 Isabel E. Faulkner
Isabel E. Faulkner Rachael Z. Pajak
Rachael Z. Pajak Michael K. Harte
Michael K. Harte Jocelyn D. Glazier
Jocelyn D. Glazier Reinmar Hager
Reinmar Hager
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 01 October 2024
Sec. Cellular Neuropathology
Volume 18 - 2024 | https://doi.org/10.3389/fncel.2024.1449151
This article is part of the Research TopicPathogenic Potassium Channel Variants in Neurological Disorders: From Functional Analysis to Personalized Pharmacological ApproachesView all 8 articles
Voltage-gated potassium channels are a widely distributed subgroup of potassium channels responsible for the efflux of potassium in the repolarisation of the cell membrane, and hence contribute to the latency and propagation of action potentials. As they are causal to synaptic transmission, alterations to the structure of these channels can lead to a variety of neurological and psychiatric diseases. The Kv3 subfamily of voltage-gated potassium channels are found on many neurons in the brain, including inhibitory interneurons where they contribute to fast-frequency firing. Changes to the firing ability of these interneurons can lead to an imbalance of inhibitory and excitatory neurotransmission. To date, we have little understanding of the mechanism by which excitatory and inhibitory inputs become imbalanced. This imbalance is associated with cognitive deficits seen across neurological and neuropsychiatric disorders, which are currently difficult to treat. In this review, we collate evidence supporting the hypothesis that voltage-gated potassium channels, specifically the Kv3 subfamily, are central to many neurological and psychiatric disorders, and may thus be considered as an effective drug target. The collective evidence provided by the studies reviewed here demonstrates that Kv3 channels may be amenable to novel treatments that modulate the activity of these channels, with the prospect of improved patient outcome.
Current treatments of psychiatric and neurological diseases with a cognitive deficit component such as schizophrenia (SCZ), autism spectrum disorder (ASD) and some forms of epilepsy, rely on symptom control, for example, antipsychotics for SCZ (Gomes and Grace, 2021). These treatments tend to alleviate acute symptoms such as psychosis for SCZ, but do not work for other symptom domains such as cognitive deficits. Thus, despite the large number of over 20 first and second generation antipsychotics available, none are effective in treating the full range of symptoms and some, such as cognitive deficits, remain difficult to treat. Our lack of understanding the mechanisms underlying in particular cognitive deficits contributes to this. Despite many drugs being developed to treat the cognitive deficits seen in SCZ, many other neurological and psychiatric diseases could also benefit from these treatments. In this review, we consider the central role that voltage-gated potassium channels play in the biological mechanisms and aetiology of many psychiatric and neurological disorders. Furthermore, we draw attention to investigations that examine modulation of voltage-gated potassium channel activity as a potential therapeutic target. Hence, accumulating evidence suggests that potassium channels should be at the centre of new research investigations, and further pre-clinical studies should focus on their potential as an efficacious drug target (Deakin et al., 2019).
Transmembrane ion channels play a fundamental role in cell physiology through the transport of ions (e.g., potassium, calcium, sodium) in or out of the cell. In particular, voltage-gated potassium (Kv) channels that open or close in response to changes in the electrical membrane potential play a critical role in excitable cells, especially in neurons, for the establishment of membrane potential and the repolarisation of the cell membrane following an action potential, ensuring the return of the membrane to the resting state (Clatot et al., 2023). The human genome contains over 70 different genes associated with Kv channels (Gunthorpe, 2022). Kv channel genes are divided into 12 families, Kv1 to Kv12 (Liang et al., 2024). Members of the Kv3 subfamily are unique in having a high activation potential (>−10 mV), contributing to high-frequency firing (>100 Hz) and rapid deactivation, which are essential properties for the function of the cells in which they are expressed (Kaczmarek and Zhang, 2017; Chen Y.-T. et al., 2023). Kv3 channels are expressed in neurons that need to fire at high frequencies, such as cerebellar Purkinje cells, hippocampal and prefrontal parvalbumin-expressing interneurons (PVI), basal ganglia nuclei, reticular thalamic nucleus and principal neurons of several auditory brainstem nuclei (Zhang et al., 2021; Clatot et al., 2023). The ability of neurons to fire rapidly plays a crucial role in mechanisms underpinning learning and memory in the hippocampus through cortical inhibition, motor co-ordination in the cerebellum and sensory processing in the auditory nuclei (Chi et al., 2022).
There are four subfamilies of Kv3 channels, Kv3.1, Kv3.2, Kv3.3 and Kv3.4, which are encoded by the KCNC1, KCNC2, KCNC3 and KCNC4 genes, respectively (Gunthorpe, 2022). The functional Kv3 channel consists of four alpha subunits which form a pore permeable to K+ ions (Grizel et al., 2014; Irie et al., 2014). Kv3 channels have either a homotetrameric (all the same Kva subunits), or a heterotetrameric (combination of Kv3 subunits), structure. All the Kv3 subunits contain a N-terminal cytoplasmic T1 domain, followed by six transmembrane segments, S1–S6 (Chi et al., 2022), as shown in Figure 1. S1–S4 make up the voltage sensing domain (VSD), with S4 being the primary voltage sensor (Liang et al., 2024). S4 has six positively charged arginine and lysine residues (represented by the “+” symbol within S4 in Figure 1), found at every third position within the amino acid sequence (Li et al., 2021). The positively charged arginine residues detect changes to membrane potential, resulting in the re-orientation allowing the channel to open (Aggarwal and MacKinnon, 1996). Between S4 and S5 is a linker domain that connects to the S6 pore-forming unit, with S5 and S6 forming the pore domain (Mukherjee et al., 2022). The domains are arranged so that S5 and S6 are in the centre of the channel and are peripherally surrounded by S1 to S4 (Grizel et al., 2014). This forms a selective filter that allows the exclusive transfer of K+ in response to changes in membrane potential (Tombola et al., 2005). The unique structure and properties of these transmembrane segments allow Kv3 channels to sustain high-frequency neuronal firing by allowing rapid repolarisation of action potentials. This rapid repolarisation is vital for neurons that require fast signalling and precise timing, such as those involved in auditory processing and fast-spiking interneurons. Therefore, any alterations in Kv3 channel structure through genetic mutations can lead to phenotypic changes affecting neuronal excitability and the overall function of neural circuits, potentially resulting in neurological and psychiatric disorders.
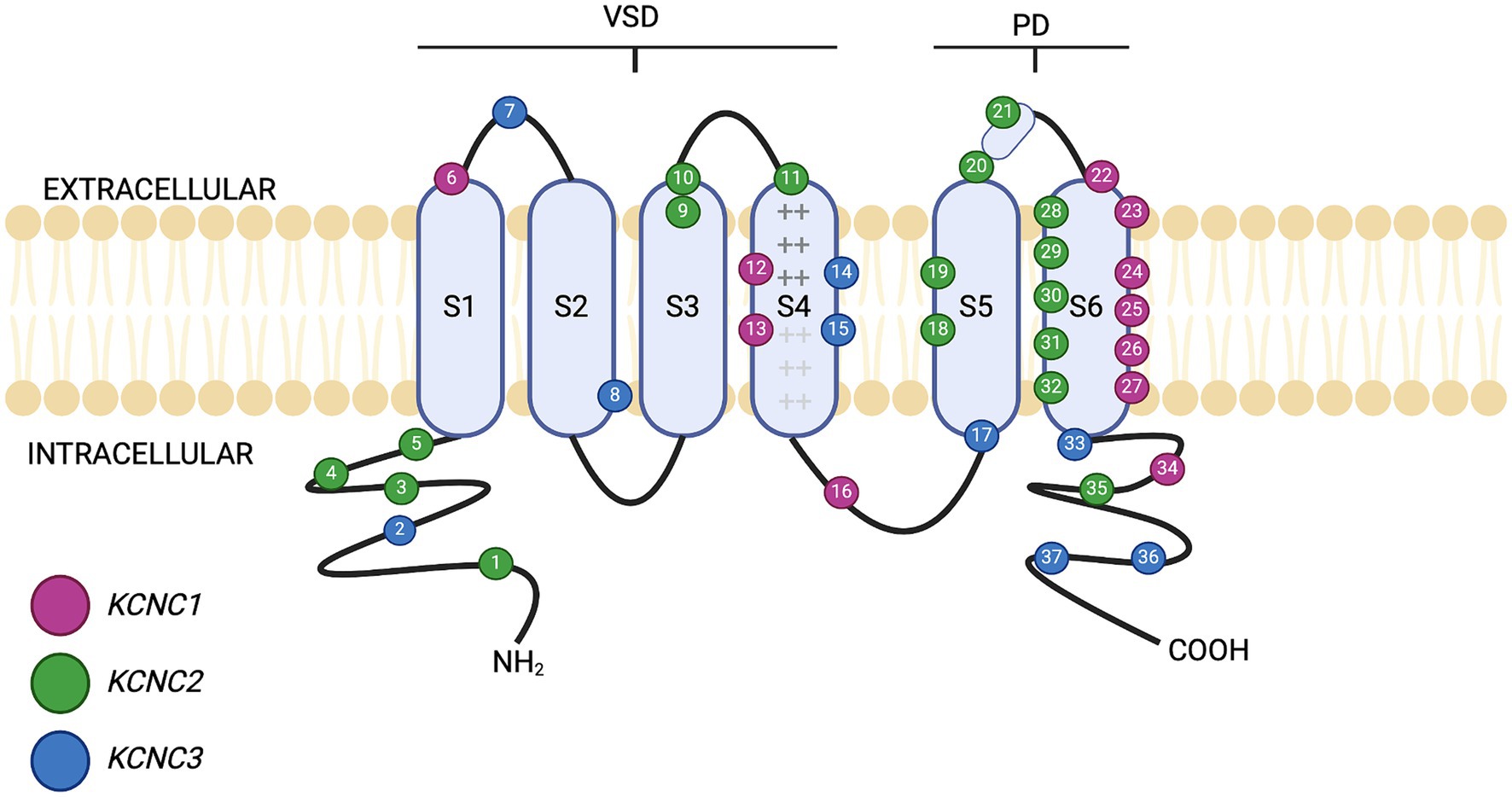
Figure 1. Schematic of Kv3 channel structure. The six transmembrane domains (S1–S6) are shown, along with the intracellular (cytosolic) distribution of the amino (–NH2) and carboxyl (–COOH) termini. The presence of basic amino acids arginine and lysine within segment S4 are represented by “+” notation. S1–S4 form the voltage sensing domain (VSD). Between S5 and S6 lies the pore domain (PD). The location of different mutations on Kv3.1, Kv3.2 and Kv3.3 is shown by numbered positions, colour-coded for each individual gene. Further detail of the clinical presentation of each mutation is given in Tables 1–3. Figure created using Biorender.com.
Mutations can affect Kv3 channel function to fire at high frequencies, altering neural physiology and brain circuitry. Mutations in KCNC1, KCNC2 and KCNC3 have been implicated in several neurological and psychiatric disorders such as epilepsy, ataxia, and neurodevelopmental disorders (NDD) such as ASD and SCZ (Tables 1–3). Currently, no known clinical disorders have been implicated with mutations in KCNC4. Known mutations associated with different transmembrane segment domains of Kv3 channels are shown in Figure 1.
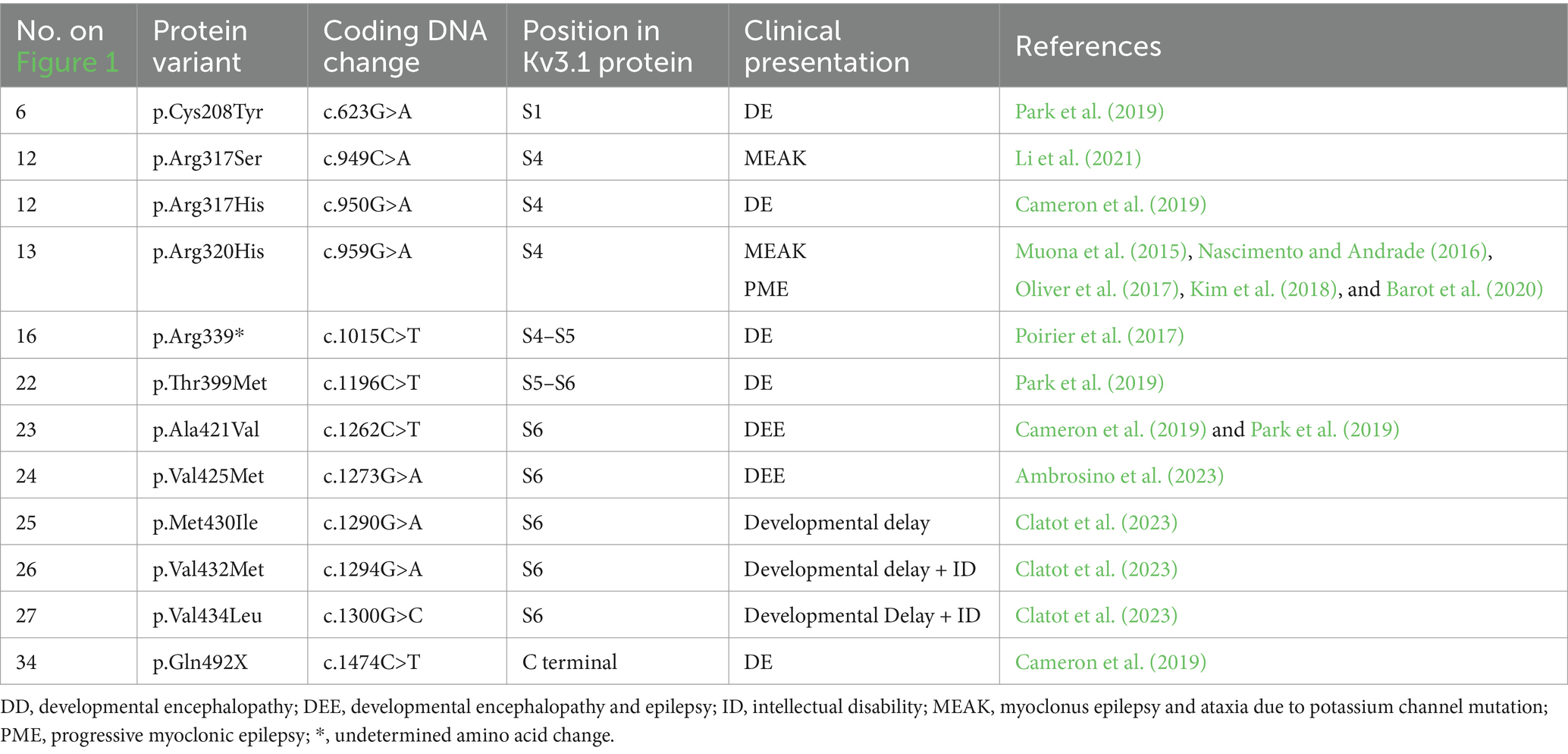
Table 1. KCNC1 channel variants and associated clinical presentation.
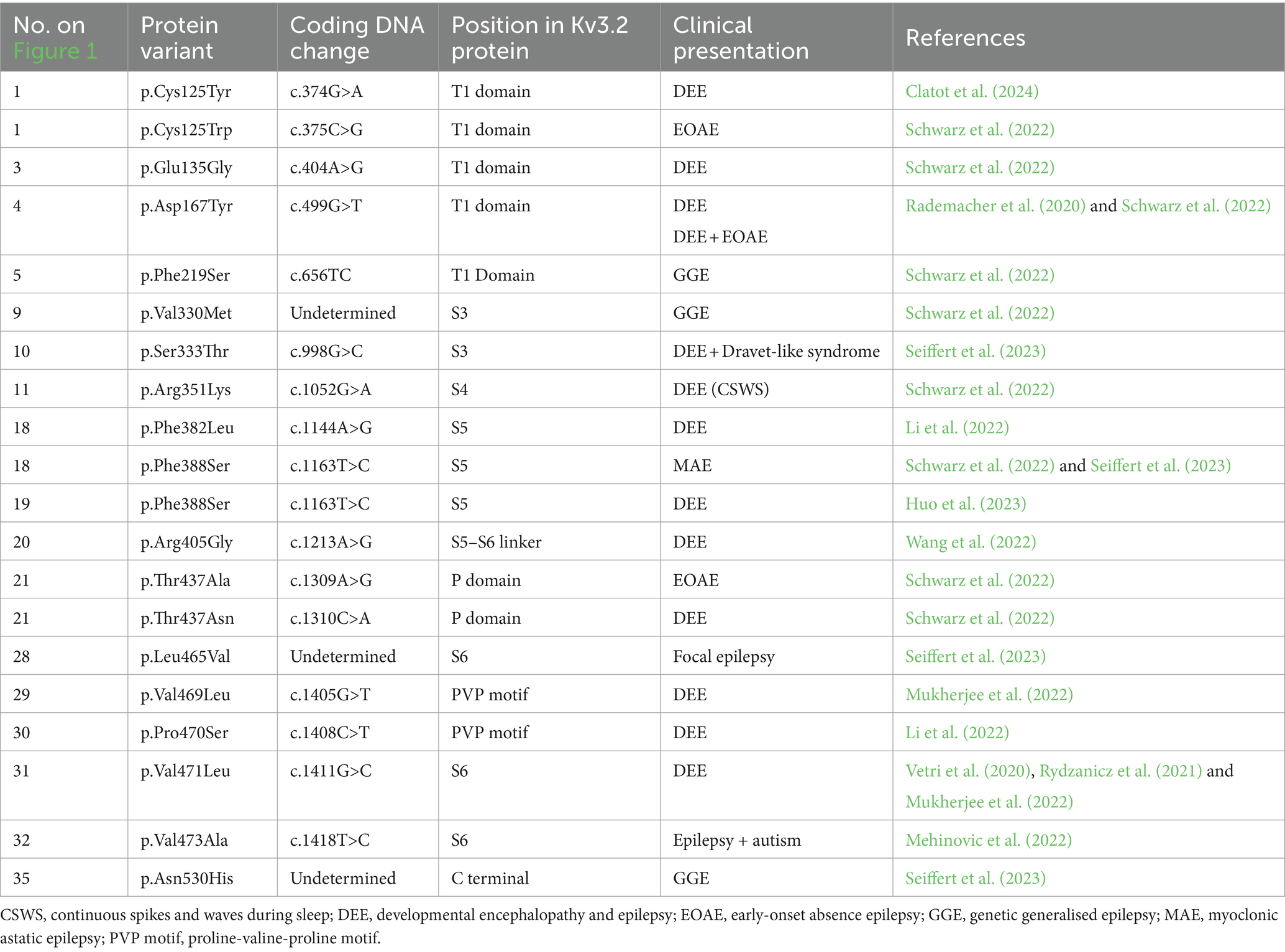
Table 2. KCNC2 channel variants and associated clinical presentation.
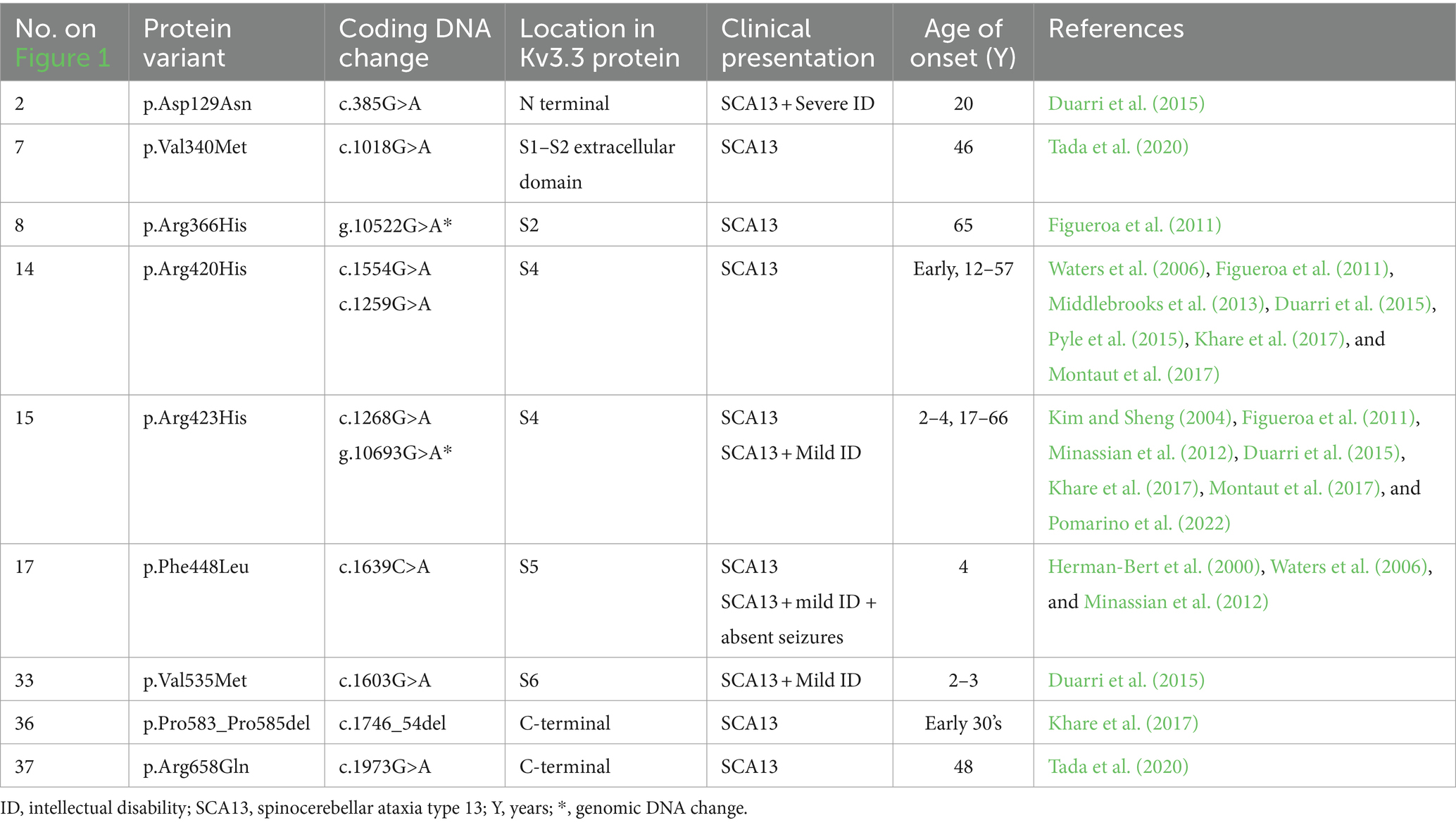
Table 3. KCNC3 channel variants and associated clinical presentation.
As highlighted in Tables 1–3, variants in Kv3 channel structure are associated with a wide range of altered functions, manifest as several neurological and psychiatric disorders, of which the major types are described below in more detail. The variants in Kv3 channel function lead to these disease states mechanistically by affecting channel function; for example, changes to the T1 domain can affect the open state stability, important for rapid activation and deactivation (Gunthorpe, 2022).
Epilepsy is a common complex group of neurological disorders that affect approximately 50 million people globally (Chen, Y.-T. et al., 2023; Chen Z. et al., 2023). Epilepsy is defined as recurrent unprovoked seizures and has many aetiologies and pathophysiologies, including traumatic brain injury, infections, tumours, and stroke (Vezzani et al., 2015). Additionally, gene mutations such as variants in ion channel genes have been associated with epilepsy (Symonds et al., 2017). The diverse aetiology of epilepsy is underpinned by its heterogeneous nature; different types of epilepsies are categorised based on the types of seizures and associated features. Seizures can occur due to the disruption in the excitatory and inhibitory (E-I) balance of brain networks, resulting in an imbalance of excitatory and inhibitory synaptic inputs and increased excitability, as well as hypersynchronisation of neuronal populations (Bertocchi et al., 2023). This synchronicity that precedes a seizure may result from a loss of inhibitory restraint, indicating that seizures can be triggered not only by hyperexcitability but also by a failure of inhibition (Jiruska et al., 2013). In around two-thirds of epileptic cases, the underlying cause is unknown, however, this number is decreasing as the number of monogenic causes discovered has increased (Stenshorne et al., 2022). Various brain areas are associated with seizures including the cerebral cortex, thalamic reticular nucleus, hippocampus, and cerebellum, which are also areas with Kv3-expressing cells (Holmes, 2020; Bernardi et al., 2023).
The voltage-gated K+ channel superfamily has been implicated in various forms of epilepsy (Allen et al., 2020). Two main subtypes of epilepsy are associated with Kv3 channel variants: progressive myoclonic epilepsy (PME) and developmental encephalopathy and epilepsy (DEE), as outlined in Tables 1, 2. Despite medical intervention, only around 75% of epileptic patients become seizure-free (McWilliam et al., 2024). However, for a large proportion of epileptic patients, there are currently no treatments available to manage their seizures. Additionally, epilepsy has many psychiatric comorbidities such as ASD and SCZ, suggesting a potential shared pathophysiology.
PME is an umbrella term for a rare collection of autosomal recessive disorders that are characterised by cerebellar ataxia, myoclonus, drug-resistant epilepsy and cognitive decline that gradually worsens over time (Nascimento and Andrade, 2016). Signs of PME tend to emerge in late childhood and early adolescence, typically between the ages of six to fourteen years but can affect any age (Holmes, 2020; Carpenter et al., 2021; Feng et al., 2024). Mutations in KCNC1 are associated with a specific type of PME, myoclonus epilepsy and ataxia due to potassium channel mutation (MEAK). Kv3.1 undertakes alternative splicing to form two isoforms of the channel, Kv3.1a and Kv3.1b. Kv3.1a expression is greatest during early embryonic development, whereas Kv3.1b is the principal isoform in the adult brain with its expression peaking during adolescence, which overlaps with the onset of PME (Carpenter et al., 2021). A recurrent heterozygous missense de novo mutation, c.959G>A (p.Arg320His), has been seen in multiple unrelated patients (Nascimento and Andrade, 2016; Carpenter et al., 2021). This mutation results in the substitution of an arginine residue for a histidine residue in the S4 segment, causing loss of function. This results in mutant channels producing more hyperpolarised potentials and creating barely detectible currents in Xenopus laevis oocytes (Muona et al., 2015). When modelled in vitro in HEK293 cells (mammalian expression system; Munch et al., 2018) and in neurons (Carpenter et al., 2021), the Kv3.1 mutant channels have a significantly decreased K+ current level, and reduced high-frequency firing of the neurons compared to wildtype Kv3.1 channels. Due to the variety of genetic mutations associated with PME, each with mechanistic differences in disease progression, treatment to target the disease mechanism, or slow disease progression, is limited. At present, treating PME is challenging, with current treatment for PME is largely symptom control. Anti-epileptic drugs like valproate are used to manage myoclonic seizures, however, there are no treatments available to manage the cognitive decline seen with the condition (Holmes, 2020). Importantly, pre-clinical research has shown that positive allosteric modulators of Kv3.1 have restored channel function in cell lines expressing the mutated Kv3.1 channel, and phase 1b trials are to continue to investigate these compounds (Autifony, 2024).
Developmental encephalopathy and DEE are a group of severe rare drug-resistant neurological syndromes that are characterised by developmental delay, with or without epilepsy, intellectual disability (ID) with stark abnormal electroencephalogram (EEG) readings (Lin et al., 2022). About 22.2% of children with ID also have some form of epilepsy (McTague et al., 2016). However, in DEE, developmental encephalopathy and epilepsy have evolved independently from each other. Signs of DEE can show within the first few months following birth (Specchio and Curatolo, 2021). Most DEE cases (30–50%) have a sporadic de novo cause with over 50 genes associated with DEE (Bertocchi et al., 2023), including genes that encode for various ion channels including potassium channels (KCNA, KCNB and KCNQ subfamilies) (Wang et al., 2022). DEE is implicated with mutations in KCNC1 and KCNC2 (Muona et al., 2015; Tables 1, 2). The KCNC1 variant, p.Ala421Val, is associated with DEE in several patients. This variant alters the function of S6, leading to a loss of whole-cell current and has a dominant negative effect (Cameron et al., 2019; Park et al., 2019). Treatment for DEE is limited to managing epileptic seizures. However, the increasing identification of gene variants associated with DEE and tools such as CRISPR/Cas9 opens the potential for gene therapy and generates preclinical models to further understand mechanisms and subsequent treatment targets (Bertocchi et al., 2023).
Spinocerebellar ataxia comprises a group of over 40 autosomal dominant neurodegenerative disorders that result in the progressive loss of voluntary movement due to cerebellar atrophy (Shakkottai and Fogel, 2013; Ashizawa et al., 2018) which can impact a patient’s speech, coordination, eye movement and balance. Spinocerebellar ataxia is a heterogeneous disease which is progressive and neurodegenerative and typically affects the cerebellum. It is inherited in an autosomal dominant pattern. Most types of spinocerebellar ataxias (60%) are caused by the abnormal expansion of CAG repeat sequences and ion channel gene mutations that are inherited de novo. The numbering of different types of spinocerebellar ataxia is chronologically based on when the associated gene was discovered (Ghanekar et al., 2022). Spinocerebellar type 13 (SCA13) is exclusively due to de novo mutations in the KCNC3 (Kaczmarek and Zhang, 2017; Table 3). Common channel variants include p.Arg420His (R420H) and p.Arg423His (R423H) where an arginine residue is substituted for a histidine in the 420 or 423-position which impacts S4 function (Figueroa et al., 2010; Khare et al., 2017). Co-expression of mutant R420H or R423H in Xenopus oocytes results in the suppression of current via a dominant negative mechanism (Irie et al., 2014). R424 mutant mice have a reduced current density, increased basal concentration and broadened action potentials, similar to observed outcomes in humans. There are two developmental time points of SCA13 onset, early onset where cerebellar degeneration is seen during early childhood, or adult-onset which presents in middle age and results in slow and progressive loss of motor co-ordination and cognitive decline (Rossi et al., 2014). Late onset is also associated with a disruption in auditory information processing such as locating sounds in space (Middlebrooks et al., 2013). Currently, outcomes are poor with no definitive treatment. There is a multidisciplinary approach to management including neurologists, physiotherapists, occupational therapists and speech and language therapists to help manage the symptoms.
ASD can be defined as a heterogeneous collection of neurodevelopmental disorders characterised by impairment in communication, social interaction, and restrictive or repetitive behaviours (NICE, 2021). ASD has an undefined aetiology, believed to be multifactorial, due to complex interactions between genes, environmental factors and epigenetics. It has a prevalence of 1 in 44 children which is increasing (Port et al., 2019; Maenner et al., 2021). ASD patients have depleted numbers of PVI and PV mRNA in post-mortem brains (Hashemi et al., 2017). Furthermore, genetic and environmental risk factor models of ASD also have reduced PVI (Bee et al., 2021). PV knock-out mice show ASD-like behaviours (Wöhr et al., 2015). There is evidence to suggest that voltage-gated K+ channels are also implicated in the pathology of ASD. Kv3.1 deficient mice have abnormal social behaviour and hyperactivity, and it has been shown that there are changes to PV in the striatum and prefrontal cortex (Parekh et al., 2018; Bee et al., 2021).
While the majority of ASD aetiology is unclear, the most common single-gene cause of ASD is believed to be due to the silencing of the fragile X messenger ribonucleoprotein 1 (FMR1) gene resulting in Fragile X syndrome (5% of ASD cases). Approximately 50–60% of Fragile X patients are also co-diagnosed with ASD (Juarez and Martínez Cerdeño, 2022). FMR1 is located on the X chromosome (Bhakar et al., 2012). The majority of mutations in FMR1 are due to an expansion of the CGG repeats in the promoter region. This results in hypermethylation and leads to transcriptional silencing of the FMR1 gene, reducing the production of Fragile X Messenger Ribonucleoprotein (FMRP), an mRNA-binding protein (Bhakar et al., 2012). FMR1 is strongly expressed in neurons and regulates the expression of various ion channels including binding to mRNA encoding Kv3.1b in brain synaptosomes (Strumbos et al., 2010; El-Hassar et al., 2019). The hallmark characteristics of Fragile X syndrome include ID, extreme hypersensitivity to sensory stimuli (including auditory stimuli), attention deficit and seizures (Strumbos et al., 2010; El-Hassar et al., 2019). Post-mortem human Fragile X brains and Fmfr1 knockout brains have a significant decrease and lower density of PVI numbers in all brain areas (medial prefrontal cortex (PFC), primary somatosensory cortex, primary motor cortex, superior temporal cortex, and anterior cingulate cortex) (Juarez and Martínez Cerdeño, 2022; Kourdougli et al., 2023). Additionally, hyperactivity of PV neurons has been found in Fmfr1 knockout mice (Gibson et al., 2008; Nomura et al., 2017; Domanski et al., 2019). There have been efforts to find a successful treatment for Fragile X (Large et al., 2017), however to date, there are no approved medicines.
SCZ is a complex disorder without a clear central pathology and is often co-morbid with other conditions such as major depressive disorder, obsessive compulsive disorder, and substance use disorders (Sharma and Reddy, 2019). SCZ exhibits a natural course of progression that often has premorbid impairments and a prodrome, that continues with a relapsing and remitting course (Tandon et al., 2024). SCZ presents with a wide range of symptomology, and due to this, and its various co-morbidities, it is often difficult to diagnose and ascertain an accurate prevalence of the disease. It is a disorder that is one of the top 15 causes of disability worldwide, and can be highly detrimental to individuals (GBD 2016 Disease and Injury Incidence and Prevalence Collaborators, 2017). The main symptoms for those afflicted with SCZ can be classified into three categories: positive symptoms, also referred to as psychotic (hallucinations, delusions, confusion), negative symptoms (alogia, anhedonia, social withdrawal), and cognitive symptoms (working memory impairment, executive function impairment, attention deficit) (Tamminga, 2008).
There is a functional loss of PVI in SCZ and these GABAergic interneurons have a decreased firing rate (Kaar et al., 2019). There are also reduced Kv3.1 containing K+ channels in human brain tissue from untreated schizophrenia patients (Yanagi et al., 2014). PVI are usually fast spiking (Nahar et al., 2021), and they fire at a rate capable of entraining gamma band oscillations (30–80 Hz). Therefore, a decreased output from these interneurons leads to gamma band abnormalities, as gamma band activity is governed by PVI (McNally et al., 2013). Indeed, a study found that only cells expressing PV saw an increase in power ratio of local field potential (LFP) in the gamma band activity, which was absent in cells not expressing PV, suggesting that LFP activity in this band frequency is dependent upon this cell type (Smausz et al., 2022). Gamma band abnormalities have been associated with SCZ, with numerous studies linking such abnormalities to the pathology of the disease. An MRI based experiment measuring cortical GABA provides evidence supporting impaired GABAergic neurotransmission in SCZ patients and importantly, correlated this with gamma band activity due to a reduced auditory steady state response in SCZ patients in the gamma band (McNally and McCarley, 2016).
Currently, antipsychotics are mainly used clinically to control the positive symptoms of SCZ, despite cognitive and negative symptoms being predictive of functional outcomes. Pre-clinical research has begun to investigate the Kv3 channels for SCZ to combat the decreased firing of the PVI (Chen, Y.-T. et al., 2023). It is imperative that this research continues so that all three symptom classes can be addressed with new therapies, leading to improved patient outcomes.
It is widely accepted that imbalances in excitatory and inhibitory synaptic currents underlie many psychiatric disorders (Shao et al., 2019; Culotta and Penzes, 2020; Liu et al., 2021). Excitatory activity is mainly glutamatergic driven, while inhibitory activity is GABAergic (Rubenstein and Merzenich, 2003). Inhibitory GABAergic interneurons and excitatory pyramidal cells have often been hypothesised to be at the centre of this imbalance (Figure 2). E-I currents are indeed in a dynamic state, with an imbalance in this, or indeed a change in the E-I ratio, underlying pathological changes relevant to specific symptom domains. Throughout this review we will refer to this imbalance/change in ratio as a balance/imbalance of synaptic E-I inputs in a given system.
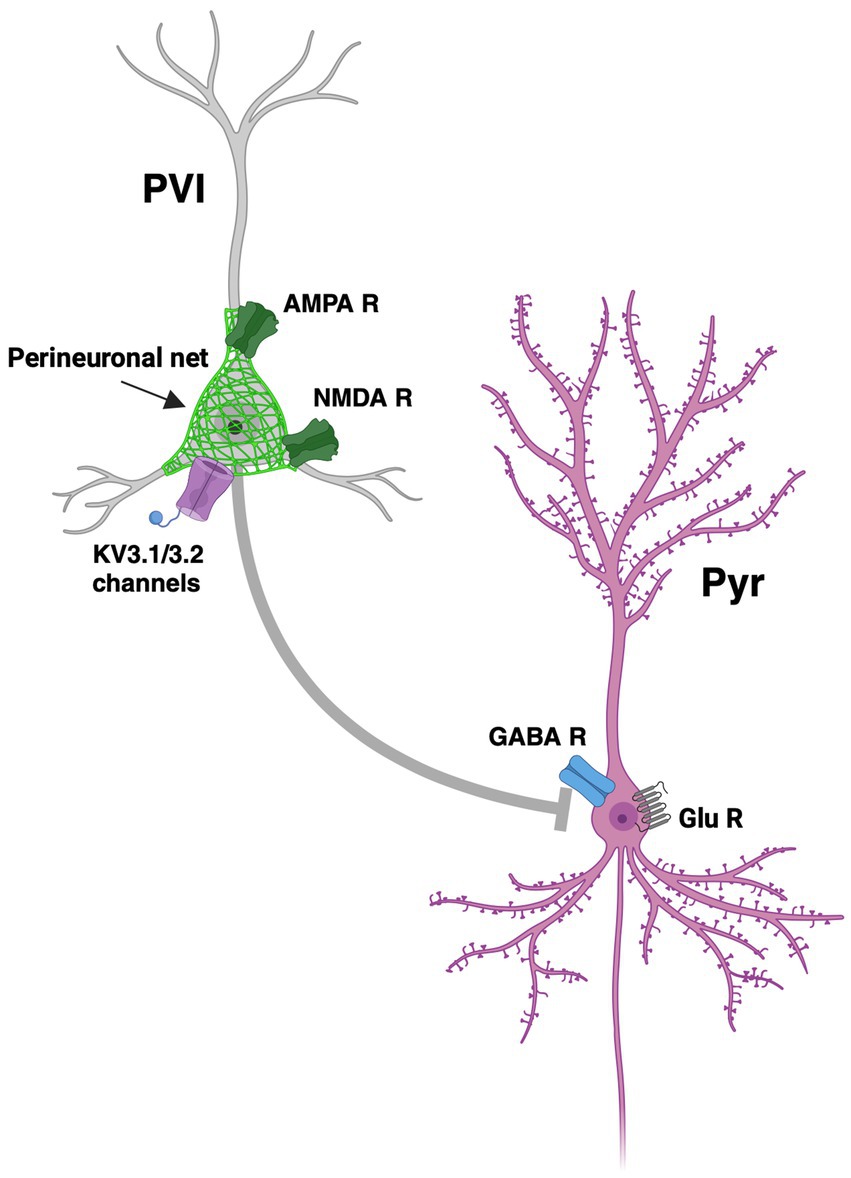
Figure 2. Dysregulation of excitatory-inhibitory imbalance. Functional loss of PVI (parvalbumin expressing interneurons) and hypofunction of NMDA (N-Methyl D-aspartate) receptors on the PVI leads to a reduction in inhibition of Pyr (pyramidal cells) and therefore an increase in glutamate release. Such disruption of excitatory and inhibitory inputs leads to excitatory-inhibitory imbalance. Pictured is a PV interneuron (PVI) covered in a perineuronal net (PNN), represented as a green matrix covering PVI, with an NMDA receptor (NMDA R), and an AMPA receptor (AMPA R), and Kv3.1 and Kv3.2 channels, inhibiting a pyramidal neuron (Pyr) with a γ-aminobutyric acid receptor (GABA R) and glutamate receptors (Glu R) and channels. Figure created using Biorender.com.
High frequency gamma oscillations were reduced in SCZ patients by EEG studies (Lett et al., 2014), and have been proposed to be one of the main driving mechanisms of social and cognitive deficits. Oscillations occur when populations of cells fire in near synchrony. In particular, gamma oscillations (30–80 Hz) are hypothesised to be generated from the pyramidal interneuron network gamma (PING) model, a relationship between pyramidal cells and PV-positive interneurons. Here, the pyramidal cells excite the interneurons using glutamatergic input in a phasic manner, and these currents are synchronised rhythmically by an inhibitory feedback loop (Gonzalez-Burgos and Lewis, 2012; Börgers, 2017). Pyramidal cell inhibition can cause a decrease in gamma oscillation amplitude, which has been linked this to social interaction deficits, implicating gamma oscillations and firing rates as a key locus for social emotion in the PFC (Chen and Hong, 2018; Sato et al., 2023).
E-I balance is underpinned by the relationship between excitatory pyramidal neurons and glutamatergic neurotransmission and the phasic inhibition of these cells by PV-positive interneurons, as shown in Figure 2. Perturbation of this balance of excitatory and inhibitory neurotransmission is central to many psychiatric disorders, underpinned by the altered development of the neurotransmitter systems that contribute to maintaining E-I balance (Pietropaolo and Provenzano, 2022). Dysfunction of this interaction in hippocampal and cortical regions is one of the more consistent alterations in SCZ (Gomes and Grace, 2021). Loss of PVI leads not only to an excitatory overdrive of pyramidal cells but also to dysregulated oscillatory activity across a broad neural network (Lewis et al., 2012). Gamma oscillations in particular depict the functional state and coordinated activity of networks that coordinate cognitive processes, dependent on PVI (Buzsáki and Wang, 2012).
Importantly, E-I imbalance affects hierarchical networks. An example of this are feed forward projections that are associated with hallucinations in SCZ and psychotic disorders. This has been demonstrated by both computational (Jardri and Deneve, 2013), and experimental (Jardri et al., 2017) approaches. Further, the E-I balance is considered to be involved in the default mode network (DMN) and extrinsic cognitive control. These are anti-correlated networks of internally focussed versus outward focussed processes, and the dynamic relationship between them is likely mediated by E-I neurotransmission (Allen et al., 2019). These perturbed network interactions are paramount to the underlying causes of psychiatric symptoms, and are often rooted in childhood trauma and early life insult (Allen et al., 2019).
Both human and animal studies have shown that E-I imbalance is perpetuated by a reduction of PVI, which is linked to oscillatory activity differences (Lodge et al., 2009). Reduced levels and expression of PV explains a decrease in PVI output and therefore a loss of the fast-spiking phenotype of these cells. Without the fast-spiking ability of the PVI they are not able to entrain gamma rhythms. Thus, PVI, and their fast-spiking ability, are a potential target for drug development. Cardin et al., 2009, demonstrated that the PV expressing cells (i.e., fast-spiking cells) are responsible for gamma rhythm generation (Cardin et al., 2009). Here, they drove a cortical network of fast spiking cells in PV-Cre mice (PVI) and regular spiking cells in αCamKII-Cre mice (including pyramidal neurons) at a range of frequencies between 8–200 Hz with 1 ms light pulses (Cardin et al., 2009). Only at the gamma range (40 Hz) did they see an increase in local field potential power ratio from the fast spiking PVI, demonstrating that gamma rhythms are dependent on PV (Cardin et al., 2009).
Furthermore, studies have shown a reduction in GAD67 mRNA, the gene responsible for the synthesis of GABA, in post-mortem human brains of patients with SCZ and mood disorders (Thompson et al., 2009). In the maternal immune activation model (mIA) model, there is a reduction in Arx gene expression, which is critical to PVI development (Nakamura et al., 2019). We also note that there are morphological and functional changes to the PVI in different types of epilepsy (Ye and Kaszuba, 2017), and a reduced number of PVI in sections of post-mortem ASD brain (Hashemi et al., 2017).
Taken together, the accumulating evidence highlights that PVI should be considered as a main target for the modulation of the E-I imbalance, as depicted in Figure 2. Therefore, in further drug development, research on voltage-dependent potassium channels has gained interest, as these channels are expressed on the PVI. Two voltage-gated potassium channels, Kv3.1 and 3.2 are co-localised with PVI (McDonald and Mascagni, 2006), as shown in Figure 2. Potentiating these channels through pharmacological intervention could hold promise for the treatment of cognitive and social deficits found in neurological disease.
Neuronal cell characteristics are shaped by the potassium channels they express. Neurons in the auditory brain stem, fast-spiking GABAergic PVI, and the Purkinje cells of the cerebellum all express potassium channels from the Kv3 family (Kaczmarek and Zhang, 2017). These channels have particularly high activation potentials and rapid kinetics, activating and deactivating to release neurotransmitters from the interneurons at rates of up to 1,000 Hz (Kaczmarek and Zhang, 2017; Chen et al., 2023a). This is achieved by detecting changes in the membrane potential and repolarising the cell. Cryo-EM structural characterisation found a binding site for positive allosteric modulators, providing the potential for a Kv3 channel modulator to be developed (Botte et al., 2022).
One of the challenges in developing a Kv3 channel modulator is conferring specificity of action to the desired channel. Lack of structural high resolution for Kv3 channels has previously hindered development of a targeted therapeutic, but recent cryo-EM studies have now characterised the Kv3.1 binding sites (Botte et al., 2022; Liang et al., 2024). Botte et al. (2022) identified a difference between the Kv1.2 and Kv3.1 structures, which are very similar, at the intracellular T1 domain, and this distinction facilitated development of a specific Kv3.1 modulator. Further, Boddum et al. (2017) used Xenopus laevis oocyte electrophysiology specificity studies to confirm that a widely used positive allosteric modulator specifically modulated Kv3.1 and partially Kv3.2. This compound has been developed further to be highly specific to Kv3.1 and Kv3.2, and is now widely used in the field (Liang et al., 2024).
Kv3 channel modulators work by lowering the voltage activation of the fast spiking GABAergic PVI closer to that of a normal action potential in an ex vivo mouse model (Brown et al., 2016). They enable the fast-spiking phenotype of the cells to be rescued, demonstrated in rat hippocampal studies, where the firing rate was increased in GABAergic interneurons (Boddum et al., 2017). This makes this class of drugs a novel avenue for restoring the E-I balance. Andrade-Talavera et al. (2020) used the neurotoxic amyloid beta protein 42 to reduce cognitive-relevant gamma oscillatory activity in fast spiking interneurons. When the Kv3 modulator was applied it resulted in faster activation kinetics and an increased firing rate, correlated with gamma normality. Autifony Therapeutics1 have been developing a Kv3.1b and Kv3.2 channel modulator compound, currently known as AUT00206, that is investigated in a number of disorders with underlying cognitive and social deficits such as Fragile X, epilepsy, hearing disorders, and SCZ, which may all have the common underlying mechanism of E-I imbalance.
Recent studies have shown that ketamine-challenged patients (an NMDA receptor antagonist) had an increased blood oxygen level dependent (BOLD) signal (an index of neuronal activity), which was subsequently reduced with Kv3 channel positive allosteric modulator (PAM) treatment (Deakin et al., 2019). Moreover, it normalised gamma oscillations in schizophrenic patients, further suggesting that this may be the underlying cause of cognitive impairment (Ben-Mabrouk et al., 2014). Kv3 modulators have been used to restore certain cognitive phenotypes such as motor function in an epilepsy model (Feng et al., 2024), and reversal learning deficits in a SCZ model (Leger et al., 2014a). A summary of in vitro, in vivo, and human studies where Kv3-modulating drugs have been used is shown below in Table 4.
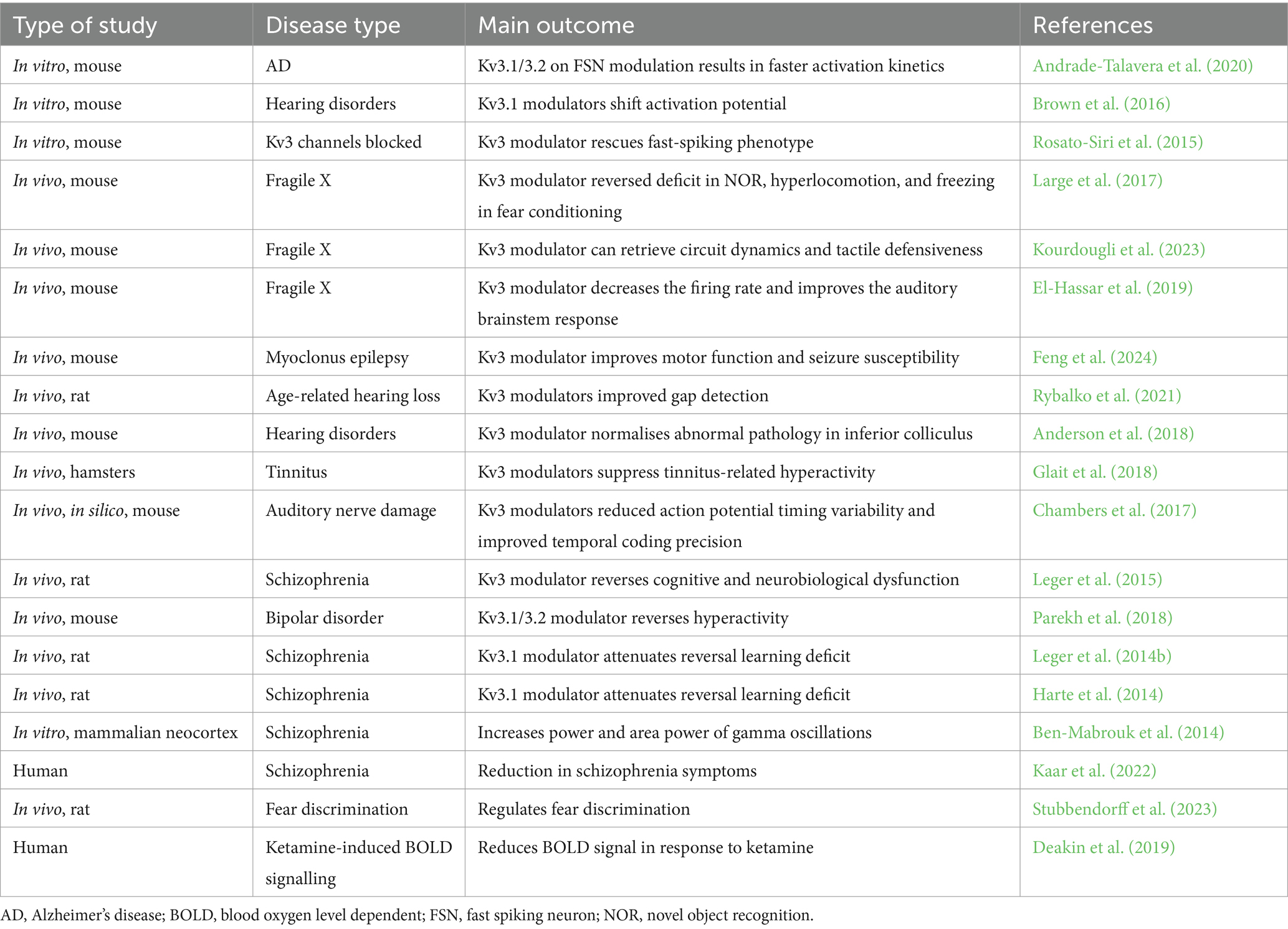
Table 4. A list of studies in which Kv3 channel modulators have been used, highlighting the disease model and the main outcome measured.
Voltage-gated potassium channels of the Kv3 subfamily are central to synaptic transmission due to their location on fast spiking neurons. These channels therefore are likely to contribute significantly to neurological and psychiatric disorders, many of which are characterised by cognitive deficits. Currently, treatment for key symptoms of many of these disorders is limited, with many therapeutics focused on symptomatic relief as opposed to directly targeting the underlying pathological mechanism. Therefore, there is an urgent need to find novel treatments for disorders with a strong cognitive component. Recent data also draw attention to the possibility that these channels can be utilised as a therapeutic avenue for treating neurological and psychiatric disorders, which would address an unmet clinical need.
Together, the collective evidence presented in this review highlights the central role of voltage-gated Kv3 channels in maintaining E-I balance. Hence, there is the exciting possibility of targeting Kv3 channel function as a new locus for novel treatment, whilst also advancing our understanding of the mechanisms by which such deficits give rise to E-I imbalance that underpin a variety of neurodevelopmental, neurological, and psychiatric disorders.
IF: Writing – review & editing, Writing – original draft. RP: Writing – review & editing, Writing – original draft. MH: Writing – review & editing. JG: Writing – review & editing. RH: Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. IF was supported by a BBSRC Doctoral Training Partnership Studentship BB/T008725/1. RP was supported by the Kennedy Trust for Rheumatology Research IMPACT Inflammation MB-PhD programme.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aggarwal, S. K., and MacKinnon, R. (1996). Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron 16, 1169–1177. doi: 10.1016/S0896-6273(00)80143-9
Allen, P., Sommer, I. E., Jardri, R., Eysenck, M. W., and Hugdahl, K. (2019). Extrinsic and default mode networks in psychiatric conditions: relationship to excitatory-inhibitory transmitter balance and early trauma. Neurosci. Biobehav. Rev. 99, 90–100. doi: 10.1016/j.neubiorev.2019.02.004
Allen, N. M., Weckhuysen, S., Gorman, K., King, M. D., and Lerche, H. (2020). Genetic potassium channel-associated epilepsies: clinical review of the Kv family. Eur. J. Paediatr. Neurol. 24, 105–116. doi: 10.1016/j.ejpn.2019.12.002
Ambrosino, P., Ragona, F., Mosca, I., Vannicola, C., Canafoglia, L., Solazzi, R., et al. (2023). A novel KCNC1 gain-of-function variant causing developmental and epileptic encephalopathy: “precision medicine” approach with fluoxetine. Epilepsia 64, e148–e155. doi: 10.1111/epi.17656
Anderson, L. A., Hesse, L. L., Pilati, N., Bakay, W. M. H., Alvaro, G., Large, C. H., et al. (2018). Increased spontaneous firing rates in auditory midbrain following noise exposure are specifically abolished by a Kv3 channel modulator. Hear. Res. 365, 77–89. doi: 10.1016/j.heares.2018.04.012
Andrade-Talavera, Y., Arroyo-García, L. E., Chen, G., Johansson, J., and Fisahn, A. (2020). Modulation of Kv3.1/Kv3.2 promotes gamma oscillations by rescuing Aβ-induced desynchronization of fast-spiking interneuron firing in an AD mouse model in vitro. J. Physiol. 598, 3711–3725. doi: 10.1113/JP279718
Ashizawa, T., Öz, G., and Paulson, H. L. (2018). Spinocerebellar ataxias: prospects and challenges for therapy development. Nat. Rev. Neurol. 14, 590–605. doi: 10.1038/s41582-018-0051-6
Autifony . (2024). About orphan neurology and our clinical programme. Available at: https://autifony.com/pipeline/orphan-epilepsy-syndromes/. (Accessed June 10, 2024)
Barot, N., Margiotta, M., Nei, M., and Skidmore, C. (2020). Progressive myoclonic epilepsy: myoclonic epilepsy and ataxia due to KCNC1 mutation (MEAK): a case report and review of the literature. Epileptic Disord. 22, 654–658. doi: 10.1684/epd.2020.1197
Bee, S., Ringland, A., and Coutellier, L. (2021). Social impairments in mice lacking the voltage-gated potassium channel Kv3.1. Behav. Brain Res. 413:113468. doi: 10.1016/j.bbr.2021.113468
Ben-Mabrouk, F., Alvaro, G., Large, C., Lebeau, F., and Cunningham, M. (2014). A novel Kv3 positive modulator augments gamma frequency oscillations in the mammalian neocortex in vitro. Schizophr. Res. 153:S130. doi: 10.1016/S0920-9964(14)70392-5
Bernardi, S., Gemignani, F., and Marchese, M. (2023). The involvement of Purkinje cells in progressive myoclonic epilepsy: focus on neuronal ceroid lipofuscinosis. Neurobiol. Dis. 185:106258. doi: 10.1016/j.nbd.2023.106258
Bertocchi, I., Cambiaghi, M., and Hasan, M. T. (2023). Advances toward precision therapeutics for developmental and epileptic encephalopathies. Front. Neurosci. 17:1140679. doi: 10.3389/fnins.2023.1140679
Bhakar, A. L., Dölen, G., and Bear, M. F. (2012). The pathophysiology of fragile X (and what it teaches us about synapses). Annu. Rev. Neurosci. 35, 417–443. doi: 10.1146/annurev-neuro-060909-153138
Boddum, K., Hougaard, C., Xiao-Ying Lin, J., von Schoubye, N. L., Jensen, H. S., Grunnet, M., et al. (2017). Kv3.1/Kv3.2 channel positive modulators enable faster activating kinetics and increase firing frequency in fast-spiking GABAergic interneurons. Neuropharmacology 118, 102–112. doi: 10.1016/j.neuropharm.2017.02.024
Börgers, C. (2017). “The PING model of gamma rhythms” in An introduction to modeling neuronal dynamics (Cham: Springer).
Botte, M., Huber, S., Bucher, D., Klint, J. K., Rodríguez, D., Tagmose, L., et al. (2022). Apo and ligand-bound high resolution Cryo-EM structures of the human Kv3.1 channel reveal a novel binding site for positive modulators. Proc. Natl. Acad. Sci. Nexus 1:83. doi: 10.1093/pnasnexus/pgac083
Brown, M. R., El-Hassar, L., Zhang, Y., Alvaro, G., Large, C. H., and Kaczmarek, L. K. (2016). Physiological modulators of Kv3.1 channels adjust firing patterns of auditory brain stem neurons. J. Neurophysiol. 116, 106–121. doi: 10.1152/jn.00174.2016
Buzsáki, G., and Wang, X.-J. (2012). Mechanisms of gamma oscillations. Annu. Rev. Neurosci. 35, 203–225. doi: 10.1146/annurev-neuro-062111-150444
Cameron, J. M., Maljevic, S., Nair, U., Aung, Y. H., Cogné, B., Bézieau, S., et al. (2019). Encephalopathies with KCNC1 variants: genotype-phenotype-functional correlations. Ann. Clin. Transl. Neurol. 6, 1263–1272. doi: 10.1002/acn3.50822
Cardin, J. A., Carlén, M., Meletis, K., Knoblich, U., Zhang, F., Deisseroth, K., et al. (2009). Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 459, 663–667. doi: 10.1038/nature08002
Carpenter, J. C., Männikkö, R., Heffner, C., Heneine, J., Sampedro-Castañeda, M., Lignani, G., et al. (2021). Progressive myoclonus epilepsy KCNC1 variant causes a developmental dendritopathy. Epilepsia 62, 1256–1267. doi: 10.1111/epi.16867
Chambers, A. R., Pilati, N., Balaram, P., Large, C. H., Kaczmarek, L. K., and Polley, D. B. (2017). Pharmacological modulation of Kv3.1 mitigates auditory midbrain temporal processing deficits following auditory nerve damage. Sci. Rep. 7:17496. doi: 10.1038/s41598-017-17406-x
Chen, Z., Brodie, M. J., Ding, D., and Kwan, P. (2023). Editorial: epidemiology of epilepsy and seizures. Front. Epidemiol. 3:1273163. doi: 10.3389/fepid.2023.1273163
Chen, P., and Hong, W. (2018). Neural circuit mechanisms of social behavior. Neuron 98, 16–30. doi: 10.1016/j.neuron.2018.02.026
Chen, Y.-T., Hong, M. R., Zhang, X.-J., Kostas, J., Li, Y., Kraus, R. L., et al. (2023). Identification, structural, and biophysical characterization of a positive modulator of human Kv3.1 channels. Proc. Natl. Acad. Sci. U.S.A. 120:e2220029120. doi: 10.1073/pnas.2220029120
Chi, G., Liang, Q., Sridhar, A., Cowgill, J. B., Sader, K., Radjainia, M., et al. (2022). Cryo-EM structure of the human Kv3.1 channel reveals gating control by the cytoplasmic T1 domain. Nat. Commun. 13:4087. doi: 10.1038/s41467-022-29594-w
Clatot, J., Currin, C. B., Liang, Q., Pipatpolkai, T., Massey, S. L., Helbig, I., et al. (2024). A structurally precise mechanism links an epilepsy-associated KCNC2 potassium channel mutation to interneuron dysfunction. Proc. Natl. Acad. Sci. U.S.A. 121:e2307776121. doi: 10.1073/pnas.2307776121
Clatot, J., Ginn, N., Costain, G., and Goldberg, E. M. (2023). A KCNC1 -related neurological disorder due to gain of Kv3.1 function. Ann. Clin. Transl. Neurol. 10, 111–117. doi: 10.1002/acn3.51707
Culotta, L., and Penzes, P. (2020). Exploring the mechanisms underlying excitation/inhibition imbalance in human iPSC-derived models of ASD. Mol. Autism. 11:32. doi: 10.1186/s13229-020-00339-0
Deakin, B., Perini, F., Nazimek, J., McKie, S., Hutchison, J. B., McFarquhar, M., et al. (2019). AUT00206, a novel Kv3 channel modulator, reduces ketamine-induced BOLD signalling in healthy male volunteers: a randomised placebo-controlled crossover trial. Schizophr. Bull. 45, S245–S246. doi: 10.1093/schbul/sbz019.388
Domanski, A. P. F., Booker, S. A., Wyllie, D. J. A., Isaac, J. T. R., and Kind, P. C. (2019). Cellular and synaptic phenotypes lead to disrupted information processing in Fmr1-KO mouse layer 4 barrel cortex. Nat. Commun. 10:4814. doi: 10.1038/s41467-019-12736-y
Duarri, A., Nibbeling, E. A. R., Fokkens, M. R., Meijer, M., Boerrigter, M., Verschuuren-Bemelmans, C. C., et al. (2015). Functional analysis helps to define KCNC3 mutational spectrum in Dutch ataxia cases. PLoS One 10:e0116599. doi: 10.1371/journal.pone.0116599
El-Hassar, L., Song, L., Tan, W. J. T., Large, C. H., Alvaro, G., Santos-Sacchi, J., et al. (2019). Modulators of Kv3 potassium channels rescue the auditory function of fragile X mice. J. Neurosci. 39, 4797–4813. doi: 10.1523/JNEUROSCI.0839-18.2019
Feng, H., Clatot, J., Kaneko, K., Flores-Mendez, M., Wengert, E. R., Koutcher, C., et al. (2024). Targeted therapy improves cellular dysfunction, ataxia, and seizure susceptibility in a model of a progressive myoclonus epilepsy. Cell Rep. Med. 5:101389. doi: 10.1016/j.xcrm.2023.101389
Figueroa, K. P., Minassian, N. A., Stevanin, G., Waters, M., Garibyan, V., Forlani, S., et al. (2010). KCNC3: phenotype, mutations, channel biophysics-a study of 260 familial ataxia patients. Hum. Mutat. 31, 191–196. doi: 10.1002/humu.21165
Figueroa, K. P., Waters, M. F., Garibyan, V., Bird, T. D., Gomez, C. M., Ranum, L. P. W., et al. (2011). Frequency of KCNC3 DNA variants as causes of spinocerebellar ataxia 13 (SCA13). PLoS One 6:e17811. doi: 10.1371/journal.pone.0017811
GBD 2016 Disease and Injury Incidence and Prevalence Collaborators (2017). Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390, 1211–1259. doi: 10.1016/S0140-6736(17)32154-2
Ghanekar, S. D., Kuo, S.-H., Staffetti, J. S., and Zesiewicz, T. A. (2022). Current and emerging treatment modalities for spinocerebellar ataxias. Expert. Rev. Neurother. 22, 101–114. doi: 10.1080/14737175.2022.2029703
Gibson, J. R., Bartley, A. F., Hays, S. A., and Huber, K. M. (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 100, 2615–2626. doi: 10.1152/jn.90752.2008
Glait, L., Fan, W., Stillitano, G., Sandridge, S., Pilati, N., Large, C., et al. (2018). Effects of AUT00063, a Kv3.1 channel modulator, on noise-induced hyperactivity in the dorsal cochlear nucleus. Hear. Res. 361, 36–44. doi: 10.1016/j.heares.2018.01.017
Gomes, F. V., and Grace, A. A. (2021). Beyond dopamine receptor antagonism: new targets for schizophrenia treatment and prevention. Int. J. Mol. Sci. 22:4467. doi: 10.3390/ijms22094467
Gonzalez-Burgos, G., and Lewis, D. A. (2012). NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr. Bull. 38, 950–957. doi: 10.1093/schbul/sbs010
Grizel, A. V., Glukhov, G. S., and Sokolova, O. S. (2014). Mechanisms of activation of voltage-gated potassium channels. Acta Nat. 6, 10–26. doi: 10.32607/20758251-2014-6-4-10-26
Gunthorpe, M. J. (2022). Timing is everything: structural insights into the disease-linked Kv3 channels controlling fast action-potential firing in the brain. Nat. Commun. 13:4086. doi: 10.1038/s41467-022-31537-4
Harte, M. K., Leger, M., Grayson, B., Marsh, S., Large, C., and Neill, J. (2014). Efficacy and relevance of the modulation of Kv3 channels to alleviate cognitive dysfunction in an animal model of schizophrenia symptamology. Schizophr. Res. 153:S273. doi: 10.1016/S0920-9964(14)70779-0
Hashemi, E., Ariza, J., Rogers, H., Noctor, S. C., and Martínez-Cerdeño, V. (2017). The number of parvalbumin-expressing interneurons is decreased in the prefrontal cortex in autism. Cereb. Cortex 27, 1931–1943. doi: 10.1093/cercor/bhw021
Herman-Bert, A., Stevanin, G., Netter, J. C., Rascol, O., Brassat, D., Calvas, P., et al. (2000). Mapping of spinocerebellar ataxia 13 to chromosome 19q13.3-q13.4 in a family with autosomal dominant cerebellar ataxia and mental retardation. Am. J. Hum. Genet. 67, 229–235. doi: 10.1086/302958
Holmes, G. L. (2020). Drug treatment of progressive myoclonic epilepsy. Pediatr. Drugs 22, 149–164. doi: 10.1007/s40272-019-00378-y
Huo, L., Wu, Q., Yang, F., Liu, X., Yang, Z., and Wang, H. (2023). Novel KCNC2 variant associated with developmental and epileptic encephalopathy. Int. J. Dev. Neurosci. 83, 357–367. doi: 10.1002/jdn.10263
Irie, T., Matsuzaki, Y., Sekino, Y., and Hirai, H. (2014). Kv3.3 channels harbouring a mutation of spinocerebellar ataxia type 13 alter excitability and induce cell death in cultured cerebellar Purkinje cells. J. Physiol. 592, 229–247. doi: 10.1113/jphysiol.2013.264309
Jardri, R., and Deneve, S. (2013). Circular inferences in schizophrenia. Brain 136, 3227–3241. doi: 10.1093/brain/awt257
Jardri, R., Duverne, S., Litvinova, A. S., and Denève, S. (2017). Experimental evidence for circular inference in schizophrenia. Nat. Commun. 8:14218. doi: 10.1038/ncomms14218
Jiruska, P., de Curtis, M., Jefferys, J. G. R., Schevon, C. A., Schiff, S. J., and Schindler, K. (2013). Synchronization and desynchronization in epilepsy: controversies and hypotheses. J. Physiol. 591, 787–797. doi: 10.1113/jphysiol.2012.239590
Juarez, P., and Martínez Cerdeño, V. (2022). Parvalbumin and parvalbumin chandelier interneurons in autism and other psychiatric disorders. Front Psychiatry 13:913550. doi: 10.3389/fpsyt.2022.913550
Kaar, S. J., Angelescu, I., Marques, T. R., and Howes, O. D. (2019). Pre-frontal parvalbumin interneurons in schizophrenia: a meta-analysis of post-mortem studies. J. Neural Transm. 126, 1637–1651. doi: 10.1007/s00702-019-02080-2
Kaar, S. J., Angelescu, I., Nour, M. M., Marques, T. R., Sharman, A., Sajjala, A., et al. (2022). The effects of AUT00206, a novel Kv3.1/3.2 potassium channel modulator, on task-based reward system activation: a test of mechanism in schizophrenia. Psychopharmacology 239, 3313–3323. doi: 10.1007/s00213-022-06216-3
Kaczmarek, L. K., and Zhang, Y. (2017). Kv3 channels: enablers of rapid firing, neurotransmitter release, and neuronal endurance. Physiol. Rev. 97, 1431–1468. doi: 10.1152/physrev.00002.2017
Khare, S., Nick, J. A., Zhang, Y., Galeano, K., Butler, B., Khoshbouei, H., et al. (2017). A KCNC3 mutation causes a neurodevelopmental, non-progressive SCA13 subtype associated with dominant negative effects and aberrant EGFR trafficking. PLoS One 12:e0173565. doi: 10.1371/journal.pone.0173565
Kim, H., Lee, S., Choi, M., Kim, H., Hwang, H., Choi, J., et al. (2018). Familial cases of progressive myoclonic epilepsy caused by maternal somatic mosaicism of a recurrent KCNC1 p.Arg320His mutation. Brain Dev. 40, 429–432. doi: 10.1016/j.braindev.2018.01.006
Kim, E., and Sheng, M. (2004). PDZ domain proteins of synapses. Nat. Rev. Neurosci. 5, 771–781. doi: 10.1038/nrn1517
Kourdougli, N., Suresh, A., Liu, B., Juarez, P., Lin, A., Chung, D. T., et al. (2023). Improvement of sensory deficits in fragile X mice by increasing cortical interneuron activity after the critical period. Neuron 111, 2863–2880.e6. doi: 10.1016/j.neuron.2023.06.009
Large, C., Pilati, N., Ga, A., Cogram, P., Srl, A., and Padova, U. (2017). AUT00206, a Kv3 channel modulator with potential for the treatment of fragile X syndrome. Fragile X conference. 10271. Available at: https://autifony.com/wp-content/uploads/2022/06/Large-et-al-2017-Fragile-X-Conference.pdf
Leger, M., Alvaro, G., Large, C., Harte, M., and Neill, J. (2015). P.3.c.003 AUT6, a novel Kv3 channel modulator, reverses cognitive and neurobiological dysfunction in a rat model of relevance to schizophrenia symptomatology. Eur. Neuropsychopharmacol. 25:S480. doi: 10.1016/S0924-977X(15)30653-2
Leger, M., Grayson, B., Marsh, S., Alvaro, G., Large, C., Harte, M., et al. (2014a). P.3.c.002 Kv3 channel modulation alleviates cognitive dysfunction and negative symptoms in an animal model of schizophrenia. Eur. Neuropsychopharmacol. 24, S515–S516. doi: 10.1016/s0924-977x(14)70825-9
Leger, M., Grayson, B., Marsh, S., Harte, M., Neill, J., and Large, C. (2014b). P.2.025 a novel KV3 ion channel modulator restores cognitive function in an animal model of cognitive impairment in schizophrenia. Eur. Neuropsychopharmacol. 24, S49–S50. doi: 10.1016/S0924-977X(14)70056-2
Lett, T. A., Voineskos, A. N., Kennedy, J. L., Levine, B., and Daskalakis, Z. J. (2014). Treating working memory deficits in schizophrenia: a review of the neurobiology. Biol. Psychiatry 75, 361–370. doi: 10.1016/j.biopsych.2013.07.026
Lewis, D. A., Curley, A. A., Glausier, J. R., and Volk, D. W. (2012). Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 35, 57–67. doi: 10.1016/j.tins.2011.10.004
Li, L., Liu, Z., Yang, H., Li, Y., Zeng, Q., Chen, L., et al. (2022). Investigation of novel de novo KCNC2 variants causing severe developmental and early-onset epileptic encephalopathy. Seizure 101, 218–224. doi: 10.1016/j.seizure.2022.09.004
Li, X., Zheng, Y., Li, S., Nair, U., Sun, C., Zhao, C., et al. (2021). Kv3.1 channelopathy: a novel loss-of-function variant and the mechanistic basis of its clinical phenotypes. Ann. Transl. Med. 9:1397. doi: 10.21037/atm-21-1885
Liang, Q., Chi, G., Cirqueira, L., Zhi, L., Marasco, A., Pilati, N., et al. (2024). The binding and mechanism of a positive allosteric modulator of Kv3 channels. Nat. Commun. 15:2533. doi: 10.1038/s41467-024-46813-8
Lin, J.-J., Meletti, S., Vaudano, A. E., and Lin, K.-L. (2022). Developmental and epileptic encephalopathies: is prognosis related to different epileptic network dysfunctions? Epilepsy Behav. 131:107654. doi: 10.1016/j.yebeh.2020.107654
Liu, Y., Ouyang, P., Zheng, Y., Mi, L., Zhao, J., Ning, Y., et al. (2021). A selective review of the excitatory-inhibitory imbalance in schizophrenia: underlying biology, genetics, microcircuits, and symptoms. Front. Cell Dev. Biol. 9:664535. doi: 10.3389/fcell.2021.664535
Lodge, D. J., Behrens, M. M., and Grace, A. A. (2009). A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J. Neurosci. 29, 2344–2354. doi: 10.1523/JNEUROSCI.5419-08.2009
Maenner, M. J., Shaw, K. A., Bakian, A. V., Bilder, D. A., Durkin, M. S., Esler, A., et al. (2021). Prevalence and characteristics of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2018. MMWR Surveill. Summ. 70, 1–16. doi: 10.15585/mmwr.ss7011a1
McDonald, A. J., and Mascagni, F. (2006). Differential expression of Kv3.1b and Kv3.2 potassium channel subunits in interneurons of the basolateral amygdala. Neuroscience 138, 537–547. doi: 10.1016/j.neuroscience.2005.11.047
McNally, J. M., and McCarley, R. W. (2016). Gamma band oscillations: a key to understanding schizophrenia symptoms and neural circuit abnormalities. Curr. Opin. Psychiatry 29, 202–210. doi: 10.1097/YCO.0000000000000244
McNally, J. M., McCarley, R. W., and Brown, R. E. (2013). Impaired GABAergic neurotransmission in schizophrenia underlies impairments in cortical gamma band oscillations. Curr. Psychiatry Rep. 15:346. doi: 10.1007/s11920-012-0346-z
McTague, A., Howell, K. B., Cross, J. H., Kurian, M. A., and Scheffer, I. E. (2016). The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 15, 304–316. doi: 10.1016/S1474-4422(15)00250-1
McWilliam, M., Asuncion, R., and Al Khalili, Y. (2024). Idiopathic (genetic) generalised epilepsy. Treasure Island, FL: StatPearls Publishing.
Mehinovic, E., Gray, T., Campbell, M., Ekholm, J., Wenger, A., Rowell, W., et al. (2022). Germline mosaicism of a missense variant in KCNC2 in a multiplex family with autism and epilepsy characterized by long-read sequencing. Am. J. Med. Genet. A 188, 2071–2081. doi: 10.1002/ajmg.a.62743
Middlebrooks, J. C., Nick, H. S., Subramony, S. H., Advincula, J., Rosales, R. L., Lee, L. V., et al. (2013). Mutation in the kv3.3 voltage-gated potassium channel causing spinocerebellar ataxia 13 disrupts sound-localization mechanisms. PLoS One 8:e76749. doi: 10.1371/journal.pone.0076749
Minassian, N. A., Lin, M.-C. A., and Papazian, D. M. (2012). Altered Kv3.3 channel gating in early-onset spinocerebellar ataxia type 13. J. Physiol. 590, 1599–1614. doi: 10.1113/jphysiol.2012.228205
Montaut, S., Apartis, E., Chanson, J.-B., Ewenczyk, C., Renaud, M., Guissart, C., et al. (2017). SCA13 causes dominantly inherited non-progressive myoclonus ataxia. Parkinsonism Relat. Disord. 38, 80–84. doi: 10.1016/j.parkreldis.2017.02.012
Mukherjee, S., Cassini, T. A., Hu, N., Yang, T., Li, B., Shen, W., et al. (2022). Personalized structural biology reveals the molecular mechanisms underlying heterogeneous epileptic phenotypes caused by de novo KCNC2 variants. Hum. Genet. Genomics Adv. 3:100131. doi: 10.1016/j.xhgg.2022.100131
Munch, A. S., Saljic, A., Boddum, K., Grunnet, M., Hougaard, C., and Jespersen, T. (2018). Pharmacological rescue of mutated Kv3.1 ion-channel linked to progressive myoclonus epilepsies. Eur. J. Pharmacol. 833, 255–262. doi: 10.1016/j.ejphar.2018.06.015
Muona, M., Berkovic, S. F., Dibbens, L. M., Oliver, K. L., Maljevic, S., Bayly, M. A., et al. (2015). A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat. Genet. 47, 39–46. doi: 10.1038/ng.3144
Nahar, L., Delacroix, B. M., and Nam, H. W. (2021). The role of parvalbumin interneurons in neurotransmitter balance and neurological disease. Front. Psych. 12:679960. doi: 10.3389/fpsyt.2021.679960
Nakamura, J. P., Schroeder, A., Hudson, M., Jones, N., Gillespie, B., Du, X., et al. (2019). The maternal immune activation model uncovers a role for the Arx gene in GABAergic dysfunction in schizophrenia. Brain Behav. Immun. 81, 161–171. doi: 10.1016/j.bbi.2019.06.009
Nascimento, F. A., and Andrade, D. M. (2016). Myoclonus epilepsy and ataxia due to potassium channel mutation (MEAK) is caused by heterozygous KCNC1 mutations. Epileptic Disord. 18, 135–138. doi: 10.1684/epd.2016.0859
NICE (2021). “Autism spectrum disorder in adults: diagnosis and management” in NICE clinical guidelines (London: National Institute for Health and Care Excellence (NICE)).
Nomura, T., Musial, T. F., Marshall, J. J., Zhu, Y., Remmers, C. L., Xu, J., et al. (2017). Delayed maturation of fast-spiking interneurons is rectified by activation of the TrkB receptor in the mouse model of fragile X syndrome. J. Neurosci. 37, 11298–11310. doi: 10.1523/JNEUROSCI.2893-16.2017
Oliver, K. L., Franceschetti, S., Milligan, C. J., Muona, M., Mandelstam, S. A., Canafoglia, L., et al. (2017). Myoclonus epilepsy and ataxia due to KCNC1 mutation: analysis of 20 cases and K+ channel properties. Ann. Neurol. 81, 677–689. doi: 10.1002/ana.24929
Parekh, P. K., Sidor, M. M., Gillman, A., Becker-Krail, D., Bettelini, L., Arban, R., et al. (2018). Antimanic efficacy of a novel Kv3 potassium channel modulator. Neuropsychopharmacology 43, 435–444. doi: 10.1038/npp.2017.155
Park, J., Koko, M., Hedrich, U. B. S., Hermann, A., Cremer, K., Haberlandt, E., et al. (2019). KCNC1-related disorders: new de novo variants expand the phenotypic spectrum. Ann. Clin. Transl. Neurol. 6, 1319–1326. doi: 10.1002/acn3.50799
Pietropaolo, S., and Provenzano, G. (2022). Editorial: targeting excitation-inhibition imbalance in neurodevelopmental and autism spectrum disorders. Front. Neurosci. 16:968115. doi: 10.3389/fnins.2022.968115
Poirier, K., Viot, G., Lombardi, L., Jauny, C., Billuart, P., and Bienvenu, T. (2017). Loss of function of KCNC1 is associated with intellectual disability without seizures. Eur. J. Hum. Genet. 25, 560–564. doi: 10.1038/ejhg.2017.3
Pomarino, D., Thren, J. R., Thren, A., Rostasy, K., and Schoenfeldt, J. (2022). Toe walking as the initial symptom of a spinocerebellar ataxia 13 in a patient presenting with a mutation in the KCNC3 gene. Glob. Med. Genet. 9, 51–53. doi: 10.1055/s-0041-1736483
Port, R. G., Dipiero, M. A., Ku, M., Liu, S., Blaskey, L., Kuschner, E. S., et al. (2019). Children with autism spectrum disorder demonstrate regionally specific altered resting-state phase–amplitude coupling. Brain Connect. 9, 425–436. doi: 10.1089/brain.2018.0653
Pyle, A., Smertenko, T., Bargiela, D., Griffin, H., Duff, J., Appleton, M., et al. (2015). Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain 138, 276–283. doi: 10.1093/brain/awu348
Rademacher, A., Schwarz, N., Seiffert, S., Pendziwiat, M., Rohr, A., van Baalen, A., et al. (2020). Whole-exome sequencing in NF1-related west syndrome leads to the identification of KCNC2 as a novel candidate gene for epilepsy. Neuropediatrics 51, 368–372. doi: 10.1055/s-0040-1710524
Rosato-Siri, M. D., Zambello, E., Mutinelli, C., Garbati, N., Benedetti, R., Aldegheri, L., et al. (2015). A novel modulator of Kv3 potassium channels regulates the firing of parvalbumin-positive cortical interneuronss. J. Pharmacol. Exp. Ther. 354, 251–260. doi: 10.1124/jpet.115.225748
Rossi, M., Perez-Lloret, S., Cerquetti, D., and Merello, M. (2014). Movement disorders in autosomal dominant cerebellar ataxias: a systematic review. Mov. Disord. Clin. Pract. 1, 154–160. doi: 10.1002/mdc3.12042
Rubenstein, J. L. R., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267. doi: 10.1034/j.1601-183X.2003.00037.x
Rybalko, N., Popelář, J., Šuta, D., Svobodová Burianová, J., Alvaro, G. S., Large, C. H., et al. (2021). Effect of Kv3 channel modulators on auditory temporal resolution in aged Fischer 344 rats. Hear. Res. 401:108139. doi: 10.1016/j.heares.2020.108139
Rydzanicz, M., Zwoliński, P., Gasperowicz, P., Pollak, A., Kostrzewa, G., Walczak, A., et al. (2021). A recurrent de novo variant supports KCNC2 involvement in the pathogenesis of developmental and epileptic encephalopathy. Am. J. Med. Genet. A 185, 3384–3389. doi: 10.1002/ajmg.a.62455
Sato, M., Nakai, N., Fujima, S., Choe, K. Y., and Takumi, T. (2023). Social circuits and their dysfunction in autism spectrum disorder. Mol. Psychiatry 28, 3194–3206. doi: 10.1038/s41380-023-02201-0
Schwarz, N., Seiffert, S., Pendziwiat, M., Rademacher, A. V., Brünger, T., Hedrich, U. B. S., et al. (2022). Spectrum of phenotypic, genetic, and functional characteristics in patients with epilepsy with KCNC2 pathogenic variants. Neurology 98, e2046–e2059. doi: 10.1212/WNL.0000000000200660
Seiffert, S., Pendziwiat, M., Hedrich, U. B. S., Helbig, I., Weber, Y., and Schwarz, N. (2023). KCNC2 variants of uncertain significance are also associated to various forms of epilepsy. Front. Neurol. 14:1212079. doi: 10.3389/fneur.2023.1212079
Shakkottai, V. G., and Fogel, B. L. (2013). Clinical neurogenetics: autosomal dominant spinocerebellar ataxia. Neurol. Clin. 31, 987–1007. doi: 10.1016/j.ncl.2013.04.006
Shao, L.-R., Habela, C. W., and Stafstrom, C. E. (2019). Pediatric epilepsy mechanisms: expanding the paradigm of excitation/inhibition imbalance. Children 6:23. doi: 10.3390/children6020023
Sharma, L. P., and Reddy, Y. C. J. (2019). Obsessive-compulsive disorder comorbid with schizophrenia and bipolar disorder. Indian J. Psychiatry 61, S140–S148. doi: 10.4103/psychiatry.IndianJPsychiatry_527_18
Smausz, R., Neill, J., and Gigg, J. (2022). Neural mechanisms underlying psilocybin’s therapeutic potential—the need for preclinical in vivo electrophysiology. J. Psychopharmacol. 36, 781–793. doi: 10.1177/02698811221092508
Specchio, N., and Curatolo, P. (2021). Developmental and epileptic encephalopathies: what we do and do not know. Brain 144, 32–43. doi: 10.1093/brain/awaa371
Stenshorne, I., Syvertsen, M., Ramm-Pettersen, A., Henning, S., Weatherup, E., Bjørnstad, A., et al. (2022). Monogenic developmental and epileptic encephalopathies of infancy and childhood, a population cohort from Norway. Front. Pediatr. 10:965282. doi: 10.3389/fped.2022.965282
Strumbos, J. G., Brown, M. R., Kronengold, J., Polley, D. B., and Kaczmarek, L. K. (2010). Fragile X mental retardation protein is required for rapid experience-dependent regulation of the potassium channel Kv3.1b. J. Neurosci. 30, 10263–10271. doi: 10.1523/JNEUROSCI.1125-10.2010
Stubbendorff, C., Hale, E., Day, H. L. L., Smith, J., Alvaro, G. S., Large, C. H., et al. (2023). Pharmacological modulation of Kv3 voltage-gated potassium channels regulates fear discrimination and expression in a response-dependent manner. Prog. Neuropsychopharmacol. Biol. Psychiatry 127:110829. doi: 10.1016/j.pnpbp.2023.110829
Symonds, J. D., Zuberi, S. M., and Johnson, M. R. (2017). Advances in epilepsy gene discovery and implications for epilepsy diagnosis and treatment. Curr. Opin. Neurol. 30, 193–199. doi: 10.1097/WCO.0000000000000433
Tada, Y., Kume, K., Matsuda, Y., Kurashige, T., Kanaya, Y., Ohsawa, R., et al. (2020). Genetic screening for potassium channel mutations in Japanese autosomal dominant spinocerebellar ataxia. J. Hum. Genet. 65, 363–369. doi: 10.1038/s10038-019-0717-y
Tamminga, C. A. (2008). Domains of dysfunction in schizophrenia: implications for diagnosis. World Psychiatry 7, 34–35. doi: 10.1002/j.2051-5545.2008.tb00147.x
Tandon, R., Nasrallah, H., Akbarian, S., Carpenter, W. T., DeLisi, L. E., Gaebel, W., et al. (2024). The schizophrenia syndrome, circa 2024: what we know and how that informs its nature. Schizophr. Res. 264, 1–28. doi: 10.1016/j.schres.2023.11.015
Thompson, M., Weickert, C. S., Wyatt, E., and Webster, M. J. (2009). Decreased glutamic acid decarboxylase(67) mRNA expression in multiple brain areas of patients with schizophrenia and mood disorders. J. Psychiatr. Res. 43, 970–977. doi: 10.1016/j.jpsychires.2009.02.005
Tombola, F., Pathak, M. M., and Isacoff, E. Y. (2005). Voltage-sensing arginines in a potassium channel permeate and occlude cation-selective pores. Neuron 45, 379–388. doi: 10.1016/j.neuron.2004.12.047
Vetri, L., Calì, F., Vinci, M., Amato, C., Roccella, M., Granata, T., et al. (2020). A de novo heterozygous mutation in KCNC2 gene implicated in severe developmental and epileptic encephalopathy. Eur. J. Med. Genet. 63:103848. doi: 10.1016/j.ejmg.2020.103848
Vezzani, A., Lang, B., and Aronica, E. (2015). Immunity and inflammation in epilepsy. Cold spring Harb. Perspect. Med. 6:a022699. doi: 10.1101/cshperspect.a022699
Wang, S., Yu, Y., Wang, X., Deng, X., Ma, J., Liu, Z., et al. (2022). Emerging evidence of genotype–phenotype associations of developmental and epileptic encephalopathy due to KCNC2 mutation: identification of novel R405G. Front. Mol. Neurosci. 15:950255. doi: 10.3389/fnmol.2022.950255
Waters, M. F., Minassian, N. A., Stevanin, G., Figueroa, K. P., Bannister, J. P. A., Nolte, D., et al. (2006). Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat. Genet. 38, 447–451. doi: 10.1038/ng1758
Wöhr, M., Orduz, D., Gregory, P., Moreno, H., Khan, U., Vörckel, K. J., et al. (2015). Lack of parvalbumin in mice leads to behavioral deficits relevant to all human autism core symptoms and related neural morphofunctional abnormalities. Transl. Psychiatry 5:e525. doi: 10.1038/tp.2015.19
Yanagi, M., Joho, R. H., Southcott, S. A., Shukla, A. A., Ghose, S., and Tamminga, C. A. (2014). Kv3.1-containing K+ channels are reduced in untreated schizophrenia and normalized with antipsychotic drugs. Mol. Psychiatry 19, 573–579. doi: 10.1038/mp.2013.49
Ye, H., and Kaszuba, S. (2017). Inhibitory or excitatory? Optogenetic interrogation of the functional roles of GABAergic interneurons in epileptogenesis. J. Biomed. Sci. 24:93. doi: 10.1186/s12929-017-0399-8
Keywords: Kv3 channels, schizophrenia, autism, epilepsy, ataxia, cognition, neurodevelopment
Citation: Faulkner IE, Pajak RZ, Harte MK, Glazier JD and Hager R (2024) Voltage-gated potassium channels as a potential therapeutic target for the treatment of neurological and psychiatric disorders. Front. Cell. Neurosci. 18:1449151. doi: 10.3389/fncel.2024.1449151
Edited by:
Luigi Catacuzzeno, University of Perugia, ItalyReviewed by:
Isabella Ferando, University of Miami, United StatesCopyright © 2024 Faulkner, Pajak, Harte, Glazier and Hager. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabel E. Faulkner, SXNhYmVsLmZhdWxrbmVyQHBvc3RncmFkLm1hbmNoZXN0ZXIuYWMudWs=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.