 Ziyi Jia
Ziyi Jia Hongtao Li2†
Hongtao Li2† Ke Xu
Ke Xu Siyu Yang
Siyu Yang Xiaowei Sun
Xiaowei Sun- 1The First Clinical Medical College, Heilongjiang University of Chinese Medicine, Harbin, China
- 2The First Affiliated Hospital, Heilongjiang University of Chinese Medicine, Harbin, China
- 3The Second Clinical Medical College, Heilongjiang University of Chinese Medicine, Harbin, China
Ischemic stroke (IS) is the predominant subtype of stroke and a leading contributor to global mortality. The mitochondrial-associated endoplasmic reticulum membrane (MAM) is a specialized region that facilitates communication between the endoplasmic reticulum and mitochondria, and has been extensively investigated in the context of neurodegenerative diseases. Nevertheless, its precise involvement in IS remains elusive. This literature review elucidates the intricate involvement of MAM in mitophagy and endoplasmic reticulum stress during IS. PINK1, FUNDC1, Beclin1, and Mfn2 are highly concentrated in the MAM and play a crucial role in regulating mitochondrial autophagy. GRP78, IRE1, PERK, and Sig-1R participate in the unfolded protein response (UPR) within the MAM, regulating endoplasmic reticulum stress during IS. Hence, the diverse molecules on MAM operate independently and interact with each other, collectively contributing to the pathogenesis of IS as the covert orchestrator.
1 Introduction
Stroke ranks as the second most common reason for disability and mortality worldwide (Saini et al., 2021), with Ischemic stroke (IS) responsible for the majority of new cases, accounting for around 87% of all instances (Kleindorfer et al., 2021). IS occurs when there is a decrease in blood flow, resulting in a shortage of oxygen and glucose in the brain tissue. This triggers a series of responses (Shi et al., 2021), including endoplasmic reticulum (ER) stress, mitochondrial autophagy, and cell ion overload, all of which can contribute to neuronal death, worsen the progression of cerebral ischemic injury, and impact the final outcome (Noh et al., 2023). At present, the primary focus of treating IS is on restoring blood flow, especially by using thrombolytic therapy within a 3–4.5 h window to reverse reversible ischemic damage. However, due to the limited treatment timeframe and the resulting reperfusion injury, patients with IS often experience unfavorable outcomes (Liu et al., 2023). After the passing of this time frame, it becomes impossible to repair damaged neurons, and reperfusion could result in cerebral ischemia/reperfusion injury. This is mainly caused by ER stress and mitochondrial autophagy (Dhapola et al., 2024), which further worsens cell death and brain damage. Hence, it is crucial to investigate effective therapeutic approaches that can prolong the treatment window and enhance results for individuals suffering from IS.
The mitochondria-associated endoplasmic reticulum (MAM) acts as a channel that facilitates communication between the ER and mitochondria. It consists of substructures within the ER, the outer mitochondrial membrane (OMM), and a set of proteins that establish a dynamic connection (Yang et al., 2020). Hung et al. identified 634 proteins on the ER and 137 proteins on mitochondria. They found that 68 of these proteins overlapped with MAM when comparing them (Hung et al., 2017). The initial estimation of the distance between the ER and the outer membrane of the mitochondria was approximately 100 nanometers. Nevertheless, a growing body of research indicates that the configuration of MAM is not fixed; instead, it is fluid and capable of adapting to various cellular physiological and pathological circumstances. The distance between the rough ER and the outer membrane of the mitochondria typically ranges from 10 to 100 nm (Giacomello and Pellegrini, 2016), with a smooth ER gap width of 10–15 nm and a rough ER gap width of 20–30 nm (Csordás et al., 2006). Artificially disrupting this contact can result in ER stress (Bravo et al., 2011).
Undoubtedly, it has been established that MAM serves as a focal point for transmitting stress signals from the ER to the mitochondria, particularly when ER protein homeostasis is compromised (van Vliet and Agostinis, 2018). Due to its role as a signal transduction platform, MAM primarily depends on proteins to carry out its diverse functions. As a result, numerous studies categorize the fundamental elements of MAM based on their primary roles. For instance, the transportation of calcium involves glucose-regulated protein 75 (GRP75), mitochondrial voltage-dependent anion channel (VDAC1), and inositol 1,4,5-trisphosphate receptor (IP3R) (Tubbs et al., 2014). Cell apoptosis involves BAP31, a protein linked to the B cell receptor, and FIS1, a homolog of mitochondrial fission 1 (FIS1) (Saito and Imaizumi, 2018). ER stress involves the sigma-1 receptor (Sig-1R), inositol-requiring enzyme 1 (IRE1), protein kinase R-like ER kinase (PERK) (Saito and Imaizumi, 2018), and glucose-regulated protein 78 (GRP78) (Eysert et al., 2020). Autophagy involves VAPB (vesicle-associated membrane protein B), PTPIP51 (protein tyrosine phosphatase interacting protein 51), MFN1/2 (mitofusin 1/2), TOM40 (translocase of OMM component), PINK1 (serine/threonine kinase), Parkin (E3 ubiquitin ligase), and FUNDC1 (protein with FUN14 domain) (Ji et al., 2022; Lv et al., 2022). The mitochondria and ER play crucial roles as cell organelles in eukaryotic cells. In mammalian cells, the surface of mitochondria is approximately 5–20% parallel to the ER (Rizzuto et al., 1998). This intimate structural connection enables the ER to respond to diverse stress stimuli and transmit stress signals to mitochondria (Iurlaro and Muñoz-Pinedo, 2016). In a similar manner, mitochondria have the ability to send signals to the ER, ensuring that compensatory responses or cell death events are effectively carried out. This system of signal transduction is typically examined as a relatively independent subcellular organelle structure referred to as the MAM (Figure 1). In conclusion, MAM plays a crucial role in maintaining cellular homeostasis and biological functions by acting as an imperceptible regulator that governs mitochondrial autophagy and ER stress.
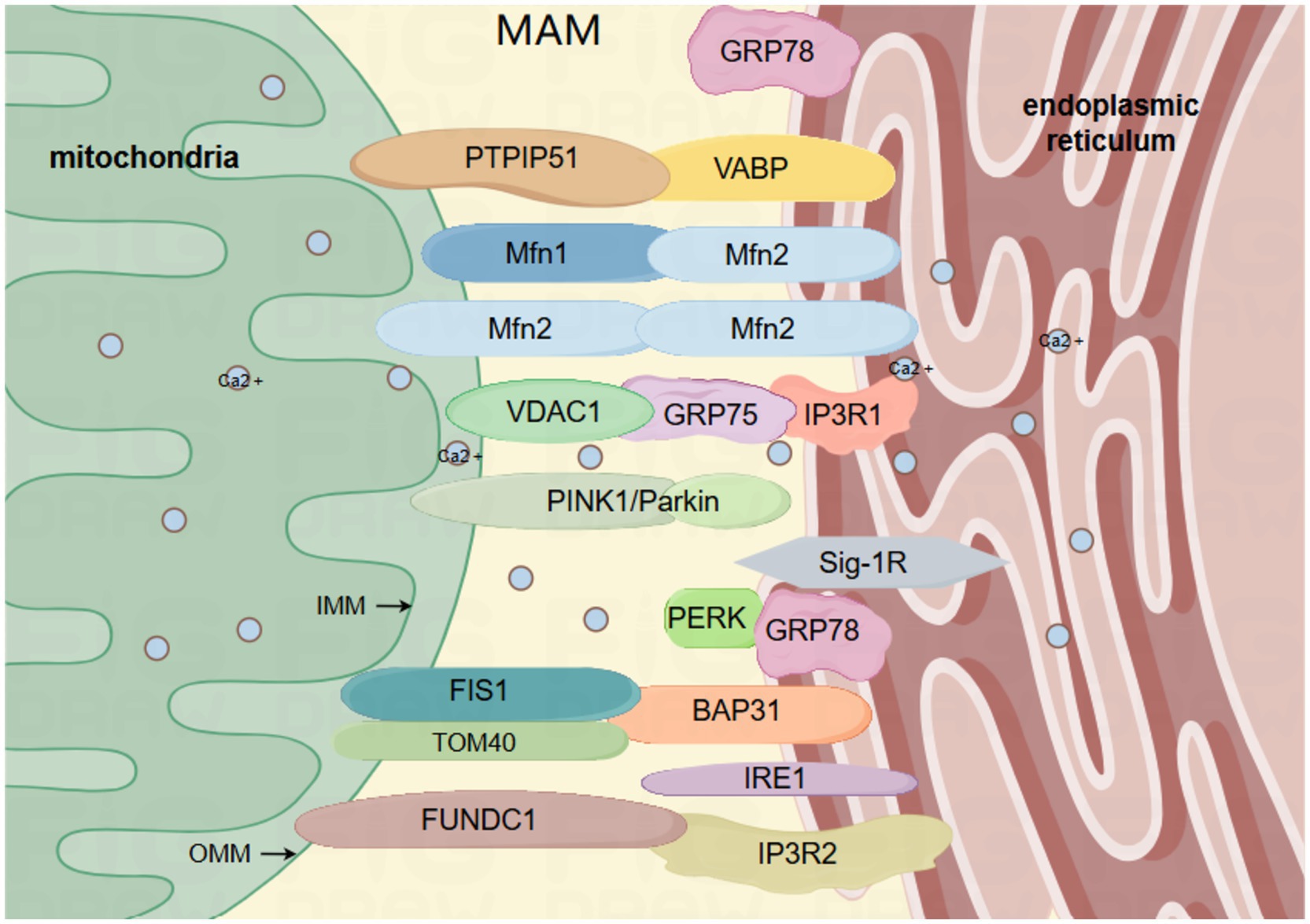
Figure 1. The primary framework of MAM: the mitochondrial associated membrane (MAM) comprises the outer and inner membranes of the mitochondria, and is abundant in diverse proteins that are interconnected by tethering proteins, such as VAPB-PTPIP51, IP3R-GRP75-VDAC1, BAP31-FIS1, FUNDC1-IP3R2, MFN2-MFN1/2. VAPB, vesicle-associated membrane protein-related protein B; PTPIP51, Protein Tyrosine Phosphatase Interacting Protein 51; IP3R, inositol 1,4,5-trisphosphate receptor; GRP75, glucose-regulated protein 75; GRP78, glucose-regulated protein 78; VDAC1, voltage-dependent anion channel 1; BAP31, B cell receptor-associated protein 31; FIS1, mitochondrial fission protein 1; FUNDC1, protein 1 with FUN14 domain; MFN1/2, mitofusin 1/2; IRE1, inositol 1,4,5-trisphosphate 3-kinase 1; Sig-1R, sigma-1 receptor; PINK1, serine/threonine kinase; Parkin, E3 ubiquitin ligase; mitochondrial outer membrane translocase component TOM40; PERK, protein kinase R-like ER kinase. By Figdraw.
2 MAM modulates mitophagy to mitigate IS
Mitochondrial autophagy is a specific type of autophagy capable of removing impaired or ineffective mitochondria. Mitochondrial autophagy is activated in the aftermath of an IS. Research has indicated that in the context of IS, mitochondrial dysfunction can result in an increased release of pro-apoptotic factors, ultimately leading to cell death within the affected area (Jürgensmeier et al., 1998). In the reperfusion phase, mitochondrial autophagy mainly plays a protective role (Kumar et al., 2016). Hence, there is a widespread belief that moderate mitochondrial autophagy provides a protective effect, whereas an excessive level of mitochondrial autophagy could potentially worsen neuronal demise (Mo Y. et al., 2020).
MAM plays a crucial role in initiating and carrying out autophagy, and it contributes to the formation of autophagosomes. Interestingly, MAM can play both positive and negative roles in autophagy under various physiological or pathological conditions, a phenomenon closely associated with the intricate composition of its constituent proteins and their multifaceted functions (Ji et al., 2022). The PINK1/Parkin pathway is widely recognized as a crucial regulator of mitochondrial autophagy. Recent studies have confirmed the accumulation of PINK1 and Parkin in MAM (Van Laar et al., 2015). In the process of mitochondrial autophagy, PINK1 and Beclin1 are observed to gather on the surface of MAM, facilitating the establishment of ER-mitochondria connections and the formation of autophagosomes (Gelmetti et al., 2017). FUNDC1 has the ability to function as a receptor for mitochondrial autophagy during hypoxia, regulating both mitochondrial fission and mitochondrial autophagy at MAMs. This ultimately strengthens its association with mitochondrial processes (Wu et al., 2016b). Moreover, prior research has demonstrated that Mfn2 depletion disrupts MAM and results in compromised autophagosome formation (Gomez-Suaga et al., 2017). All of these observations indicate an indisputable connection between MAM and mitophagy. Hence, considering the diverse roles of mitophagy at various stages of IS, it is imperative to target MAM at the appropriate time for intervention in stroke regulation (Table 1).
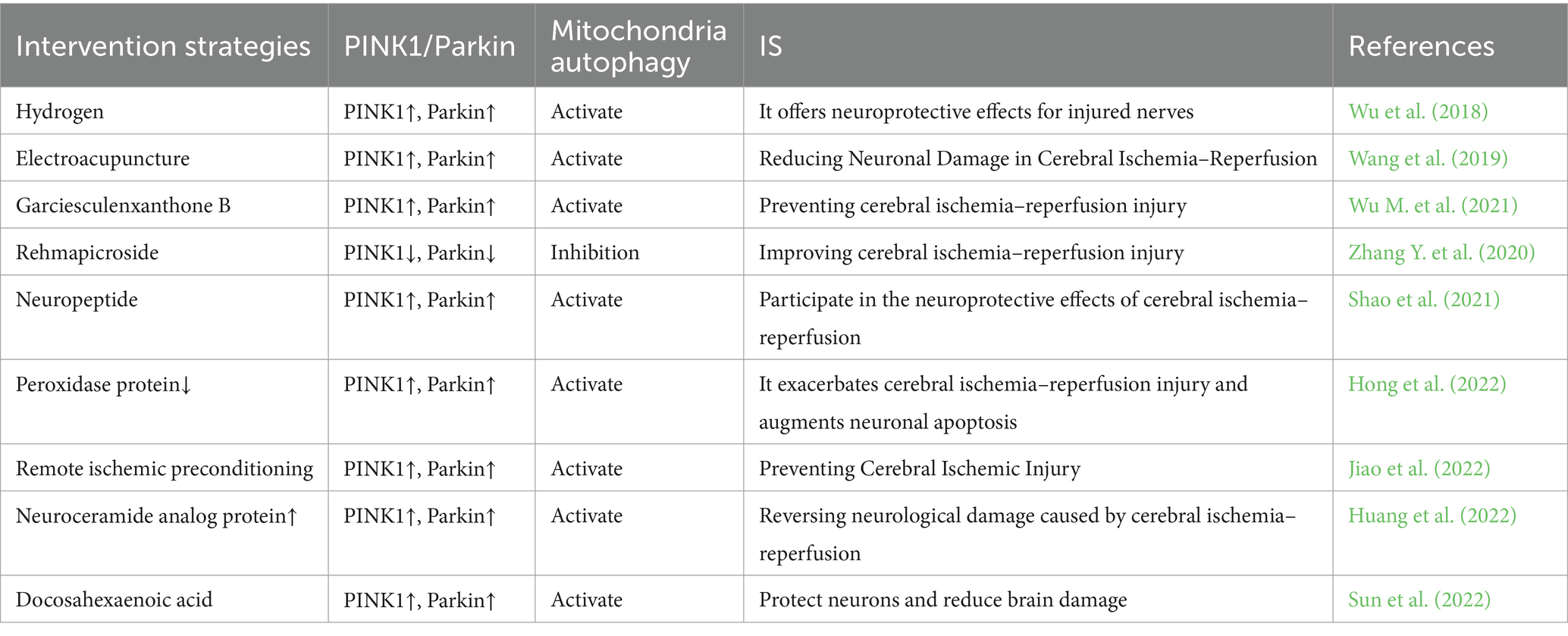
Table 1. The role of PINK1/Parkin in IS.
2.1 PINK1/Parkin in MAM and mitophagy
The PINK1/Parkin pathway is a highly intricate regulatory mechanism, and studies have shown that both PINK1 and Parkin are situated in the MAM under normal physiological conditions as well as during cellular stress (Barazzuol et al., 2020). Following an IS, detrimental stimuli such as ROS and hypoxia facilitate the initiation of PINK1 via phosphorylation. Phosphorylated PINK1 subsequently attaches to the surface of mitochondria, leading to the binding and phosphorylation of Parkin on the outer membrane of mitochondria. Upon activation, Parkin will partially attach ubiquitin to the OMM protein, thereby triggering mitophagy. Certain mitochondrial proteins undergo ubiquitination and subsequent degradation, a process crucial for mitophagy (Tanaka et al., 2010). Other ubiquitinated proteins are recruited by autophagy adaptor proteins, such as optineurin (OPTN) and nuclear dot protein 52 (NDP52), which anchor marked mitochondria to autophagosomes via their LC3-interacting region (LIR) motif, thereby initiating mitophagy (Lazarou et al., 2015). The main site where LC3-II is locally accumulated and the membrane origin of the mitochondria has been identified as MAM (Yang and Yang, 2013). Post-ischemic reperfusion therapy enhances mitochondrial autophagy through the induction of mitochondrial ubiquitination mediated by PINK1/Parkin, thereby mitigating transient global cerebral ischemia and ameliorating neuronal injury (Wen et al., 2021). Following mitochondrial depolarization, PINK1 and Parkin are recruited to the MAM, and it seems that the translocation of PINK1 to MAM is crucial for recruiting the autophagy machinery to this region (Gelmetti et al., 2017). When mitophagy is triggered, the amount of PINK1 rises in a manner dependent on calcium, and the expression of PINK1 is stimulated while mitophagy is initiated when there is a local increase in calcium levels during the exchange of calcium between cellular organelles in MAM (Gómez-Sánchez et al., 2014). Under typical conditions, there exists a delicate equilibrium between the generation and elimination of ROS within the human body. However, this balance is disrupted in the event of an IS. ROS serves as the primary initiator of mitochondrial autophagy, with excessive ROS triggering this process. The PINK1/Parkin pathway has the capacity to impede ROS accumulation and promptly eliminate damaged mitochondria, thereby regulating IS and substantially mitigating oxidative harm (Wen et al., 2023). IS-induced neuronal injury is partly due to the excitatory toxicity of glutamate (L-Glu), and it has been observed that the PINK1/Parkin pathway safeguards neurons against L-Glu-induced damage by maintaining mitochondrial function (Dou et al., 2022). It is important to mention that the recently identified extended form of PTEN (PTEN-L) has been demonstrated to function as a suppressor of mitophagy by obstructing Parkin’s movement to mitochondria and restraining its E3 ligase activity. Some PTEN has been identified to be localized on the ER membrane and in MAM (Wang et al., 2018a). Prior research has indicated that the activation of mitophagy occurs via the PINK1/Parkin pathway in instances of brain ischemia and reperfusion-induced brain injury (Lan et al., 2018). Therefore, the regulation of mitochondrial autophagy mediated by PINK1/Parkin in MAM may represent a promising approach for the treatment of IS.
2.2 FUNDC1 in MAM and mitophagy
FUNDC1 functions as a newly discovered MAM protein essential for triggering mitochondrial autophagy under hypoxic conditions. Research has indicated that FUNDC1 has the ability to interact with MAM-specific proteins, facilitate the generation of MAM, and influence mitochondrial dynamics (Wu et al., 2017). Under typical oxygen levels, a minimal quantity of FUNDC1 can be found within the MAM (Wu et al., 2016a). In the absence of oxygen, FUNDC1 accumulates at the MAM through its interaction with the ER resident protein CANX (calreticulin). The regulation of FUNDC1 mitochondrial autophagy is influenced by a variety of stress factors and cellular proteins. The phosphorylation of FUNDC1 at Tyr18 and Ser13 is carried out by Src and casein kinase 2 (CK2), respectively, which serves to inhibit its interaction with LC3 and the initiation of autophagy (Chen et al., 2016). In neurons undergoing ischemic reperfusion, excessive activation of Src due to ischemic injury leads to the phosphorylation of Tyr18 on FUNDC1. This results in the impairment of FUNDC1’s ability to bind with LC3, ultimately hindering its participation in neuronal mitochondrial autophagy. Repressing Src activity can preserve the function of FUNDC1 and initiate neuroprotective effects mediated by mitochondrial autophagy through FUNDC1 (Tang et al., 2023). FUNDC1 has the ability to engage with OPA1, a mitochondrial fusion protein, as well as Drp1, a mitochondrial fission protein (Chen et al., 2016). The breakdown of OPA1 at the S1 site caused by cerebral ischemia worsens the splitting of mitochondria and reperfusion damage in neurons (Li et al., 2022). Inhibition of Drp1 may restrict brain injury following IS and avert neuronal impairment and mortality (Grohm et al., 2012). FUNDC1 may enhance mitophagy and ameliorate neuronal damage following IS through its interaction with OPA1 and Drp1. Prior research has indicated a clear link between MAM and FUNDC1 in facilitating the process of mitochondrial autophagy, FUNDC1 interacts with another MAM protein, IP3R2, to mediate IP3R2-dependent calcium signaling from the ER to the mitochondria (Wu et al., 2017), thereby enhancing calcium transport in MAM and impacting mitophagy. An increasing number of studies are demonstrating the potential for regulating FUNDC1-mediated mitochondrial autophagy to improve IS (refer to Table 2). These findings indicate that targeting FUNDC1-mediated mitochondrial autophagy in MAM may hold promise for the treatment of IS.
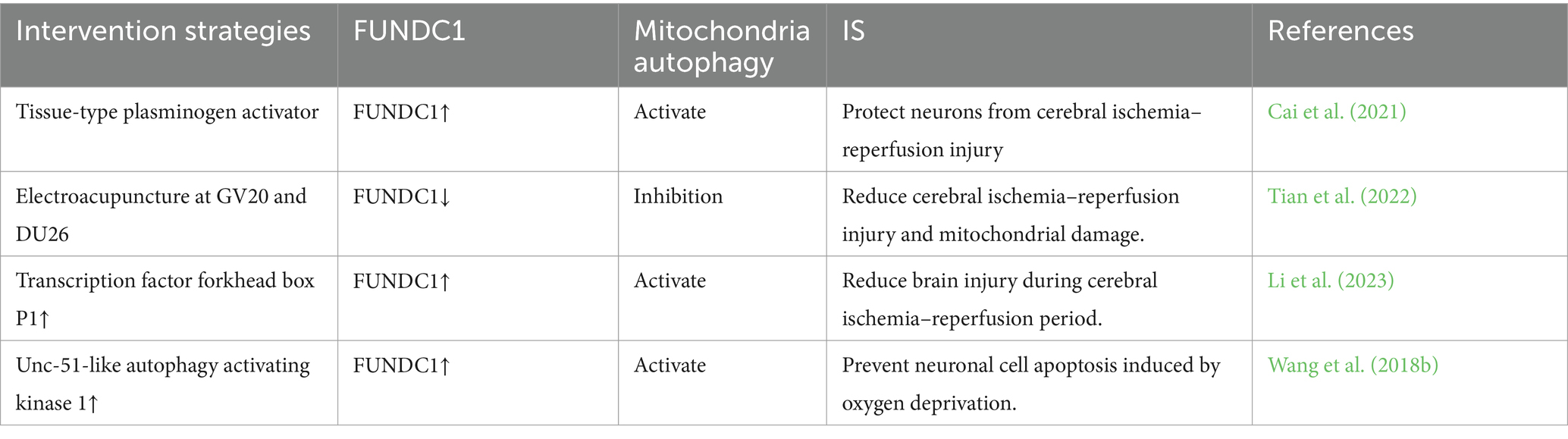
Table 2. The role of FUNDC1 in IS.
2.3 Beclin1 in MAM and mitophagy
While Beclin 1 was initially investigated for its role in regulating autophagy, it also contributes to the regulation of mitochondrial autophagy. After stimulating mitochondrial autophagy, it was translocated to the MAM, facilitating the crosstalk between the ER and mitochondria and boosting the efficacy of mitochondrial autophagy (Gelmetti et al., 2017). Beclin1’s localization on MAM ensures that the initiation of autophagosome formation occurs in close proximity to impaired mitochondria, facilitating their efficient engulfment. Beclin 1 collaborates with different cofactors to regulate the function of the Vps-34 protein and promote the formation of the Beclin 1-Vps34-Vps15 core complex, thus initiating autophagy (Kang et al., 2011). Furthermore, Beclin1 initiates mitophagy by promoting the translocation of Parkin from the cytosol to the mitochondria. It is noteworthy that Beclin1 is significantly upregulated by ischemic preconditioning (Xie et al., 2018), whereas its expression is downregulated following reperfusion injury (Dai et al., 2017). After cerebral ischemia, the primary localization of Ulk1 is in the small glial cells within the infarcted region, where it facilitates the development of anti-inflammatory pathways in these cells (Xiong et al., 2024). Beclin1 ensures the accurate targeting of autophagosomes to mitochondria via ULK1-dependent serine phosphorylation (Quiles et al., 2023), and Beclin1 may facilitate mitochondrial autophagy, thereby promoting neuronal repair following ischemic brain injury. Mouse studies have demonstrated that Beclin1 safeguards mitochondria by preferentially triggering a distinct mitophagy pathway. The protective role of Beclin1 in this process may potentially extend to MAM involvement, possibly contributing to the formation or maintenance of MAM (Sun et al., 2021). A recent investigation has presented fresh findings indicating that Beclin1 and Beclin2 exhibit around 57% sequence similarity and possess several common structural domains. However, it was observed that Beclin2 does not contain the Ulk1 phosphorylation site found in Beclin1 and is not localized to MAM during mitophagy (Quiles et al., 2023). These results provide additional evidence that the distinctive N-terminal domain of Beclin1 and its precise positioning in MAM could account for its distinct role in mitophagy. Research has demonstrated that the downregulation of Beclin1 can attenuate cell death in the ischemic hemisphere, facilitate neurogenesis, and diminish infarct size (Zheng et al., 2009). Repressing Beclin1 can impede the formation of autophagosomes, leading to a notable decrease in neuronal loss, glial proliferation, and cell death (Xing et al., 2012). In conclusion, Beclin1-mediated mitochondrial autophagy in MAM represents a promising therapeutic target for the management of IS.
2.4 Mfn2 in MAM and mitophagy
Mfn2 can be found in the MAM and serves as a prototypical functional tethering protein within this region. Its presence in the MAM allows for interaction with mitochondria, facilitating the formation of an organelle bridge within the cell (McFie et al., 2016). In the context of cerebral ischemia–reperfusion, the lack of Mfn2 hinders platelet activation, formation of prothrombotic platelets, and diminishes the extent of the infarct (Jacob et al., 2024). Mfn2 is essential for the proper functioning of mitochondria, encompassing fusion, axonal transport, inter-organelle communication, and mitophagy (Stuppia et al., 2015). Mfn2’s regulation of mitochondrial function can attenuate hypoxia-induced cell apoptosis (Peng et al., 2015). Downregulation of Mfn2 results in impaired mitochondrial function, disrupting calcium homeostasis and ultimately leading to delayed neuronal cell death. Studies have indicated a notable decline in Mfn2 levels 90 min after 6 h of middle cerebral artery occlusion. As a result, the decrease in Mfn2 is considered to occur later during reperfusion. Targeting this reduction may contribute to minimizing ischemic damage and widening the current limited stroke treatment window (Martorell-Riera et al., 2014). In order for mitophagy to take place, mitochondria need to undergo fission initially. This process involves the separation of dysfunctional mitochondria through mitochondrial fission and selective fusion, enabling their removal via mitophagy (Twig et al., 2008). Mfn2 is involved in the regulation of the structure of MAM and mitochondrial function, which is essential for protecting neurons. Decreased expression of Mfn2 results in impaired MAM function and mitochondrial activity, ultimately leading to increased fragmentation of mitochondria (Zhu et al., 2024). Ubiquitination of Mfn2 could potentially inhibit mitochondrial fusion by facilitating the breakdown of these proteins through proteasomes or by obstructing the formation of MFN dimers between mitochondria (Joaquim and Escobar-Henriques, 2020). Cerebral ischemia–reperfusion injury results from inadequate autophagy, and Mfn2 has the potential to enhance ischemia–reperfusion injury by boosting the generation of autophagosomes and facilitating their fusion with lysosomes (Peng et al., 2018). Additionally, the interaction between the ER and mitochondria occurs as an initial stage in the process of mitophagy, preceding the engulfment of cellular organelles by the autophagosome. Mfn2, a protein that tethers mitochondria to the ER, is essential for the formation of autophagosomes during mammalian starvation (Naon et al., 2016). Knockout or silencing of Mfn2 results in the separation of ER and mitochondria (de Brito and Scorrano, 2008). Studies have indicated a connection between global cerebral ischemia and the swift migration of Mfn2 from the mitochondria to the cytosol (Klacanova et al., 2019). Additionally, Mfn2 serves as a substrate for parkin, and during the initial phases of mitophagy, parkin may attach to ubiquitinated Mfn2 by interacting with the pUb domain. The ubiquitination of Mfn2’s HR1 domain by parkin is essential for efficient mitophagy (McLelland et al., 2018). Recent research has shown that Mfn2 is directly involved in inhibiting mitochondrial autophagy by connecting mitochondria to the ER (Basso et al., 2018). These studies indicate that targeting Mfn2-mediated mitochondrial autophagy in MAM may be an effective approach for regulating IS.
3 MAM modulates endoplasmic reticulum stress in IS
The ER plays a crucial role as a cellular organelle, overseeing the synthesis, transportation, and regulation of calcium levels within the cell. Following an IS, the regular function of the ER is disturbed, potentially leading to the onset of ER stress. During cerebral ischemia, the occurrence of ER stress is associated with local acute ischemia and neuronal damage in brain tissue. The stress in the ER leads to an adaptive reaction known as the unfolded protein response (UPR), which serves primarily as a survival-promoting mechanism aimed at reinstating ER homeostasis. Nevertheless, when cells undergo extended and persistent ER stress, the UPR directs them toward cellular apoptosis (Lenna et al., 2014). During the initial phase of UPR, MAM’s dynamic assembly takes place, a process typically viewed as supportive of cell survival. This is linked to heightened mitochondrial calcium absorption and improved respiration (Bravo et al., 2011). The MAM component has a strong connection with UPR, and essential elements of UPR like PERK and IRE1α have been identified within MAM. When the ER is under stress, there is an accumulation of IRE1α in MAM (Carreras-Sureda et al., 2017). Hence, targeting MAM-mediated ER stress could be a promising therapeutic strategy for IS (Tables 3, 4).
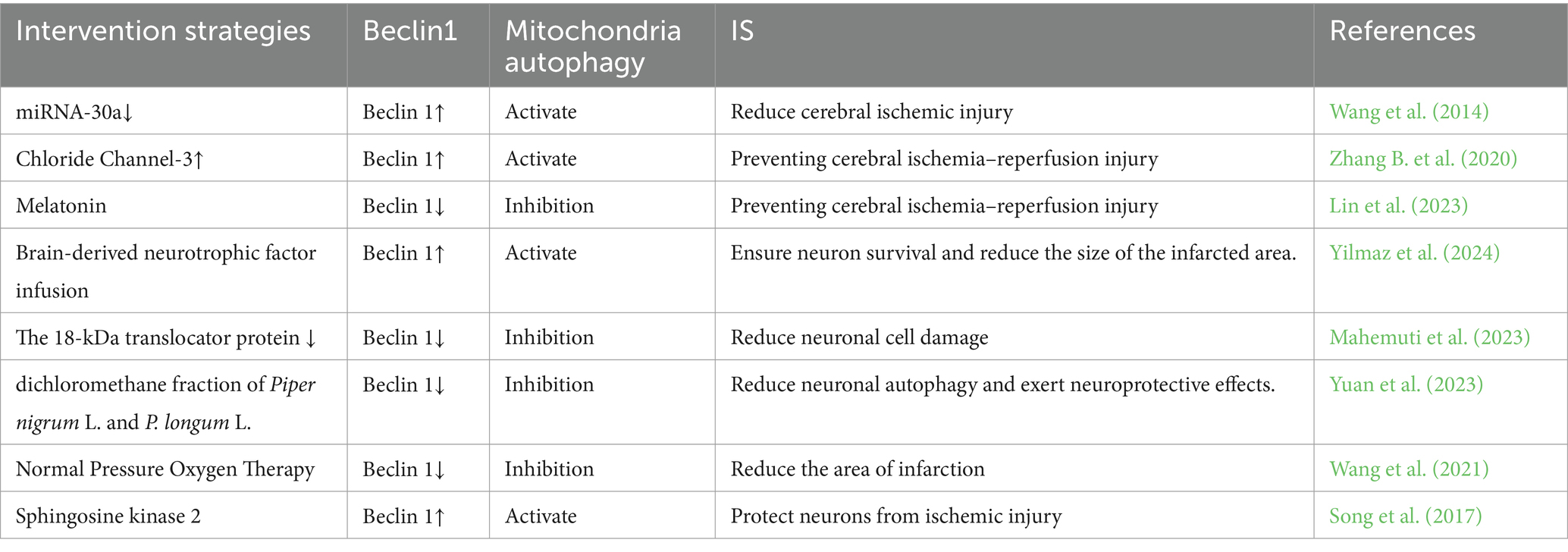
Table 3. The role of Beclin1 in IS.
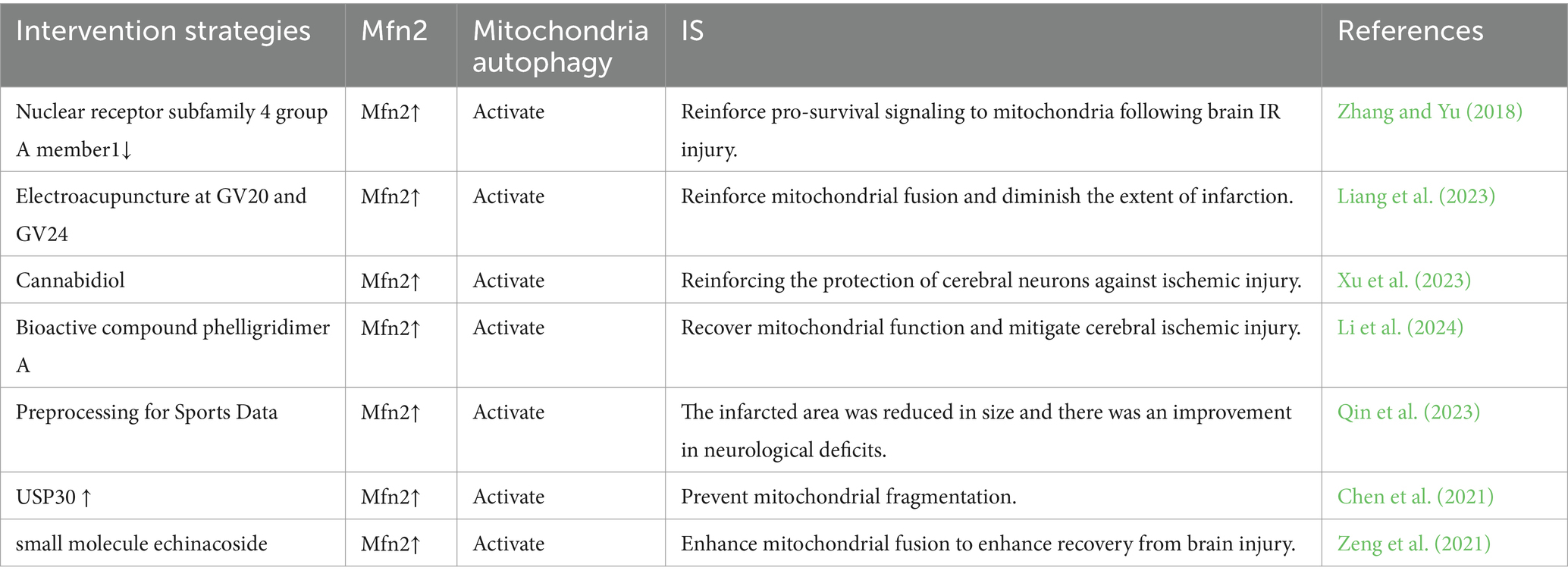
Table 4. The role of Mfn2 in IS.
3.1 GRP78 in MAM and endoplasmic reticulum stress
Glucose regulation protein 78 (GRP78) functions as an ER chaperone located within the intraluminal space and is a significant constituent of MAM (Prasad et al., 2017; Mahamed et al., 2023). The upregulation of GRP78 serves as an indicator of ER stress, regulating the activation of transmembrane ER stress sensors by means of binding and release mechanisms (Lee, 2005). GRP78 serves as the primary controller of the UPR, facilitating proper protein folding and inhibiting the accumulation of protein aggregates within the ER. UPR is controlled by three nearby detectors IRE1, PERK, and ATF6. GRP78 interacts with these three sensors via its peptide-binding domain, maintaining them in a state of inactivity. When cells accumulate misfolded proteins, they attach to GRP78 and interfere with its communication with the stress sensors located upstream. Moreover, GRP78 is a protein that is activated under stress and has broad expression in the cells of animals (Chang et al., 1987). When cells experience ER stress, particularly during the depletion of stored calcium and the buildup of irregular proteins, there is an increase in the transcription rate of GRP78 (Li et al., 1994). IS can result in the disruption of ER calcium homeostasis, damage to UPR, and impairment of proteasome function, thereby leading to secondary ER dysfunction. The sole method to break free from this potentially life-threatening pattern is to initiate UPR, thus prompting the production of GRP78 at a degree adequate for reconfiguring misshapen proteins (Paschen, 2004). The serine protease tPA is considered the standard treatment for cerebral ischemia, and prior research has identified Grp78 as a novel neural receptor for tPA. Activation of tPA by Grp78 on cell surfaces triggers a negative feedback loop that inhibits the activation of the PERK branch, leading to decreased phosphorylation of eIF2α and ultimately reducing neuronal death in cerebral ischemia (Louessard et al., 2017). Moreover, a research study indicates that GRP78 plays a role in facilitating the movement of peptides through the ER membrane and serves to safeguard cells against cell death induced by ER stress (Rao et al., 2002). An increasing number of research findings indicate the essential involvement of GRP78 in ER stress (refer to Table 5). In conclusion, GRP78 located in the mitochondrial associated membrane (MAM) is capable of regulating neuronal death induced by ER stress. Targeting GRP78 in MAM may represent a promising therapeutic approach for managing ER stress following IS (Figure 2).
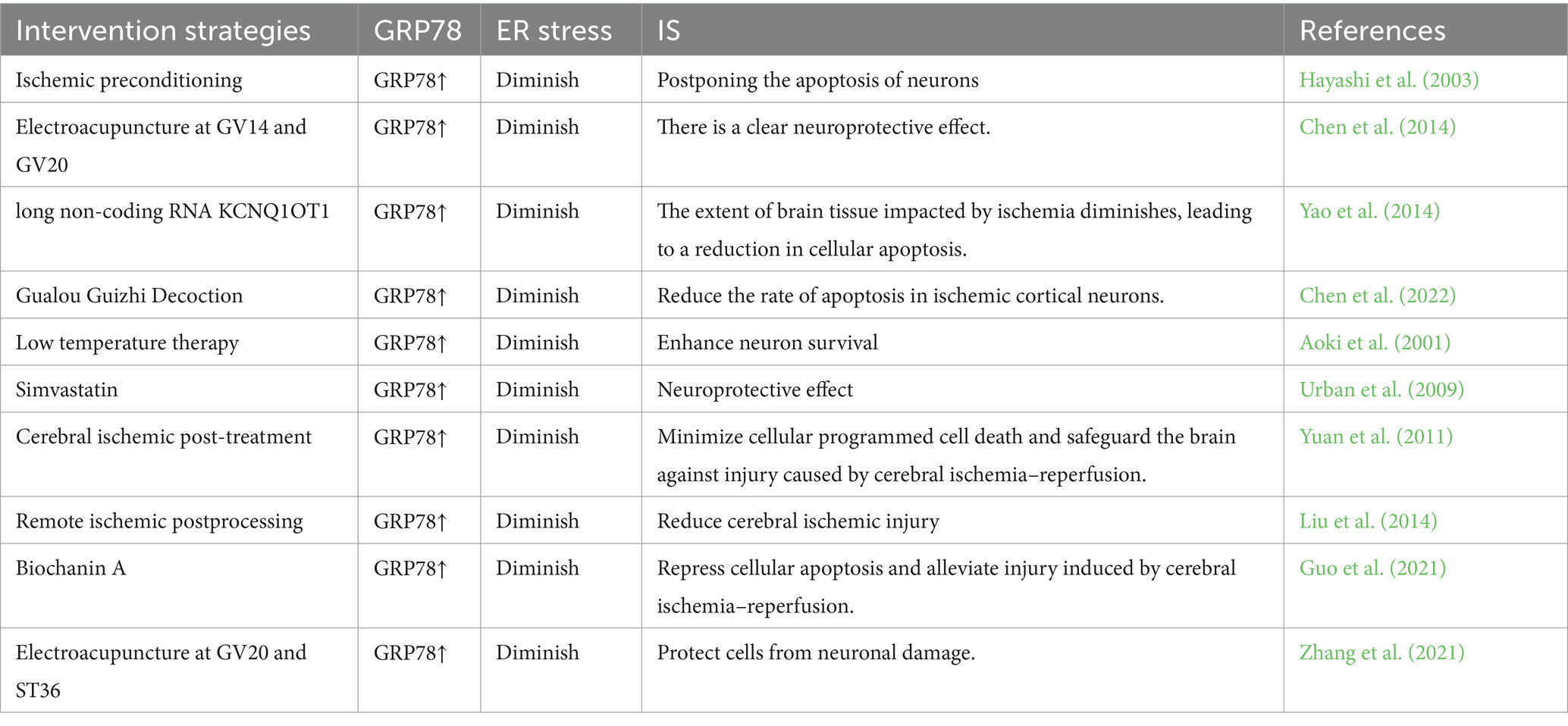
Table 5. The role of GRP78 in IS.
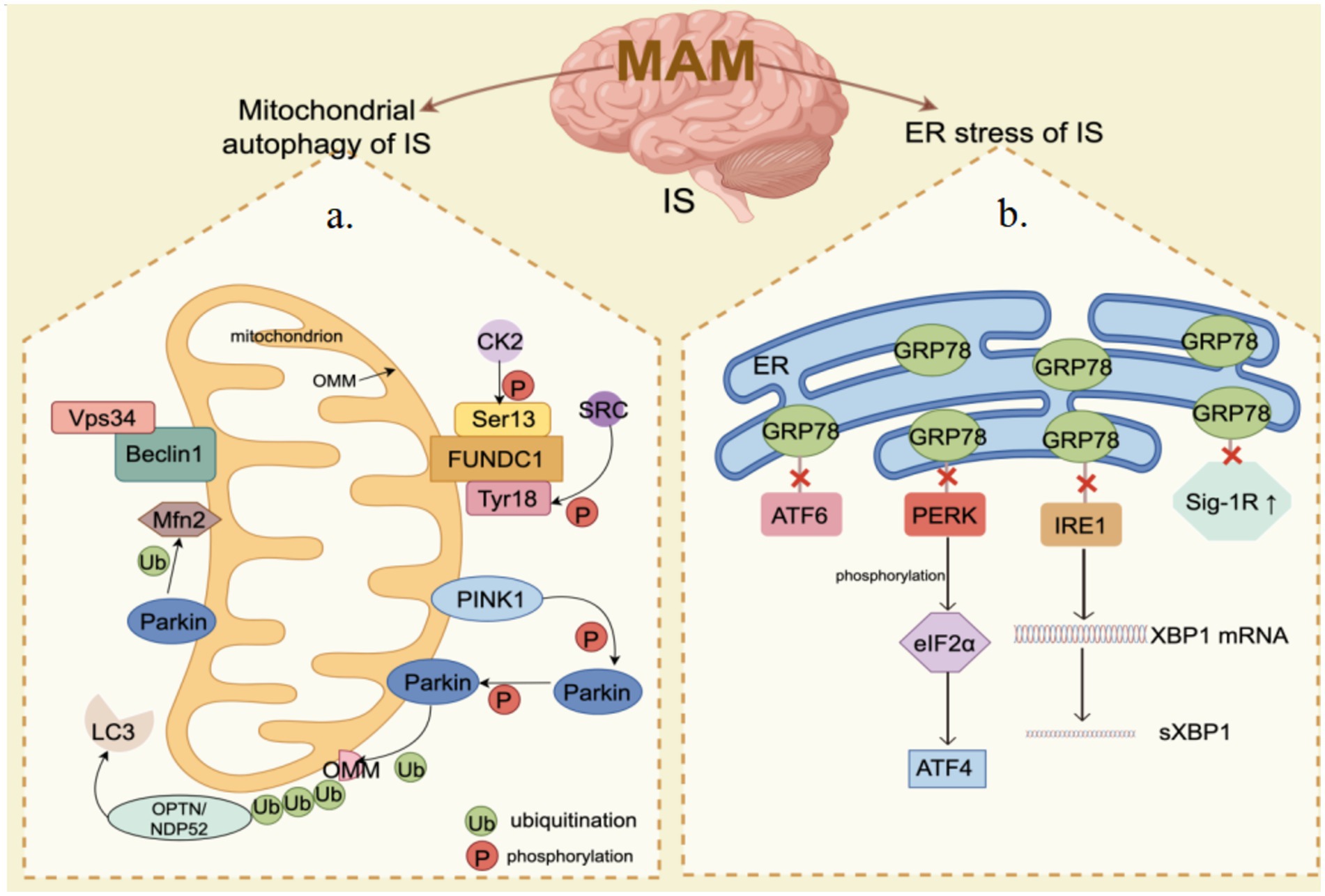
Figure 2. MAM mediates mitochondrial autophagy and endoplasmic reticulum (ER) stress in ischemia–reperfusion (IS). (a) Mitochondrial Autophagy: PINK1/Parkin, FUNDC1, Beclin1, and Mfn2 are significantly enriched in the MAM. When IS happens, the PINK1/Parkin pathway is activated, inducing mitochondrial autophagy through the ubiquitination of Mfn2 and other substrates. Beclin1 facilitates mitochondrial autophagy by interacting with Vps34. The phosphorylation status of FUNDC1 critically influences mitochondrial autophagy under hypoxic conditions. (b) ER Stress: The core components of the UPR, including IRE1, PERK, and ATF6, are also enriched in the MAM. Under ER stress conditions, dissociation of GRP78 from UPR leads to activation of PERK and IRE1 as well as upregulation of Sig-1R, which subsequently impacts ER function and cell survival. By Figdraw.
3.2 IRE1 in MAM and endoplasmic reticulum stress
The transmembrane protein IRE1, which is involved in the UPR, possesses both a kinase domain and an RNAse domain (RD) and is highly conserved across species (Nikawa and Yamashita, 1992). As part of the MAM complex, the accumulation of IRE1α in MAM leads to either cell survival or cell death through the facilitation of excessive mitochondrial calcium levels (Shuda et al., 2003). Under normal conditions, the ER resident chaperone Bip is attached to the luminal domain of IRE1α, exerting negative regulation on it. During ER stress, Bip separates from IRE1, leading to the homodimerization and self-phosphorylation of IRE1. This is followed by the activation of its RNase domain. The activated IRE1 can then cleave the XBP1 precursor mRNA to generate the active spliced form of XBP1 (sXBP1). Once inside the nucleus, the sXBP1 mRNA is translated into the mature protein, which in turn stimulates the expression of genes related to protein folding and ultimately helps alleviate ER stress (Wang et al., 2022a). The likelihood of patients surviving IS is associated with the promotion of angiogenesis, and XBP1 plays a positive role in controlling the growth and development of brain microvascular endothelial cells following brain ischemia (Shi et al., 2019). A different research indicates that the IRE1/XBP1 pathway of the UPR becomes active during IS, and the lack of Xbp1 results in deteriorating stroke consequences (Jiang et al., 2017). Hence, IRE1 may modulate the occurrence of IS through the mediation of XBP1. Nevertheless, persistent and elevated levels of self-phosphorylation result in the formation of higher-order IRE1α oligomers, leading to a relaxation in the specificity of IRE1α. This subsequently triggers the degradation of numerous mRNAs encoding secretory proteins on the ER membrane through an intramolecular process known as regulated IRE1α-dependent decay (RIDD) (Oakes, 2017). RIDD plays a role in the degradation of various mRNAs and microRNAs, thereby regulating pathological processes such as inflammation and cell apoptosis (Wang et al., 2022a). Studies have demonstrated that continuous activation of IRE1α leads to neuronal death by reducing the expression of 14-3-3θ mRNA through IRE1α RIDD activity. Furthermore, IRE1α also serves as a scaffold to regulate the localization of inositol 1,4,5-trisphosphate receptors (IP3Rs) on MAM (Carreras-Sureda et al., 2019). Following an IS, the maintenance of intracellular calcium balance is essential for regulating neuronal function, with inositol 1,4,5-trisphosphate receptors (IP3Rs) serving as crucial channels for this purpose (Kesherwani and Agrawal, 2012). During cerebral ischemia, the ATP consumption is also linked to the continuous decrease in levels of inositol 1,4,5-trisphosphate, potentially resulting in neuronal function impairment and influencing cerebral ischemic injury (Sun and Hsu, 1996). IRE1α’s regulation of IP3Rs receptors has the potential to decrease neuronal mortality and subsequently control IS. In conclusion, the involvement of IRE1 in MAM is crucial for the regulation of ER stress, suggesting that targeting IRE1 in MAM could be a promising therapeutic strategy for IS (Table 6).
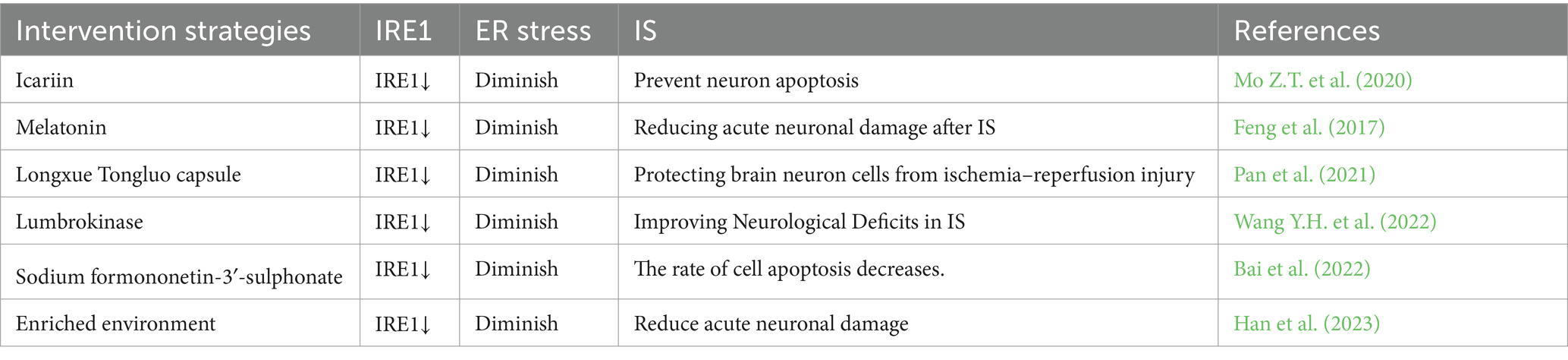
Table 6. The role of IRE1 in IS.
3.3 PERK in MAM and endoplasmic reticulum stress
The ER stress sensor PERK, a protein kinase R-like kinase, plays a crucial role in the UPR and is specifically abundant in MAM (Verfaillie et al., 2012). PERK plays a crucial role in MAM and has the ability to create a compound with S1R and Mfn2, contributing to the formation of MAM and enhancing the stability of ER-mitochondria interaction (Mao et al., 2022). One key sign of UPR activation following cerebral ischemia reperfusion involves the detection of PERK activation, which becomes fully active in the early stage of cerebral reperfusion (DeGracia and Montie, 2004). As a result of intracellular stress, unstructured or incorrectly folded proteins vie with GRP78 for binding, resulting in the dissociation of GRP78 from PERK. This leads to the release of inhibition on PERK and its activation through dimerization and self-phosphorylation (McQuiston and Diehl, 2017). Upon activation, PERK phosphorylates eukaryotic translation initiation factor 2 (eIF2α), leading to the inhibition of protein translation and a reduction in the aggregation of unfolded proteins within the ER lumen (Harding et al., 2000). PERK, a protein, is accountable for reducing mRNA translation during ER stress. This action helps to block the flow of newly produced proteins into the already stressed ER compartment. The process of translation attenuation is facilitated by the phosphorylation of eIF2α (Sano and Reed, 2013). During the early ischemia/reperfusion period, PERK significantly enhances the phosphorylation of eIF2α (Owen et al., 2005). In addition to its role in inhibiting protein translation, phosphorylated eIF2α is also capable of activating the expression of activated transcription factor 4 (ATF4) (Harding et al., 2000). ATF4 has the potential to influence cerebral ischemic injury through the regulation of cell apoptosis and neuronal function (Wu et al., 2022). Therefore, the PERK-ATF4 signaling pathway may play a role in neuronal apoptosis subsequent to IS. Apart from elF2α, PERK has the ability to phosphorylate nuclear red cell 2 p45-related factor 2 (NRF2) as well. The activation of NRF2 by PERK contributes to the maintenance of oxidation–reduction balance and promotes cell survival following ER stress (Cullinan and Diehl, 2004). During the occurrence of IS, NRF2 is involved in reducing oxidative stress, combating inflammation, regulating mitochondrial balance, and safeguarding the integrity of the blood–brain barrier to mitigate cerebral ischemic damage (Wang et al., 2022b). PERK may modulate IS through the phosphorylation of NRF2. Currently, there is a growing body of research investigating the role of PERK in the regulation of IS (refer to Table 7), and targeting PERK in MAM may hold promise as an approach for treating IS.
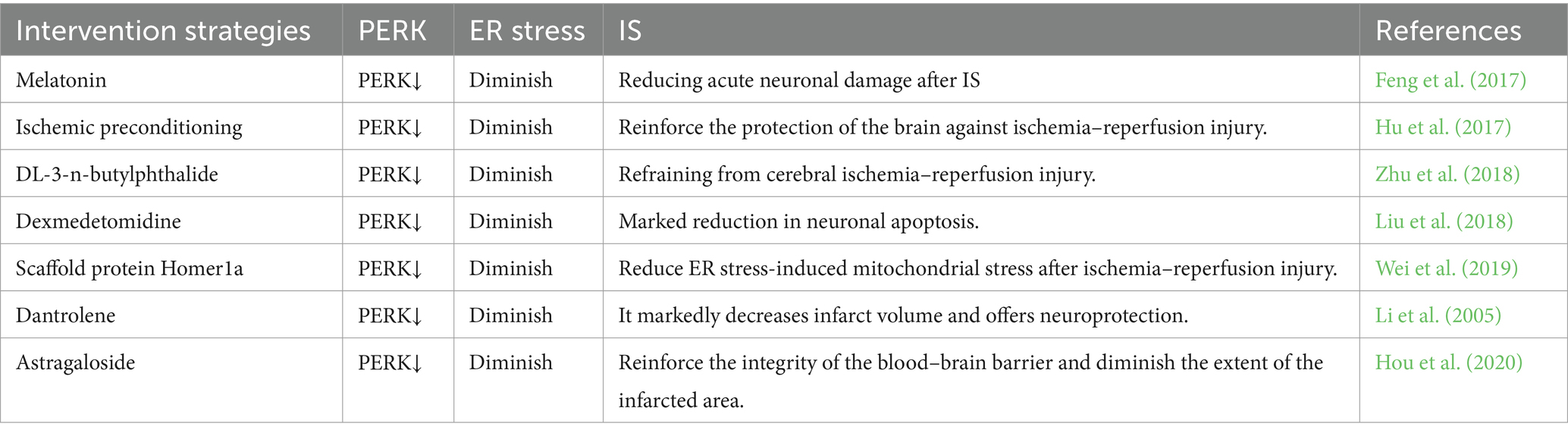
Table 7. The role of PERK in IS.
3.4 Sig-1R in MAM and endoplasmic reticulum stress
The sigma-1 receptor (Sig-1R) is a widely distributed co-factor found in the ER, specifically located in the MAM of the central nervous system. It plays a role in mediating signal transduction between the ER and mitochondria. Under normal conditions, Sig-1R is in a dormant state in MAM and forms a complex with another ER partner, BiP. During periods of ER stress, Sig-1R separates from BiP and controls the function of the three primary pathways of the UPR (PERK, IRE1a, ATF6) (Penke et al., 2018). The expression of Sig-1R is elevated in response to ER stress (Mitsuda et al., 2011). Overexpression of Sig-1R suppresses the activation of PERK and ATF6 signals, leading to enhanced cell survival (Hall et al., 2009). The decreased activity of Sig-1R destabilizes the conformation of IRE1a and diminishes cell viability (Mori et al., 2013). Agonists targeting Sig-1R have demonstrated protective effects on cells during stroke, and these effects are associated with the alleviation of ER stress (Morihara et al., 2018). It is evident that the upregulation of Sig-1R serves as a defensive mechanism for cell survival in response to ER stress. Moreover, Sig-1R has the capability to engage with inositol 1,4,5-trisphosphate receptor 3 (IP3R3) in order to stabilize its association with voltage-dependent anion channel 1 (VDAC1), thereby ensuring appropriate calcium transfer between the two cellular organelles (Morihara et al., 2018). After an IS, the downregulation of VDAC1 expression leads to intracellular calcium overload. Stabilizing the expression of VDAC1 can alleviate neuronal loss following cerebral ischemia (Yao et al., 2018). Sig-1R may stabilize VDAC1 through its interaction with IP3R3, thereby modulating the regulation of IS. It is important to mention that Sig-1R shows significant expression in the central nervous system, leading to its crucial involvement in functions such as cell differentiation, synapse growth, and the stimulation of microglia cells (Wu N.H. et al., 2021). The polarization of microglia/macrophages is a critical factor in the damage to tissues and the recovery of function following an IS. Additionally, Sig-1R serves as a crucial regulatory element in the macrophage-mediated clearance of deceased cells. The complete transplantation of Sig-1R macrophages has been shown to significantly decrease tissue damage and neurological impairments associated with IS (Zhang et al., 2023). In conclusion, targeting Sig-1R in MAM shows great promise for the treatment of IS (Table 8).

Table 8. The role of Sig-1R in IS.
4 Discussion
IS has been one of the leading causes of death worldwide, and to date, there is no effective treatment to stop the progression of the disease. As the most direct interaction bridge between the ER and mitochondria, MAM plays a crucial role in regulating the activities carried out by these two organelles. In ubiquitin-dependent mitochondrial autophagy, PINK1/Parkin is enriched in MAM. After IS, PINK1/Parkin pathway is activated to induce mitochondrial autophagy through Mfn2 ubiquitination. Beclin1 promotes mitochondrial autophagy by promoting the translocation of Parkin. As one of the non-ubiquitin-dependent mitochondrial autophagy, FUNDC1 is enriched in MAM, and phosphorylation of FUNDC1 under hypoxia affects its binding to LC3, thereby mediating mitochondrial autophagy. ER stress induced by IS can enhance the transcriptional rate of GRP78, dissociation of GRP78 from UPR leads to activation of PERK and IRE1, and upregulation of Sig-1R, thus affecting ER function and cell survival. In addition, more and more studies have shown that MAM plays an important role in regulating inflammation, oxidative stress, lipid metabolism, membrane dynamics, calcium signaling, etc. Thus, as an interface between energy metabolism, protein homeostasis, and cell fate control, MAM-mediated mitochondrial autophagy and ER stress play an important role in cell survival in the brain. MAM may be an important target for regulating IS and has important significance for improving the survival rate of patients.
However, despite the summary of many relevant molecules involved in the regulation of IS in the MAM, many questions remain to be addressed. The role of MAM in IS has not received enough attention, and the signaling pathway of MAM regulating IS has not been studied. Metabolic syndrome (MetS) is a series of interrelated vascular risk factors (including insulin resistance/diabetes, hypertension, central obesity, dyslipidemia) that are positively associated with adverse outcomes of IS and may lead to cerebrovascular damage and deterioration of neurofunction (Zhang et al., 2016). It is worth noting that numerous studies have found that MAM is involved in the regulation of metabolic syndrome: 1. mTOR, the mammalian target of rapamycin, is involved in a series of physiological and pathological processes of insulin signal transduction, and mTOR is located in MAM (Xu et al., 2017). Disruption of MAM integrity affects insulin resistance (Tubbs et al., 2018). 2. Gopinath et al. confirmed direct evidence that MAM is involved in the development of hypertension, and the Nogo-B family of reticulum proteins after pulmonary hypertension destroys MAM and inhibits apoptosis (Sutendra et al., 2011). 3. Enzymes involved in lipid metabolism are the most abundant proteins on MAM (Vance, 2014). 4. MAM plays a potential role in fat storage by transporting sufficient lipids to mitochondria for β-oxidation during fasting (Theurey et al., 2016). 5. PINK1/Parkin and FUNDC1 regulate the development of MetS by exerting anti-inflammatory and antioxidant stress effects through mitochondrial autophagy (Miao et al., 2023). But the link between MAM and IS and MetS has not been thoroughly studied. In the future, it may be a new research direction to explore the mechanism that can improve metabolic function, reduce ischemic injury and promote the recovery of neurological function.
Author contributions
ZJ: Writing – original draft. HL: Writing – review & editing. KX: Investigation, Writing – original draft. RL: Data curation, Writing – original draft. SY: Conceptualization, Writing – review & editing. LC: Investigation, Writing – review & editing. QZ: Data curation, Writing – review & editing. SL: Writing – review & editing. XS: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The National Nature Science Foundation of China (No. 81503669), Heilongiiang Natural Science Foundation (H2015031), the outstanding Training Foundation of Heilongjiang University of Chinese Medicine (2019JC05), the outstanding Innovative Talents Support Plan of Heilongjiang University of Chinese Medicine (2018RCD11), 2023 young Qhuang Scholars Training project in Heilongjiang Province. Heilongjiang Traditional Chinese Medicine Research Project (ZHY2020-125).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
IS, Ischemic stroke; MAM, mitochondrial-associated endoplasmic reticulum membrane; ER, endoplasmic reticulum; UPR, unfolded protein response; OMM, outer mitochondrial membrane; VAPB, vesicle-associated membrane protein-related protein B; PTPIP51, Protein Tyrosine Phosphatase Interacting Protein 51; IP3R, 1,4,5-trisphosphate receptor; GRP75, glucose-regulated protein 75; GRP78, glucose-regulated protein 78; VDAC1, voltage-dependent anion channel 1; BAP31, B cell receptor-associated protein 31; FIS1, mitochondrial fission protein 1; FUNDC1, protein 1 with FUN14 domain; Mfn1/2, mitofusin 1/2; IRE1, inositol 1,4,5-trisphosphate 3-kinase 1; Sig-1R, sigma-1 receptor; PINK1, serine/threonine kinase; Parkin, E3 ubiquitin ligase; TOM40, mitochondrial outer membrane translocase component; PERK, protein kinase R-like endoplasmic reticulum kinase; RIDD, regulated IRE1αdependent decay; eIF2α, eukaryotic translation initiation factor 2; ATF4, transcription factor 4; NRF2, nuclear red cell 2 p45-related factor 2.
References
Aoki, M., Tamatani, M., Taniguchi, M., Yamaguchi, A., Bando, Y., Kasai, K., et al. (2001). Hypothermic treatment restores glucose regulated protein 78 (GRP78) expression in ischemic brain. Brain Res. Mol. Brain Res. 95, 117–128. doi: 10.1016/s0169-328x(01)00255-8
Bai, Y., He, Z., Duan, W., Gu, H., Wu, K., Yuan, W., et al. (2022). Sodium formononetin-3′-sulphonate alleviates cerebral ischemia-reperfusion injury in rats via suppressing endoplasmic reticulum stress-mediated apoptosis. BMC Neurosci. 23:74. doi: 10.1186/s12868-022-00762-4
Barazzuol, L., Giamogante, F., Brini, M., and Calì, T. (2020). PINK1/Parkin mediated Mitophagy, ca(2+) Signalling, and ER-mitochondria contacts in Parkinson's disease. Int. J. Mol. Sci. 21:1772. doi: 10.3390/ijms21051772
Basso, V., Marchesan, E., Peggion, C., Chakraborty, J., von Stockum, S., Giacomello, M., et al. (2018). Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 138, 43–56. doi: 10.1016/j.phrs.2018.09.006
Bravo, R., Vicencio, J. M., Parra, V., Troncoso, R., Munoz, J. P., Bui, M., et al. (2011). Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 124, 2143–2152. doi: 10.1242/jcs.080762
Cai, Y., Yang, E., Yao, X., Zhang, X., Wang, Q., Wang, Y., et al. (2021). FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol. 38:101792. doi: 10.1016/j.redox.2020.101792
Carreras-Sureda, A., Jaña, F., Urra, H., Durand, S., Mortenson, D. E., Sagredo, A., et al. (2019). Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 21, 755–767. doi: 10.1038/s41556-019-0329-y
Carreras-Sureda, A., Pihán, P., and Hetz, C. (2017). The unfolded protein response: at the intersection between endoplasmic reticulum function and mitochondrial bioenergetics. Front. Oncol. 7:55. doi: 10.3389/fonc.2017.00055
Chang, S. C., Wooden, S. K., Nakaki, T., Kim, Y. K., Lin, A. Y., Kung, L., et al. (1987). Rat gene encoding the 78-kDa glucose-regulated protein GRP78: its regulatory sequences and the effect of protein glycosylation on its expression. Proc. Natl. Acad. Sci. USA 84, 680–684. doi: 10.1073/pnas.84.3.680
Chen, Y., Chen, Z., Cheng, W., Cao, Y., Xu, W., Lai, W., et al. (2022). The role of the GRP78/PERK/ATF4 pathway in the ability of Gua Lou Gui Zhi decoction to attenuate apoptosis by inhibiting endoplasmic reticulum stress after ischemia-reperfusion injury. Front. Biosci. (Landmark Ed.) 27:296. doi: 10.31083/j.fbl2710296
Chen, M., Chen, Z., Wang, Y., Tan, Z., Zhu, C., Li, Y., et al. (2016). Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12, 689–702. doi: 10.1080/15548627.2016.1151580
Chen, H. L., Qi, H., Liu, X. J., and Wang, M. S. (2014). Effect of electroacupuncture pretreatment on expression of glucose-regulated protein 78 in hippocampus of rats with cerebral ischemia/reperfusion. Zhongguo Zhen Jiu 34, 889–893
Chen, C., Qin, H., Tang, J., Hu, Z., Tan, J., and Zeng, L. (2021). USP30 protects against oxygen-glucose deprivation/reperfusion induced mitochondrial fragmentation and ubiquitination and degradation of MFN2. Aging (Albany NY) 13, 6194–6204. doi: 10.18632/aging.202629
Csordás, G., Renken, C., Várnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921. doi: 10.1083/jcb.200604016
Cullinan, S. B., and Diehl, J. A. (2004). PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 279, 20108–20117. doi: 10.1074/jbc.M314219200
Dai, S., Xu, Q., Liu, S., Yu, B., Liu, J., and Tang, J. (2017). Role of autophagy and its signaling pathways in ischemia/reperfusion injury. Am. J. Transl. Res. 9, 4470–4480
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
DeGracia, D. J., and Montie, H. L. (2004). Cerebral ischemia and the unfolded protein response. J. Neurochem. 91, 1–8. doi: 10.1111/j.1471-4159.2004.02703.x
Dhapola, R., Medhi, B., and HariKrishnaReddy, D. (2024). Insight into the pathophysiological advances and molecular mechanisms underlying cerebral stroke: current status. Mol. Biol. Rep. 51:649. doi: 10.1007/s11033-024-09597-0
Dou, Y. N., Wu, X., Fei, X., and Fei, Z. (2022). The neuroprotective effect of increased PINK1 expression following glutamate excitotoxicity in neuronal cells. Neuroscience 480, 97–107. doi: 10.1016/j.neuroscience.2021.11.020
Eysert, F., Kinoshita, P. F., Mary, A., Vaillant-Beuchot, L., Checler, F., and Chami, M. (2020). Molecular dysfunctions of mitochondria-associated membranes (MAMs) in Alzheimer's disease. Int. J. Mol. Sci. 21:9521. doi: 10.3390/ijms21249521
Feng, D., Wang, B., Wang, L., Abraham, N., Tao, K., Huang, L., et al. (2017). Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE1 signalings. J. Pineal Res. 62. doi: 10.1111/jpi.12395
Gelmetti, V., De Rosa, P., Torosantucci, L., Marini, E. S., Romagnoli, A., Di Rienzo, M., et al. (2017). PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 13, 654–669. doi: 10.1080/15548627.2016.1277309
Giacomello, M., and Pellegrini, L. (2016). The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ. 23, 1417–1427. doi: 10.1038/cdd.2016.52
Gómez-Sánchez, R., Gegg, M. E., Bravo-San Pedro, J. M., Niso-Santano, M., Alvarez-Erviti, L., Pizarro-Estrella, E., et al. (2014). Mitochondrial impairment increases FL-PINK1 levels by calcium-dependent gene expression. Neurobiol. Dis. 62, 426–440. doi: 10.1016/j.nbd.2013.10.021
Gomez-Suaga, P., Paillusson, S., and Miller, C. C. J. (2017). ER-mitochondria signaling regulates autophagy. Autophagy 13, 1250–1251. doi: 10.1080/15548627.2017.1317913
Grohm, J., Kim, S. W., Mamrak, U., Tobaben, S., Cassidy-Stone, A., Nunnari, J., et al. (2012). Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 19, 1446–1458. doi: 10.1038/cdd.2012.18
Guo, M. M., Qu, S. B., Lu, H. L., Wang, W. B., He, M. L., Su, J. L., et al. (2021). Biochanin a alleviates cerebral ischemia/reperfusion injury by suppressing endoplasmic reticulum stress-induced apoptosis and p38MAPK signaling pathway in vivo and in vitro. Front. Endocrinol. (Lausanne) 12:646720. doi: 10.3389/fendo.2021.646720
Hall, A. A., Herrera, Y., Ajmo, C. T. Jr., Cuevas, J., and Pennypacker, K. R. (2009). Sigma receptors suppress multiple aspects of microglial activation. Glia 57, 744–754. doi: 10.1002/glia.20802
Han, Y., Shen, X. Y., Gao, Z. K., Ping Han, P., and Bi, X. (2023). Pre-ischaemic treatment with enriched environment alleviates acute neuronal injury by inhibiting endoplasmic reticulum stress-dependent autophagy and apoptosis. Neuroscience 513, 14–27. doi: 10.1016/j.neuroscience.2022.12.014
Harding, H. P., Zhang, Y., Bertolotti, A., Zeng, H., and Ron, D. (2000). Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 5, 897–904. doi: 10.1016/s1097-2765(00)80330-5
Hayashi, T., Saito, A., Okuno, S., Ferrand-Drake, M., and Chan, P. H. (2003). Induction of GRP78 by ischemic preconditioning reduces endoplasmic reticulum stress and prevents delayed neuronal cell death. J. Cereb. Blood Flow Metab. 23, 949–961. doi: 10.1097/01.Wcb.0000077641.41248.Ea
Hong, T., Zhou, Y., Peng, L., Wu, X., Li, Y., Li, Y., et al. (2022). Knocking down peroxiredoxin 6 aggravates cerebral ischemia-reperfusion injury by enhancing mitophagy. Neuroscience 482, 30–42. doi: 10.1016/j.neuroscience.2021.11.043
Hou, B., Liu, R., Wu, Y., and Huang, S. (2020). Astragaloside IV reduces cerebral ischemia/reperfusion-induced blood-brain barrier permeability in rats by inhibiting ER stress-mediated apoptosis. Evid. Based Complement. Alternat. Med. 2020:9087873. doi: 10.1155/2020/9087873
Hu, Y. Q., Chen, W., Yan, M. H., Lai, J. J., Tang, N., and Wu, L. (2017). Ischemic preconditioning protects brain from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress-induced apoptosis through PERK pathway. Eur. Rev. Med. Pharmacol. Sci. 21, 5736–5744. doi: 10.26355/eurrev_201712_14020
Huang, S., Hong, Z., Zhang, L., Guo, J., Li, Y., and Li, K. (2022). CERKL alleviates ischemia reperfusion-induced nervous system injury through modulating the SIRT1/PINK1/Parkin pathway and mitophagy induction. Biol. Chem. 403, 691–701. doi: 10.1515/hsz-2021-0411
Hung, V., Lam, S. S., Udeshi, N. D., Svinkina, T., Guzman, G., Mootha, V. K., et al. (2017). Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. eLife 6:e24463. doi: 10.7554/eLife.24463
Iurlaro, R., and Muñoz-Pinedo, C. (2016). Cell death induced by endoplasmic reticulum stress. FEBS J. 283, 2640–2652. doi: 10.1111/febs.13598
Jacob, S., Kosaka, Y., Bhatlekar, S., Denorme, F., Benzon, H., Moody, A., et al. (2024). Mitofusin-2 regulates platelet mitochondria and function. Circ. Res. 134, 143–161. doi: 10.1161/circresaha.123.322914
Ji, C., Zhang, Z., Li, Z., She, X., Wang, X., Li, B., et al. (2022). Mitochondria-associated endoplasmic reticulum membranes: inextricably linked with autophagy process. Oxidative Med. Cell. Longev. 2022, 7086807–7086811. doi: 10.1155/2022/7086807
Jiang, M., Yu, S., Yu, Z., Sheng, H., Li, Y., Liu, S., et al. (2017). XBP1 (X-box-binding protein-1)-dependent O-GlcNAcylation is neuroprotective in ischemic stroke in young mice and its impairment in aged mice is rescued by Thiamet-G. Stroke 48, 1646–1654. doi: 10.1161/strokeaha.117.016579
Jiao, Y., Wang, J., Jia, Y., and Xue, M. (2022). Remote ischemic preconditioning protects against cerebral ischemia injury in rats by upregulating miR-204-5p and activating the PINK1/Parkin signaling pathway. Metab. Brain Dis. 37, 945–959. doi: 10.1007/s11011-022-00910-z
Joaquim, M., and Escobar-Henriques, M. (2020). Role of mitofusins and mitophagy in life or death decisions. Front. Cell Dev. Biol. 8:572182. doi: 10.3389/fcell.2020.572182
Jürgensmeier, J. M., Xie, Z., Deveraux, Q., Ellerby, L., Bredesen, D., and Reed, J. C. (1998). Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA 95, 4997–5002. doi: 10.1073/pnas.95.9.4997
Kang, R., Zeh, H. J., Lotze, M. T., and Tang, D. (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580. doi: 10.1038/cdd.2010.191
Kesherwani, V., and Agrawal, S. K. (2012). Regulation of inositol 1,4,5-triphosphate receptor, type 1 (IP3R1) in hypoxic/reperfusion injury of white matter. Neurol. Res. 34, 504–511. doi: 10.1179/1743132812y.0000000038
Klacanova, K., Kovalska, M., Chomova, M., Pilchova, I., Tatarkova, Z., Kaplan, P., et al. (2019). Global brain ischemia in rats is associated with mitochondrial release and downregulation of Mfn2 in the cerebral cortex, but not the hippocampus. Int. J. Mol. Med. 43, 2420–2428. doi: 10.3892/ijmm.2019.4168
Kleindorfer, D. O., Towfighi, A., Chaturvedi, S., Cockroft, K. M., Gutierrez, J., Lombardi-Hill, D., et al. (2021). 2021 guideline for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline from the American Heart Association/American Stroke Association. Stroke 52, e364–e467. doi: 10.1161/str.0000000000000375
Kumar, R., Bukowski, M. J., Wider, J. M., Reynolds, C. A., Calo, L., Lepore, B., et al. (2016). Mitochondrial dynamics following global cerebral ischemia. Mol. Cell. Neurosci. 76, 68–75. doi: 10.1016/j.mcn.2016.08.010
Lan, R., Wu, J. T., Wu, T., Ma, Y. Z., Wang, B. Q., Zheng, H. Z., et al. (2018). Mitophagy is activated in brain damage induced by cerebral ischemia and reperfusion via the PINK1/Parkin/p62 signalling pathway. Brain Res. Bull. 142, 63–77. doi: 10.1016/j.brainresbull.2018.06.018
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Lee, A. S. (2005). The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 35, 373–381. doi: 10.1016/j.ymeth.2004.10.010
Lenna, S., Han, R., and Trojanowska, M. (2014). Endoplasmic reticulum stress and endothelial dysfunction. IUBMB Life 66, 530–537. doi: 10.1002/iub.1292
Li, Y., Changhong, Y., Liyu, Y., Changchang, M., Zeng, L., Yue, L., et al. (2023). Transcription factor Forkhead box P (Foxp) 1 reduces brain damage during cerebral ischemia-reperfusion injury in mice through FUN14 domain-containing protein 1. Neuroscience 530, 1–16. doi: 10.1016/j.neuroscience.2023.07.029
Li, F., Hayashi, T., Jin, G., Deguchi, K., Nagotani, S., Nagano, I., et al. (2005). The protective effect of dantrolene on ischemic neuronal cell death is associated with reduced expression of endoplasmic reticulum stress markers. Brain Res. 1048, 59–68. doi: 10.1016/j.brainres.2005.04.058
Li, X., Li, H., Xu, Z., Ma, C., Wang, T., You, W., et al. (2022). Ischemia-induced cleavage of OPA1 at S1 site aggravates mitochondrial fragmentation and reperfusion injury in neurons. Cell Death Dis. 13:321. doi: 10.1038/s41419-022-04782-0
Li, W. W., Sistonen, L., Morimoto, R. I., and Lee, A. S. (1994). Stress induction of the mammalian GRP78/BiP protein gene: in vivo genomic footprinting and identification of p70CORE from human nuclear extract as a DNA-binding component specific to the stress regulatory element. Mol. Cell. Biol. 14, 5533–5546. doi: 10.1128/mcb.14.8.5533-5546.1994
Li, X., Xu, B., Long, L., Li, Y., Xiao, X., Qiu, S., et al. (2024). Phelligridimer a enhances the expression of mitofusin 2 and protects against cerebral ischemia/reperfusion injury. Chem. Biol. Interact. 398:111090. doi: 10.1016/j.cbi.2024.111090
Liang, H., Ruan, S., Wang, F., Yan, N. W., Wang, Y. X., Chen, B., et al. (2023). Electroacupuncture alleviates neurological function via activating the Yap-OPA1 axis and mitochondrial fusion in rats with cerebral ischemia-reperfusion injury. Zhen Ci Yan Jiu 48, 1088–1094. doi: 10.13702/j.1000-0607.20230345
Lin, C., Li, L., Xu, Q., Xu, S., and Tang, C. (2023). Yap1-Usp14 Axis inhibits neuronal mitophagy during neonatal hypoxia-ischemia encephalopathy by regulation of Beclin-1 ubiquitination in mouse. Mol. Neurobiol. 60, 4273–4287. doi: 10.1007/s12035-023-03344-5
Liu, X., Du, Y., Liu, J., Cheng, L., He, W., and Zhang, W. (2023). Ferrostatin-1 alleviates cerebral ischemia/reperfusion injury through activation of the AKT/GSK3β signaling pathway. Brain Res. Bull. 193, 146–157. doi: 10.1016/j.brainresbull.2022.12.009
Liu, C., Fu, Q., Mu, R., Wang, F., Zhou, C., Zhang, L., et al. (2018). Dexmedetomidine alleviates cerebral ischemia-reperfusion injury by inhibiting endoplasmic reticulum stress dependent apoptosis through the PERK-CHOP-Caspase-11 pathway. Brain Res. 1701, 246–254. doi: 10.1016/j.brainres.2018.09.007
Liu, X., Zhao, S., Liu, F., Kang, J., Xiao, A., Li, F., et al. (2014). Remote ischemic postconditioning alleviates cerebral ischemic injury by attenuating endoplasmic reticulum stress-mediated apoptosis. Transl. Stroke Res. 5, 692–700. doi: 10.1007/s12975-014-0359-5
Louessard, M., Bardou, I., Lemarchand, E., Thiebaut, A. M., Parcq, J., Leprince, J., et al. (2017). Activation of cell surface GRP78 decreases endoplasmic reticulum stress and neuronal death. Cell Death Differ. 24, 1518–1529. doi: 10.1038/cdd.2017.35
Lv, Y., Cheng, L., and Peng, F. (2022). Compositions and functions of mitochondria-associated endoplasmic reticulum membranes and their contribution to cardioprotection by exercise preconditioning. Front. Physiol. 13:910452. doi: 10.3389/fphys.2022.910452
Mahamed, Z., Shadab, M., Najar, R. A., Millar, M. W., Bal, J., Pressley, T., et al. (2023). The protective role of mitochondria-associated endoplasmic reticulum membrane (MAM) protein Sigma-1 receptor in regulating endothelial inflammation and permeability associated with acute lung injury. Cells 13:5. doi: 10.3390/cells13010005
Mahemuti, Y., Kadeer, K., Su, R., Abula, A., Aili, Y., Maimaiti, A., et al. (2023). TSPO exacerbates acute cerebral ischemia/reperfusion injury by inducing autophagy dysfunction. Exp. Neurol. 369:114542. doi: 10.1016/j.expneurol.2023.114542
Mao, H., Chen, W., Chen, L., and Li, L. (2022). Potential role of mitochondria-associated endoplasmic reticulum membrane proteins in diseases. Biochem. Pharmacol. 199:115011. doi: 10.1016/j.bcp.2022.115011
Martorell-Riera, A., Segarra-Mondejar, M., Muñoz, J. P., Ginet, V., Olloquequi, J., Pérez-Clausell, J., et al. (2014). Mfn2 downregulation in excitotoxicity causes mitochondrial dysfunction and delayed neuronal death. EMBO J. 33, 2388–2407. doi: 10.15252/embj.201488327
McFie, P. J., Ambilwade, P., Vu, H., and Stone, S. J. (2016). Endoplasmic reticulum-mitochondrial interaction mediated by mitofusin-1 or mitofusin-2 is not required for lipid droplet formation or adipocyte differentiation. Biochem. Biophys. Res. Commun. 478, 392–397. doi: 10.1016/j.bbrc.2016.07.040
McLelland, G. L., Goiran, T., Yi, W., Dorval, G., Chen, C. X., Lauinger, N. D., et al. (2018). Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 7:e32866. doi: 10.7554/eLife.32866
McQuiston, A., and Diehl, J. A. (2017). Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000Res 6:1897. doi: 10.12688/f1000research.12138.1
Miao, M. Q., Han, Y. B., and Liu, L. (2023). Mitophagy in metabolic syndrome. J. Clin. Hypertens. (Greenwich) 25, 397–403. doi: 10.1111/jch.14650
Mitsuda, T., Omi, T., Tanimukai, H., Sakagami, Y., Tagami, S., Okochi, M., et al. (2011). Sigma-1Rs are upregulated via PERK/eIF2α/ATF4 pathway and execute protective function in ER stress. Biochem. Biophys. Res. Commun. 415, 519–525. doi: 10.1016/j.bbrc.2011.10.113
Mo, Z. T., Liao, Y. L., Zheng, J., and Li, W. N. (2020). Icariin protects neurons from endoplasmic reticulum stress-induced apoptosis after OGD/R injury via suppressing IRE1α-XBP1 signaling pathway. Life Sci. 255:117847. doi: 10.1016/j.lfs.2020.117847
Mo, Y., Sun, Y. Y., and Liu, K. Y. (2020). Autophagy and inflammation in ischemic stroke. Neural Regen. Res. 15, 1388–1396. doi: 10.4103/1673-5374.274331
Mori, T., Hayashi, T., Hayashi, E., and Su, T. P. (2013). Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS One 8:e76941. doi: 10.1371/journal.pone.0076941
Morihara, R., Yamashita, T., Liu, X., Nakano, Y., Fukui, Y., Sato, K., et al. (2018). Protective effect of a novel sigma-1 receptor agonist is associated with reduced endoplasmic reticulum stress in stroke male mice. J. Neurosci. Res. 96, 1707–1716. doi: 10.1002/jnr.24270
Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., et al. (2016). Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 113, 11249–11254. doi: 10.1073/pnas.1606786113
Nikawa, J., and Yamashita, S. (1992). IRE1 encodes a putative protein kinase containing a membrane-spanning domain and is required for inositol phototrophy in Saccharomyces cerevisiae. Mol. Microbiol. 6, 1441–1446. doi: 10.1111/j.1365-2958.1992.tb00864.x
Noh, B., McCullough, L. D., and Moruno-Manchon, J. F. (2023). Sex-biased autophagy as a potential mechanism mediating sex differences in ischemic stroke outcome. Neural Regen. Res. 18, 31–37. doi: 10.4103/1673-5374.340406
Oakes, S. A. (2017). Endoplasmic reticulum proteostasis: a key checkpoint in cancer. Am. J. Physiol. Cell Physiol. 312, C93–c102. doi: 10.1152/ajpcell.00266.2016
Omi, T., Tanimukai, H., Kanayama, D., Sakagami, Y., Tagami, S., Okochi, M., et al. (2014). Fluvoxamine alleviates ER stress via induction of Sigma-1 receptor. Cell Death Dis. 5:e1332. doi: 10.1038/cddis.2014.301
Owen, C. R., Kumar, R., Zhang, P., McGrath, B. C., Cavener, D. R., and Krause, G. S. (2005). PERK is responsible for the increased phosphorylation of eIF2alpha and the severe inhibition of protein synthesis after transient global brain ischemia. J. Neurochem. 94, 1235–1242. doi: 10.1111/j.1471-4159.2005.03276.x
Pan, B., Sun, J., Liu, Z., Wang, L., Huo, H., Zhao, Y., et al. (2021). Longxuetongluo capsule protects against cerebral ischemia/reperfusion injury through endoplasmic reticulum stress and MAPK-mediated mechanisms. J. Adv. Res. 33, 215–225. doi: 10.1016/j.jare.2021.01.016
Paschen, W. (2004). Endoplasmic reticulum dysfunction in brain pathology: critical role of protein synthesis. Curr. Neurovasc. Res. 1, 173–181. doi: 10.2174/1567202043480125
Peng, C., Rao, W., Zhang, L., Gao, F., Hui, H., Wang, K., et al. (2018). Mitofusin 2 exerts a protective role in ischemia reperfusion injury through increasing autophagy. Cell. Physiol. Biochem. 46, 2311–2324. doi: 10.1159/000489621
Peng, C., Rao, W., Zhang, L., Wang, K., Hui, H., Wang, L., et al. (2015). Mitofusin 2 ameliorates hypoxia-induced apoptosis via mitochondrial function and signaling pathways. Int. J. Biochem. Cell Biol. 69, 29–40. doi: 10.1016/j.biocel.2015.09.011
Penke, B., Fulop, L., Szucs, M., and Frecska, E. (2018). The role of Sigma-1 receptor, an intracellular chaperone in neurodegenerative diseases. Curr. Neuropharmacol. 16, 97–116. doi: 10.2174/1570159x15666170529104323
Prasad, M., Pawlak, K. J., Burak, W. E., Perry, E. E., Marshall, B., Whittal, R. M., et al. (2017). Mitochondrial metabolic regulation by GRP78. Sci. Adv. 3:e1602038. doi: 10.1126/sciadv.1602038
Qin, Y. Y., Pan, S. Y., Dai, J. R., Wang, Q. M., Luo, X., Qin, Z. H., et al. (2023). Alleviation of ischemic brain injury by exercise preconditioning is associated with modulation of autophagy and mitochondrial dynamics in cerebral cortex of female aged mice. Exp. Gerontol. 178:112226. doi: 10.1016/j.exger.2023.112226
Quiles, J. M., Najor, R. H., Gonzalez, E., Jeung, M., Liang, W., Burbach, S. M., et al. (2023). Deciphering functional roles and interplay between Beclin1 and Beclin2 in autophagosome formation and mitophagy. Sci. Signal. 16:eabo4457. doi: 10.1126/scisignal.abo4457
Rao, R. V., Peel, A., Logvinova, A., del Rio, G., Hermel, E., Yokota, T., et al. (2002). Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett. 514, 122–128. doi: 10.1016/s0014-5793(02)02289-5
Rizzuto, R., Pinton, P., Carrington, W., Fay, F. S., Fogarty, K. E., Lifshitz, L. M., et al. (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766. doi: 10.1126/science.280.5370.1763
Saini, V., Guada, L., and Yavagal, D. R. (2021). Global epidemiology of stroke and access to acute ischemic stroke interventions. Neurology 97, S6–s16. doi: 10.1212/wnl.0000000000012781
Saito, A., and Imaizumi, K. (2018). Unfolded protein response-dependent communication and contact among endoplasmic reticulum, mitochondria, and plasma membrane. Int. J. Mol. Sci. 19:3215. doi: 10.3390/ijms19103215
Sano, R., and Reed, J. C. (2013). ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 1833, 3460–3470. doi: 10.1016/j.bbamcr.2013.06.028
Shao, Z., Dou, S., Zhu, J., Wang, H., Xu, D., Wang, C., et al. (2021). Apelin-36 protects HT22 cells against oxygen-glucose deprivation/reperfusion-induced oxidative stress and mitochondrial dysfunction by promoting SIRT1-mediated PINK1/Parkin-dependent mitophagy. Neurotox. Res. 39, 740–753. doi: 10.1007/s12640-021-00338-w
Shi, Q., Cheng, Q., and Chen, C. (2021). The role of autophagy in the pathogenesis of ischemic stroke. Curr. Neuropharmacol. 19, 629–640. doi: 10.2174/1570159x18666200729101913
Shi, S., Tang, M., Li, H., Ding, H., Lu, Y., Gao, L., et al. (2019). X-box binding protein l splicing attenuates brain microvascular endothelial cell damage induced by oxygen-glucose deprivation through the activation of phosphoinositide 3-kinase/protein kinase B, extracellular signal-regulated kinases, and hypoxia-inducible factor-1α/vascular endothelial growth factor signaling pathways. J. Cell. Physiol. 234, 9316–9327. doi: 10.1002/jcp.27614
Shuda, M., Kondoh, N., Imazeki, N., Tanaka, K., Okada, T., Mori, K., et al. (2003). Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 38, 605–614. doi: 10.1016/s0168-8278(03)00029-1
Song, D. D., Zhang, T. T., Chen, J. L., Xia, Y. F., Qin, Z. H., Waeber, C., et al. (2017). Sphingosine kinase 2 activates autophagy and protects neurons against ischemic injury through interaction with Bcl-2 via its putative BH3 domain. Cell Death Dis. 8:e2912. doi: 10.1038/cddis.2017.289
Stuppia, G., Rizzo, F., Riboldi, G., Del Bo, R., Nizzardo, M., Simone, C., et al. (2015). MFN2-related neuropathies: clinical features, molecular pathogenesis and therapeutic perspectives. J. Neurol. Sci. 356, 7–18. doi: 10.1016/j.jns.2015.05.033
Sun, Y., Cai, Y., Qian, S., Chiou, H., and Zang, Q. S. (2021). Beclin-1 improves mitochondria-associated membranes in the heart during endotoxemia. FASEB Bioadv. 3, 123–135. doi: 10.1096/fba.2020-00039
Sun, G. Y., and Hsu, C. Y. (1996). Poly-phosphoinositide-mediated messengers in focal cerebral ischemia and reperfusion. J. Lipid Mediat. Cell Signal. 14, 137–145. doi: 10.1016/0929-7855(96)00519-6
Sun, E., Zhang, J., Deng, Y., Wang, J., Wu, Q., Chen, W., et al. (2022). Docosahexaenoic acid alleviates brain damage by promoting mitophagy in mice with ischaemic stroke. Oxidative Med. Cell. Longev. 2022, 3119649–3119615. doi: 10.1155/2022/3119649
Sutendra, G., Dromparis, P., Wright, P., Bonnet, S., Haromy, A., Hao, Z., et al. (2011). The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci. Transl. Med. 3:88ra55. doi: 10.1126/scitranslmed.3002194
Szabó, Í., Varga, V., Dvorácskó, S., Farkas, A. E., Körmöczi, T., Berkecz, R., et al. (2021). N,N-dimethyltryptamine attenuates spreading depolarization and restrains neurodegeneration by sigma-1 receptor activation in the ischemic rat brain. Neuropharmacology 192:108612. doi: 10.1016/j.neuropharm.2021.108612
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D. F., Karbowski, M., et al. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380. doi: 10.1083/jcb.201007013
Tang, T., Hu, L. B., Ding, C., Zhang, Z., Wang, N., Wang, T., et al. (2023). Src inhibition rescues FUNDC1-mediated neuronal mitophagy in ischaemic stroke. Stroke Vasc. Neurol. 9, 367–379. doi: 10.1136/svn-2023-002606
Theurey, P., Tubbs, E., Vial, G., Jacquemetton, J., Bendridi, N., Chauvin, M. A., et al. (2016). Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell Biol. 8, 129–143. doi: 10.1093/jmcb/mjw004
Tian, W., Zhu, M., Zhou, Y., Mao, C., Zou, R., Cui, Y., et al. (2022). Electroacupuncture pretreatment alleviates cerebral ischemia-reperfusion injury by regulating Mitophagy via mTOR-ULK1/FUNDC1 Axis in rats. J. Stroke Cerebrovasc. Dis. 31:106202. doi: 10.1016/j.jstrokecerebrovasdis.2021.106202
Tubbs, E., Chanon, S., Robert, M., Bendridi, N., Bidaux, G., Chauvin, M. A., et al. (2018). Disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes 67, 636–650. doi: 10.2337/db17-0316
Tubbs, E., Theurey, P., Vial, G., Bendridi, N., Bravard, A., Chauvin, M. A., et al. (2014). Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63, 3279–3294. doi: 10.2337/db13-1751
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
Urban, P., Pavlíková, M., Sivonová, M., Kaplán, P., Tatarková, Z., Kaminska, B., et al. (2009). Molecular analysis of endoplasmic reticulum stress response after global forebrain ischemia/reperfusion in rats: effect of neuroprotectant simvastatin. Cell. Mol. Neurobiol. 29, 181–192. doi: 10.1007/s10571-008-9309-7
Urfer, R., Moebius, H. J., Skoloudik, D., Santamarina, E., Sato, W., Mita, S., et al. (2014). Phase II trial of the Sigma-1 receptor agonist cutamesine (SA4503) for recovery enhancement after acute ischemic stroke. Stroke 45, 3304–3310. doi: 10.1161/strokeaha.114.005835
Van Laar, V. S., Roy, N., Liu, A., Rajprohat, S., Arnold, B., Dukes, A. A., et al. (2015). Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and, in the presence of N-acetyl cysteine, mitophagy. Neurobiol. Dis. 74, 180–193. doi: 10.1016/j.nbd.2014.11.015
van Vliet, A. R., and Agostinis, P. (2018). Mitochondria-associated membranes and ER stress. Curr. Top. Microbiol. Immunol. 414, 73–102. doi: 10.1007/82_2017_2
Vance, J. E. (2014). MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim. Biophys. Acta 1841, 595–609. doi: 10.1016/j.bbalip.2013.11.014
Verfaillie, T., Rubio, N., Garg, A. D., Bultynck, G., Rizzuto, R., Decuypere, J. P., et al. (2012). PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 19, 1880–1891. doi: 10.1038/cdd.2012.74
Wang, H., Chen, S., Zhang, Y., Xu, H., and Sun, H. (2019). Electroacupuncture ameliorates neuronal injury by Pink1/Parkin-mediated mitophagy clearance in cerebral ischemia-reperfusion. Nitric Oxide 91, 23–34. doi: 10.1016/j.niox.2019.07.004
Wang, L., Cho, Y. L., Tang, Y., Wang, J., Park, J. E., Wu, Y., et al. (2018a). PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res. 28, 787–802. doi: 10.1038/s41422-018-0056-0
Wang, M., Geng, X., Dandu, C., Patel, R., and Ding, Y. (2021). Normobaric oxygen (NBO) therapy reduces cerebral ischemia/reperfusion injury through inhibition of early autophagy. Evid. Based Complement. Alternat. Med. 2021, 7041290–7041211. doi: 10.1155/2021/7041290
Wang, P., Liang, J., Li, Y., Li, J., Yang, X., Zhang, X., et al. (2014). Down-regulation of miRNA-30a alleviates cerebral ischemic injury through enhancing Beclin 1-mediated autophagy. Neurochem. Res. 39, 1279–1291. doi: 10.1007/s11064-014-1310-6
Wang, Y. H., Liao, J. M., Chen, K. M., Su, H. H., Liu, P. H., Chen, Y. H., et al. (2022). Lumbrokinase regulates endoplasmic reticulum stress to improve neurological deficits in ischemic stroke. Neuropharmacology 221:109277. doi: 10.1016/j.neuropharm.2022.109277
Wang, L., Liu, Y., Zhang, X., Ye, Y., Xiong, X., Zhang, S., et al. (2022a). Endoplasmic reticulum stress and the unfolded protein response in cerebral ischemia/reperfusion injury. Front. Cell. Neurosci. 16:864426. doi: 10.3389/fncel.2022.864426
Wang, L., Wang, P., Dong, H., Wang, S., Chu, H., Yan, W., et al. (2018b). Ulk1/FUNDC1 prevents nerve cells from hypoxia-induced apoptosis by promoting cell autophagy. Neurochem. Res. 43, 1539–1548. doi: 10.1007/s11064-018-2568-x
Wang, L., Zhang, X., Xiong, X., Zhu, H., Chen, R., Zhang, S., et al. (2022b). Nrf2 regulates oxidative stress and its role in cerebral ischemic stroke. Antioxidants (Basel) 11:2377. doi: 10.3390/antiox11122377
Wei, J., Wu, X., Luo, P., Yue, K., Yu, Y., Pu, J., et al. (2019). Homer1a attenuates endoplasmic reticulum stress-induced mitochondrial stress after ischemic reperfusion injury by inhibiting the PERK pathway. Front. Cell. Neurosci. 13:101. doi: 10.3389/fncel.2019.00101
Wen, H., Li, L., Zhan, L., Zuo, Y., Li, K., Qiu, M., et al. (2021). Hypoxic postconditioning promotes mitophagy against transient global cerebral ischemia via PINK1/Parkin-induced mitochondrial ubiquitination in adult rats. Cell Death Dis. 12:630. doi: 10.1038/s41419-021-03900-8
Wen, X., Tang, L., Zhong, R., Liu, L., Chen, L., and Zhang, H. (2023). Role of Mitophagy in regulating intestinal oxidative damage. Antioxidants (Basel) 12:480. doi: 10.3390/antiox12020480
Wu, W., Li, W., Chen, H., Jiang, L., Zhu, R., and Feng, D. (2016a). FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy 12, 1675–1676. doi: 10.1080/15548627.2016.1193656
Wu, X., Li, X., Liu, Y., Yuan, N., Li, C., Kang, Z., et al. (2018). Hydrogen exerts neuroprotective effects on OGD/R damaged neurons in rat hippocampal by protecting mitochondrial function via regulating mitophagy mediated by PINK1/Parkin signaling pathway. Brain Res. 1698, 89–98. doi: 10.1016/j.brainres.2018.06.028
Wu, W., Lin, C., Wu, K., Jiang, L., Wang, X., Li, W., et al. (2016b). FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 35, 1368–1384. doi: 10.15252/embj.201593102
Wu, M., Lu, G., Lao, Y. Z., Zhang, H., Zheng, D., Zheng, Z. Q., et al. (2021). Garciesculenxanthone B induces PINK1-Parkin-mediated mitophagy and prevents ischemia-reperfusion brain injury in mice. Acta Pharmacol. Sin. 42, 199–208. doi: 10.1038/s41401-020-0480-9
Wu, S., Lu, Q., Wang, Q., Ding, Y., Ma, Z., Mao, X., et al. (2017). Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation 136, 2248–2266. doi: 10.1161/circulationaha.117.030235
Wu, N. H., Ye, Y., Wan, B. B., Yu, Y. D., Liu, C., and Chen, Q. J. (2021). Emerging benefits: pathophysiological functions and target drugs of the Sigma-1 receptor in neurodegenerative diseases. Mol. Neurobiol. 58, 5649–5666. doi: 10.1007/s12035-021-02524-5
Wu, G., Zhang, X., Li, S., Wang, L., Bai, J., Wang, H., et al. (2022). Silencing ATF4 inhibits JMJD3-dependent JUNB/ETS1 axis and mitigates cerebral ischemic injury. J. Biochem. Mol. Toxicol. 36:e23070. doi: 10.1002/jbt.23070
Xie, Y., Jiang, D., Xiao, J., Fu, C., Zhang, Z., Ye, Z., et al. (2018). Ischemic preconditioning attenuates ischemia/reperfusion-induced kidney injury by activating autophagy via the SGK1 signaling pathway. Cell Death Dis. 9:338. doi: 10.1038/s41419-018-0358-7
Xing, S., Zhang, Y., Li, J., Zhang, J., Li, Y., Dang, C., et al. (2012). Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy 8, 63–76. doi: 10.4161/auto.8.1.18217
Xiong, Y., Cui, M. Y., Li, Z. L., Fu, Y. Q., Zheng, Y., Yu, Y., et al. (2024). ULK1 confers neuroprotection by regulating microglial/macrophages activation after ischemic stroke. Int. Immunopharmacol. 127:111379. doi: 10.1016/j.intimp.2023.111379
Xu, B. T., Li, M. F., Chen, K. C., Li, X., Cai, N. B., Xu, J. P., et al. (2023). Mitofusin-2 mediates cannabidiol-induced neuroprotection against cerebral ischemia in rats. Acta Pharmacol. Sin. 44, 499–512. doi: 10.1038/s41401-022-01004-3
Xu, Z. H., Liu, C. H., Hang, J. B., Gao, B. L., and Hu, J. A. (2017). Rituximab effectively reverses tyrosine kinase inhibitors (TKIs) resistance through inhibiting the accumulation of rictor on mitochondria-associated ER-membrane (MAM). Cancer Biomark. 20, 581–588. doi: 10.3233/cbm-170575
Yang, M., Li, C., Yang, S., Xiao, Y., Xiong, X., Chen, W., et al. (2020). Mitochondria-associated ER membranes - the origin site of autophagy. Front. Cell Dev. Biol. 8:595. doi: 10.3389/fcell.2020.00595
Yang, J. Y., and Yang, W. Y. (2013). Bit-by-bit autophagic removal of Parkin-labelled mitochondria. Nat. Commun. 4:2428. doi: 10.1038/ncomms3428
Yao, C., Zhang, J., Liu, G., Chen, F., and Lin, Y. (2014). Neuroprotection by (−)-epigallocatechin-3-gallate in a rat model of stroke is mediated through inhibition of endoplasmic reticulum stress. Mol. Med. Rep. 9, 69–72. doi: 10.3892/mmr.2013.1778
Yao, G. Y., Zhu, Q., Xia, J., Chen, F. J., Huang, M., Liu, J., et al. (2018). Ischemic postconditioning confers cerebroprotection by stabilizing VDACs after brain ischemia. Cell Death Dis. 9:1033. doi: 10.1038/s41419-018-1089-5
Yilmaz, U., Tanbek, K., Gul, S., Koc, A., Gul, M., and Sandal, S. (2024). Intracerebroventricular BDNF infusion may reduce cerebral ischemia/reperfusion injury by promoting autophagy and suppressing apoptosis. J. Cell. Mol. Med. 28:e18246. doi: 10.1111/jcmm.18246
Yuan, Y., Guo, Q., Ye, Z., Pingping, X., Wang, N., and Song, Z. (2011). Ischemic postconditioning protects brain from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress-induced apoptosis through PI3K-Akt pathway. Brain Res. 1367, 85–93. doi: 10.1016/j.brainres.2010.10.017
Yuan, Q., Ren, H., Lu, J., Yang, M., Xie, Z., Ma, B., et al. (2023). Effects of dichloromethane extraction from Piper nigrum L. and P. longum L. on the expression of autophagy-related proteins in ischemic stroke. J. Chem. Neuroanat. 127:102201. doi: 10.1016/j.jchemneu.2022.102201
Zeng, K. W., Wang, J. K., Wang, L. C., Guo, Q., Liu, T. T., Wang, F. J., et al. (2021). Small molecule induces mitochondrial fusion for neuroprotection via targeting CK2 without affecting its conventional kinase activity. Signal Transduct. Target. Ther. 6:71. doi: 10.1038/s41392-020-00447-6
Zhai, M., Liu, C., Li, Y., Zhang, P., Yu, Z., Zhu, H., et al. (2019). Dexmedetomidine inhibits neuronal apoptosis by inducing Sigma-1 receptor signaling in cerebral ischemia-reperfusion injury. Aging (Albany NY) 11, 9556–9568. doi: 10.18632/aging.102404
Zhang, B., Deng, F., Zhou, C., and Fang, S. (2020). ClC-3 induction protects against cerebral ischemia/reperfusion injury through promoting Beclin1/Vps34-mediated autophagy. Hum. Cell 33, 1046–1055. doi: 10.1007/s13577-020-00406-x
Zhang, Y., He, Y., Wu, M., Chen, H., Zhang, L., Yang, D., et al. (2020). Rehmapicroside ameliorates cerebral ischemia-reperfusion injury via attenuating peroxynitrite-mediated mitophagy activation. Free Radic. Biol. Med. 160, 526–539. doi: 10.1016/j.freeradbiomed.2020.06.034
Zhang, G., Li, Q., Tao, W., Qin, P., Chen, J., Yang, H., et al. (2023). Sigma-1 receptor-regulated efferocytosis by infiltrating circulating macrophages/microglial cells protects against neuronal impairments and promotes functional recovery in cerebral ischemic stroke. Theranostics 13, 543–559. doi: 10.7150/thno.77088
Zhang, X., Sun, Z., Ding, C., Tang, Y., Jiang, X., Xie, Y., et al. (2016). Metabolic syndrome augments the risk of early neurological deterioration in acute ischemic stroke patients independent of inflammatory mediators: a hospital-based prospective study. Oxidative Med. Cell. Longev. 2016:8346301. doi: 10.1155/2016/8346301
Zhang, Y. M., Xu, H., Chen, S. H., and Sun, H. (2021). Electroacupuncture regulates endoplasmic reticulum stress and ameliorates neuronal injury in rats with acute ischemic stroke. Evid. Based Complement. Alternat. Med. 2021, 9912325–9912311. doi: 10.1155/2021/9912325
Zhang, Z., and Yu, J. (2018). NR4A1 promotes cerebral ischemia reperfusion injury by repressing Mfn2-mediated mitophagy and inactivating the MAPK-ERK-CREB signaling pathway. Neurochem. Res. 43, 1963–1977. doi: 10.1007/s11064-018-2618-4
Zheng, Y. Q., Liu, J. X., Li, X. Z., Xu, L., and Xu, Y. G. (2009). RNA interference-mediated downregulation of Beclin1 attenuates cerebral ischemic injury in rats. Acta Pharmacol. Sin. 30, 919–927. doi: 10.1038/aps.2009.79
Zhu, R., Liu, L., Mao, T., Wang, X., Li, Y., Li, T., et al. (2024). Mfn2 regulates mitochondria and mitochondria-associated endoplasmic reticulum membrane function in neurodegeneration induced by repeated sevoflurane exposure. Exp. Neurol. 377:114807. doi: 10.1016/j.expneurol.2024.114807
Keywords: mitochondrial-associated endoplasmic reticulum membrane (MAM), mitophagy, endoplasmic reticulum stress, ischemic stroke (IS), unfolded protein response (UPR)
Citation: Jia Z, Li H, Xu K, Li R, Yang S, Chen L, Zhang Q, Li S and Sun X (2024) MAM-mediated mitophagy and endoplasmic reticulum stress: the hidden regulators of ischemic stroke. Front. Cell. Neurosci. 18:1470144. doi: 10.3389/fncel.2024.1470144
Edited by:
Gunnar P.H. Dietz, University Medical Center Göttingen, GermanyReviewed by:
Ganapathi Kandasamy, University of Miami, United StatesYanet Karina Gutierrez-Mercado, University of Guadalajara, Mexico
Copyright © 2024 Jia, Li, Xu, Li, Yang, Chen, Zhang, Li and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaowei Sun, Z2VtaW5pMTk3OTA1MzBAMTYzLmNvbQ==; Shulin Li, bGlzbDAwMjJAMTYzLmNvbQ==
†These authors share first authorship