 Ravi Philip Rajkumar
Ravi Philip Rajkumar- Department of Psychiatry, Jawaharlal Institute of Postgraduate Medical Education and Research, Pondicherry, India
The significance of potassium channels in neuropsychiatric disorders
Potassium (K+) channels are a diverse group of ion channels which regulate the excitability and stability of biological membranes through their effects on inward K+ flux, leading to reduced excitability and hyperpolarization. Contemporary classifications recognize anywhere from three to five subtypes of K+ channels, based on their subunit composition, number of transmembrane domains, and functional properties. The four most widely recognized subtypes are: (a) voltage-gated K channels (KV), (b) calcium (Ca++)-activated K channels (KCa), (c) inward-rectifying K channels (KIR), and (d) two-pore domain K channels (K2P). Apart from these, there are ligand-gated K channels that are activated by specific molecules, such as cyclic nucleotides (Kuang et al., 2015).
K+ channels are highly expressed in several brain regions, including the frontal cortex, basal ganglia, hippocampus and amygdala, where they influence neuronal firing, transmitter release, and neural plasticity. Mendelian disorders involving K+ channel-related mutations in the brain have been associated with developmental delays, epilepsy, and symptoms suggestive of anxiety, hyperactivity, and autism spectrum disorder (Alam et al., 2023). This has led researchers to investigate the possible contribution of K+ channel functioning to non-Mendelian psychiatric syndromes. Such research has found possible evidence of altered K+ channel activity in schizophrenia, depression, and autism spectrum disorders. This raises the possibility of novel therapeutic approaches aimed at modulating the functioning of these channels (Vukadinovic and Rosenzweig, 2012; Cheng et al., 2021; Meshkat et al., 2024). More recent research has highlighted the importance of K+ channels in anxiety- and fear-related processes. In animal models, KV channels have been found to play a key role in fear conditioning and anxiety-like behaviors (Stubbendorff et al., 2023; Page and Coutellier, 2024). In humans, polymorphisms in genes encoding KV and KIR channel subunits have been associated with vulnerability to anxiety disorders in youth (Thapaliya et al., 2023). This paper examines recent translational evidence implicating changes in K+ channel functioning in the pathogenesis of post-traumatic stress disorder.
Understanding the neurobiology of post-traumatic stress disorder
Post-traumatic stress disorder (PTSD) is a chronic psychiatric syndrome caused by exposure either to a single, overwhelming traumatic stressor or chronic traumatic stress. PTSD affects about 6–8% of the global population, and is characterized by intrusive “re-experiencing” of the traumatic event, increased arousal, avoidance of trauma-related cues, and associated changes in thought and mood, which can persist for months or years after a traumatic exposure. Currently approved treatments for PTSD include serotonergic antidepressants and specific types of psychotherapy, but their efficacy is often limited (Lee et al., 2016).
The neurobiology of PTSD is complex and involves dysregulation of neurotransmitter, neuroendocrine and immune-inflammatory pathways (Ressler et al., 2022). The core features of PTSD probably reflect dysregulation of fear-related processes, involving neural circuits connecting the ventromedial prefrontal and anterior cingulate cortices to the insula and limbic structures such as the hippocampus and the central and basolateral regions of the amygdala (France and Jovanovic, 2023). Distinct symptom domains of PTSD may reflect alterations in discrete neural circuits. For example, hypervigilance may reflect increased amygdala activity, while alterations in sleep may result from additional alterations in the functioning of the insula, hippocampus, and dorsal anterior cingulate cortex (Ressler et al., 2022).
This neuroanatomical diversity reflects a diversity in pathogenic cellular mechanisms. In a recent genome-wide association study (GWAS) of over 150,000 patients with PTSD, 43 genes coding for neurotransmitter receptors, ion channels, neural development, synaptic structure and function, and the regulation of endocrine and immune responses were all associated with vulnerability to this disorder. A common thread that unites this diverse set of genes is that they all are involved in fear-, threat- and stress-related psychophysiological responses (Nievergelt et al., 2024). A smaller GWAS of trauma-exposed adults found a suggestive association between a polymorphism of the DPP6 gene and certain specific symptoms of PTSD, such as experiences of unreality and detachment from one's surroundings (Wolf et al., 2014). DPP6 codes for a protein that is associated with a particular subtype of voltage-gated (KV) K+ channel, which is involved in the regulation of dendritic excitability and synaptic integration of information (Sun et al., 2011). This raises the possibility that K+ channel subtypes play a role in the development of specific PTSD symptoms, and may even represent potential therapeutic targets.
Potassium channels in animal models of fear-related phenomena
Research involving rodent models of fear memory, conditioning and extinction has found that at least three types of K+ channels are involved in these processes. Kir3 channels, also known as GIRK channels, are tetrameric, G-protein-gated inwardly rectifying (KIR) K+ channels, which exist in four varieties labeled GIRK1 through GIRK4. GIRK1 and GIRK2 are more highly expressed in the brain. Activation of these channels with an experimental agent has been found to facilitate the extinction of conditioned fear responses in mice, probably through increased GIRK inhibitory tone in the basolateral amygdala (BLA) (Xu et al., 2020). GIRK channels can be activated by the neurotransmitter gamma-aminobutyric acid (GABA) through GABAB receptors, and it has been found that GIRK-mediated currents in the prelimbic area of the medial prefrontal cortex are stronger in male than in female mice. These gender differences in GIRK-mediated fear extinction may account for the increased susceptibility of women to PTSD following exposure to trauma (Fernandez de Velasco et al., 2015).
KCNQ, a type of KV K+ channel, appears to inhibit the consolidation of fear-related memories in the BLA. The use of a KCNQ agonist was associated with impaired fear consolidation in mice. Inhibition of the KCNQ-mediated current through direct antagonism, or through the activation of muscarinic M1, adrenergic β2, or dopaminergic D5 receptors, had the opposite effect, leading to enhanced fear consolidation. This suggests that monoamine transmitters may influence fear memory consolidation through their effects on this channel (Young and Thomas, 2014). In primates, activation of the α1 adrenergic receptor in the prefrontal cortex has complex effects: postsynaptic α1 receptors on dendritic spines lead to KCNQ opening and reduced cortical activity, while presynaptic α1 receptors increase cortical activity. It has been hypothesized that the former mechanism is operative at high levels of stress, leading to reduced cortical regulation of subcortical fear and stress responses (Datta et al., 2019). This may explain why the α1-receptor antagonist prazosin is effective in some patients with PTSD. More importantly, these results highlight the importance of considering the localization of K+ channels when evaluating their effects on fear-related disorders. In this case, KCNQ activation in the amygdala appears to protect against PTSD, but may have the opposite effect in the prefrontal cortex.
SK channels, which are KCa-type K+ channels, may also have significant effects on fear conditioning in mice. More specifically, they may inhibit the activity of infralimbic cortical neurons involved in fear extinction. In rats undergoing fear conditioning, blocking SK channels had no immediate effect on fear responses, but increased the extinction of fear responses on the next day; activation of SK channels led to hyperpolarization of infralimbic neurons and reduced fear extinction (Criado-Marrero et al., 2014). A somewhat different picture was obtained in the mouse amygdala, where fear conditioning was associated with reduced SK2 channel expression in the BLA, while fear extinction was associated with increased numbers of synaptic SK2 channels, an effect which appeared to be mediated by the synaptic regulator protein membrane palmitoylated protein 2 (MPP2) (Peng et al., 2023). KCa channel activation may also inhibit the increased excitability of the lateral amygdala caused by chronic stress in rats (Rosenkranz et al., 2010). These results are remarkably similar to those observed with KCNQ. Overall, potassium channel activation in the BLA may inhibit the consolidation of fear memories, while activation of the same channels in adjacent regions of the medial prefrontal cortex may inhibit their extinction (Criado-Marrero et al., 2014).
Potassium channels in animal models of PTSD
PTSD-like behavior can be induced in animals through exposure to experimental trauma, which involves prolonged immobilization and forced swimming (single prolonged stress, SPS) either alone or in combination with electric foot shock (single prolonged stress and shock, SPS&S) (Zhang et al., 2019). These animal models of PTSD have also been studied in relation to potential changes in K+ channel expression and functioning. Hyperpolarization-activated cyclic nucleotide-gated channel 1 (HCN1), a six-transmembrane domain K+ channel highly expressed in brain regions such as the frontal cortex and hippocampus (Zhao et al., 2023), was examined in rats exposed to SPS&S. It was found that inhibition of HCN1 alleviated PTSD-like behaviors, while administration of a HCN1 activator increased them. These behavioral changes appeared to be related to the brain-derived neurotrophic factor (BDNF)-mTOR signaling pathway, which is involved in synaptic plasticity, and activation of HCN1 appears to antagonize this pathway (Ni et al., 2020). In a separate study of mice, prenatal exposure to alcohol increased the likelihood of PTSD-like behavior in offspring exposed to electric foot shock. This susceptibility was associated with increased expression of HCN1 in the prefrontal cortex, but not the hippocampus, and administration of a HCN1 antagonist increased fear extinction and reduced PTSD- and depressive-like behavior (Yao et al., 2023). In an independent study of the SPS&S model of PTSD in rats, exposure to SPS&S was associated with increased expression of HCN1 and reduced expression of BDNF. Administration of ketamine, an antagonist of the N-methyl d-aspartate (NMDA) glutamate receptor, ameliorated PTSD-like behavior and led to reduced HCN1 expression and increased BDNF levels. Overall, a negative correlation was observed between prefrontal BDNF and HCN1 expression (Hou et al., 2018). Similar results were found in an SPS model of PTSD in mice, where the administration of ketamine reduced PTSD-like behavior and normalized stress-induced elevations in HCN1 in the prefrontal cortex, but not in the hippocampus (Zhang X. et al., 2021). The consistency and replicability of these findings suggest that prefrontal HCN1 K+ channel expression increases after traumatic stress, and may contribute to PTSD symptoms through an apparently antagonistic effects on the neurotrophic and neuroplasticity-enhancing effects of BDNF.
Two other K+ channel subtypes have been tentatively implicated in mouse models of PTSD. In the first, PTSD-like behavior and hippocampal expression of the KIR channel Kir4.1 were reduced by the administration of a ginsenoside, and increased by intra-cerebroventricular injection of the pro-inflammatory cytokine TNFα in mice exposed to SPS (Zhang Z. et al., 2021). In the second, apparent improvements in PTSD-like behavior in an SPS&S model were observed with a polyherbal extract, and these beneficial changes were associated with increased phosphorylation of the KV channel Kv4.2 in the hippocampus (Park et al., 2023). As these results have not yet been replicated, their significance is uncertain.
Other possible links between potassium channels and PTSD
There are other indirect sources of evidence linking altered K+ channel functioning to PTSD, derived from research not directly involving animal models of this disorder. Increasing the expression of the outward rectifying KV channel Kv1.1 through a viral vector is associated with reduced BLA firing and reduced hippocampal neurogenesis in rats, though the relevance of these changes to PTSD is uncertain (Kirby et al., 2012). Animals exposed to chronic stress exhibit increased K+ channel opening in the prefrontal cortex, which appears to be mediated by the activation of α1-adrenergic receptors and D1 dopamine receptors (Datta and Arnsten, 2019). Conversely, the “anti-stress” peptide transmitter neuropeptide Y (NPY) has been associated with activation of GIRK channels, leading to reduced activity of the BLA which may protect against the development of PTSD (Tasan et al., 2016).
The evidence reviewed above is summarized in Figure 1 below.
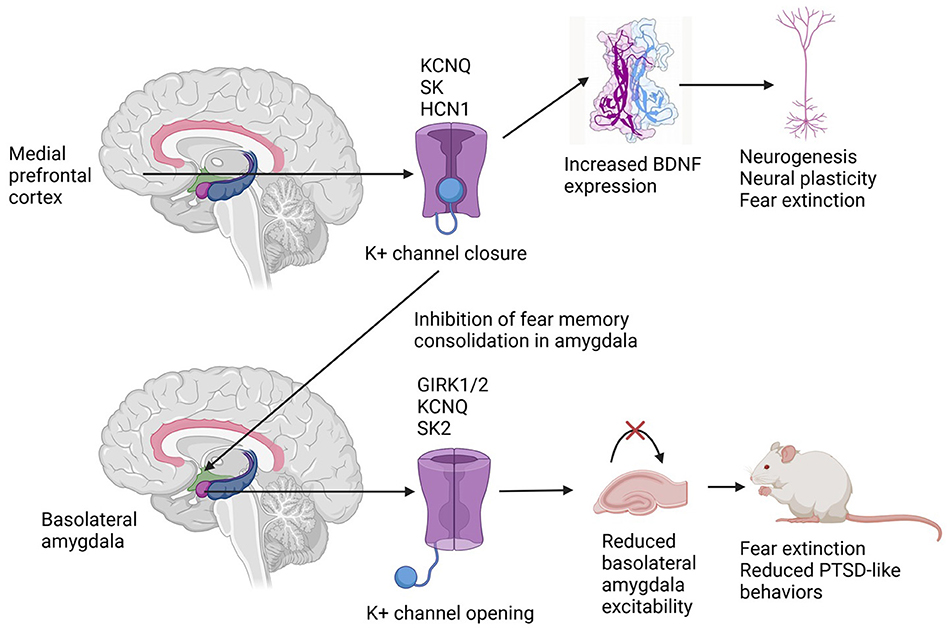
Figure 1. Differential effects of potassium channel subtype activation in the medial prefrontal cortex and the basolateral amygdala in rodent models of post-traumatic stress disorder. See the text for explanations of potassium channel nomenclature.
Limitations of the available evidence
The realistic appraisal of the evidence presented above requires an acknowledgment of both conceptual and methodological limitations. First, it is not clear to what extent animal models of PTSD, such as SPS and SPS&S, truly overlap with the syndrome of PTSD in trauma-exposed humans. Second, findings of K+ channel alterations in murine and primate brains may not “translate” directly to human brain physiology: therefore, these results require replication in humans. Third, the relationship between K+ channel activity and PTSD-like phenomena is complex, and depends crucially on factors such as the specific model of PTSD, the species being studied, the brain region being studied, and the pharmacological agents used to activate or block specific K+ channels. Finally, it is likely that specific K+ channel subtypes represent only one of many molecular mechanisms involved in PTSD, and that their activity depends crucially on levels of specific neurotransmitters, hormones, and even immune-inflammatory regulatory proteins. These limitations highlight the need for research on altered K+ channel functioning in humans with PTSD and other disorders related to traumatic stress.
Summary and conclusions
The evidence available from animal models suggests that K+ channels from several families—voltage-gated, inward rectifying and calcium-activated—could play a role in the pathogenesis of PTSD-like phenomena following exposure to traumatic stress. At the cortical level, K+ channel activation may maintain PTSD symptoms by interfering with neural plasticity and fear extinction; at the limbic level, and particularly in specific regions of the amygdala, K+ channel activation may help in fear extinction and reduce PTSD symptoms. Pharmacological therapies aimed at “balancing” or “stabilizing” K+ channel activity between these brain regions may offer advantages over existing treatments for PTSD, and it is possible that some emerging treatments for this disorder, such as ketamine and prazosin, may act partly through their effects on K+ flux through specific channel types. The development of clinically effective and safe activators or antagonists of these channel subtypes may represent a significant step forward in the management of this chronic and disabling condition.
Author contributions
RR: Conceptualization, Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
Figure 1 was created with BioRender.com.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alam, K. A., Svalastoga, P., Martinez, A., Glennon, J. C., and Haavik, J. (2023). Potassium channels in behavioral brain disorders. Molecular mechanisms and therapeutic potential: a narrative review. Neurosci. Biobehav. Rev. 152:105301. doi: 10.1016/j.neubiorev.2023.105301
Cheng, P., Qiu, Z., and Du, Y. (2021). Potassium channels and autism spectrum disorder: an overview. Int. J. Dev. Neurosci. 81, 479–491. doi: 10.1002/jdn.10123
Criado-Marrero, M., Santini, E., and Porter, J. T. (2014). Modulating fear extinction memory by manipulating SK potassium channels in the infralimbic cortex. Front. Behav. Neurosci. 8:96. doi: 10.3389/fnbeh.2014.00096
Datta, D., and Arnsten, A. F. T. (2019). Loss of prefrontal cortical higher cognition with uncontrollable stress: molecular mechanisms, changes with age, and relevance to treatment. Brain Sci. 9:113. doi: 10.3390/brainsci9050113
Datta, D., Yang, S. T., Galvin, V. C., Solder, J., Luo, F., Morozov, Y. M., et al. (2019). Noradrenergic α1-adrenoceptor actions in the primate dorsolateral prefrontal cortex. J. Neurosci. 39, 2722–2734. doi: 10.1523/JNEUROSCI.2472-18.2019
Fernandez de Velasco, E. M., Hearing, M., Xia, Z., Victoria, N. C., Lujan, R., and Wickman, K. (2015). Sex differences in GABAB-GIRK signaling in layer 5/6 pyramidal neurons of the mouse prelimbic cortex. Neuropharmacology 95, 353–360. doi: 10.1016/j.neuropharm.2015.03.029
France, J. M., and Jovanovic, T. (2023). Human fear neurobiology reimagined: can brain-derived biotypes predict fear-based disorders after trauma? Neurosci. Biobehav. Rev. 144:104988. doi: 10.1016/j.neubiorev.2022.104988
Hou, L., Qi, Y., Sun, H., Wang, G., Li, Q., Wang, Y., et al. (2018). Applying ketamine to alleviate the PTSD-like effects by regulating the HCN1-related BDNF. Prog. Neuropsychopharmacol. Biol. Psychiatry 86, 313–321. doi: 10.1016/j.pnpbp.2018.03.019
Kirby, E. D., Friedman, A. R., Covarrubias, D., Ying, C., Sun, W. G., Goosens, K. A., et al. (2012). Basolateral amygdala regulation of adult hippocampal neurogenesis and fear-related activation of newborn neurons. Mol. Psychiatry 17, 527–536. doi: 10.1038/mp.2011.71
Kuang, Q., Purhonen, P., and Hebert, H. (2015). Structure of potassium channels. Cell. Mol. Life Sci. 72, 3677–3693. doi: 10.1007/s00018-015-1948-5
Lee, D. J., Schnitzlein, C. W., Wolf, J. P., Vythilingam, M., Rasmusson, A. M., and Hoge, C. W. (2016). Psychotherapy versus pharmacotherapy for posttraumatic stress disorder: systematic review and meta-analyses to determine first-line treatments. Depress. Anxiety 33, 792–806. doi: 10.1002/da.22511
Meshkat, S., Kwan, A. T. H., Le, G. H., Wong, S., Rhee, T. G., Ho, R., et al. (2024). The role of KCNQ channel activators in management of major depressive disorder. J. Affect. Disord. 359, 364–372. doi: 10.1016/j.jad.2024.05.067
Ni, L., Xu, Y., Dong, S., Kong, Y., Wang, H., and Lu, G. (2020). The potential role of the HCN1 ion channel and BDNF-mTOR signaling pathways and synaptic transmission in the alleviation of PTSD. Transl. Psychiatry 10:101. doi: 10.1038/s41398-020-0782-1
Nievergelt, C. M., Maihofer, A. X., Atkinson, E. G., Chen, C. Y., Choi, K. W., Coleman, J. R. I., et al. (2024). Genome-wide association analyses identify 95 risk loci and provide insights into the neurobiology of post-traumatic stress disorder. Nat. Genet. 56, 792–808. doi: 10.1038/s41588-024-01707-9
Page, C. E., and Coutellier, L. (2024). Kv3.1 voltage-gated potassium channels modulate anxiety-like behaviors in female mice. Neuroscience 538, 68–79. doi: 10.1016/j.neuroscience.2023.12.011
Park, H. R., Cai, M., and Yang, E. J. (2023). Novel psychopharmacological herbs relieve behavioral abnormalities and hippocampal dysfunctions in an animal model of post-traumatic stress disorder. Nutrients 15:3815. doi: 10.3390/nu15173815
Peng, X., Chen, P., Zhang, Y., Wu, K., Ji, N., Gao, J., et al. (2023). MPP2 interacts with SK2 to rescue the excitability of glutamatergic neurons in the BLA and facilitate the extinction of conditioned fear in mice. CNS Neurosci. Ther. 30:e14362. doi: 10.1111/cns.14362
Ressler, K. J., Berretta, S., Bolshakov, V. Y., Rosso, I. M., Meloni, E. G., Rauch, S. L., et al. (2022). Post-traumatic stress disorder: clinical and translational neuroscience from cells to circuits. Nat. Rev. Neurol. 18, 273–288. doi: 10.1038/s41582-022-00635-8
Rosenkranz, J. A., Venheim, E. R., and Padival, M. (2010). Chronic stress causes amygdala hyperexcitability in rodents. Biol. Psychiatry 67, 1128–1136. doi: 10.1016/j.biopsych.2010.02.008
Stubbendorff, C., Hale, E., Day, H. L. L., Smith, J., Alvaro, G. S., Large, C. H., et al. (2023). Pharmacological modulation of Kv3 voltage-gated potassium channels regulates fear discrimination and expression in a response-dependent manner. Prog. Neuropsychopharmacol. Biol. Psychiatry 127:110829. doi: 10.1016/j.pnpbp.2023.110829
Sun, W., Maffie, J. K., Lin, L., Petralia, R. S., Rudy, B., and Hoffman, D. A. (2011). DPP6 establishes the A-type K(+) current gradient critical for the regulation of dendritic excitability in CA1 hippocampal neurons. Neuron 71, 1102–1115. doi: 10.1016/j.neuron.2011.08.008
Tasan, R. O., Verma, D., Wood, J., Lach, G., Horner, B., de Lima, T. C. M., et al. (2016). The role of neuropeptide Y in fear conditioning and extinction. Neuropeptides 55, 111–126. doi: 10.1016/j.npep.2015.09.007
Thapaliya, B., Ray, B., Farahdel, B., Suresh, P., Sapkota, R., IMAGEN Consortium, et al. (2023). Cross-continental environmental and genome-wide association study on children and adolescent anxiety and depression. medRxiv. [preprint]. doi: 10.1101/2023.02.06.23285530
Vukadinovic, Z., and Rosenzweig, I. (2012). Abnormalities in thalamic neurophysiology in schizophrenia: could psychosis be a result of potassium channel dysfunction? Neurosci. Biobehav. Rev. 36, 960–968. doi: 10.1016/j.neubiorev.2011.11.005
Wolf, E. J., Rasmusson, A. M., Mitchell, K. S., Logue, M. W., Baldwin, C. T., and Miller, M. W. (2014). A genome-wide association study of clinical symptoms of dissociation in a trauma-exposed sample. Depress. Anxiety 31, 352–360. doi: 10.1002/da.22260
Xu, Y., Cantwell, L., Molosh, A. I., Plant, L. D., Gazgalis, D., Fitz, S. D., et al. (2020). The small molecule GAT1508 activates brain-specific GIRK1/2 channel heteromers and facilitates conditioned fear extinction in rodents. J. Biol. Chem. 295, 3614–3634. doi: 10.1074/jbc.RA119.011527
Yao, H., Wang, C., and Xia, Z. (2023). Prenatal alcohol exposure enhanced alcohol preference and susceptibility to PTSD in a sex-dependent manner through the synaptic HCN1 channel. J. Affect. Disord. 324, 143–152. doi: 10.1016/j.jad.2022.12.069
Young, M. B., and Thomas, S. A. (2014). M1-muscarinic receptors promote fear memory consolidation via phospholipase C and the M-current. J. Neurosci. 354, 1570–1578. doi: 10.1523/JNEUROSCI.1040-13.2014
Zhang, L., Hu, X. Z., Li, H., Li, X., Yu, T., Dohl, J., et al. (2019). Updates in PTSD animal models characterization. Methods Mol. Biol. 2011, 331–344. doi: 10.1007/978-1-4939-9554-7_19
Zhang, X., Zhao, Y., Du, Y., Sun, H., Zhang, W., Wang, A., et al. (2021). Effect of ketamine on mood dysfunction and spatial cognition deficits in PTSD mouse models via HCN1–BDNF signaling. J. Affect. Disord. 286, 248–258. doi: 10.1016/j.jad.2021.02.058
Zhang, Z., Song, Z., Shen, F., Xie, P., Wang, J., Zhu, A., et al. (2021). Ginsenoside Rg1 prevents PTSD-like behaviors in mice through promoting synaptic proteins, reducing Kir4.1 and TNF-α in the hippocampus. Mol. Neurobiol. 58, 1550–1563. doi: 10.1007/s12035-020-02213-9
Keywords: post-traumatic stress disorder, potassium channels, HCN1, KCNQ, GIRK, amygdala, medial prefrontal cortex
Citation: Rajkumar RP (2024) Potassium channels in animal models of post-traumatic stress disorder: mechanistic and therapeutic implications. Front. Cell. Neurosci. 18:1441514. doi: 10.3389/fncel.2024.1441514
Received: 31 May 2024; Accepted: 17 July 2024;
Published: 30 July 2024.
Edited by:
Ilenio Servettini, University of Naples Federico II, ItalyReviewed by:
Niels Decher, University of Marburg, GermanyCopyright © 2024 Rajkumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ravi Philip Rajkumar, cmF2aS5wc3ljaEBnbWFpbC5jb20=