 Yanyun Wu
Yanyun Wu Qianqian Wei
Qianqian Wei Junyu Lin
Junyu Lin Huifang Shang
Huifang Shang Ruwei Ou
Ruwei Ou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 04 April 2024
Sec. Cellular Neuropathology
Volume 18 - 2024 | https://doi.org/10.3389/fncel.2024.1369332
Myotonic dystrophy (DM) encompasses a spectrum of neuromuscular diseases characterized by myotonia, muscle weakness, and wasting. Recent research has led to the recognition of DM as a neurological disorder. Cognitive impairment is a central nervous system condition that has been observed in various forms of DM. Neuroimaging studies have increasingly linked DM to alterations in white matter (WM) integrity and highlighted the relationship between cognitive impairment and abnormalities in WM structure. This review aims to summarize investigations into cognitive impairment and brain abnormalities in individuals with DM and to elucidate the correlation between these factors and the potential underlying mechanisms contributing to these abnormalities.
Myotonic dystrophy (DM) is a neuromuscular disease characterized by autosomal dominant inheritance, with a prevalence of 9.99 per 100,000 (Liao et al., 2022). DM is generally acknowledged as a condition that primarily affects skeletal muscle, leading to myotonia, progressive muscle weakness, and wasting (Vydra and Rayi, 2023). Additionally, it is known to cause abnormalities in multiple systems. Patients with DM may experience complications such as arrhythmia, cataracts, abnormal glucose tolerance, and gastrointestinal dysfunction (Rodbard et al., 2023).
DM is commonly categorized into two subtypes based on distinct genetic mutation sites: DM type 1 (DM1) and DM type 2 (DM2). DM1 and DM2 share numerous similarities but exhibit notable distinctions (Vydra and Rayi, 2023). DM1, also known as Steinert disease, is caused by an unstable CTG trinucleotide repeat expansion in the 3′ untranslated region (UTR) of the myotonic dystrophy protein kinase (DMPK) gene located on chromosome 19q13.3. This gene encodes DMPK, which is expressed in skeletal muscle and functions as a myosin kinase (Brook et al., 1992). DM2 results from an unstable tetranucleotide CCTG repeat expansion in intron 1 of the nucleic acid-binding protein (CNBP) gene (previously known as zinc finger 9 gene, ZNF9) on chromosome 3q21, consisting of a complex repeat motif (TG)n (TCTG)n (CCTG) n (Ranum et al., 1998). The expansion of the CCTG repeats, ranging from 75 to 10,000 times, leads to the manifestation of DM2 (Liquori et al., 2001).
DM1 can be classified into five distinct forms: congenital, childhood-onset, juvenile-onset, adult-onset (also known as classic form), and late-onset (Meola and Sansone, 2007). The congenital form is the most severe and is characterized by CTG extensions of more than 1,000 repeats. The resulting hypotonia and respiratory distress have a detrimental effect on both survival and development (Ho et al., 2015). Childhood-onset or juvenile-onset describes children who develop symptoms after the age of 1 year. Both of them develop symptoms similar to those of adults. The main difference between them is that childhood-onset is initially clinically apparent between the ages of 1 and 10 years, while juvenile-onset is apparent between 10 and 20 years (Stokes et al., 2019). The adult or classic form is the most common type of DM1. Additionally, the adult-onset form exhibits high variability. It can vary in presentation and severity among different individuals, even within the same family. The classic presentation is characterized by the presence of myotonia and muscle weakness (Joosten et al., 2023). The late-onset subtype, which typically manifests after the age of 40, is challenging to diagnose due to its subtle symptoms and is often associated with the development of cataracts (Yum et al., 2017).
The available evidence suggests the involvement of the central nervous system (CNS) in patients with DM. The recognition of cognitive impairment is increasingly gaining importance due to its significant impact on overall quality of life (Wenninger et al., 2018). However, the heterogeneity of clinical manifestations makes it challenging to identify specific features of cognitive impairment. Current cognitive assessments predominantly rely on scales like the Mini-Mental State Examination (MMSE), which may be influenced by subjective factors. Evidence has demonstrated that changes in imaging can provide insights into the location and extent of CNS involvement (Angelini and Pinzan, 2019). Moreover, numerous studies have established a correlation between cognitive impairment and neuroimaging abnormalities (Cabada et al., 2021; Labayru et al., 2022; Koscik et al., 2023). Finally, current research on mechanisms falls short of providing a comprehensive explanation for the observed abnormalities in the CNS and neuroimaging changes, as well as their interrelationship.
Therefore, this review investigates cognitive impairment and abnormalities in neuroimaging among patients with DM. It also explores their interrelationships to establish them as reliable markers of disease severity in cognitive impairment.
Ample evidence suggests cerebral involvement in DM (see Supplementary Table S1). Patients with DM may experience cognitive impairment, personality disorders, sleep disorders, fatigue, social difficulties, and other related issues, which are more pronounced in DM1 (Hamel, 2022). Among them, cognitive impairment is frequently observed in individuals with DM, expecially in those with DM1 (Table 1).
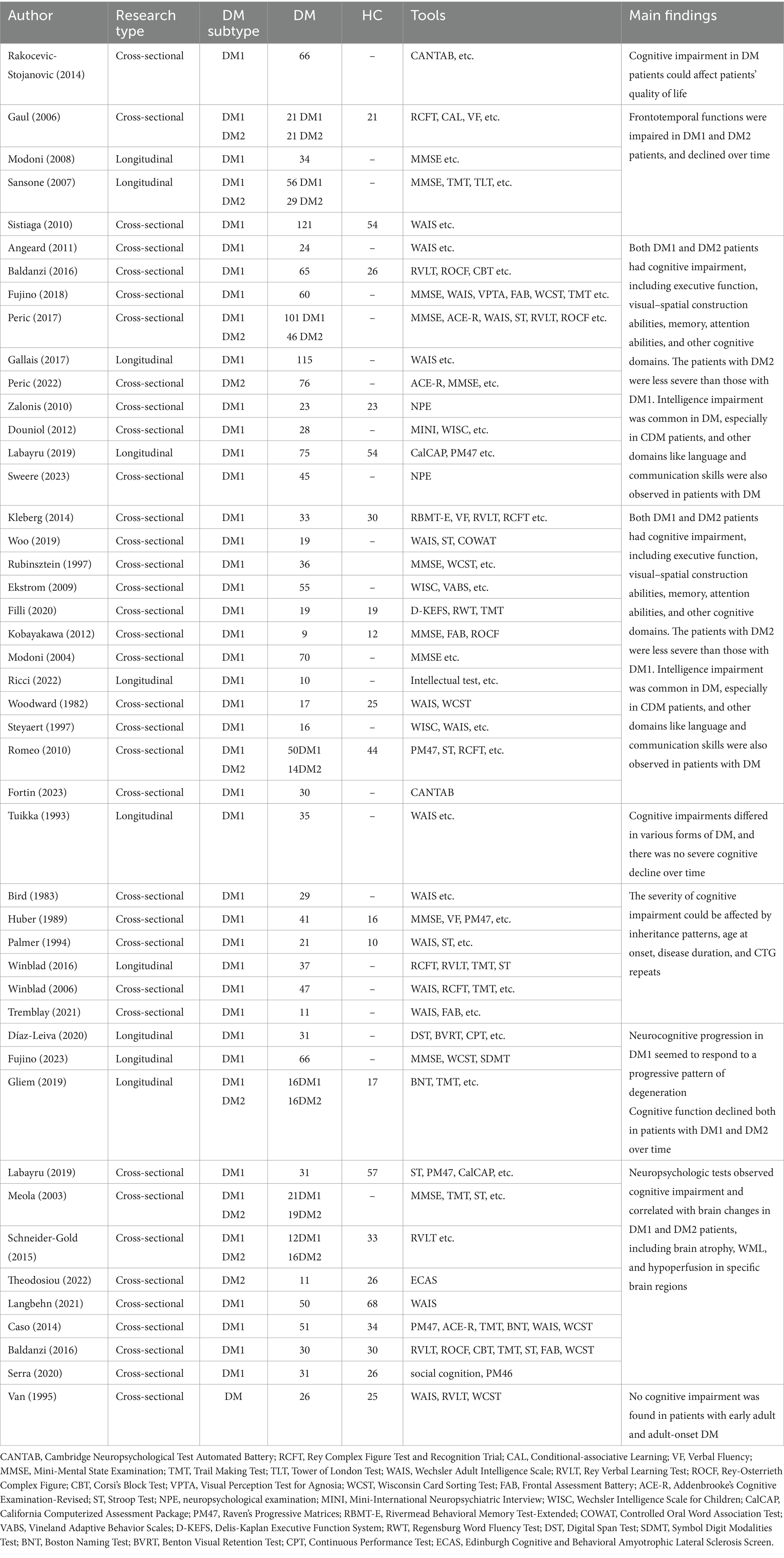
Table 1. Summary of literature on cognitive impairment in DM.
A previous review summarized that cognitive impairment was observed in over 50% of DM1 patients, which manifests as a variable combination of global cognitive deficits across various domains, including social cognition, memory, executive function and visuospatial function (Rosado Bartolomé et al., 2022). It explicitly affects frontotemporal functions (Gaul et al., 2006; Sansone et al., 2007; Modoni et al., 2008; Sistiaga et al., 2010), the prominent characteristic of which is executive dysfunction (Baldanzi et al., 2016; Gallais et al., 2017; Peric et al., 2017; Fujino et al., 2018; Peric et al., 2022). This manifests as difficulties in planning, organizing, initiating tasks, and making decisions. Consequently, it can significantly impact the ability to effectively carry out daily activities, resulting in apathy and reduced physical activity. Additionally, visuospatial processing difficulties exist, encompassing the comprehension and interpretation of visual information about spatial connections among objects (Angeard et al., 2011; Douniol et al., 2012; Baldanzi et al., 2016; Gallais et al., 2017; Peric et al., 2017; Fujino et al., 2018; Labayru et al., 2020a; Peric et al., 2022; Sweere et al., 2023). Memory deficits, particularly in working memory and short-term memory domains, are prevalent among patients with DM1, leading to difficulties in recalling recent events and acquiring new knowledge (Gallais et al., 2017; Fujino et al., 2018; Woo et al., 2019; Sweere et al., 2023). Some patients with DM1 may also experience challenges in intelligence, which can impact their social interactions, language, and communication skills (Kobayakawa et al., 2012; Filli et al., 2020; Ricci et al., 2022; Fortin et al., 2023). It may manifest as difficulties with word retrieval, comprehension, and expression. These investigations enhance our understanding of the involvement of the central nervous system in DM1 and facilitate the formulation of corresponding therapeutic strategies.
The cognitive symptoms in DM1 patients exhibit significant variations due to the diverse onset patterns, encompassing a spectrum from mild to severe (Table 2). Congenital myotonic dystrophy (CDM) is a fetal disease that is characterized by CTG repeat extensions longer than 1,000 repeats. Typically, patients have an average intelligence quotient (IQ) below 70 and experience mental retardation, which may precede the development of muscle symptoms (Reardon et al., 1993; Spranger, 1997; Cascais et al., 2024). Apart from lower IQ, adaptive skills, executive dysfunction, sleep disorders, and autistic symptoms can be observed in CDM (Patel et al., 2024). These defects deeply affected the quality of life, even extending into adolescence for affected patients. It’s disheartening to know that despite multiple efforts, numerous patients are still unable to achieve a satisfactory life. Childhood-onset and juvenile-onset forms can display attention deficit hyperactivity disorder (ADHD) and autism spectrum disorder (ASD), along with difficulties in learning and other aspects, resulting in poor interpersonal relationships. Conversely, the adult-onset form can exhibit normal intelligence. As time passes, they may experience declines in various cognitive domains. The late-onset subtype, typically occurring after the age of 40, presents challenges in diagnosis due to its subtle symptoms and is frequently associated with the involvement of other systems. Cognitive impairment often occurs in numerous patients with late-onset DM1, although it is generally less severe compared to other forms.
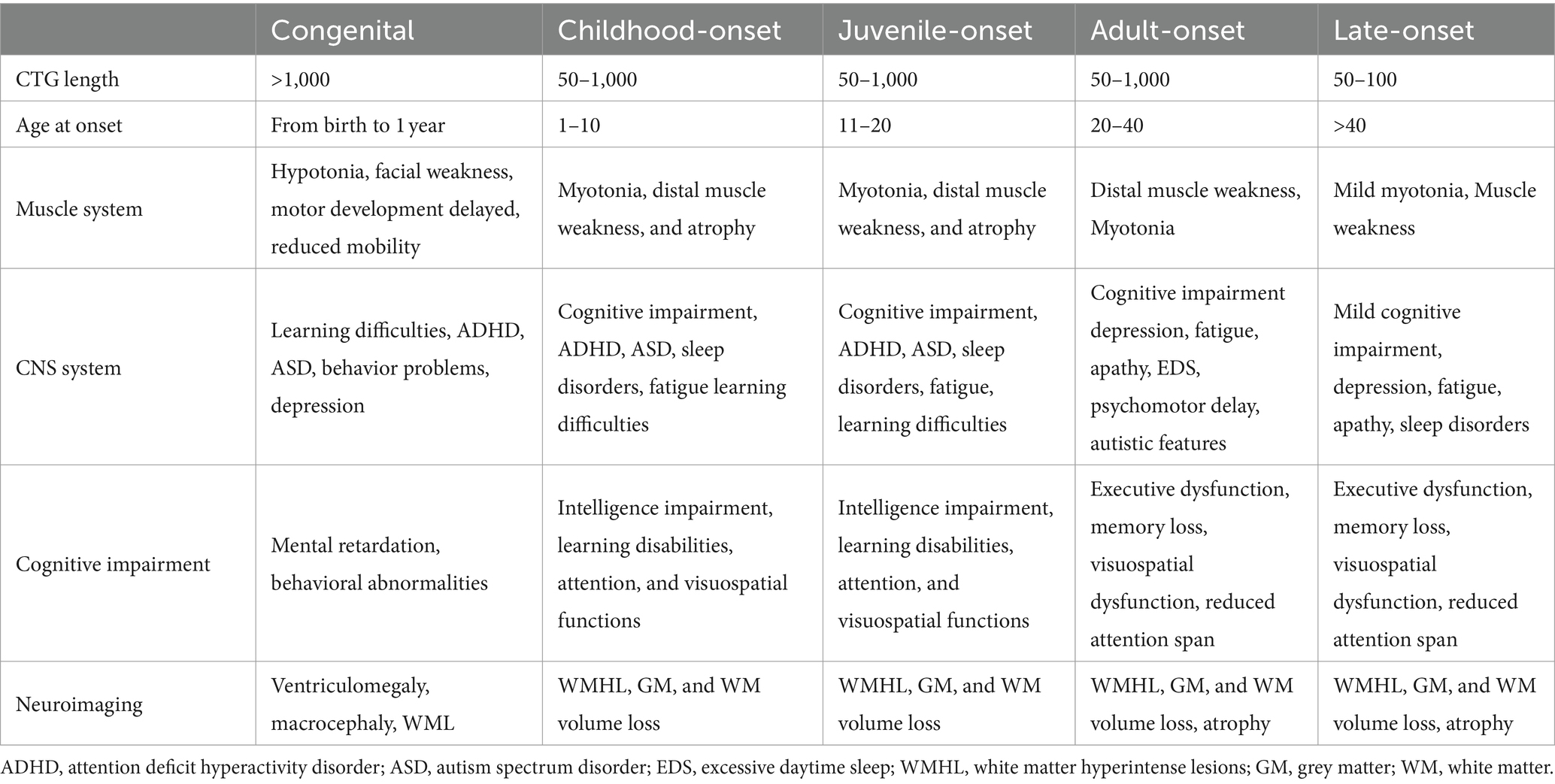
Table 2. Differences between all forms of DM1.
Multiple factors play an essential role in contributing to the variations in cognitive status among these forms, including the length of the CTG repeat expansion in the DMPK gene, age at onset, and inheritance pattern (Palmer et al., 1994; Winblad et al., 2006, 2016; Tremblay et al., 2021). An earlier onset and longer disease duration both indicate more severe cognitive deficits. Moreover, cognitive impairment tends to worsen progressively over time in adult-onset and late-onset forms, suggesting that there may be a positive correlation between cognitive decline and disease duration (Maresca et al., 2020; De Serres-Bérard et al., 2021). It provides evidence that brain changes in DM1 are more likely to be neurodegenerative (Labayru et al., 2019; Díaz-Leiva et al., 2020; Fujino et al., 2023).
The manifestation of DM2 is similar to DM1 in both the muscular and central nervous systems but less severe (Table 3) (Johnson et al., 2021). For example, almost all patients with DM1 experience cognitive impairment in multiple domains, while only one-third of DM2 patients may encounter cognitive difficulties (Sansone et al., 2007; Romeo et al., 2010; Gallais et al., 2017). Unfortunately, the amount of research on cognitive impairment in DM2 is not comparable to that conducted on DM1. Several studies suggest that patients with DM2 may exhibit better neuropsychological outcomes than those with DM1, implying potentially milder and less pervasive cognitive deficits (Peric et al., 2015; Gliem et al., 2019; Theodosiou et al., 2022). They indicate executive dysfunction associated with frontal and temporal lobe dysfunction, along with memory deficits and other cognitive challenges among individuals with DM2. However, it is mainly observed in older patients during testing or disease onset and those experiencing more pronounced muscle weakness (Peric et al., 2015).
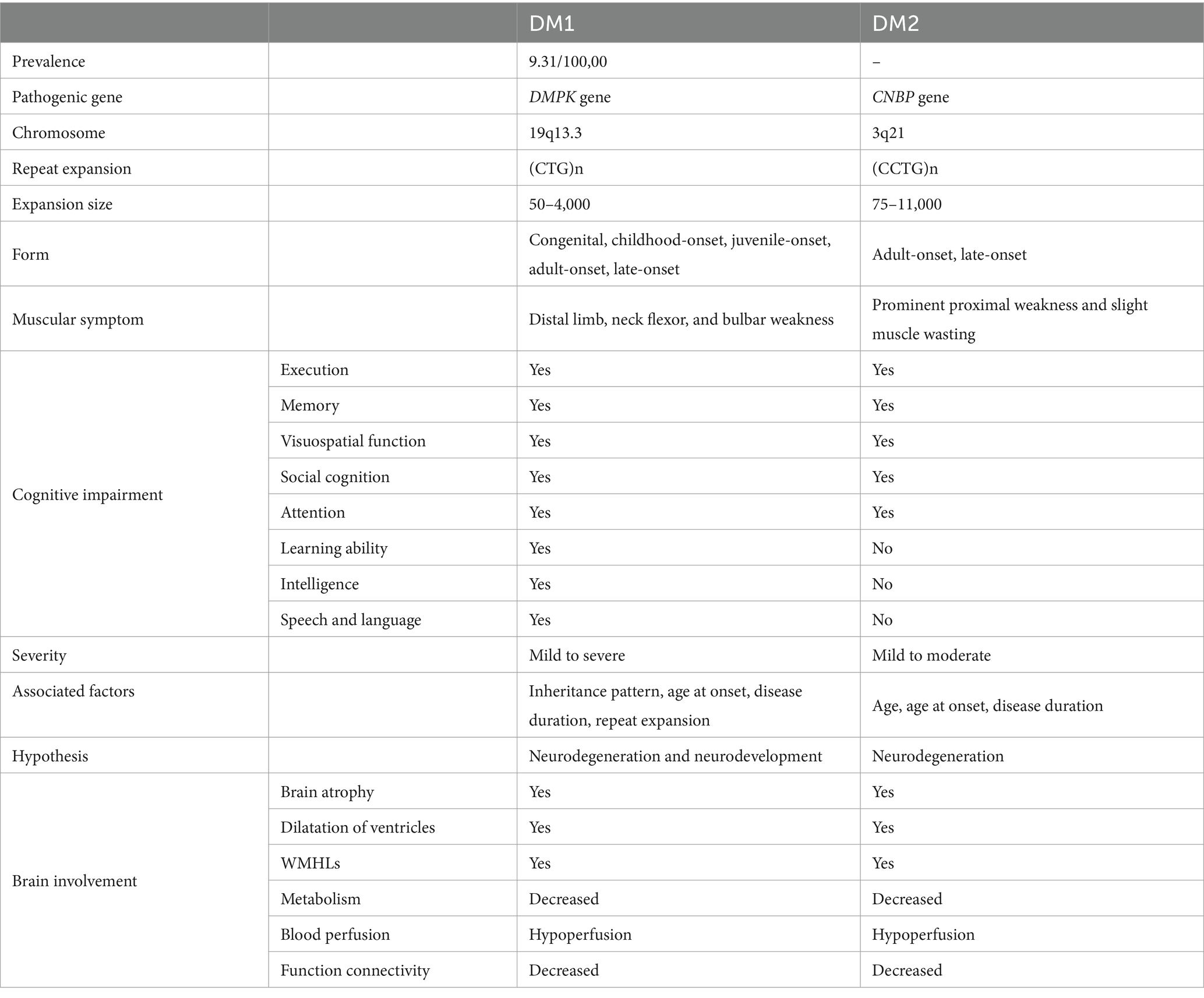
Table 3. Comparisons for clinical features between DM1 and DM2.
It is obvious that in various forms of DM, cognitive impairment becomes a significant problem, which increases the burden of life and healthcare. The exact reasons behind the diverse forms of cognitive impairment are not entirely understood. The different subtypes show varying disease progressions. Cognitive impairment is present from birth in CDM, while cognitive function in childhood, adult, and late-onset forms can develop normally but tends to decline over time. There is a need for a thorough investigation into cognitive impairment and its dynamic changes in different forms and subtypes of DM.
The cerebral involvement in DM1 and DM2 has been demostrated in vivo using diverse neuroimaging techniques (Table 4). Advancements in neuroradiological techniques have facilitated the potential for neuroimaging to identify DM (Minnerop et al., 2018). It entails employing various magnetic resonance imaging (MRI) techniques, such as traditional MRI to depict the basic brain structure and lesions, diffusion tensor imaging (DTI) to illustrate the microstructure in brain tissue, functional MRI (fMRI) to evaluate the metabolism, Fluorodeoxyglucose positron emission tomography (FDG-PET) to exam cerebral glucose metabolism, and single-photon emission computed tomography (SPECT) to evaluate cerebral perfusion. The neuroimaging features of DM exhibit variability and are not as pronounced as the clinical symptoms. These features include structural changes such as brain atrophy, ventricular enlargement, and white matter lesions (WML), as well as functional abnormalities such as hypoperfusion, decreased metabolism, and altered brain connectivity (see Supplementary Table S2).
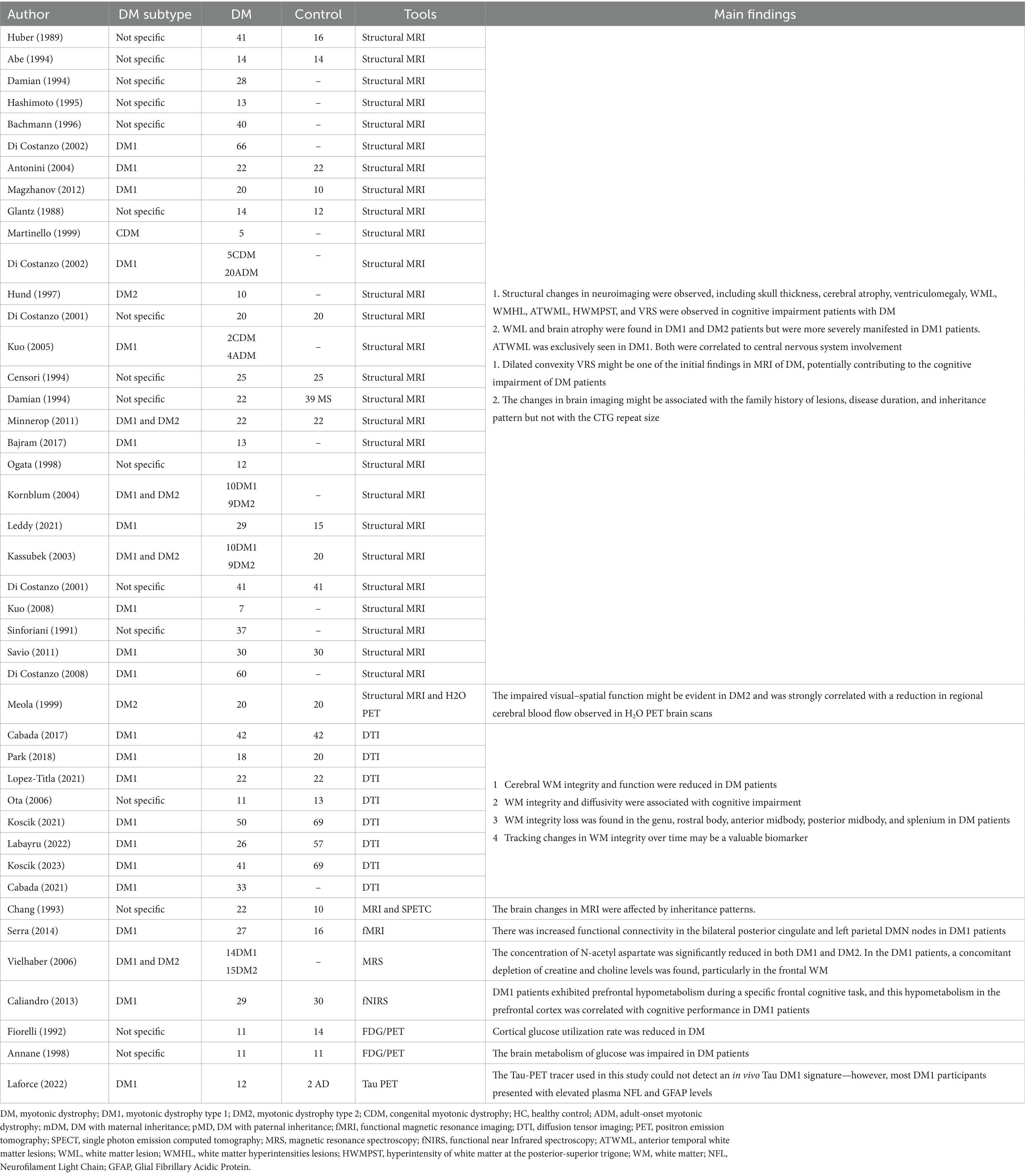
Table 4. Summary of neuroimaging studies on DM.
Traditional structural MRI is the most frequently used neuroimaging technique to detect brain involvement in patients with DM1. Several studies have reported that patients with DM1 could suffer from widespread brain atrophy, resulting in a reduction in specific brain regions (Di Costanzo et al., 2002b; Antonini et al., 2004; Magzhanov et al., 2012). Shang et al. summarized that the reduction of GM1 volume involved the bilateral Rolandic operculum, bilateral posterior central gyrus, bilateral precentral gyrus, right insula, right Heschel gyrus, right superior temporal gyrus, bilateral supplementary motor area, bilateral middle cingulate gyrus/paracingulate gyrus, left paracentral lobule, and bilateral caudate nucleus (Jiang et al., 2022). Additionally, enlargement of the ventricles may occur due to grey matter (GM) and white matter (WM) volume reduction, a condition referred to as ventriculomegaly (Glantz et al., 1988; Martinello et al., 1999; Di Costanzo et al., 2002a). The findings of a study demonstrated the potential occurrence of multifocal cortical and thalamic atrophy in individuals with DM2 as well (Theodosiou et al., 2022).
The neuroimaging of patients with DM1 frequently reveals WM abnormalities. Conventional MRI in DM1 patients often detects WML or white matter hyperintensity lesions (WMHL) on T2-weighted imaging and fluid-attenuated inversion-recovery (FLAIR) pulse sequences (Di Costanzo et al., 2001a, 2002a; Kuo et al., 2005; Magzhanov et al., 2012), which may indicate a decrease in myelin sheaths. Studies have demonstrated that WMHL can be localized or widespread (Huber et al., 1989; Gliem et al., 2019; Garmendia et al., 2023). In patients with DM1, WMHL predominantly occur in the brain’s anterior temporal, frontal, parieto-occipital, and periventricular regions (Censori et al., 1994; Damian et al., 1994; Minnerop et al., 2011; Bajrami et al., 2017). Anterior temporal white matter lesions (ATWML) are more prevalent among patients with DM1 than those with DM2 (Ogata et al., 1998; Kornblum et al., 2004; Leddy et al., 2021). Although research on WML in DM2 may be limited compared to DM1, there are still reports indicating the presence of diffuse periventricular WMHL (Meola et al., 1999; Kassubek et al., 2003).
Over the past two decades, advanced MRI techniques, such as DTI, have been extensively utilized to investigate alterations in WM (Cabada et al., 2017; Park et al., 2018; Koscik et al., 2021; Lopez-Titla et al., 2021; Labayru et al., 2022). The use of DTI has also revealed alterations in WM microstructure among patients with DM1, indicating potential changes in the integrity of WM. Studies using region-of-interest (ROI) approach and tract-based spatial statistics (TBSS) have demonstrated a widespread decrease in fractional anisotropy (FA) and an increase in mean diffusivity (MD) in DM1 (Koscik et al., 2021; Laforce et al., 2022). It refers to the axons damage and myelin loss. Similarly, other investigations have consistently observed a symmetrical reduction in FA across various significant connections, projections, and commissural fibers (Wozniak et al., 2013; van Dorst et al., 2019). The proposed pattern of heightened impairment, predominantly affecting the fronto-temporo-parietal and subcortical regions, along with a loss of GM-related WM integrity, suggests that disruptions to neuronal networks may underlie the development of central nervous system symptoms in DM1 (De Antonio et al., 2016; van Dorst et al., 2019).
Dilated Virchow-Robin spaces (VRS) are reported to occur at a high frequency in patients with DM1 and may precede the development of WML (Bachmann et al., 1996; Di Costanzo et al., 2001b, 2002b). The presence of VRS is commonly observed in association with aging, vascular risk factors, neurodegenerative disorders, and neuroinflammatory diseases. However, the role of dilated VRS in DM1 remains inadequately understood.
Functional MRI has shown that there are more cerebral dysfunctions than just structural changes. Research findings have indicated that individuals with DM1 may exhibit cerebral hypoperfusion in perfusion MRI, which refers to a reduction in blood flow to the brain (Chang et al., 1993; Meola et al., 1999). Furthermore, patients with DM1 have been observed to display altered connectivity patterns in fMRI, particularly involving the default mode network (DMN), which is associated with self-referential thinking and internal mental processes (Serra et al., 2014; Okkersen et al., 2017; Minnerop et al., 2018; Serra et al., 2020). Additionally, decreased levels of N-acetyl aspartate (NAA) and hypometabolism were reported in patients with DM1 (Fiorelli et al., 1992; Annane et al., 1998; Vielhaber et al., 2006; Caliandro et al., 2013). At the same time, there is a lack of comprehensive research on functional MRI.
Nevertheless, these modifications may present distinctly across different subcategories of individuals with DM1. Evidence supports that ventriculomegaly or macrocephaly regularly occurs in CDM (Garcia-Alix et al., 1991). Neuroimaging studies in these patients reveal that more than 50% of them exhibit ventricular dilatation, which is present at birth. This finding supports the hypothesis of a prenatal origin for mental retardation (Meola and Sansone, 2007). A thorough review has made a comprehensive summary of them (Angelini and Pinzan, 2019). Diffuse microstructural WM damage with widespread FA reduction and increased mean diffusivity in fibers in noncongenital patients with DM1 associated with GM volume reduction were reported in numerous studies (Wozniak et al., 2013). The magnetic resonance spectroscopy (MRS) investigation revealed decreased NAA concentrations in both the grey and WM regions of DM1 patients, specifically those affected by congenital and juvenile forms, providing evidence of potential disruptions in neuronal maturation within the brain (Evangelisti et al., 2022).
In summary, various neuroimaging techniques, including the identification of both traditional structural modifications and functional changes, indicate that distinct mechanisms play a role in brain alterations in DM1 patients and suggest the presence of microstructural abnormalities.
The presentation of DM2 exhibits similarities to DM1 in both the muscular and central nervous systems, albeit with a milder manifestation. In DM2 patients, conventional brain MRI findings may appear within normal limits. However, diffuse WM changes can be observed in advanced stages or more severe cases, although they may exhibit less pronounced characteristics or differ from those seen in DM1. Surprisingly, a limited number of studies have reported that a subset of DM2 patients with diffuse and confluent WMHL similar to those found in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) (Renard and Menjot De Champfleur, 2018).
Evidence also indicates a correlation between various factors and the abnormalities observed in brain imaging. Muscular and genetic features are associated with brain abnormalities in DM1, as evidenced by the changes in different forms (Labayru et al., 2019). Individuals with extended CTG repeats, prolonged disease duration, and specific inheritance patterns are more likely to experiencing sifnificant structural and functional changes in DM1 (Di Costanzo et al., 2001a, 2002a, 2008).
A 10-year longitudinal study reported a comparable decline in GM and WM volume loss and an increase in WML among individuals with childhood-onset, adult-onset, and late-onset DM1 (Labayru et al., 2020b). Furthermore, it was discovered that new GM and WM areas were impacted by disease progression in both childhood and adult-onset DM1 patients, in addition to the initially affected regions. The childhood-onset presented a reduction in both GM and WM volumes at baseline and during follow-up. The longitudinal analyses revealed that only adult and late-onset patients experienced a significant decline in WM volume over time (Garmendia et al., 2023). Other longitudinal studies have provided novel insights into the prevalence of WML in adult-onset DM1 and the progression of brain atrophy over time (Cabada et al., 2021; Koscik et al., 2021). However, a recent 5-year longitudinal study revealed no significant progression in the changes of GM and WM in patients with DM1 and DM2, suggesting either a slowly progressive process or a stable course of cerebral changes in middle-aged and adult-onset patients (Gliem et al., 2019). Microstructural WM changes might be attributed to development changes in congenital or child-onset forms, while GM degeneration may be more closely associated with a degenerative process related to aging. Additionally, WM abnormalities in the juvenile form may contribute to GM degeneration. Moreover, research using magnetization transfer imaging on DM indicated reduced magnetization transfer (MT) ratios in WMHL compared to normal-appearing white matter (NAWM), showing that structural changes in the WM may progress during the clinical course of DM. The independence of WM and GM neurodegenerative processes is suggested by the presence of WM T2 hyperintense lesions, and no correlation with GM loss is observed (Naka et al., 2002). It is generally suggested that both developmental and neurodegenerative changes are observed in DM. Cognitive impairment in DM is more likely to be neurodegenerative due to natural aging processes, despite differing results.
Extensive research has been conducted to better understand the cerebral changes associated with cognitive impairment in DM (Labayru et al., 2019).
Numerous studies have consistently reported that cognitive decline among patients with DM is closely associated with the degree of brain involvement, which can be found in neuroimaging (Cabada et al., 2021; Koscik et al., 2021; Lopez-Titla et al., 2021; Labayru et al., 2022). Specific brain regions, such as the anterior temporal lobes and basal ganglia, were significantly affected in social cognitive dysfunction. Atrophy and microstructural damage were found in DM1 patients in these regions. Bilateral ATWML may be a contributing factor to intellectual impairment in DM1, as it was observed in these patients. According to previous pathological findings, the loss and disordered arrangement of myelin sheaths and axons, along with heterotopic neurons raised the possibility that the ATWML is compatible with focal dysplasia of WM (Ogata et al., 1998). A negative correlation was observed between hippocampal volume and the Perceptual Reasoning Index (PRI) as well as Processing Speed Index (PSI) scores in patients with DM1 (Langbehn et al., 2021). Individuals with WM damages experienced more severe cognitive impairments, especially those with a higher burden of WMHL. This was associated with impairments in memory, executive function, reasoning, and visuospatial abilities (Bajrami et al., 2017).
Multiple studies have investigated the associations between cognitive performance and functional brain changes in patients with DM1 (Sinforiani et al., 1991; Bajrami et al., 2017; Lopez-Titla et al., 2021; Labayru et al., 2022). A DTI study in childhood-onset DM1 revealed abnormal WM integrity, demonstrating a strong correlation between FA, intelligence, and executive functioning (Koscik et al., 2023). Another analysis focusing on specific ROI in patients with DM1 found a significant association between FA and MD, particularly impacting working memory in the frontal and temporal lobes (Wozniak et al., 2013). Additionally, a significant correlation was observed between the decline in FA in the frontal, temporomedial, and parietal lobes and specific deficits in memory impairments (Lopez-Titla et al., 2021). Furthermore, studies using FDG-PET in DM1 have demonstrated an association between hypometabolism in brain lobes and executive dysfunction as well as attention deficit (Weber et al., 2010; Weijs et al., 2021; Sweere et al., 2023).
Longitudinal studies that track these changes over time have provided valuable insights into the progressive impact on cognitive function and WM. The degree of WM involvement is associated with declining cognitive function (Sansone et al., 2007). A longitudinal study found that the Rey-Osterrieth Complex Figure (ROCF) and two California Computerized Assessment Package (CALCAP) measures were positively correlated with the percentage of WM volume loss in adult and late-onset DM1 (Labayru et al., 2020b). Lower scores on neuropsychological tests were associated with higher percentage of total WM volume loss. Another longitudinal study using DTI in DM1 patients revealed a significant deterioration in WM integrity over time compared to healthy controls (Labayru et al., 2022). The FA values exhibited positive correlations with the visuo-construction domain and intellectual functioning. However, another longitudinal study indicated that the functional performance of middle-aged adult-onset patients improved at a slower rate (Gliem et al., 2019). In conjunction with a Bayesian stacked random forest model, a diffusion-based model using WM fiber tracts as predictors was employed to diagnose and predict outcomes in DM (Kamali et al., 2021). This work highlights the potential of neuroimaging in comprehending and monitoring clinical performance, especially cognitive impairment.
Although genetically distinct, DM1 and DM2 share a common pathogenic mechanism characterized by the formation of nuclear inclusions, sequestration of regulatory proteins, defects in alternative splicing, and repeat-associated non-ATG (RAN) translation (Liu et al., 2021).
A toxic RNA-mediated mechanism has been proposed based on the detection of abnormal RNA aggregates in muscle neurons obtained from post-mortem biopsies of patients with DM (Wheeler and Thornton, 2007). The aberrant RNAs are widely expressed in cortical and subcortical neurons, forming imperfect double-stranded hairpin structures that result in the deregulation of RNA-binding proteins (RBPs) responsible for mRNA splicing, such as muscleblind-like (MBNL) proteins and CUG RNA-binding protein (CUGBP1) (Giménez-Bejarano et al., 2023). The accumulation of these aberrant RNAs in the neuronal nucleus compromises a specific developmental program of alternative splicing (Lin et al., 2006; López-Martínez et al., 2020; Soltanzadeh, 2022). The dysregulation leads to a range of symptoms in patients with DM1. The RNA gain-of-function mechanism involves the accumulation of expanded CUG or CCUG transcripts within the cell nuclei, leading to the formation of ribonuclear inclusions.
The MBNL proteins play a prominent role in DM pathogenesis, as evidenced by the sequestration of each of the three MBNL isoforms (MBNL1, MBNL2, and MBNL3) by CUG RNAs in the cell nuclei (Meola and Cardani, 2015). The expression of MBNL1 is most prominent in the heart and muscles, indicating its predominant role in regulating alternative splicing. The expression of MBNL2 is prominently observed in the brain, while its levels decrease in muscle during postnatal development. It serves a related function in the central nervous system. Conversely, MBNL3 is expressed in the placenta and muscle satellite cells, although its functions in vivo remain unknown. Both MBNL1 and MBNL2 play essential roles in neuronal maturation. The MBNL1 is crucial for promoting the growth of neuronal dendrites, whereas MBNL2 is involved in overseeing developmental transitions related to alternative splicing and polyadenylation. Dysfunction of MBNL1 and MBNL2 may result in disrupted neuronal transmission and cognitive behavior, underscoring their significance in modulating synaptic transmission. There used to be MBNL2 knock-out rodent models that exhibited cognitive impairment and depressive-like behavior (Wang et al., 2018; Ramon-Duaso et al., 2019). An increased nuclear level of calpain-2 and a reduced MBNL2 level were observed in DM1 (Wang et al., 2022). Moreover, inhibiting the nuclear translocation of calpain-2 effectively prevented the degradation of MBNL2 and preserved its function in regulating RNA processing in the adult-onset form. Goodwin et al. found that MBNL2 protein also exhibits binding affinity for the CCUG RNA repeats in DM2 brain tissue (Goodwin et al., 2015). Therefore, MBNL2 might play an essential role in the CNS involvement of patients with DM.
Moreover, the discovery of repeat-associated non-ATG (RAN) translation and its increasing correlation with neurodegenerative disorders resulting from microsatellite expansions indicates that RAN proteins play a pivotal role in the neurological manifestations of DM (Zu et al., 2011). The study demonstrated that the CCTG expansion in DM2 expresses sense and antisense tetrapeptide poly-(LPAC) and poly-(QAGR) RAN proteins, which are modulated by MBNL through nuclear sequestration and are also found in neurons and accumulate within WM (Zu et al., 2017). However, the connection between the toxic RNA hypothesis and CNS involvement remains incomplete.
The RNA-seq analysis of both GM and WM tissue samples from the brains of patients with DM1 revealed a potential correlation between mis-spliced events and cerebral changes. Surprisingly, these events dominantly occurred in astrocytes and oligodendrocytes, which might be related to substantial WM damage (Yoshizumi et al., 2023). Another study found that the splicing abnormalities tended to be more prominent in the WM glial cells than in the GM neurons (Nakamori et al., 2022). Masayuki et al. used a particular approach to detect the neuronal cells in the cortex and glial cells in the WM (Nakamori et al., 2022). They found that cortical neurons had more unstable repeats and higher methylation compared to WM glial cells. These findings indicate that GM and WM are involved in patients with DM.
A study investigated the association between DMPK blood DNA methylation (DNAm) and cognitive functions in DM1 patients, revealing its potential role in predicting future cognitive decline at baseline (Breton et al., 2020). However, no methylation was observed in the CNBP gene of DM2 patients, neither in their blood cells nor muscles (Santoro et al., 2018). Furthermore, there is no evidence indicating methylation occurring in brain tissue among both DM1 and DM2 patients. The discussion about its association with neuroimaging changes was omitted.
According to the most recent systematic review, the primary neuropathological findings include WM changes, cellular changes, and deposition of protein and nucleotide (Weijs et al., 2021). In deep and subcortical WM, there was myelin loss with varying axonal loss, dilation of perivascular spaces, gliosis, and capillary hyalinization. Additionally, widened perivascular spaces were consistent with the observed features on MRI scans. One study proposed a hypothesis suggesting that the increased burden caused by microvascular changes, inadequate drainage of interstitial fluid, and degradation of protein products may serve as mechanisms for anterior temporal WML in DM1 (Renard and Menjot De Champfleur, 2018). However, it is worth noting that these changes may not be observed in all individuals with DM. It is essential to distinguish between DM and CADASIL, as both conditions can present with anterior temporal lobe hyperintensities and WMHL (Liu et al., 2022). Moreover, this suggests that the occurrence of these changes depends on the disconnection of the cortical regions due to the breakdown of the WM tract (Simoncini et al., 2020). A further study has demonstrated that cerebrospinal fluid (CSF) and blood neurofilament light chain (NFL) may serve as biomarkers of CNS involvement (Rossi and Silvestri, 2023). Neuronal loss was not observed in patients with CMD, but it was demonstrated in a subset of patients with adult-onset. However, these results were inconclusive. Several studies have shown that it may be related to clinical features of excessive daytime sleepiness and ventilatory dysfunction (Ono et al., 1996; Zalonis et al., 2010). Neurofibrillary tangles (NFTs), consisting of mis-spliced tau protein, were observed in the entorhinal to transentorhinal cortices in all patients with DM1 and exhibited a generally widespread distribution (Vermersch et al., 1996; Maurage et al., 2005; Iwasaki, 2018). However, limited neuropathological data is available for DM2; similar brain tau pathology has also been found in DM2 cases (Maurage et al., 2005). Nonetheless, no evidence of amyloid plaques associated with DM was found. Whether the cognitive impairment reported in DM is related to the development of tau pathology remains unclear. In a review of the pathogenesis and pathology of CNS deficits in DM1, the authors summarized the possible causes that may contribute to CNS damage (Liu et al., 2021).
The evidence strongly suggests that the brain manifests various pathologies that significantly impact its function, mainly cognitive function. However, further investigation is required to elucidate the underlying mechanisms in DM.
This review provides a comprehensive overview of cognitive impairment and brain changes in patients with DM, emphasizing the importance of neuroimaging for early detection and management. Future prospective cohort studies with larger sample sizes are warranted to elucidate its progression. At the same time, further investigation into underlying mechanisms is crucial for developing targeted therapeutic interventions that can improve patient outcomes.
YW: Conceptualization, Writing – review & editing, Writing – original draft, Data curation, Investigation, Visualization, Methodology. QW: Conceptualization, Supervision, Validation, Writing – original draft, Writing – review & editing, Data curation. JL: Data curation, Investigation, Supervision, Validation, Writing – original draft, Writing – review & editing. HS: Conceptualization, Supervision, Writing – review & editing. RO: Conceptualization, Investigation, Writing – original draft, Writing – review & editing, Supervision, Methodology.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China (No. 82271272) and the Sichuan Science and Technology Program (Nos. 2022ZDZX0023 and 2023YFS0265).
The authors thank all subjects for their participation in the study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2024.1369332/full#supplementary-material
Angeard, N., Jacquette, A., Gargiulo, M., Radvanyi, H., Moutier, S., Eymard, B., et al. (2011). A new window on neurocognitive dysfunction in the childhood form of myotonic dystrophy type 1 (DM1). Neuromuscul. Disord. NMD 21, 468–476. doi: 10.1016/j.nmd.2011.04.009
Angelini, C., and Pinzan, E. (2019). Advances in imaging of brain abnormalities in neuromuscular disease. Ther. Adv. Neurol. Disord. 12:175628641984556. doi: 10.1177/1756286419845567
Annane, D., Fiorelli, M., Mazoyer, B., Pappata, S., Eymard, B., Radvanyi, H., et al. (1998). Impaired cerebral glucose metabolism in myotonic dystrophy: a triplet-size dependent phenomenon. Neuromuscul. Disord. 8, 39–45. doi: 10.1016/S0960-8966(97)00144-2
Antonini, G., Mainero, C., Romano, A., Giubilei, F., Ceschin, V., Gragnani, F., et al. (2004). Cerebral atrophy in myotonic dystrophy: a voxel based morphometric study. J. Neurol. Neurosurg. Psychiatry 75, 1611–1613. doi: 10.1136/jnnp.2003.032417
Bachmann, G., Damian, M. S., Koch, M., Schilling, G., Fach, B., and Stöppler, S. (1996). The clinical and genetic correlates of MRI findings in myotonic dystrophy. Neuroradiology 38, 629–635. doi: 10.1007/s002340050322
Bajrami, A., Azman, F., Yayla, V., Cagirici, S., Keskinkiliç, C., and Sozer, N. (2017). MRI findings and cognitive functions in a small cohort of myotonic dystrophy type 1: retrospective analyses. Neuroradiol. J. 30, 23–27. doi: 10.1177/1971400916678223
Baldanzi, S., Bevilacqua, F., Lorio, R., Volpi, L., Simoncini, C., Petrucci, A., et al. (2016). Disease awareness in myotonic dystrophy type 1: an observational cross-sectional study. Orphanet J. Rare Dis. 11:34. doi: 10.1186/s13023-016-0417-z
Breton, É., Légaré, C., Overend, G., Guay, S. P., Monckton, D., Mathieu, J., et al. (2020). DNA methylation at the DMPK gene locus is associated with cognitive functions in myotonic dystrophy type 1. Epigenomics 12, 2051–2064. doi: 10.2217/epi-2020-0328
Brook, J. D., McCurrach, M. E., Harley, H. G., Buckler, A. J., Church, D., Aburatani, H., et al. (1992). Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68, 799–808. doi: 10.1016/0092-8674(92)90154-5
Cabada, T., Díaz, J., Iridoy, M., López, P., Jericó, I., Lecumberri, P., et al. (2021). Longitudinal study in patients with myotonic dystrophy type 1: correlation of brain MRI abnormalities with cognitive performances. Neuroradiology 63, 1019–1029. doi: 10.1007/s00234-020-02611-9
Cabada, T., Iridoy, M., Jericó, I., Lecumberri, P., Seijas, R., Gargallo, A., et al. (2017). Brain involvement in myotonic dystrophy type 1: a morphometric and diffusion tensor imaging study with neuropsychological correlation. Arch. Clin. Neuropsychol. Off. J. Natl. Acad. Neuropsychol. 32, 401–412. doi: 10.1093/arclin/acx0008
Caliandro, P., Silvestri, G., Padua, L., Bianchi, M. L., Simbolotti, C., Russo, G., et al. (2013). fNIRS evaluation during a phonemic verbal task reveals prefrontal hypometabolism in patients affected by myotonic dystrophy type 1. Clin. Neurophysiol. 124, 2269–2276. doi: 10.1016/j.clinph.2013.05.010
Cascais, I., Garrido, C., Morais, L., Amorim, R., Lima, R., Mansilha, H. F., et al. (2024). Myotonic dystrophy type 1 (Steinert disease): 29 years of experience at a tertiary pediatric hospital. Europ. J. Paediat. Neurol. 48, 85–90. doi: 10.1016/j.ejpn.2023.12.001
Censori, B., Provinciali, L., Danni, M., Chiaramoni, L., Maricotti, M., Foschi, N., et al. (1994). Brain involvement in myotonic dystrophy: MRI features and their relationship to clinical and cognitive conditions. Acta Neurol. Scand. 90, 211–217. doi: 10.1111/j.1600-0404.1994.tb02708.x
Chang, L., Anderson, T., Migneco, O. A., Boone, K., Mehringer, C. M., Villanueva-Meyer, J., et al. (1993). Cerebral abnormalities in myotonic dystrophy. Cerebral blood flow, magnetic resonance imaging, and neuropsychological tests. Arch. Neurol. 50, 917–923. doi: 10.1001/archneur.1993.00540090024006
Damian, M. S., Schilling, G., Bachmann, G., Simon, C., Stöppler, S., and Dorndorf, W. (1994). White matter lesions and cognitive deficits: relevance of lesion pattern? Acta Neurol. Scand. 90, 430–436. doi: 10.1111/j.1600-0404.1994.tb02753.x
De Antonio, M., Dogan, C., Hamroun, D., Mati, M., Zerrouki, S., Eymard, B., et al. (2016). Unravelling the myotonic dystrophy type 1 clinical spectrum: a systematic registry-based study with implications for disease classification. Rev. Neurol. (Paris) 172, 572–580. doi: 10.1016/j.neurol.2016.08.003
De Serres-Bérard, T., Pierre, M., Chahine, M., and Puymirat, J. (2021). Deciphering the mechanisms underlying brain alterations and cognitive impairment in congenital myotonic dystrophy. Neurobiol. Dis. 160:105532. doi: 10.1016/j.nbd.2021.105532
Di Costanzo, A., Di Salle, F., Santoro, L., Bonavita, V., and Tedeschi, G. (2001a). T2 relaxometry of brain in myotonic dystrophy. Neuroradiology 43, 198–204. doi: 10.1007/s002340000459
Di Costanzo, A., Di Salle, F., Santoro, L., Bonavita, V., and Tedeschi, G. (2001b). Dilated Virchow-Robin spaces in myotonic dystrophy: frequency, extent and significance. Eur. Neurol. 46, 131–139. doi: 10.1159/000050786
Di Costanzo, A., Di Salle, F., Santoro, L., Bonavita, V., and Tedeschi, G. (2002a). Brain MRI features of congenital- and adult-form myotonic dystrophy type 1: case-control study. Neuromuscul. Disord. 12, 476–483. doi: 10.1016/S0960-8966(01)00324-8
Di Costanzo, A., Di Salle, F., Santoro, L., Tessitore, A., Bonavita, V., and Tedeschi, G. (2002b). Pattern and significance of white matter abnormalities in myotonic dystrophy type 1: an MRI study. J. Neurol. 249, 1175–1182. doi: 10.1007/s00415-002-0796-z
Di Costanzo, A., Santoro, L., de Cristofaro, M., Manganelli, F., Di Salle, F., and Tedeschi, G. (2008). Familial aggregation of white matter lesions in myotonic dystrophy type 1. Neuromuscul Disord. 18, 299–305. doi: 10.1016/j.nmd.2008.01.008
Díaz-Leiva, J., Cabada-Giadás, T., Seijas-Gómez, R., Jericó-Pascual, I., López-Sala, P., and Iridoy-Zulet, M. (2020). Neuropsychological profile in patients with myotonic dystrophy type 1: a four-year follow-up study. Rev. Neurol. 70, 406–412. doi: 10.33588/rn.7011.2019455
Douniol, M., Jacquette, A., Cohen, D., Bodeau, N., Rachidi, L., Angeard, N., et al. (2012). Psychiatric and cognitive phenotype of childhood myotonic dystrophy type 1. Dev. Med. Child Neurol. 54, 905–911. doi: 10.1111/j.1469-8749.2012.04379.x
Evangelisti, S., Gramegna, L. L., De Pasqua, S., Rochat, M. J., Morandi, L., Mitolo, M., et al. (2022). In vivo Parieto-occipital white matter metabolism is correlated with visuospatial deficits in adult DM1 patients. Diagnostics 12:2305. doi: 10.3390/diagnostics12102305
Filli, L., Schwegler, S., Meyer, C., Killeen, T., Easthope, C. S., Broicher, S. D., et al. (2020). Characterizing cognitive-motor impairments in patients with myotonic dystrophy type 1. Neuromuscul. Disord. 30, 510–520. doi: 10.1016/j.nmd.2020.04.005
Fiorelli, M., Duboc, D., Mazoyer, B. M., Blin, J., Eymard, B., Fardeau, M., et al. (1992). Decreased cerebral glucose utilization in myotonic dystrophy. Neurology 42, 91–94. doi: 10.1212/WNL.42.1.91
Fortin, J., Côté, I., Gagnon, C., and Gallais, B. (2023). Do classical and computerized cognitive tests have equal intrarater reliability in myotonic dystrophy type 1? Neuromuscul. Disord. 33, 490–497. doi: 10.1016/j.nmd.2023.04.008
Fujino, H., Shingaki, H., Suwazono, S., Ueda, Y., Wada, C., Nakayama, T., et al. (2018). Cognitive impairment and quality of life in patients with myotonic dystrophy type 1. Muscle Nerve 57, 742–748. doi: 10.1002/mus.26022
Fujino, H., Suwazono, S., Ueda, Y., Kobayashi, M., Nakayama, T., Imura, O., et al. (2023). Longitudinal changes in neuropsychological functioning in Japanese patients with myotonic dystrophy type 1: a five year follow-up study. J. Neuromuscul. Dis. 10, 1083–1092. doi: 10.3233/JND-230083
Gallais, B., Gagnon, C., Mathieu, J., and Richer, L. (2017). Cognitive decline over time in adults with myotonic dystrophy type 1: a 9-year longitudinal study. Neuromuscul. Disord. 27, 61–72. doi: 10.1016/j.nmd.2016.10.003
Garcia-Alix, A., Cabañas, F., Morales, C., Pellicer, A., Echevarria, J., Paisan, L., et al. (1991). Cerebral abnormalities in congenital myotonic dystrophy. Pediatr. Neurol. 7, 28–32. doi: 10.1016/0887-8994(91)90102-Q
Garmendia, J., Labayru, G., Zulaica, M., Villanúa, J., López de Munain, A., and Sistiaga, A. (2023). Shedding light on motor premanifest myotonic dystrophy type 1: a molecular, muscular and central nervous system follow-up study. Eur. J. Neurol. 30, 215–223. doi: 10.1111/ene.15604
Gaul, C., Schmidt, T., Windisch, G., Wieser, T., Müller, T., Vielhaber, S., et al. (2006). Subtle cognitive dysfunction in adult onset myotonic dystrophy type 1 (DM1) and type 2 (DM2). Neurology 67, 350–352. doi: 10.1212/01.wnl.0000225180.27833.c1
Giménez-Bejarano, A., Alegre-Cortés, E., Yakhine-Diop, S. M. S., Gómez-Suaga, P., and Fuentes, J. M. (2023). Mitochondrial dysfunction in repeat expansion diseases. Antioxidants 12:1593. doi: 10.3390/antiox12081593
Glantz, R. H., Wright, R. B., Huckman, M. S., Garron, D. C., and Siegel, I. M. (1988). Central nervous system magnetic resonance imaging findings in myotonic dystrophy. Arch. Neurol. 45, 36–37. doi: 10.1001/archneur.1988.00520250042017
Gliem, C., Minnerop, M., Roeske, S., Gärtner, H., Schoene-Bake, J. C., Adler, S., et al. (2019). Tracking the brain in myotonic dystrophies: a 5-year longitudinal follow-up study. PloS One 14:e0213381. doi: 10.1371/journal.pone.0213381
Goodwin, M., Mohan, A., Batra, R., Lee, K. Y., Charizanis, K., Fernández Gómez, F. J., et al. (2015). MBNL sequestration by toxic RNAs and RNA misprocessing in the myotonic dystrophy brain. Cell Rep. 12, 1159–1168. doi: 10.1016/j.celrep.2015.07.029
Hamel, J. I. (2022). Myotonic dystrophy. Contin Minneap Minn. 28, 1715–1734. doi: 10.1212/CON.0000000000001184
Ho, G., Cardamone, M., and Farrar, M. (2015). Congenital and childhood myotonic dystrophy: current aspects of disease and future directions. World J. Clin. Pediatr. 4, 66–80. doi: 10.5409/wjcp.v4.i4.66
Huber, S. J., Kissel, J. T., Shuttleworth, E. C., Chakeres, D. W., Clapp, L. E., and Brogan, M. A. (1989). Magnetic resonance imaging and clinical correlates of intellectual impairment in myotonic dystrophy. Arch. Neurol. 46, 536–540. doi: 10.1001/archneur.1989.00520410070026
Iwasaki, Y. (2018). “Brain pathology in myotonic dystrophy” in Myotonic dystrophy. eds. M. P. Takahashi and T. Matsumura (Singapore: Springer Singapore), 95–113.
Jiang, Q., Lin, J., Li, C., Hou, Y., and Shang, H. (2022). Gray matter abnormalities in myotonic dystrophy type 1: a voxel-wise Meta-analysis. Front. Neurol. 13:891789. doi: 10.3389/fneur.2022.891789
Johnson, N. E., Butterfield, R. J., Mayne, K., Newcomb, T., Imburgia, C., Dunn, D., et al. (2021). Population-based prevalence of myotonic dystrophy type 1 using genetic analysis of statewide blood screening program. Neurology 96, e1045–e1053. doi: 10.1212/WNL.0000000000011425
Joosten, I. B. T., Horlings, C. G. C., Vosse, B. A. H., Wagner, A., Bovenkerk, D. S. H., Evertz, R., et al. (2023). Myotonic dystrophy type 1: a comparison between the adult- and late-onset subtype. Muscle Nerve 67, 130–137. doi: 10.1002/mus.27766
Kamali, T., Parker, D., Day, J. W., Sampson, J., Deutsch, G. K., and Wozniak, J. R. (2021). Toward developing robust myotonic dystrophy brain biomarkers using white matter tract profiles sub-band energy and a framework of ensemble predictive learning. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2021, 3838–3841. doi: 10.1109/EMBC46164.2021.9630544
Kassubek, J., Juengling, F. D., Hoffmann, S., Rosenbohm, A., Kurt, A., Jurkat-Rott, K., et al. (2003). Quantification of brain atrophy in patients with myotonic dystrophy and proximal myotonic myopathy: a controlled 3-dimensional magnetic resonance imaging study. Neurosci. Lett. 348, 73–76. doi: 10.1016/S0304-3940(03)00740-7
Kobayakawa, M., Tsuruya, N., and Kawamura, M. (2012). Theory of mind impairment in adult-onset myotonic dystrophy type 1. Neurosci. Res. 72, 341–346. doi: 10.1016/j.neures.2012.01.005
Kornblum, C., Reul, J., Kress, W., Grothe, C., Amanatidis, N., Klockgether, T., et al. (2004). Cranial magnetic resonance imaging in genetically proven myotonic dystrophy type 1 and 2. J. Neurol. 251, 710–714. doi: 10.1007/s00415-004-0408-1
Koscik, T. R., Van Der Plas, E., Gutmann, L., Cumming, S. A., Monckton, D. G., Magnotta, V., et al. (2021). White matter microstructure relates to motor outcomes in myotonic dystrophy type 1 independently of disease duration and genetic burden. Sci. Rep. 11:4886. doi: 10.1038/s41598-021-84520-2
Koscik, T. R., Van Der Plas, E., Long, J. D., Cross, S., Gutmann, L., Cumming, S. A., et al. (2023). Longitudinal changes in white matter as measured with diffusion tensor imaging in adult-onset myotonic dystrophy type 1. Neuromuscul. Disord. 33, 660–669. doi: 10.1016/j.nmd.2023.05.010
Kuo, H. C., Hsiao, K. M., Chen, C. J., Hsieh, Y. C., and Huang, C. C. (2005). Brain magnetic resonance image changes in a family with congenital and classic myotonic dystrophy. Brain and Development 27, 291–296. doi: 10.1016/j.braindev.2004.09.002
Labayru, G., Aliri, J., Zulaica, M., López de Munain, A., and Sistiaga, A. (2020a). Age-related cognitive decline in myotonic dystrophy type 1: an 11-year longitudinal follow-up study. J. Neuropsychol. 14, 121–134. doi: 10.1111/jnp.12192
Labayru, G., Camino, B., Jimenez-Marin, A., Garmendia, J., Villanua, J., Zulaica, M., et al. (2022). White matter integrity changes and neurocognitive functioning in adult-late onset DM1: a follow-up DTI study. Sci. Rep. 12:3988. doi: 10.1038/s41598-022-07820-1
Labayru, G., Diez, I., Sepulcre, J., Fernández, E., Zulaica, M., Cortés, J. M., et al. (2019). Regional brain atrophy in gray and white matter is associated with cognitive impairment in myotonic dystrophy type 1. Neuroimage Clin. 24:102078. doi: 10.1016/j.nicl.2019.102078
Labayru, G., Jimenez-Marin, A., Fernández, E., Villanua, J., Zulaica, M., Cortes, J. M., et al. (2020b). Neurodegeneration trajectory in pediatric and adult/late DM1: a follow-up MRI study across a decade. Ann. Clin. Transl. Neurol. 7, 1802–1815. doi: 10.1002/acn3.51163
Laforce, R. J., Dallaire-Théroux, C., Racine, A. M., Dent, G., Salinas-Valenzuela, C., Poulin, E., et al. (2022). Tau positron emission tomography, cerebrospinal fluid and plasma biomarkers of neurodegeneration, and neurocognitive testing: an exploratory study of participants with myotonic dystrophy type 1. J. Neurol. 269, 3579–3587. doi: 10.1007/s00415-022-10970-x
Langbehn, K. E., van der Plas, E., Moser, D. J., Long, J. D., Gutmann, L., and Nopoulos, P. C. (2021). Cognitive function and its relationship with brain structure in myotonic dystrophy type 1. J. Neurosci. Res. 99, 190–199. doi: 10.1002/jnr.24595
Leddy, S., Serra, L., Esposito, D., Vizzotto, C., Giulietti, G., Silvestri, G., et al. (2021). Lesion distribution and substrate of white matter damage in myotonic dystrophy type 1: comparison with multiple sclerosis. Neuroimage Clin. 29:102562. doi: 10.1016/j.nicl.2021.102562
Liao, Q., Zhang, Y., He, J., and Huang, K. (2022). Global prevalence of myotonic dystrophy: an updated systematic review and Meta-analysis. Neuroepidemiology 56, 163–173. doi: 10.1159/000524734
Lin, X., Miller, J. W., Mankodi, A., Kanadia, R. N., Yuan, Y., Moxley, R. T., et al. (2006). Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 15, 2087–2097. doi: 10.1093/hmg/ddl132
Liquori, C. L., Ricker, K., Moseley, M. L., Jacobsen, J. F., Kress, W., Naylor, S. L., et al. (2001). Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293, 864–867. doi: 10.1126/science.1062125
Liu, J., Guo, Z. N., Yan, X. L., Yang, Y., and Huang, S. (2021). Brain pathogenesis and potential therapeutic strategies in myotonic dystrophy type 1. Front. Aging Neurosci. 13:755392. doi: 10.3389/fnagi.2021.755392
Liu, B., Yang, C. L., Li, X. L., Zhang, M., Li, Y. B., and Duan, R. S. (2022). CADASIL-like leukodystrophy and symptomatic cerebral infarction in myotonic dystrophy type 1. CNS Neurosci. Ther. 28, 1655–1657. doi: 10.1111/cns.13908
Lopez-Titla, M. M., Chirino, A., Cruz Solis, S. V., Hernandez-Castillo, C. R., Diaz, R., Márquez-Quiroz, L. D. C., et al. (2021). Cognitive decline and white matter integrity degradation in myotonic dystrophy type I. J. Neuroimaging 31, 192–198. doi: 10.1111/jon.12786
López-Martínez, A., Soblechero-Martín, P., de-la-Puente-Ovejero, L., Nogales-Gadea, G., and Arechavala-Gomeza, V. (2020). An overview of alternative splicing defects implicated in myotonic dystrophy type I. Genes 11:1109. doi: 10.3390/genes11091109
Magzhanov, R. V., Saĭfullina, E. V., Mukhametova, R. R., and Mukhamedrakhimov, R. R. (2012). Cognitive disorders in patients with myotonic dystrophy type I: a clinical and magnetic resonance study. Zh Nevrol. Psikhiatr. Im Korsakova. 112, 18–22.
Maresca, G., Portaro, S., Naro, A., Scarcella, I., Bramanti, P., Militi, D., et al. (2020). Look at the cognitive deficits in patients with myotonic dystrophy type 1: an exploratory research on the effects of virtual reality. Int. J. Rehabil. Res. 43, 90–94. doi: 10.1097/MRR.0000000000000384
Martinello, F., Piazza, A., Pastorello, E., Angelini, C., and Trevisan, C. P. (1999). Clinical and neuroimaging study of central nervous system in congenital myotonic dystrophy. J. Neurol. 246, 186–192. doi: 10.1007/s004150050332
Maurage, C. A., Udd, B., Ruchoux, M. M., Vermersch, P., Kalimo, H., Krahe, R., et al. (2005). Similar brain tau pathology in DM2/PROMM and DM1/Steinert disease. Neurology 65, 1636–1638. doi: 10.1212/01.wnl.0000184585.93864.4e
Meola, G., and Cardani, R. (2015). Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta. BBA-Mol. Basis Dis. 1852, 594–606. doi: 10.1016/j.bbadis.2014.05.019
Meola, G., and Sansone, V. (2007). Cerebral involvement in myotonic dystrophies. Muscle Nerve 36, 294–306. doi: 10.1002/mus.20800
Meola, G., Sansone, V., Perani, D., Colleluori, A., Cappa, S., Cotelli, M., et al. (1999). Reduced cerebral blood flow and impaired visual-spatial function in proximal myotonic myopathy. Neurology 53, 1042–1050. doi: 10.1212/WNL.53.5.1042
Minnerop, M., Gliem, C., and Kornblum, C. (2018). Current Progress in CNS imaging of myotonic dystrophy. Front. Neurol. 9:646. doi: 10.3389/fneur.2018.00646
Minnerop, M., Weber, B., Schoene-Bake, J. C., Roeske, S., Mirbach, S., Anspach, C., et al. (2011). The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain J. Neurol. 134, 3530–3546. doi: 10.1093/brain/awr299
Modoni, A., Silvestri, G., Vita, M. G., Quaranta, D., Tonali, P. A., and Marra, C. (2008). Cognitive impairment in myotonic dystrophy type 1 (DM1): a longitudinal follow-up study. J. Neurol. 255, 1737–1742. doi: 10.1007/s00415-008-0017-5
Naka, H., Imon, Y., Ohshita, T., Honjo, K., Kitamura, T., Mimori, Y., et al. (2002). Magnetization transfer measurements of cerebral white matter in patients with myotonic dystrophy. J. Neurol. Sci. 193, 111–116. doi: 10.1016/S0022-510X(01)00652-9
Nakamori, M., Shimizu, H., Ogawa, K., Hasuike, Y., Nakajima, T., Sakurai, H., et al. (2022). Cell type-specific abnormalities of central nervous system in myotonic dystrophy type 1. Brain Commun. 4:fcac 154. doi: 10.1093/braincomms/fcac154
Ogata, A., Terae, S., Fujita, M., and Tashiro, K. (1998). Anterior temporal white matter lesions in myotonic dystrophy with intellectual impairment: an MRI and neuropathological study. Neuroradiology 40, 411–415. doi: 10.1007/s002340050613
Okkersen, K., Monckton, D. G., Le, N., Tuladhar, A. M., Raaphorst, J., and van Engelen, B. G. M. (2017). Brain imaging in myotonic dystrophy type 1: a systematic review. Neurology 89, 960–969. doi: 10.1212/WNL.0000000000004300
Ono, S., Kanda, F., Takahashi, K., Fukuoka, Y., Jinnai, K., Kurisaki, H., et al. (1996). Neuronal loss in the medullary reticular formation in myotonic dystrophy: a clinicopathological study. Neurology 46, 228–231. doi: 10.1212/WNL.46.1.228
Palmer, B. W., Boone, K. B., Chang, L., Lee, A., and Black, S. (1994). Cognitive deficits and personality patterns in maternally versus paternally inherited myotonic dystrophy. J. Clin. Exp. Neuropsychol. 16, 784–795. doi: 10.1080/01688639408402692
Park, J. S., Song, H., Jang, K. E., Cha, H., Lee, S. H., Hwang, S. K., et al. (2018). Diffusion tensor imaging and voxel-based morphometry reveal corticospinal tract involvement in the motor dysfunction of adult-onset myotonic dystrophy type 1. Sci. Rep. 8:15592. doi: 10.1038/s41598-018-34048-9
Patel, N., Berggren, K. N., Hung, M., Bates, K., Dixon, M. M., Bax, K., et al. (2024). Neurobehavioral phenotype of children with congenital myotonic dystrophy. Neurology 102:e208115. doi: 10.1212/WNL.0000000000208115
Peric, S., Brajkovic, L., Belanovic, B., Ilic, V., Salak-Djokic, B., Basta, I., et al. (2017). Brain positron emission tomography in patients with myotonic dystrophy type 1 and type 2. J. Neurol. Sci. 378, 187–192. doi: 10.1016/j.jns.2017.05.013
Peric, S., Gunjic, I., Delic, N., Stojiljkovic Tamas, O., Salak-Djokic, B., Pesovic, J., et al. (2022). Cognitive assessment in patients with myotonic dystrophy type 2. Neuromuscul. Disord. 32, 743–748. doi: 10.1016/j.nmd.2022.06.011
Peric, S., Mandic-Stojmenovic, G., Stefanova, E., Savic-Pavicevic, D., Pesovic, J., Ilic, V., et al. (2015). Frontostriatal dysexecutive syndrome: a core cognitive feature of myotonic dystrophy type 2. J. Neurol. 262, 142–148. doi: 10.1007/s00415-014-7545-y
Ramon-Duaso, C., Gener, T., Consegal, M., Fernández-Avilés, C., Gallego, J. J., Castarlenas, L., et al. (2019). Methylphenidate attenuates the cognitive and mood alterations observed in Mbnl 2 knockout mice and reduces microglia overexpression. Cereb Cortex 29, 2978–2997. doi: 10.1093/cercor/bhy164
Ranum, L. P., Rasmussen, P. F., Benzow, K. A., Koob, M. D., and Day, J. W. (1998). Genetic mapping of a second myotonic dystrophy locus. Nat. Genet. 19, 196–198. doi: 10.1038/570
Reardon, W., Newcombe, R., Fenton, I., Sibert, J., and Harper, P. S. (1993). The natural history of congenital myotonic dystrophy: mortality and long term clinical aspects. Arch. Dis. Child. 68, 177–181. doi: 10.1136/adc.68.2.177
Renard, D., and Menjot De Champfleur, N. (2018). MRI hydrographic 3D sequences: myotonic dystrophy type 1 meets CADASIL. Acta Neurol. Belg. 118, 307–308. doi: 10.1007/s13760-017-0859-7
Ricci, F. S., Vacchetti, M., Brusa, C., D’Alessandro, R., La Rosa, P., Martone, G., et al. (2022). Cognitive, neuropsychological and emotional-behavioural functioning in a sample of children with myotonic dystrophy type 1. Eur. J. Paediatr. Neurol. 39, 59–64. doi: 10.1016/j.ejpn.2022.05.008
Rodbard, G., Kishi, N., Ducci, R., Cirino, R., Kay, C., Lorenzoni, P., et al. (2023). Non-motor symptoms and signs of myotonic dystrophy type 1. J. Neurol. Sci. 455:122046. doi: 10.1016/j.jns.2023.122046
Romeo, V., Pegoraro, E., Ferrati, C., Squarzanti, F., Sorarù, G., Palmieri, A., et al. (2010). Brain involvement in myotonic dystrophies: neuroimaging and neuropsychological comparative study in DM1 and DM2. J. Neurol. 257, 1246–1255. doi: 10.1007/s00415-010-5498-3
Rosado Bartolomé, A., Puertas Martín, V., Domínguez González, C., and Ramos, M. M. (2022). Cognitive impairment in myotonic dystrophy type 1 (Steinert’s disease). SEMERGEN 48, 208–213. doi: 10.1016/j.semerg.2022.01.013
Rossi, S., and Silvestri, G. (2023). Fluid biomarkers of central nervous system (CNS) involvement in myotonic dystrophy type 1 (DM1). Int. J. Mol. Sci. 24:2204. doi: 10.3390/ijms24032204
Sansone, V., Gandossini, S., Cotelli, M., Calabria, M., Zanetti, O., and Meola, G. (2007). Cognitive impairment in adult myotonic dystrophies: a longitudinal study. Neurol. Sci. 28, 9–15. doi: 10.1007/s10072-007-0742-z
Santoro, M., Fontana, L., Maiorca, F., Centofanti, F., Massa, R., Silvestri, G., et al. (2018). Expanded [CCTG] n repetitions are not associated with abnormal methylation at the CNBP locus in myotonic dystrophy type 2 (DM2) patients. Biochim. Biophys. Acta Mol. basis Dis. 1864, 917–924. doi: 10.1016/j.bbadis.2017.12.037
Serra, L., Scocchia, M., Meola, G., D’Amelio, M., Bruschini, M., Silvestri, G., et al. (2020). Ventral tegmental area dysfunction affects decision-making in patients with myotonic dystrophy type-1. Cortex 128, 192–202. doi: 10.1016/j.cortex.2020.03.022
Serra, L., Silvestri, G., Petrucci, A., Basile, B., Masciullo, M., Makovac, E., et al. (2014). Abnormal functional brain connectivity and personality traits in myotonic dystrophy type 1. JAMA Neurol. 71, 603–611. doi: 10.1001/jamaneurol.2014.130
Simoncini, C., Spadoni, G., Lai, E., Santoni, L., Angelini, C., Ricci, G., et al. (2020). Central nervous system involvement as outcome measure for clinical trials efficacy in myotonic dystrophy type 1. Front. Neurol. 11:624. doi: 10.3389/fneur.2020.00624
Sinforiani, E., Sandrini, G., Martelli, A., Mauri, M., Uggetti, C., Bono, G., et al. (1991). Cognitive and neuroradiological findings in myotonic dystrophy. Funct. Neurol. 6, 377–384.
Sistiaga, A., Urreta, I., Jodar, M., Cobo, A. M., Emparanza, J., Otaegui, D., et al. (2010). Cognitive/personality pattern and triplet expansion size in adult myotonic dystrophy type 1 (DM1): CTG repeats, cognition and personality in DM1. Psychol. Med. 40, 487–495. doi: 10.1017/S0033291709990602
Soltanzadeh, P. (2022). Myotonic dystrophies: a genetic overview. Genes 13:367. doi: 10.3390/genes13020367
Spranger, M. (1997). Myotonic dystrophy: the role of large triplet repeat length in the development of mental retardation. Arch. Neurol. 54:251. doi: 10.1001/archneur.1997.00550150017009
Stokes, M., Varughese, N., Iannaccone, S., and Castro, D. (2019). Clinical and genetic characteristics of childhood-onset myotonic dystrophy. Muscle Nerve 60, 732–738. doi: 10.1002/mus.26716
Sweere, D. J. J., Moelands, S. V. L., Klinkenberg, S., Leenen, L., Hendriksen, J. G. M., and Braakman, H. M. H. (2023). Cognitive phenotype of childhood myotonic dystrophy type 1: a multicenter pooled analysis. Muscle Nerve 68, 57–64. doi: 10.1002/mus.27836
Theodosiou, T., Christidi, F., Xirou, S., Karavasilis, E., Bede, P., Papadopoulos, C., et al. (2022). Executive dysfunction, social cognition impairment, and gray matter pathology in myotonic dystrophy type 2: a pilot study. Neuropsychiatry Neuropsychol. Behav. Neurol. 35, 204–211. doi: 10.1097/WNN.0000000000000314
Tremblay, M., Muslemani, S., Côté, I., Gagnon, C., Fortin, J., and Gallais, B. (2021). Accomplishment of instrumental activities of daily living and its relationship with cognitive functions in adults with myotonic dystrophy type 1 childhood phenotype: an exploratory study. BMC Psychol. 9:56. doi: 10.1186/s40359-021-00562-1
van Dorst, M., Okkersen, K., Kessels, R. P. C., Meijer, F. J. A., Monckton, D. G., van Engelen, B. G. M., et al. (2019). Structural white matter networks in myotonic dystrophy type 1. NeuroImage Clin. 21:101615. doi: 10.1016/j.nicl.2018.101615
Vermersch, P., Sergeant, N., Ruchoux, M. M., Hofmann-Radvanyi, H., Wattez, A., Petit, H., et al. (1996). Specific tau variants in the brains of patients with myotonic dystrophy. Neurology 47, 711–717. doi: 10.1212/WNL.47.3.711
Vielhaber, S., Jakubiczka, S., Gaul, C., Schoenfeld, M. A., Debska-Vielhaber, G., Zierz, S., et al. (2006). Brain 1H magnetic resonance spectroscopic differences in myotonic dystrophy type 2 and type 1. Muscle Nerve 34, 145–152. doi: 10.1002/mus.20565
Vydra, D. G., and Rayi, A. Myotonic dystrophy. In: Stat pearls. Treasure Island, FL: Stat Pearls Publishing; (2023)
Wang, P. Y., Chang, K. T., Lin, Y. M., Kuo, T. Y., and Wang, G. S. (2018). Ubiquitination of MBNL1 is required for its cytoplasmic localization and function in promoting neurite outgrowth. Cell Rep. 22, 2294–2306. doi: 10.1016/j.celrep.2018.02.025
Wang, L. H., Lin, C. Y., Lin, Y. M., Buée, L., Sergeant, N., Blum, D., et al. (2022). Calpain-2 mediates MBNL2 degradation and a developmental RNA processing program in neurodegeneration. J. Neurosci. 42, 5102–5114. doi: 10.1523/JNEUROSCI.2006-21.2022
Weber, Y. G., Roebling, R., Kassubek, J., Hoffmann, S., Rosenbohm, A., Wolf, M., et al. (2010). Comparative analysis of brain structure, metabolism, and cognition in myotonic dystrophy 1 and 2. Neurology 74, 1108–1117. doi: 10.1212/WNL.0b013e3181d8c35f
Weijs, R., Okkersen, K., van Engelen, B., Küsters, B., Lammens, M., Aronica, E., et al. (2021). Human brain pathology in myotonic dystrophy type 1: a systematic review. Neuropathol. Off. J. Jpn Soc. Neuropathol. 41, 3–20. doi: 10.1111/neup.12721
Wenninger, S., Montagnese, F., and Schoser, B. (2018). Core clinical phenotypes in myotonic dystrophies. Front. Neurol. 9:303. doi: 10.3389/fneur.2018.00303
Wheeler, T. M., and Thornton, C. A. (2007). Myotonic dystrophy: RNA-mediated muscle disease. Curr. Opin. Neurol. 20, 572–576. doi: 10.1097/WCO.0b013e3282ef6064
Winblad, S., Lindberg, C., and Hansen, S. (2006). Cognitive deficits and CTG repeat expansion size in classical myotonic dystrophy type 1 (DM1). Behav. Brain Funct. BBF. 2:16. doi: 10.1186/1744-9081-2-16
Winblad, S., Samuelsson, L., Lindberg, C., and Meola, G. (2016). Cognition in myotonic dystrophy type 1: a 5-year follow-up study. Eur. J. Neurol. 23, 1471–1476. doi: 10.1111/ene.13062
Woo, J., Lee, H. W., and Park, J. S. (2019). Differences in the pattern of cognitive impairments between juvenile and adult onset myotonic dystrophy type 1. J. Clin. Neurosci. 68, 92–96. doi: 10.1016/j.jocn.2019.07.029
Wozniak, J. R., Mueller, B. A., Bell, C. J., Muetzel, R. L., Lim, K. O., and Day, J. W. (2013). Diffusion tensor imaging reveals widespread white matter abnormalities in children and adolescents with myotonic dystrophy type 1. J. Neurol. 260, 1122–1131. doi: 10.1007/s00415-012-6771-4
Yoshizumi, K., Nishi, M., Igeta, M., Nakamori, M., Inoue, K., Matsumura, T., et al. (2023). Analysis of splicing abnormalities in the white matter of myotonic dystrophy type 1 brain using RNA sequencing. Neurosci. Res. 200, 48–56. doi: 10.1016/j.neures.2023.10.002
Yum, K., Wang, E. T., and Kalsotra, A. (2017). Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr. Opin. Genet. Dev. 44, 30–37. doi: 10.1016/j.gde.2017.01.007
Zalonis, I., Bonakis, A., Christidi, F., Vagiakis, E., Papageorgiou, S. G., Kalfakis, N., et al. (2010). Toward understanding cognitive impairment in patients with myotonic dystrophy type 1. Arch. Clin. Neuropsychol. 25, 303–313. doi: 10.1093/arclin/acq016
Zu, T., Cleary, J. D., Liu, Y., Bañez-Coronel, M., Bubenik, J. L., Ayhan, F., et al. (2017). RAN translation regulated by Muscleblind proteins in myotonic dystrophy type 2. Neuron 95, 1292–1305.e5. doi: 10.1016/j.neuron.2017.08.039
Keywords: myotonic dystrophy, cognitive impairment, neuroimaging abnormalities, mechanism, comprehensive review
Citation: Wu Y, Wei Q, Lin J, Shang H and Ou R (2024) Cognitive impairment, neuroimaging abnormalities, and their correlations in myotonic dystrophy: a comprehensive review. Front. Cell. Neurosci. 18:1369332. doi: 10.3389/fncel.2024.1369332
Edited by:
Mario Gomes-Pereira, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Giovanni Meola, University of Milan, ItalyCopyright © 2024 Wu, Wei, Lin, Shang and Ou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruwei Ou, b3VydXdlaUBhbGl5dW4uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.