 Keyuan Ren1†
Keyuan Ren1† Qinglong Wang1†
Qinglong Wang1† Douglas Jiang1,2
Douglas Jiang1,2 Ethan Liu1Julie Alsmaan1,3Rui Jiang1,3
Ethan Liu1Julie Alsmaan1,3Rui Jiang1,3 Seward B. Rutkove1*
Seward B. Rutkove1* Feng Tian1*
Feng Tian1*- 1Department of Neurology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
- 2Scripps Institution of Oceanography, San Diego, CA, United States
- 3School of Arts and Science, Harvard College, Cambridge, MA, United States
Amyotrophic lateral sclerosis (ALS), a devastating neurodegenerative disease, is characterized by progressive motor neuron degeneration, leading to widespread weakness and respiratory failure. While a variety of mechanisms have been proposed as causes of this disease, a full understanding remains elusive. Electrophysiological alterations, including increased motor axon excitability, likely play an important role in disease progression. There remains a critical need for non-animal disease models that can integrate electrophysiological tools to better understand underlying mechanisms, track disease progression, and evaluate potential therapeutic interventions. This review explores the integration of electrophysiological technologies with ALS disease models. It covers cellular and clinical electrophysiological tools and their applications in ALS research. Additionally, we examine conventional animal models and highlight advancements in humanized models and 3D organoid technologies. By bridging the gap between these models, we aim to enhance our understanding of ALS pathogenesis and facilitate the development of new therapeutic strategies.
1 Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterized by the degeneration of both upper motor neurons (UMNs) in the motor cortex and lower motor neurons (LMNs) in the brainstem and spinal cord. This progressive neuronal loss results in widespread muscle weakness and atrophy, ultimately leading to death due to respiratory failure. ALS has a global incidence of approximately 2 cases per 100,000 person-years (Hardiman et al., 2017). Approximately 5–10% of ALS cases are familial, inherited typically in an autosomal dominant manner, while the majority of cases are sporadic, arising without a clear familial history (Kim et al., 2020; Akcimen et al., 2023). Several factors have been implicated in increasing the likelihood of disease, including sex (men have a moderately higher risk), exposure to certain environmental factors, and military service. However, the disease often appears unexpectedly in otherwise healthy middle-aged individuals (Kiernan et al., 2011; Grad et al., 2017). The progression rate of ALS varies among patients, but most succumb to respiratory failure within approximately 2–5 years of symptom onset. Current therapies of ALS remain very limited, and none of them can fundamentally halt motor neuron loss.
The pathophysiology of ALS remains complex and likely multifactorial. Researchers have observed extensive structural and functional damage to motor neurons, including the degeneration of axons and dendrites. In nearly 97% of patients, ALS is associated with TAR DNA-binding protein 43 (TDP-43) pathology. Affected motor neurons exhibit a shift in TDP-43 protein from cell nuclei to the cytoplasm, where they form dense, skein-like aggregates (Wijesekera and Nigel Leigh, 2009; Grad et al., 2017). However, there is considerable heterogeneity in the pathology of the disease. Mutations in the SOD1 (superoxide dismutase 1) and FUS (fused in sarcoma) genes also cause ALS with very dissimilar pathology (Saccon et al., 2013). Additionally, the most common genetic subtype of ALS is caused by the expansion of a hexanucleotide repeat (GGGGCC) within the C9orf72 (chromosome 9 open reading frame 72) gene, which has also been associated with TDP-43 abnormalities (DeJesus-Hernandez et al., 2011; Renton et al., 2011). This diversity suggests that ALS pathogenesis likely involves multiple pathways, including abnormal protein aggregation within cells and the integrated stress response (Costa-Mattioli and Walter, 2020). These findings underscore the complexity of ALS pathology and may provide valuable directions for potential therapeutic approaches.
One broader pathophysiological concept that has been of interest for some time is excitotoxicity. Excitotoxicity has been proposed as a key underlying mechanism in ALS pathogenesis. For example, spinal motor neurons in mSOD1 mouse embryos demonstrate hyperexcitability (Gunes et al., 2020), which precedes subsequent hypoexcitability and neurodegeneration. This fluctuating excitability pattern is observed in both UMNs and LMNs, highlighting the intricate dynamics throughout the course of the disease. Additionally, there is ongoing debate regarding cortical neuronal hyperexcitability in ALS. It remains unclear whether this hyperexcitability contributes to excitotoxicity or functions as some form of neuroprotective mechanism. Simultaneously, peripheral axonal hyperexcitability has also been described (Bostock et al., 1995), suggesting that this may be a generalized phenomenon contributing to ALS pathogenesis. Denervation, particularly characterized by neuromuscular junction (NMJ) deficits, is another critical pathological mechanism in ALS (Tallon et al., 2022; Verma et al., 2022).
A complex network of pathological changes shapes the progression of ALS. Within the cortical circuitry, there is heightened interconnectivity among motor cortex regions and a concurrent reduction in cortical inhibitory function (Van den Bos et al., 2018). Simultaneously, spinal circuit pathology is characterized by dysregulated neuronal activity mediated by neurotransmitters such as glutamate and GABA/glycine (Ramírez-Jarquín et al., 2014; Brownstone and Lancelin, 2018; Ramírez-Jarquín and Tapia, 2018; Falgairolle and O'Donovan, 2020). Despite extensive research, the specific input circuits primarily contributing to ALS pathology remain uncertain, presenting a crucial area for further investigation. Recent studies have identified glial cells as significant contributors to the disease process, challenging the conventional belief in the intrinsic susceptibility of motoneurons in ALS. The recognition of glial cells as significant contributors to ALS pathogenesis suggests new avenues for therapeutic interventions and further complicates our understanding of ALS (Lasiene and Yamanaka, 2011; Ban et al., 2019; Henstridge et al., 2019).
To investigate the complex pathophysiology of ALS, researchers have developed various experimental models, including both animal and humanized models. Animal models, such as transgenic mice expressing mutant forms of human genes (e.g., SOD1, TDP-43, FUS, and C9orf72), have been instrumental in studying disease mechanisms and testing potential therapies (Philips and Rothstein, 2015). These models mimic many aspects of ALS pathology, including motor neuron degeneration, protein aggregation, and neuroinflammation. However, they also have limitations, such as differences in disease progression and pathology compared to human ALS, prompting a shift towards humanized models like human-induced pluripotent stem cells (iPSCs), which can differentiate into motor neurons and other relevant cell types, thereby providing a more accurate system to study ALS (Bhargava et al., 2022). The advent of iPSC technology enables investigation of patient-specific genetic background and their impact on ALS pathology, thereby offering broad applications in disease modeling, cell therapy, and drug screening and aiding in the identification of patient-specific therapeutic targets (Fujimori et al., 2018; Du et al., 2023). More recently, 3D organoid models have emerged as a promising tool for ALS research. These models offer a more physiologically relevant environment compared to conventional 2D cultures, allowing the study of complex cell–cell interactions and tissue architecture (Sun X. et al., 2018). Organoids derived from ALS patient iPSCs can recapitulate key features of the disease, such as TDP-43 aggregation and neuronal hyperexcitability, providing valuable insights into ALS mechanisms and potential therapeutic targets (Tamaki et al., 2023).
Another important challenge in ALS research is effectively tracking disease deterioration in both the laboratory and clinical settings so as to understand progression rates and the effects of potential therapies (Chipika et al., 2019). A variety of approaches and biomarkers have been suggested over the years. Clinically, these include imaging studies, such as magnetic resonance imaging (MRI) to evaluate brain, spinal cord, or muscle volume, and ultrasound to measure muscle thickness (Grolez et al., 2018; Leoni et al., 2022). Serological assays such as assessment of serum neurofilament heavy, neurofilament light, and cystatin C, have also been advocated (Steffen et al., 2022; Zhu et al., 2023b). In the laboratory, such tools are less critical since cells can be directly visualized and characterized, and animals can be euthanized and disease status easily quantified. Still, animal biomarkers that are relatively translatable to humans can be helpful in a variety of ways, including therapy validation. Among these tools, electrophysiological techniques are especially useful due to their flexibility and relevance. Indeed, given the profound alterations in motor neuron function and number and the consequent alterations in the NMJ and myofiber, electrophysiologic approaches have remained a foundational approach for assessing basic pathophysiology, disease progression and potential response to new therapies.
In this review, we explore the range of electrophysiological tools available for studying ALS. We discuss the integration of these tools with various ALS models, including conventional animal models and emerging humanized and 3D organoid models, highlighting their important role in tracking disease progression and investigating underlying mechanisms.
2 The electrophysiological tools to synergize disease modeling of ALS
Electrophysiological assessments can be generally categorized into cellular and organismal techniques. Cellular electrophysiology includes tools such as patch clamp, calcium imaging, and multielectrode arrays (MEAs) (Neher and Sakmann, 1992; Grienberger and Konnerth, 2012; Soscia et al., 2020). These tools have been used to elucidate many primary features of ALS, including abnormalities in single motor neuron resting membrane potential and specific channel dysfunctions (Wainger et al., 2014). Tools for organismal assessment can be applied to humans as well as animal models and include entities such as motor unit number estimation (MUNE), excitability testing, and electrical impedance myography (EIM) (Shefner et al., 2011; Sanchez and Rutkove, 2017; Wainger et al., 2021). Organoid technology represents a unique intersection in this classification. While organoids can be studied using cellular electrophysiological techniques to explore cellular responses and interactions, they also bridge the gap to organismal-level studies. This is due to their ability to mimic tissue and organ structures, allowing for complex tissue-level electrophysiological investigations (Qian et al., 2019).
2.1 Cellular electrophysiological tools
2.1.1 Patch clamp
Ion channels, embedded in the cell membrane, are responsible for signal transmission between and within cells (Neher and Sakmann, 1992). These transmissions are typically fostered by the movement of cations such as Na+, K+, and Ca2+ (Kornreich, 2007). The patch clamp technique involves creating a high-resistance seal on a patch of the cell membrane with a glass micropipette, allowing for the precise measurement of electrical activity of a single cell, even across a single ion channel (Figure 1A). This technique provides exceptional temporal resolution, making it ideal for capturing rapid changes in ion channel activity (Passaro and Stice, 2020). For example, Yang et al. used whole-cell patch clamp on LMNs derived from iPSCs from day 18 to 28 of differentiation and measured significantly more action potentials on day 28 (Yang et al., 2023). Wainger et al. also used patch clamp on iPSC-derived motor neurons on day 28, observing a significant increase of minimal current step that triggers action potential with retigabine, which reduced hyperexcitability (Wainger et al., 2014). Despite its many benefits, achieving low-noise measurements with the patch clamp can be extremely laborious, requiring delicate manipulation of the pipette and detailed fabrication of the electrodes (Zhao et al., 2008). While providing exceptional temporal resolution, patch clamp offers little spatial resolution, making it less suitable for studying large networks of neurons (Passaro and Stice, 2020).
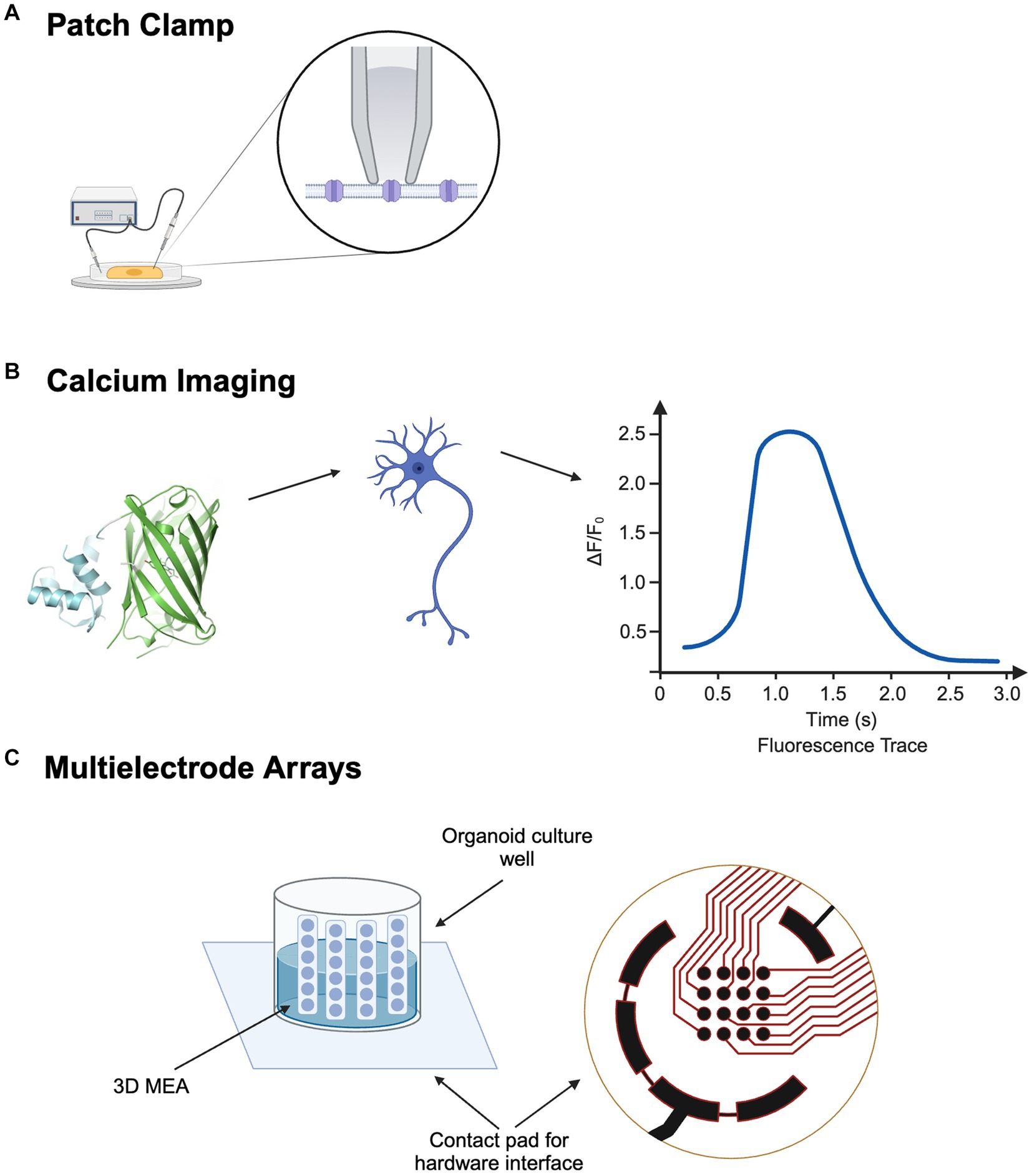
Figure 1. Electrophysiological Approaches for Cellular and Network Analysis in ALS: (A) Patch Clamp for ion channel profiling. (B) Calcium Imaging for neuronal network activity. (C) 3D Multi-electrode Array. Created with BioRender.com.
2.1.2 Calcium imaging
Calcium ions play an essential role in a myriad of cellular activities across virtually all cell types (Berridge et al., 2000). By leveraging numerous fluorescent calcium indicators, calcium imaging allows the visualization and measurement of the status of calcium cations in cells (Figure 1B) (Grienberger and Konnerth, 2012). This technique is particularly instrumental in mapping neural activities, as neuronal firing is closely linked to fluctuations in intracellular Ca2+ levels. While calcium imaging offers compromised temporal resolution, it compensates with higher spatial resolution, providing large-scale activity information (Passaro and Stice, 2020). When integrated with optogenetics, calcium imaging significantly enhances the analysis of neural circuits within organoids, mapping and elucidating interactions among different pathways by manipulating ion channels (Stamatakis et al., 2018). This combined methodology has shown potential in drug discovery and screening by enabling precise observation of drug effects on neural activity and calcium dynamics. However, despite these advantages, calcium imaging’s less optimal temporal resolution can be a limitation for capturing rapid changes in cellular activity (Peterka et al., 2011).
2.1.3 MEAs for detection of neuronal and myofiber action potentials
Neurons can be plated onto MEAs, where microelectrodes are embedded on the bottom of the dish (Figure 1C). After axons and dendrites form uncountable synaptic connections, the MEA acts as a neural interface where network activity can be manipulated by artificial electrical stimulations (Hales et al., 2010). While effective for 2D cultures, conventional MEAs face limitations with 3D organoids (Passaro and Stice, 2020). To combat this limitation, 3D MEAs have been developed. With the addition of implantable MEA probes, vertical cross-sections of the 3D organoid can be measured with increasing spatial and temporal precision (Lam et al., 2021). For example, Soscia et al. developed a polyimide-based 3D MEA, where the vertical positions of the probes were held by the mechanical forces of plastic deformation. Recordings were conducted over 38 days on a human iPSC neuronal culture. Electrodes began to pick up electrophysiological signals by day 15, and 70 out of 80 electrodes across three arrays were active at the end of 38 days (Soscia et al., 2020). While the insertion of probes allows MEA to measure electrical activity across 3D cultures, it may also inflict considerable harm to the neural network. Recent advancements in flexible bioelectronics have led to the creation of soft, conformable electronic devices, where a 2D electronic device can be folded into a 3D structure, allowing it to delicately wrap around 3D brain organoids and facilitate a less invasive electrical interface (Tasnim and Liu, 2022). Wainger et al. used a 64-electrode MEA to compare action potentials in iPSC-derived motor neurons from ALS and control subjects, revealing more spontaneous action potentials in ALS samples (Wainger et al., 2014).
2.2 Clinical electrophysiological tools
On the clinical side, both hyperexcitability and hypoexcitability have been described as features of ALS, with corresponding tools, including peripheral nerve excitability testing and transcranial magnetic stimulation (TMS) being extensively studied (Suzuki et al., 2022; Verma et al., 2022; De Carvalho and Swash, 2023). The assessment of motor neuron number has been pursued for even longer, utilizing methods such as motor unit number estimation (MUNE) and motor unit number index (MUNIX) (Shefner et al., 2011; Jacobsen et al., 2018). NMJ abnormalities can be quantified via repetitive nerve stimulation or jitter analysis via single fiber electromyography (Sanders et al., 2022; Zhu et al., 2023a). And finally, the consequent atrophied state of skeletal muscle can be quantified using EIM (Sanchez and Rutkove, 2017).
2.2.1 MUNE/MUNIX
Reliable outcome measurement tools are needed to accurately diagnose ALS and assess therapy effectiveness. Assessment tools such as the ALS Functional Rating Scale-Revised (ALSFRS-R) or handheld dynamometry have been used in many clinical trials but are limited by variability between research subjects and reliance on patient motivation and mood (Benatar et al., 2016). MUNE and MUNIX offer interesting alternatives, since they provide quantitative assessments of collective motor unit number and health that are entirely independent of patient mood or motivation (Figure 2A) (Gawel, 2019). A study comparing ALSFRS-R to MUNE demonstrated a greater mean rate of decline per year as well as a lower coefficient of variation for MUNE, supporting its value over ALSFRS-R in future clinical trials (Shefner et al., 2011). The individual motor unit potential (MUP) amplitude or area is another measure that shows changes with reinnervation in ALS. However, evaluating the MUP does not provide information about motor unit loss and only MUPs produced by early recruited motor units can be measured (Jacobsen et al., 2018). MUNIX, offers a simpler, more automated approach for obtaining an index (versus an estimation) of the number of functioning motor units and continues to be studied as a useful alternative approach (Fatehi et al., 2018). Finally, the compound motor action potential (CMAP) itself is sensitive to decline and can be followed over time (Figure 2A) (Shefner et al., 2011).
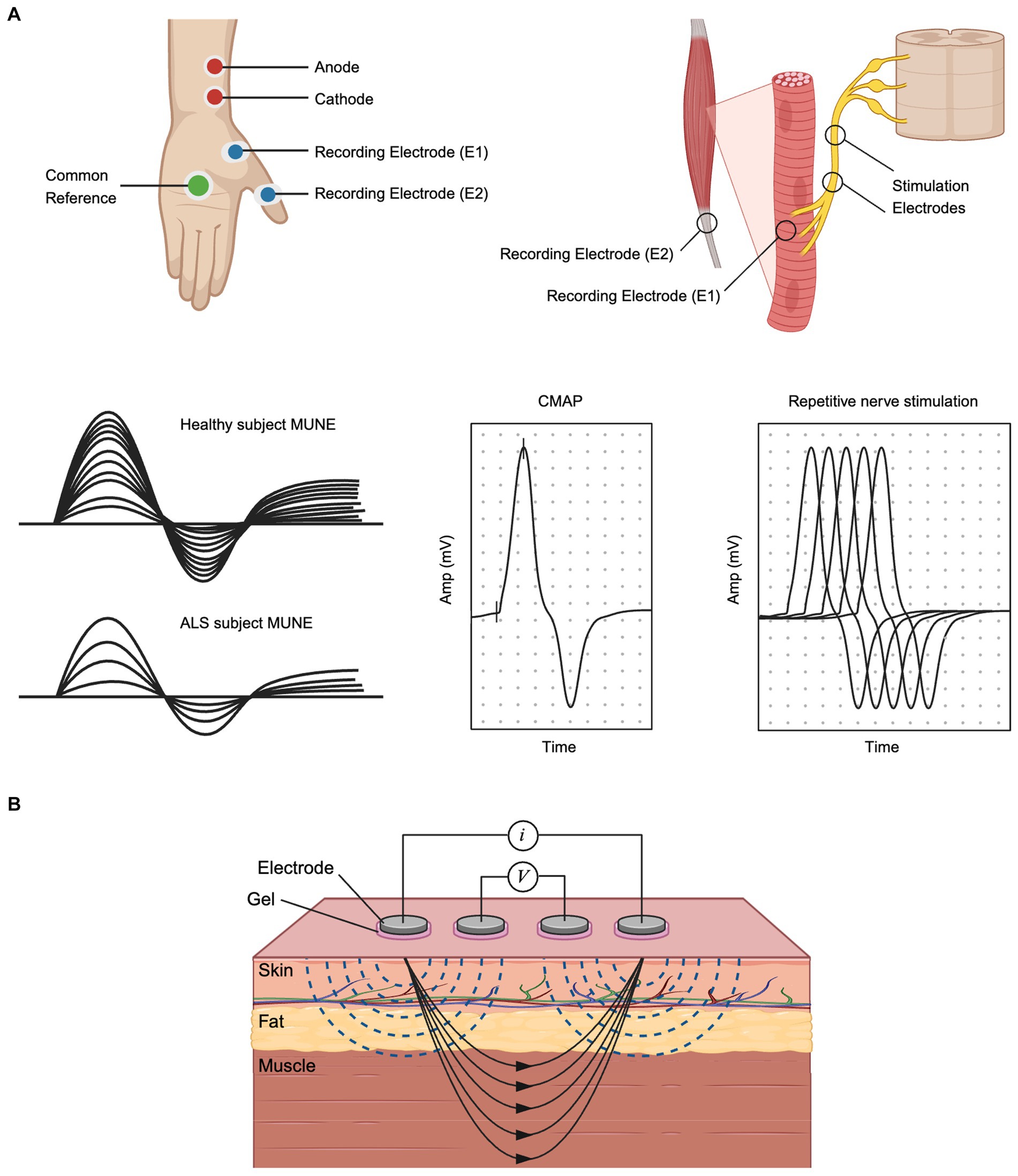
Figure 2. Electrophysiological Approaches for Structural and Functional Analysis in ALS: (A) MUNE and CMAP recording setup and typical MUNE, CMAP and RNS waveforms. Stimulation electrodes are placed on the nerve, while recording electrodes are placed on the muscle. (B) Electrical Impedance Myography. Created with BioRender.com.
2.2.2 Excitability testing
Both central and peripheral hyperexcitability and hypoexcitability are characteristic features of ALS; therefore, tools such as TMS and neuronal excitability testing (NET) are valuable for understanding ALS pathogenesis, progression, and therapy impact. TMS techniques such as cortical motor threshold (CMT) and the motor evoked potential amplitude (MEP) have been used to study disease progression in ALS (Wang et al., 2017; Yu et al., 2021). However, the sample size of these TMS studies is small and lack inter-test comparisons and multi-center reproducibility (De Carvalho and Swash, 2023). TMS measurements are also limited by progressive muscle degeneration and increasing cortical inexcitiability as ALS progresses. Therefore, TMS studies, such as peristimulus time histogram (PSTH), cortical silent period (CSP), and short-interval intracortical inhibition (SICI), which investigate cortical inhibition evaluate early disease-course abnormalities, show promise (Wainger et al., 2021; De Carvalho and Swash, 2023). Studies using TMS and median nerve excitability testing have revealed that patients with ALS exhibit both cortical and peripheral hyperexcitability compared to controls; additionally, only peripheral hyperexcitability and not cortical hyperexcitability is associated with the extent of peripheral burden (Suzuki et al., 2022).
2.2.3 NMJ testing: repetitive stimulation and single fiber electromyography
Repetitive nerve stimulation (RNS) is a commonly applied assessment of NMJs (Figure 2A). It has been known since 1959 that decremental responses in low-frequency RNS (LF-RNS) are a common phenotype in ALS patients (Sun X. S. et al., 2018). Studies applying LF-RNS to study regions and patterns of decremental response have found that degeneration of NMJ could occur before that of motor neurons, and damage of the NMJ occurred at both post-and pre-synaptic membranes (Sun X. S. et al., 2018). Similarly, single fiber electromyography has long been known to be an early finding of denervation with resulting NMJ instability (described as increased jitter) (Liu et al., 2013). However, given the time intensive nature of this procedure, it is not typically used in the clinical setting to diagnosis ALS or evaluate its progression.
2.2.4 Electrical impedance myography (EIM)
EIM is a non-invasive bioimpedance-based technique that evaluates neuromuscular health by recording voltages from muscle tissue in response to a weak, high-frequency electrical current (Figure 2B) (Sanchez and Rutkove, 2017). Changes in the composition and histology of muscle, which are common to nearly all neuromuscular disorders, including ALS, impact the impedance properties of the tissue, which can be readily captured by technology. Indeed, EIM is especially sensitive to denervation-associated atrophy of ALS, making it a biomarker uniquely sensitive to disease progression in ALS (Shefner et al., 2018). Critically, unlike nearly all standard electrophysiologic measures, EIM does not measure the innate electrical properties of the muscle (e.g., myofiber depolarization). Accordingly, EIM can be readily performed on ex vivo muscle tissue, which has been a major area of study, showing alterations in muscular dystrophy animal models (Li et al., 2014; Pandeya et al., 2021), disuse/microgravity analogues (Semple et al., 2020), and sciatic crush (Ahad et al., 2009). It can also even be performed on very small animals, such as zebrafish (Rutkove et al., 2023b). Given the simplicity of measurement, EIM can be readily adapted to be used on human organoids that include myofibers as a component, providing a rapid assessment of the integrity and health of the tissue. Indeed, the same MEAs that are used to perform standard electrophysiological analysis can also be used for EIM, with 2 (or more) electrodes being selected for current application and a separate 2 (or more) electrodes selected for voltage measurement. Finally, we note that while in the context of ALS, impedance techniques have been used predominantly on muscle, there is nothing precluding their use on brain organoids as well.
3 Conventional animal models of ALS
With the identification of multiple ALS susceptibility genes, various gene-edited animal models have been employed for investigating ALS pathology. While these animal models offer certain research advantages, they also possess inherent limitations (Table 1). Several animal models, from the most primitive to most complex, commonly used in ALS research and the potential applications of neurophysiology are described here.

Table 1. Comparative overview of animal and humanized models in ALS research.
3.1 Caenorhabditis elegans
The complete anatomical structure of the C. elegans cell lineage and gene sequence has been deciphered, revealing a highly conserved gene sequence with humans. As a disease model, C. elegans possesses characteristics such as a short life cycle, simple gene editing capabilities, and a transparent whole life cycle, making it an ideal simplified model for long-term observation research on chronic diseases. A variety of ALS models have been established in C. elegans, including C9orf72, SOD1, TDP-43, and FUS models (Roussos et al., 2023). Based on the C. elegans model, Yoshifumi Sonobe and his colleagues demonstrated a conserved role for eif-2D/elF2D in dipeptide repeats expression (Sonobe et al., 2021). Additionally, axonal degeneration and NMJ pathology are also reproduced in the C. elegans model (Markert et al., 2020; Baskoylu et al., 2022; Tossing et al., 2022). Despite its advantages, the nematode model has significant limitations, such as the absence of visceral metabolic pathways, a blood–brain barrier, and myelinated neurons.
A variety of electrophysiological tools have been employed to unravel the complexities of neural and muscular functions in C. elegans. Patch-clamp techniques have been pivotal in studying ion channel functions and electrophysiological properties of neurons and muscle cells, providing insights into the modulation of neuronal function and mechanotransduction (Ronan and Gillespie, 2005). Calcium imaging is another critical tool, used to visualize intracellular calcium dynamics and understand neuronal signaling and muscle contractions, illuminating the role of calcium channels in these processes (Goodman et al., 2012). Other electrophysiological approaches, such as MEA, EIM, or MUNE have yet to be attempted given the organism’s very small size.
3.2 Drosophila melanogaster
Drosophila, commonly known as the fruit fly, emerges as a pivotal genetic model, offering profound insights into disease mechanisms. The utility of Drosophila is anchored in its amenability to straightforward genetic manipulation, diverse methodologies for drug administration, which includes feeding, injection, as well as various assays that could be used to evaluate disease progression, including climbing and crawling tests, eclosion rate, phenotypical and electrophysiological observations and screenings. These methodologies facilitate a comprehensive understanding of ALS pathogenesis, closely mirroring human symptomatology in aspects of survival, motor function, neuron degeneration, and cellular and mitochondrial anomalies. A unique feature of Drosophila in ALS studies are its visual system. Approximately two-thirds of vital genes in the Drosophila genome play roles in eye development, rendering the fly’s eye a potent tool for studying neurodegeneration. The eye’s structured array of approximately 800 ommatidia, each comprising photoreceptor neurons and pigment cells, allows for the discernment of subtle genetic alterations manifesting as morphological aberrations, thus enabling the investigation of potential genetic modifiers, including C9orf72, SOD1, FUS, TDP-43 and Ataxin-2 (Casci and Pandey, 2015).
However, limitations in this model must be acknowledged. The fruit fly’s brain and major organ anatomy markedly differ from humans, and it lacks an adaptive immune system. Furthermore, the absence of complex cognitive abilities in flies can constrain behavioral studies. Additionally, divergent drug responses between Drosophila and humans necessitate comparative analyses across various animal models. Despite these constraints, Drosophila remains an invaluable organism for rapid, cost-effective genetic screening, and elucidating ALS’s genetic and mechanistic pathways, thus bolstering the search for novel diagnostics and therapeutics for ALS (Liguori et al., 2021).
Patch-clamp techniques have been refined for Drosophila to allow recordings from specific neuronal types, enabling a detailed study of the electrical properties of fly neurons. For example, Fernández-Chiappe and Muraro provided a comprehensive protocol for mastering this technique in Drosophila brain neurons, highlighting its utility in neuroscientific research (Fernandez-Chiappe and Muraro, 2022). Calcium imaging is another critical tool in Drosophila research, used to observe intracellular calcium dynamics. Maimon et al. adapted this technique for tethered flying and walking Drosophila, revealing how the gain of visual motion–processing interneurons changes with locomotor state (Maimon et al., 2010). As with C. elegans, the small size of Drosophila makes it challenging to study with techniques typically geared for larger animals.
3.3 Danio rerio (zebrafish)
As vertebrates, zebrafish are phylogenetically closer to humans than nematodes or Drosophila, and can be easily subjected to simple behavioral tests (Oliveira et al., 2023). As an emerging ALS model organism, zebrafish offer advantages such as relatively easy genetic regulation, highly conserved disease-related genes, and efficient drug screening compared to other vertebrate models. Additionally, the transparency of zebrafish embryos provides a significant advantage for observing ALS progression during development (Morrice et al., 2018). The zebrafish genome and connectomes have been completely mapped, making them an ideal simplified model for studying signaling pathways (Howe et al., 2013; Kunst et al., 2019; Zhang et al., 2021). Currently, commonly used zebrafish ALS models include SOD1, TDP-43, FUS, C9orf72, and other environmental neurotoxins exposure models (Chia et al., 2022). ALS models in zebrafish can be achieved through chemical induced pathological alterations (Powers et al., 2017; Roy et al., 2017; Martin et al., 2021) or through the expression of disease-related gene mutations in systematic pathways such as the integrated stress pathway (Fortier et al., 2020; Campanari et al., 2021; Petel Legare et al., 2023). These models have revealed mechanisms underlying multiple signaling pathways (Shaw et al., 2018; Swinnen et al., 2018; Pant and Nazarko, 2021). However, zebrafish lack a clear connection between cortical and spinal motor neurons; thus, while very effective at mirroring lower motor neuron aspects of the disease, their relevance to upper motor neuron pathology is far less certain.
Patch-clamp recordings have been effectively used in zebrafish to study spinal neurons and their development. For example, Saint-Amant and Drapeau described a preparation for obtaining patch-clamp recordings from embryonic spinal cord interneurons, motoneurons, and sensory neurons in zebrafish, allowing the study of spontaneous and touch-evoked electrical activity during spinal cord development (Saint-Amant and Drapeau, 2003). Calcium imaging is another significant tool; Brette et al. presented a method for isolating ventricular myocytes from zebrafish heart and used calcium imaging to study their electrophysiological characteristics, which is pertinent for cardiac alteration research in ALS (Brette et al., 2008). EIM has now been successfully applied to zebrafish to evaluate the effects of aging (Rutkove et al., 2023a,b), and studies of ALS progression mutant SOD1 zebrafish are currently ongoing (Rutkove et al., unpublished results). Other standard clinical neurophysiological studies may have potential to be applied to zebrafish, such as obtaining motor unit potentials, MUNE, and single fiber electromyography (EMG).
3.4 Mus musculus (mouse)
The mouse remains the most commonly utilized animal model for studying ALS due to its neuroanatomical and cardiovascular similarities with humans, making it extensively employed in ALS pathological investigations, drug screening, and clinical trials (Morrice et al., 2018). The optimization of gene editing technology and the emergence of tools such as circuit-specific virus labeling have enabled more efficient investigation into the contribution of specific signaling pathways to ALS disease progression in mouse models (Lee et al., 2020; Rodriguez et al., 2021; Van Schoor et al., 2022; Ziff et al., 2022). With the intact brain-spine-muscle axis preserved in the mouse model, further investigations can systematically elucidate the underlying mechanisms involved in the pathological development of ALS (Quarta et al., 2018; Haney et al., 2019; Bayer et al., 2021; Amalyan et al., 2022). Although studies in mice have provided insights into various mechanisms underlying ALS, treatments developed based on mouse models often fail to demonstrate efficacy in human disease (Bonifacino et al., 2021). Part of the reason for this is that most of these models rely on over expression of toxic molecular components, which may not accurately mimic human disease.
The patch-clamp technique is a cornerstone in this research, with automated systems developed for in vivo applications in the mouse cortex and hippocampus (Kodandaramaiah et al., 2012). Calcium imaging is also instrumental; for example, Ladewig and Keller used it to study calcium oscillations in motoneurons, which is important for understanding ALS (Ladewig and Keller, 2000). Given this animal’s larger size than those listed earlier, MUNE (Shefner et al., 2006) and EIM (Li et al., 2013) have both been applied.
3.5 Other animal models
Including rats, dogs, and non-human primates, have significantly contributed to our comprehension of ALS pathophysiology and the evaluation of potential therapeutic avenues. Rats, with their larger body size and greater tissue availability, have served as valuable models in ALS research. A recent study showcased a robust correlation between EIM and MUNE data with ALS progression in rat models (Wang et al., 2011). Notably, both EIM and MUNE exhibited strong associations with survival, with EIM phase-slope emerging as the most reliable indicator of disease progression. Degenerative myelopathy (DM) is a late adult-onset, progressive neurodegenerative condition in dogs that shares similarities with some forms of SOD1-associated human ALS. Studies on DM in dogs explore the utility of EIM in assessing muscle pathology, demonstrating that EIM, particularly 100 kHz phase values, offers sensitive detection of muscle pathology in DM-affected dogs. This highlights the potential of EIM as a valuable tool for monitoring disease progression and evaluating therapeutic interventions in veterinary medicine (Kowal et al., 2022). Another study examines the feasibility of MUNE in canine neurology, measuring the number of motor units in healthy dogs and those with various neurological disorders (Kauder et al., 2015). Non-human primates have also been utilized as animal models in studying ALS, as primates share many genetic, developmental, and physiological similarities with humans (Bang et al., 2022). Researchers using Cynomolgus monkeys have been able to create a primate model of C9orf72 related dipeptide repeats (DPRs) and show that poly (PR) DPRs are sufficient to induce C9-ALS/frontotemporal dementia (FTD)-like symptoms in primates (Xu L. et al., 2023). Additionally, researchers using Cynomolgus monkeys with overexpressed human wild-type TDP-43 in their spinal motoneurons have been able to detect TDP-43 mislocalization in almost all the large motoneurons of the lateral nuclear group at the early or even the presymptomatic stage. This finding is consistent with the observation that, in human ALS patients, the highest percentage of neurons with TDP-43 mislocalization is found in the early stages of the disease. In contrast, mouse models seem to show that overexpression of wild-type TDP-43 in the nuclei is sufficient to be toxic to spinal motoneurons (Uchida et al., 2012). These inconsistent results across different animal models may indicate that TDP-43 mediated pathology is variable across animal species and that human-relevant pathology may be more accurately modeled in species more closely related to humans. However, it is important to note the limited published data on using standard electrophysiological tools in non-human primates. A recent thesis has detailed a novel approach using rhesus macaques, where the overexpression of human TDP-43 in selective spinal motoneuron populations induced motor neuron disease (MND)-like pathogenesis (Jones, 2022). The study used clinical monitoring techniques such as MRI, EMG, and nerve stimulation, revealing a progression of changes analogous to those observed in human MND. While these findings provide compelling evidence of shared MND-like pathogenesis in primates, further research is needed to comprehensively evaluate the utility of these electrophysiological tools in non-human primate models.
4 Humanized models of ALS and translational applications of 3D organoids
In short, the expression of ALS mutations in animal models often fails to accurately replicate the diverse phenotypes observed in human disease. Given the limitations of animal models in disease treatment, there has been a growing emphasis on utilizing humanized models for research purposes (Bhargava et al., 2022). Differences in epigenetics between animal models and the human brain, as well as variations in cell types and neural networks, contribute to the limited predictive ability of animal models for drug efficacy. Additionally, the human brain exhibits longer axons and a greater number of synaptic connections, which pose challenges when attempting to replicate these features in animal models (Okano and Morimoto, 2022).
4.1 iPSC and patient derived-iPSC for disease modeling
iPSCs are pluripotent stem cells generated by introducing specific transcription factors into somatic cells. This reprogramming process allows the cells to acquire the morphology and growth properties of embryonic stem cells (ESCs) (Takahashi and Yamanaka, 2006). iPSCs are invaluable for disease research due to their capacity for continuous differentiation and self-renewal (Chehelgerdi et al., 2023). Before iPSC-based models were developed, ESCs were the primary pluripotent stem cells used in research. However, ESCs are challenging to obtain and present ethical concerns regarding the use of embryos. iPSCs circumvent these ethical issues and offer a robust, patient-specific platform for disease modeling, drug screening, and cell therapy development (Cerneckis et al., 2024). This is particularly advantageous for studying diseases like ALS, where patient-derived iPSCs can be differentiated into motor neurons, enabling precise disease modeling and therapeutic testing.
iPSC models hold significant potential for advancing pathological research and facilitating drug prediction. Electrophysiological characterization of human iPSC-derived motor neurons with SOD1 mutations and C9orf72 repeat expansions have revealed a membrane hyperexcitability phenotype (Burley et al., 2022). Moreover, research conducted on human iPSC-derived neurons, specifically motor neurons carrying the TDP-43 Q331K mutation associated with ALS, has shown a decrease in the firing rate of action potentials (Ronchi et al., 2021). The current state of research demonstrates the successful generation of phenotypes associated with ALS mutations using iPSC models, leading to the elucidation of multiple signaling pathways and biomarkers in patients (Badu-Mensah et al., 2020; Li et al., 2022; Zhang et al., 2022).
Following the introduction of 3D organoid models, iPSCs have gained some utility in the intersection of organoids and iPSCs. With recent advances with the application of iPSCs in disease research, iPSCs have been incorporated in the generation of 3D organoid models to form iPSC-derived 3D organoid models. These organ-specific iPSC-derived 3D organoid models capture more in vivo characteristics of cells than conventional two-dimensional in vitro models while removing limitations of imaging difficulty, confounding variables, and availability constraints that animal models often have (Xu Z. et al., 2023).
4.2 iPSC-based 2D models
iPSC-based 2D models are highly valued by researchers for their efficiency, cost-effectiveness, and ethical benefits. These models are particularly useful for high-throughput screening and detailed cellular analysis. However, they have notable limitations, including reduced cell–cell interactions and the inability to mimic the complex three-dimensional structure of tissues. This simplification can result in altered cellular behaviors, such as changes in apoptosis pathways (Yamada and Cukierman, 2007). Despite these challenges, iPSC-based 2D models continue to be instrumental in early-stage disease modeling and drug discovery, providing critical insights into cellular responses and disease mechanisms.
4.3 3D organoid models
Compared to 2D iPSC models, 3D organoids offer a more faithful representation of the physiological architecture of in vivo tissues, exhibiting not only different cell types but also structural characteristics (Figure 3). For instance, 3D organoids can replicate human cortical folds. Leveraging the structural advantage of organoids, an increasing number of studies have been dedicated to investigating neuron–neuron and neuron-astrocyte communication in organoid models (Meijboom et al., 2022; Wang et al., 2022; de Majo et al., 2023).
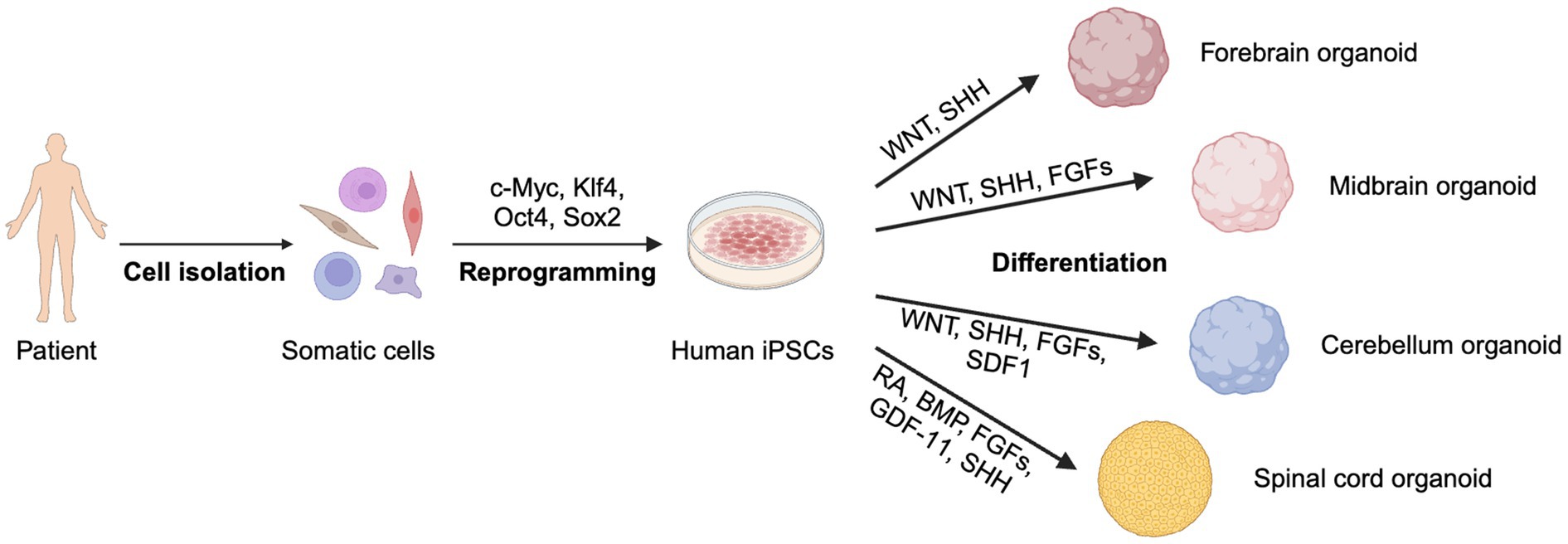
Figure 3. Generation of 3D organoids from patient-derived iPSCs. Somatic cells are reprogrammed into iPSCs, which differentiate into forebrain, midbrain, cerebellum, and spinal cord organoids using specific signaling factors. Created with BioRender.com.
Recent research has demonstrated the successful generation of long-term cultures of cortical brain organoid slices (cBOS), revealing mature neurons and astrocytes organized in complex architecture. Employing whole-cell patch-clamp and calcium imaging techniques, researchers observed subthreshold synaptic inputs, action potential firing, and synchronous large-scale oscillations in cBOS (Petersilie et al., 2024). This method offers the potential to investigate the hyperexcitability and hypoexcitability characteristics of motor neurons in ALS.
In addition to cBOS, recent advancements in intact 3D organoid models have shown significant promise. Intact 3D organoids maintain their structural integrity without being sectioned into slices, allowing for more comprehensive studies of tissue-level electrophysiological properties. For example, human cortical spheroids, a type of intact 3D organoid, sustain spontaneous excitatory postsynaptic currents (sEPSCs), exhibit action potentials, and display complex network activities, making them valuable for modeling cortical development and neurological diseases (Pasca et al., 2015). These studies have demonstrated that intact organoids can exhibit more complex and physiologically relevant behaviors compared to cultured slices, such as long-range neuronal connectivity and three-dimensional tissue interactions.
Moreover, recent advancements in MEAs have overcome limitations posed by organoid complexity, allowing precise detection of neuronal and myofiber action potentials (Soscia et al., 2020). These innovations enable less invasive interfaces with 3D brain organoids, highlighting differences in spontaneous activity between ALS patients and controls (Tasnim and Liu, 2022). Particularly sensitive to denervation-associated atrophy in ALS, EIM serves as a unique biomarker for disease progression. Its simplicity suggests potential for future implementation in human organoids containing myofibers, enabling rapid assessments of tissue health and integrity. Overall, organoid models hold the potential to uncover how ALS impacts neuronal electrophysiology, aiding in understanding disease progression and developing targeted interventions. These advancements highlight organoids’ transformative role in elucidating disease pathophysiology and advancing novel therapies.
4.4 Translational applications of 3D organoid
3D organoids offer significant advantages in translational research due to their ability to replicate the complex architecture and cellular diversity of human tissues (Velasco et al., 2019). These models provide a more physiologically relevant environment compared to conventional 2D cultures, enabling the study of intricate cell–cell interactions, tissue organization, and functional properties (Lancaster and Knoblich, 2014; Quadrato et al., 2017; Levy and Pasca, 2023). 3D organoids derived from patient-specific iPSCs can recapitulate key pathological features of ALS, such as protein mislocalization and aggregation (Ratti et al., 2020). These models facilitate the testing of new therapeutic compounds and enhance the translational relevance of preclinical findings, thus improving drug discovery efforts (Keller and Frega, 2019; Burley et al., 2022).
4.4.1 Personalized medicine
In personalized medicine, 3D organoids derived from ALS patient iPSCs can be used to test specific therapeutic approaches tailored to individual genetic and phenotypic profiles. These patient-specific organoids closely mimic the patient’s unique disease pathology, allowing researchers to evaluate the efficacy and safety of potential treatments in a more precise and personalized manner (Wichterle and Przedborski, 2010; Smits et al., 2019). For example, a study leveraged 3D spinal cord organoids derived from ALS patient iPSCs and electrophysiological tools to model disease-specific phenotypes and assess the efficacy of potential therapeutic compounds, identifying already-approved drugs with targets that modulate TDP-43 aggregates (Burkhardt et al., 2013). Additionally, studies utilizing motor neuron organoids derived from ALS patient iPSCs to conduct drug screening, concluding that patient-specific 3D organoid research, with the help of bioinformatics, has the potential to circumnavigate the limitations of ALS research and give rise to new drug and treatment discoveries (Amoros et al., 2022). This approach not only enhances the likelihood of therapeutic success but also minimizes adverse effects, leading to more effective and personalized healthcare solutions (Burley et al., 2022).
4.4.2 Drug discovery and testing
3D organoids serve as powerful platforms for drug discovery and testing, providing a scalable and reproducible system for high-throughput screening of therapeutic compounds (Clevers, 2016). Organoids’ ability to mimic the complex cellular environment of human tissues allows for more accurate predictions of drug efficacy and toxicity (Fatehullah et al., 2016). In ALS research, organoids can be used to identify compounds that modulate neuronal excitability, reduce protein aggregation, or enhance cell survival. Additionally, organoid models can facilitate the discovery of novel biomarkers for disease progression and treatment response, further advancing the development of targeted therapies (Wainger et al., 2021).
4.4.3 Transplantation of organoids
The transplantation of organoids offers significant potential in the treatment of ALS. Recent advancements have demonstrated that 3D organoids derived from human iPSCs can successfully integrate with tissue and contribute to functional recovery when implanted into animal models (Revah et al., 2022; Jgamadze et al., 2023). For instance, human-derived 3D cortical and spinal cord organoids formed synaptic connections and promoted neural regeneration when transplanted in mouse models with spinal cord injuries (Xu J. et al., 2023). However, several limitations related to vascularized structure formation, cell viability, and tissue integration remain before clinical applications of organoid transplantation can occur (Sagrac et al., 2021). Optimizing organoid transplantation involves integrating bioengineering technologies into the culturing process. For example, the vascularization of human brain organoids was shown to enhance the survival, integration, and functionality of transplanted organoids (Pham et al., 2018). To translate these organoids into clinical practice for treating patients with intractable diseases, challenges such as precise identification of the composition and size of cells forming the 3D structure, quality assurance, and mass production need to be addressed (Choi et al., 2023).
While 3D organoid models offer significant advantages in replicating human tissue complexity and cellular diversity, they also face limitations such as variability in organoid formation, lack of vascularization, and challenges in long-term culture (Huch and Koo, 2015; Lancaster et al., 2017). Standardization of organoid protocols and incorporation of vascular components are critical for improving the consistency and physiological relevance of these models (Pham et al., 2018). Future advancements should focus on integrating advanced imaging techniques, microfluidic systems, and bioengineering approaches to enhance the functionality and applicability of organoid models (Takebe et al., 2017). Additionally, combining organoids with other innovative technologies, such as single-cell RNA sequencing and CRISPR-based gene editing, will provide deeper insights into ALS pathogenesis and facilitate the development of more effective therapies (Sato et al., 2023).
5 Discussion
Electrophysiology has been essential in elucidating ALS mechanisms, revealing motor neuron dysfunction, ion channel changes, and network activity through techniques such as patch clamp, calcium imaging, and MEAs (Brette et al., 2008; Wainger et al., 2014; Yang et al., 2023). Complementary clinical techniques like MUNE and EIM further enrich our understanding by assessing motor neuron loss and muscle health (Shefner et al., 2011; Shefner et al., 2018). Recent advances allow electrophysiology techniques to be applied to organoids, bridging cellular-level insights with more complex, tissue-level analyses (Soscia et al., 2020).
The application of organoid research in the study of ALS pathology offers both promise and challenges (Chow et al., 2022). A critical challenge lies in the standardization and reproducibility of organoid protocols. Organoids differ in structure and activity patterns, and even organoids produced by the same iPS cell line may not be consistent. The existence of “batch syndrome,” where there is significant variability in both regional features and overall quality among produced organoids, poses a significant hindrance (Renner et al., 2017; Ilieva et al., 2018). Moreover, limitations in the progression and maturation of current brain organoid models during differentiation present another hurdle (Lancaster et al., 2013).
Human brain organoids take longer to produce specific cell types, such as upper-layer neurons and astrocytes, and often lack certain cell types and structures, such as the blood and immune systems. These might be minor concerns for organoid application, but the lack of a complete input/output system can be an important limitation in organoid research, although this can also be advantageous for dissecting intricate mechanisms in simplified systems. Another major constraint is the inability to stably observe and manipulate organoids over long periods, which hinders their application in ALS research. Conducting pathological research on organoids for extended periods, on a larger scale, and with more diverse methods remains a significant challenge for their effective use as ALS models.
Another challenge of organoid electrophysiology stems from current limitations in comprehensively probing structural, genetic, and functional properties, as well as heterogeneities across organoids over time. While bioelectronics enable long-term, stable recording of single-cell activities, there are constraints in specifically probing neuronal (or myofiber) activities with cell-type specificity and simultaneously recording from a large number of neurons within the 3D volume. Future advancements should prioritize developing technologies that enhance the analyses of organoid electrophysiological properties. This necessitates multimodal recording approaches integrating bioelectronics with in situ sequencing or cell-type-specific imaging to overcome existing limitations, allowing specific probing of neuronal activities and simultaneous recording from a larger neuron population within the 3D structure of brain organoids (Wang et al., 2018; Li et al., 2023).
One can envision applying some or ultimately all the electrophysiological techniques discussed here to organoids. For the more clinically relevant techniques, it would be relatively simple to perform EIM on the organoids. Indeed, there are impedance electrode-based cell culture dishes expressly for the purpose of measuring impedance of cells (Canali et al., 2015). Similarly, using an MEA would be possible in organoids using a similar concept (Tasnim and Liu, 2022). Performing other more clinically focused methods in organoids would depend on the complexity and organization of the organoid. For example, MUNE or MUNIX would require actual motor neurons and functioning synapses (Shin-Yi Lin et al., 2024). The same would also be true for NMJ assessments (Wang and Frank, 2024). More basic techniques such as patch clamp or calcium imaging, could be easily applied to organoids without significant challenges (Cakir et al., 2019; Sakaguchi et al., 2019). Additionally, integrating advanced imaging techniques and microfluidic systems into organoid studies could offer more detailed analyses and dynamic studies, respectively, thereby enhancing the physiological relevance of organoid models for understanding ALS pathology.
In conclusion, electrophysiological assessment of ALS, from single cells to humans, offers considerable potential value and remarkable flexibility. However, the application of these established techniques to organoid research in ALS will require a concerted effort to achieve standardization, reproducibility, and a more comprehensive representation of all cell types. Addressing issues such as batch variability, refining differentiation processes, and enhancing the ability to probe neuronal and myofiber activities within the 3D volume of tissue will significantly contribute to advancing our understanding of ALS pathology. These efforts pave the way for nuanced investigations into electrical signals, network-scale recordings, and excitability patterns, offering promising new avenues for assessing targeted therapeutic developments in ALS.
Author contributions
KR: Writing – original draft, Writing – review & editing. QW: Writing – original draft, Writing – review & editing. DJ: Writing – original draft, Writing – review & editing. EL: Writing – review & editing. JA: Writing – review & editing. RJ: Writing – review & editing. SR: Writing – review & editing. FT: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was support by National Institutes of Health (R01 NS055099 and R00 EY032181).
Acknowledgments
We are grateful for the feedback from Eva (Yiqi) Wang, and Isha Nagireddy on this manuscript. Figures were created with BioRender.com.
Conflict of interest
SR has equity in and serves as a consultant and scientific advisor to Myolex, Inc and Haystack Diagnostics, companies that design impedance devices for clinical and research use; he is also a member of Myolex’s Board of Directors. The companies also have an option to license patented impedance technology of which SR is named as an inventor.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahad, M. A., Fogerson, P. M., Rosen, G. D., Narayanaswami, P., and Rutkove, S. B. (2009). Electrical characteristics of rat skeletal muscle in immaturity, adulthood and after sciatic nerve injury, and their relation to muscle fiber size. Physiol. Meas. 30, 1415–1427. doi: 10.1088/0967-3334/30/12/009
Akcimen, F., Lopez, E. R., Landers, J. E., Nath, A., Chio, A., Chia, R., et al. (2023). Amyotrophic lateral sclerosis: translating genetic discoveries into therapies. Nat. Rev. Genet. 24, 642–658. doi: 10.1038/s41576-023-00592-y
Amalyan, S., Tamboli, S., Lazarevich, I., Topolnik, D., Bouman, L. H., and Topolnik, L. (2022). Enhanced motor cortex output and disinhibition in asymptomatic female mice with C9orf72 genetic expansion. Cell Rep. 40:111043. doi: 10.1016/j.celrep.2022.111043
Amoros, M. A., Choi, E. S., Cofre, A. R., Dokholyan, N. V., and Duzzioni, M. (2022). Motor neuron-derived induced pluripotent stem cells as a drug screening platform for amyotrophic lateral sclerosis. Front. Cell Dev. Biol. 10:962881. doi: 10.3389/fcell.2022.962881
Badu-Mensah, A., Guo, X., McAleer, C. W., Rumsey, J. W., and Hickman, J. J. (2020). Functional skeletal muscle model derived from SOD1-mutant ALS patient iPSCs recapitulates hallmarks of disease progression. Sci. Rep. 10:14302. doi: 10.1038/s41598-020-70510-3
Ban, J., Samano, C., Mladinic, M., and Munitic, I. (2019). Glia in amyotrophic lateral sclerosis and spinal cord injury: common therapeutic targets. Croat. Med. J. 60, 109–120. doi: 10.3325/cmj.2019.60.109
Bang, L., Da-Jian, H., Xiao-Jiang, L., and Xiang-Yu, G. (2022). Modeling neurodegenerative diseases using non-human primates: advances and challenges. Ageing Neurodegenerative Dis. 2:12. doi: 10.20517/and.2022.14
Baskoylu, S. N., Chapkis, N., Unsal, B., Lins, J., Schuch, K., Simon, J., et al. (2022). Disrupted autophagy and neuronal dysfunction in C. elegans knockin models of FUS amyotrophic lateral sclerosis. Cell Rep. 38:110195. doi: 10.1016/j.celrep.2021.110195
Bayer, D., Antonucci, S., Müller, H.-P., Saad, R., Dupuis, L., Rasche, V., et al. (2021). Disruption of orbitofrontal-hypothalamic projections in a murine ALS model and in human patients. Transl. Neurodegener. 10:17. doi: 10.1186/s40035-021-00241-6
Benatar, M., Boylan, K., Jeromin, A., Rutkove, S. B., Berry, J., Atassi, N., et al. (2016). ALS biomarkers for therapy development: state of the field and future directions. Muscle Nerve 53, 169–182. doi: 10.1002/mus.24979
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21. doi: 10.1038/35036035
Bhargava, A., Sandoval Castellanos, A. M., Shah, S., and Ning, K. (2022). An insight into the iPSCs-derived two-dimensional culture and three-dimensional organoid models for neurodegenerative disorders. Interface Focus. 12:20220040. doi: 10.1098/rsfs.2022.0040
Bonifacino, T., Zerbo, R. A., Balbi, M., Torazza, C., Frumento, G., Fedele, E., et al. (2021). Nearly 30 years of animal models to study amyotrophic lateral sclerosis: a historical overview and future perspectives. Int. J. Mol. Sci. 22:12236. doi: 10.3390/ijms222212236
Bostock, H., Sharief, M. K., Reid, G., and Murray, N. M. F. (1995). Axonal ion channel dysfunction in amyotrophic lateral sclerosis. Brain 118, 217–225. doi: 10.1093/brain/118.1.217
Brette, F., Luxan, G., Cros, C., Dixey, H., Wilson, C., and Shiels, H. A. (2008). Characterization of isolated ventricular myocytes from adult zebrafish (Danio rerio). Biochem. Biophys. Res. Commun. 374, 143–146. doi: 10.1016/j.bbrc.2008.06.109
Brownstone, R. M., and Lancelin, C. (2018). Escape from homeostasis: spinal microcircuits and progression of amyotrophic lateral sclerosis. J. Neurophysiol. 119, 1782–1794. doi: 10.1152/jn.00331.2017
Burkhardt, M. F., Martinez, F. J., Wright, S., Ramos, C., Volfson, D., Mason, M., et al. (2013). A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell. Neurosci. 56, 355–364. doi: 10.1016/j.mcn.2013.07.007
Burley, S., Beccano-Kelly, D. A., Talbot, K., Llana, O. C., and Wade-Martins, R. (2022). Hyperexcitability in young iPSC-derived C9ORF72 mutant motor neurons is associated with increased intracellular calcium release. Sci. Rep. 12:7378. doi: 10.1038/s41598-022-09751-3
Bykhovskaia, M., and Vasin, A. (2017). Electrophysiological analysis of synaptic transmission in Drosophila. Wiley Interdiscip. Rev. Dev. Biol. 6:10.1002/wdev.277. doi: 10.1002/wdev.277
Cakir, B., Xiang, Y., Tanaka, Y., Kural, M. H., Parent, M., Kang, Y. J., et al. (2019). Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 16, 1169–1175. doi: 10.1038/s41592-019-0586-5
Campanari, M.-L., Marian, A., Ciura, S., and Kabashi, E. (2021). TDP-43 regulation of AChE expression can mediate ALS-like phenotype in zebrafish. Cells 10:221. doi: 10.3390/cells10020221
Canali, C., Heiskanen, A., Muhammad, H. B., Hoyum, P., Pettersen, F. J., Hemmingsen, M., et al. (2015). Bioimpedance monitoring of 3D cell culturing--complementary electrode configurations for enhanced spatial sensitivity. Biosens. Bioelectron. 63, 72–79. doi: 10.1016/j.bios.2014.07.020
Casci, I., and Pandey, U. B. (2015). A fruitful endeavor: modeling ALS in the fruit fly. Brain Res. 1607, 47–74. doi: 10.1016/j.brainres.2014.09.064
Cerneckis, J., Cai, H., and Shi, Y. (2024). Induced pluripotent stem cells (iPSCs): molecular mechanisms of induction and applications. Signal Transduct. Target. Ther. 9:112. doi: 10.1038/s41392-024-01809-0
Chehelgerdi, M., Behdarvand Dehkordi, F., Chehelgerdi, M., Kabiri, H., Salehian-Dehkordi, H., Abdolvand, M., et al. (2023). Exploring the promising potential of induced pluripotent stem cells in cancer research and therapy. Mol. Cancer 22:189. doi: 10.1186/s12943-023-01873-0
Chia, K., Klingseisen, A., Sieger, D., and Priller, J. (2022). Zebrafish as a model organism for neurodegenerative disease. Front. Mol. Neurosci. 15:940484. doi: 10.3389/fnmol.2022.940484
Chipika, R. H., Finegan, E., Shing, L. H., Hardiman, O., and Bede, P. (2019). Tracking a fast-moving disease: longitudinal markers, monitoring, and clinical trial endpoints in ALS. Front. Neurol. 10:229. doi: 10.3389/fneur.2019.00229
Choi, W. H., Bae, D. H., and Yoo, J. (2023). Current status and prospects of organoid-based regenerative medicine. BMB Rep. 56, 10–14. doi: 10.5483/BMBRep.2022-0195
Chow, S. Y. A., Hu, H., Osaki, T., Levi, T., and Ikeuchi, Y. (2022). Advances in construction and modeling of functional neural circuits in vitro. Neurochem. Res. 47, 2529–2544. doi: 10.1007/s11064-022-03682-1
Clevers, H. (2016). Modeling development and disease with organoids. Cell 165, 1586–1597. doi: 10.1016/j.cell.2016.05.082
Costa-Mattioli, M., and Walter, P. (2020). The integrated stress response: from mechanism to disease. Science 368:eaat5314. doi: 10.1126/science.aat5314
De Carvalho, M., and Swash, M. (2023). Transcranial magnetic stimulation to monitor disease progression in ALS: a review. Amyotroph. Lateral Scler Frontotemporal. Degener. 24, 362–368. doi: 10.1080/21678421.2022.2160649
de Majo, M., Koontz, M., Marsan, E., Salinas, N., Ramsey, A., Kuo, Y. M., et al. (2023). Granulin loss of function in human mature brain organoids implicates astrocytes in TDP-43 pathology. Stem Cell Reports 18, 706–719. doi: 10.1016/j.stemcr.2023.01.012
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Du, H., Huo, Z., Chen, Y., Zhao, Z., Meng, F., Wang, X., et al. (2023). Induced pluripotent stem cells and their applications in amyotrophic lateral sclerosis. Cells 12:971. doi: 10.3390/cells12060971
Eremeev, A. V., Lebedeva, O. S., Bogomiakova, M. E., Lagarkova, M. A., and Bogomazova, A. N. (2021). Cerebral organoids-challenges to establish a brain prototype. Cells 10:1790. doi: 10.3390/cells10071790
Falgairolle, M., and O'Donovan, M. J. (2020). Motoneuronal spinal circuits in degenerative Motoneuron disease. Front. Mol. Neurosci. 13:74. doi: 10.3389/fnmol.2020.00074
Fatehi, F., Grapperon, A. M., Fathi, D., Delmont, E., and Attarian, S. (2018). The utility of motor unit number index: a systematic review. Neurophysiol. Clin. 48, 251–259. doi: 10.1016/j.neucli.2018.09.001
Fatehullah, A., Tan, S. H., and Barker, N. (2016). Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 18, 246–254. doi: 10.1038/ncb3312
Fernandez-Chiappe, F., and Muraro, N. I. (2022). Patch-clamping drosophila brain neurons. Cold Spring Harb. Protoc. 2022:pdb.prot107936. doi: 10.1101/pdb.prot107936
Fortier, G., Butti, Z., and Patten, S. A. (2020). Modelling C9orf72-related amyotrophic lateral sclerosis in zebrafish. Biomedicines 8:440. doi: 10.3390/biomedicines8100440
Fujimori, K., Ishikawa, M., Otomo, A., Atsuta, N., Nakamura, R., Akiyama, T., et al. (2018). Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 24, 1579–1589. doi: 10.1038/s41591-018-0140-5
Gawel, M. (2019). Electrodiagnostics: MUNE and MUNIX as methods of estimating the number of motor units - biomarkers in lower motor neurone disease. Neurol. Neurochir. Pol. 53, 251–257. doi: 10.5603/PJNNS.a2019.0026
Goodman, M. B., Lindsay, T. H., Lockery, S. R., and Richmond, J. E. (2012). Electrophysiological methods for Caenorhabditis elegans neurobiology. Methods Cell Biol. 107, 409–436. doi: 10.1016/B978-0-12-394620-1.00014-X
Grad, L. I., Rouleau, G. A., Ravits, J., and Cashman, N. R. (2017). Clinical Spectrum of amyotrophic lateral sclerosis (ALS). Cold Spring Harb. Perspect. Med. 7:a024117. doi: 10.1101/cshperspect.a024117
Grienberger, C., and Konnerth, A. (2012). Imaging calcium in neurons. Neuron 73, 862–885. doi: 10.1016/j.neuron.2012.02.011
Grolez, G., Kyheng, M., Lopes, R., Moreau, C., Timmerman, K., Auger, F., et al. (2018). MRI of the cervical spinal cord predicts respiratory dysfunction in ALS. Sci. Rep. 8:1828. doi: 10.1038/s41598-018-19938-2
Gunes, Z. I., Kan, V. W. Y., Ye, X., and Liebscher, S. (2020). Exciting complexity: the role of motor circuit elements in ALS pathophysiology. Front. Neurosci. 14:573. doi: 10.3389/fnins.2020.00573
Hales, C. M., Rolston, J. D., and Potter, S. M. (2010). How to culture, record and stimulate neuronal networks on micro-electrode arrays (MEAs). J. Vis. Exp. 30:2056. doi: 10.3791/2056
Haney, M. M., Sinnott, J., Osman, K. L., Deninger, I., Andel, E., Caywood, V., et al. (2019). Mice lacking brain-derived serotonin have altered swallowing function. Otolaryngol. Head Neck Surg. 161, 468–471. doi: 10.1177/0194599819846109
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., et al. (2017). Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 3:17071. doi: 10.1038/nrdp.2017.71
Henstridge, C. M., Tzioras, M., and Paolicelli, R. C. (2019). Glial contribution to excitatory and inhibitory synapse loss in neurodegeneration. Front. Cell. Neurosci. 13:63. doi: 10.3389/fncel.2019.00063
Howe, K., Clark, M. D., Torroja, C. F., Torrance, J., Berthelot, C., Muffato, M., et al. (2013). The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503. doi: 10.1038/nature12111
Howland, D. S., Liu, J., She, Y., Goad, B., Maragakis, N. J., Kim, B., et al. (2002). Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. USA 99, 1604–1609. doi: 10.1073/pnas.032539299
Huch, M., and Koo, B. K. (2015). Modeling mouse and human development using organoid cultures. Development 142, 3113–3125. doi: 10.1242/dev.118570
Ilieva, M., Fex Svenningsen, Å., Thorsen, M., and Michel, T. M. (2018). Psychiatry in a dish: stem cells and brain organoids modeling autism Spectrum disorders. Biol. Psychiatry 83, 558–568. doi: 10.1016/j.biopsych.2017.11.011
Jacobsen, A. B., Kristensen, R. S., Witt, A., Kristensen, A. G., Duez, L., Beniczky, S., et al. (2018). The utility of motor unit number estimation methods versus quantitative motor unit potential analysis in diagnosis of ALS. Clin. Neurophysiol. 129, 646–653. doi: 10.1016/j.clinph.2018.01.002
Jgamadze, D., Lim, J. T., Zhang, Z., Harary, P. M., Germi, J., Mensah-Brown, K., et al. (2023). Structural and functional integration of human forebrain organoids with the injured adult rat visual system. Cell Stem Cell 30:e7, 137–152.e7. doi: 10.1016/j.stem.2023.01.004
Jones, R. H. A. (2022). A macaque model of motor Neurone disease. Newcastle upon Tyne: Newcastle University.
Kauder, J., Petri, S., Tipold, A., and Stein, V. M. (2015). The potential role of motor unit number estimation as an additional diagnostic and prognostic value in canine neurology. Front. Vet. Sci. 2:53. doi: 10.3389/fvets.2015.00053
Keller, J. M., and Frega, M. (2019). Past, present, and future of neuronal models in vitro. Adv. Neurobiol. 22, 3–17. doi: 10.1007/978-3-030-11135-9_1
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophic lateral sclerosis. Lancet 377, 942–955. doi: 10.1016/S0140-6736(10)61156-7
Kim, G., Gautier, O., Tassoni-Tsuchida, E., Ma, X. R., and Gitler, A. D. (2020). ALS genetics: gains, losses, and implications for future therapies. Neuron 108, 822–842. doi: 10.1016/j.neuron.2020.08.022
Kodandaramaiah, S. B., Franzesi, G. T., Chow, B. Y., Boyden, E. S., and Forest, C. R. (2012). Automated whole-cell patch-clamp electrophysiology of neurons in vivo. Nat. Methods 9, 585–587. doi: 10.1038/nmeth.1993
Kornreich, B. G. (2007). The patch clamp technique: principles and technical considerations. J. Vet. Cardiol. 9, 25–37. doi: 10.1016/j.jvc.2007.02.001
Kowal, J. B., Verga, S. A., Pandeya, S. R., Cochran, R. J., Sabol, J. C., Rutkove, S. B., et al. (2022). Electrical impedance Myography in dogs with degenerative myelopathy. Front. Vet. Sci. 9:874277. doi: 10.3389/fvets.2022.874277
Kunst, M., Laurell, E., Mokayes, N., Kramer, A., Kubo, F., Fernandes, A. M., et al. (2019). A cellular-resolution atlas of the larval zebrafish brain. Neuron 103:e5, 21–38.e5. doi: 10.1016/j.neuron.2019.04.034
Ladewig, T., and Keller, B. U. (2000). Simultaneous patch-clamp recording and calcium imaging in a rhythmically active neuronal network in the brainstem slice preparation from mouse. Pflugers Arch. 440, 322–332. doi: 10.1007/s004240000277
Lagier-Tourenne, C., and Cleveland, D. W. (2009). Rethinking ALS: the FUS about TDP-43. Cell 136, 1001–1004. doi: 10.1016/j.cell.2009.03.006
Lam, D., Fischer, N. O., and Enright, H. A. (2021). Probing function in 3D neuronal cultures: a survey of 3D multielectrode array advances. Curr. Opin. Pharmacol. 60, 255–260. doi: 10.1016/j.coph.2021.08.003
Lancaster, M. A., Corsini, N. S., Wolfinger, S., Gustafson, E. H., Phillips, A. W., Burkard, T. R., et al. (2017). Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol. 35, 659–666. doi: 10.1038/nbt.3906
Lancaster, M. A., and Knoblich, J. A. (2014). Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 9, 2329–2340. doi: 10.1038/nprot.2014.158
Lancaster, M. A., Renner, M., Martin, C.-A., Wenzel, D., Bicknell, L. S., Hurles, M. E., et al. (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. doi: 10.1038/nature12517
Lasiene, J., and Yamanaka, K. (2011). Glial cells in amyotrophic lateral sclerosis. Neurol. Res. Int. 2011:718987, 1–7. doi: 10.1155/2011/718987
Lee, S., Jeon, Y.-M., Cha, S. J., Kim, S., Kwon, Y., Jo, M., et al. (2020). PTK2/FAK regulates UPS impairment via SQSTM1/p62 phosphorylation in TARDBP/TDP-43 proteinopathies. Autophagy 16, 1396–1412. doi: 10.1080/15548627.2019.1686729
Leoni, T. B., Rezende, T. J. R., Peluzzo, T. M., Martins, M. P., Neto, A. R. C., Gonzalez-Salazar, C., et al. (2022). Structural brain and spinal cord damage in symptomatic and pre-symptomatic VAPB-related ALS. J. Neurol. Sci. 434:120126. doi: 10.1016/j.jns.2021.120126
Levy, R. J., and Pasca, S. P. (2023). What have organoids and Assembloids taught us about the pathophysiology of neuropsychiatric disorders? Biol. Psychiatry 93, 632–641. doi: 10.1016/j.biopsych.2022.11.017
Li, Q., Feng, Y., Xue, Y., Zhan, X., Fu, Y., Gui, G., et al. (2022). Edaravone activates the GDNF/RET neurotrophic signaling pathway and protects mRNA-induced motor neurons from iPS cells. Mol. Neurodegener. 17:8. doi: 10.1186/s13024-021-00510-y
Li, J., Geisbush, T. R., Rosen, G. D., Lachey, J., Mulivor, A., and Rutkove, S. B. (2014). Electrical impedance myography for the in vivo and ex vivo assessment of muscular dystrophy (mdx) mouse muscle. Muscle Nerve 49, 829–835. doi: 10.1002/mus.24086
Li, J., Sung, M., and Rutkove, S. B. (2013). Electrophysiologic biomarkers for assessing disease progression and the effect of riluzole in SOD1 G93A ALS mice. PLoS One 8:e65976. doi: 10.1371/journal.pone.0065976
Li, Q., Lin, Z., Liu, R., Tang, X., Huang, J., He, Y., et al. (2023). Multimodal charting of molecular and functional cell states via in situ electro-sequencing. Cell. 186:2002-2017e21. doi: 10.1016/j.cell.2023.03.023
Liguori, F., Amadio, S., and Volonté, C. (2021). Fly for ALS: Drosophila modeling on the route to amyotrophic lateral sclerosis modifiers. Cell. Mol. Life Sci. 78, 6143–6160. doi: 10.1007/s00018-021-03905-8
Liu, M., Cui, L., Guan, Y., Li, B., and Du, H. (2013). Single-fiber electromyography in amyotrophic lateral sclerosis and cervical spondylosis. Muscle Nerve 48, 137–139. doi: 10.1002/mus.23767
Lutz, C. (2018). Mouse models of ALS: past, present and future. Brain Res. 1693, 1–10. doi: 10.1016/j.brainres.2018.03.024
Maimon, G., Straw, A. D., and Dickinson, M. H. (2010). Active flight increases the gain of visual motion processing in Drosophila. Nat. Neurosci. 13, 393–399. doi: 10.1038/nn.2492
Markert, S. M., Skoruppa, M., Yu, B., Mulcahy, B., Zhen, M., Gao, S., et al. (2020). Overexpression of an ALS-associated FUS mutation in C. elegans disrupts NMJ morphology and leads to defective neuromuscular transmission. Biol Open 9:bio055129. doi: 10.1242/bio.055129
Martin, R. M., Bereman, M. S., and Marsden, K. C. (2021). BMAA and MCLR interact to modulate behavior and exacerbate molecular changes related to neurodegeneration in larval zebrafish. Toxicol. Sci. 179, 251–261. doi: 10.1093/toxsci/kfaa178
Meijboom, K. E., Abdallah, A., Fordham, N. P., Nagase, H., Rodriguez, T., Kraus, C., et al. (2022). CRISPR/Cas9-mediated excision of ALS/FTD-causing hexanucleotide repeat expansion in C9ORF72 rescues major disease mechanisms in vivo and in vitro. Nat. Commun. 13:6286. doi: 10.1038/s41467-022-33332-7
Morrice, J. R., Gregory-Evans, C. Y., and Shaw, C. A. (2018). Animal models of amyotrophic lateral sclerosis: a comparison of model validity. Neural Regen. Res. 13, 2050–2054. doi: 10.4103/1673-5374.241445
Neher, E., and Sakmann, B. (1992). The patch clamp technique. Sci. Am. 266, 44–51. doi: 10.1038/scientificamerican0392-44
Okano, H., and Morimoto, S. (2022). iPSC-based disease modeling and drug discovery in cardinal neurodegenerative disorders. Cell Stem Cell 29, 189–208. doi: 10.1016/j.stem.2022.01.007
Oliveira, N. A. S., Pinho, B. R., and Oliveira, J. M. A. (2023). Swimming against ALS: how to model disease in zebrafish for pathophysiological and behavioral studies. Neurosci. Biobehav. Rev. 148:105138. doi: 10.1016/j.neubiorev.2023.105138
Pandeya, S. R., Nagy, J. A., Riveros, D., Semple, C., Taylor, R. S., Mortreux, M., et al. (2021). Estimating myofiber cross-sectional area and connective tissue deposition with electrical impedance myography: a study in D2-mdx mice. Muscle Nerve 63, 941–950. doi: 10.1002/mus.27240
Pant, D. C., and Nazarko, T. Y. (2021). Selective autophagy: the rise of the zebrafish model. Autophagy 17, 3297–3305. doi: 10.1080/15548627.2020.1853382
Pasca, A. M., Sloan, S. A., Clarke, L. E., Tian, Y., Makinson, C. D., Huber, N., et al. (2015). Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 12, 671–678. doi: 10.1038/nmeth.3415
Passaro, A. P., and Stice, S. L. (2020). Electrophysiological analysis of brain organoids: current approaches and advancements. Front. Neurosci. 14:622137. doi: 10.3389/fnins.2020.622137
Petel Legare, V., Rampal, C. J., Gurberg, T. J. N., Aaltonen, M. J., Janer, A., Zinman, L., et al. (2023). Loss of mitochondrial Chchd10 or Chchd2 in zebrafish leads to an ALS-like phenotype and complex I deficiency independent of the mitochondrial integrated stress response. Dev. Neurobiol. 83, 54–69. doi: 10.1002/dneu.22909
Peterka, D. S., Takahashi, H., and Yuste, R. (2011). Imaging voltage in neurons. Neuron 69, 9–21. doi: 10.1016/j.neuron.2010.12.010
Petersilie, L., Heiduschka, S., Nelson, J. S. E., Neu, L. A., Le, S., Anand, R., et al. (2024). Cortical brain organoid slices (cBOS) for the study of human neural cells in minimal networks. iScience 27:109415. doi: 10.1016/j.isci.2024.109415
Pham, M. T., Pollock, K. M., Rose, M. D., Cary, W. A., Stewart, H. R., Zhou, P., et al. (2018). Generation of human vascularized brain organoids. Neuroreport 29, 588–593. doi: 10.1097/WNR.0000000000001014
Philips, T., and Rothstein, J. D. (2015). Rodent models of amyotrophic lateral sclerosis. Curr. Protoc. Pharmacol. 69:5.67.1. doi: 10.1002/0471141755.ph0567s69
Powers, S., Kwok, S., Lovejoy, E., Lavin, T., and Sher, R. B. (2017). Editor's highlight: embryonic exposure to the environmental neurotoxin BMAA negatively impacts early neuronal development and progression of neurodegeneration in the Sod1-G93R zebrafish model of amyotrophic lateral sclerosis. Toxicol. Sci. 157, 129–140. doi: 10.1093/toxsci/kfx020
Qian, X., Song, H., and Ming, G. L. (2019). Brain organoids: advances, applications and challenges. Development 146:dev166074. doi: 10.1242/dev.166074
Quadrato, G., Nguyen, T., Macosko, E. Z., Sherwood, J. L., Min Yang, S., Berger, D. R., et al. (2017). Cell diversity and network dynamics in photosensitive human brain organoids. Nature 545, 48–53. doi: 10.1038/nature22047
Quarta, E., Fulgenzi, G., Bravi, R., Cohen, E. J., Yanpallewar, S., Tessarollo, L., et al. (2018). Deletion of the endogenous TrkB.T1 receptor isoform restores the number of hippocampal CA1 parvalbumin-positive neurons and rescues long-term potentiation in pre-symptomatic mSOD1(G93A) ALS mice. Mol. Cell. Neurosci. 89, 33–41. doi: 10.1016/j.mcn.2018.03.010
Ramírez-Jarquín, U. N., Lazo-Gómez, R., Tovar-y-Romo, L. B., and Tapia, R. (2014). Spinal inhibitory circuits and their role in motor neuron degeneration. Neuropharmacology 82, 101–107. doi: 10.1016/j.neuropharm.2013.10.003
Ramírez-Jarquín, U. N., and Tapia, R. (2018). Excitatory and inhibitory neuronal circuits in the spinal cord and their role in the control of motor neuron function and degeneration. ACS Chem. Neurosci. 9, 211–216. doi: 10.1021/acschemneuro.7b00503
Ratti, A., Gumina, V., Lenzi, P., Bossolasco, P., Fulceri, F., Volpe, C., et al. (2020). Chronic stress induces formation of stress granules and pathological TDP-43 aggregates in human ALS fibroblasts and iPSC-motoneurons. Neurobiol. Dis. 145:105051. doi: 10.1016/j.nbd.2020.105051
Renner, M., Lancaster, M. A., Bian, S., Choi, H., Ku, T., Peer, A., et al. (2017). Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 36, 1316–1329. doi: 10.15252/embj.201694700
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Revah, O., Gore, F., Kelley, K. W., Andersen, J., Sakai, N., Chen, X., et al. (2022). Maturation and circuit integration of transplanted human cortical organoids. Nature 610, 319–326. doi: 10.1038/s41586-022-05277-w
Rodriguez, S., Sahin, A., Schrank, B. R., Al-Lawati, H., Costantino, I., Benz, E., et al. (2021). Genome-encoded cytoplasmic double-stranded RNAs, found in C9ORF72 ALS-FTD brain, propagate neuronal loss. Sci. Transl. Med. 13:eaaz4699. doi: 10.1126/scitranslmed.aaz4699
Ronan, D., and Gillespie, P. (2005). Metazoan mechanotransduction mystery finally solved. Nat. Neurosci. 8, 7–8. doi: 10.1038/nn0105-7
Ronchi, S., Buccino, A. P., Prack, G., Kumar, S. S., Schroter, M., Fiscella, M., et al. (2021). Electrophysiological phenotype characterization of human iPSC-derived neuronal cell lines by means of high-density microelectrode arrays. Adv. Biol. (Weinh) 5:e2000223. doi: 10.1002/adbi.202000223
Roussos, A., Kitopoulou, K., Borbolis, F., and Palikaras, K. (2023). Caenorhabditis elegans as a model system to study human neurodegenerative disorders. Biomolecules 13:478. doi: 10.3390/biom13030478
Roy, U., Conklin, L., Schiller, J., Matysik, J., Berry, J. P., and Alia, A. (2017). Metabolic profiling of zebrafish (Danio rerio) embryos by NMR spectroscopy reveals multifaceted toxicity of β-methylamino-L-alanine (BMAA). Sci. Rep. 7:17305. doi: 10.1038/s41598-017-17409-8
Rutkove, S. B., Callegari, S., Concepcion, H., Mourey, T., Widrick, J., Nagy, J. A., et al. (2023a). Electrical impedance myography detects age-related skeletal muscle atrophy in adult zebrafish. Sci. Rep. 13:7191. doi: 10.1038/s41598-023-34119-6
Rutkove, S. B., Chen, Z. Z., Pandeya, S., Callegari, S., Mourey, T., Nagy, J. A., et al. (2023b). Surface electrical impedance Myography detects skeletal muscle atrophy in aged wildtype zebrafish and aged gpr27 knockout zebrafish. Biomedicines 11:1938. doi: 10.3390/biomedicines11071938
Saccon, R. A., Bunton-Stasyshyn, R. K., Fisher, E. M., and Fratta, P. (2013). Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136, 2342–2358. doi: 10.1093/brain/awt097
Sagrac, D., Sisli, H. B., Senkal, S., Hayal, T. B., Sahin, F., and Dogan, A. (2021). Organoids in tissue transplantation. Adv. Exp. Med. Biol. 1347, 45–64. doi: 10.1007/5584_2021_647
Saint-Amant, L., and Drapeau, P. (2003). Whole-cell patch-clamp recordings from identified spinal neurons in the zebrafish embryo. Methods Cell Sci. 25, 59–64. doi: 10.1023/B:MICS.0000006896.02938.49
Sakaguchi, H., Ozaki, Y., Ashida, T., Matsubara, T., Oishi, N., Kihara, S., et al. (2019). Self-organized synchronous calcium transients in a cultured human neural network derived from cerebral organoids. Stem Cell Reports 13, 458–473. doi: 10.1016/j.stemcr.2019.05.029
Sanchez, B., and Rutkove, S. B. (2017). Electrical impedance Myography and its applications in neuromuscular disorders. Neurotherapeutics 14, 107–118. doi: 10.1007/s13311-016-0491-x
Sanders, D. B., Kouyoumdjian, J. A., and Stalberg, E. V. (2022). Single fiber electromyography and measuring jitter with concentric needle electrodes. Muscle Nerve 66, 118–130. doi: 10.1002/mus.27573
Sato, Y., Asahi, T., and Kataoka, K. (2023). Integrative single-cell RNA-seq analysis of vascularized cerebral organoids. BMC Biol. 21:245. doi: 10.1186/s12915-023-01711-1
Semple, C., Riveros, D., Sung, D. M., Nagy, J. A., Rutkove, S. B., and Mortreux, M. (2020). Using electrical impedance Myography as a biomarker of muscle deconditioning in rats exposed to Micro-and partial-gravity analogs. Front. Physiol. 11:557796. doi: 10.3389/fphys.2020.557796
Shaw, M. P., Higginbottom, A., McGown, A., Castelli, L. M., James, E., Hautbergue, G. M., et al. (2018). Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol. Commun. 6:125. doi: 10.1186/s40478-018-0629-7
Shefner, J. M., Cudkowicz, M., and Brown, R. H. Jr. (2006). Motor unit number estimation predicts disease onset and survival in a transgenic mouse model of amyotrophic lateral sclerosis. Muscle Nerve 34, 603–607. doi: 10.1002/mus.20628
Shefner, J. M., Rutkove, S. B., Caress, J. B., Benatar, M., David, W. S., Cartwright, M. S., et al. (2018). Reducing sample size requirements for future ALS clinical trials with a dedicated electrical impedance myography system. Amyotroph. Lateral Scler. Frontotemporal. Degener. 19, 555–561. doi: 10.1080/21678421.2018.1510008
Shefner, J. M., Watson, M. L., Simionescu, L., Caress, J. B., Burns, T. M., Maragakis, N. J., et al. (2011). Multipoint incremental motor unit number estimation as an outcome measure in ALS. Neurology 77, 235–241. doi: 10.1212/WNL.0b013e318225aabf
Shin-Yi Lin, C., Howells, J., Rutkove, S., Nandedkar, S., Neuwirth, C., Noto, Y. I., et al. (2024). Neurophysiological and imaging biomarkers of lower motor neuron dysfunction in motor neuron diseases/amyotrophic lateral sclerosis: IFCN handbook chapter. Clin. Neurophysiol. 162, 91–120. doi: 10.1016/j.clinph.2024.03.015
Smits, L. M., Reinhardt, L., Reinhardt, P., Glatza, M., Monzel, A. S., Stanslowsky, N., et al. (2019). Modeling Parkinson's disease in midbrain-like organoids. NPJ Parkinsons Dis. 5:5. doi: 10.1038/s41531-019-0078-4
Sonobe, Y., Aburas, J., Krishnan, G., Fleming, A. C., Ghadge, G., Islam, P., et al. (2021). A C. elegans model of C9orf72-associated ALS/FTD uncovers a conserved role for eIF2D in RAN translation. Nature Commun. 12:6025. doi: 10.1038/s41467-021-26303-x
Soscia, D. A., Lam, D., Tooker, A. C., Enright, H. A., Triplett, M., Karande, P., et al. (2020). A flexible 3-dimensional microelectrode array for in vitro brain models. Lab Chip 20, 901–911. doi: 10.1039/c9lc01148j
Stamatakis, A. M., Schachter, M. J., Gulati, S., Zitelli, K. T., Malanowski, S., Tajik, A., et al. (2018). Simultaneous Optogenetics and cellular resolution calcium imaging during active behavior using a miniaturized microscope. Front. Neurosci. 12:496. doi: 10.3389/fnins.2018.00496
Steffen, H., Petra, S., Federico, V., Jochen, W., Patrick, O., Christine von, A., et al. (2022). Comparison of CSF and serum neurofilament light and heavy chain as differential diagnostic biomarkers for ALS. J. Neurol. Neurosurg. Psychiatry 93, 68–74. doi: 10.1136/jnnp-2021-327129
Sun, X. S., Liu, W. X., Chen, Z. H., Ling, L., Yang, F., Wang, H. F., et al. (2018). Repetitive nerve stimulation in amyotrophic lateral sclerosis. Chin. Med. J. 131, 2146–2151. doi: 10.4103/0366-6999.240798
Sun, X., Song, J., Huang, H., Chen, H., and Qian, K. (2018). Modeling hallmark pathology using motor neurons derived from the family and sporadic amyotrophic lateral sclerosis patient-specific iPS cells. Stem Cell Res Ther 9:315. doi: 10.1186/s13287-018-1048-1
Suzuki, Y. I., Shibuya, K., Misawa, S., Suichi, T., Tsuneyama, A., Kojima, Y., et al. (2022). Relationship between motor cortical and peripheral axonal hyperexcitability in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 93, 1074–1079. doi: 10.1136/jnnp-2021-328550
Swinnen, B., Bento-Abreu, A., Gendron, T. F., Boeynaems, S., Bogaert, E., Nuyts, R., et al. (2018). A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. 135, 427–443. doi: 10.1007/s00401-017-1796-5
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Takebe, T., Sekine, K., Kimura, M., Yoshizawa, E., Ayano, S., Koido, M., et al. (2017). Massive and reproducible production of liver buds entirely from human pluripotent stem cells. Cell Rep. 21, 2661–2670. doi: 10.1016/j.celrep.2017.11.005
Tallon, C., Sharma, A., Zhang, Z., Thomas, A. G., Ng, J., Zhu, X., et al. (2022). Dendrimer-2PMPA delays muscle function loss and denervation in a murine model of amyotrophic lateral sclerosis. Neurotherapeutics 19, 274–288. doi: 10.1007/s13311-021-01159-7
Tamaki, Y., Ross, J. P., Alipour, P., Castonguay, C. E., Li, B., Catoire, H., et al. (2023). Spinal cord extracts of amyotrophic lateral sclerosis spread TDP-43 pathology in cerebral organoids. PLoS Genet. 19:e1010606. doi: 10.1371/journal.pgen.1010606
Tasnim, K., and Liu, J. (2022). Emerging bioelectronics for brain organoid electrophysiology. J. Mol. Biol. 434:167165. doi: 10.1016/j.jmb.2021.167165
Tossing, G., Livernoche, R., Maios, C., Bretonneau, C., Labarre, A., and Parker, J. A. (2022). Genetic and pharmacological PARP inhibition reduces axonal degeneration in C. elegans models of ALS. Hum. Mol. Genet. 31, 3313–3324. doi: 10.1093/hmg/ddac116
Uchida, A., Sasaguri, H., Kimura, N., Tajiri, M., Ohkubo, T., Ono, F., et al. (2012). Non-human primate model of amyotrophic lateral sclerosis with cytoplasmic mislocalization of TDP-43. Brain 135, 833–846. doi: 10.1093/brain/awr348
Van den Bos, M. A. J., Higashihara, M., Geevasinga, N., Menon, P., Kiernan, M. C., and Vucic, S. (2018). Imbalance of cortical facilitatory and inhibitory circuits underlies hyperexcitability in ALS. Neurology 91, e1669–e1676. doi: 10.1212/WNL.0000000000006438
Van Schoor, E., Ospitalieri, S., Moonen, S., Tomé, S. O., Ronisz, A., Ok, O., et al. (2022). Increased pyroptosis activation in white matter microglia is associated with neuronal loss in ALS motor cortex. Acta Neuropathol. 144, 393–411. doi: 10.1007/s00401-022-02466-9
Velasco, S., Kedaigle, A. J., Simmons, S. K., Nash, A., Rocha, M., Quadrato, G., et al. (2019). Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 570, 523–527. doi: 10.1038/s41586-019-1289-x
Verma, S., Khurana, S., Vats, A., Sahu, B., Ganguly, N. K., Chakraborti, P., et al. (2022). Neuromuscular junction dysfunction in amyotrophic lateral sclerosis. Mol. Neurobiol. 59, 1502–1527. doi: 10.1007/s12035-021-02658-6
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S., Sandoe, J., et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11. doi: 10.1016/j.celrep.2014.03.019
Wainger, B. J., Macklin, E. A., Vucic, S., McIlduff, C. E., Paganoni, S., Maragakis, N. J., et al. (2021). Effect of Ezogabine on cortical and spinal motor neuron excitability in amyotrophic lateral sclerosis: a randomized clinical trial. JAMA Neurol. 78, 186–196. doi: 10.1001/jamaneurol.2020.4300
Wang, T., and Frank, C. A. (2024). Eliciting presynaptic homeostatic potentiation at the Drosophila larval neuromuscular junction. Cold Spring Harb. Protoc. doi: 10.1101/pdb.prot108424
Wang, L. L., Spieker, A. J., Li, J., and Rutkove, S. B. (2011). Electrical impedance myography for monitoring motor neuron loss in the SOD1 G93A amyotrophic lateral sclerosis rat. Clin. Neurophysiol. 122, 2505–2511. doi: 10.1016/j.clinph.2011.04.021
Wang, X., Takaki, S., and Yamagishi, J. (2018). Investigating very deep highway networks for parametric speech synthesis. Speech Comm. 96, 1–9. doi: 10.1016/j.specom.2017.11.002
Wang, T., Tian, X., Kim, H. B., Jang, Y., Huang, Z., Na, C. H., et al. (2022). Intracellular energy controls dynamics of stress-induced ribonucleoprotein granules. Nat. Commun. 13:5584. doi: 10.1038/s41467-022-33079-1
Wang, Y., Xiao, Z., Chu, H., Liang, J., Wu, X., Dong, H., et al. (2017). Correlations between slow-rate repetitive nerve stimulation and characteristics associated with amyotrophic lateral sclerosis in Chinese patients. J. Phys. Ther. Sci. 29, 737–743. doi: 10.1589/jpts.29.737
Wichterle, H., and Przedborski, S. (2010). What can pluripotent stem cells teach us about neurodegenerative diseases? Nat. Neurosci. 13, 800–804. doi: 10.1038/nn.2577
Wijesekera, L. C., and Nigel Leigh, P. (2009). Amyotrophic lateral sclerosis. Orphanet J. Rare Dis. 4:3. doi: 10.1186/1750-1172-4-3
Xu, J., Fang, S., Deng, S., Li, H., Lin, X., Huang, Y., et al. (2023). Generation of neural organoids for spinal-cord regeneration via the direct reprogramming of human astrocytes. Nat. Biomed. Eng. 7, 253–269. doi: 10.1038/s41551-022-00963-6
Xu, L., Wang, D., Zhao, L., Yang, Z., Liu, X., Li, X., et al. (2023). C9orf72 poly(PR) aggregation in nucleus induces ALS/FTD-related neurodegeneration in cynomolgus monkeys. Neurobiol. Dis. 184:106197. doi: 10.1016/j.nbd.2023.106197
Xu, Z., Yang, J., Xin, X., Liu, C., Li, L., Mei, X., et al. (2023). Merits and challenges of iPSC-derived organoids for clinical applications. Front. Cell Dev. Biol. 11:1188905. doi: 10.3389/fcell.2023.1188905
Yamada, K. M., and Cukierman, E. (2007). Modeling tissue morphogenesis and cancer in 3D. Cell 130, 601–610. doi: 10.1016/j.cell.2007.08.006
Yang, M., Liu, M., Sanchez, Y. F., Avazzadeh, S., Quinlan, L. R., Liu, G., et al. (2023). A novel protocol to derive cervical motor neurons from induced pluripotent stem cells for amyotrophic lateral sclerosis. Stem Cell Reports 18, 1870–1883. doi: 10.1016/j.stemcr.2023.07.004
Yu, H., Chen, L., Zhang, S., He, J., and Fan, D. (2021). Early axonal dysfunction of the peripheral nervous system influences disease progression of ALS: evidence from clinical Neuroelectrophysiology. Front. Neurol. 12:574919. doi: 10.3389/fneur.2021.574919
Zhang, S., Cooper-Knock, J., Weimer, A. K., Shi, M., Moll, T., Marshall, J. N. G., et al. (2022). Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis. Neuron 110:e11, 992–1008.e11. doi: 10.1016/j.neuron.2021.12.019
Zhang, H., Wang, H., Shen, X., Jia, X., Yu, S., Qiu, X., et al. (2021). The landscape of regulatory genes in brain-wide neuronal phenotypes of a vertebrate brain. eLife 10:e68224. doi: 10.7554/eLife.68224
Zhao, Y., Inayat, S., Dikin, D. A., Singer, J. H., Ruoff, R. S., and Troy, J. B. (2008). Patch clamp technique: review of the current state of the art and potential contributions from nanoengineering. Proc. Inst. Mech. Eng., Part N 222, 1–11. doi: 10.1243/17403499JNN149
Zhu, Y., Bai, J., Li, M., Wang, H., Wang, J., and Huang, X. (2023a). Repetitive nerve stimulation on survival in amyotrophic lateral sclerosis. Front. Neurol. 14:1244385. doi: 10.3389/fneur.2023.1244385
Zhu, Y., Huo, Y., Bai, J., Li, M., Wang, H., Wang, J., et al. (2023b). Serum cystatin C is a potential biomarker for predicting amyotrophic lateral sclerosis survival. Neurol. Sci. 45, 197–201. doi: 10.1007/s10072-023-06957-9
Keywords: amyotrophic lateral sclerosis, electrophysiology, excitability, excitotoxicity, organoid, disease modeling, electrical impedance myography
Citation: Ren K, Wang Q, Jiang D, Liu E, Alsmaan J, Jiang R, Rutkove SB and Tian F (2024) A comprehensive review of electrophysiological techniques in amyotrophic lateral sclerosis research. Front. Cell. Neurosci. 18:1435619. doi: 10.3389/fncel.2024.1435619
Edited by:
Jin Hao, Stanford University, United StatesReviewed by:
Chunyang Dong, Stanford University, United StatesStefania Marcuzzo, IRCCS Carlo Besta Neurological Institute Foundation, Italy
Copyright © 2024 Ren, Wang, Jiang, Liu, Alsmaan, Jiang, Rutkove and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seward B. Rutkove, c3J1dGtvdmVAYmlkbWMuaGFydmFyZC5lZHU=; Feng Tian, ZnRpYW5AYmlkbWMuaGFydmFyZC5lZHU=
†These authors have contributed equally to this work and share first authorship