeneous water oxidation catalysis: confirmation that [Co4(H2O)2(α-PW9O34)2]10– is a molecular water oxidation catalyst. J. Am. Chem. Soc. 135 (38), 14110–14118. doi:10.1021/ja4024868
Vinyard, D. J., Ananyev, G. M., and Charles Dismukes, G. (2013). Photosystem II: the reaction center of oxygenic photosynthesis. Annu. Rev. Biochem. 82 (1), 577–606. doi:10.1146/annurev-biochem-070511-100425
Vinyard, D. J., Khan, S., and Brudvig, G. W. (2015). Photosynthetic water oxidation: binding and activation of substrate waters for O–O bond formation. Faraday Discuss. 185, 37–50. doi:10.1039/C5FD00087D
Wang, C., O’Hagan, M., Willner, B., and Willner, I. (2022). Bioinspired artificial photosynthetic systems. Chem. Weinheim der Bergstrasse, Ger. 28 (9), e202103595. doi:10.1002/chem.202103595
Wang, M., Zhong, W., Zhang, S., Liu, R., Xing, J., and Zhang, G. (2018). An overall water-splitting polyoxometalate catalyst for the electromicrobial conversion of CO2 in neutral water. J. Mat. Chem. A 6 (21), 9915–9921. doi:10.1039/c8ta01902a
Wang, S. M., Hwang, J., and Kim, E. (2019). Polyoxometalates as promising materials for electrochromic devices. J. Mater. Chem. C 7 (26), 7828–7850. doi:10.1039/c9tc01722d
Wang, S.-S., and Yang, G.-Y. (2015). Recent advances in polyoxometalate-catalyzed reactions. Chem. Rev. 115 (11), 4893–4962. doi:10.1021/cr500390v
Wang, Z., Hu, Y., Zhang, S., and Sun, Y. (2022). Artificial photosynthesis systems for solar energy conversion and storage: platforms and their realities. Chem. Soc. Rev. 51 (15), 6704–6737. doi:10.1039/d1cs01008e
Weakley, T. J. R., Evans, H. T., Showell, J. S., Tourné, G. F., and Tourné, C. M. (1973). 18-Tungstotetracobalto(II)diphosphate and related anions: a novel structural class of heteropolyanions. J. Chem. Soc. Chem. Commun. (4), 139–140. doi:10.1039/c39730000139
Wei, J., Feng, Y., Zhou, P., Liu, Y., Xu, J., Xiang, R., et al. (2015). A bioinspired molecular polyoxometalate catalyst with two cobalt(II) oxide cores for photocatalytic water oxidation. ChemSusChem 8 (16), 2630–2634. doi:10.1002/cssc.201500490
Weinstock, I. A., Cowan, J. J., Barbuzzi, E. M., Zeng, H., and Hill, C. L. (1999). Equilibria between α and β isomers of Keggin heteropolytungstates. J. Am. Chem. Soc. 121 (19), 4608–4617. doi:10.1021/ja982908j
Wiechen, M., Najafpour, M. M., Allakhverdiev, S. I., and Spiccia, L. (2014). Water oxidation catalysis by manganese oxides: learning from evolution. Energy Environ. Sci. 7 (7), 2203. doi:10.1039/c4ee00681j
Wu, Y., Pei, J., Yu, X., and Bi, L. (2022). Study on catalytic water oxidation properties of polynuclear manganese containing polyoxometalates. Catalysts 12 (2), 160. doi:10.3390/catal12020160
Xiang, R., Ding, Y., and Zhao, J. (2014). Visible-light-induced water oxidation mediated by a mononuclear-cobalt(II)-Substituted silicotungstate. Chem. – Asian J. 9 (11), 3228–3237. doi:10.1002/asia.201402483
Xinrong, L., Jinyu, X., Huizhang, L., Bin, Y., Songlin, J., and Gaoyang, X. (2000). Studies on styrene oxidation reaction catalyzed by ruthenium substituted polyoxotungstates: kinetics and phase transfer effect. J. Mol. Catal. A Chem. 161 (1-2), 163–169. doi:10.1016/s1381-1169(00)00331-9
Yagi, M., and Kaneko, M. (2000). Molecular catalysts for water oxidation. Chem. Rev. 101 (1), 21–36. doi:10.1021/cr980108l
Yao, R., Li, Y., Chen, Y., Xu, B., Chen, C., and Zhang, C. (2021). Rare-earth elements can structurally and energetically replace the calcium in a synthetic Mn4CaO4-cluster mimicking the oxygen-evolving center in photosynthesis. J. Am. Chem. Soc. 143 (42), 17360–17365. doi:10.1021/jacs.1c09085
Ye, T., Wang, J., Dong, G., Jiang, Y., Feng, C., and Yang, Y. (2016). Recent progress in the application of polyoxometalates for dye-sensitized/organic solar cells. Chin. J. Chem. 34 (8), 747–756. doi:10.1002/cjoc.201600231
Yin, Q., Tan, J. M., Besson, C., Geletii, Y. V., Musaev, D. G., Kuznetsov, A. E., et al. (2010). A fast soluble carbon-free molecular water oxidation catalyst based on abundant metals. Science 328 (5976), 342–345. doi:10.1126/science.1185372
Yoro, K. O., and Daramola, M. O. (2020). “CO2 emission sources, greenhouse gases, and the global warming effect,” in Advances in carbon capture (Woodhead Publishing), 3–28. doi:10.1016/b978-0-12-819657-1.00001-3
Young, I. D., Ibrahim, M., Chatterjee, R., Gul, S., Fuller, F. D., Koroidov, S., et al. (2016). Structure of photosystem II and substrate binding at room temperature. Nature 540 (7633), 453–457. doi:10.1038/nature20161
Youngblood, W. J., Lee, S.-H. A., Kobayashi, Y., Hernandez-Pagan, E. A., Hoertz, P. G., Moore, T. A., et al. (2009). Photoassisted overall water splitting in a visible light-absorbing dye-sensitized photoelectrochemical cell. J. Am. Chem. Soc. 131 (3), 926–927. doi:10.1021/ja809108y
Yu, L., Ding, Y., and Zheng, M. (2017). Polyoxometalate-based manganese clusters as catalysts for efficient photocatalytic and electrochemical water oxidation. Appl. Catal. B Environ. 209, 45–52. doi:10.1016/j.apcatb.2017.02.061
Yu, L., Ding, Y., Zheng, M., Chen, H., and Zhao, J. (2016). [{β-SiNi2W10O36(OH)2(H2O)}4]24−: a new robust visible light-driven water oxidation catalyst based on nickel-containing polyoxometalate. Chem. Commun. 52 (100), 14494–14497. doi:10.1039/c6cc02728h
Yu, L., Du, X., Ding, Y., Chen, H., and Zhou, P. (2015). Efficient visible light-driven water oxidation catalyzed by an all-inorganic copper-containing polyoxometalate. Chem. Commun. 51 (98), 17443–17446. doi:10.1039/c5cc07119d
Yu, L., Lin, J., Zheng, M., Chen, M., and Ding, Y. (2018). Homogeneous electrocatalytic water oxidation at neutral pH by a robust trinuclear copper(II)-substituted polyoxometalate. Chem. Commun. 54 (4), 354–357. doi:10.1039/C7CC08301G
Zhang, B., and Sun, L. (2018). Why nature chose the Mn4CaO5 cluster as water-splitting catalyst in photosystem II: a new hypothesis for the mechanism of O–O bond formation. Dalton Trans. 47 (41), 14381–14387. doi:10.1039/C8DT01931B
Zhang, C., Chen, C., Dong, H., Shen, J.-R., Dau, H., and Zhao, J. (2015). A synthetic Mn 4 Ca-cluster mimicking the oxygen-evolving center of photosynthesis. Science 348 (6235), 690–693. doi:10.1126/science.aaa6550
Zhang, L., Mathew, S., Hessels, J., Reek, J. N. H., and Yu, F. (2021). Homogeneous catalysts based on first-row transition-metals for electrochemical water oxidation. ChemSusChem 14 (1), 234–250. doi:10.1002/cssc.202001876
Zhang, L.-Z., Gu, W., Liu, X., Dong, Z., Yang, Y.-S., Li, B., et al. (2007). K10[Co4(H2O)2(B-α-SiW9O34H)2]·21H2O: a sandwich polyoxometalate based on the magnetically interesting element cobalt. Inorg. Chem. Commun. 10 (11), 1378–1380. doi:10.1016/j.inoche.2007.08.025
Zhang, Y., Li, Y., Guo, H., Guo, Y., and Song, R. (2024). Recent advances in polyoxometalate-based materials and their derivatives for electrocatalysis and energy storage. Mat. Chem. Front. 8, 732–768. doi:10.1039/d3qm01000g
Zheng, S.-T., and Yang, G.-Y. (2012). Recent advances in paramagnetic-TM-substituted polyoxometalates (TM = Mn, Fe, Co, Ni, Cu). Chem. Soc. Rev. 41 (22), 7623. doi:10.1039/c2cs35133a
Zhu, G., Geletii, Y. V., Kögerler, P., Schilder, H., Song, J., Lense, S., et al. (2012a). Wateroxidation catalyzed by a new tetracobalt-substituted polyoxometalate complex: [{Co4(μ-OH)(H2O)3}(Si2W19O70)]11−. Dalton Trans. 41 (7), 2084–2090. doi:10.1039/c1dt11211b
Zhu, G., Glass, E. N., Zhao, C., Lv, H., Vickers, J. W., Geletii, Y. V., et al. (2012b). A nickel containing polyoxometalate water oxidation catalyst. Dalton Trans. 41 (42), 13043–13049. doi:10.1039/C2DT30331K
Keywords: molecular water oxidation catalysts, photosystem II, polyoxometalate-based materials, earth-abundant metals, catalysis
Citation: Sánchez-Lara E, Favela R and Castillo I (2024) Bioinspired polyoxometalates as light-driven water oxidation catalysts. Front. Chem. Biol 3:1373647. doi: 10.3389/fchbi.2024.1373647
Received: 20 January 2024; Accepted: 26 February 2024;
Published: 11 March 2024.
Edited by:
Lukáš Krivosudský, Comenius University, SlovakiaReviewed by:
Rami Oweini, American University of Beirut, LebanonCopyright © 2024 Sánchez-Lara, Favela and Castillo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eduardo Sánchez-Lara, ZXNsXzI0QGhvdG1haWwuY29t; Ivan Castillo, am9zZWl2YW5AdW5hbS5teA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.