
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. Biol. , 11 October 2023
Sec. Bioinorganic Chemistry
Volume 2 - 2023 | https://doi.org/10.3389/fchbi.2023.1243564
This article is part of the Research Topic New Developments in Bioinorganic and Bioorganic Chemistry View all 5 articles
 Noemi Carosella1†
Noemi Carosella1† Kelly P. Brock2†
Kelly P. Brock2† Barbara Zambelli1†
Barbara Zambelli1† Francesco Musiani1†Chris Sander2,3,4†
Francesco Musiani1†Chris Sander2,3,4† Stefano Ciurli1*†
Stefano Ciurli1*†Introduction: Urease is an enzyme exploited by many virulent bacteria and fungi to infect the host and exert their virulence. The Gram-negative bacterium Helicobacter pylori relies on the activity of urease to infect the highly acidic human stomach. The activity of urease depends on the presence of a catalytic site containing two Ni(II) ions. In vivo, urease is initially synthesized as an inactive apo-enzyme and requires a post-translational activation process that involves the incorporation of the metal ions into its buried active site. In H. pylori, as well as in other bacteria, this activation process is mediated by four accessory proteins, named UreD, UreF, UreG, and UreE. Targeting the interactions between urease chaperones could potentially inhibit the activation of urease through blocking the Ni(II) ions incorporation, providing a route for the development of antimicrobial strategies against ureolytic pathogens.
Methods: In this paper, an evolutionary couplings (EC) approach was adopted to determine the interaction surface between urease and UreD, the first protein that binds the enzyme, preparing it for the subsequent activation steps. Site-directed mutagenesis and an in-cell assay were used to detect urease activity in recombinant bacteria expressing the mutated operon. The obtained data were used to drive a protein-protein docking computational approach.
Results and Discussion: The EC prediction retrieved ten pairs of residues lying at the interface between UreD and the urease subunit UreB, likely involved in contacts essential to build the protein complex. These contacts were largely confirmed experimentally, leading to the obtainment of a model for the urease-UreD complex that agrees well with the recently reported experimental cryo-EM structure. This work represents a proof of concept for the calculation of reliable models of protein interaction surfaces in the absence of experimental structures of critical assemblies.
Urease is a Ni(II) ion-dependent enzyme involved in essential biological processes of bacteria, archaea, unicellular eukaryotes, and plants (Zambelli et al., 2011; Mazzei et al., 2017; Mazzei et al., 2019). The enzymatic hydrolysis of urea leads to the production of ammonia and carbamate, the latter spontaneously decomposing into ammonia and bicarbonate, with a rise of the environmental pH that is exploited by many bacteria and fungi to infect the host and exert their virulence (Rutherford, 2014). Some pathogenic agents, such as the Gram-negative bacterium Helicobacter pylori that infects the highly acidic human stomach, use the pH buffering outcome of urea hydrolysis to colonize specific niches and to survive in the host environment (Savoldi et al., 2018), thus making Ni(II) a virulence factor for this WHO Class I bacterial carcinogen (Maroney and Ciurli, 2021). Therefore, urease represents a target to develop antibiotics for infections by ureolytic bacteria. The activity of urease relies on the presence of an active site containing two Ni(II) ions bridged by the carboxylate group of a carbamylated lysine and by a hydroxide ion, the nucleophile of the hydrolysis reaction (Mazzei et al., 2020; Mazzei and Ciurli, 2021). While several enzyme inhibitors that target the catalytic mechanism have been proposed to this task, the enzyme activation process could be another way to modulate its activity.
In vivo, urease is initially synthesized as an inactive apo-enzyme, and its activation requires a post-translational process that involves the incorporation of the metal ions, concomitantly with carbamylation of the lysine, both located in its buried active site. The accepted general view of this process, which is specifically discussed here for the case of H. pylori urease (HPU), involves four accessory proteins, named UreD, UreF, UreG, and UreE (Zambelli et al., 2011; Mazzei et al., 2017; Nim and Wong, 2019). The genes encoding these chaperones are in the same operon as the structural genes ureA and ureB coding for the β and α subunits respectively, which assemble into the enzyme. Indeed, while bacterial ureases are generally hetero oligomers of the (αβγ)3 type, the Helicobacter genus represents an exception in which the β and γ subunits are fused to yield a [(αβ)3]4 quaternary structure (Ha et al., 2001b; Cunha et al., 2021). The activation of urease requires the formation and dissociation of protein complexes with various combinations of the chaperones, this process being regulated by Ni(II) ions, GTP hydrolysis, CO2 uptake for lysine carbamylation, as well as conformational changes in flexible regions at the protein-protein and protein-metal interfaces (Zambelli et al., 2020). In particular, the UreDFG complex plays a critical role by binding and pre-activating urease, a modification needed for urease to receive Ni(II) ions (Fong et al., 2013; Musiani et al., 2017). Nickel trafficking is effected by UreE2, a dimeric metallochaperone (Soriano et al., 2000; Bellucci et al., 2009; Merloni and Ciurli, 2014). The Ni(II) ion-binding site in UreE2 is situated at the edge of the dimerization interface, on the surface of the protein, and involves two conserved His residues (Remaut et al., 2001; Song et al., 2001; Shi et al., 2010; Banaszak et al., 2012; Zambelli et al., 2013). Some UreE2 proteins contain His-rich C-terminal tails that bind additional Ni(II) ions acting as a Ni(II) ion deposit (Grossoehme et al., 2007). In all structurally characterized UreE2 proteins, these sequences show intrinsic flexibility and contain at least one His residue involved in the Ni(II) ion coordination, acting as “arms” that take and release the metal ions to other protein chaperones such as UreG (Zambelli et al., 2020).
Urease activation requires the binding of UreD to urease, which was proposed to induce a conformational change preparing the enzyme for the subsequent steps (Park et al., 1994; Chang et al., 2004; Farrugia et al., 2013; Eschweiler et al., 2018). The formation of the urease-UreD complex recruits UreF (Moncrief and Hausinger 1996; Farrugia et al., 2013) in a UreDF complex, whose crystal structure has been reported (Fong et al., 2011). Structural analysis of this complex revealed extended contacts between an α-helical segment of UreF, which is degraded by proteolysis when the protein is expressed alone (Lam et al., 2010; Zambelli et al., 2014). This segment is required for the UreDF interaction (Fong et al., 2011; Tarsia et al., 2018). The UreDF complex interacts with UreG to form the UreDFG super-complex (Fong et al., 2013). UreE2 has been shown to deliver Ni(II) ions to the pre-activation assembly through the interaction with UreG (Bellucci et al., 2009). UreG thus switches between the UreDFG and the UreEG complex, depending on the nucleotide and metal ion-bound states (Yuen et al., 2017; Pierro et al., 2020). In this mechanism, a role is played also by HypA and HypB, two Ni-chaperones involved in the maturation of (Ni-Fe)-hydrogenase (Tsang and Wong, 2022). In particular, HypA provides Ni(II) ions to the urease system through its interaction with UreE2 in a Ni-UreE2-HypA assembly (Spronk et al., 2018; Zambelli et al., 2023). Targeting the interactions between urease chaperones could potentially inhibit the activation of urease through blocking the Ni(II) ions incorporation, providing a route for the development of antimicrobial strategies against ureolytic pathogens (Tarsia et al., 2018). A model for the structure of K. aerogenes urease bound to its accessory proteins was proposed based on crosslinking, mass spectrometry experiments, and small angle X-ray scattering studies; this model suggested the occurrence of specific interactions between UreD and UreB in the proximity of the active site, which caused a conformational change with better access to the nascent active site (Quiroz-Valenzuela et al., 2008; Farrugia et al., 2013; Ligabue-Braun et al., 2013; Eschweiler et al., 2018).
The mechanism of Ni(II) ion transfer from the UreDFG complex to urease has been only partly elucidated (Zambelli et al., 2011; Zambelli et al., 2020). The binding site of the catalytic Ni(II) ions in urease is buried in its structure (Zambelli et al., 2011; Maroney and Ciurli, 2014; Mazzei et al., 2017). Furthermore, while UreD is the chaperone that directly contacts urease, the interaction surface is far away from the Ni(II) ion binding site of UreG. Thus, the question arises as to how Ni(II) ions can be transported from one side of the UreDFG chaperone complex to the other. The answer to this question was provided by the X-ray structure of UreDFG, which revealed the presence of intra-molecular tunnels as viable routes for Ni(II) ions translocation (Zambelli et al., 2014). This hypothesis, initially supported by site-directed mutagenesis studies on UreF (Zambelli et al., 2014) and UreD (Farrugia et al., 2015), as well as molecular dynamics simulations (Musiani et al., 2017; Masetti et al., 2020; Masetti et al., 2021), was then confirmed experimentally by the cryo-electron microscopy (cryo-EM) structure of the UreDF-urease complex (Nim et al., 2023).
In this paper, an evolutionary couplings (EC) approach (Marks et al., 2011; Hopf et al., 2014) was adopted to investigate the interaction surface between urease and UreD. The EC predictions were experimentally evaluated by site-directed mutagenesis and subsequently used to drive a protein-protein docking computational approach that led to the obtainment of a model for the urease-UreD complex that agrees well with the experimental cryo-EM structure. This study thus represents a proof-of-concept indicating that only a minimum number of experimentally verified contacts predicted using evolutionary couplings is sufficient to build reliable structural models of protein-protein complexes.
Multiple sequence alignments (MSAs) were generated for UreA, UreB, and UreD by running the Jackhmmer software (Johnson et al., 2010) against the Uniref100 dataset (Suzek et al., 2015) downloaded on 24 July 2020 for bitscore thresholds (normalized to sequence length) between 0.1 and 0.9. From these MSAs, sequences (rows) were dropped if they contained more than 30% gaps, a threshold that was used to exclude fragmental hits and improve the quality of the alignment. Similarly, MSA columns—each representing an individual amino acid—were retained for further analysis if they contained fewer than 30% gaps, which is the default in the EVcouplings software and limits our predictions only to residue positions that align to a high proportion of sequences. One MSA was manually chosen for each UreA, UreB, and UreD subunit based on optimizing for maximum number of sequences in the alignment while maintaining a high number of residues meeting our column coverage threshold. The UreA MSA contained 4,663 sequences and 93.7% sequence coverage, the UreB MSA contained 23,307 sequences with 99.3% sequence coverage, and the UreD MSA contained 22,907 sequences with 93.2% sequence coverage.
These chosen alignments were then run through the EVcouplings software package, using both the monomer (Marks et al., 2011; Hopf et al., 2019) and complex (Hopf et al., 2014; Green et al., 2021) pipelines for these chosen alignments. For all complex runs to investigate inter-protein interactions between the three subunits, theta (a clustering threshold for down-weighting redundant sequences) was set to 0.9 and the row and column maximum gap thresholds were retained at 30%. Sequences from the two subunits per pairing were concatenated based on the “best hits” protocol with only reciprocal best hits retained. The resulting concatenated alignment contained 10,723 sequences with 97.7% sequence coverage across both sequences.
Top contacts contained within each subunit were compared to the following PDB structures detected through homology search: for UreB, PDB IDs 6QSU and 6ZJA (Cunha et al., 2021) and 1E9Z (Ha et al., 2001b); for UreD, PDB ID 3SF5 (Fong et al., 2011) and 4HI0 (Fong et al., 2013), and for UreA, PDB IDs 1E9Y and 1E9Z (Ha et al., 2001a), as well as 6ZJA and 6QSU (Cunha et al., 2021). The top predicted inter-protein contacts (i.e., between subunits) for UreB-UreD were then used to design mutations in these subunits for further experimental testing and for protein-protein docking. The multiple sequence alignments are freely available at https://site.unibo.it/bioinorgchem/en/downloads.
The pGEM-ureOP plasmid, carrying the H. pylori strain G27 urease operon (Zambelli et al., 2014) was used as PCR template for site-directed mutagenesis using the Quikchange mutagenesis kit (Agilent), according to the manufacturer’s instructions. The vectors containing the mutated operons were amplified in E. coli TOP10 cells and sequenced on both strands to confirm the mutations.
TOP10 bacterial cells harboring the pGEM-ureOP plasmid, or its variants, were tested for urease activity by modifying a previously reported method (Tarsia et al., 2018). Briefly, cells were pre-cultured at 37°C in 5 mL of lysogeny broth (LB) containing 50 μg/mL of carbenicillin (Cb). After 16 h, 2 mL of this culture were used to inoculate 10 mL of LB containing the same concentration of Cb and kept at 37°C until OD600 reached 0.5–0.6; subsequently, 2 mM NiSO4 and 320 mM urea were added to each culture to 2 and 320 mM respectively, together with 10 μg/L of cresol red as pH indicator. The color change of the cresol red indicator was monitored over time by measuring the absorbance at 430 nm and at 580 nm. All experiments were performed in triplicate.
The H. pylori 26695 UreD structure from PDB ID 4HI0 (Fong et al., 2013) together with one UreA subunit and two UreB subunits from the urease structures from the same bacterium [PDB IDs 6ZJA and 6QSU (Cunha et al., 2021)] were used for the docking calculations by using the data-driven docking webserver Haddock 2.2 (Dominguez et al., 2003; van Zundert et al., 2016) with default parameters. Haddock’s algorithm uses biochemical and/or biophysical information to drive the docking process in the form of “active residues,” that are supposed to be at the protein-protein interface. For the present calculations, the residues identified through the evolutionary couplings analysis were used as “active residues.” A Haddock computation is composed of three rounds: 1) a rigid-docking stage that produces 1,000 putative docking complexes that are ranked according to an apt scoring function called “Haddock score”; 2) a semiflexible simulated annealing performed on the best 200 solutions calculated in the first round, and 3) an explicit water refinement performed on the same 200 structures resulting from the second round. The achieved docking poses are then clustered using a fraction of common native contacts cutoff of 0.6. The best urease-UreD complex was selected based on the cluster population and the Haddock score. The urease-UreDFG model complex was then reconstructed through structure superimposition between the best docked structure (i.e., the structure with the lowest Haddock score in the most populated cluster) and the whole H. pylori 26695 urease (HPU) structure together with the UreDFG complex. The results were visualized and analyzed with UCSF ChimeraX 1.6 (Goddard et al., 2018; Pettersen et al., 2021). The model structure was compared with the HPU-UreDF cryo-EM structure (PDB ID: 8HC1) (Nim et al., 2023). The calculated model is freely available at https://site.unibo.it/bioinorgchem/en/downloads.
Because selective pressures can act to maintain interactions between protein complex subunits, protein evolutionary information can be used to identify co-evolving residues that are critical for maintaining protein-protein interactions across species. The EVcouplings framework (Marks et al., 2011; Hopf et al., 2014) has been used successfully to predict the interaction interface for two proteins involved in bacterial cell wall synthesis before the crystal structure of the complex was determined (Sjodt et al., 2018; Sjodt et al., 2020). Here, we used this same in silico framework to predict which residues are important for binding between UreB and UreD.
Both intra- and inter-protein contacts were predicted using a single complex pipeline EVcouplings run. The resultant predicted intra-protein contact maps for the UreA, UreB and UreD monomers matched well overall with homologous cryo-EM and crystal structures (Figures 1–3). In addition, contacts between different chains of the same protein, occurring in the urease topological assembly, are well predicted for UreA and UreB proteins (Figure 1). These observations are a primary measure of how well this method can predict unknown interacting residues between proteins. As a proof of principle, the predicted contact maps for the UreA-UreB interactions (Figure 1), matching very well with the experimental structure, confirm the utility of this approach. The overall H. pylori urease structure [PDB ID 6ZJA, (Cunha et al., 2021)] made of UreA and UreB subunits, indicates that 12 out of the top 15 predicted inter-protein contacts via EC analysis are observed within 5 Å minimum atom distance in the structure. The overall precision, including intra-protein, monomeric predicted contacts at this level, is 0.8.
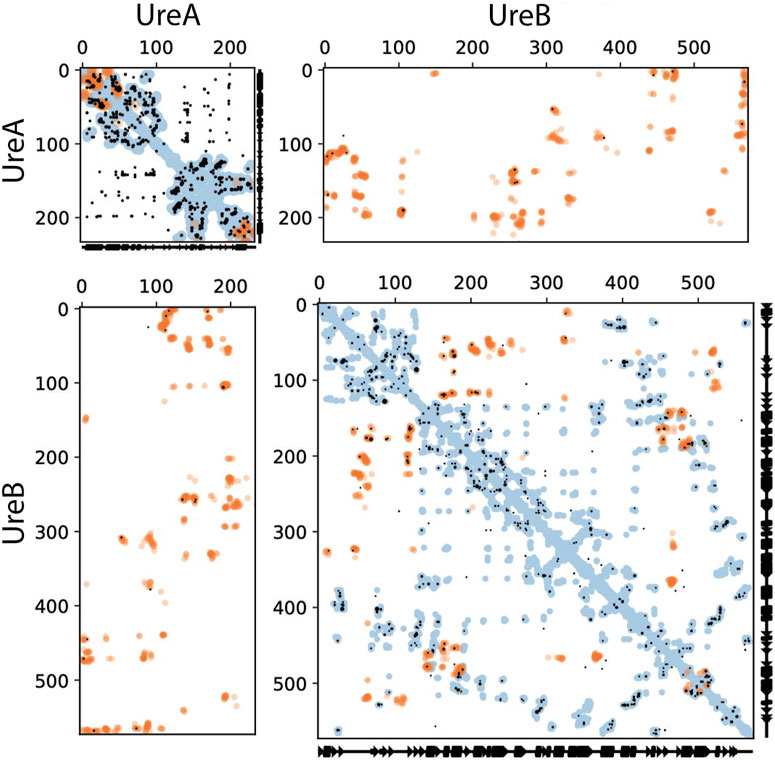
FIGURE 1. Intra- and inter-protein predicted contacts using evolutionary couplings for HPU UreA and UreB. The complex pipeline was used to compute evolutionary coupled residues for a compound alignment of UreA and UreB, shown in these contact maps. In all cases, axes correspond to residue indices. Predicted contacts (black dots) are shown that are entirely within UreA (top left), UreB (bottom right), or inter-protein between UreA and UreB (top right and bottom left, with axes flipped between the two versions). Residue pairs with EC scores at or above the 15th top ranked inter-EC are shown. For comparison, known contacts from the monomeric (blue dots) and homo-multimeric (e.g., different chains of the same protein, orange dots) are shown, taken from PDB IDs 1E9Y, 1E9Z, 6ZJA, and 6QSU for UreA and PDB IDs 6QSU, 6ZJA, and 1E97 for UreB. Orange dots in the top right and bottom left panels are the known interacting residues between UreA and UreB, taken from PDB ID 6ZJA. Where the black dots (evolutionary coupled residue pairs) overlay the orange dots (contacts from experimentally determined structure), our predictions are accurate. All these structural contacts are shown if they are within 5 Å minimum atom distance between residues.
The ECs for the UreA-UreD interaction were then identified and shown in Figure 2; the overall precision of monomer contacts above or at the level of the top three inter-protein residue pairs was 0.49, with one predicted contact having a relative monomer precision above 0.8. However, the UreD-UreB complex run outperformed the UreA-UreD run; ECs calculated for the UreB-UreD pair revealed the presence of up to ten strong correlations (Figure 3; Table 1). For the top-ranked residue pair, involving UreB Asn87 and UreD Asn539, the overall precision of monomer contacts up to that point is 0.98. Similarly, for the top ten inter-protein residue pairs, the overall monomer precision is 0.72. These predicted contacts do not appear to introduce any inconsistent clashes with each other, further giving confidence in the predictions.
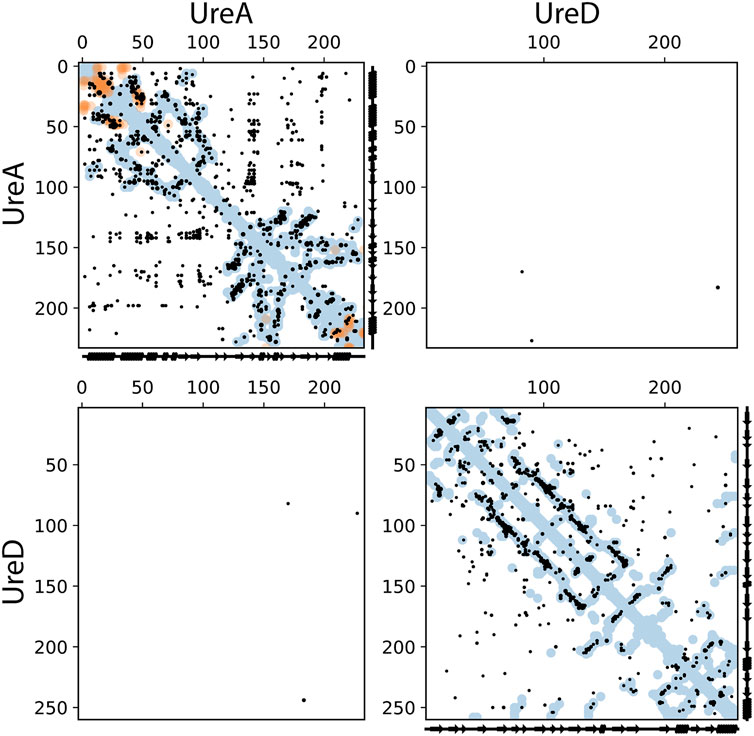
FIGURE 2. Intra- and inter-protein predicted contacts using evolutionary couplings for UreA and UreD. The complex pipeline was used to compute evolutionary coupled residues for a compound alignment of UreA and UreD, shown in these contact maps. In all cases, axes correspond to residue indices. Predicted contacts (black dots) are shown that are entirely within UreA (top left), UreD (bottom right), or inter-protein between UreA and UreD (top right and bottom left, with axes flipped between the two versions). EC pairs with scores at or above the level of the top 3 inter-EC pairs are shown. For comparison, known contacts from the monomeric (blue dots) and homo-multimeric (e.g., different chains of the same protein, orange dots) are shown, taken from PDB IDs 1E9Y, 1E9Z, 6ZJA, and 6QSU for UreA and PDB IDs 3SF5 and 4HI0 for Ure D. These structural contacts are shown if they are within 5 Å minimum atom distance between residues.
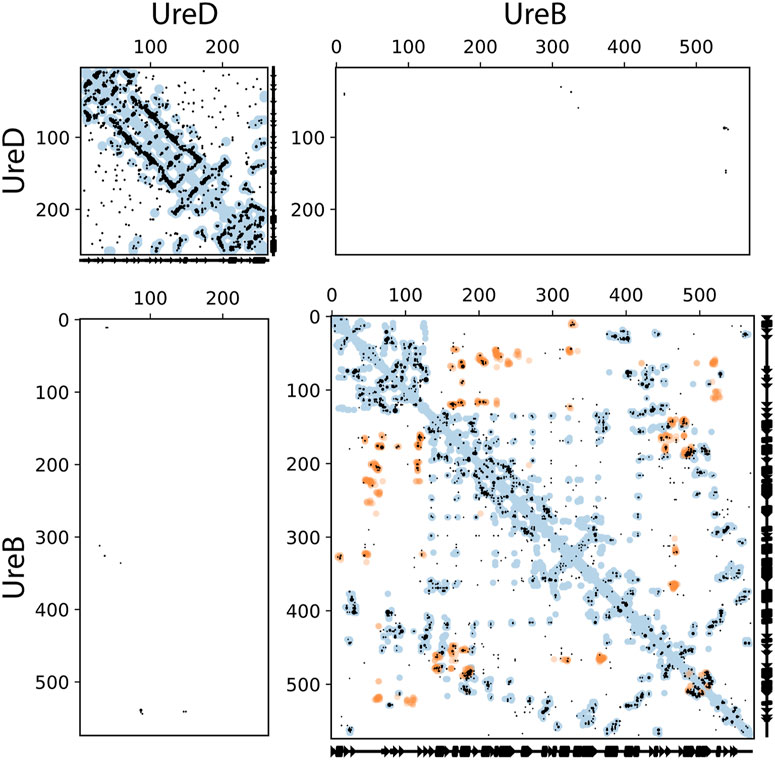
FIGURE 3. Intra- and inter-protein predicted contacts using evolutionary couplings for UreD and UreB. The complex pipeline was used to compute evolutionary coupled residues for a compound alignment of UreD and UreB, shown in these contact maps. In all cases, axes correspond to residue indices. Predicted contacts (black dots) are shown that are entirely within UreD (top left), UreB (bottom right), or inter-protein between UreD and UreB (top right and bottom left, with axes flipped between the two versions). The top 10 inter-EC pairs are shown, along with all monomeric ECs that are at or above the same score. For comparison, known contacts from the monomeric (blue dots) and homo-multimeric (e.g., different chains of the same protein, orange dots) are shown, taken from PDB IDs 3SF5 and 4HI0 for UreD and PDB IDs 6QSU, 6ZJA, and 1E97 for UreB. These structural contacts are shown if they are within 5 Å minimum atom distance between residues.
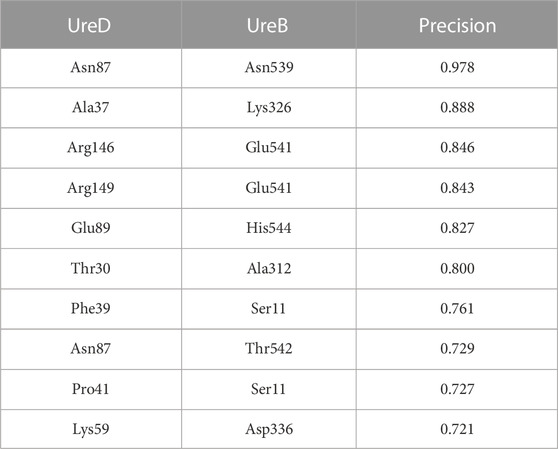
TABLE 1. The identified ECs for the interaction between the UreB subunit in urease and the UreD chaperone, ranked according to the overall precision of monomer contacts.
To experimentally validate the protein-protein interaction (PPI) contacts derived by EC analysis, in-cell experiments were performed heterologously, according to a method previously described (Tarsia et al., 2018). The plasmid, expressing both H. pylori urease enzyme and its accessory genes, was inserted in E. coli TOP10 cells to produce a holo-enzyme, whose activity was evaluated in living bacteria, using a colorimetric assay that determined the pH increase in the bacterial environment caused by urea hydrolysis by urease as a change of absorbance of a pH indicator (Figure 4). The sequence of the plasmid was used as a template for site-directed mutagenesis of the amino acids of UreB and UreD found at the protein interface and involved in ionic interactions in EC analysis. If the protein contacts are correctly predicted, mutations of these residues would break the interaction between UreD and UreB, which is necessary for Ni(II) ion loading into urease active site. This would be reflected in lower or absent enzyme activity. Specifically, from the list of identified residues (Table 1), the charged residues were chosen and mutated to alanine, in the hypothesis that the replacement of a charged residue with a hydrophobic one has a higher impact on PPI. Lys326, Asp336, Glu541, and His544 were mutated to alanine in the UreB sequence, and Lys59, Glu89, Arg146, and Arg149 were mutated to alanine in the UreD sequence, likely breaking five essential contacts between UreB and UreD, i.e., Lys326/Ala37; Glu541/Arg146; Glu541/Arg149; His544/Glu89; Asp336/Lys59. In addition, the double mutants Arg146Glu/Arg149Glu and Lys59Ala/Arg149Ala of UreD were produced to evaluate possible additive effects of the residue variations. The triple mutant Arg146Glu/Arg149Glu in UreD and Glu541Arg in UreB was also produced to evaluate the possible restorative effect of charge exchange. Ser163Ala and Asp167Ala in UreD, not involved in PPI according to the EC analysis, were produced as negative controls.
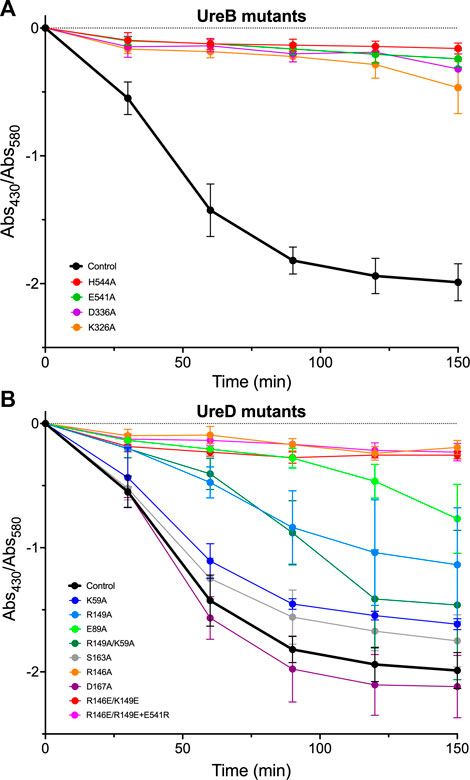
FIGURE 4. Urease activity, measured as the ratio of absorbance at 430 nm and at 580 nm for the cresol red indicator, added to the E. coli cultures harboring the pGEM-ureOP plasmid with site-directed mutations in ureB (A) and ureD (B) genes, as indicated in the figure. The urease activity of the E. coli strain, transformed with the wild-type urease operon, is shown for reference. Every measurement was performed in triplicate and the error bars represent the standard deviations of the average values. The single-letter amino acid nomenclature is used.
All UreB mutations fully inactivated the urease activity (Figure 4A), confirming the importance of the selected residues either for PPI or for the enzymatic activity. The negative controls, with UreD mutated as Ser163Ala and Asp167Ala, presented an activity similar to that of the wild-type operon, indicating that these residues do not impact on the PPI, as predicted by EC analysis (Figure 4B). Similarly, Lys59Ala mutation did not significantly influence the in-cell measure of urease activity (Figure 4B). Arg149Ala showed a partial increase of pH over time, with no additive effect observed for Lys59Ala/Arg149Ala double mutant, coherently with the absence of influence of Lys59 (Figure 4B). Both Glu89Ala and Arg146Ala single mutations, as well as Arg146Glu/Arg149Glu double mutation, fully switched off the urease activation, coherently with their predicted importance for UreD contacts with residues His144 and Glu541 of UreB. Charge inversion of Glu541Arg was not able to restore the activity of Arg146Glu/Arg149Glu double mutant, indicating either that one of the two Arg residues in UreD might be involved in an additional electrostatic interaction that was not predicted by EC analysis, or that this asymmetric interaction, showing two positively charged residues on UreD contacting a single negatively charged residue on UreB, cannot be compensated using simple charge inversion.
These results indicate that altering most of the charged residues predicted to be involved in UreB-UreD contacts strongly impacts the in-cell urease activity. The only exception is Lys59 in the UreD sequence, whose mutation to Ala shows negligible effect on urea hydrolysis rate, suggesting that its interaction with Asp336 in UreB is not required for in-cell UreB-UreD functional interaction. On the other hand, mutation to Ala of Asp336 in the UreB sequence fully abrogates the urease activity, indicating that this residue might be important to contact other residues of UreD, still to be identified. To confirm that the identified UreB-UreD interactions functional to Ni(II) ion delivery are close on the protein-protein interface in the quaternary structure, a model of the protein complex is necessary and complements the experimental observations.
All but one of the five EC-predicted residue pairs experimentally tested were confirmed by the enzymatic assay. In addition, the very good match between contacts for the structural protein monomers predicted and experimentally observed (see above) confirms the reliability of the data deriving from the EC analysis. For this reason, all UreB-UreD contacts derived from EC were included in the procedure to build up our model for UreB-UreD complex. The UreB and UreD residues identified through the EC analysis (Table 1) were mapped on the surface of urease and UreD in the UreDFG complex, respectively (Figures 5A, B).
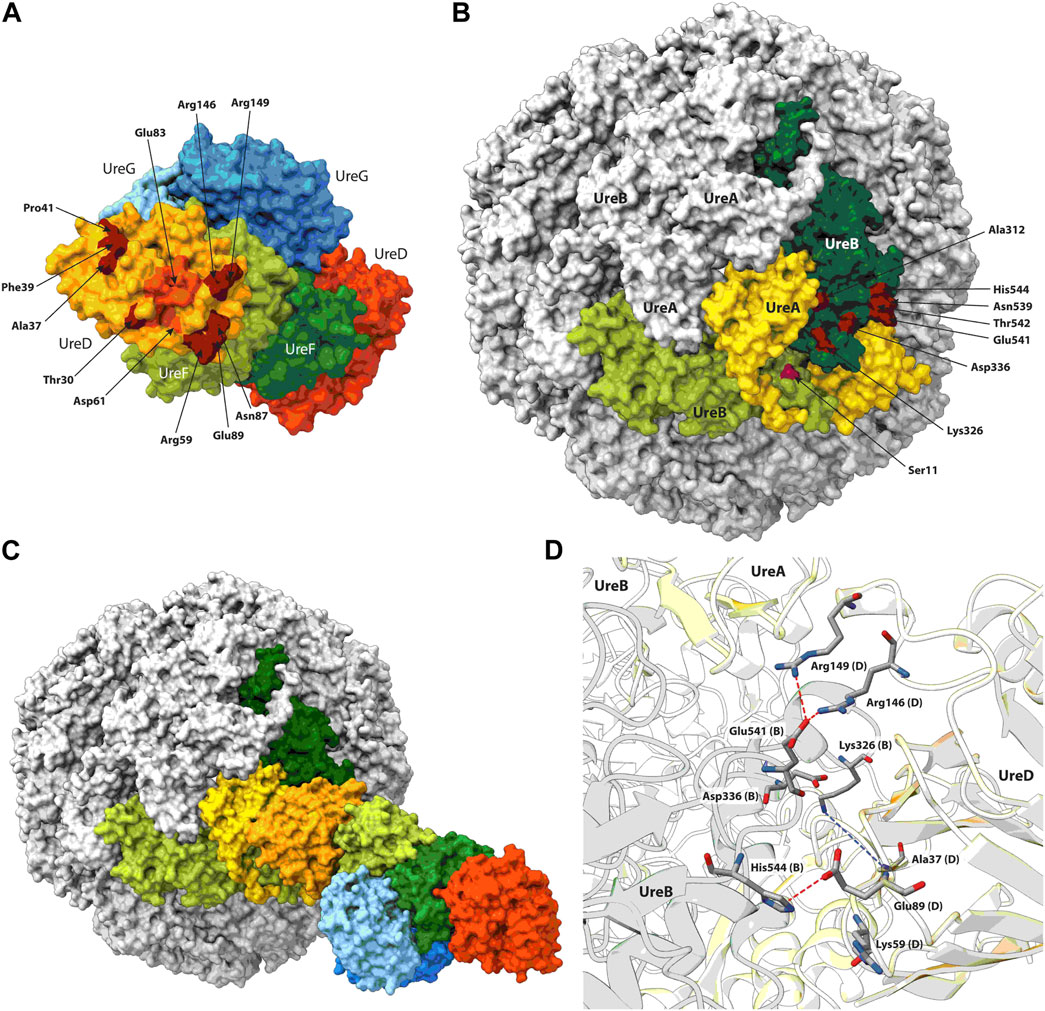
FIGURE 5. UreD2-UreF2-UreG2 (A) and urease (B) molecular surfaces reporting in dark red the position of the residues identified through the EC analysis. The residues identified through MD simulations to be at the exit of one of the UreD tunnels are in dark orange. For urease, the subunits used in the docking calculations have been colored, while the other subunits are in grey. The urease-UreDFG complex resulting from the docking is reported in (C), while the residues found at the urease-UreD interface are shown in (D). The residues cited in the text are in sticks colored according to the atom type, proposed H-bonds are indicated using dashed red lines, while possible contacts are indicated using dashed blue lines.
On UreD, the identified residues are localized in a specific region of the protein surface. Interestingly, the residues found by EC analysis surround the exit of one of the protein tunnels, identified through molecular dynamics (MD) calculations on the UreDFG complex, that was proposed to bring the Ni(II) ions from UreG to UreD, and eventually to the urease active site, passing through UreF (Musiani et al., 2017; Masetti et al., 2020; Masetti et al., 2021). Among the residues composing the tunnel exit, Asp61 and Glu83 are among a list of residues required for urease activation and identified in a mutagenesis/MD study conducted on Klebsiella aerogenes UreD (Farrugia et al., 2015).
While most of the identified residues on UreB are located on a limited area on the surface of the protein, Ser11 appears to be placed in a completely different region. However, in HPU each UreB subunit is part of a large, nearly spherical quaternary structure formed by twelve UreB and twelve UreA subunits, so that if one considers Ser11 from a subunit of UreB adjacent to the one to which all other residues are mapped, then all residues localize in a well-defined region. Thus, the minimal set of protein subunits for protein docking should include at least two UreB subunits. Moreover, in the region comprising the selected residues of UreB, a UreA subunit is also present. For these reasons, and to reduce the computational cost of the docking calculations, a UreD monomer was docked on a UreA-(UreB)2 urease portion. The knowledge-based docking software Haddock (Dominguez et al., 2003; van Zundert et al., 2016) used for the calculations allows the inclusion of, upstream of the calculation, biochemical and/or biophysical interaction data as ambiguous interaction restraints to drive the docking process. The list of UreB and UreD residues predicted by the EC analysis was provided to the docking software without imposing any residue pairing. The best docking model was selected as the best scoring protein complex in the most densely populated cluster of model structures. The latter cluster comprised 50 structures against a total of 200 models, with an average Haddock score of −152 ± 8, and an average buried surface area of 2,745 ± 124 Å2. The use of different urease structures (PDB IDs 6ZJA and 6QSU) did not have any relevant effect on the result of the calculation.
The analysis of the UreD-UreB interface showed that several of the interactions predicted by the EC analysis are present in the model structure (Figures 5C, D). In particular, UreB Glu541 is forming two H-bonds with UreD Arg146 and Arg149. UreD Glu89 is at H-bond distance with UreB His544, even if the side chain of the latter is not in the ideal conformation. Also, UreB Lys326 is in the vicinity of UreD Ala37 and, considering the flexibility of lysine side chain, it is easy to imagine how a H-bond could be formed between the side chain of the former and the alanine backbone. The only exception is Lys59 in the UreD sequence, which is far from Asp336 in UreB, which is consistent with the experimental observation that Lys59Ala does not have any impact on urease activation.
A comparison of the calculated docked urease-UreD complex with that determined using the recently reported cryo-EM structure [PDB ID 8HC1, (Nim et al., 2023)] indicates that the interaction surface has been correctly identified by the EC analysis. In fact, the superimposition of UreD structure as found in the cryo-EM structure with that coming from the docking calculations (Figure 6) shows how in the computational model the urease-UreD interacting surfaces are well modelled. Although there is a rotation of about 30° and a small translation in the orientation of UreD on the urease surface, the model achieved is in good agreement with the experimental structure. After superimposition of the UreA-(UreB)2 portion, the root mean square deviation of the Cα atoms in the UreD subunit is 11.1 Å. However, if one considers only the UreD residues found at the UreB-UreD interface in the experimental cryo-EM structure and identified using the PDBSum server (Laskowski et al., 2018), the Cα RMSD value is reduced to 6.6 Å. This result indicates that, although the orientation is perfectible, the calculated interaction interface is in fair agreement with the experimental structure. Moreover, the Cα-Cα distances calculated for the residue pairs identified through the EC analysis in the cryo-EM structure and in the modelled structure are in good agreement (Table 2). Finally, an estimation of the binding affinity based on the network of interfacial contacts and done by using the PRODIGY webserver (Vangone and Bonvin, 2015; Xue et al., 2016) resulted in comparable values (ΔG = −16.0 and −13.5 kcal·mol−1 for the experimental and the modelled complex, respectively), showing that the achieved model structure is reliable.
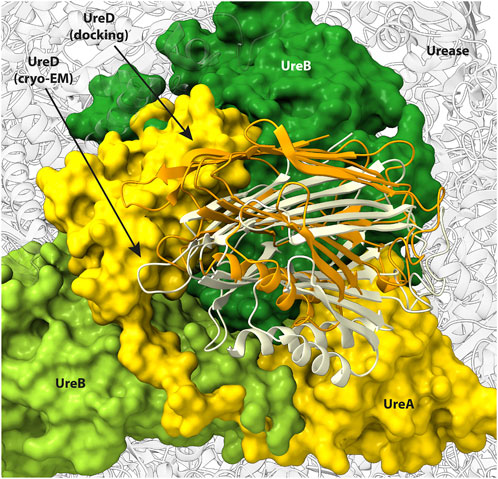
FIGURE 6. Urease surface oriented as in Figure 5B and comparison of UreD position as found in the cryo-EM structure (light yellow ribbons) and in the docking model (orange ribbons). Urease subunits in contact with UreD are reported in green for UreB and in yellow for UreA. The remaining urease subunits are reported as transparent grey ribbons.
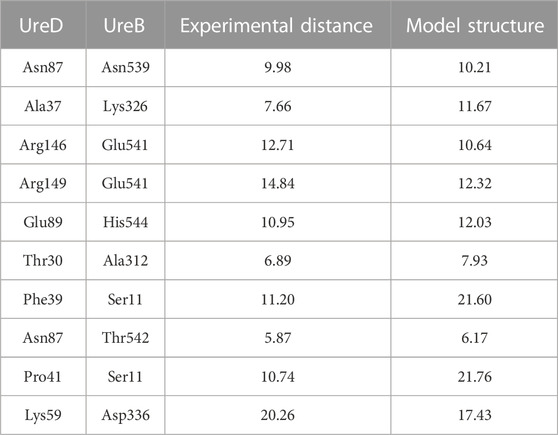
TABLE 2. Cα-Cα distances (in Å) calculated from the cryo-EM urease-UreDF structure (PDB ID 8HC1) and in best docked structure for the UreD-UreB residues pairs identified with the EC analysis.
An EC analysis has been shown to correctly predict the interaction surface between urease and its accessory protein UreD, as demonstrated through the experimental validation via point mutations and the evaluation of in-cell urease activation. The correct mapping of the residues identified by EC analysis on the urease multimer proved crucial to obtain a reasonable model by macromolecular docking. The availability of the cryo-EM structure of the protein complex confirms the accuracy of the predicted model. This work represents a proof of concept of a protocol exportable to other protein systems, potentially making up for the lack of experimental structures to generate realistic models of interaction surfaces. The result of this approach predicts a position and orientation of the proteins in the urease-UreDF complex that is largely consistent with the experimental cryo-EM structure. In particular, the interaction patch is correctly predicted to be located on the large and fairly flat surface of an oblate triangular prism that approximates the shape of each (αβ)3 trimer that makes up the nearly spherical [(αβ)3]4 aggregate in HPU, in the proximity of the active site channel. The results of such methodology, applied to structurally undetermined protein complexes, can be used to provide new information on the structure-function relationships of such quaternary contacts or for the design of molecules capable of interfering with the protein assembly.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
NC and BZ carried out the experimental in cell enzymatic activation assays; KB performed the evolutionary couplings calculations; FM carried out the computational structural modelling; BZ, FM, CS, and SC are responsible for conceptualization and design of the study, analysis, and interpretation of data. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Banaszak, K., Martin-Diaconescu, V., Bellucci, M., Zambelli, B., Rypniewski, W., Maroney, M. J., et al. (2012). Crystallographic and X-ray absorption spectroscopic characterization of Helicobacter pylori UreE bound to Ni²⁺ and Zn²⁺ reveals a role for the disordered C-terminal arm in metal trafficking. Biochem. J. 441, 1017–1026. doi:10.1042/BJ20111659
Bellucci, M., Zambelli, B., Musiani, F., Turano, P., and Ciurli, S. (2009). Helicobacter pylori UreE, a urease accessory protein: specific Ni2+- and Zn2+-binding properties and interaction with its cognate UreG. Biochem. J. 422, 91–100. doi:10.1042/bj20090434
Chang, Z., Kuchar, J., and Hausinger, R. P. (2004). Chemical cross-linking and mass spectrometric identification of sites of interaction for UreD, UreF, and urease. J. Biol. Chem. 279, 15305–15313. doi:10.1074/jbc.M312979200
Cunha, E. S., Chen, X., Sanz-Gaitero, M., Mills, D. J., and Luecke, H. (2021). Cryo-EM structure of Helicobacter pylori urease with an inhibitor in the active site at 2.0 A resolution. Nat. Commun. 12, 230. doi:10.1038/s41467-020-20485-6
Dominguez, C., Boelens, R., and Bonvin, A. M. (2003). Haddock: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737. doi:10.1021/ja026939x
Eschweiler, J. D., Farrugia, M. A., Dixit, S. M., Hausinger, R. P., and Ruotolo, B. T. (2018). A structural model of the urease activation complex derived from ion mobility-mass spectrometry and integrative modeling. Structure 26, 599–606. doi:10.1016/j.str.2018.03.001
Farrugia, M. A., Macomber, L., and Hausinger, R. P. (2013). Biosynthesis of the urease metallocenter. J. Biol. Chem. 288, 13178–13185. doi:10.1074/jbc.R112.446526
Farrugia, M. A., Wang, B., Feig, M., and Hausinger, R. P. (2015). Mutational and computational evidence that a nickel-transfer tunnel in UreD is used for activation of Klebsiella aerogenes urease. Biochemistry 54, 6392–6401. doi:10.1021/acs.biochem.5b00942
Fong, Y. H., Wong, H. C., Chuck, C. P., Chen, Y. W., Sun, H., and Wong, K. B. (2011). Assembly of preactivation complex for urease maturation in Helicobacter pylori: crystal structure of UreF-UreH protein complex. J. Biol. Chem. 286, 43241–43249. doi:10.1074/jbc.m111.296830
Fong, Y. H., Wong, H. C., Yuen, M. H., Lau, P. H., Chen, Y. W., and Wong, K. B. (2013). Structure of UreG/UreF/UreH complex reveals how urease accessory proteins facilitate maturation of Helicobacter pylori urease. PLoS Biol. 11, e1001678. doi:10.1371/journal.pbio.1001678
Goddard, T. D., Huang, C. C., Meng, E. C., Pettersen, E. F., Couch, G. S., Morris, J. H., et al. (2018). UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25. doi:10.1002/pro.3235
Green, A. G., Elhabashy, H., Brock, K. P., Maddamsetti, R., Kohlbacher, O., and Marks, D. S. (2021). Large-scale discovery of protein interactions at residue resolution using co-evolution calculated from genomic sequences. Nat. Commun. 12, 1396. doi:10.1038/s41467-021-21636-z
Grossoehme, N. E., Mulrooney, S. B., Hausinger, R. P., and Wilcox, D. E. (2007). Thermodynamics of Ni2+, Cu2+, and Zn2+ binding to the urease metallochaperone UreE. Biochemistry 46, 10506–10516. doi:10.1021/bi700171v
Ha, N. C., Oh, S. T., Sung, J. Y., Cha, K. A., Lee, M. H., and Oh, B. H. (2001a). Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat. Struct. Biol. 8, 505–509. doi:10.1038/88563
Ha, N. C., Oh, S. T., Sung, J. Y., Cha, K. A., Lee, M. H., and Oh, B. H. (2001b). Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat. Struct. Biol. 8, 505–509. doi:10.1038/88563
Hopf, T. A., Green, A. G., Schubert, B., Mersmann, S., Scharfe, C. P. I., Ingraham, J. B., et al. (2019). The EVcouplings Python framework for coevolutionary sequence analysis. Bioinformatics 35, 1582–1584. doi:10.1093/bioinformatics/bty862
Hopf, T. A., Scharfe, C. P., Rodrigues, J. P., Green, A. G., Kohlbacher, O., Sander, C., et al. (2014). Sequence co-evolution gives 3D contacts and structures of protein complexes. Elife 3, e03430. doi:10.7554/elife.03430
Johnson, L. S., Eddy, S. R., and Portugaly, E. (2010). Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinforma. 11, 431. doi:10.1186/1471-2105-11-431
Lam, R., Romanov, V., Johns, K., Battaile, K. P., Wu-Brown, J., Guthrie, J. L., et al. (2010). Crystal structure of a truncated urease accessory protein UreF from Helicobacter pylori. Proteins 78, 2839–2848. doi:10.1002/prot.22802
Laskowski, R. a.-O., Jabłońska, J., Pravda, L., Vařeková, R. S., and Thornton, J. M. (2018). PDBsum: structural summaries of PDB entries. Protein Sci. 27, 129–134. doi:10.1002/pro.3289
Ligabue-Braun, R., Real-Guerra, R., Carlini, C. R., and Verli, H. (2013). Evidence-based docking of the urease activation complex. J. Biomol. Struct. Dyn. 31, 854–861. doi:10.1080/07391102.2012.713782
Marks, D. S., Colwell, L. J., Sheridan, R., Hopf, T. A., Pagnani, A., Zecchina, R., et al. (2011). Protein 3D structure computed from evolutionary sequence variation. PLoS One 6, e28766. doi:10.1371/journal.pone.0028766
Maroney, M. J., and Ciurli, S. (2021). Nickel as a virulence factor in the Class I bacterial carcinogen, Helicobacter pylori. Seminars Cancer Biol. 76, 143–155. doi:10.1016/j.semcancer.2021.04.009
Maroney, M. J., and Ciurli, S. (2014). Nonredox nickel enzymes. Chem. Rev. 114, 4206–4228. doi:10.1021/cr4004488
Masetti, M., Bertazzo, M., Recanatini, M., Ciurli, S., and Musiani, F. (2021). Probing the transport of Ni(II) ions through the internal tunnels of the Helicobacter pylori UreDFG multimeric protein complex. J. Inorg. Biochem. 223, 111554. doi:10.1016/j.jinorgbio.2021.111554
Masetti, M., Falchi, F., Gioia, D., Recanatini, M., Ciurli, S., and Musiani, F. (2020). Targeting the protein tunnels of the urease accessory complex: A theoretical investigation. Molecules 25, 2911. doi:10.3390/molecules25122911
Mazzei, L., Cianci, M., Benini, S., and Ciurli, S. (2019). The structure of the elusive urease-urea complex unveils the mechanism of a paradigmatic nickel-dependent enzyme. Angew. Chem. Int. Ed. 58, 7415–7419. doi:10.1002/anie.201903565
Mazzei, L., and Ciurli, S. (2021). “Urease,” in Encycl. Inorg. Bioinorg. Chem. (Chichester (UK): John Wiley & Sons, Ltd), 1–17.
Mazzei, L., Musiani, F., and Ciurli, S. (2020). The structure-based reaction mechanism of urease, a nickel dependent enzyme: tale of a long debate. J. Biol. Inorg. Chem. 25, 829–845. doi:10.1007/s00775-020-01808-w
Mazzei, L., Musiani, F., and Ciurli, S. (2017). “Urease,” in The biological Chemistry of nickel. Editors D. Zamble, M. RowińSka-ŻYrek, and H. Kozłowski (Royal Society of Chemistry), 60–97.
Moncrief, M. C., and Hausinger, R. P. (1996). Purification and activation properties of UreD-UreF-urease apoprotein complexes. J. Bacteriol. 178, 5417–5421. doi:10.1128/jb.178.18.5417-5421.1996
Musiani, F., Gioia, D., Masetti, M., Falchi, F., Cavalli, A., Recanatini, M., et al. (2017). Protein tunnels: the case of urease accessory proteins. J. Chem. Theory Comput. 13, 2322–2331. doi:10.1021/acs.jctc.7b00042
Nim, Y. S., Fong, I. Y. H., Deme, J., Tsang, K. L., Caesar, J., Johnson, S., et al. (2023). Delivering a toxic metal to the active site of urease. Sci. Adv. 9, eadf7790. doi:10.1126/sciadv.adf7790
Nim, Y. S., and Wong, K.-B. (2019). The maturation pathway of nickel urease. Inorganics 7, 85. doi:10.3390/inorganics7070085
Park, I. S., Carr, M. B., and Hausinger, R. P. (1994). In vitro activation of urease apoprotein and role of UreD as a chaperone required for nickel metallocenter assembly. Proc. Natl. Acad. Sci. U.S.A. 91, 3233–3237. doi:10.1073/pnas.91.8.3233
Pettersen, E. F., Goddard, T. D., Huang, C. C., Meng, E. C., Couch, G. S., Croll, T. I., et al. (2021). UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82. doi:10.1002/pro.3943
Pierro, A., Etienne, E., Gerbaud, G., Guigliarelli, B., Ciurli, S., Belle, V., et al. (2020). Nickel and GTP modulate Helicobacter pylori UreG structural flexibility. Biomolecules 10, 1062. doi:10.3390/biom10071062
Quiroz-Valenzuela, S., Sukuru, S. C., Hausinger, R. P., Kuhn, L. A., and Heller, W. T. (2008). The structure of urease activation complexes examined by flexibility analysis, mutagenesis, and small-angle X-ray scattering. Arch. Biochem. Biophys. 480, 51–57. doi:10.1016/j.abb.2008.09.004
Remaut, H., Safarov, N., Ciurli, S., and Van Beeumen, J. (2001). Structural basis for Ni2+ transport and assembly of the urease active site by the metallochaperone UreE from Bacillus pasteurii. J. Biol. Chem. 276, 49365–49370. doi:10.1074/jbc.M108304200
Rutherford, J. C. (2014). The emerging role of urease as a general microbial virulence factor. PLoS Pathog. 10, e1004062. doi:10.1371/journal.ppat.1004062
Savoldi, A., Carrara, E., Graham, D. Y., Conti, M., and Tacconelli, E. (2018). Prevalence of antibiotic resistance in Helicobacter pylori: A systematic review and meta-analysis in world health organization regions. Gastroenterology 155, 1372–1382.e17. doi:10.1053/j.gastro.2018.07.007
Shi, R., Munger, C., Asinas, A., Benoit, S. L., Miller, E., Matte, A., et al. (2010). Crystal structures of apo and metal-bound forms of the UreE protein from Helicobacter pylori: role of multiple metal binding sites. Biochemistry 49, 7080–7088. doi:10.1021/bi100372h
Sjodt, M., Brock, K., Dobihal, G., Rohs, P. D. A., Green, A. G., Hopf, T. A., et al. (2018). Structure of the peptidoglycan polymerase RodA resolved by evolutionary coupling analysis. Nature 556, 118–121. doi:10.1038/nature25985
Sjodt, M., Rohs, P. D. A., Gilman, M. S. A., Erlandson, S. C., Zheng, S., Green, A. G., et al. (2020). Structural coordination of polymerization and crosslinking by a SEDS-bPBP peptidoglycan synthase complex. Nat. Microbiol. 5, 813–820. doi:10.1038/s41564-020-0687-z
Song, H.-K., Mulrooney, S. B., Huber, R., and Hausinger, R. P. (2001). Crystal structure of Klebsiella aerogenes UreE, a nickel-binding metallochaperone for urease activation. J. Biol. Chem. 276, 49359–49364. doi:10.1074/jbc.M108619200
Soriano, A., Colpas, G. J., and Hausinger, R. P. (2000). UreE stimulation of GTP-dependent urease activation in the UreD-UreF-UreG-urease apoprotein complex. Biochemistry 39, 12435–12440. doi:10.1021/bi001296o
Spronk, C., Zerko, S., Gorka, M., Kozminski, W., Bardiaux, B., Zambelli, B., et al. (2018). Structure and dynamics of Helicobacter pylori nickel-chaperone HypA: an integrated approach using NMR spectroscopy, functional assays and computational tools. J. Biol. Inorg. Chem. 23, 1309–1330. doi:10.1007/s00775-018-1616-y
Suzek, B. E., Wang, Y., Huang, H., Mcgarvey, P. B., Wu, C. H., and Uniprot, C. (2015). UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932. doi:10.1093/bioinformatics/btu739
Tarsia, C., Danielli, A., Florini, F., Cinelli, P., Ciurli, S., and Zambelli, B. (2018). Targeting Helicobacter pylori urease activity and maturation: in-cell high-throughput approach for drug discovery. Biochimica Biophysica Acta (BBA) - General Subj. 1862, 2245–2253. doi:10.1016/j.bbagen.2018.07.020
Tsang, K. L., and Wong, K. B. (2022). Moving nickel along the hydrogenase-urease maturation pathway. Metallomics 14, mfac003. doi:10.1093/mtomcs/mfac003
Van Zundert, G. C. P., Rodrigues, J., Trellet, M., Schmitz, C., Kastritis, P. L., Karaca, E., et al. (2016). The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 428, 720–725. doi:10.1016/j.jmb.2015.09.014
Vangone, A., and Bonvin, A. M. J. J. (2015). Contacts-based prediction of binding affinity in protein–protein complexes. eLife 4, e07454. doi:10.7554/elife.07454
Xue, L. C., Rodrigues, J. P., Kastritis, P. L., Bonvin, A. M., and Vangone, A. (2016). Prodigy: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 32, 3676–3678. doi:10.1093/bioinformatics/btw514
Yuen, M. H., Fong, Y. H., Nim, Y. S., Lau, P. H., and Wong, K. B. (2017). Structural insights into how GTP-dependent conformational changes in a metallochaperone UreG facilitate urease maturation. Proc. Natl. Acad. Sci. U. S. A. 114, E10890–E10898. doi:10.1073/pnas.1712658114
Zambelli, B., Banaszak, K., Merloni, A., Kiliszek, A., Rypniewski, W., and Ciurli, S. (2013). Selectivity of Ni(II) and Zn(II) binding to sporosarcina pasteurii UreE, a metallochaperone in the urease assembly: A calorimetric and crystallographic study. J. Biol. Inorg. Chem. 18, 1005–1017. doi:10.1007/s00775-013-1049-6
Zambelli, B., Berardi, A., Martin-Diaconescu, V., Mazzei, L., Musiani, F., Maroney, M. J., et al. (2014). Nickel binding properties of Helicobacter pylori UreF, an accessory protein in the nickel-based activation of urease. J. Biol. Inorg. Chem. 19, 319–334. doi:10.1007/s00775-013-1068-3
Zambelli, B., Basak, P., Hu, H., Piccioli, M., Musiani, F., Broll, V., et al. (2023). The structure of the high-affinity nickel-binding site in the Ni,Zn-HypA*UreE2 complex. Metallomics 15, mfad003. doi:10.1093/mtomcs/mfad003
Zambelli, B., Mazzei, L., and Ciurli, S. (2020). Intrinsic disorder in the nickel-dependent urease network. Prog. Mol. Biol. Transl. Sci. 174, 307–330. doi:10.1016/bs.pmbts.2020.05.004
Keywords: urease (urea amidohydrolase EC 3.5.1.5), enzyme activation, Ni(II) transport, evolutionary couplings, in cell enzymatic assay, molecular modelling
Citation: Carosella N, Brock KP, Zambelli B, Musiani F, Sander C and Ciurli S (2023) Functional contacts for activation of urease from Helicobacter pylori: an integrated approach using evolutionary couplings, in-cell enzymatic assays, and computational docking. Front. Chem. Biol 2:1243564. doi: 10.3389/fchbi.2023.1243564
Received: 20 June 2023; Accepted: 20 September 2023;
Published: 11 October 2023.
Edited by:
Debbie C. Crans, Colorado State University, United StatesReviewed by:
Craig C. McLauchlan, Illinois State University, United StatesCopyright © 2023 Carosella, Brock, Zambelli, Musiani, Sander and Ciurli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Ciurli, c3RlZmFuby5jaXVybGlAdW5pYm8uaXQ=
†ORCID: Noemi Carosella, orcid.org/0009-0007-1232-1484; Kelly P. Brock, orcid.org/0000-0002-5236-3773; Barbara Zambelli, orcid.org/0000-0002-3876-0051; Francesco Musiani, orcid.org/0000-0003-0200-1712; Chris Sander, orcid.org/0000-0001-6059-6270; Stefano Ciurli, orcid.org/0000-0001-9557-926X
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.