 Emanuela Paldino
Emanuela Paldino Giorgia Migliorato
Giorgia Migliorato Francesca R. Fusco
Francesca R. Fusco- 1Laboratory of Neuroanatomy, Fondazione Santa Lucia IRCCS, Rome, Italy
- 2Department of Life Sciences, University of Trieste, Trieste, Italy
Mechanisms of tissue damage in Huntington’s disease (HD) involve excitotoxicity, mitochondrial damage, and neuroinflammation, including microglia activation. CD47 is a membrane protein that interacts with the inhibitory immunoreceptor SIRPα. Engagement of SIRPα by CD47 provides a downregulatory signal that inhibits host cell phagocytosis, promoting a “don’t-eat-me” signal. These proteins are involved in the immune response and are downmodulated in inflammatory diseases. The involvement of inflammation and of the inflammasome in HD has already been described. In this study, we focused on other factors that can be involved in the unregulated inflammatory response that accelerates and exacerbate the neurodegenerative process in HD. Our results show that CD47 on striatal neurons decreased in HD mice, while it increased in wild type mice with age. SIRPα, on the other hand, was present in neurons in the wild type and increases in the R6/2 mice at all stages. Recruitment of SIRPα and binding to CD47 promotes the activation through phosphorylating events of non-receptor protein tyrosine phosphatase SHP-1 and SHP-2 in neurons and microglia. SHP phosphatases are able to curb the activity of NLRP3 inflammasome thereby reducing the detrimental effect of neuroinflammation. Such activity is mediated by the inhibition (dephosphorylation) of the proteins signal transducer and activator of transcription (STAT). We found that activated SHP-1 was present in microglia and neurons of WT mice at 5 and 13 weeks, increasing with time; while in R6/2 it was not localized in neurons but only in microglia, where it decreases with time. Consequently, STAT1 was overexpressed in neurons of R6/2 mice, as an effect of lack of modulation by SHP-1. Thus, our results shed light on the pathophysiology of neuronal damage, on one hand, paving the way toward a modulation of signal transducer proteins by specific inhibitors to achieve neuroprotection in HD, on the other.
1 Introduction
Huntington’s disease (HD) is a devastating neurodegenerative disorder that affects millions of people worldwide. This inherited genetic disorder is characterized by the progressive degeneration of nerve cells in the brain, leading to a wide range of motor, cognitive, and psychiatric symptoms. While the genetic underpinnings of HD have been extensively studied, recent research has shed light on the role of neuroinflammation, on one hand, and of phagocytosis, on the other. Phagocytosis is a fundamental cellular process in the pathogenesis of this disease. Indeed, phagocytosis, the process by which cells engulf and digest foreign particles or cellular debris, plays a crucial role in maintaining brain homeostasis. In the context of Huntington’s disease, aberrations in phagocytic activity within the central nervous system are emerging as a critical factor in disease progression. Neuroinflammation, on the other hand, includes several pathways and cytokines, and it is plausible that several of them play an important role in HD pathology (Han et al., 2012). Recently, focus has turned to the NLRP3 inflammasome, that we studied in the R6/2 mouse model of HD, thereby contributing to the understanding of the involvement of inflammatory processes in neurodegenerative disorders (Paldino et al., 2020; Paldino and Fusco, 2022).
Both phagocytosis and neuroinflammation involve the activity of the microglia. Microglia are immune cells that reside in the central nervous system (CNS) where they exert an increased phagocytosis, particularly in periods of neuronal growth and remodeling (Silverman and Wong, 2018; Wilton et al., 2019). The regulation of microglial phagocytosis is important to understand neurodegenerative disorders, where dysregulated microglia phagocytosis is involved in the process of tissue damage and neuronal death (Perry et al., 2010; Sellgren et al., 2019). Microglial cell surface receptors have been identified to regulate phagocytosis over development (Fu et al., 2014) as well as neuron-derived signal receptors. One of cell surface receptor expressed by phagocytic cells, which acts as “don’t eat me” signal to reduce phagocytosis is the signal-regulatory protein alpha (SIRPα) (Elward and Gasque, 2003; Jiang et al., 2020).
In the present study, we directed our interest in exploring several factors involved in phagocytosis such as the expression of SIRPα and its receptor CD47, in the mouse model of HD, to better understand the mechanisms that are at the foundation of neuronal death. The SIRPα-CD47 system activates the non-receptor protein tyrosine phosphatases, namely Src homology region 2 domain-containing phosphatase-1 (SHP-1) and Src homology 2 (SH2) domain-containing tyrosine phosphatase-2 (SHP-2), at the level of cell membrane, in response to extracellular stimuli, exerting its action of inhibiting phagocytosis (Khanna et al., 2007). SHP-1 belongs to the protein tyrosine phosphatase (PTP) family and plays a crucial role in the regulation of signaling pathways involved in immune response and cell growth. Dysregulation of SHP-1 has been implicated in various diseases, including cancers and autoimmune disorders (Chong and Maiese, 2007; Murata et al., 2018). SHP-2 is a non-receptor protein tyrosine phosphatase (PTP) ubiquitously expressed. It has important functions in cell proliferation and differentiation (Agazie et al., 2003) particularly as a response to factors such as epidermal growth factor and platelet-derived growth factor-induced Ras-Raf-Erk cascade (De Rocca Serra-Nédélec et al., 2012). Compelling evidence shows that SHP-2 is also implicated in immune signaling and inflammatory response; indeed, SHP-2 activation prevents overactivation of NLRP3 inflammasome, and therefore, in normal conditions, modulates inflammation and controls tissue damage (Guo et al., 2017).
With respect to the activity of SHP-1 and SHP-2, in this study we have also considered the signal-transducers and activators of transcription (STAT). STATs are important signaling molecules that can activate neuroinflammation. Activation and inactivation of STATs are regulated by phosphorylation and dephosphorylation of tyrosine residues of STATs (Shuai et al., 1993). SHP-1 negatively regulates STAT signaling pathways induced by interferon. It has strong anti-inflammatory roles, as evidenced by the fact that IFN-γ- and LPS-induced iNOS (inducible nitric oxide synthase) expression is higher in SHP-1-deficient macrophages than in wild-type (WT) cells (Hardin and Yu, 2006; Christophi et al., 2009). Thus, we investigated the expression of SHP-1 and SHP-2 and of STAT1/3 to better understand their importance in relation with neuroinflammation in the pathology of HD, in the effort to find a possible therapeutic strategy to fight HD.
2 Materials and methods
2.1 Animals and tissue processing
All animal experiments, which satisfied ARRIVE guidelines, were performed in accordance with European Communities Council Directive (2010/63 EU) as adopted by the Santa Lucia Foundation Animal Care and Use and approved by Italian Ministry of Health. Transgenic R6/2 mice were obtained by ovarian transplant of hemi zygote females × B6CBAF1/J males, all obtained from Jackson Laboratories (Bar Harbor, ME). F1 mice were used to perform all experiments. Eight mice per group were used. No randomization was performed. The study groups included: R6/2 mice at 5 weeks of age, R6/2 mice at 13 weeks of age, Wild-type mice at 5 and 13 weeks. Mice were handled under the same conditions by one investigator at the same day and time. Genotyping was performed at 21 days of age, and all mice were weaned and housed four in each cage under standard conditions (room temperature: 20 ± 2 °C; humidity: 60%) and a 12/12-h light/dark cycle (7:00 am–7:00 pm) with ad libitum access to food and water. Animals (8 R6/2 5 weeks, 8 R6/2 13 weeks, 8 wild type 5 weeks and 8 wild type 13 weeks) were transcardially perfused under deep anesthesia with saline solution containing 0.01 ml heparin, brains were removed and cut in half. One half brain was post-fixed in 14 ml of 4% paraformaldehyde, 10% sucrose and 20% glycerol in 0.1 M phosphate buffer (PB) at + 4 °C for 48 h. Sectioning was performed on a sliding frozen cryostat at 30 μm thickness. Observers who were in blind, collected primary data.
2.2 Immunohistochemical studies of mice brain tissue
2.2.1 Evaluation of CD47 neuronal expression
Coronal brain sections were incubated with the marker CD47 (BS-21460R, rabbit polyclonal anti-CD47, Bioss, USA) at 1:200 dilution in a 0.1 M PB solution containing 0.3% Triton X-100 for 72 h at 4°C. Sections were rinsed three times for 5 min at room temperature and subsequently incubated with the secondary biotinylated antibody, 1:200 (biotinylated goat anti-rabbit, Jackson Immunoresearch, West Grove, PA, USA). The immunohistochemical staining was completed with the Streptavidin-Cy3 (Sigma-Aldrich, Merck). All sections were rinsed three times for 5 min at room temperature and subsequently counterstained with Neurotrace™ (Nissl-like fluorescent marker, Jackson, MS, USA) to visualize the number and the immunofluorescence intensity of neurons expressing CD47. Sections were mounted on specimen slides; cover slipped with Anti Fade™ Mounting Medium (Immunological Sciences, Italy) and a confocal laser-scanning microscope (Zeiss LSM 800) was used to acquire all images. The single plane confocal images were acquired under no saturation conditions, with a × 20 objective gaining a × 1 zoom with value 0 of Offset and producing images in the format 1024 × 1024, Airy Units 1.0. A sample of about 100 neurons for each of four sections obtained by the 8 mice per group were analyzed, in order to determine the distribution and the immunoreaction product of CD47 in the striatum of R6/2 and wild-type mice at the different time points 5 and 13 weeks.
2.2.2 Immunohistochemistry for SIRPα
To evaluate the involvement of SIRPα/CD47 signaling pathway, we performed an immunohistochemistry for SIRPα. Peroxidase-antiperoxidase diaminobenzidine tetrahydrochloride single-label immunohistochemistry for Sirpα was performed to identification and distribution of this phagocytosis marker in the mice striatum. Serial sections from rostral neostriatum through the level of anterior commissure (interaural 4.66 mm/Bregma 0.86 mm to interaural 3.34 mm/Bregma-0.46 mm) for three animals per groups, were incubated with mouse anti-SIRPα at 1:200 dilution in 0.1 M PB solution containing 0.3% Triton X-100 for 72 h at 4 °C (NB100-65530 mouse anti-SIRPα, Novus Biologicals, Italy). Subsequently, sections were incubated with mouse peroxidase–antiperoxidase complex diluted 1:100 in 0.1 M PB solution with 0.3% Triton X-100 at room temperature for 1 h. After peroxidase–antiperoxidase incubation, sections were incubated in Tris–HCl buffer containing 10 mg diaminobenzidine tetrahydrochloride for 2 min, adding 15 μl of 3% hydrogen peroxidase. The peroxidase–antiperoxidase diaminobenzidine tetrahydrochloride-labeled sections were then washed in distillated water, placed in 0.1 M PB, mounted on gelatin-coated slides, dried, dehydrated and coverslipped. SIRPα positive cells visualization was performed on collected images with a × 40 and × 100 objective, obtained by Neurolucida™ Stereo Investigator software (Zeiss, Rochester, NY, USA). To analyze the expression of SIRPα in microglia and astrocyte cells, double immunofluorescence were performed with the primary antibodies at 1:500 dilution against IBA-1 (NB100-1028, goat anti-IBA1, Abcam, Novus Biologicals, Italy) and GFAP (mouse anti-GFAP, Immunological Sciences, Italy) in a 0.1 M PB solution containing 0.3% Triton X-100 for 72 h at 4 °C. Microglia and astrocytes in the area of interest were captured performing Z-stacks acquisitions using a × 20 objective increasing the × 1.5 zoom and images in the format 1024 × 1024 were produced.
2.2.3 Analysis of the Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2 in R6/2 mice brain
Double-label immunofluorescences were carried out to evaluate the distribution of activated SHP-1 and SHP2 in the mice striatum. Coronal brain sections of mice were incubated with specific antibodies for the phosphorylated proteins: SHP1 (ABP11214 rabbit anti phospho SHP1, Immunological Sciences, Italy) or SHP2 (ABP11215 rabbit anti phospho SHP2, Immunological Sciences, Italy) at 1:200 dilution and the primary antibodies against IBA-1 and GFAP protein at 1:500 dilution. Mice brain sections were, also, incubated with the primary antibody pSTAT1 (ABP-0453 rabbit anti pSTAT-1, Immunological Sciences, Italy). After that, sections were rinsed three times for 5 min at room temperature and subsequently incubated with the Alexa Fluor 555 and 488 secondary antibodies (Immunological Sciences, Italy) for 2 h at room temperature at 1:200 dilution in a 0.1 M PB solution containing 0.3% Triton X-100. Sections were then mounted on slides cover slipped with Anti Fade™ Mounting Medium (Immunological Sciences, Italy). A confocal laser scanning microscope (Zeiss LSM 800) was used to acquire all the images. Three separate fields (dorsolateral, central and medial each 1 mm in diameter) in each of three rostro caudally spaced sections of eight mice per group were examined. pSHP1 e pSHP2 confocal images were acquired under no saturation conditions performing Z-stacks acquisitions using a × 20 objective increasing the × 1.0 zoom and images in the format 1024 × 1024 were produced. pSTAT1 single plane images were acquired with a × 20 objective gaining a × 0,8 zoom with value 0 of Offset and producing images in the format 1024 × 1024 and Airy Units 1.0. The same set configuration was performed for all samples.
2.3 Western blotting
Dissected striata from the Wt and R6/2 half brains were homogenized with the RIPA lysis buffer containing a protease and phosphatase inhibitor cocktail (Sigma Aldrich, USA) and centrifuged at 13,000 × g for 20 min, to obtain a total lysate containing neurons and microglial proteins. Equal amounts of protein were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene fluoride membranes, and incubated with CD47 (1:500), SIRPα (1:500), pSHP-1 (1:1000), total STAT-1 (AB-81448 rabbit anti STAT-1, Immunological Sciences, Italy) (1:1000), pSTAT-1 (1:1000), pSTAT3 (ABP-5041 rabbit anti pSTAT3, Immunological Sciences, Italy) (1:1000) and mouse GAPDH-HRP (1:10.000, Immunological Sciences, Italy) antibodies, overnight at 4 °C. After being washed with Tris-buffered saline (TBS)/Tween 20, membranes were incubated with HRP-labeled secondary antibodies. Proteins signal was visualized using the Invitrogen iBright CL 1500 Imaging system.
2.4 Statistical analysis
All the collected images have been quantified by using the Java image processing and analysis program Fiji ImageJ. Cells of interest were selected using the freehand tool. From the Analyze menu, Set measurements Mean “Gray Value,” “Area,” and “Min and Max Gray Value” were selected. The region characterized by absence of fluorescence was considered in the background and it was subtracted. Finally, the mean values with SEM were obtained for all measures. ANOVA analysis available in the software GraphPad Prism version 8.0 was performed. P-values < 0.05 were considered statistically significant.
3 Results
In order to evaluate the possible dysfunction of the phagocytosis mechanism, the expression of the main players of this pathway was evaluated in two age stages of R6/2 mice: pre-symptomatic (corresponding to 5 weeks) and symptomatic represented by 13 weeks of age.
3.1 Modulation of CD47 protein expression levels in R6/2 mice striatum
CD47 is a transmembrane protein widely expressed on most mammalian cells, including neurons. The function of this protein in regulating phagocytosis has been established, more specifically by inhibiting it through the recruitment and bonding to SIRPα, expressed on phagocytes (Brown and Frazier, 2001; Gardai et al., 2005). We investigated the expression of CD47 in the mice striatum of pre and fully symptomatic R6/2 compared to their Wt littermates. CD47 was preferentially expressed in the striatal neurons as evidenced by immunofluorescence staining performed on brain tissue of R6/2 and Wt mice at 5 and 13 weeks of age (Figures 1A–D). In the 13 weeks old Wt mice the CD47 puncta expression and cytoplasmic membrane distribution appeared more intense compared to 13 weeks old R6/2 mice in which CD47 expression was significantly decreased and its striatal neuron distribution appeared not sharp (Figures 1E, F). The immunofluorescence intensity of CD47-positive striatal neurons was higher in 13 weeks old Wt mice p < 0.0001, while a statistically significant reduction of CD47-positive neurons was observed in R6/2 mice at 13 weeks (Figure 1G). Data was confirmed with molecular experiments (Figures 1H, I).
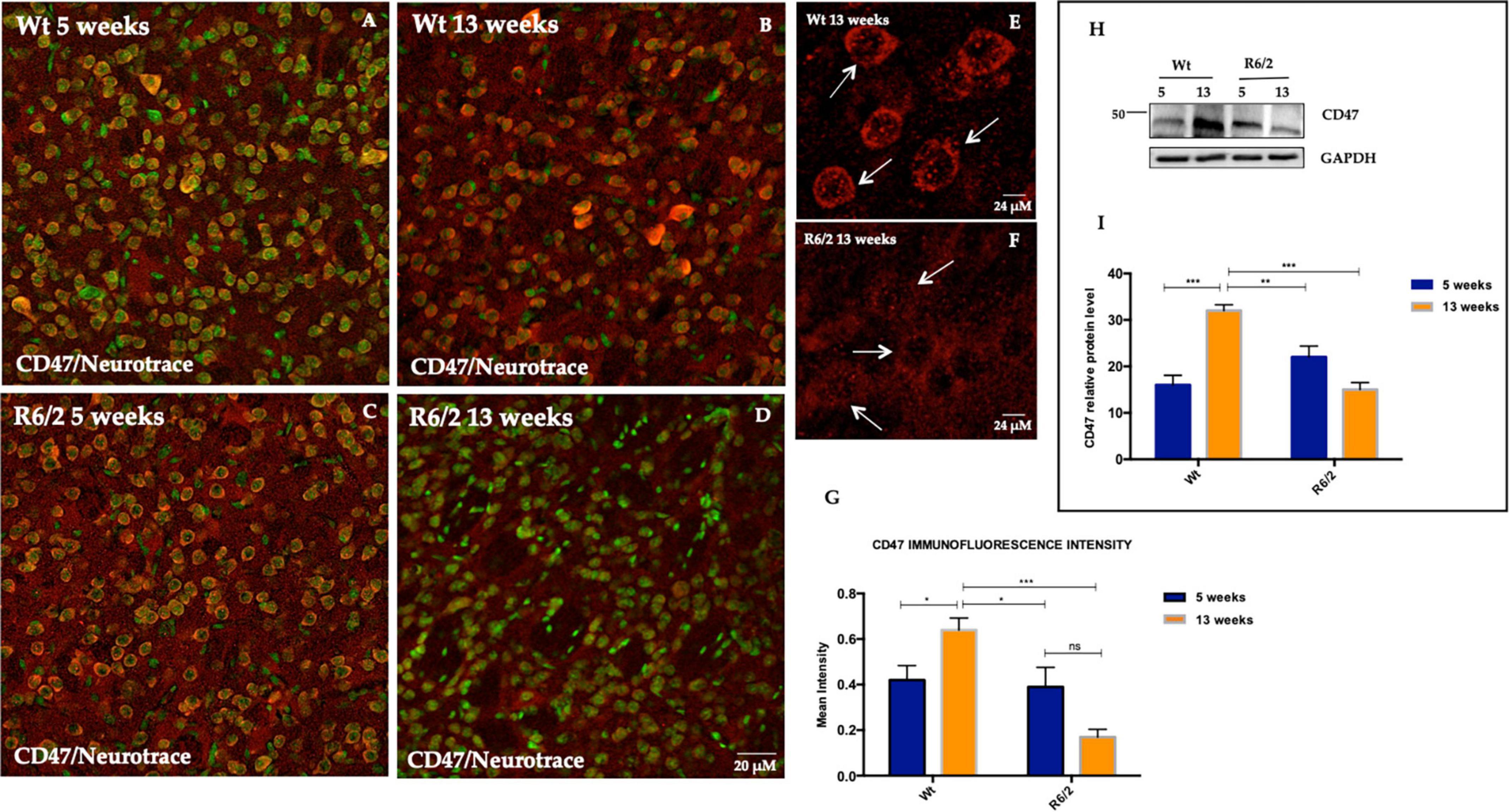
Figure 1. CD47 expression in striatal neurons of R6/2 mice. Confocal laser scanning microscopy images of double-label immunofluorescence for CD47 (visualized in red fluorescence) and the Nissl-like fluorescent marker Neurotrace (visualized in green fluorescence). (A–D) Images show the expression pattern of CD47 in the striatum of 5 and 13 weeks-old wild-type mice and in 5 and 13 weeks-old R6/2 mice. (E,F) Detail of cytoplasmic membrane CD47 expression in 13 weeks-old Wt mice. In 13 weeks-old R6/2 mice CD47 is lower due to cell death process (G) Histogram shows CD47 intensity quantification which is significantly decreased in the striatal neurons of 13 weeks-old R6/2 mice [Genotype effect F(1,153) = 11.97, ***P = 0.0007]. (H,I) Tukey’s multiple comparisons analysis revealed a significant protein decrease of striatal neuronal CD47, as evaluated by western blotting in R6/2 mice [Genotype effect F(1,20) = 8.667, **P = 0.0080; Time effect F(1,20) = 5.802, **P = 0.0258; Genotype X Time Interaction F(1,20) = 37.89, ***P < 0,0001].
3.2 Neuronal signal-regulatory protein alpha (SIRPα) upregulation during neurodegeneration of the R6/2 mice striatum
We performed immunohistochemistry experiments to evaluate SIRPα protein expression in the R6/2 mice striatum and to investigate its expression pattern. Surprisingly, DAB (diaminobenzidine tetrahydrochloride) immunochemistry highlighted a basal neuronal levels of SIRPα protein in Wt animals (Figures 2A, B) and a significant and persistent SIRPα expression in the striatal neurons of R6/2 mice at 13 weeks of age (Figures 2C, D). The characteristic membrane cytoplasmic distribution is referred to presynaptic SIRPα localization which promotes binding with postsynaptic CD47 (Jiang et al., 2022; Figure 2E), however, we cannot rule out the possibility of a postsynaptic localization SIRPα (Ding et al., 2021) (Supplementary Figure 1). DAB immunohistochemistry data were confirmed by molecular experiments performed on striata half-brain lysates derived from the same animals groups on which immunohistochemistry results were obtained (Figure 2F). Since SIRPα was highly expressed in 5 weeks old R6/2 mice striatal neurons and its persisting expression was observed in the surviving neurons of 13 weeks old R6/2 (Figure 2G), we evaluated the possibility that SIRPα protein may be sequestered by the neuronal nuclear inclusions (NIIs), a hallmark of HD. As the antibody against ubiquitin labels NIIs in R6/2 mice, we examined whether SIRPα was ubiquitin immunoreactive in each group. Ubiquitin immunolabeling of striata from wild-type animals did not show any immunoreactive inclusions (Figures 2H, I) but rather a very light diffuse immunolabeling of cell bodies and neuropil. By contrast, and as expected from previous studies in other cell lines and tissues (Giampà et al., 2006), striatal brain sections isolated from 5/13-week-old R6/2 transgenic mice showed the NIIs marked by ubiquitin which did not co localize with SIRPα protein (Figures 2J, K). This ruled out the possibility that SIRPα could be sequestered in NIIs.
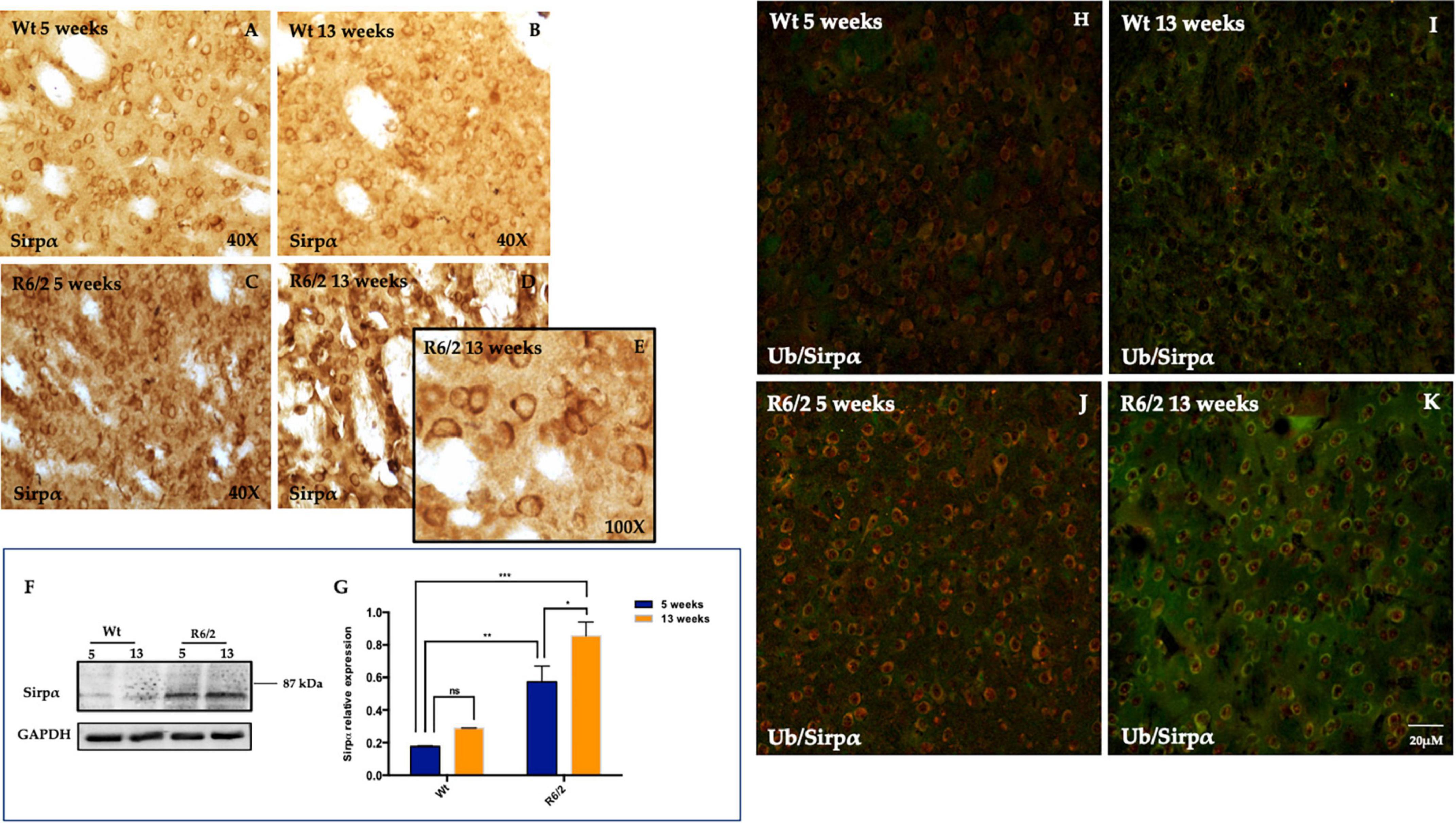
Figure 2. Signal-regulatory protein alpha (SIRPα) distribution in striatal neurons. (A–D) Representative transmitted light microscope images showing Dab staining for SIRPα counterstained with Hematoxylin in the striatum of 5 and 13 weeks-old experimental animal groups. (E) Higher magnification detail of presynaptic SIRPα expression in 13 weeks-old R6/2 mice. (F,G) Representative image of Western Blot and densitometry analysis of SIRPα. Tukey’s multiple comparisons analysis performed on probed membranes revealed a significant increase of the striatal SIRPα protein expression in the 5 and 13 weeks-old R6/2 mice [Genotype effect F(1,16) = 56.23, ***P < 0.0001; Time effect F(1,16) = 9.423, **P = 0.0073] compared to 5 and 13 week-old Wt. (H–K) Representative confocal microscopy images showed the ubiquitin (visualized in red fluorescence), which did not colocalize with SIRPα protein (visualized in green fluorescence).
Signal-regulatory protein alpha was not detectable in microglia (Figures 3A–D) and GFAP positive cells (astrocytes) were completely devoid of it (Figures 3E–H).
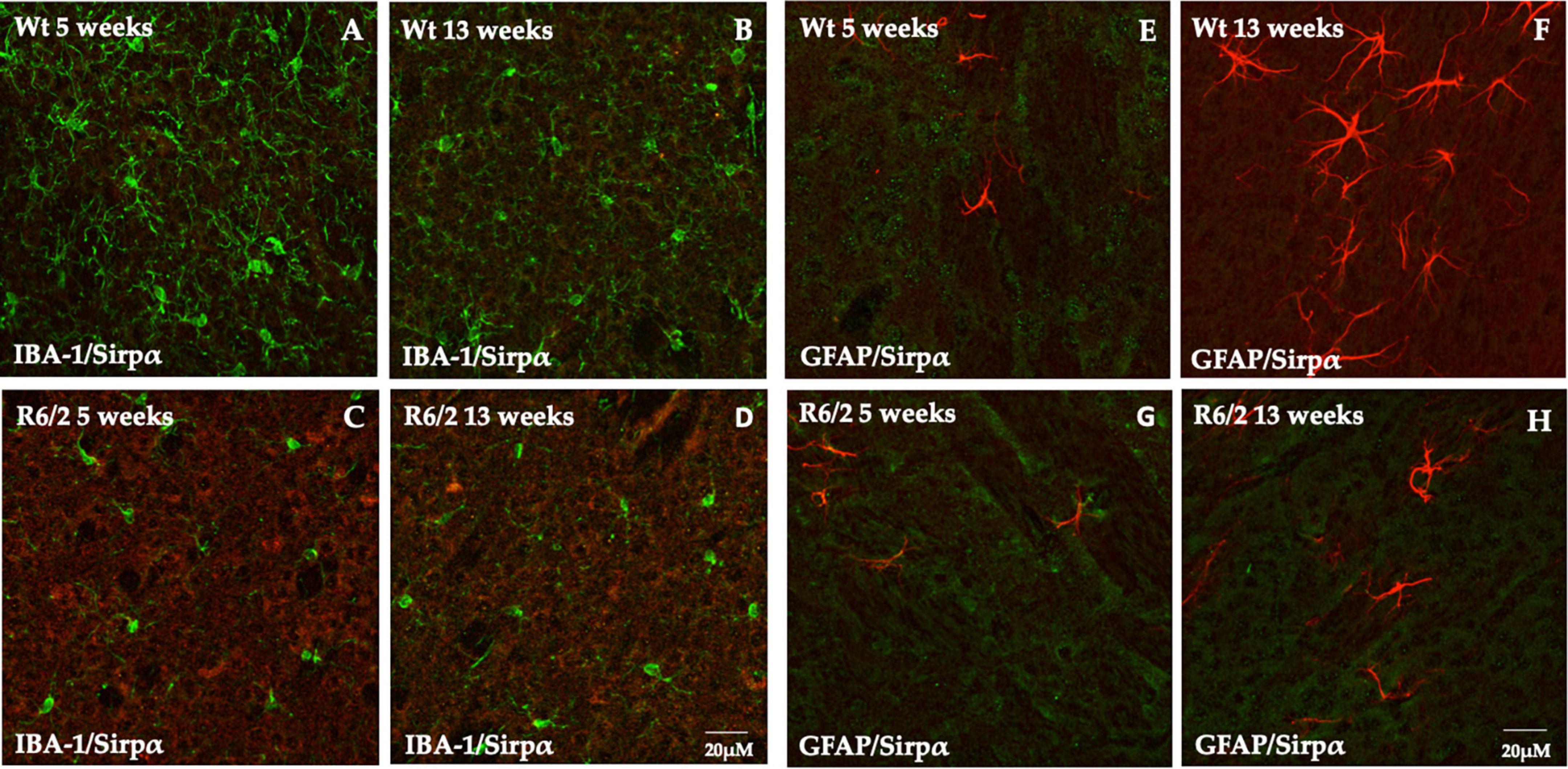
Figure 3. Study of SIRPα expression in microglia and astrocytes cells. (A–D) Z-stack confocal laser scanning microscopy images of double-label immunofluorescence for SIRPα (visualized in red fluorescence) and IBA-1 (visualized in green fluorescence). Representative images of mice striatum from all experimental groups show the presence of phenotypically activated microglia in 5 and 13 weeks old R6/2 mice. In all animal groups the colocalization of SIRPα and IBA-1 was undetectable. (E–H) Z-stack confocal laser scanning microscopy images of double-label immunofluorescence for SIRPα (in green) and GFAP (in red) of dorsal mice striatum where a higher number of astrocyte is generally observed. In all experimental animals, there was no evidence of colocalization of SIRPα with GFAP positive cells.
3.3 The PTPs activation pattern
In the PTP family, a subgroup of cytoplasmic PTPs characterized by containing two Src homology 2 (SH2) NH2-terminal domains and a C-terminal protein-tyrosine phosphatase domain is referred to as SHP.
We investigated the expression pattern of phosphorylated SHP-1 and SHP-2 in the mouse brain of R6/2 mice. R6/2 mice showed a lower expression of neuronal phosphorylated SHP-1 protein from pre-symptomatic to late stages compared to WT control group (Figures 4A–D), in which pSHP-1 protein expression increased during the time (Figure 4I). The presence of pSHP-1 was, also, observed in cells whose morphology can be traced back to the microglial one (Supplementary Figure 2). The immunoreaction product of pSHP-2 was not detected in the striatal neurons neither in R6/2 nor in the wild type mice. However, its expression was specifically localized on microglial cells as represented in Figures 4E–H. The immunofluorescence of phosphorylated SHP-2 intensity analysis performed on microglia cells was statistically significant lower in the R6/2 mice at 13 weeks of age (Figure 4J).
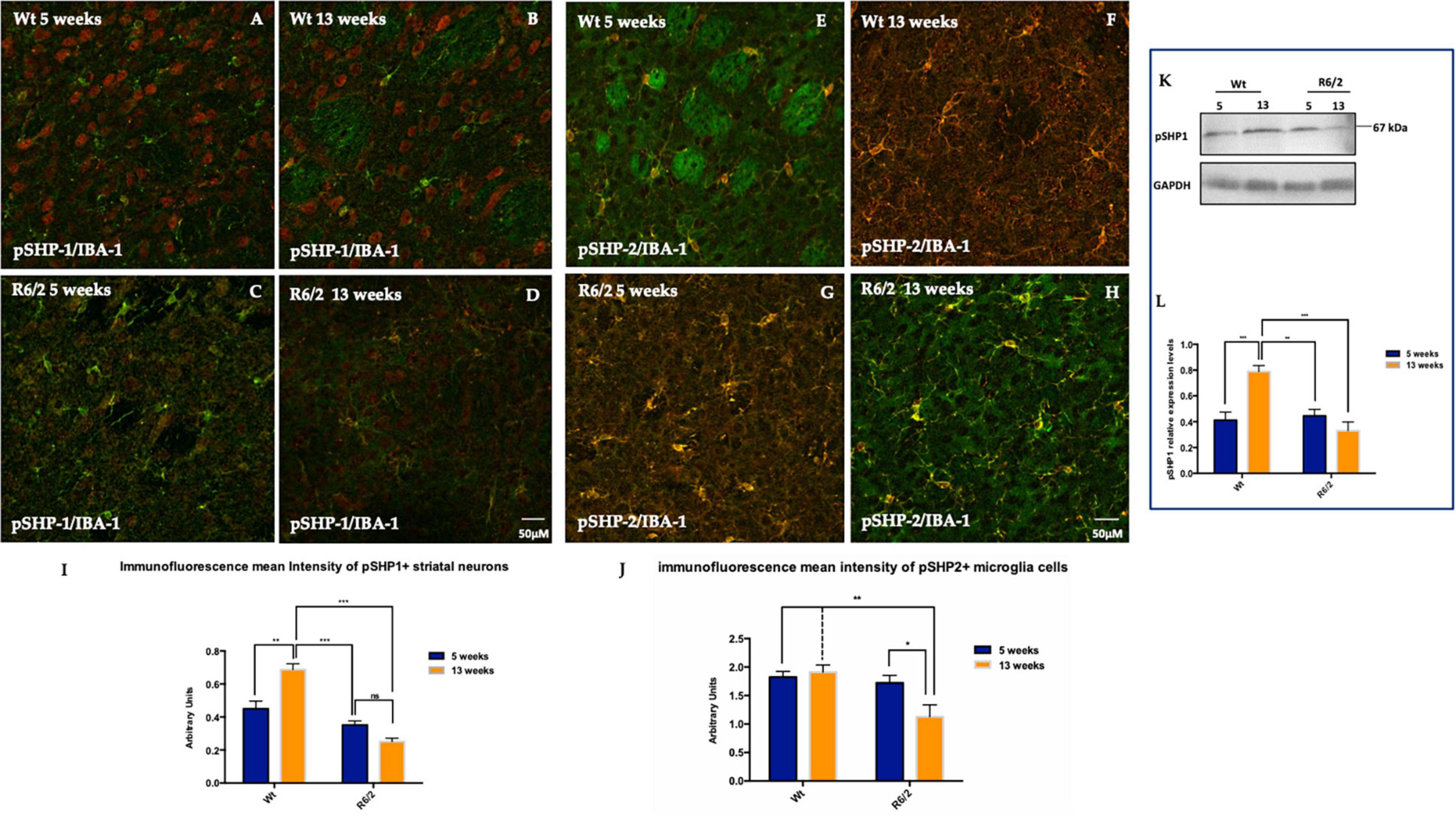
Figure 4. Distribution pattern of phosphorylated SHP-1 and SHP-2. (A–D) pSHP-1 is showed in red fluorescence, IBA-1 in green. Microglia expression of pSHP-2 in the R6/2 mice striatum. (E–H) pSHP-2 (visualized in red fluorescence) and IBA-1 (visualized in green fluorescence). (I) Two-way ANOVA analysis performed on data obtained by 5 and 13 weeks-old mice showed a statistically significant genotype effect of pSHP-1 positive striatal neurons [F(1,44) = 65.85, ***P < 0.0001; Time F(1,44) = 4.374, **P = 0.00423; Genotype X Interaction F(1,44) = 26.35, ***P < 0.0001]. (J) Tukey’s multiple comparisons test showed a statistically significant Genotype Effect on microglial pSHP-2 reduction expression in the R6/2 mice at 13 weeks-old of age [F(1,36) = 9.094, **P = 0.0047]. (K,L) Molecular data analysis of pSHP-1 revealed a significantly decreased in the striatal neurons of 5 and 13 week-old R6/2 mice with a genotype effect [F(1,28) = 13.74, ***P = 0.0009; Time F(1,28) = 5.283, **P = 0.00292; Genotype X Time Interaction F(1,28) = 18.57, ***P = 0.0002].
Since pSHP1 plays an anti-inflammatory role by dephosphorylating pro inflammatory transcription factors role, in this study we evaluated STAT1 activation in the R6/2 mice striatum. In the animals control the expression levels of activated STAT1 are mostly undetectable (Figures 5A–F). Fluorescence images revealed a significant activation of STAT1 expression in 5 weeks-old R6/2 mice that statistically increased in 13 weeks-old R6/2 mice (Figures 5G–M). Molecular biology experiments confirmed what we observed on mice brain tissue, showing also the activation of STAT3 exclusively in the R6/2 mice (Figure 5N).
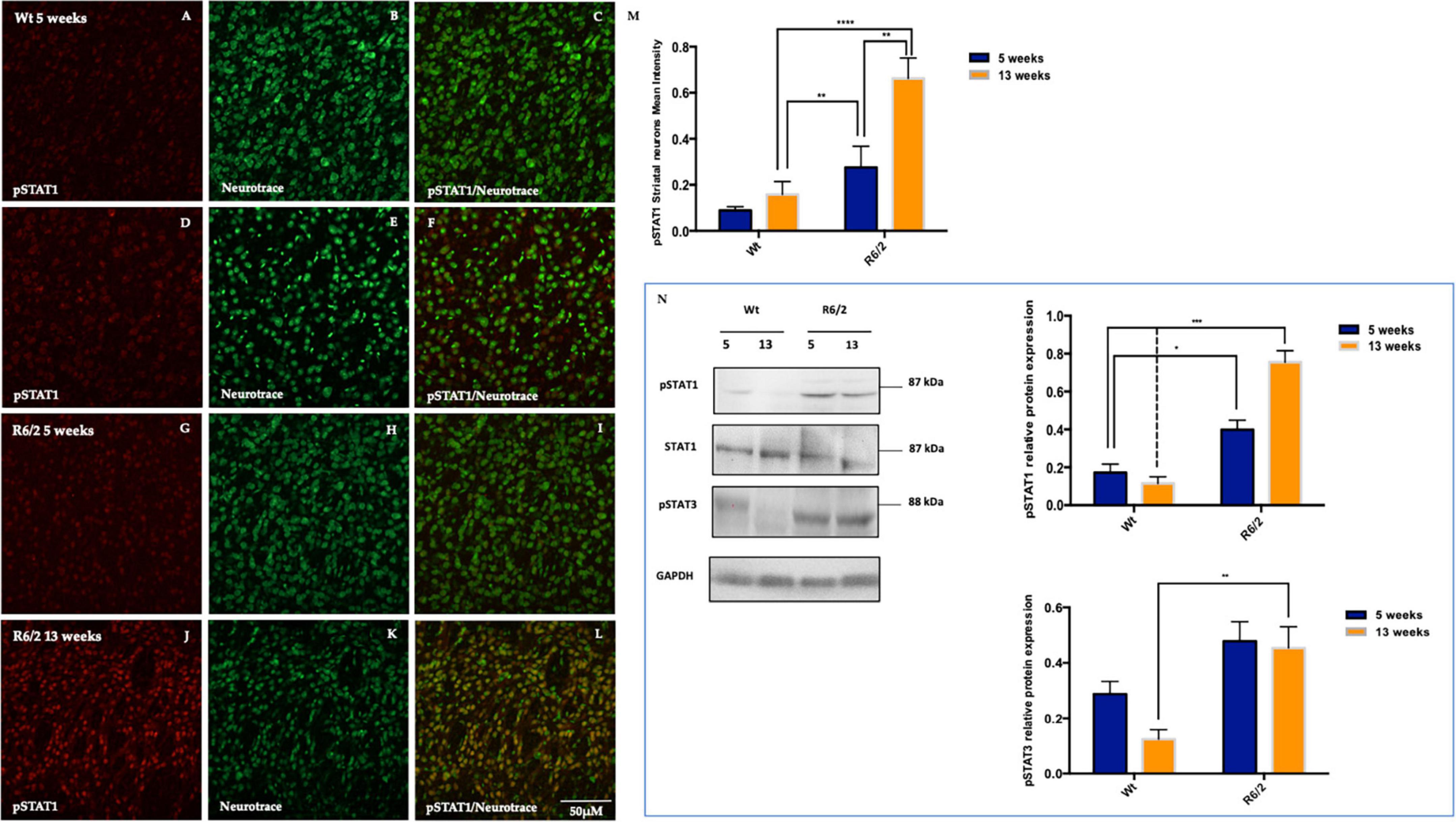
Figure 5. Representative confocal microscopy images of pSTAT1 immunostaining in R6/2 mice striatum. (A–L) Phosphorylated STAT1 is showed in red fluorescence, counterstained with Nissl-like fluorescent marker Neurotrace visualized in green. (M) Two-way ANOVA analysis performed on 5 and 13 weeks-old animals group showed a statistically significant effect of genotype of pSTAT1 positive striatal neurons [F(1,32) = 24.26, ***P < 0.0001, Time F(1,32) = 10.58, **P = 0.0027 and Genotype X Time Interaction F(1,36) = 5.102, *P = 0.0309]. Protein quantification analysis of pSTAT1 revealed a significantly increment in the striatal neurons of 5 and 13 week-old R6/2 mice with genotype effect [F(1,12) = 80.35 ***P < 0.0001; Time F(1,12) = 9.655; **P = 0.0091, Genotype X Time Interaction F(1,12) = 18.27; **P = 0.0011]. (N) Moreover, molecular experiments data analysis revealed a statistical significant high protein expression of pSTAT3 in the R6/2 mice with a genotype effect [F(1,12) = 19.26; **P = 0.009; Time F(1,12) = 2.537; *P = 0.1372, Genotype X Time Interaction F(1,12) = 1.378; *P = 0.2632].
4 Discussion
The injured CNS (e.g., stroke, HD, Alzheimer’s disease, HD) requires an innate immune response in order to curb the damage and trigger the repair of tissue by clearance of apoptotic cells and other toxic debris (Gasque et al., 2000; Barnum, 2002; Nguyen et al., 2002).
To this end, it is crucial that both glia and neuronal cells can recognize non-self, toxic materials, and self, so that they are able to remove apoptotic bodies and further restrict inflammation. In some occasions glia will respond with massive recruitment of peripheral macrophages and reactive microglia, which can represent a threat for CNS (Wyss-Coray and Mucke, 2002). However, neuronal cells can express specific “don’t eat me” signals to diminish an adverse phagocytosis and to induce a restorative inflammatory response by anti-inflammatory cytokines and growth factors that are involved in tissue repair.
The importance of microglia in the health of central nervous system and its involvement in its disease has been the object of many studies. Microglial activation is protective for the injured tissue because of its ability to remove cell debris and to reconstruct the integrity of damaged tissue (Hanisch and Kettenmann, 2007; Lalancette-Hébert et al., 2007; Perry et al., 2010). However, the excessive microglial activation could lead to CNS tissue damage and interfere with CNS repairing mechanisms by releasing toxic substances, such as reactive oxygen species (ROS), pro-inflammatory cytokines (Thored et al., 2009) and nitric oxide (NO).
Several receptors have been described on the surface of microglia to detect the “turning on” and “turning off” signals (“eat me/don’t eat me”) released from other cells in the CNS and play critical role in modulating microglial responses in many CNS injuries and neurodegenerative diseases (Hu et al., 2014). One of these signaling proteins, namely, SIRPα and its ligand CD47, are expressed on the surface of microglia as well as on other types of CNS cells (Gaikwad et al., 2009; Hu et al., 2015). Several studies have documented the importance of the interaction between SIRP and CD47 in mediating the intercellular communication in the CNS. In particular, SIRPα activation has been related to the inhibition of several cell functions, such as cytokine production (Latour et al., 2001; Kong et al., 2007; Gitik et al., 2011), monocyte adhesion to the extracellular matrix, and phagocytosis (Oldenborg et al., 2000; Smith et al., 2003; Liu et al., 2005).
On the other hand, SIRPα has been described on neuronal surfaces, where it seems to exert quite a different function than that on microglia. Indeed, while SIRPα on microglia displays an inhibitory effect on phagocytosis, neuronal SIRPα allows microglial phagocytosis and the fine-tuning of synapses (Ide et al., 2007; Janssen et al., 2008). In our study, we have investigated the expression of signals of phagocytosis in the R6/2 mouse model of HD. We found changes in the SIRPα/CD47 signaling system throughout the course of the disease. Indeed, in the R6/2 mouse we found the expression of SIRPα predominantly on neurons of the striatum, rather than on microglial cells. This expression was constantly higher compared to that of wild type animals at all stages. The relative lower levels of SIRP observed in Wt mice, may suggest that animals are not at the peak of synaptic pruning during which increased microglia activation is observed. This finding is consistent with the effects of neuronal SIRPα mentioned above. In fact, the activity of SIRPα is expected to be higher in case of a disease where an active process of neuroinflammation and tissue damage evokes higher level of phagocytosis.
The immunoreaction of CD47, known to be localized post-synaptically on neurons (Jiang et al., 2022), became less intense in the R6/2 mouse brain, as to indicate that this signaling factor has a somewhat independent function from SIRPα. This is not so surprising if one considers that the mechanism by which CD47 would inhibit phagocytosis depends on the presence of SIRPα on microglial cells. We found, instead, that SIRPα is localized on neurons and exerts its functions as a promoter of phagocytosis. This is also consistent with the findings of Jiang and co-workers that observed that a reduction of CD47 caused and increased microglial phagocytosis, while its overexpression decreased it.
Our study also investigated SHP-1 and SHP- 2 proteins. The Protein tyrosine phosphatase (PTP) family, a subgroup of cytoplasmic PTPs characterized by containing two Src homology 2 (SH2) NH2-terminal domains and a C-terminal protein-tyrosine phosphatase domain is referred to as SHP. They are involved in several cellular activities, such as cytoskeletal maintenance, cell division, and cell differentiation (Bialy and Waldmann, 2005). Of these, two particular SHP proteins known as SHP-1 (SH-PTP1, PTP1C, HCP, and SHP) and SHP-2 (SH-PTP2, PTP1D, Syp, PTP2C, and SH-PTP3) have been linked to trophic factor signaling (You and Zhao, 1997), cell growth, mitogen-activated protein kinase (MAPK) activity and chemotactic responses. SHP-1 was believed to play a negative role in modulating cell function, whereas SHP-2 was believed to up-regulate a variety of cell signal transduction processes. However, given the complex functions performed by SHP-1 and SHP-2, their role has not been completely defined, especially in the field of neurodegeneration. The function of SHP-1 and SHP-2 during cell injury and inflammation are not easily defined, since these PTPs appear to possess dual roles to either enhance or inhibit cell survival under a variety of environmental conditions.
Here, we showed that SHP-1 in its phosphorylated from was expressed both in neurons and the microglia of wild type mice. On the contrary, SHP-1 was only expressed in microglia with no signal of immunoreaction in the neurons of R6/2 mice, as an effect of genotype, even in the early stages of the disease. Such activity is mediated by the activation (phosphorylation) of the protein signal transducer and activator of transcription (STAT1). STATs are important signaling molecules that can activate brain inflammation. Activation and inactivation of STATs are both regulated by phosphorylation and dephosphorylation of tyrosine residues of STATs (You and Zhao, 1997). SHP-1 inhibits STAT signaling pathways induced by interferon.
In our study, in fact, we found that pSTAT1 was overexpressed in the neurons of R6/2 mice brain, as an effect of the lack of inhibition exerted by SHP-1 of which R6/2 mice neurons were devoid. Interestingly, STAT1 inhibition is able to reduce apoptotic cell death (Wu et al., 2016). On the contrary, SHP-2 intensity of immunoreaction decreased with disease progression in R6/2. Interestingly, the activation of SHP-2 is known to modulate inflammation. In fact, Guo and coworkers showed that NLRP3 activation causes SHP-2 to migrate to the mitochondria and prevent the release of proinflammatory mitochondrial DNA and Reactive oxygen species. Thus, such increase in SHP-2 could be interpreted as an attempt to counterbalance the ongoing inflammatory reaction occurring in HD.
Taken together, our data emphasize the importance of phagocytosis and inflammation signals in HD pathology, indicating the possibilities of its modulation as a therapeutic strategy for such a devastating disease. In a recent study, Morozko et al. (2021) showed that knocking down the gene for protein inhibitor of activated STAT1 in HD mice had a normalizing effect on HD transcriptional dysregulation. Thus, our results pave the way toward a modulation of STAT-1 through specific inhibitors that are expected to exert a neuroprotection in HD.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Dr. Vincenzo Ugo SANTUCCI, Italian Health Minestery. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
EP: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Resources, Supervision, Writing – original draft. GM: Data curation, Resources, Writing – original draft. FF: Data curation, Funding acquisition, Project administration, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2024.1360066/full#supplementary-material
Supplementary Figure 1 | (A–C) Confocal laser scanning microscopy of Z-stack images of double-label immunofluorescence for SIRPα (visualized in red fluorescence) and Synaptophysin (visualized in green fluorescence). Representative images of mice striatum from Wt mice show the colocalization of SIRPα with a presynaptic marker, Synaptophysin. (D,E) Confocal laser scanning microscopy of Z-stacks images of double-label immunofluorescence for CD47 (in red) and PSD95 (in green) of Wt mice striatum show the colocalization of CD47 with a postsynaptic marker, PSD95.
Supplementary Figure 2 | (A–H) pSHP-1 is showed in red fluorescence, IBA-1 in green. Representative confocal laser scanning microscopy images show phosphorylated SHP1 in the striatal neurons of WT animals at 5 and 13 weeks. Furthermore, it is possible to observe pSHP1 immunofluorescence staining in cells whose morphology can be traced back to microglia as highlighted by the arrows.
References
Agazie, Y., Movilla, N., Ischenko, I., and Hayman, M. (2003). The phosphotyrosine phosphatase SHP2 is a critical mediator of transformation induced by the oncogenic fibroblast growth factor receptor 3. Oncogene 22, 6909–6918. doi: 10.1038/sj.onc.1206798
Bialy, L., and Waldmann, H. (2005). Inhibitors of protein tyrosine phosphatases: Next-generation drugs? Angew. Chem. Int. Ed. Engl. 44, 3814–3839. doi: 10.1002/anie.200461517
Brown, E., and Frazier, W. (2001). Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 11, 130–135. doi: 10.1016/s0962-8924(00)01906-1
Chong, Z., and Maiese, K. (2007). The Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2: Diversified control of cell growth, inflammation, and injury. Histol. Histopathol. 22, 1251–1267. doi: 10.14670/HH-22.1251
Christophi, G., Panos, M., Hudson, C., Christophi, R., Gruber, R., Mersich, A., et al. (2009). Macrophages of multiple sclerosis patients display deficient SHP-1 expression and enhanced inflammatory phenotype. Lab. Invest. 89, 742–759. doi: 10.1038/labinvest.2009.32
De Rocca Serra-Nédélec, A., Edouard, T., Tréguer, K., Tajan, M., Araki, T., Dance, M., et al. (2012). Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature. Proc. Natl. Acad. Sci. U. S. A. 109, 4257–4262. doi: 10.1073/pnas.1119803109
Ding, X., Wang, J., Huang, M., Chen, Z., Liu, J., Zhang, Q., et al. (2021). Loss of microglial SIRPα promotes synaptic pruning in preclinical models of neurodegeneration. Nat. Commun. 12:2030. doi: 10.1038/s41467-021-22301-1
Elward, K., and Gasque, P. (2003). “Eat me” and “don’t eat me” signals govern the innate immune response and tissue repair in the CNS: Emphasis on the critical role of the complement system. Mol. Immunol. 40, 85–94. doi: 10.1016/s0161-5890(03)00109-3
Fu, R., Shen, Q., Xu, P., Luo, J., and Tang, Y. (2014). Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 49, 1422–1434. doi: 10.1007/s12035-013-8620-6
Gaikwad, S., Larionov, S., Wang, Y., Dannenberg, H., Matozaki, T., Monsonego, A., et al. (2009). Signal regulatory protein-beta1: A microglial modulator of phagocytosis in Alzheimer’s disease. Am. J. Pathol. 175, 2528–2539. doi: 10.2353/ajpath.2009.090147
Gardai, S., McPhillips, K., Frasch, S., Janssen, W., Starefeldt, A., Murphy-Ullrich, J., et al. (2005). Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334. doi: 10.1016/j.cell.2005.08.032
Gasque, P., Dean, Y. D., McGreal, E. P., VanBeek, J., and Morgan, B. P. (2000). Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology 49, 171–186.
Giampà, C., DeMarch, Z., D’Angelo, V., Morello, M., Martorana, A., Sancesario, G., et al. (2006). Striatal modulation of cAMP-response-element-binding protein (CREB) after excitotoxic lesions: Implications with neuronal vulnerability in Huntington’s disease. Eur. J. Neurosci. 23, 11–20. doi: 10.1111/j.1460-9568.2005.04545.x
Gitik, M., Liraz-Zaltsman, S., Oldenborg, P., Reichert, F., and Rotshenker, S. (2011). Myelin down-regulates myelin phagocytosis by microglia and macrophages through interactions between CD47 on myelin and SIRPα (signal regulatory protein-α) on phagocytes. J. Neuroinflamm. 8:24. doi: 10.1186/1742-2094-8-24
Guo, W., Liu, W., Chen, Z., Gu, Y., Peng, S., Shen, L., et al. (2017). Tyrosine phosphatase SHP2 negatively regulates NLRP3 inflammasome activation via ANT1-dependent mitochondrial homeostasis. Nat. Commun. 8:2168. doi: 10.1038/s41467-017-02351-0
Han, M., Lundgren, D., Jaiswal, S., Chao, M., Graham, K., Garris, C., et al. (2012). Janus-like opposing roles of CD47 in autoimmune brain inflammation in humans and mice. J. Exp. Med. 209, 1325–1334. doi: 10.1084/jem.20101974
Hanisch, U., and Kettenmann, H. (2007). Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Hardin, P., and Yu, W. (2006). Circadian transcription: Passing the HAT to CLOCK. Cell 125, 424–426. doi: 10.1016/j.cell.2006.04.010
Hu, X., Leak, R., Shi, Y., Suenaga, J., Gao, Y., Zheng, P., et al. (2015). Microglial and macrophage polarization—new prospects for brain repair. Nat. Rev. Neurol. 11, 56–64. doi: 10.1038/nrneurol.2014.207
Hu, X., Liou, A., Leak, R., Xu, M., An, C., Suenaga, J., et al. (2014). Neurobiology of microglial action in CNS injuries: Receptor-mediated signaling mechanisms and functional roles. Prog. Neurobiol. 11, 60–84. doi: 10.1016/j.pneurobio.2014.06.002
Ide, K., Wang, H., Tahara, H., Liu, J., Wang, X., Asahara, T., et al. (2007). Role for CD47-SIRPalpha signaling in xenograft rejection by macrophages. Proc. Natl. Acad. Sci. U. S. A. 104, 5062–5066. doi: 10.1073/pnas.0609661104
Janssen, W., McPhillips, K., Dickinson, M., Linderman, D., Morimoto, K., Xiao, Y., et al. (2008). Surfactant proteins A and D suppress alveolar macrophage phagocytosis via interaction with SIRP alpha. Am. J. Respir. Crit. Care Med. 178, 158–167. doi: 10.1164/rccm.200711-1661OC
Jiang, D., Burger, C., Akhanov, V., Liang, J., Mackin, R., Albrecht, N., et al. (2022). Neuronal signal-regulatory protein alpha drives microglial phagocytosis by limiting microglial interaction with CD47 in the retina. Immunity 55, 2318–2335.e7. doi: 10.1016/j.immuni.2022.10.018
Jiang, N., Zhang, L., Zhao, G., Lin, J., Wang, Q., Xu, Q., et al. (2020). Indoleamine 2,3-dioxygenase regulates macrophage recruitment, polarization and phagocytosis in aspergillus fumigatus keratitis. Invest. Ophthalmol. Vis. Sci. 61:28. doi: 10.1167/iovs.61.8.28
Khanna, S., Roy, S., Park, H., and Sen, C. (2007). Regulation of c-Src activity in glutamate-induced neurodegeneration. J. Biol. Chem. 282, 23482–23490. doi: 10.1074/jbc.M611269200
Kong, X., Yan, H., Chen, L., Dong, L., Yang, W., Liu, Q., et al. (2007). LPS-induced down-regulation of signal regulatory protein {alpha} contributes to innate immune activation in macrophages. J. Exp. Med. 204, 2719–2731. doi: 10.1084/jem.20062611
Lalancette-Hébert, M., Gowing, G., Simard, A., Weng, Y., and Kriz, J. (2007). Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 27, 2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007
Latour, S., Tanaka, H., Demeure, C., Mateo, V., Rubio, M., Brown, E., et al. (2001). Bidirectional negative regulation of human T and dendritic cells by CD47 and its cognate receptor signal-regulator protein-alpha: Down-regulation of IL-12 responsiveness and inhibition of dendritic cell activation. J. Immunol. 167, 2547–2554.
Liu, S., Alkema, P., Tieché, C., Tefft, B., Liu, D., Li, Y., et al. (2005). Negative regulation of monocyte adhesion to arterial elastic laminae by signal regulatory protein alpha and Src homology 2 domain-containing protein-tyrosine phosphatase-1. J. Biol. Chem. 280, 39294–39301. doi: 10.1074/jbc.M503866200
Morozko, E., Smith-Geater, C., Monteys, A., Pradhan, S., Lim, R., Langfelder, P., et al. (2021). PIAS1 modulates striatal transcription, DNA damage repair, and SUMOylation with relevance to Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A. 118:e2021836118. doi: 10.1073/pnas.2021836118
Murata, Y., Saito, Y., Kotani, T., and Matozaki, T. (2018). CD47-signal regulatory protein α signaling system and its application to cancer immunotherapy. Cancer Sci. 109, 2349–2357. doi: 10.1111/cas.13663
Nguyen, M. D., Julien, J. P., and Rivest, S. (2002). Innate immunity: The missing link in neuroprotection and neurodegeneration? Nat. Rev. Neurosci. 3, 216–227.
Oldenborg, P., Zheleznyak, A., Fang, Y., Lagenaur, C., Gresham, H., and Lindberg, F. (2000). Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054. doi: 10.1126/science.288.5473.2051
Paldino, E., D’Angelo, V., Sancesario, G., and Fusco, F. (2020). Pyroptotic cell death in the R6/2 mouse model of Huntington’s disease: New insight on the inflammasome. Cell Death Discov. 6:69. doi: 10.1038/s41420-020-00293-z
Paldino, E., and Fusco, F. (2022). Emerging role of NLRP3 inflammasome/pyroptosis in Huntington’s disease. Int. J. Mol. Sci. 23:8363. doi: 10.3390/ijms23158363
Perry, V., Nicoll, J., and Holmes, C. (2010). Microglia in neurodegenerative disease. Nat. Rev. Neurol. 6, 193–201. doi: 10.1038/nrneurol.2010.17
Sellgren, C., Gracias, J., Watmuff, B., Biag, J., Thanos, J., Whittredge, P., et al. (2019). Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 22, 374–385. doi: 10.1038/s41593-018-0334-7
Shuai, K., Stark, G., Kerr, I., and Darnell, J. Jr. (1993). A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science 261, 1744–1746. doi: 10.1126/science.7690989
Silverman, S., and Wong, W. (2018). Microglia in the retina: Roles in development, maturity, and disease. Annu. Rev. Vis. Sci. 4, 45–77. doi: 10.1146/annurev-vision-091517-034425
Smith, R., Patel, V., Seatter, S., Deehan, M., Brown, M., Brooke, G., et al. (2003). A novel MyD-1 (SIRP-1alpha) signaling pathway that inhibits LPS-induced TNFalpha production by monocytes. Blood 102, 2532–2540. doi: 10.1182/blood-2002-11-3596
Thored, P., Heldmann, U., Gomes-Leal, W., Gisler, R., Darsalia, V., Taneera, J., et al. (2009). Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia 57, 835–849. doi: 10.1002/glia.20810
Wilton, D., Dissing-Olesen, L., and Stevens, B. (2019). Neuron-glia signaling in synapse elimination. Annu. Rev. Neurosci. 42, 107–127. doi: 10.1146/annurev-neuro-070918-050306
Wu, Y., Gao, C., Fan, K., Yang, L., and Mei, X. (2016). STAT1 inhibitor alleviates spinal cord injury by decreasing apoptosis. Genet. Mol. Res. 15:gmr.15017271. doi: 10.4238/gmr.15017271
Wyss-Coray, T., and Mucke, L. (2002). Inflammation in neurodegenerative disease—a double-edged sword. Neuron 35, 419–432.
Keywords: pSHP-1, SIRPα, CD47, pSTAT1, microglia, neurodegeneration, Huntington’s disease
Citation: Paldino E, Migliorato G and Fusco FR (2024) Neuroimmune pathways involvement in neurodegeneration of R6/2 mouse model of Huntington’s disease. Front. Cell. Neurosci. 18:1360066. doi: 10.3389/fncel.2024.1360066
Received: 22 December 2023; Accepted: 23 January 2024;
Published: 20 February 2024.
Edited by:
Dirk M. Hermann, University of Duisburg-Essen, GermanyReviewed by:
Tiffany Jo Petrisko, University of California, Irvine, United StatesBijay Parajuli, University of Yamanashi, Japan
Copyright © 2024 Paldino, Migliorato and Fusco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca R. Fusco, Zi5mdXNjb0Boc2FudGFsdWNpYS5pdA==; orcid.org/0000-0003-2226-523X