 Claire Ward
Claire Ward Lucas Sjulson
Lucas Sjulson Renata Batista-Brito
Renata Batista-Brito- 1Dominick P. Purpura Department of Neuroscience, Albert Einstein College of Medicine, Bronx, NY, United States
- 2Department of Psychiatry and Behavioral Sciences, Albert Einstein College of Medicine, Bronx, NY, United States
- 3Department of Genetics, Albert Einstein College of Medicine, Bronx, NY, United States
Neurodevelopmental disorders (NDDs) are caused by abnormal brain development, leading to altered brain function and affecting cognition, learning, self-control, memory, and emotion. NDDs are often demarcated as discrete entities for diagnosis, but empirical evidence indicates that NDDs share a great deal of overlap, including genetics, core symptoms, and biomarkers. Many NDDs also share a primary sensitive period for disease, specifically the last trimester of pregnancy in humans, which corresponds to the neonatal period in mice. This period is notable for cortical circuit assembly, suggesting that deficits in the establishment of brain connectivity are likely a leading cause of brain dysfunction across different NDDs. Regulators of gene programs that underlie neurodevelopment represent a point of convergence for NDDs. Here, we review how the transcription factor MEF2C, a risk factor for various NDDs, impacts cortical development. Cortical activity requires a precise balance of various types of excitatory and inhibitory neuron types. We use MEF2C loss-of-function as a study case to illustrate how brain dysfunction and altered behavior may derive from the dysfunction of specific cortical circuits at specific developmental times.
Introduction
Neurodevelopmental disorders (NDDs), such as autism spectrum disorder (ASD), intellectual disability, and schizophrenia, represent a leading cause of neuropsychiatric illness, affecting one in eight children in the US (Bitta et al., 2017). We remain far from understanding the complex biology underlying NDDs; however, recent human genetic studies have consistently shown large degrees of genetic overlap between distinct NDDs (International Schizophrenia Consortium et al., 2009; Williams et al., 2011; Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013; Insel and Landis, 2013; Ruderfer et al., 2014; Cross-Disorder Group of the Psychiatric Genomics Consortium, 2019). Recent work shows that subtle genetic changes, especially during critical developmental periods, can cause diverse impairments in brain activity and behavior. NDD risk-associated genes are highly expressed during neonatal development, a sensitive period for NDDs notable for cortical circuit assembly and synaptic maturation (LeBlanc and Fagiolini, 2011; Willsey et al., 2013; Meredith, 2015; Marin, 2016; Satterstrom et al., 2020). These findings have introduced the possibility that changes to a small set of common signaling pathways might result in various NDD phenotypes. The identification of these common signaling pathways is a fundamental aim of NDD research. Consistent with this idea, NDDs often present with a shared set of core symptoms and feature common biomarkers, such as changes in brain synchrony and cortical oscillations (Bartos et al., 2007; Milh et al., 2007; Lakatos et al., 2008; Allene and Cossart, 2010; Tan et al., 2013; Fryer et al., 2016; Hwang et al., 2016; Seibt et al., 2016).
Using bioinformatics and ever-growing genetic data, many known NDD-associated genes have been classified according to cellular and circuit functions. Numerous cellular processes are impaired across NDDs, including transcriptional and epigenetic regulation, neuronal proliferation, neuronal migration, neuronal survival, connectivity of both excitatory and inhibitory circuits (Rossignol, 2011; Meredith, 2015; Marin, 2016; Krol and Feng, 2018), and synaptic transmission (Chattopadhyaya and Cristo, 2012; Clement et al., 2012; Meredith et al., 2012; Estes et al., 2015; Meredith, 2015; Canitano and Pallagrosi, 2017; Krol and Feng, 2018; Parenti et al., 2020). Transcription factors, a group of proteins that interact with DNA and regulate gene expression through modulation of RNA synthesis, represent a major point of convergence for neurodevelopmental and neuropsychiatric diseases (Santos-Terra et al., 2021). One of such transcription factors is the myocyte enhancement factor 2c protein (MEF2C), which is encoded by the MEF2C gene in humans and the orthologous Mef2c gene in mice. MEF2C has been widely associated with various NDDs such as autism spectrum disorder (ASD), schizophrenia, bipolar disorder (Harrington et al., 2016; Rajkovich et al., 2017; Tu et al., 2017; Assali et al., 2019). In this review we will treat Mef2c as a test case for how changes in one gene can impact excitatory and inhibitory cortical circuits, and will focus on how dysfunction of Mef2c in different cortical circuits relates to pathological pattens of activity and altered behavior.
Mef2c and neurodevelopmental disorders
Human genome-wide association studies (GWAS) reveal that MEF2C is a common genetic risk factor for various neurodevelopmental and neuropsychiatric disorders (Gandal et al., 2018; Harrington et al., 2020; Fahey et al., 2023). Microdeletions or coding-region missense or nonsense mutations in MEF2C during development can lead to MEF2C haploinsufficiency syndrome, which often features intellectual disability, epilepsy, and autism spectrum disorder (Borlot et al., 2019; Chaudhary et al., 2021; Cooley Coleman et al., 2021; Cooley Coleman et al., 2022), and loss of Mef2c in rodent models leads to profound changes in behaviors associated with NDDs (Barbosa et al., 2008; Li H. et al., 2008; Adachi et al., 2016; Harrington et al., 2016; Deczkowska et al., 2017; Tu et al., 2017; Bjorness et al., 2020; Harrington et al., 2020; Li et al., 2023; Cho et al., 2024; Ward et al., 2024) (Figure 1A).
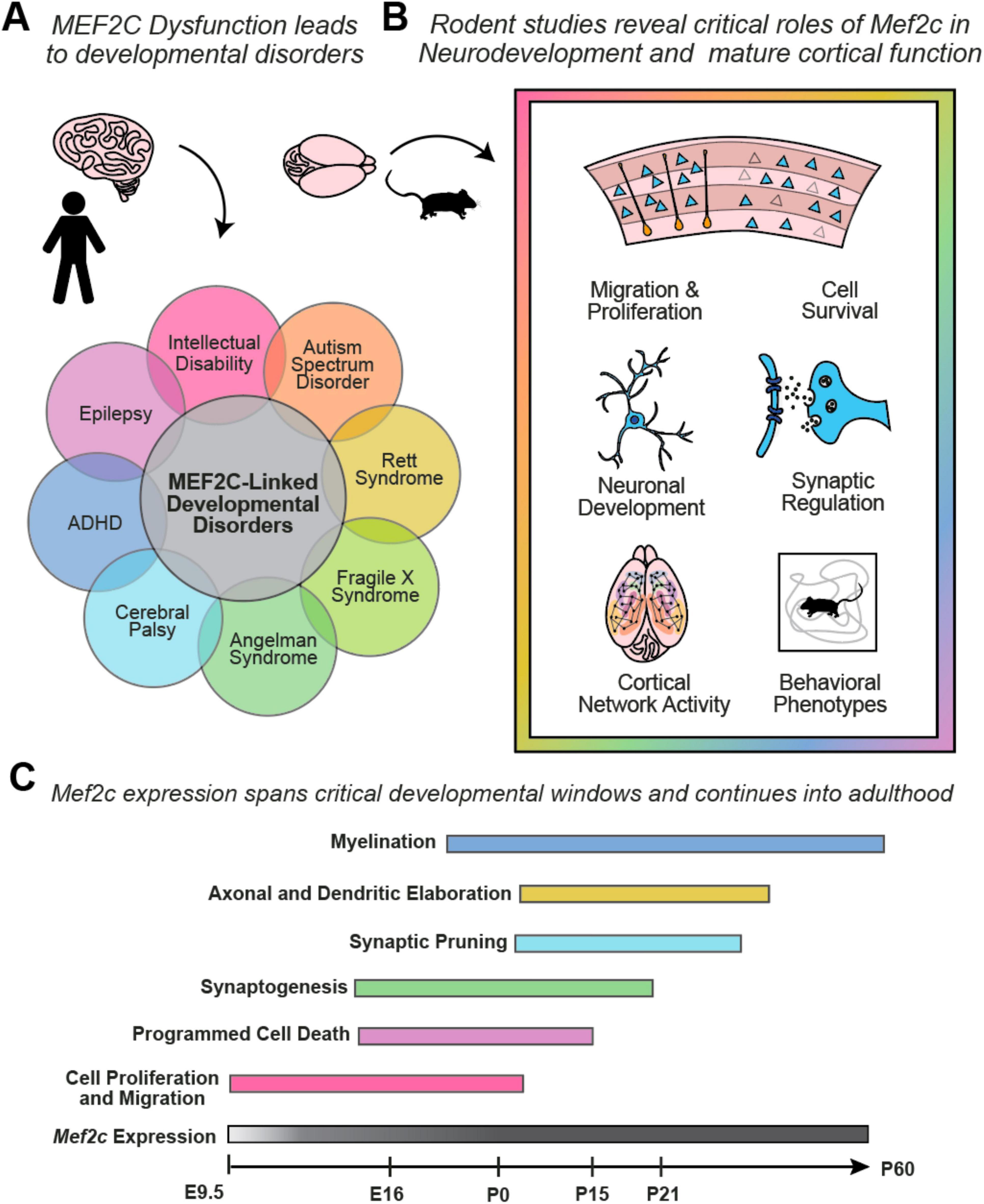
Figure 1. (A) Dysfunction of the MEF2C gene in humans can give rise to many neurodevelopmental disorders, which often are characterized by overlapping behavioral phenotypes. (B) Transgenic mouse studies of the Mef2c gene have identified MEF2C as a transcription factor that regulates gene programs that are critical to many cellular processes. Loss of Mef2c expression can give rise to impaired neuronal activity across cell populations and leads to behavioral phenotypes typical of mouse models of neurodevelopmental disorders, such as hyperactivity in an open field arena. (C) Mef2c is expressed from early embryonic development through adulthood, with roles in shaping gene expression during critical neurodevelopmental windows and regulating synapses in mature cortical circuits.
The symptom severity associated with MEF2C loss-of-function, along with the number of NDDs associated with it, underscore its necessity in regulating genetic programs during neurodevelopment. Gene ontology studies in mice revealed that Mef2c regulates processes such as neurogenesis, neuronal differentiation and morphogenesis, cell survival, and synapse development (Harrington et al., 2016; Tu et al., 2017; Harrington et al., 2020; Allaway et al., 2021). Indeed, Mef2c knockdown mice exhibit many cellular deficits, such as reduced neurogenesis, increased cell death, and increased excitatory to inhibitory (E/I) neurotransmission (Tu et al., 2017) (Figure 1B). In addition to the many cellular phenotypes, Mef2c knockdown mice exhibit behavioral phenotypes common to mouse models of NDDs, such as hyperactivity and deficits in social interactions (Tu et al., 2017; Harrington et al., 2020; Li et al., 2023), enabling us to investigate the circuits involved in Mef2c-related pathophysiology and behavior.
Mef2c regulates the cellular and synaptic development of excitatory and inhibitory neurons
Mef2c is highly expressed within the cerebral cortex throughout the life of an animal, including during embryonic development. Mef2c is broadly expressed in pyramidal cells, the principal excitatory cells of the cortex, and subsets of inhibitory interneurons (Assali et al., 2019). Within GABAergic cortical interneurons Mef2c is expressed in virtually all parvalbumin-expressing INs (PV-INs) and a subset of somatostatin expressing INs (SST-INs) (Mayer et al., 2018; Ward et al., 2024). Using conditional genetics, and with the increasing availability of mouse Cre-driver lines that can be used to target specific cell types across various stages of development, we have gained a granularity that allows for better understanding of how Mef2c-mediated gene regulation is critical for the development of excitatory and inhibitory circuits, as detailed in Table 1.
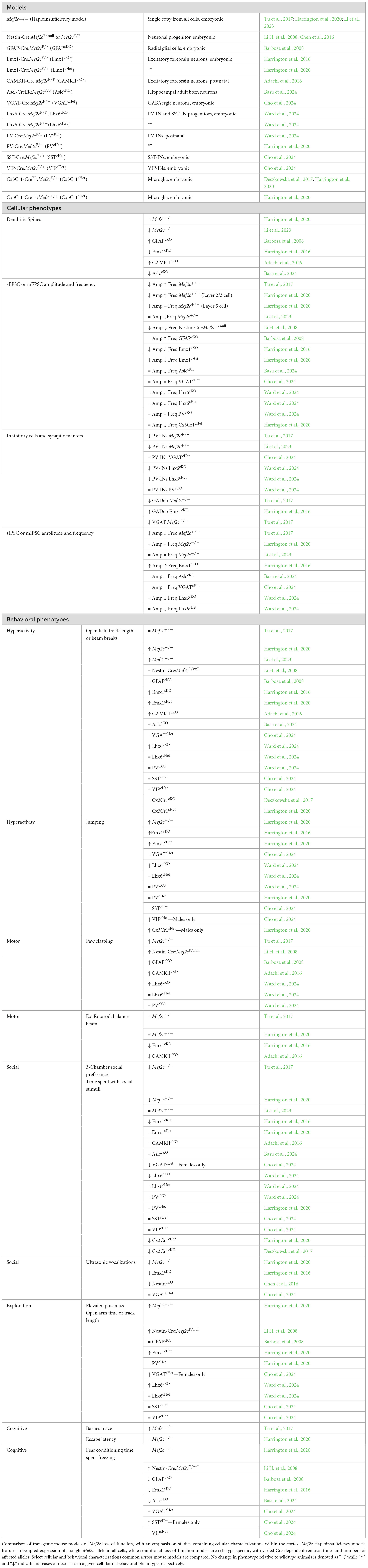
Table 1. Cellular and behavioral phenotypes in Mef2c mouse models.
Neurogenesis, neuronal differentiation, and survival
The MEF2C transcription factor is critical during development and regulates neurogenesis, neuronal differentiation, and survival (Mao et al., 1999; Li H. et al., 2008; Li Z. et al., 2008) (Figure 1C). Removal of the Mef2c gene in Nestin-expressing neural stem/progenitor cells impairs neuronal proliferation and results in disrupted layer formation at embryonic day 18.5 (E18.5) and disorganized cortical plate at postnatal day 7 (P7) (Li H. et al., 2008). Deficits in neuronal migration may be a common feature in ASDs, and despite the distinct progenitor pools and migratory streams of excitatory and inhibitory neurons, deficits in Mef2c expression have been shown to affect both cell types (Li H. et al., 2008; Reiner et al., 2016). This effect appears to be dependent on factors such as cell type, dose, and developmental stage, as we highlight across many molecular and behavioral phenotypes in this review. Embryonic removal of Mef2c from excitatory neurons alone does not lead to gross changes in cortical structure, while overexpression of Mef2c in adult born neurons leads to mislocalization in the hippocampus (Harrington et al., 2016; Basu et al., 2024). MEF2C continues to be expressed in postmitotic pyramidal cells, where it promotes their survival by activating anti-apoptotic genes and inhibiting pro-apoptotic pathways, thus highlighting its protective role in the nervous system (Li Z. et al., 2008; Tu et al., 2017). MEF2C promotes differentiation by regulating the expression of genes involved in neuronal differentiation and maturation, such as Bdnf (Lyons et al., 2012; Harrington et al., 2016; Tu et al., 2017; Avarlaid et al., 2024), and has been shown to regulate excitatory neurons’ dendritic arborization, and axonal guidance (Ma and Telese, 2015; Harrington et al., 2016; Tu et al., 2017; Basu et al., 2024). Embryonic overexpression of Mef2c increases basal dendritic arborization (Jung et al., 2016), while reduced Mef2c results in decreased arborization (Tu et al., 2017). MEF2C has been shown to positively modulate the expression of MECP2 (Zweier et al., 2010; Paciorkowski et al., 2013), a transcriptional regulator that facilitates expression of the KCC2 ion channel that is critical for developmental transition from the excitatory to inhibitory of action GABA (Tang et al., 2016; Oyarzabal et al., 2020), and it is possible that this aspect of cellular development could be affected by Mef2c loss of function as well. In addition, depletion of MEF2C leads to the defective activation of the mTOR pathway (Pereira et al., 2009) and mTOR positively regulates MEF2C (Wu et al., 2015). Furthermore, members of the MEF2C interactome are strongly linked to mTOR pathway activity, such as MECP2 (Ricciardi et al., 2011; Tsujimura et al., 2015; Rangasamy et al., 2016; Olson et al., 2018; Zhang et al., 2018) and FMR1 (Sharma et al., 2010; Casingal et al., 2020; Zhang et al., 2024). These serve as examples of the many gene regulatory networks implicated in neurodevelopmental disorders that are upstream or downstream of Mef2c (D’Haene et al., 2019; Zhang and Zhao, 2022). Mef2c is also expressed in cortical interneurons originating in the medial ganglionic eminence (MGE) (Mayer et al., 2018; Ward et al., 2024). Mef2c is expressed in cortical parvalbumin interneuron (PV-IN) precursors during embryonic development, where it functions as the earliest indicator of PV-IN fate and is necessary for the survival and molecular maturation of the PV-IN lineage (Mayer et al., 2018). Mef2c continues to be expressed in PV-INs throughout the animal’s life, but postnatal removal of Mef2c using the PVCre mouse line does not impact PV-IN survival or differentiation (Ward et al., 2024). Mef2c is also expressed by roughly 30% of SST-INs, however, its loss does not impact SST-IN survival or differentiation (Ward et al., 2024).
Synapse development and function
Mef2c regulates synaptic transmission and plays a critical role in synapse development and plasticity of cortical pyramidal cells (Barbosa et al., 2008; Li H. et al., 2008; Adachi et al., 2016; Harrington et al., 2016; Rajkovich et al., 2017; Tu et al., 2017; Harrington et al., 2020; Basu et al., 2024). Mef2c influences the expression of synaptic proteins, such as Arc and SynGAP, which are involved in synaptic strength and plasticity (Flavell et al., 2006; Harrington et al., 2016; Tu et al., 2017; Puang et al., 2020); however, the role of Mef2c in synaptic development is complex. Selective loss of Mef2c in cortical pyramidal cells can result in either an increased (Barbosa et al., 2008; Adachi et al., 2016) or decreased (Harrington et al., 2016; Tu et al., 2017; Li et al., 2023) density of dendritic spines, with various changes in electrophysiological synaptic properties (Barbosa et al., 2008; Li Z. et al., 2008; Adachi et al., 2016; Harrington et al., 2016; Tu et al., 2017; Harrington et al., 2020; Ward et al., 2024). These seemingly opposing roles underscore the nuanced role of Mef2c in synapse maturation and/or maintenance, which is input- (local versus long-range) and activity-dependent (Rajkovich et al., 2017). Mef2c strengthens long-range inputs from the contralateral cortex and weakens local inputs (Rajkovich et al., 2017). Interestingly, Mef2c only results in weakened synaptic strength in the presence of sensory experience, highlighting the role of activity in Mef2c-dependent function. In addition to the role of sensory experience on Mef2c function, Mef2c activity is also dependent on sleep (Bjorness et al., 2020). Sleep deprivation leads to Mef2c-dependent upregulation of synapse-weakening genes and a reduction in synapse-strengthening genes, as well as changes in glutamatergic synaptic transmission and sleep-dependent synaptic remodeling (Bjorness et al., 2020). Mef2c is also necessary for the synaptic maturation of PV-INs (Ward et al., 2024). Embryonic loss of Mef2c in MGE INs (Lhx6-Cre:Mef2cF/F) causes a massive reduction in glutamatergic synaptic transmission onto INs, but postnatal loss of Mef2c by using the line PVCre does not impact synaptic transmission onto PV-INs (Ward et al., 2024), thus highlighting the critical role of Mef2c during development.
E/I imbalance
Imbalances in excitatory and inhibitory synaptic transmission, particularly increased E/I balance, have been proposed to cause various NDDs, including ASD and schizophrenia (Rubenstein and Merzenich, 2003; Gao and Penzes, 2015; Sohal and Rubenstein, 2019; Liu et al., 2021). The E/I imbalance hypothesis was originally based on the observation that patients with NDDs have increased rates of epilepsy, which is true for MEF2C haploinsufficiency patients (Cooley Coleman et al., 2021; Wan et al., 2021; Cooley Coleman et al., 2022). However, GABAA agonists or positive allosteric modulators, which reduce E/I balance, do not alleviate the core symptoms of NDDs (Lingjaerde, 1991), and conditions that increase E/I balance, such as withdrawal from alcohol or sedative/hypnotics (Hendricson et al., 2007), do not cause syndromes resembling NDDs, calling this hypothesis into question. Increasingly, the field is moving away from the overly simplistic one-dimensional E/I imbalance hypothesis to more sophisticated models of cortical circuit dysfunction (O’Donnell et al., 2017; Sohal and Rubenstein, 2019) that incorporate multiple distinct excitatory and inhibitory cell types. The complex cell-type-dependent and temporally-specific effects of MEF2C haploinsufficiency provide a compelling illustration of why this more nuanced approach is necessary.
Impact of Mef2c on behavior
Despite their complex etiologies, NDDs feature overlapping behavioral traits. Most Mef2c loss-of-function mouse models include assays for behavioral domains affected in various NDDs (Table 1). While there is no set battery of tests conducted across NDD studies, there are several phenotypes that stand out across studies. Loss or reduction of Mef2c in both pyramidal cells and MGE interneurons leads to hyperactivity (Adachi et al., 2016; Harrington et al., 2016; Harrington et al., 2020; Li et al., 2023; Cho et al., 2024; Ward et al., 2024), paw clasping (Barbosa et al., 2008; Li H. et al., 2008; Adachi et al., 2016; Tu et al., 2017; Ward et al., 2024), and social deficits (Harrington et al., 2016; Deczkowska et al., 2017; Tu et al., 2017; Harrington et al., 2020; Li et al., 2023; Basu et al., 2024; Cho et al., 2024; Ward et al., 2024). Mef2c haploinsufficiency mice (Mef2c+/–) exhibited hyperactivity and social deficit phenotypes; however, not all assays were in agreement (Tu et al., 2017; Harrington et al., 2020; Li et al., 2023). One study reported that Mef2c+/– animals exhibit impaired spatial memory in the Barnes maze and Morris water maze (Tu et al., 2017), however, a separate study of Mef2c+/– animals revealed deficits in a battery of cognitive tests including fear conditioning, Barnes maze, y-maze, and an operant reward learning test (Harrington et al., 2020), thus illustrating the challenge comparing behavioral domains across studies.
An advantage of mouse models and the advent of conditional genetics is the ability to investigate how specific brain circuits contribute to pathological neuronal activity and altered behavior (see behavior phenotypes resulting from reduced Mef2c in various cell types in Table 1). For instance, Harrington and colleagues observed that both Mef2c+/– animals and animals with embryonic loss of Mef2c specifically in pyramidal cells (Emx1-Cre:Mef2cF /F animals) exhibit a significant reduction in the frequency of ultrasonic vocalizations in pups, with adult animals emitting fewer and less complex vocalizations (Harrington et al., 2016; Harrington et al., 2020). On the other hand, Emx1-Cre:Mef2cF /F animals exhibited cognitive deficits in contextual memory fear conditioning while none were observed in Mef2c+/– animals when tested on the same paradigm (Harrington et al., 2016; Harrington et al., 2020). Another important factor in considering how the loss of a gene impacts behavior is the age at which the loss occurred. For instance, early (embryonic) but not late (postnatal) removal of Mef2c from MGE interneurons leads to hyperactivity (Ward et al., 2024). Furthermore, some studies have found that some behavioral phenotypes are sex specific (Harrington et al., 2020; Cho et al., 2024). There is evidence to suggest that anxiety-like behavior correlates with Mef2c expression in a way that is influenced by sex and estrous cycle phase (Jaric et al., 2019; Hong et al., 2024), further underscoring the importance of considering sex as a biological variable. These findings provide an example of how relating gene dysfunction to behavior is extremely challenging; however, investment in relating changes in brain activity with target behaviors holds the promise of revealing the circuits underlying neurodevelopmental behaviors in NDDs.
Discussion
Brain development is a pivotal and meticulously orchestrated process, governed by numerous neurobiological pathways that lay the foundation for essential brain functions. It is likely that various NDDs share pathways and circuits that become altered during prenatal or early postnatal stages. A main challenge in NDD treatment is the fact that diagnosis often occurs after the most effective time for treatment. Early identification of biomarkers for NDDs is critical given its potential for early intervention. Another big challenge is that despite significant advancements in understanding the pathophysiology of NDDs, targeted and effective treatments are still rare. Identification of common NDD molecular pathways and circuit dysfunction could potentially serve as an early prognostic indicator for NDDs and allow for targeted therapies.
The convergence of genetic factors and the existence of critical periods of vulnerability for NDDs underscores the potential for drugs to target fundamental networks as soon as possible in order to prevent or mitigate clinical manifestations of NDDs. However, more work needs to be done to establish a causal link between molecular and circuit abnormalities, disease pathology, and abnormal behavior. Animal models offer the opportunity to comprehensively unravel the molecular, circuit, and temporal intricacies of NDDs, pinpointing potential therapeutic targets, and ultimately informing us about new treatment modalities. In particular, the mouse as a disease model can provide valuable tools in deciphering the intricate genetic landscape of NDDs as well as elucidating the molecular, cellular, and circuit impacts of diverse mutations toward brain development and disease physiology. Interestingly, pharmacological and gene therapy approaches have both been applied in Mef2c+/– models, rescuing both cellular and behavioral phenotypes (Tu et al., 2017; Li et al., 2023). Nonetheless, for animal models to be useful it is important to consider the fundamental differences between animal models and humans, especially during development.
Single-cell omics techniques, alongside non-invasive brain activity measures like EEGs, computational models, and bioinformatics network analysis, offer a pathway to delineate parallels between animal models and humans. These methodologies have the potential to unravel the intricate relationship between gene expression alterations during human brain neuronal development and the manifestation of behaviors associated with NDDs. This understanding holds promise for developing tailored therapeutic interventions, catalyzing a transition from the prevailing symptom-based approach toward more proactive, targeted, and effective treatments.
Author contributions
CW: Conceptualization, Writing – original draft, Writing – review & editing. LS: Writing – original draft, Writing – review & editing. RB-B: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by a Ruth L. Kirschstein Predoctoral Fellowship F31HD101360 to CW; a NARSAD Young Investigator Award, a Simons Bridge to Independence Award, a Whitehall Award, and National Institutes of Health (NIH) grants R01EY034617, R21MH133097, and R01EY034310 to RB-B; DA051608 to LS.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adachi, M., Lin, P. Y., Pranav, H., and Monteggia, L. M. (2016). Postnatal loss of Mef2c results in dissociation of effects on synapse number and learning and memory. Biol. Psychiatry 80, 140–148. doi: 10.1016/j.biopsych.2015.09.018
Allaway, K. C., Gabitto, M. I., Wapinski, O., Saldi, G., Wang, C. Y., Bandler, R. C., et al. (2021). Genetic and epigenetic coordination of cortical interneuron development. Nature 597, 693–697. doi: 10.1038/s41586-021-03933-1
Allene, C., and Cossart, R. (2010). Early NMDA receptor-driven waves of activity in the developing neocortex: Physiological or pathological network oscillations? J. Physiol. 588, 83–91. doi: 10.1113/jphysiol.2009.178798
Assali, A., Harrington, A. J., and Cowan, C. W. (2019). Emerging roles for MEF2 in brain development and mental disorders. Curr. Opin. Neurobiol. 59, 49–58. doi: 10.1016/j.conb.2019.04.008
Avarlaid, A., Falkenberg, K., Lehe, K., Mudò, G., Belluardo, N., Di Liberto, V., et al. (2024). An upstream enhancer and MEF2 transcription factors fine-tune the regulation of the Bdnf gene in cortical and hippocampal neurons. J. Biol. Chem. 300:107411. doi: 10.1016/j.jbc.2024.107411
Barbosa, A. C., Kim, M. S., Ertunc, M., Adachi, M., Nelson, E. D., McAnally, J., et al. (2008). MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc. Natl. Acad. Sci. U.S.A. 105, 9391–9396. doi: 10.1073/pnas.0802679105
Bartos, M., Vida, I., and Jonas, P. (2007). Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. Rev. Neurosci. 8, 45–56. doi: 10.1038/nrn2044
Basu, S., Ro, E. J., Liu, Z., Kim, H., Bennett, A., Kang, S., et al. (2024). The Mef2c gene dose-dependently controls hippocampal neurogenesis and the expression of autism-like behaviors. J. Neurosci. 44:1058232023. doi: 10.1523/jneurosci.1058-23.2023
Bitta, M., Kariuki, S. M., Abubakar, A., and Newton, C. (2017). Burden of neurodevelopmental disorders in low and middle-income countries: A systematic review and meta-analysis. Wellcome Open Res. 2:121. doi: 10.12688/wellcomeopenres.13540.3
Bjorness, T. E., Kulkarni, A., Rybalchenko, V., Suzuki, A., Bridges, C., Harrington, A. J., et al. (2020). An essential role for MEF2C in the cortical response to loss of sleep in mice. Elife 9:e58331. doi: 10.7554/eLife.58331
Borlot, F., Whitney, R., Cohn, R. D., and Weiss, S. K. (2019). MEF2C-related epilepsy: Delineating the phenotypic spectrum from a novel mutation and literature review. Seizure 67, 86–90. doi: 10.1016/j.seizure.2019.03.015
Canitano, R., and Pallagrosi, M. (2017). Autism spectrum disorders and schizophrenia spectrum disorders: Excitation/inhibition imbalance and developmental trajectories. Front. Psychiatry 8:69. doi: 10.3389/fpsyt.2017.00069
Casingal, C. R., Kikkawa, T., Inada, H., Sasaki, Y., and Osumi, N. (2020). Identification of FMRP target mRNAs in the developmental brain: FMRP might coordinate Ras/MAPK, Wnt/β-catenin, and mTOR signaling during corticogenesis. Mol. Brain 13:167. doi: 10.1186/s13041-020-00706-1
Chattopadhyaya, B., and Cristo, G. D. (2012). GABAergic circuit dysfunctions in neurodevelopmental disorders. Front. Psychiatry 3:51. doi: 10.3389/fpsyt.2012.00051
Chaudhary, R., Agarwal, V., Kaushik, A. S., and Rehman, M. (2021). Involvement of myocyte enhancer factor 2c in the pathogenesis of autism spectrum disorder. Heliyon 7:e06854. doi: 10.1016/j.heliyon.2021.e06854
Chen, Y. C., Kuo, H. Y., Bornschein, U., Takahashi, H., Chen, S. Y., Lu, K. M., et al. (2016). Foxp2 controls synaptic wiring of corticostriatal circuits and vocal communication by opposing Mef2c. Nat. Neurosci. 19, 1513–1522. doi: 10.1038/nn.4380
Cho, J. Y., Rumschlag, J. A., Tsvetkov, E., Proper, D. S., Lang, H., Berto, S., et al. (2024). MEF2C hypofunction in GABAergic cells alters sociability and prefrontal cortex inhibitory synaptic transmission in a sex-dependent manner. Biol. Psychiatry Glob. Open Sci. 4:100289. doi: 10.1016/j.bpsgos.2024.100289
Clement, J. P., Aceti, M., Creson, T. K., Ozkan, E. D., Shi, Y., Reish, N. J., et al. (2012). Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151, 709–723. doi: 10.1016/j.cell.2012.08.045
Cooley Coleman, J. A., Sarasua, S. M., Boccuto, L., Moore, H. W., Skinner, S. A., and DeLuca, J. M. (2021). Comprehensive investigation of the phenotype of MEF2C-related disorders in human patients: A systematic review. Am. J. Med. Genet. A 185, 3884–3894. doi: 10.1002/ajmg.a.62412
Cooley Coleman, J. A., Sarasua, S. M., Moore, H. W., Boccuto, L., Cowan, C. W., Skinner, S. A., et al. (2022). Clinical findings from the landmark MEF2C-related disorders natural history study. Mol. Genet. Genom. Med. 10:e1919. doi: 10.1002/mgg3.1919
Cross-Disorder Group of the Psychiatric Genomics Consortium (2013). Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet 381, 1371–1379. doi: 10.1016/S0140-6736(12)62129-1
Cross-Disorder Group of the Psychiatric Genomics Consortium (2019). Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 179:1469–1482.e1411. doi: 10.1016/j.cell.2019.11.020
D’Haene, E., Bar-Yaacov, R., Bariah, I., Vantomme, L., Van Loo, S., Cobos, F. A., et al. (2019). A neuronal enhancer network upstream of MEF2C is compromised in patients with Rett-like characteristics. Hum. Mol. Genet. 28, 818–827. doi: 10.1093/hmg/ddy393
Deczkowska, A., Matcovitch-Natan, O., Tsitsou-Kampeli, A., Ben-Hamo, S., Dvir-Szternfeld, R., Spinrad, A., et al. (2017). Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat. Commun. 8:717. doi: 10.1038/s41467-017-00769-0
Estes, A., Zwaigenbaum, L., Gu, H., St John, T., Paterson, S., Elison, J. T., et al. (2015). Behavioral, cognitive, and adaptive development in infants with autism spectrum disorder in the first 2 years of life. J. Neurodev. Disord. 7: 24. doi: 10.1186/s11689-015-9117-6
Fahey, L., Ali, D., Donohoe, G., and Morris, D. W. (2023). Genes positively regulated by Mef2c in cortical neurons are enriched for common genetic variation associated with IQ and educational attainment. Hum. Mol. Genet. 32, 3194–3203. doi: 10.1093/hmg/ddad142
Flavell, S. W., Cowan, C. W., Kim, T. K., Greer, P. L., Lin, Y., Paradis, S., et al. (2006). Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311, 1008–1012. doi: 10.1126/science.1122511
Fryer, S. L., Roach, B. J., Wiley, K., Loewy, R. L., Ford, J. M., and Mathalon, D. H. (2016). Reduced amplitude of low-frequency brain oscillations in the psychosis risk syndrome and early illness schizophrenia. Neuropsychopharmacology 41, 2388–2398. doi: 10.1038/npp.2016.51
Gandal, M. J., Zhang, P., Hadjimichael, E., Walker, R. L., Chen, C., Liu, S., et al. (2018). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362:aat8127. doi: 10.1126/science.aat8127
Gao, R., and Penzes, P. (2015). Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 15, 146–167. doi: 10.2174/1566524015666150303003028
Harrington, A. J., Bridges, C. M., Berto, S., Blankenship, K., Cho, J. Y., Assali, A., et al. (2020). MEF2C hypofunction in neuronal and neuroimmune populations produces MEF2C haploinsufficiency syndrome-like behaviors in mice. Biol. Psychiatry 88, 488–499. doi: 10.1016/j.biopsych.2020.03.011
Harrington, A. J., Raissi, A., Rajkovich, K., Berto, S., Kumar, J., Molinaro, G., et al. (2016). MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. Elife 5:e20059. doi: 10.7554/eLife.20059
Hendricson, A. W., Maldve, R. E., Salinas, A. G., Theile, J. W., Zhang, T. A., Diaz, L. M., et al. (2007). Aberrant synaptic activation of N-methyl-D-aspartate receptors underlies ethanol withdrawal hyperexcitability. J. Pharmacol. Exp. Ther. 321, 60–72. doi: 10.1124/jpet.106.111419
Hong, Y., Hu, J., Zhang, S., Liu, J., Yan, F., Yang, H., et al. (2024). Integrative analysis identifies region- and sex-specific gene networks and Mef2c as a mediator of anxiety-like behavior. Cell Rep. 43:114455. doi: 10.1016/j.celrep.2024.114455
Hwang, K., Ghuman, A. S., Manoach, D. S., Jones, S. R., and Luna, B. (2016). Frontal preparatory neural oscillations associated with cognitive control: A developmental study comparing young adults and adolescents. Neuroimage 136, 139–148. doi: 10.1016/j.neuroimage.2016.05.017
Insel, T. R., and Landis, S. C. (2013). Twenty-five years of progress: The view from NIMH and NINDS. Neuron 80, 561–567. doi: 10.1016/j.neuron.2013.09.041
International Schizophrenia Consortium, Purcell, S. M., Wray, N. R., Stone, J. L., Visscher, P. M., O’Donovan, M. C., et al. (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752. doi: 10.1038/nature08185
Jaric, I., Rocks, D., Greally, J. M., Suzuki, M., and Kundakovic, M. (2019). Chromatin organization in the female mouse brain fluctuates across the oestrous cycle. Nat. Commun. 10:2851. doi: 10.1038/s41467-019-10704-0
Jung, Y., Hsieh, L. S., Lee, A. M., Zhou, Z., Coman, D., Heath, C. J., et al. (2016). An epigenetic mechanism mediates developmental nicotine effects on neuronal structure and behavior. Nat. Neurosci. 19, 905–914. doi: 10.1038/nn.4315
Krol, A., and Feng, G. (2018). Windows of opportunity: Timing in neurodevelopmental disorders. Curr. Opin. Neurobiol. 48, 59–63. doi: 10.1016/j.conb.2017.10.014
Lakatos, P., Karmos, G., Mehta, A. D., Ulbert, I., and Schroeder, C. E. (2008). Entrainment of neuronal oscillations as a mechanism of attentional selection. Science 320, 110–113. doi: 10.1126/science.1154735
LeBlanc, J. J., and Fagiolini, M. (2011). Autism: A “critical period” disorder? Neural Plast. 2011:921680. doi: 10.1155/2011/921680
Li, H., Radford, J. C., Ragusa, M. J., Shea, K. L., McKercher, S. R., Zaremba, J. D., et al. (2008). Transcription factor MEF2C influences neural stem/progenitor cell differentiation and maturation in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 9397–9402. doi: 10.1073/pnas.0802876105
Li, Z., McKercher, S. R., Cui, J., Nie, Z., Soussou, W., Roberts, A. J., et al. (2008). Myocyte enhancer factor 2C as a neurogenic and antiapoptotic transcription factor in murine embryonic stem cells. J. Neurosci. 28, 6557–6568. doi: 10.1523/jneurosci.0134-08.2008
Li, W.-K., Zhang, S.-Q., Peng, W.-L., Shi, Y.-H., Yuan, B., Yuan, Y.-T., et al. (2023). Whole-brain in vivo base editing reverses behavioral changes in Mef2c-mutant mice. Nat. Neurosci. 27, 116–128. doi: 10.1038/s41593-023-01499-x
Lingjaerde, O. (1991). Benzodiazepines in the treatment of schizophrenia: An updated survey. Acta Psychiatr. Scand. 84, 453–459. doi: 10.1111/j.1600-0447.1991.tb03177.x
Liu, Y., Ouyang, P., Zheng, Y., Mi, L., Zhao, J., Ning, Y., et al. (2021). A selective review of the excitatory-inhibitory imbalance in schizophrenia: Underlying biology, genetics, microcircuits, and symptoms. Front. Cell. Dev. Biol. 9:664535. doi: 10.3389/fcell.2021.664535
Lyons, M. R., Schwarz, C. M., and West, A. E. (2012). Members of the myocyte enhancer factor 2 transcription factor family differentially regulate Bdnf transcription in response to neuronal depolarization. J. Neurosci. 32, 12780–12785. doi: 10.1523/jneurosci.0534-12.2012
Ma, Q., and Telese, F. (2015). Genome-wide epigenetic analysis of MEF2A and MEF2C transcription factors in mouse cortical neurons. Commun. Integr. Biol. 8:e1087624. doi: 10.1080/19420889.2015.1087624
Mao, Z., Bonni, A., Xia, F., Nadal-Vicens, M., and Greenberg, M. E. (1999). Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 286, 785–790. doi: 10.1126/science.286.5440.785
Marin, O. (2016). Developmental timing and critical windows for the treatment of psychiatric disorders. Nat. Med. 22, 1229–1238. doi: 10.1038/nm.4225
Mayer, C., Hafemeister, C., Bandler, R. C., Machold, R., Batista Brito, R., Jaglin, X., et al. (2018). Developmental diversification of cortical inhibitory interneurons. Nature 555, 457–462. doi: 10.1038/nature25999
Meredith, R. M. (2015). Sensitive and critical periods during neurotypical and aberrant neurodevelopment: A framework for neurodevelopmental disorders. Neurosci. Biobehav. Rev. 50, 180–188. doi: 10.1016/j.neubiorev.2014.12.001
Meredith, R. M., Dawitz, J., and Kramvis, I. (2012). Sensitive time-windows for susceptibility in neurodevelopmental disorders. Trends Neurosci. 35, 335–344. doi: 10.1016/j.tins.2012.03.005
Milh, M., Kaminska, A., Huon, C., Lapillonne, A., Ben-Ari, Y., and Khazipov, R. (2007). Rapid cortical oscillations and early motor activity in premature human neonate. Cereb. Cortex 17, 1582–1594. doi: 10.1093/cercor/bhl069
O’Donnell, C., Gonçalves, J. T., Portera-Cailliau, C., and Sejnowski, T. J. (2017). Beyond excitation/inhibition imbalance in multidimensional models of neural circuit changes in brain disorders. Elife 6:e26724. doi: 10.7554/eLife.26724
Olson, C. O., Pejhan, S., Kroft, D., Sheikholeslami, K., Fuss, D., Buist, M., et al. (2018). MECP2 Mutation Interrupts Nucleolin-mTOR-P70S6K Signaling in Rett Syndrome Patients. Front. Genet. 9:635. doi: 10.3389/fgene.2018.00635
Oyarzabal, A., Xiol, C., Castells, A. A., Grau, C., O’Callaghan, M., Fernández, G., et al. (2020). Comprehensive analysis of GABA(A)-A1R developmental alterations in Rett syndrome: Setting the focus for therapeutic targets in the time frame of the disease. Int. J. Mol. Sci. 21:518. doi: 10.3390/ijms21020518
Paciorkowski, A. R., Traylor, R. N., Rosenfeld, J. A., Hoover, J. M., Harris, C. J., Winter, S., et al. (2013). MEF2C Haploinsufficiency features consistent hyperkinesis, variable epilepsy, and has a role in dorsal and ventral neuronal developmental pathways. Neurogenetics 14, 99–111. doi: 10.1007/s10048-013-0356-y
Parenti, I., Rabaneda, L. G., Schoen, H., and Novarino, G. (2020). Neurodevelopmental disorders: From genetics to functional pathways. Trends Neurosci. 43, 608–621. doi: 10.1016/j.tins.2020.05.004
Pereira, A. H., Clemente, C. F., Cardoso, A. C., Theizen, T. H., Rocco, S. A., Judice, C. C., et al. (2009). MEF2C silencing attenuates load-induced left ventricular hypertrophy by modulating mTOR/S6K pathway in mice. PLoS One 4:e8472. doi: 10.1371/journal.pone.0008472
Puang, S. J., Elanggovan, B., Ching, T., and Sng, J. C. G. (2020). MEF2C and HDAC5 regulate Egr1 and Arc genes to increase dendritic spine density and complexity in early enriched environment. Neuronal. Signal. 4:NS20190147. doi: 10.1042/NS20190147
Rajkovich, K. E., Loerwald, K. W., Hale, C. F., Hess, C. T., Gibson, J. R., and Huber, K. M. (2017). Experience-dependent and differential regulation of local and long-range excitatory neocortical circuits by postsynaptic Mef2c. Neuron 93, 48–56. doi: 10.1016/j.neuron.2016.11.022
Rangasamy, S., Olfers, S., Gerald, B., Hilbert, A., Svejda, S., and Narayanan, V. (2016). Reduced neuronal size and mTOR pathway activity in the Mecp2 A140V Rett syndrome mouse model. F1000Res 5:2269. doi: 10.12688/f1000research.8156.1
Reiner, O., Karzbrun, E., Kshirsagar, A., and Kaibuchi, K. (2016). Regulation of neuronal migration, an emerging topic in autism spectrum disorders. J. Neurochem. 136, 440–456. doi: 10.1111/jnc.13403
Ricciardi, S., Boggio, E. M., Grosso, S., Lonetti, G., Forlani, G., Stefanelli, G., et al. (2011). Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum. Mol. Genet. 20, 1182–1196. doi: 10.1093/hmg/ddq563
Rossignol, E. (2011). Genetics and function of neocortical GABAergic interneurons in neurodevelopmental disorders. Neural Plast. 2011:649325. doi: 10.1155/2011/649325
Rubenstein, J. L., and Merzenich, M. M. (2003). Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267. doi: 10.1034/j.1601-183x.2003.00037.x
Ruderfer, D. M., Fanous, A. H., Ripke, S., McQuillin, A., Amdur, R. L., Schizophrenia Working Group of the Psychiatric Genomics Consortium, et al. (2014). Polygenic dissection of diagnosis and clinical dimensions of bipolar disorder and schizophrenia. Mol. Psychiatry 19, 1017–1024. doi: 10.1038/mp.2013.138
Santos-Terra, J., Deckmann, I., Fontes-Dutra, M., Schwingel, G. B., Bambini-Junior, V., and Gottfried, C. (2021). Transcription factors in neurodevelopmental and associated psychiatric disorders: A potential convergence for genetic and environmental risk factors. Int. J. Dev. Neurosci. 81, 545–578. doi: 10.1002/jdn.10141
Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., De Rubeis, S., An, J. Y., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 56:e523. doi: 10.1016/j.cell.2019.12.036
Seibt, J., Timofeev, I., Carrier, J., and Peyrache, A. (2016). Role of spindle oscillations across lifespan in health and disease. Neural Plast. 2016:8103439. doi: 10.1155/2016/8103439
Sharma, A., Hoeffer, C. A., Takayasu, Y., Miyawaki, T., McBride, S. M., Klann, E., et al. (2010). Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 30, 694–702. doi: 10.1523/jneurosci.3696-09.2010
Sohal, V. S., and Rubenstein, J. L. R. (2019). Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 24, 1248–1257. doi: 10.1038/s41380-019-0426-0
Tan, H. R., Lana, L., and Uhlhaas, P. J. (2013). High-frequency neural oscillations and visual processing deficits in schizophrenia. Front. Psychol. 4:621. doi: 10.3389/fpsyg.2013.00621
Tang, X., Kim, J., Zhou, L., Wengert, E., Zhang, L., Wu, Z., et al. (2016). KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 113, 751–756. doi: 10.1073/pnas.1524013113
Tsujimura, K., Irie, K., Nakashima, H., Egashira, Y., Fukao, Y., Fujiwara, M., et al. (2015). miR-199a links MeCP2 with mTOR signaling and its dysregulation leads to Rett syndrome phenotypes. Cell Rep. 12, 1887–1901. doi: 10.1016/j.celrep.2015.08.028
Tu, S., Akhtar, M. W., Escorihuela, R. M., Amador-Arjona, A., Swarup, V., Parker, J., et al. (2017). NitroSynapsin therapy for a mouse MEF2C haploinsufficiency model of human autism. Nat. Commun. 8:1488. doi: 10.1038/s41467-017-01563-8
Wan, L., Liu, X., Hu, L., Chen, H., Sun, Y., Li, Z., et al. (2021). Genotypes and phenotypes of MEF2C haploinsufficiency syndrome: New cases and novel point mutations. Front. Pediatr. 9:664449. doi: 10.3389/fped.2021.664449
Ward, C., Nasrallah, K., Tran, D., Sabri, E., Vazquez, A., Sjulson, L., et al. (2024). Developmental disruption of Mef2c in medial ganglionic eminence-derived cortical inhibitory interneurons impairs cellular and circuit function. Biol. Psychiatry doi: 10.1016/j.biopsych.2024.05.021 [Epub ahead of print].
Williams, H. J., Norton, N., Dwyer, S., Moskvina, V., Nikolov, I., Carroll, L., et al. (2011). Fine mapping of ZNF804A and genome-wide significant evidence for its involvement in schizophrenia and bipolar disorder. Mol. Psychiatry 16, 429–441. doi: 10.1038/mp.2010.36
Willsey, A. J., Sanders, S. J., Li, M., Dong, S., Tebbenkamp, A. T., Muhle, R. A., et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. doi: 10.1016/j.cell.2013.10.020
Wu, H., Ren, Y., Pan, W., Dong, Z., Cang, M., and Liu, D. (2015). The mammalian target of rapamycin signaling pathway regulates myocyte enhancer factor-2C phosphorylation levels through integrin-linked kinase in goat skeletal muscle satellite cells. Cell Biol. Int. 39, 1264–1273. doi: 10.1002/cbin.10499
Zhang, B., Zhang, J., Chen, H., Qiao, D., Guo, F., Hu, X., et al. (2024). Role of FMRP in AKT/mTOR pathway-mediated hippocampal autophagy in fragile X syndrome. Prog. Neuropsychopharmacol. Biol. Psychiatry 134:111036. doi: 10.1016/j.pnpbp.2024.111036
Zhang, W., Feng, G., Wang, L., Teng, F., Wang, L., Li, W., et al. (2018). MeCP2 deficiency promotes cell reprogramming by stimulating IGF1/AKT/mTOR signaling and activating ribosomal protein-mediated cell cycle gene translation. J. Mol. Cell Biol. 10, 515–526. doi: 10.1093/jmcb/mjy018
Zhang, Z., and Zhao, Y. (2022). Progress on the roles of MEF2C in neuropsychiatric diseases. Mol. Brain 15:8. doi: 10.1186/s13041-021-00892-6
Keywords: cortical development, MEF2C haploinsufficiency syndrome, autism spectrum disorder, GABA, interneurons
Citation: Ward C, Sjulson L and Batista-Brito R (2024) The function of Mef2c toward the development of excitatory and inhibitory cortical neurons. Front. Cell. Neurosci. 18:1465821. doi: 10.3389/fncel.2024.1465821
Received: 16 July 2024; Accepted: 05 September 2024;
Published: 23 September 2024.
Edited by:
Anirban Paul, The Pennsylvania State University, United StatesReviewed by:
Andrzej W. Cwetsch, Instituto de Biotecnología y Biomedicina Universidad de Valencia, SpainDhananjay Huilgol, Duke University, United States
Copyright © 2024 Ward, Sjulson and Batista-Brito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renata Batista-Brito, cmVuYXRhLmJyaXRvQGVpbnN0ZWlubWVkLmVkdQ==