 Xinhang Yang1
Xinhang Yang1 Callie Miller
Callie Miller Brian Bennett
Brian Bennett Richard C. Holz
Richard C. Holz- 1Department of Chemistry, Marquette University, Milwaukee, WI, United States
- 2Department of Chemistry, Colorado School of Mines, Golden, CO, United States
- 3Department of Physics, Milwaukee, WI, United States
The catalytically competent Co(II)-loaded form of the chlorothalonil dehalogenase from Pseudomonas sp. CTN-3 (Chd, EC 3.8.1.2) was characterized by kinetic and spectroscopic methods. Maximum chlorothalonil (TPN; 2,4,5,6-tetrachloroisophtalonitrile) dehalogenase activity was observed in the presence of one Co(II) ion per monomer with kcat and Km values of 12 ± 3 s−1 and 130 ± 10 μM, respectively, providing a catalytic efficiency (kcat/Km) of ∼9.2 × 104 M−1s−1. The dissociation constant (Kd) for Co(II) was determined to be 0.29 µM, and UV-Vis spectroscopy indicated the active site Co(II) ion resides in a penta-coordinate environment. EPR spectra of Co1-Chd contain at least three distinct signals, an MS = ± 1/2 signal with a ∼94 G 59Co hyperfine pattern centered at g1’ ≅ 6.7, a broader MS = ± 1/2 signal with g1’ ≅ 5.7, an MS = ± 3/2 signal with tentatively estimated parameters of g1’ ≅ 10.5 (gz = 2.75), A1(59Co) ≅ 110 G, and a high-field broad resonance at g3’ ≅ 1.8. Four substrate-analog inhibitors with IC50 values ranging from 110 μM to 19 mM were also identified and characterized. Upon the addition of each of the substrate-like inhibitors to Co1-Chd, changes in the EPR spectrum were observed that, in all cases, were simpler than that of Co1-Chd in the absence of inhibitors and could be simulated as either a single species or a mixture of two. Simulation of these data indicate that the corresponding EPR signals are each due to a ground state MS = 1/2 Kramers’ doublet and are consistent with pentacoordinate Co(II) with a relatively constrained coordination sphere. These data suggest that the nitrile moiety of TPN may not directly coordinate to the active site metal ion, providing new insight into the catalytic mechanism for Chd.
Introduction
Chlorothalonil (TPN; 2,4,5,6-tetrachloroisophthalonitrile) is one of the most commonly used fungicides in the US with more than five million kilograms sprayed on crops and fruits each year (Caux et al., 1996; Sakkas et al., 2002; Carlo-Rojas et al., 2004; Mozzachio et al., 2008). TPN has low solubility in water (100 mg L−1) but is strongly absorbed in soil, particularly soil with high organic matter such as those found in aquatic environments. TPN is highly toxic to fish and aquatic species as well as birds and invertebrates. It is quite resistant to hydrolysis between pH 5 to 7, with a half-life of 30–60 days, and it can remain in soil for over a year (Kwon and Armbrust, 2006). As such, TPN is emerging as a major environmental issue (Vickers et al., 1985; Sims et al., 1991; Wang et al., 2010). TPN is also a human skin and eye irritant that can cause severe gastrointestinal issues. Animal studies involving mice have shown that TPN can cause kidney cancer, so it has been classified by the U.S. Environmental Protection Agency (EPA) as a probable human carcinogen (Mozzachio et al., 2008). Given the widespread use of TPN and its human toxicity, its biodegradation and environmental clean-up has become a topic of significant importance (Viciu et al., 2001).
The chlorothalonil dehalogenase from Pseudomonas sp.
CTN-3 (Chd, EC 3.8.1.2) is a Zn(II)-dependent hydrolytic dehalogenase that selectively substitutes an aromatic chlorine with an aromatic alcohol (yielding 4-hydroxytrichloro-isophthalonitrile, 4-dOH-TPN, from TPN) (Figure 1) (Wang et al., 2010). Several bacterial strains harbor Chd genes that exhibit remarkable sequence identity (>95%) (Liang et al., 2011; Ren et al., 2011; Yue et al., 2015; Yang et al., 2019). Chd was recently structurally characterized (PDB: 6UXU at 1.96 Å) revealing an αββα-sandwich fold that is commonly observed in the β-lactamase superfamily (Catlin et al., 2020). Chd is a “head-to-tail” homodimer, formed between two α-helices from each monomer with a Zn(II) ion at the dimer interface that likely functions as a structural site. The catalytic mononuclear Zn(II) center in Chd resides in a slightly distorted trigonal bipyramid (TBP) geometry with His117, His257, Asp116, Asn216, and a water/hydroxide as ligands (Figure 1B). The X-ray crystal structure of Chd, in combination with kinetic data allowed us to propose a catalytic mechanism for Chd (Figure 2A) (Catlin et al., 2020). However, several aspects of this proposed mechanism are unknown.
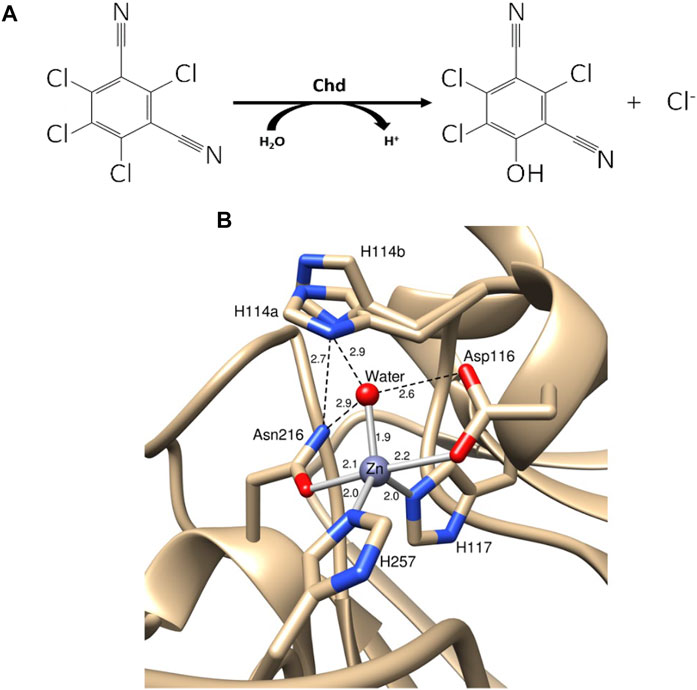
FIGURE 1. (A) Hydrolysis of TPN to 4-OH-TPN and chloride by Chd. (B) Chd active site with Zn(II) bonds shown as gray sticks and hydrogen bonds shown as black dashed lines (PDB: 6UXU). Key distances are shown in angstroms and the two alternate conformations of His114 are labeled as His114a and His114b.
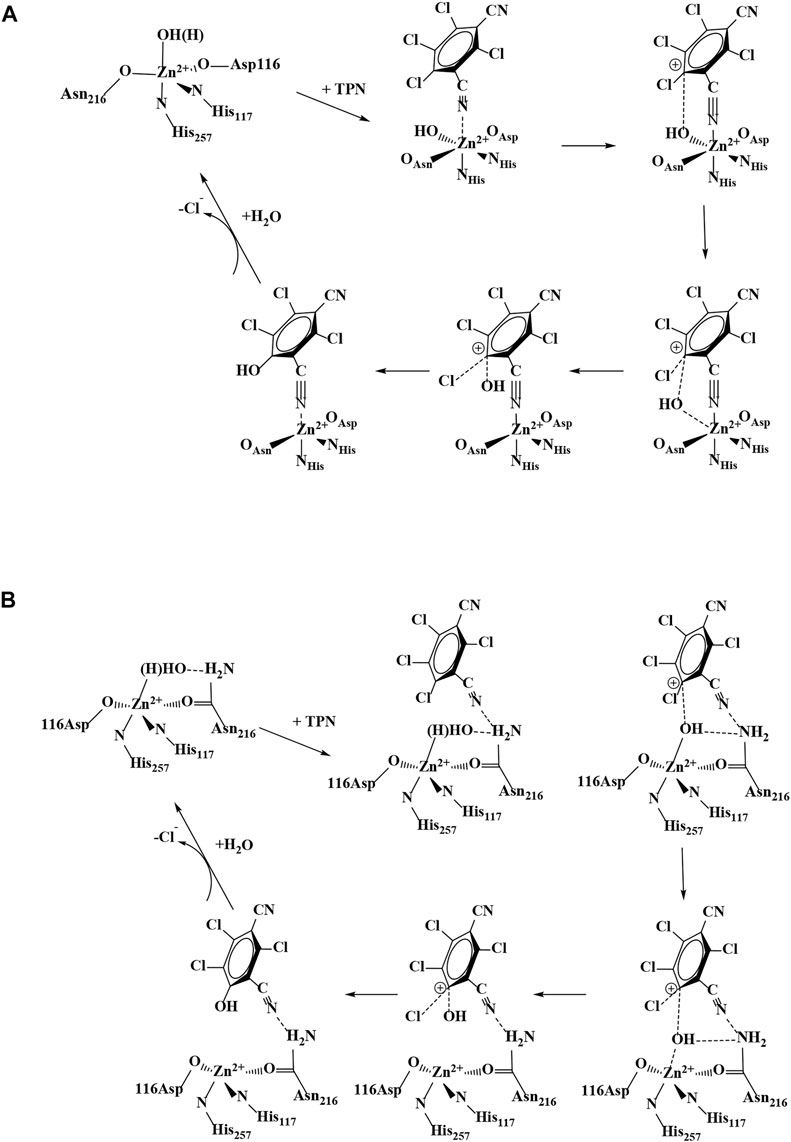
FIGURE 2. Proposed catalytic mechanisms for TPN hydrolysis by Chd: (A) direct coordination of TPN and (B) TPN binding near the Zn(II) active site.
One major mechanistic question involves whether the substrate, TPN, binds directly to the active site metal ion or simply binds near the metal ion in the active site. To date, no X-ray structure exists with Chd bound by either a substrate or an inhibitor. However, substrate docking was recently utilized to examine possible substrate binding poses (Catlin et al., 2020). The best pose suggested a binding interaction between the nitrogen atom of a cyano group of TPN and the active site Zn ion (∼2.3 Å), along with an energetically favorable π- π interaction between the TPN aromatic ring and that of an active site residue, Trp227. This pose pre-organizes a possible transition state of TPN hydrolysis by placing the ortho carbon of TPN within ∼3.0 Å of the O atom of the bound water/hydroxide. In an effort to gain insight into the substrate binding step of the catalytic mechanism of Chd, several aromatic compounds were tested as inhibitors of Chd (Figure 3). Each of these compounds were found to inhibit Chd activity with IC50 values ranging from 110 μM to 19 mM. Utilizing the catalytically competent Co(II)-substituted form of Chd (Chen et al., 2016), EPR spectra were obtained in the absence and presence of each newly identified Chd inhibitor. The combination of these data with the previously reported kinetic (Yang et al., 2019) and X-ray crystallographic (Catlin et al., 2020) data, provides new insight into the substrate binding step of the Chd catalytic mechanism.
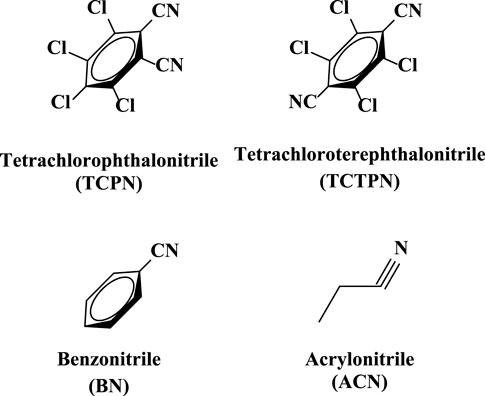
FIGURE 3. Structures of the four Chd inhibitors examined.
Materials and methods
Materials
4-(2-hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES), sodium chloride, PD-Minitrap G10 desalting column, potassium bromide, potassium sodium tartrate, chlorothalonil, Chelex 100 resin, Slide-A-Lyzer dialysis cassettes, benzonitrile, ethylenediaminetetraacetic acid (EDTA), isopropyl-β-D-1- thiogalactopyranoside (IPTG), kanamycin, imidazole, Coomassie protein assay reagent, Amicon Ultra-15 centrifugal filters, glycerol, tris(2-carboxyethyl)phosphine (TCEP), tetrachlorophthalonitrile (TCPN), tetrachloroterephthalonitrile (TCTPN), acrylonitrile (ACN), benzonitrile (BN), and BL21(DE3) competent Escherichia coli cells were purchased from commercial sources, and reagents were of the highest quality available. TEV protease (EC 3.4.22.44) was obtained via previously published methods (Tropea et al., 2009).
Expression and purification of Chd
A 15 residue N-terminally-truncated form of Chd was expressed and purified as previously described (Catlin et al., 2020). The purified enzyme exhibited a single band on 12% SDS-PAGE, consistent with its calculated Mr of 36,107 Da. Chd protein concentrations were determined via the Bradford assay. The rate of TPN dehalogenation by Chd was determined by continuously monitoring the formation of 4-OH-TPN at 345 nm (Δε345 = 3.5 mM−1 cm−1) as previously reported (Kwon and Armbrust, 2006). Chd exhibited a kcat of 24 ± 2 s−1 and a Km value of 110 ± 30 µM in 50 mM HEPES buffer, pH 7.0, at 25°C. IC50 values were determined for BN, TCPN, TCTPN, and ACN. Initial rates were determined at a minimum of five inhibitor concentrations, providing a standard deviation of ±10%, and a dose-response curve yielded the IC50 value where the maximum inhibitor concentration was at least 2X the IC50 value.
Cobalt-substituted Chd
Apo-Chd was obtained by anaerobic incubation of as-purified Chd, for 24 h at 4°C, in buffer containing ∼15 mM 1,10-phenanthroline and 40 mM EDTA. The metal chelators were removed via a PD-Minitrap G10 desalting column followed by dialysis for 16 h with Chelex-100 treated 50 mM HEPES at pH 7.0. The resulting enzyme exhibited no detectable activity, and inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis indicated <0.05 eq Zn per mole of Chd. Titration of Chd with Co(II) was carried out by incubation of Chd, for 30 min at 25°C, with 2–10 μL of 8–32 mM Co(II)aq (99.999% CoCl2, Strem Chemicals, Newburyport, MA, United States) to 200 μM apo-Chd, and was monitored spectrophotometrically between 400 and 650 nm and the rate of hydrolysis of TPN (0.25 mM) was measured as a function of [Co(II)] (D'Souza et al., 2000).
Spectroscopic measurements
Electronic absorption spectra were recorded on a Shimadzu UV-2450 spectrophotometer equipped with a TCC-240A temperature-controlled cell holder. All solutions were degassed prior to performing an experiment.
Samples for EPR titration of Co(II) were prepared by direct addition of 0.5–3.0 eq Co(II) per monomer Chd to a solution of apo-Chd in a 1.5 mL microcentrifuge tube, with immediate subsequent mixing by multiple inversions of the closed tube, and incubation for 30 min at 25°C. EPR samples of Chd with substrate-like inhibitors were prepared by direct addition of an aliquot of the substrate-like inhibitor (1–5 eq.) to a solution of Chd previously incubated with 1 eq Co(II) and the sample again mixed and incubated for 10 min. Each sample was then clarified by centrifugation for 30 s at 2,200 × g (Fischer Mini Centrifuge), and the supernatant was transferred to an EPR tube and immediately frozen in liquid nitrogen.
Experimental (∂χ″/∂B) EPR spectra were recorded at 9.468 GHz, 12 K, 2 mW microwave power, and 12 G (1.2 mT) magnetic field modulation, using instrumentation described in earlier work (Karunagala Pathiranage et al., 2021). Second derivative (∂2χ″/∂B2) EPR spectra were generated by pseudomodulation (Hyde et al., 1990) of the experimental spectra with a 22 G pseudomodulation amplitude (Xenon, Bruker). EPR simulations were carried out using EasySpin (Stoll and Schweiger, 2006). Where only a single Kramers’ doublet is observed in the spectrum for an S > 1/2 system, a unique solution to the phenomenological EPR parameters g’1, g’2, and g’3 in terms of spin Hamiltonian parameters is not possible for a rhombic g tensor (Bennett et al., 2010) and an axial g tensor is, instead, assumed with gx = gy (= g⊥) ≠ gz (= g||). For S = 3/2 Co(II) in proteins and enzymes, the magnitude of the axial zero field splitting (ZFS), |D|, is typically much greater than the Zeeman interaction and the spectrum at low temperatures (i.e., when kT << Δ, where Δ = 2D[1 + 3(E/D)2]1/2 and E is the rhombic ZFS term) therefore exhibits a signal only from the ground state Kramers’ doublet and is otherwise insensitive to |D|. Values of 1 × 106 MHz (33 cm−1; for S = 3/2 signal) and 2 × 106 MHz (66 cm−1; for S = 3) were assumed here, corresponding to interdoublet splittings, Δ, of ≥66 cm−1 and ≥132 cm−1, respectively, and excited state population temperatures of ≥92 and ≥184 K. The salient spin Hamiltonian parameters are then g|| and g⊥; E/D; the sign of D, which is indicative of the signal-exhibiting ground state Kramers’ doublet (MS = ± 1/2 or ± 3/2); and any 59Co I = 7/2 hyperfine interaction (hfi). The hfi was calculated in frequency (energy equivalent) terms, which is related to the phenomenological hyperfine splitting (hfs) in field terms by A (MHz) = g(μB/h) × A (G) and, therefore, knowledge of the g tensor is required to interconvert these values. Where simulations indicated the presence of more than one chemical species, the relative contributions to the total spin densities were obtained by double integration of the simulations of the contributory signals. Supplementary information includes all parameters used (Supplementary Tables S1–S6) to simulate all EPR spectra reported in the manuscript for the inhibited complexes using EasySpin.
Results and discussion
Cobalt(II) binding to Chd
Apo-Chd was prepared by adding a 15 mM 1,10-Phenanthroline/40 mM EDTA solution to as-purified Chd under anaerobic conditions for ∼24 h followed by desalting via column chromatography and dialysis against 50 mM HEPES, pH 7.0 (Chen et al., 2016). Chd containing 1 eq. of Co(II) per monomer (Co1-Chd) exhibited a kcat of 12 ± 3 s−1 and a Km value of 130 ± 10 µM with TPN as the substrate providing a catalytic efficiency (kcat/Km) of ∼9.2 × 104 M−1s−1. Compared to the Zn(II) bound Chd enzyme, which exhibited a kcat value of 24 s−1, a Km value of 110 μM, and kcat/Km ∼2.2 × 105 M−1s−1 (Yang et al., 2019), the Co(II) form of Chd exhibits a 2-fold decrease in kcat, in line with typical Zn(II) enzymes substituted with Co(II).
The intrinsic dissociation constant (Kd) for Co(II) binding to Chd was determined by titrating 0.25 µM apo-Chd at pH 7.0 in 50 mM HEPES buffer at 25°C with Co(II)aq and monitoring the catalytic activity as a function of [Co(II)], as previously described (Yang et al., 2019). Briefly, Kd, and the number of binding sites, p, were determined by fitting these titration data to Eq. 1 (D'Souza et al., 2000), where r, the binding function, is defined by the fractionation saturation fa and number of binding sites (Eq. 2) (Winzor and Sawyer, 1995). In Eq. 1, Cs is the concentration of free Co(II).
Cs was calculated from the total concentration of zinc added to the reaction (CTS) by Eq. 3 (Watterson et al., 2008), where CA is the total molar concentration of enzyme (0.25 µM).
These data are plotted as r vs. [Co(II)] (Figure 4). The best fits indicated a single Co(II) binding site per monomer (p = 0.91 ± 0.01), with an intrinsic Kd value of 0.29 µM. This is comparable to Zn(II) binding to Chd, where a single Zn(II) bound per monomer (p = 1.03) with an intrinsic Kd value of 0.17 µM (Yang et al., 2019). Attempts to fit these data with a cooperative model were inconclusive as the resulting goodness-of-fit did not improve compared to the non-cooperative model. As previously reported, removal of the structural Zn(II) site ligands had no apparent effect on Chd activity (Catlin et al., 2020), so these data reflect only the binding to the Chd active site. Occupation of the structural site would affect the calculation in as far as it would affect the value of CS but additional data indicated that, under the experimental conditions employed, the structural site is not occupied but that Chd exists in solution primarily as a dimer (Catlin et al., 2020). In particular, inductively coupled atomic emission spectroscopy (ICP-AES) data obtained on Chd after the addition of either 4 eq of Zn(II) or Co(II) to Chd followed by dialysis for 24 h indicated that ∼0.9 eq of Zn(II) and ∼1.0 eq. Co(II), respectively, were tightly bound per Chd monomer. No other first-row transition metal ions were detected via ICP-MS (<10 ppb).
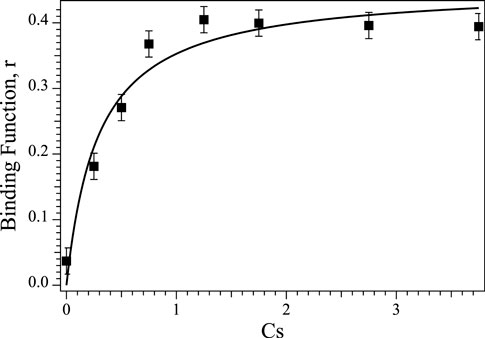
FIGURE 4. Plot of Binding Function, r vs. the concentration of free Co(II) ions (Cs) in solution for Co(II) titration into apo-Chd at 25°C in Chelex-100 treated 50 mM HEPES buffer, pH 7.0.
Chd inhibitor kinetic studies
Inhibition data for the compounds in Figure 3 are summarized in Table 1. TCPN and TCTPN are structurally similar to TPN, with ortho and para-CN groups, respectively, rather than meta-CN groups in TPN (Figure 1). These two substrate-analog inhibitors exhibited IC50 values of 0.11 ± 0.01 mM and 0.15 ± 0.02 mM, respectively. Surprisingly, Chd was found to be capable of hydrolyzing TCPN, albeit very slowly, with a kcat value of 0.50 ± 0.02 s−1. The fact that both of these compounds inhibit Chd activity, but TCPN can be slowly hydrolyzed indicates the importance of the nitrile position in catalysis and also establishes them as likely substrate-analog inhibitors. On the other hand, simple nitriles such as acrylonitrile (ACN) and benzonitrile (BN) are weak inhibitors of Chd with IC50 values in the region of 19 mM for both. It should also be noted that Chd activity was unaffected by the presence of 2 M NaCl or NaBr, suggesting that halide anions do not bind to the active site Zn(II) ion. The stronger interaction between Chd and TCPN and TCTPN vs. BN and ACN is likely due to the energetically favorable π—π interaction with Trp227, based on docking studies of TPN into the hydrophobic channel of Chd, which also placed a CN nitrogen atom within bonding distance to the active site Zn(II) (∼2.3 A) (Catlin et al., 2020).
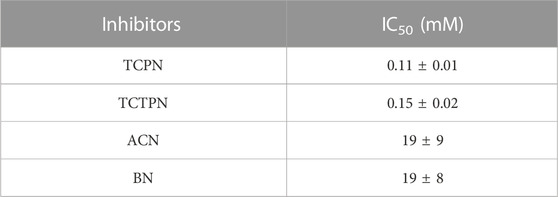
TABLE 1. IC50 values of the substrate analog Chd inhibitors examined.
Spectroscopic studies on Co(II)-substituted Chd
Electronic absorption spectra were obtained from buffered solutions of Chd at varying concentrations at which the dimer form predominated. The Co(II)-substituted Chd enzyme containing one eq. of Co(II) per monomer (Co1-Chd), exhibited a broad absorption band with a maximum at 535 nm (ε535 = ∼95 M−1 cm−1) along with shoulders at 500 nm (ε500 = ∼70 M−1 cm−1) and 580 nm (ε580 = ∼30 M−1 cm−1) (Figure 5). These data suggest that the Co(II) ions in Co1-Chd reside in pentacoordinate environments (Bennett et al., 2010), consistent with X-ray crystallographic data for Chd, which indicates a pentacoordinate, mononuclear Zn(II) active site (Catlin et al., 2020).
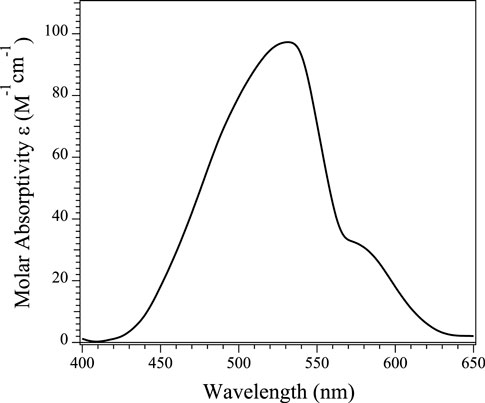
FIGURE 5. UV-Vis absorption spectra of a 1 mM sample of Chd in the presence of one equivalent of Co(II) per monomer at 25°C in Chelex-100 treated 50 mM HEPES buffer, pH 7.0.
The EPR spectrum obtained for as-prepared Co1-Chd at pH 7.0 in 50 mM HEPES buffer is shown in Figure 6 (top trace, top panel). The lowest field region of the spectrum, from 550 to 1,300 G (this “g1'-region” of an S = 3/2, MS = ± 1/2 spectrum is due to the gy resonance whereas for MS = ± 3/2 it is due to gz) is characterized by a 59Co hyperfine splitting (hfs) pattern with A1 ≅ 94 G (a tentative estimate for the corresponding hyperfine (hfi) value was ∼300 MHz), centered around g1’ ≅ 6.7, where the effective g-values g1,2,3’ = (h/μB)νB1,2,3, i.e., the apparent g-values of the lowest-field, middle, and highest-field resonances of a signal derived from the resonant fields (in contrast to gx,y,z, the principal values of the g tensor in the S = 3/2 spin Hamiltonian). The shape of the overall signal in this region, however, strongly suggests the presence of an additional underlying EPR absorption, with g’ around 5.7 that is devoid of resolved 59Co hfs. Close examination of the ∂2χ″/∂B2 spectrum (top trace, bottom panel) suggests yet another overlapping hfi pattern that is responsible for features at 750 and 860 G that are distinct from the 94 G hfs pattern, and for the “lumpy” absorption in the 300–600 G region; a tentative estimate for g’ for this signal is ∼10.5, which would indicate MS = ± 3/2 and a high gz value of ∼2.75. The g2'-region of the spectrum, from 1,405 to 2,405 G, contains multiple features. Spectral simulations proved unable to unambiguously assign features in the g2’ region to corresponding features in the g1’ region and sets of principal g’-values and, therefore, g and E/D, could not be reliably estimated for individual signals. The highest field g3'-region of the spectrum exhibited one clear feature at 2,760 G and two additional broad resonances at 3,230 and 3,790 G. These observations taken together indicate that the EPR spectrum of Co1-Chd contains at least three distinct signals, an MS = ± 1/2 signal with a ∼94 G 59Co hfs pattern centered at g1’ ≅ 6.7, a broader MS = ± 1/2 signal with g1’ ≅ 5.7, and an MS = ± 3/2 signal with tentatively estimated parameters of g1’ ≅ 10.5 (gz = 2.75), A1(59Co) ≅ 110 G, and a high-field broad resonance at g3’ ≅ 1.8.
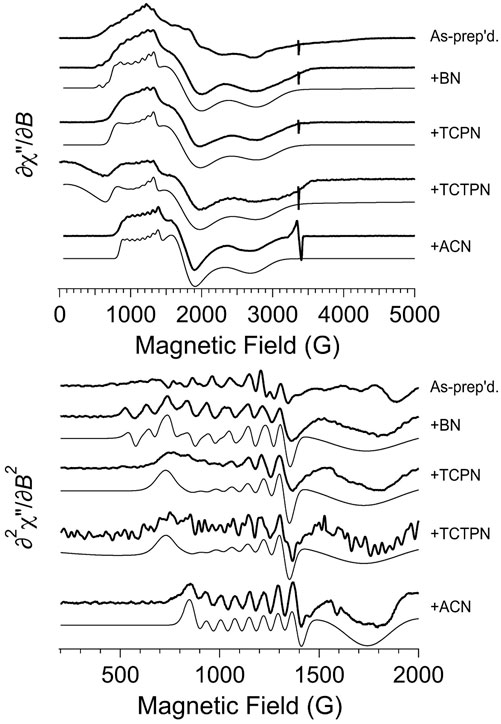
FIGURE 6. EPR spectroscopy of as-prepared (Co1-Chd) and upon addition of the inhibitors BN, TCPN, TCTPN, and ACN. The top panel shows the first-derivative spectra and the bottom panel shows an expanded region of the derived second-derivative spectra. In each panel, the top trace is the experimental data from naked (Co1-Chd) and, beneath that, are the data for the inhibited signals. For each of the inhibited signal, a computer simulation is shown directly beneath the experimental data. The individual signals used for the simulations are shown in Figure 8, and the EPR parameters and proportions are given in Table 2. Experimental data were recorded at 12 K, 2 mW microwave power, 9.45 GHz.
An EPR titration of apo-Chd with Co(II)aq is presented in Figure 7 and the spectra are shown normalized in intensity for the amount of added Co(II). EPR spectra of Chd to which Co(II) was added up to 2 eq are indistinguishable within the observed signal-to-noise. These data do not provide a binding constant for Co(II) to Chd but are consistent with Co(II) binding in a cooperative fashion to the two active sites of a Chd dimer until they are saturated. It should be noted that while the 59Co hfs pattern, which is typical for Co-substituted metallohydrolases (Bennett et al., 2010), is not easily discernible in the experimental ∂χ″/∂B spectra with 0.5 and 1 eq. Co(II), the pattern is very clear in the ∂2χ″/∂B2 spectra and the normalized intensities of the hyperfine lines do not change appreciably with added Co(II). The most noticeable Co-dependent change in the experimental ∂χ″/∂B spectra was a diminution, or broadening, of the g3’ feature at 3,230 G with successively higher amounts of Co(II). Additionally, there was a Co-dependent diminution of a sharp derivative-shaped feature in the ∂2χ″/∂B2 spectra at 1,170 G. This latter feature most markedly diminished upon the addition of a third eq. of Co(II). Also, the resolution of the g2’ region was noticeably reduced with 3 eq of Co(II) compared to the other spectra.
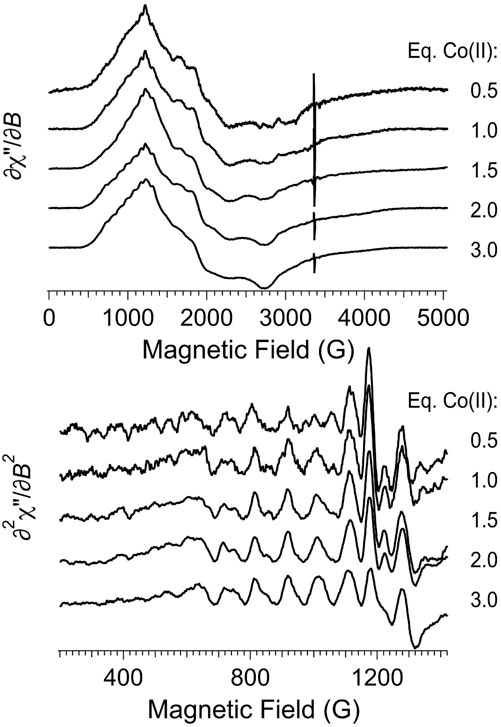
FIGURE 7. EPR titration of apo-Chd with Co(II). The top panel shows the first-derivative experimental spectra, and the bottom panel shows an expanded region of the second-derivative spectra. Experimental data were recorded at 12 K, 2 mW microwave power, 9.45 GHz.
The relationship between the observed EPR features and the structural aspects of Co(II)-bound to Chd was explored by examining the X-ray crystal structure of Chd (Figure 1B), which suggests two possible unique binding modalities for pentacoordinate Co(II) in the catalytic site (Catlin et al., 2020). His114, which was suggested to be the catalytic base (Catlin et al., 2020), can adopt two distinct conformations, one in which the Nδ1 atom forms a hydrogen bond with the Zn(II)-bound water molecule and the other where it is rotated perpendicular to the imidazole ring, likely resulting in the loss of hydrogen bonding interactions with the coordinated water/hydroxide oxygen atom. It seems logical that these two structural conformations of the pentacoordinate Co(II) ions are responsible for the two distinct S = 3/2, MS = 1/2 EPR signals. The conformation in which Nδ1 hydrogen bonds to the Co(II)-bound water/hydroxide would be expected to provide higher resolution of 59Co hyperfine structure than the rotated conformation in which the hydrogen bonding connection to the protein lattice is diminished or absent (Bennett et al., 2010). An alternative explanation for the observation of two distinct MS = 1/2 EPR signals is water vs. hydroxyl coordination. However, given the clear evidence from the structural model for two H114 conformations, that is the favored explanation. The presence of a third, MS = 3/2 signal is most simply explained as being due to Co(II) in the tetrahedral structural zinc site, although only one equivalent of Co(II) was found per monomer, so it is quite possible that the structural site is at least partially occupied at the enzyme concentrations used. Nevertheless, a third conformation of the catalytic Co(II) in addition to the two His114 conformations, perhaps due to alternative protonation states, cannot be ruled out.
It should be noted that in some cases, centrifugation of EPR samples resulted in a visible precipitate, indicating aggregation of the protein. Even where no precipitate was visible to the naked eye, some precipitation could not be ruled out. Consequently, the final amount of Chd in the sample is unclear. Furthermore, the fate of the Co(II) bound by Chd that precipitated is unknown, i.e., whether it remained bound to the precipitated Chd or was released into solution. No attempt, therefore, was made to quantify the EPR signals in terms of absolute [Co(II)]. However, Co(II)aq. EPR signals in a wide range of buffers have not been hitherto observed to exhibit EPR spectra with 59Co (I = 7/2) hfs that is resolved at the X-band frequency. Therefore, any signals exhibiting 59Co hfs do not arise from uncomplexed Co(II)aq and must be due to Co(II)-bound to Chd in the absence of other Co-binding entities (Bennett et al., 2010).
EPR studies of inhibitor bound Co(II)-substituted Chd
EPR spectra obtained for Co1-Chd in the presence of each inhibitor studied (Figure 3), at pH 7.0 in 50 mM HEPES buffer are shown in Figure 6. Upon the addition of each of the substrate-like inhibitors BN, TCPN, TCTPN or ACN to Co1-Chd, changes in the EPR spectrum were observed that, in most cases, were characteristic of the specific inhibitor employed. In each case, the spectra were simpler than that of Co1-Chd in the absence of inhibitors and could be simulated as either a single signal or a mixture of two. The spin Hamiltonian parameters for the simulated signal, and their relative contributions where two species were required, are given in Table 2 with the individual simulated signals presented in Figure 8.
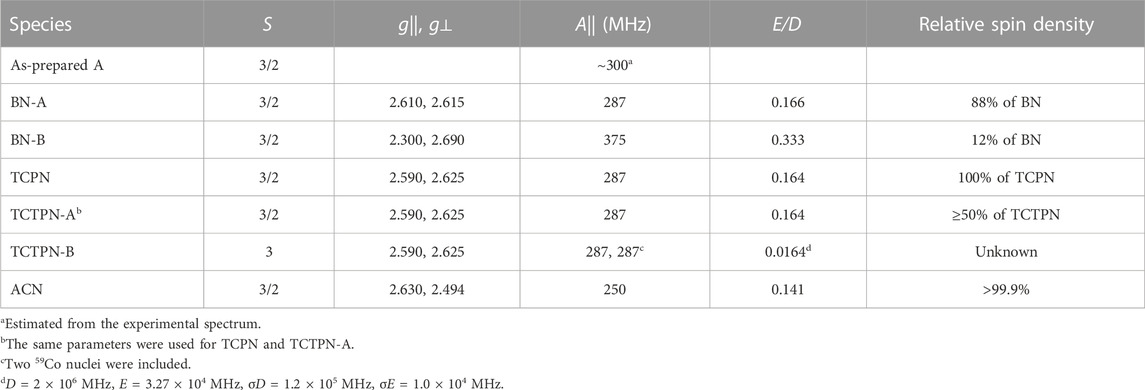
TABLE 2. EPR Parameters from Computer Simulations. All EasySpin parameters are provided in Supplementary Tables S1–S6 in the supporting information.
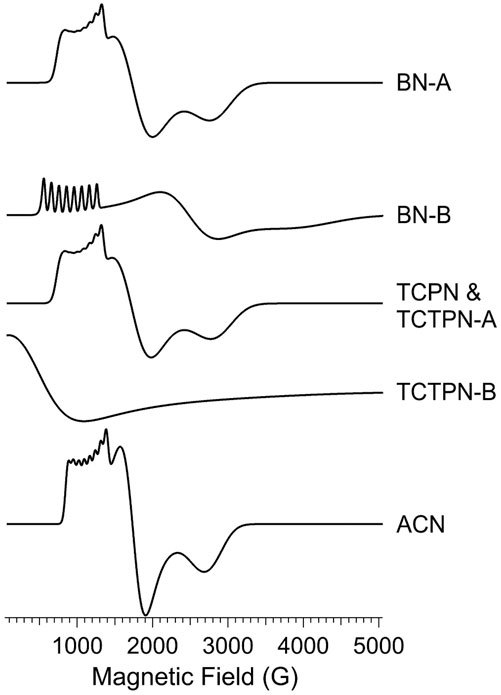
FIGURE 8. EPR simulations of Co-substituted Chd signals. The simulations shown in Figure 6 were generated using combinations of the computed individual species shown here. The spin Hamiltonian parameters and proportions of each used to generate the simulations in Figure 6 are given in Table 2.
The observed EPR spectrum of Co1-Chd with BN present was observably different from that of the unbound enzyme and was characterized by a hfs pattern in the g1’ region that was unusual in that the number of lines was 10, rather than the expected 8, and the splitting itself ranged from ∼102 G on the low-field side of the pattern to ∼78 G on the high-field side. This is particularly evident in the ∂2χ″/∂B2 spectrum and clearly indicated the presence of more than one contributory signal to the spectrum. The spectrum was simulated assuming two signals with the dominant signal (∼88%; denoted BN-A in Figure 8; Table 2) exhibiting an only slightly axial g tensor with values around 2.6, a moderately rhombic ZFS of E/D ≅ 0.17, and 59Co hfi along gy (g1) corresponding to a hfs of 78 G (Table 2). The second, minor signal (∼12%; BN-B) exhibited a more markedly anisotropic g tensor and a significantly larger 59Co hfi than BN-A. The g|| and g⊥ values pertain to an MS = ± 1/2 ground state Kramers’ doublet but with an E/D value of 1/3, the theoretical maximum, suggesting that the sign of D is not well-determined and the corresponding solution in terms of an MS = ± 3/2 signal is equally valid.
The spectrum of Chd with TCPN was simulated as a single species, with spin Hamiltonian parameters very similar to those of BN-A. The EPR spectrum with TCTPN bound to Co1-Chd, as expected, is similar to that of TCPN and consisted of one rhombic MS = ± 1/2 signal (TCTPN-A) that was indeed simulated using the same parameters used for TCPN. However, there was also a very broad EPR absorption that extended out of zero field, suggestive of an integer spin signal (TCTPN-B), which was verified by parallel mode EPR. TCTPN-B was simulated as an S = 3 system, to model a ferromagnetically-coupled pair of S = 3/2 Co(II) ions, with g values around 2.6, an effective D = 66 cm-1, and a small but necessary rhombic component with E/D = 0.0164. These parameters by no means represent a unique solution but serve to illustrate that the TCTPN-B signal is at least consistent with a model for two exchange-coupled Co(II) ions. Quantification of integer-spin systems is fraught with uncertainty and multiple caveats so was not attempted, if for no other reason than the multiple experimental frequencies required were not available. Estimating the relative content of TCTPN-A relied on absolute quantitation based on the amount of total Co(II) assumed to be in the sample which, in turn, assumed no loss due to aggregation and precipitation. The calculated estimate of 50% represents a lower limit.
The EPR spectrum of Chd with ACN was modeled extremely well as a single species with moderate axial anisotropy of g, a moderate rhombic ZFS of E/D = 0.141, and a relatively small 59Co hfi of 250 MHz (∼70 G). There was an additional narrow signal in the spectrum with g = 2.055. Double integration of this signal compared with the overall spectrum indicated that the g = 2.055 signal accounted for <0.1% of the total spin density and, therefore, no attempt was made to further characterize the signal.
The EPR spectra obtained after the addition of the substrate-like inhibitors provides some noteworthy information about possible substrate-Chd interactions. First, and in contrast to the unbound enzyme, the observed EPR spectrum of Chd with each inhibitor indicated that either all or a large majority of the S = 3/2 active site Co(II) ions in the sample exist as a single chemical species. The lack of multiple similar, yet distinct, MS = 1/2 signals is presumed to be due to inhibition of the rotation of His114 Nδ1 by the presence of the inhibitor in the active site. Second, the corresponding EPR signals are all due to a ground state MS = 1/2 Kramers’ doublet and are consistent with pentacoordinate Co(II) with a relatively constrained coordination sphere; they exhibit moderately rhombic ZFS (E/D = 0.159 ± 0.012), g values of around 2.6 (g|| = 2.605 ± 0.019; g⊥ = 2.590 ± 0.064), and a resolved hfs pattern indicative of a modest 59Co hfi along the gy (g1’) axis (A = 278 ± 19 MHz). In contrast to the unbound enzyme, depending on the origin of the MS = 3/2 signal in the uninhibited enzyme (Co1-Chd), any evidence for Co(II) occupation of the structural tetrahedral site was lacking in all but one case. With BN, a minor component, BN-B, with E/D = 1/3, leaves open the possibility that this signal is due to occupation of the structural site in about 10% of the Chd population. Other assignments for this E/D = 1/3 signal can be imagined but would require some flexibility in the coordination of the catalytic Co(II) ions that is not observed with any of the other inhibitors.
Finally, it would be remiss not to address the exhibition of an apparent integer-spin signal observed for TCTPN bound to Co1-Chd. The appearance of this signal is, frankly, baffling. The pentacoordinate catalytic Co(II) ions are deeply buried within the protein and, therefore, exchange coupling between another protein-bound Co(II) to a catalytic Co(II) ion is impossible. Two hypotheses are possible with the first involving an adventitious Co(II) ion, perhaps liberated from precipitated Chd upon TCTPN binding, being chaperoned into the active site by TCTPN itself. However, the catalytic pocket is only electrostatically negative around the active site metal ion while the substrate entry channel is lined with hydrophobic residues and is largely positive, therefore, not a good cation environment. The second is that, perhaps as a preliminary step to aggregation and precipitation, two dimers interact at the interface containing the structural Co(II) ion, which is close to the surface. Exchange coupling of a magnitude sufficient to be consistent with the TCTPN-B EPR signal would require a covalent bridging atom between the Co(II) ions and the para nitrile groups on TCTPN may facilitate this link, explaining the specificity of this signal for TCTPN. If either of these possibilities is correct, there is no evidence that either has any catalytic relevance or would significantly impact proposed environmental clean-up applications for Chd.
In conclusion, the combination of the kinetic and spectroscopic data presented herein coupled with the previously reported kinetic and X-ray crystal structure of Chd, provides additional insight into the catalytic mechanism of Chd (Figure 2B) (Yang et al., 2019; Catlin et al., 2020). The proposed first step in catalysis is likely recognition of TPN by the large hydrophobic channel that leads to the Zn(II) active site. Consistent with the observed IC50 values for TCPN and TCTPN, the substrate, TPN, is likely positioned by a π-π interaction with an active site Trp227 residue, that was also shown to be important for catalytic turnover (Wang et al., 2010). The next step in catalysis is likely the interaction of a CN nitrile nitrogen atom to the active site, either through direct, weak (∼2.3 Å bond distance based on substrate docking studies) (Yang et al., 2019; Catlin et al., 2020) coordination with the Zn(II) (Pathway A; Figure 2) or through hydrogen bond formation between the nitrogen atom of a TPN cyano group and the amide nitrogen of Asn216 (Pathway B; Figure 2). This hydrogen bonding interaction was also suggested as a possible pose based on substrate docking studies, which placed the ortho carbon of TPN within 3.1 Å of the O atom of the bound water/hydroxide (Yang et al., 2019; Catlin et al., 2020). Both of these TPN binding modes are consistent with the EPR data reported herein; however, the latter is somewhat favored as the inhibitor-bound cobalt(II)-substituted Chd EPR data indicate a five-coordinate site, which is inconsistent with CN binding directly to the active site as direct binding would likely necessitate expansion of the inner-coordination sphere to six. Moreover, direct coordination of the CN moiety to the active site divalent metal ion would result in electron delocalization from the chlorinated aromatic rings of TCPN and TCTPN, which would be expected to differ from that of the unchlorinated BN and, therefore, exhibit differences in EPR parameters, particularly the 59Co hfi. Either way, interaction of the substrate nitrile nitrogen within the active site, likely through hydrogen bond formation, has a two-fold impact. First, it removes electron density from the aromatic ring activating the ortho carbon for nucleophilic attack (Peat et al., 2015; Hu et al., 2019) and second, it activates the coordinated water/hydroxide for nucleophilic attack. Deprotonation of the metal-bound water molecule by His114, with assistance from the Zn(II) ligands Asp116 and Asn227, forms a nucleophilic hydroxide moiety, which is consistent with the postulated pKa of the zinc-bound water molecule (Sakurai et al., 1995). Once the zinc-bound hydroxide is formed, it can attack the activated ortho carbon of TPN, forming an η-1-μ-transition-state complex (Cosper et al., 2003). Kinetic isotope effect studies indicate that one proton is transferred in the transition-state, likely due to the breaking of a water O-H bond and the formation of a protonated His114 (Yang et al., 2019). Once the products are released, the Zn(II)-bound water molecule is replaced.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author contributions
XY, KD, and CM prepared expression plasmids, carried out protein expression, purification, and enzymatic assays. XY recorded the EPR spectra and simulated these data with BB. RH, and BB conceived of the idea and wrote the paper with XY, KD, and CM. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Science Foundation (CHE-2003861, RH; CHE-1808711, RH and BB; CHE-1532168 BB and RH), the Todd Wehr Foundation, Bruker Biospin, and the National Institutes of Health/NIBIB National Biomedical EPR Center (P41-EB001980).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchbi.2023.1105607/full#supplementary-material
References
Bennett, B. (2010) EPR of cobalt-substituted zinc enzymes. In Metals in Biology: Applications of high resolution EPR to metalloenzymes in Biol. Magn. Reson., metals in Biology. Editors G. Hanson, and L. Berliner (New York: Springer). 345–370.
Carlo-Rojas, Z., Bello-Mendoza, R., Figueroa, M. S., and Sokolov, M. Y. (2004). Chlorothalonil degradation under anaerobic conditions in an agricultural tropical soil. Water, Air, & Soil Pollut. 151, 397–409. doi:10.1023/b:wate.0000009918.70113.c5
Catlin, D. S., Yang, X., Bennett, B., Holz, R. C., and Liu, D. (2020). Structural basis for the hydrolytic dehalogenation of the fungicide chlorothalonil. J. Biol. Chem. 295, 8668–8677. doi:10.1074/jbc.ra120.013150
Caux, P. Y., Kent, R. A., Fan, G. T., and Stephenson, G. L. (1996). Environmental fate and effects of chlorothalonil: A Canadian perspective. Crit. Rev. Environ. Sci. Technol. 26, 45–93. doi:10.1080/10643389609388486
Chen, H., Wang, H., Wang, T., Huang, S., Zang, X., Li, S., et al. (2016). Identification of the metal center of chlorothalonil hydrolytic dehalogenase and enhancement of catalytic efficiency by directed evolution. Appl. Environ. Biotechnol. 1, 30–37. doi:10.18063/aeb.2016.01.003
Cosper, N. J., Bienvenue, D. L., Shokes, J. E., Gilner, D. M., Tsukamoto, T., Scott, R. A., et al. (2003). The dapE-encoded N-succinyl-L,L-Diaminopimelic acid desuccinylase from Haemophilus influenzae is a dinuclear metallohydrolase. J. Am. Chem. Soc. 125, 14654–14655. doi:10.1021/ja036650v
D'Souza, V, M., Bennett, B., Copik, A. J., and Holz, R. C. (2000). Divalent metal binding properties of the methionyl aminopeptidase from Escherichia coli. Biochemistry 39, 3817–3826. doi:10.1021/bi9925827
Hu, J., Su, Q., Schlessman, J. L., and Rokita, S. E. (2019). Redox control of iodotyrosine deiodinase. Protein Sci. 28, 68–78. doi:10.1002/pro.3479
Hyde, J. S., Pasenkiewicz-Gierula, M., Jesmanowicz, A., and Antholine, W. E. (1990). Pseudo field modulation in EPR spectroscopy. Appl. Magn. Reson. 1, 483. doi:10.1007/bf03166028
Karunagala Pathiranage, W. L., Gumataotao, N., Fiedler, A. T., Holz, R. C., and Bennett, B. (2021). Identification of an intermediate species along the nitrile hydratase reaction pathway by EPR spectroscopy. Biochemistry 60, 3771–3782. doi:10.1021/acs.biochem.1c00574
Kwon, J. W., and Armbrust, K. L. (2006). Degradation of chlorothalonil in irradiated water/sediment systems. J. Agric. food Chem. 54, 3651–3657. doi:10.1021/jf052847q
Liang, B., Wang, G., Zhao, Y., Chen, K., Li, S., and Jiang, J. (2011). Facilitation of bacterial adaptation to chlorothalonil-contaminated sites by horizontal transfer of the chlorothalonil hydrolytic dehalogenase gene. Appl. Environ. Microbiol. 77, 4268–4272. doi:10.1128/aem.02457-10
Mozzachio, A. M., Rusiecki, J. A., Hoppin, J. A., Mahajan, R., Patel, R., Beane-Freeman, L., et al. (2008). Chlorothalonil exposure and cancer incidence among pesticide applicator participants in the agricultural health study. Environ. Res. 108, 400–403. doi:10.1016/j.envres.2008.07.018
Peat, T. S., Newman, J., Balotra, S., Lucent, D., Warden, A. C., and Scott, C. (2015). The structure of the hexameric atrazine chlorohydrolase AtzA. Acta Crystallogr. Sect. D. Biol. Crystallogr. 71, 710–720. doi:10.1107/s1399004715000619
Ren, X., Li, H., and Chen, S. (2011). Cloning of the chlorothalonil-degrading gene cluster and evidence of its horizontal transfer. Curr. Microbiol. 62, 1068–1073. doi:10.1007/s00284-010-9824-x
Sakkas, V. A., Lambropoulou, D. A., and Albanis, T. A. (2002). Study of chlorothalonil photodegradation in natural waters and in the presence of humic substances. Chemosphere 48, 939–945. doi:10.1016/s0045-6535(02)00121-2
Sakurai, M., Furuki, T., and Inoue, Y. (1995). The pKa of the zinc-bound water in carbonic anhydrase and its model compounds as studied by the AM1 calculation coupled with a reaction field theory. J. Phys. Chem. 99, 17789–17794. doi:10.1021/j100050a019
Sims, J. L., Suflita, J. M., and Russell, H. H. (1991). “Reductive dehalogenation of organic contaminants in soils and ground water,” in Superfund Technology support center for ground water, 1–12. Agency, U. S. E. P.
Stoll, S., and Schweiger, A. (2006). EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magnetic Reson. 178, 42–55. doi:10.1016/j.jmr.2005.08.013
Tropea, J. E., Cherry, S., and Waugh, D. S. (2009). Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol. Biol. 498, 297–307. doi:10.1007/978-1-59745-196-3_19
Viciu, M. S., Grasa, G. A., and Nolan, S. P. (2001). Catalytic dehalogenation of aryl halides mediated by a palladium/imidazolium salt system. Organometallics 20, 3607–3612. doi:10.1021/om010332s
Vickers, A. E. M., Sloop, T. C., and Lucier, G. W. (1985). Mechanism of action of toxic halogenated aromatics. Environ. Health Perspect. 59, 121–126. doi:10.2307/3429884
Wang, G., Li, R., Li, S., and Jiang, J. (2010). A novel hydrolytic dehalogenase for the chlorinated aromatic compound chlorothalonil. J. Bacteriol. 192, 2737–2745. doi:10.1128/jb.01547-09
Watterson, S. J., Mitra, S., Swierczek, S. I., Bennett, B., and Holz, R. C. (2008). Kinetic and spectroscopic analysis of the catalytic role of H79 in the methionine aminopeptidase from Escherichia coli. Biochemistry 47, 11885–11893. doi:10.1021/bi801499g
Winzor, D. J., and Sawyer, W. H. (1995). Quantitative characterization of ligand binding. New York: Wiley-Liss.
Yang, X., Bennett, B., and Holz, R. C. (2019). Insights into the catalytic mechanism of a bacterial hydrolytic dehalogenase that degrades the fungicide chlorothalonil. J. Biol. Chem. 294, 13411–13420. doi:10.1074/jbc.ra119.009094
Keywords: electron paramagnetic resonance, cobalt, CHD, bioremediation, chlorothalonil, enzyme catalysis, enzyme mechanism, chlorinated aromatic hydrocarbons
Citation: Yang X, Diviesti K, Miller C, Bennett B and Holz RC (2023) Insights into the catalytic mechanism of the chlorothalonil dehalogenase from Pseudomonas sp. CTN-3. Front. Chem. Biol 2:1105607. doi: 10.3389/fchbi.2023.1105607
Received: 22 November 2022; Accepted: 25 January 2023;
Published: 08 February 2023.
Edited by:
Debbie C. Crans, Colorado State University, United StatesReviewed by:
Sofia R. Pauleta, New University of Lisbon, PortugalMarie Heffern, University of California, Davis, United States
Katlyn Meier, University of Miami, United States
Copyright © 2023 Yang, Diviesti, Miller, Bennett and Holz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian Bennett, YnJpYW4uYmVubmV0dEBtYXJxdWV0dGUuZWR1; Richard C. Holz, cmhvbHpAbWluZXMuZWR1