 Jack Butler
Jack Butler Nicholas Dale
Nicholas Dale
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci., 21 December 2023
Sec. Non-Neuronal Cells
Volume 17 - 2023 | https://doi.org/10.3389/fncel.2023.1330983
This article is part of the Research TopicThe Legacy of Sherrington and Adrian Nobel Prize: Non-Neuronal Cells in Information ProcessingView all 4 articles
Connexin32 (Cx32) is expressed in myelinating Schwann cells. It forms both reflexive gap junctions, to facilitate transfer of molecules from the outer to the inner myelin layers and hemichannels at the paranode to permit action potential-evoked release of ATP into the extracellular space. Loss of function mutations in Cx32 cause X-linked Charcot Marie Tooth disease (CMTX), a slowly developing peripheral neuropathy. The mechanistic links between Cx32 mutations and CMTX are not well understood. As Cx32 hemichannels can be opened by increases in PCO2, we have examined whether CMTX mutations alter this CO2 sensitivity. By using Ca2+ imaging, dye loading and genetically encoded ATP sensors to measure ATP release, we have found 5 CMTX mutations that abolish the CO2 sensitivity of Cx32 hemichannels (A88D, 111–116 Del, C179Y, E102G, V139M). Others cause a partial loss (L56F, R220Stop, and R15W). Some CMTX mutations have no apparent effect on CO2 sensitivity (R15Q, L9F, G12S, V13L, V84I, W133R). The mutation R15W alters multiple additional aspects of hemichannel function including Ca2+ and ATP permeability. The mutations that abolish CO2 sensitivity are transdominant and abolish CO2 sensitivity of co-expressed Cx32WT. We have shown that Schwannoma RT4 D6P2T cells can release ATP in response to elevated PCO2 via the opening of Cx32. This is consistent with the hypothesis that the CO2 sensitivity of Cx32 may be important for maintenance of healthy myelin. Our data, showing a transdominant effect of certain CMTX mutations on CO2 sensitivity, may need to be taken into account in any future gene therapies for this condition.
Connexin32 (Cx32) is expressed in oligodendrocytes and Schwann cells of the central and peripheral nervous systems, respectively. These cells generate the myelin sheath around the central and peripheral axons that is required for high speed saltatory conduction. Charcot Marie Tooth disease is a slowly developing peripheral neuropathy that involves loss of the integrity of peripheral myelin (Murakami et al., 1996). Typical symptoms include slowed peripheral conduction, peripheral numbness and tingling, muscle wasting and excessive arching of feet. There is an X linked version of this neuropathy (CMTX) that is associated with mutations of Cx32 (Bergoffen et al., 1993). Strong evidence indicates that CMTX is caused by loss of function of Cx32 (Shy et al., 2007; Sargiannidou et al., 2009): a CMTX phenotype is present in Cx32-null mice (Scherer et al., 1998); and this can be rescued by re-expression of Cx32 targeted only to Schwann cells (Scherer et al., 2005; Sargiannidou et al., 2015).
Connexin32 is a beta connexin and is present in Schwann cells both as a hemichannel and a gap junction. In the paranode Cx32 acts as a plasma membrane channel opening from the paranode to the extracellular space. The opening of Cx32 during action potential propagation allows the release of ATP from the paranode into the extracellular space (Nualart-Marti et al., 2013). However, Cx32 gap junctions are also present at the Incisures of Lantermann. Here, they form “reflexive” gap junctions to permit a fast radial pathway of intracellular diffusion from the outermost to the inner most layer of myelin that is estimated to be 106 times faster than the circumferential pathway via the spiral of the myelin sheath (Balice-Gordon et al., 1998). Strikingly, radial diffusion in myelin is not reduced in Cx32–/– mice (Balice-Gordon et al., 1998), suggesting other connexins are able to play this role. The other major connexin expressed in Schwann cells is Cx29 (also known as Cx31.3).
The effect of CMTX mutations on the permeability and gating properties of Cx32 hemichannels and gap junctions have been studied (Omori et al., 1996; Oh et al., 1997; Ressot et al., 1998; Abrams et al., 2000, 2001; Wang et al., 2004; Sargiannidou et al., 2009). Various effects have been reported such as a deficiency in the Ca2+ triggered opening of Cx32 hemichannels in R220Stop (Carrer et al., 2018). For some mutations there is evidence of altered permeability of the gap junction to molecules (Oh et al., 1997; Bicego et al., 2006), which could reduce permeation of intracellular signaling molecules such as IP3 or cAMP through the gap junction. Nevertheless, the precise mechanistic reasons for why particular Cx32 mutations lead to CMTX are not well understood. In this study we have chosen 14 mutations that are associated with CMTX that occur in different regions of the molecule, including the N-terminus (important for gating), the transmembrane regions TM2 and TM3, the cytoplasmic loop and the C-terminus. Ten of these mutations are deemed by multiple sources to be pathogenic, while the remaining four are currently of uncertain status with regard to the pathology of CMTX (Table 1).

Table 1. CMTX-associated mutations analyzed in this study.
Connexin32 is a beta connexin, closely related to Cx26 and Cx30. Hemichannels of these three connexins can be opened by changes in PCO2 at constant extracellular pH and normal physiological concentrations of extracellular Ca2+ (Huckstepp et al., 2010a; Dospinescu et al., 2019). The CO2 sensitivity of the beta connexins is imparted by the presence of a “carbamylation motif” and involves carbamylation of a specific lysine residue within this motif, which then interacts with an Arg/Lys residue on the neighboring subunit in the hexamer (Meigh et al., 2013; Dospinescu et al., 2019; van de Wiel et al., 2020; Nijjar et al., 2021). In Cx32 the critical residues are K124, which we hypothesize to be carbamylated and K104 in the neighboring subunit which could act as the interacting partner (Dospinescu et al., 2019). Cx32 differs from Cx26, the connexin in which the mechanism of CO2 sensitivity has been best studied, in requiring markedly higher concentrations of CO2 to open (an increase of PCO2 to 55 mmHg or greater) (Huckstepp et al., 2010a; Dospinescu et al., 2019). In Cx26, several mutations that cause non-syndromic hearing loss or the keratitis ichthyosis deafness syndrome (KIDS) also abolish CO2 sensitivity (Meigh et al., 2014; de Wolf et al., 2016; Cook et al., 2019). In this paper, we address whether a range of CMTX mutations might affect the CO2 sensitivity of Cx32. By using a range of assays (Ca2+ imaging, dye loading and imaging of ATP release), we show that several CMTX mutations abolish the CO2 sensitivity of Cx32, but others do not affect it. Our data suggests that loss of CO2 sensitivity of Cx32 in certain CMTX mutations should be investigated further as a potential contributing mechanism to the development of the pathology.
The Cx32 gene sequences were synthesized by Genscript and subcloned into the pCAG-GS mCherry vector prior to mammalian cell transfection. Plasmids were generated using Gibson assembly. DNA fragments were generated using PCR amplification with primers (IDT). The presence of the correct mutation was confirmed by DNA sequencing (GATC Biotech). The dnCx32 plasmid was cloned using successive Gibson assemblies to incorporate both K104A and K124R mutations. All Cx32 constructs were inserted upstream of mCherry, with a short 12 amino acid linker (GVPRARDPPVAT).
Parental HeLa DH cells (ECACC Cat# 96112022, RRID:CVCL_2483) were grown in Low-glucose Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 10% fetal bovine serum, 50 μg/mL penicillin/streptomycin. HeLa DH cells that stably expressed Cx32 (gift from Dr K. Willecke) were cultured in a similar manner, but with puromycin to select the expressing cells.
RT4-D6P2T cells were obtained from ECACC (ECACC Cat# 93011415, RRID:CVCL_4006). Cell culture was carried out in a modified medium; Dulbecco’s Modified Eagle’s Medium (DMEM) modified to contain 4 mM L-glutamine, 4500 mg/L glucose, 1 mM sodium pyruvate, and 1500 mg/L sodium bicarbonate and supplemented with 10% FBS, 5% Penicillin-Streptomycin.
Parental HeLa DH cells were plated onto coverslips at a density of 7.5 × 104 cells per well of a 6 well plate, and transiently transfected with the Cx32 expression constructs following the PEI Transfection Reagent protocol. Cells were transfected using a mixture containing 1 μg DNA and 3 μg PEI for 24 h and imaged 48 h after transfection. For transfection of dnCx32, cells (either the RT4-D6P2T, or HeLa cells stably expressing Cx32) were seeded at a known density (104 cells per well). These cells were then transfected with dnCx32. Recordings were obtained 5–6 days post-transfection to allow co-assembly with endogenous Cx32WT to occur.
Control (35 mmHg PCO2) aCSF: 124 mM NaCl, 3 mM KCl, 2 mM CaCl2, 26 mM NaHCO3, 1.25 mM NaH2PO4, 1 mM MgSO4, 10 mM D-glucose saturated with 95% O2/5% CO2, pH 7.4, PCO2 35 mmHg.
A total of 70 mmHg aCSF: 73 mM NaCl, 3 mM KCl, 2 mM CaCl2, 80 mM NaHCO3, 1.25 mM NaH2PO4, 1 mM MgSO4, 10 mM D-glucose, saturated with ∼12% CO2 (with the balance being O2) to give a pH of 7.4 and a PCO2 of 70 mmHg, respectively.
High K+ aCSF: 77 mM NaCl, 50 mM KCl, 2mM CaCl2, 26 mM NaHCO3, 1.25 mM NaH2PO4, 1 mM MgSO4, 10 mM D-glucose saturated with 95% O2/5% CO2, pH 7.4, PCO2 35 mmHg.
HeLa-DH cells were transfected with the desired pCAG-Connexin-mCherry construct as detailed in methods of transfection. These cells were then incubated in 2 ml DMEM containing 5 mM Fluo-4 AM (Invitrogen) dissolved in 2.5 μL of Pluronic-127 (Invitrogen) for 20 min. Cells were then washed in 2 ml serum free DMEM for 20 min. Coverslips were then placed in a perfusion chamber. Cells were perfused with control 35 mmHg aCSF until a stable baseline is reached and maintained, at which point the cells were perfused with hypercapnic 70 mmHg aCSF. Once a stable baseline is reached the cells were then again perfused with control 35 mmHg aCSF. Following loading with Fluo-4 AM cells were imaged by epifluorescence (Scientifica Slice Scope, Cairn Research OptoLED illumination, 60x water Olympus immersion objective, NA 1.0, Hamamatsu ImagEM EM-SSC camera, Metafluor software). Fluo-4 was excited using 470 nm LED, with fluorescent emission being recorded between 507 and 543 nm. The Cx32 constructs utilized here yielded fusion proteins with a C-terminal mCherry tag. mCherry was excited with the 535 nm LED, with emission being recorded between 570 and 640 nm.
Analysis of Ca2+ signals was performed in ImageJ. For cells that had loaded with Fluo-4 and were positive for mCherry, an ROI was manually drawn round the cell body and the median pixel intensity within the ROI measured for each image. The fluorescence pixel intensities (F) were normalized to a baseline period (F0), and the difference in F/F0 evoked by the CO2 stimulus measured for each cell. This change in fluorescence was measured by taking the median of the F/F0 baseline from the 60–120 s immediately before the CO2 stimulus and subtracting this from the median F/F0 value measured over 60–120 s during the stimulus. Statistical comparisons were performed considering each cell as an independent measurement. Five transfections were performed for each variant of Cx32.
We used a dye loading protocol that has been developed and extensively described in our prior work (Dospinescu et al., 2019). HeLa cells expressing each Cx32 construct were initially washed with control solution. They were then exposed to either control or hypercapnic solution containing 200 μM 5(6)-carboxyfluorescein (CBF) for 10 min. Subsequently, cells were returned to control solution with 200 μM CBF for 5 min, before being washed in control solution without CBF for 30 min to remove excess extracellular dye. A replacement coverslip of HeLa cells was used for each condition. For each coverslip, mCherry fluorescence was imaged to verify Cx32 expression. The experiments were replicated independently (independent transfections) at least five times to give n = 5 for each species.
Following dye loading, HeLa cells were imaged by epifluorescence (Scientifica Slice Scope, Cairn Research OptoLED illumination, 60x water Olympus immersion objective, NA 1.0, Hamamatsu ImagEM EM-CCD camera, Metafluor software). Following acquisition of the images, subsequent analysis was performed blind to Cx32 variant and treatment. ImageJ was used to measure the extent of dye loading by drawing a region of interest (ROI) around each cell, and subsequently, the mean pixel intensity of the ROI was determined. The mean pixel intensity of a representative background ROI for each image was subtracted from each cell measurement from the same image. At least 40 cells were measured for each condition per experiment, and at least five repetitions of independently transfected HeLa cells were completed. Statistical comparisons were performed on the median values obtained from each transfection.
pDisplay-GRAB_ATP1.0-IRES-mCherry-CAAX was a gift from Yulong Li (Addgene plasmid #167582; RRID:Addgene_167582).1
pDisplay-GRAB_ATP1.0 mut-IRES-mCherry-CAAX was a gift from Yulong Li (Addgene plasmid #167583; RRID:Addgene_157583).2
Following transfection with GRABATP cells were imaged by epifluorescence (Scientifica Slice Scope, Cairn Research OptoLED illumination, 60x water Olympus immersion objective, NA 1.0, Hamamatsu ImagEM EM-SSC camera, Metafluor software). cpGFP was excited using 470 nm LED, with fluorescent emission being recorded between 507 and 543 nm. The Cx32 constructs utilized here yielded fusion proteins with a C-terminal mCherry tag. mCherry was excited with the 535 nm LED, with emission being recorded between 570 and 640 nm.
Analysis of GRABATP signals was performed in ImageJ. For cells that expressed both the Cx32 variant and GRABATP, one ROI was drawn around the region of GRABATP expression per cell and the median pixel intensity within the ROIs measured for each image. The fluorescence pixel intensities (F) were normalized to a baseline period (F0), and the difference in F/F0 (ΔF/F0) evoked by the CO2 stimulus measured for each cell. Use of the GRABATP biosensor to detect ATP release via Cx32 was validated by demonstrating that an increase in fluorescence to CO2 or 50 mM KCl was only seen in HeLa cells that expressed both Cx32 and GRABATP (Supplementary Figure 1). No fluorescence changes to CO2 or 50 mM KCl were seen with either GRABATP alone or Cx32 plus GRABmut, an ATP insensitive mutated control (Supplementary Figure 1). As the dose response for GRABATP was approximately linear over the range 0–3 μM (Supplementary Figure 1), and most of the recorded changes in ATP concentration fell into this range, we converted changes of fluorescence evoked by 70 mmHg PCO2 and 50 mM KCl into ATP concentration by normalizing them to the ΔF/F0 produced by a 3 μM calibration dose of ATP in each experiment. Statistical comparisons were performed considering each cell as an independent measurement. Five transfections were performed for each variant of Cx32.
Coverslips were first washed with PBS three times, before being fixed in 4% PFA for 30 min. Coverslips were then washed in PBS three times and blocked using PBS containing 4% BSA and 0.1% Triton X-100 for 24 h. Cx32 primary antibody (1:250 dilution, Thermo Fisher Scientific Cat# 13-8200, RRID:AB_2533037) in PBS containing 4% BSA and 0.1% Triton X-100 was added to coverslips and left to incubate, constantly moving, for 3 h at room temperature. Coverslips were then washed using PBS containing 0.1% Triton X-100 six times at 10 min intervals. Anti-mouse secondary antibody (1:250, Thermo Fisher Scientific Cat# A-11032, RRID:AB_2534091) in PBS containing 4% BSA and 0.1% Triton X-100 and added to coverslips and left to incubate, constantly moving, for 2.5 h. The secondary antibody was washed using PBS containing 0.1% Triton X-100 six times at 10 min intervals. Coverslips where then mounted inverted on glass slides using Fluorshield™ with DAPI mounting medium (Sigma-Aldrich, Cat# F6057).
Cells were subsequently imaged using the Zeiss-880 confocal LSM, specifically using the 488 and 561 nm lasers. FIJI software was used for further analysis.
All quantitative data are presented as box and whisker plots where the box represents the interquartile range, the bar represents the median, and the whiskers represent 1.5 times the interquartile range. Individual data points are superimposed. Statistical analysis was via the Kruskal Wallis one-way ANOVA (KW test) followed by pairwise Mann Whitney U-tests with correction for multiple comparisons via the false discovery method (Curran-Everett, 2000) with the maximum rate of false discovery set at 0.05. For analysis of the GRABATP recordings in which the CO2 and 50 mM KCl stimuli were applied to the same cell, these data were considered to be paired. Comparisons of the amount of ATP released by each stimulus was therefore performed with the Wilcoxon Matched Pairs Signed Rank test. All pairwise tests were two sided and all calculations performed with GraphPad PRISM.
To test whether CMTX mutations might alter the CO2 sensitivity of Cx32, we first used Fluo4 to measure intracellular Ca2+ in HeLa cells expressing WT Cx32. We found that a change in PCO2 from 35 to 70 mmHg reliably evoked a change in Fluo4 fluorescence (Figures 1A, B). This was not seen in parental HeLa cells that did not express Cx32 (Supplementary Figure 2). Thus, Cx32 hemichannels are permeable to Ca2+. A similar permeability to Ca2+ has previously been reported for Cx26 hemichannels (Fiori et al., 2012).
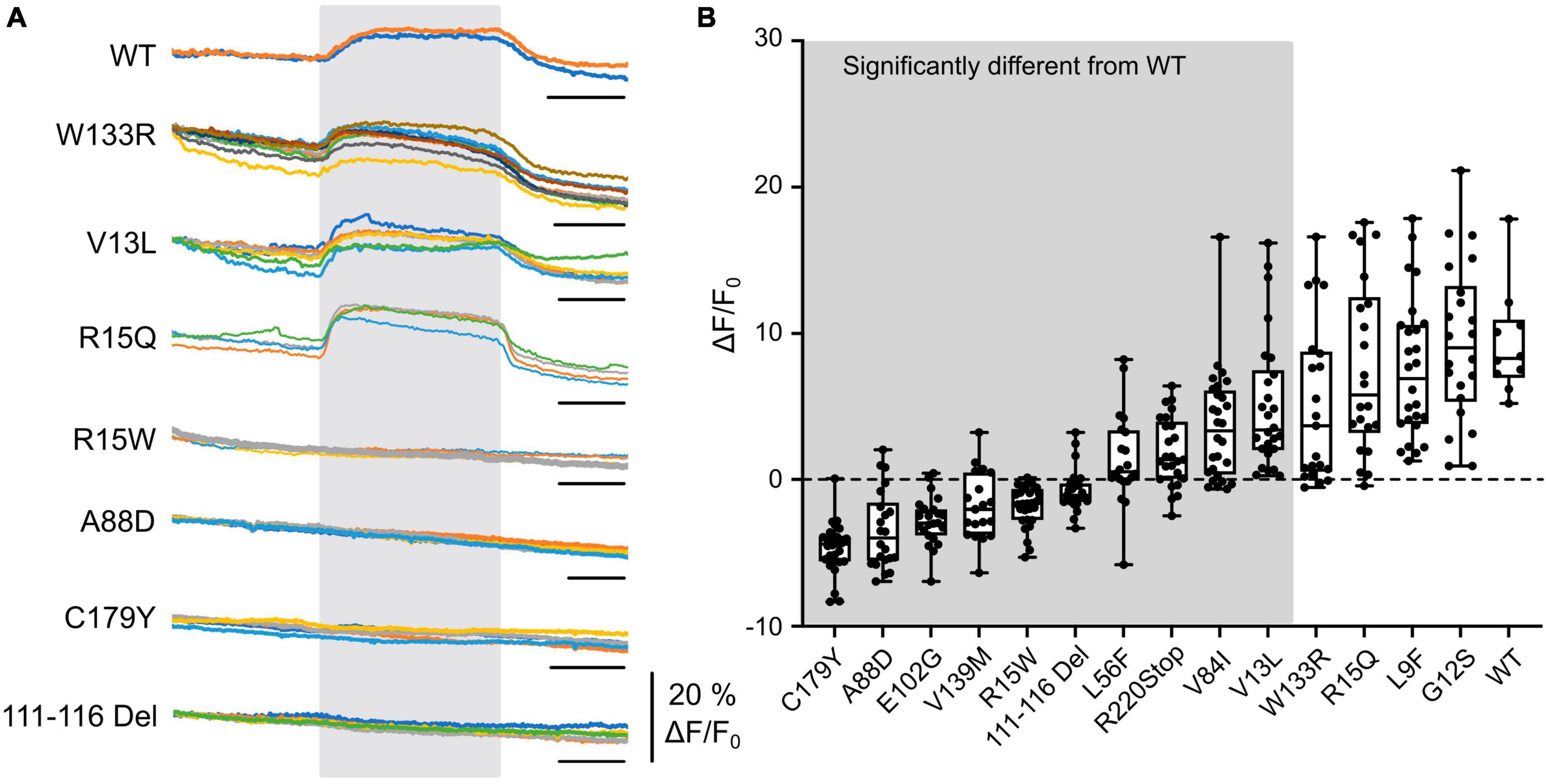
Figure 1. CO2-evoked increases in intracellular Ca2+ are mediated via Cx32 and sensitive to CMTX mutations. (A) Changes in Fluo4 fluorescence evoked by an increase of PCO2 from 35 to 70 mmHg (during gray rectangle) in HeLa cells expressing Cx32 WT and various CMTX mutations. Each colored line represents a recording from a different cell. Time scale bar 200 s. (B) Summary data for 14 CMTX mutations and WT Cx32, ordered from left to right by median change in fluorescence to CO2. A Kruskal Wallis ANOVA shows that these samples are not drawn from the same population (p < 0.0001). Pairwise testing relative to WT Cx32 (corrected for a maximum false discovery rate of 0.05) show that the mutations from the left up to and including V13L are significantly different from the WT (details in text).
We next selected a panel of mutations that affected different regions of the Cx32 subunit, including the N-terminus (important in channel gating) and the cytoplasmic loop (the location of the carbamylation motif) and various transmembrane regions. Out of these 14 selected mutations, 10 affected the CO2-evoked Ca2+ signal via Cx32 (Figure 1). While 4 mutations (L56F, R220Stop, V84I (all p < 0.0001 compared to WT) and V13L, p = 0.0042 compared to WT) caused a partial reduction of the Ca2+ signal, the remainder (covering all portions of the molecule) caused an apparent complete loss of CO2 -dependent Ca2+ signal (Figure 1, p < 0.0001 compared to WT). Interestingly, the mutation R15Q had no effect on the CO2 mediated increase in intracellular Ca2+ whereas R15W caused its complete abolition. We have previously demonstrated in Cx26 that introduction of large residues at the N terminus (N14K and N14Y) abolished its CO2 sensitivity (de Wolf et al., 2016).
An alternative interpretation of the above results is that some of these mutations might alter Ca2+ permeability of Cx32 rather than its sensitivity to CO2. We therefore further checked the effects of the six CMTX mutations that appeared to completely abolish CO2 sensitivity of Cx32, by using an established dye loading assay of hemichannel gating (Meigh et al., 2013; de Wolf et al., 2017; Dospinescu et al., 2019). We found that the mutations V139M, 111-116 Del, C179Y, E103G, and A88D completely abolished CO2-dependent dye loading (Figure 2) in agreement with the results from the Ca2+ measurements. This was not because there were no functional hemichannels, because the positive control of removing extracellular Ca2+ to unblock the hemichannels, gave robust dye loading for all 6 mutations (Figure 2 and Supplementary Figure 3). By contrast the mutation R15W only partially reduced the extent of CO2 dependent dye loading (Figure 2). Because the zero Ca2+ stimulus still gave robust dye loading (Supplementary Figure 3) the reduced dye loading in response to CO2 suggests a direct effect of R15W on CO2 sensitivity of the hemichannel. Nevertheless, the apparent complete abolition of a response to CO2 in the Ca2+ measurements indicates that this mutation may also greatly reduce the permeability of Cx32 hemichannels to Ca2+.
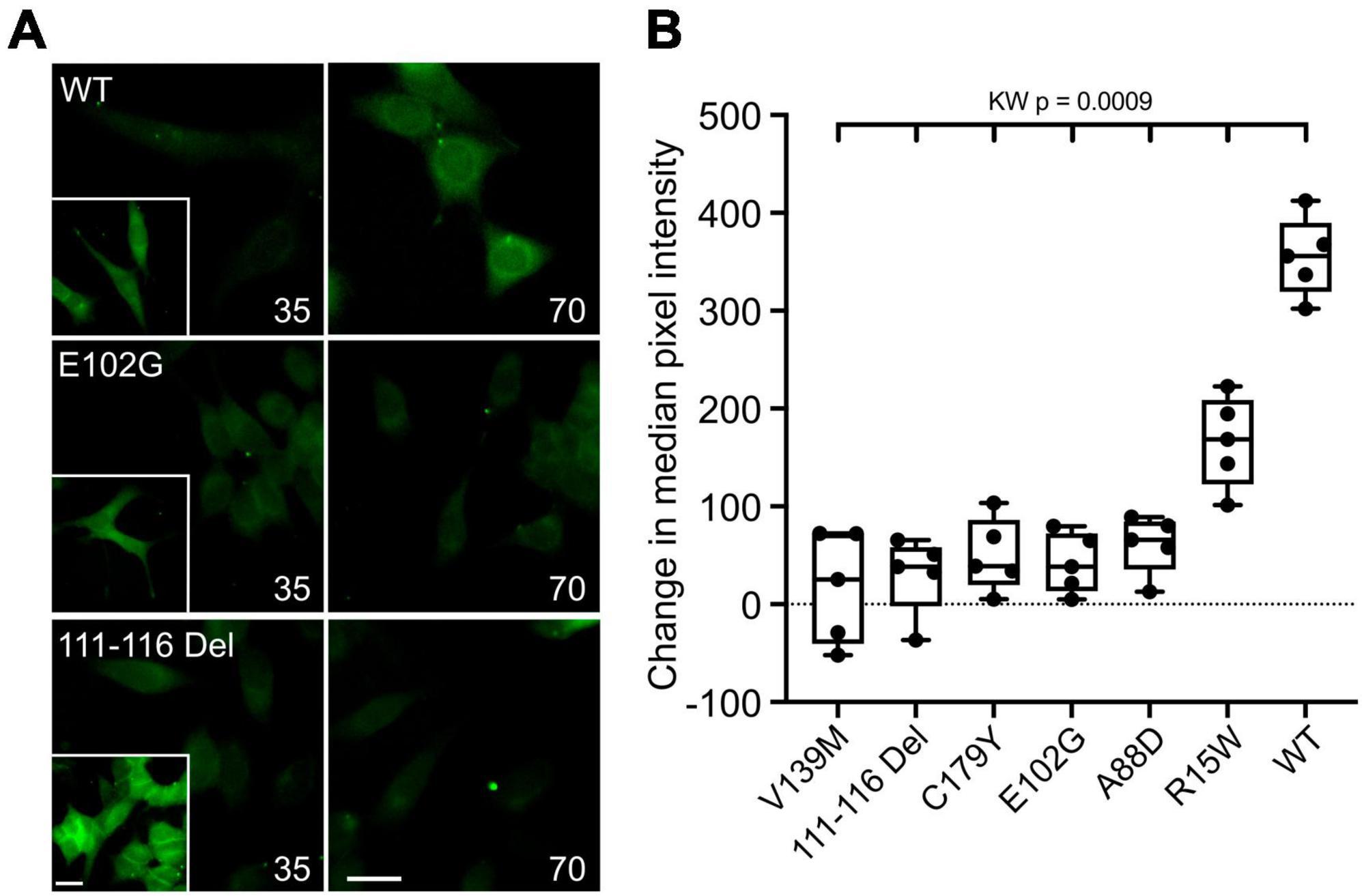
Figure 2. CMTX mutations in Cx32 can alter CO2-dependent dye loading. (A) Example images for HeLa cells expressing Cx32WT, Cx32E102G, and Cx32111–116 Del of dye loading at 35 and 70 mmHg. The insets are a positive control showing dye loading in response to removal of extracellular Ca2+. Scale bars 20 μm. (B) Summary data for the 6 CMTX mutations that completely abolished CO2-evoked Ca2+ signals in HeLa cells, ordered by median change from left to right. A Kruskal Wallis ANOVA shows that these are not drawn from the same population. All the mutations are significantly different from the WT. While 5 of the mutations cause a complete loss of CO2-dependent dye loading, the effect of R15W is only partial.
The gating of connexin hemichannels has several developmental and physiological roles e.g., (Weissman et al., 2004; Pearson et al., 2005; Huckstepp et al., 2010b; Moore et al., 2014; van de Wiel et al., 2020). In many instances, hemichannel opening permits release of ATP which then mediates these physiological effects via P2 receptors. We therefore examined whether CMTX mutations altered CO2-dependent release of ATP via Cx32 hemichannels measured by co-expression of GRABATP. As the mutations might themselves alter the permeability to ATP, we used membrane depolarization (50 mM K+) as a positive control to trigger hemichannel opening independently of changes in PCO2. As might be expected Cx32WT expressing HeLa cells released ATP in response to both CO2 and 50 mM K+ (Figures 3A, B). If HeLa cells were transfected only with GRABATP no release of ATP was evoked by either stimulus (Figures 3A, B). Six CMTX mutations (R15Q, V13L, G12S, W133R, L9F, and V84I) gave ATP release to 70 mmHg PCO2 that was not significantly different from Cx32WT (Figures 3A, B). However, the mutation L9F appeared to slightly reduce the voltage sensitivity of the hemichannel, as significantly less ATP was released by 50 mM KCl than Cx32WT (p = 0.0028, Figures 3A, B). By contrast, the mutations 111–116 Del, A88D, C179Y, E102G, and V139M completely abolished CO2 dependent ATP release (all p = 0.0001 compared to Cx32WT) but did not affect the release of ATP evoked by 50 mM K+, suggesting a selective abolition of CO2 sensitivity in these mutants (Figures 3C, D). The mutations L56F and R220Stop had apparently normal depolarization evoked ATP release (compared to Cx32WT), but reduced CO2 dependent release suggesting a partial effect of these mutations on CO2 sensitivity (p = 0.0001 and p = 0.0078 compared to Cx32WT, respectively, Figures 3C, D). The mutation R15W very greatly reduced both CO2- and depolarization-evoked ATP release compared to Cx32WT (p = 0.0001 and p < 0.0001, respectively, Figures 3C, D). The simplest interpretation is that permeability of the hemichannel to ATP release is greatly reduced but we cannot exclude additional effects of this mutation on voltage sensitivity or CO2 sensitivity, the latter being supported by the dye loading results.
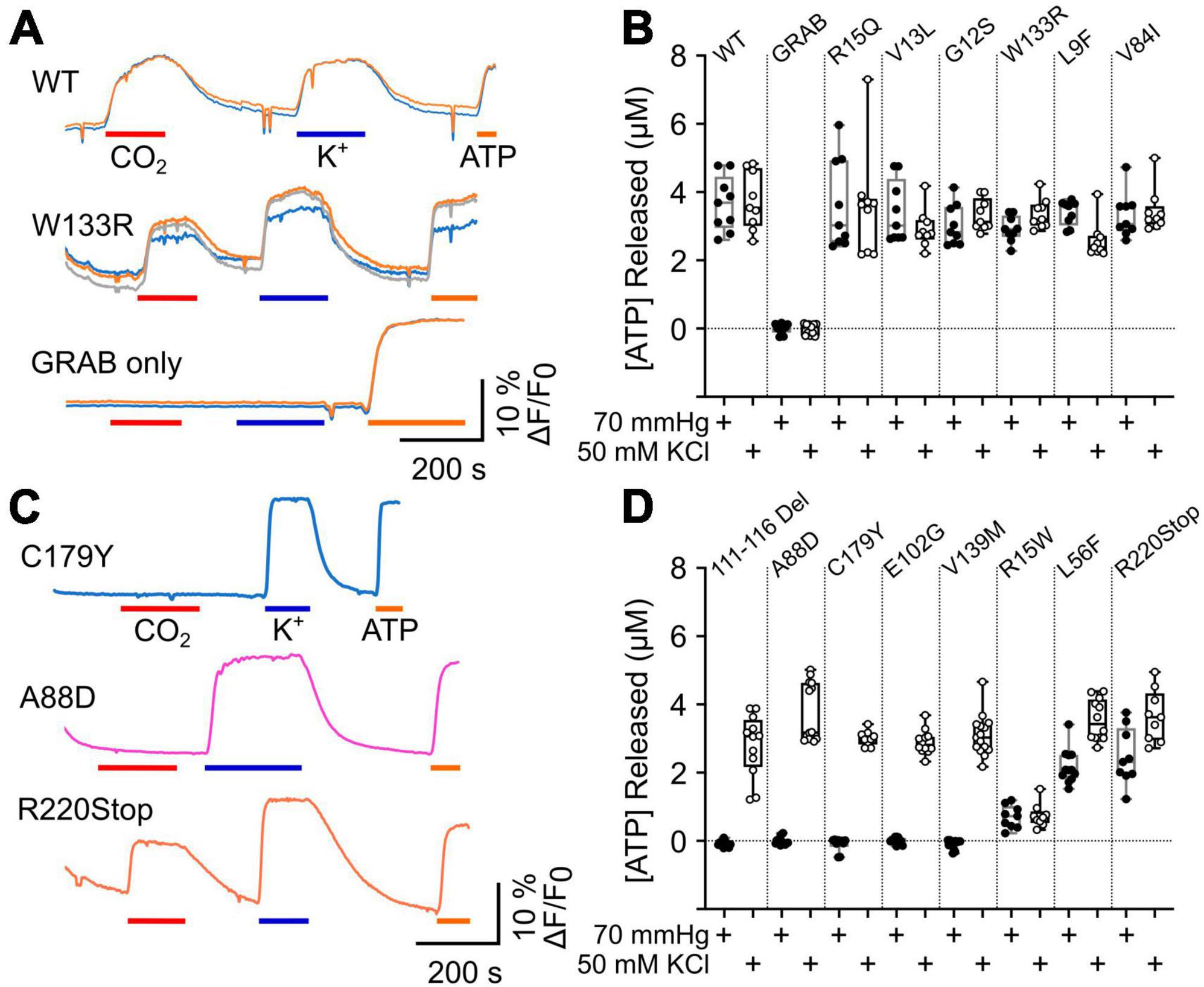
Figure 3. CMTX mutations in Cx32 can alter CO2 dependent ATP release. (A) Traces of changes in GRABATP fluorescence to 70 mmHg PCO2 (red bar), 50 mM KCl (blue bar) and 3 μM ATP (calibration, orange bar) for HeLa cells expressing Cx32WT, Cx32W133R, and just GRABATP. (B) Summary data for CMTX mutations that do not alter CO2 sensitivity of Cx32 hemichannels. (C) Traces of changes in GRABATP fluorescence to 70 mmHg PCO2 (red bar), 50 mM KCl (blue bar) and 3 μM ATP (calibration, orange bar) for HeLa cells expressing Cx32C179Y, Cx32A88D, and Cx32R220Stop. (D) Summary data for the CMTX mutations that alter CO2-dependent ATP release. Filled circles indicate ATP release to the CO2 stimulus, and open circles to the depolarizing stimulus in (B,D).
We also compared the amount of ATP released from the CO2 stimulus and the depolarizing stimulus for each variant of Cx32. For the WT, V13L, W133R, V84I, R15Q, and R15W the amount of ATP released by the two stimuli was not significantly different. As might be expected from causal inspection of Figure 3, for the mutations 111–116 Del (p = 0.0005), A88D (p = 0.0001), C179Y (p = 0.002), E102G (p = 0.0005), V139M (p < 0.0001), L56F (p = 0.0005), and R220XStop (p = 0.0391) CO2 triggered significantly less ATP release than the depolarizing stimulus. For the mutation G12S, which was difficult to express, CO2 also caused slightly less ATP release compared to depolarization (p = 0.0117) whereas for L9F, CO2 caused slightly more ATP release compared to depolarization (p = 0.0273).
Previously, we have generated a dominant negative subunit of Cx26 (dnCx26) by mutating the two residues involved in binding CO2 in that connexin (R104 and K125) (van de Wiel et al., 2020). As expected, dnCx26 is not sensitive to CO2. However, dnCx26 subunits can coassemble with those of WT Cx26 and remove CO2 sensitivity from the resulting heteromeric hexamer. We have shown that this is an effective tool in vivo to demonstrate the key role of Cx26 in respiratory chemosensing (van de Wiel et al., 2020). As the equivalent residues in Cx32 are K104 and K124, we, respectively, mutated them to Ala and Arg, to produce an equivalent dominant negative subunit for Cx32 (Cx32K104A,K124R, or dnCx32). Interestingly, individual mutations of K104 and K124 occur in patients with CMTX (Bone et al., 1997; Williams et al., 1999; Wang et al., 2015; Fattahi et al., 2017).
We first used the dye loading assay to confirm that homomeric assemblies of dnCx32 are insensitive to CO2 (Supplementary Figure 4). We then found that when transfected into HeLa cells that stably expressed Cx32, the dnCx32 subunit was able to act in a dominant manner to abolish CO2-dependent dye loading (Figures 4A, B). As with dnCx26, it required 6 days of culture post-transfection for the dominant negative effect to become fully apparent.
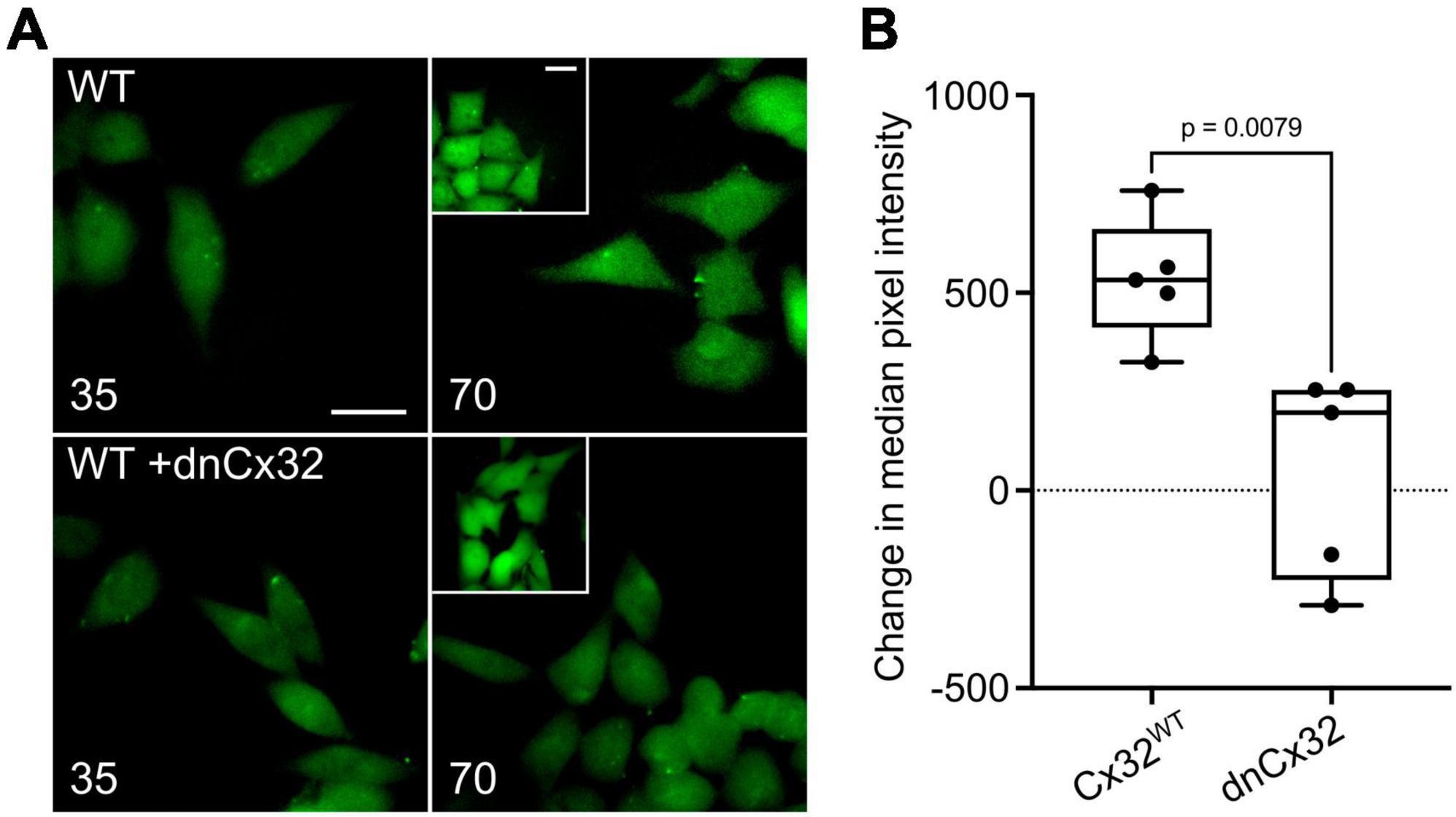
Figure 4. Characterization of a dominant negative Cx32 (dnCx32) that can remove CO2 sensitivity from Cx32WT. (A) Images of the HeLa cells stably expressing Cx32WT (WT) with and without transient transfection of dnCx32, showing the loading of dye in response to increasing PCO2 from 35 to 70 mmHg. The inset shows the positive control of dye loading to a zero Ca2+ stimulus. Note that the transfection of the cells with dnCx32 abolishes CO2 dependent dye loading. Scale bars 20 μm. (B) Summary graph of the change in median pixel intensity in response to CO2. A total of 5 independent repeats for both the Cx32WT and Cx32WT + dnCx32 cells. Mann Whitney U-test, p = 0.0079 WT Cx32 vs. WT Cx32 + dnCx32.
RT4 D6P2T Rat Schwannoma cells are a good model of Schwann cells and have previously been shown release ATP via the voltage-dependent opening of Cx32 hemichannels (Nualart-Marti et al., 2013). We therefore tested whether CO2 opened Cx32 hemichannels in this model system and whether this could be blocked by dnCx32 6 days after transfection.
Utilizing the dye-loading assay we demonstrated that the RT4 D6P2T cells robustly loaded with dye in response to both a zero Ca2+ challenge and an increase in PCO2 from 35 to 70 mmHg (Figure 5). However, transfection of the RT4 P6D2T cells with dnCx32 completely blocked their ability to load dyes in response to the CO2 challenge. Dye loading still occurred in response to the zero Ca2+ stimulus (Figure 5).
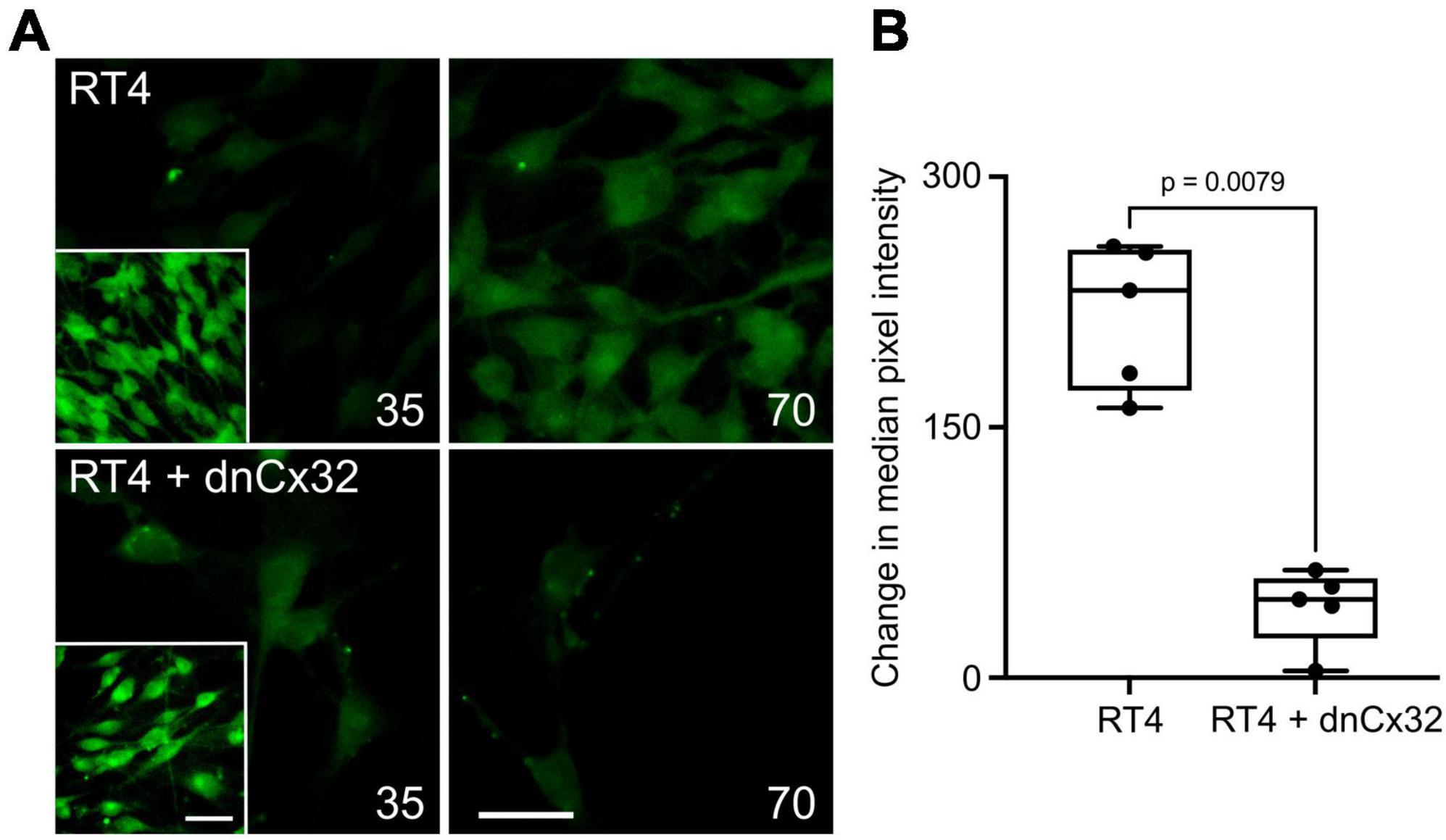
Figure 5. Cx32 mediates CO2-dependent dye loading in RT4 D6P2T Schwannoma cells. (A) Images of the Schwannoma cells loading with dye in response to increasing PCO2 from 35 to 70 mmHg. The inset shows the positive control of dye loading to a zero Ca2+ stimulus. Note that the transfection of the cells with dnCx32 abolishes CO2 dependent dye loading. Scale bars 20 μm. (B) Summary graph of the change in median pixel intensity in response to CO2. A total of 5 independent repeats for both the RT4 and RT4 + dnCx32 cells. Mann Whitney U-test, p = 0.0079 RT4 vs. RT4 + dnCx32.
We also used co-expression of GRABATP to measure ATP release from the RT4 D6P2T cells with and without expression of dnCx32 (Figure 6). In the parental RT4 D6P2T cells, 70 mmHg PCO2 and 50 mM KCl were equally effective at evoking ATP release (Figure 6). However, RT4 D6P2T cells that had been transfected with dnCx32 did not release ATP to the CO2 stimulus but displayed robust release of ATP to depolarization evoked by 50 mM KCl (Figure 6).
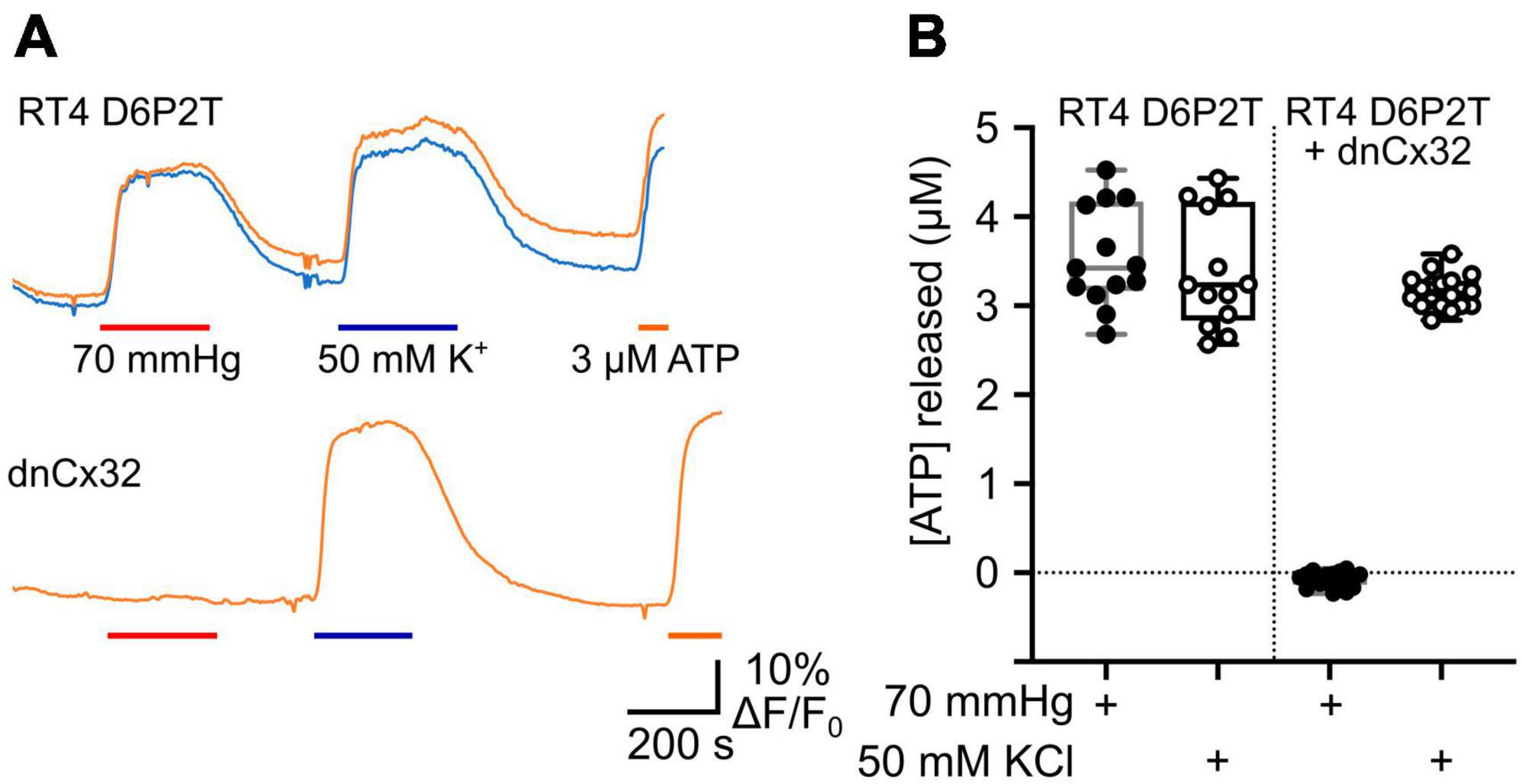
Figure 6. Cx32 mediates CO2-dependent ATP release from RT4 D6P2T Schwannoma cells. (A) Traces showing changes in GRABATP fluorescence in response to a change in PCO2 (red bar), 50 mM KCl (blue bar) and 3 μM ATP (orange bar) for parental RT4 D6P2T cells and RT4 D6P2T cells transfected with dnCx32. Both sets of cells were tested after 7 days in culture. (B) Summary data showing that dnCx32 completely abolishes CO2-dependent ATP release (filled circles) from the RT4 D6P2T cells but does not affect their ability to release ATP to a depolarizing stimulus (50 mM KCl, open circles).
As Cx32 is expressed on the X chromosome, only one copy of the gene is ever expressed in a cell. For males this is obviously because they have only one X chromosome. However, in females one X chromosome is inactivated. In somatic tissues the selection of the X chromosome for inactivation occurs at random in the stem cell population and is conserved for all the subsequent progeny of the original parental cell (Riggs and Pfeifer, 1992). Thus, females will have stochastic and chimeric expression of their two X chromosomes. CMTX is generally less severe in females presumably because, if they are heterozygous for a CMTX mutation, sufficient Schwann cells will still express the wild type allele (Scherer et al., 1998). This means that potential transdominant effects of CMTX mutations have only been occasionally studied (Omori et al., 1996; Jeng et al., 2006). Nevertheless, in the context of genetic therapies, where expression of an additional wild type allele will be potentially used to cure the disease, transdominant effects may influence the outcome of such an intervention. Given that some syndromic mutations of Cx26 remove CO2 sensitivity and have a transdominant effect on the WT Cx26 allele (Meigh et al., 2014; de Wolf et al., 2016), at least some CMTX mutations could plausibly have a similar effect on the CO2 sensitivity of the WT Cx32 allele.
We expressed Cx32 with the mutations V139M, 111–116 Del, C179Y, E103G, and A88D in the RT4 D6P2T cells, to see whether these mutated subunits could remove CO2 sensitivity from the endogenously expressed Cx32WT. At the same time, we co-expressed GRABATP to assay ATP release from these cells. We found that all 5 mutations completely prevented any CO2 dependent ATP release from the cells. However, in all 5 cases depolarization of the RT4 D6P2T cells with 50 mM K+ reliably evoked ATP release (Figure 7). This suggests that the mutant subunits co-assemble with Cx32WT to make a heteromeric hexamer that is insensitive to CO2.
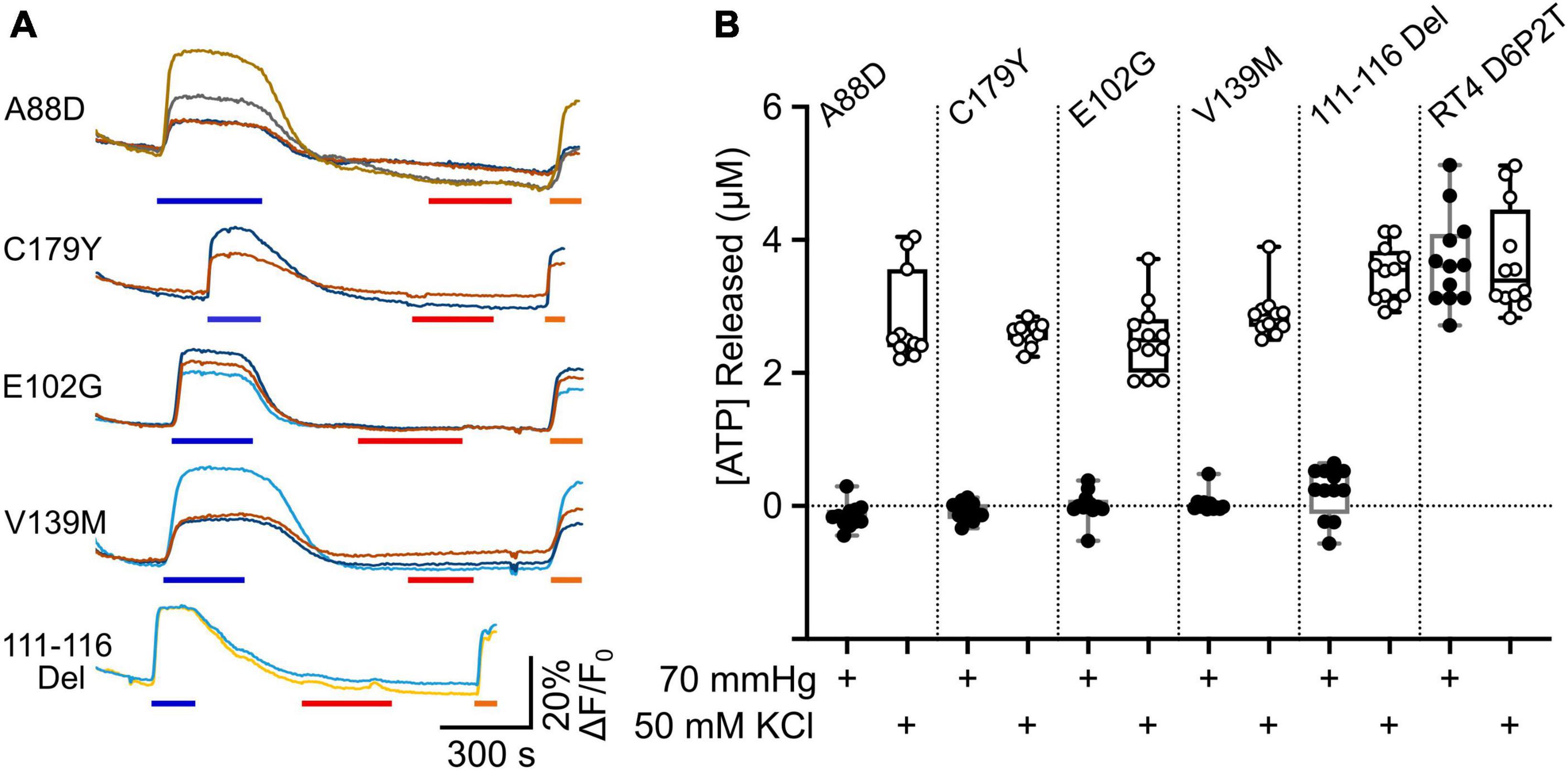
Figure 7. Transdominant effects of CMTX mutations on CO2 dependent ATP release from RT4 D6P2T cells. (A) Traces showing changes in GRABATP fluorescence to 50 mM KCl (blue bar), 70 mmHg PCO2 (red bar) and 3 μM ATP (orange bar) for RT4 D6P2T cells expressing Cx32 carrying CMTX mutations that remove CO2 sensitivity. (B) Summary data showing the transdominant effect of these CMTX mutations on CO2 evoked ATP release (filled circles). The RT4 D6P2T cells express Cx32WT which is able to release ATP both to depolarization (open circles) and an increase in PCO2 (see also Figure 6).
An alternative hypothesis is that expression of the mutant subunit suppresses expression of the endogenous wild type connexin. As our Cx32 antibody does not recognize the 111–116 Del mutant (Figure 8A), we stained RT4 D6P2T cells that coexpressed Cx32111–116 Del and found that even after 6 days in vitro Cx32WT was still expressed at levels that were indistinguishable the parental RT4 D6P2T cells and strongly colocalized with the mutant Cx32 as indicated by overlap of the immunofluorescence with that of the mCherry tag (Figures 8B, C). This supports our hypothesis that the transdominant action is exerted by coassembly of the mutant subunit into heteromeric hexamers.
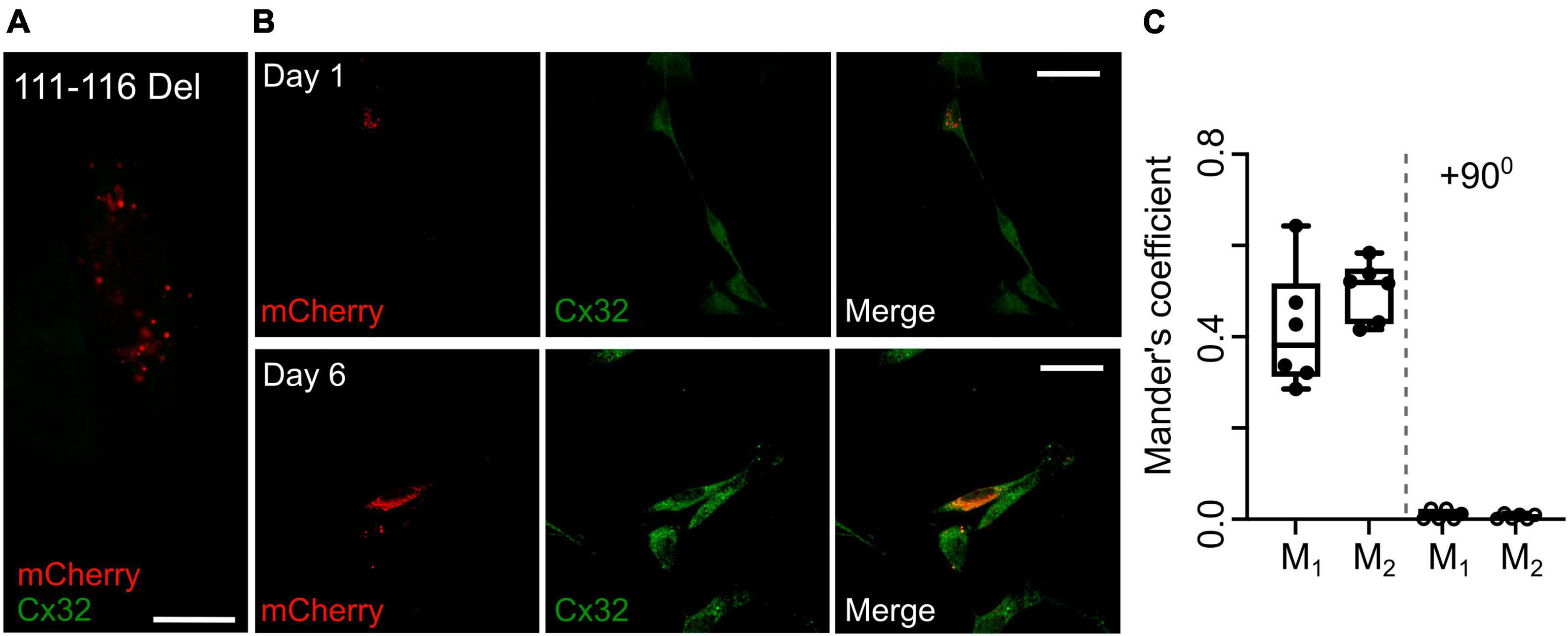
Figure 8. Expression of Cx32111–116 Del does not alter expression of Cx32WT in RT4 D6P2T cells. (A) Demonstration that the Cx32 antibody does not recognize Cx32111–116 Del. HeLa cells were transfected with this mutant tagged with mCherry and stained for Cx32: no staining is evident. Scale bar 15 μm. (B) Expression of Cx32111–116 Del in RT4 D6P2T cells imaged 1 and 6 days after transfection. Note how the mCherry fluorescence colocalizes with the Cx32WT expressed by these cells 6 days after transfection and visualized via the Cx32 immunostaining. (C) Analysis gives Mander’s coefficients that indicate substantial colocalization at day 6. The control was performed with one of the color channels rotated by 900 with respect to the other. Each dot represents analysis performed on one RT4 D6P2T cell.
The key result we report is that a series of CMTX mutations affecting different regions of Cx32 abolish or greatly reduce CO2-dependent opening of the hemichannel. We used three different assays of hemichannel function: Ca2+ imaging to measure influx through Cx32; dye loading of a membrane impermeant fluorescent dye; and ATP release measured via GRABATP. All three assays provide results that indicate that the mutations 111–116 Del, A88D, C179Y, E102G, and V139M completely abolish CO2 sensitivity of the hemichannel. The Ca2+ imaging and ATP release assays both indicate that L56F and R220Stop have a partial effect on CO2 sensitivity. The Ca2+ imaging assays suggest that V84I and V13L have a small effect on the CO2-dependent Ca2+ influx. As these mutations have no effect on CO2-dependent ATP release, we suggest that this small effect does not arise from an alteration of the CO2 sensitivity of the hemichannel but may indicate a small alteration of Ca2+ permeability or a statistical fluke from multiple comparisons even although these are corrected to a maximum false discovery rate of 0.05. Overall, the consensus of our data is that 8/14 CMTX mutations tested (including R15W, discussed below) reduce or abolish the sensitivity of Cx32 hemichannels to CO2. All constructs in this study were tagged at the C-terminus with mCherry to allow visualization of expression. This C-terminal tag is unlikely to alter channel function [e.g., the CO2 sensitivity of tagged Cx32 described here seems identical to that of untagged Cx32 stably expressed in HeLa cells (Huckstepp et al., 2010a)], but we cannot exclude it might modify trafficking properties of some of the mutants.
CO2 dependent opening of Cx32 may have some physiological importance as we find that Cx32 acts as a CO2 sensitive conduit for ATP release from RT4 D6P2T cells, a rat Schwannoma cell line. We also found that the CMTX mutations that abolish CO2 sensitivity have a transdominant effect on Cx32WT and abolish CO2 dependent ATP release from the Schwannoma cells.
The mutation R15W appears to have multiple effects on hemichannel properties: it alters Ca2+ permeability of the hemichannel (Figure 1) and most likely reduces ATP permeability (Figure 3). The dye loading data also suggest a substantial reduction in CO2 sensitivity as the fluorescent dye can still permeate the channel in the zero Ca2+ positive control for this assay (Figure 2 and Supplementary Figure 3). Our data cannot exclude that this mutation could also affect the voltage dependence of the hemichannel.
Mutations of Cx26 are the commonest cause of congenital non-syndromic sensorineural hearing loss. In addition, there are a small number of mutations that cause syndromic hearing loss. We have found that 4 of 9 mutations (A88V, N14K, N14Y, and A40V) that cause keratitis ichthyosis deafness syndrome abolish CO2 sensitivity of Cx26 gap junctions and hemichannels in a transdominant manner (Meigh et al., 2014; de Wolf et al., 2016; Cook et al., 2019; Nijjar et al., 2021). None of these mutations directly affect the CO2 sensing residues but are instead thought to decrease the flexibility of the molecule thereby preventing its reaction to changes in PCO2 (Brotherton et al., 2022). The effects on CO2 sensitivity for a range of CMTX mutations in Cx32 have striking similarities to the syndromic mutations we have studied in Cx26. Although the mutations are different, like Cx26 they do not affect the CO2 binding motif directly and they have transdominant effects on the CO2 sensitivity of Cx32WT. Given the similarity in sequence and structure of Cx32 to Cx26, it is plausible that the effects of the CMTX mutations on the CO2 sensitivity of Cx32 may also originate from the mutations restricting the flexibility of the molecule. It is important to note that the transdominant effects on CO2 sensitivity that we describe for some of the Cx32 mutations remain compatible with the strong evidence that shows CMTX being much less severe in females compared to males. As mentioned above, X chromosome inactivation ensures that in females each cell expresses genes from only one copy of the X chromosome (Gartler and Riggs, 1983; Lyon, 1988; Schwämmle and Schulz, 2023). For the same reason, as X chromosome inactivation occurs at random, in a nerve bundle of a heterozygous female (with one wild type and one mutant Cx32 allele) around half of the Schwann cells will express only the wild type gene and the remainder the mutant gene (Scherer et al., 1998). This presumably is enough to lessen the severity of CMTX in females compared to males carrying the same mutant (in whom, every Schwann cell will express the mutant gene).
As the symptoms of CMTX are recapitulated in Cx32-null mice and rescued by selective expression of Cx32 in Schwann cells, this progressive neuropathy is thought to arise from loss of Cx32 function. As 8/14 CMTX mutations tested cause partial or total loss of CO2 sensitivity, the question arises as to whether this could be a contributing mechanism to the etiology of CMTX. Furthermore, there are additional reports of CMTX-associated mutations affecting the CO2 sensing residues, K124 and K104 (Bone et al., 1997; Williams et al., 1999; Wang et al., 2015; Fattahi et al., 2017), which we predict (and indicated by dnCx32) would abolish CO2 sensitivity.
To examine whether there is any clinical evidence that supports loss of CO2 sensitivity as a contributor to CMTX, we have restricted our discussion to those mutations that are listed within the OMIM database3 as pathogenic and supported by evidence from multiple providers: R15Q, R15W, E102G, V139M, and R220Stop. Of these mutations R15W and V139M do not form gap junctions (Abrams et al., 2001). Our data show that R15W also lacks permeability to Ca2+ has greatly reduced permeability to ATP but may retain some reduced sensitivity to CO2. Given that these two mutations have multiple effects on the gap junction and hemichannel, they do not provide a test of whether loss of CO2 sensitivity is a sufficient contributor to CMTX. The mutations R15Q (Wang et al., 2004), E102G (Oh et al., 1997; Abrams et al., 2003) and R220Stop (Bicego et al., 2006) do not prevent gap junction formation. While R220Stop does cause a partial loss of CO2 sensitivity it also changes the sensitivity of Cx32 hemichannel-opening to intracellular Ca2+ (Carrer et al., 2018). Therefore, this mutation also cannot provide a test of our hypothesis. E102G stands out as a mutation that causes moderate severity CMTX while nevertheless forming gap junctions and, from the data reported here, having hemichannels with apparently normal voltage dependence and ATP permeability. Because E102G involves the loss of CO2 sensitivity of the hemichannel in the absence of other known functional effects on the hemichannel it lends some support to the hypothesis that the CO2 dependence of Cx32 may be important for the health of myelin. Set against this, is the fact that R15Q which has normal CO2 dependence, voltage dependence and ATP permeability and forms gap junctions, can cause CMTX. Almost certainly, there are likely to be several underlying mechanistic causes for CMTX, and loss of any one property of Cx32 may contribute to the origins of the neuropathy.
As CMTX is caused by lack of expression of functional Cx32 (e.g., defective trafficking mutants) or alterations of the properties of the mutant Cx32, gene therapy where a copy of the WT gene can be expressed to compensate for the missing function is a potential treatment under active development (Sargiannidou et al., 2015; Schiza et al., 2015; Kagiava et al., 2016, 2018, 2021a,2021b). In this context it is important to know whether the mutant copy of Cx32 could have transdominant effects as these will affect the success of the genetic therapy. Here we show that the CMTX mutations that abolish CO2 sensitivity also do this for RT4 D6P2T cells which express Cx32WT. If the CO2 sensitivity of Cx32 is a contributory factor in the development of CMTX, our data would suggest that expression of an additional copy of Cx32WT may be an ineffective genetic treatment for these mutations.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
JB: Conceptualization, Data curation, Investigation, Writing – review and editing. ND: Conceptualization, Supervision, Writing – original draft, Writing – review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) and University of Warwick funded Midlands Integrative Biosciences Training Partnership (MIBTP) grant number BB/T00746X/1.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2023.1330983/full#supplementary-material
Abrams, C. K., Freidin, M., Bukauskas, F., Dobrenis, K., Bargiello, T. A., Verselis, V. K., et al. (2003). Pathogenesis of X-linked Charcot-Marie-tooth disease: Differential effects of two mutations in connexin 32. J. Neurosci. 23, 10548–10558. doi: 10.1523/JNEUROSCI.23-33-10548.2003
Abrams, C. K., Freidin, M. M., Verselis, V. K., Bennett, M. V., and Bargiello, T. A. (2001). Functional alterations in gap junction channels formed by mutant forms of connexin 32: Evidence for loss of function as a pathogenic mechanism in the X-linked form of Charcot-Marie-tooth disease. Brain Res. 900, 9–25. doi: 10.1016/s0006-8993(00)03327-8
Abrams, C. K., Oh, S., Ri, Y., and Bargiello, T. A. (2000). Mutations in connexin 32: The molecular and biophysical bases for the X-linked form of Charcot-Marie-tooth disease. Brain Res. Rev. 32, 203–214. doi: 10.1016/s0165-0173(99)00082-x
Balice-Gordon, R. J., Bone, L. J., and Scherer, S. S. (1998). Functional gap junctions in the schwann cell myelin sheath. J. Cell Biol. 142, 1095–1104. doi: 10.1083/jcb.142.4.1095
Bergoffen, J., Scherer, S. S., Wang, S., Scott, M. O., Bone, L. J., Paul, D. L., et al. (1993). Connexin mutations in X-linked Charcot-Marie-tooth disease. Science 262, 2039–2042. doi: 10.1126/science.8266101
Bicego, M., Morassutto, S., Hernandez, V. H., Morgutti, M., Mammano, F., D’Andrea, P., et al. (2006). Selective defects in channel permeability associated with Cx32 mutations causing X-linked Charcot-Marie-tooth disease. Neurobiol. Dis. 21, 607–617. doi: 10.1016/j.nbd.2005.09.005
Bone, L. J., Deschenes, S. M., Balice-Gordon, R. J., Fischbeck, K. H., and Scherer, S. S. (1997). Connexin32 and X-linked Charcot-Marie-tooth disease. Neurobiol. Dis. 4, 221–230. doi: 10.1006/nbdi.1997.0152
Brotherton, D. H., Savva, C. G., Ragan, T. J., Dale, N., and Cameron, A. D. (2022). Conformational changes and CO2-induced channel gating in connexin26. Structure 30, 697–706. doi: 10.1016/j.str.2022.02.010
Brozková, D., Mazanec, R., Haberlová, J., Sakmaryová, I., Subrt, I., and Seeman, P. (2010). Six new gap junction beta 1 gene mutations and their phenotypic expression in Czech patients with Charcot-Marie-tooth disease. Genet. Test Mol. Biomarkers 14, 3–7. doi: 10.1089/gtmb.2009.0093
Carrer, A., Leparulo, A., Crispino, G., Ciubotaru, C. D., Marin, O., Zonta, F., et al. (2018). Cx32 hemichannel opening by cytosolic Ca2+ is inhibited by the R220X mutation that causes Charcot-Marie-tooth disease. Hum. Mol. Genet. 27, 80–94. doi: 10.1093/hmg/ddx386
Casasnovas, C., Banchs, I., Corral, J., Martínez-Matos, J. A., and Volpini, V. (2006). Clinical and molecular analysis of X-linked Charcot-Marie-tooth disease type 1 in Spanish population. Clin. Genet. 70, 516–523. doi: 10.1111/j.1399-0004.2006.00724.x
Cook, J., de Wolf, E., and Dale, N. (2019). Cx26 keratitis ichthyosis deafness syndrome mutations trigger alternative splicing of Cx26 to prevent expression and cause toxicity in vitro. R. Soc. Open Sci. 6:191128. doi: 10.1098/rsos.191128
Curran-Everett, D. (2000). Multiple comparisons: Philosophies and illustrations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1–R8.
de Wolf, E., Cook, J., and Dale, N. (2017). Evolutionary adaptation of the sensitivity of connexin26 hemichannels to CO2. Proc. R. Soc. B 284:20162723. doi: 10.1098/rspb.2016.2723
de Wolf, E., van de Wiel, J., Cook, J., and Dale, N. (2016). Altered CO2 sensitivity of connexin26 mutant hemichannels in vitro. Physiol. Rep. 4:e13038. doi: 10.14814/phy2.13038
Dospinescu, V.-M., Nijjar, S., Spanos, F., Cook, J., de Wolf, E., Biscotti, M. A., et al. (2019). Structural determinants of CO2-sensitivity in the β connexin family suggested by evolutionary analysis. Commun. Biol. 2:331. doi: 10.1038/s42003-019-0576-2
Fairweather, N., Bell, C., Cochrane, S., Chelly, J., Wang, S., Mostacciuolo, M. L., et al. (1994). Mutations in the connexin 32 gene in X-linked dominant Charcot-Marie-tooth disease (CMTX1). Hum. Mol. Genet. 3, 29–34. doi: 10.1093/hmg/3.1.29
Fattahi, Z., Kalhor, Z., Fadaee, M., Vazehan, R., Parsimehr, E., Abolhassani, A., et al. (2017). Improved diagnostic yield of neuromuscular disorders applying clinical exome sequencing in patients arising from a consanguineous population. Clin. Genet. 91, 386–402. doi: 10.1111/cge.12810
Fiori, M. C., Figueroa, V., Zoghbi, M. E., Saez, J. C., Reuss, L., and Altenberg, G. A. (2012). Permeation of calcium through purified connexin 26 hemichannels. J. Biol. Chem. 287, 40826–40834. doi: 10.1074/jbc.M112.383281
Gartler, S. M., and Riggs, A. D. (1983). Mammalian X-chromosome inactivation. Annu. Rev. Genet. 17, 155–190. doi: 10.1146/annurev.ge.17.120183.001103
Huckstepp, R. T., Eason, R., Sachdev, A., and Dale, N. (2010a). CO2-dependent opening of connexin 26 and related beta connexins. J. Physiol. 588(Pt 20), 3921–3931. doi: 10.1113/jphysiol.2010.192096
Huckstepp, R. T., id Bihi, R., Eason, R., Spyer, K. M., Dicke, N., Willecke, K., et al. (2010b). Connexin hemichannel-mediated CO2-dependent release of ATP in the medulla oblongata contributes to central respiratory chemosensitivity. J. Physiol. 588(Pt 20), 3901–3920. doi: 10.1113/jphysiol.2010.192088
Ionasescu, V., Searby, C., Ionasescu, R., and Meschino, W. (1995). New point mutations and deletions of the connexin 32 gene in x-linked charcot-marie-tooth neuropathy. Neuromuscul. Disord. 5, 297–299. doi: 10.1016/0960-8966(94)00077-M
Jeng, L. J., Balice-Gordon, R. J., Messing, A., Fischbeck, K. H., and Scherer, S. S. (2006). The effects of a dominant connexin32 mutant in myelinating Schwann cells. Mol. Cell Neurosci. 32, 283–298. doi: 10.1016/j.mcn.2006.05.001
Kagiava, A., Karaiskos, C., Richter, J., Tryfonos, C., Jennings, M. J., Heslegrave, A. J., et al. (2021a). AAV9-mediated Schwann cell-targeted gene therapy rescues a model of demyelinating neuropathy. Gene Ther. 28, 659–675. doi: 10.1038/s41434-021-00250-0
Kagiava, A., Richter, J., Tryfonos, C., Leal-Julià, M., Sargiannidou, I., Christodoulou, C., et al. (2021b). Efficacy of AAV serotypes to target Schwann cells after intrathecal and intravenous delivery. Sci. Rep. 11:23358. doi: 10.1038/s41598-021-02694-1
Kagiava, A., Karaiskos, C., Richter, J., Tryfonos, C., Lapathitis, G., Sargiannidou, I., et al. (2018). Intrathecal gene therapy in mouse models expressing CMT1X mutations. Hum. Mol. Genet. 27, 1460–1473. doi: 10.1093/hmg/ddy056
Kagiava, A., Sargiannidou, I., Theophilidis, G., Karaiskos, C., Richter, J., Bashiardes, S., et al. (2016). Intrathecal gene therapy rescues a model of demyelinating peripheral neuropathy. Proc. Natl. Acad. Sci. U.S.A. 113, E2421–E2429. doi: 10.1073/pnas.1522202113
Latour, P., Lévy, N., Paret, M., Chapon, F., Chazot, G., Clavelou, P., et al. (1997). Mutations in the X-linked form of Charcot-Marie-tooth disease in the French population. Neurogenetics 1, 117–123. doi: 10.1007/s100480050017
Lu, Y. Y., Lyu, H., Jin, S. Q., Zuo, Y. H., Liu, J., Wang, Z. X., et al. (2017). Clinical and genetic features of Chinese X-linked Charcot-Marie-tooth type 1 disease. Chin. Med. J. 130, 1049–1054. doi: 10.4103/0366-6999.204925
Lyon, M. F. (1988). The william allan memorial award address: X-chromosome inactivation and the location and expression of X-linked genes. Am. J. Hum. Genet. 42, 8–16.
Meigh, L., Greenhalgh, S. A., Rodgers, T. L., Cann, M. J., Roper, D. I., and Dale, N. (2013). CO2 directly modulates connexin 26 by formation of carbamate bridges between subunits. eLife 2:e01213. doi: 10.7554/eLife.01213
Meigh, L., Hussain, N., Mulkey, D. K., and Dale, N. (2014). Connexin26 hemichannels with a mutation that causes KID syndrome in humans lack sensitivity to CO2. eLife 3:e04249. doi: 10.7554/eLife.04249
Moore, A. R., Zhou, W. L., Sirois, C. L., Belinsky, G. S., Zecevic, N., and Antic, S. D. (2014). Connexin hemichannels contribute to spontaneous electrical activity in the human fetal cortex. Proc. Natl. Acad. Sci. U.S.A. 111, E3919–E3928. doi: 10.1073/pnas.1405253111
Murakami, T., Garcia, C. A., Reiter, L. T., and Lupski, J. R. (1996). Charcot-Marie-tooth disease and related inherited neuropathies. Medicine 75, 233–250. doi: 10.1097/00005792-199609000-00001
Nijjar, S., Maddison, D., Meigh, L., de Wolf, E., Rodgers, T., Cann, M. J., et al. (2021). Opposing modulation of Cx26 gap junctions and hemichannels by CO2. J. Physiol. 599, 103–118. doi: 10.1113/JP280747
Nualart-Marti, A., del Molino, E. M., Grandes, X., Bahima, L., Martin-Satue, M., Puchal, R., et al. (2013). Role of connexin 32 hemichannels in the release of ATP from peripheral nerves. Glia 61, 1976–1989. doi: 10.1002/glia.22568
Oh, S., Ri, Y., Bennett, M. V., Trexler, E. B., Verselis, V. K., and Bargiello, T. A. (1997). Changes in permeability caused by connexin 32 mutations underlie X-linked Charcot-Marie-tooth disease. Neuron 19, 927–938. doi: 10.1016/s0896-6273(00)80973-3
Omori, Y., Mesnil, M., and Yamasaki, H. (1996). Connexin 32 mutations from X-linked Charcot-Marie-tooth disease patients: Functional defects and dominant negative effects. Mol. Biol. Cell 7, 907–916. doi: 10.1091/mbc.7.6.907
Pearson, R. A., Dale, N., Llaudet, E., and Mobbs, P. (2005). ATP released via gap junction hemichannels from the pigment epithelium regulates neural retinal progenitor proliferation. Neuron 46, 731–744. doi: 10.1016/j.neuron.2005.04.024
Record, C. J., Skorupinska, M., Laura, M., Rossor, A. M., Pareyson, D., Pisciotta, C., et al. (2023). Genetic analysis and natural history of Charcot-Marie-tooth disease CMTX1 due to GJB1 variants. Brain 146, 4336–4349. doi: 10.1093/brain/awad187
Ressot, C., Gomes, D., Dautigny, A., Pham-Dinh, D., and Bruzzone, R. (1998). Connexin32 mutations associated with X-linked Charcot-Marie-tooth disease show two distinct behaviors: Loss of function and altered gating properties. J. Neurosci. 18, 4063–4075. doi: 10.1523/JNEUROSCI.18-11-04063.1998
Riggs, A. D., and Pfeifer, G. P. (1992). X-chromosome inactivation and cell memory. Trends Genet. 8, 169–174. doi: 10.1016/0168-9525(92)90219-t
Rouger, H., LeGuern, E., Birouk, N., Gouider, R., Tardieu, S., Plassart, E., et al. (1997). Charcot-Marie-tooth disease with intermediate motor nerve conduction velocities: Characterization of 14 Cx32 mutations in 35 families. Hum. Mutat. 10, 443–452. doi: 10.1002/(sici)1098-1004199710:6<443:Aid-humu5<3.0.Co;2-e
Sargiannidou, I., Kagiava, A., Bashiardes, S., Richter, J., Christodoulou, C., Scherer, S. S., et al. (2015). Intraneural GJB1 gene delivery improves nerve pathology in a model of X-linked Charcot-Marie-tooth disease. Ann. Neurol. 78, 303–316. doi: 10.1002/ana.24441
Sargiannidou, I., Vavlitou, N., Aristodemou, S., Hadjisavvas, A., Kyriacou, K., Scherer, S. S., et al. (2009). Connexin32 mutations cause loss of function in Schwann cells and oligodendrocytes leading to PNS and CNS myelination defects. J. Neurosci. 29, 4736–4749. doi: 10.1523/jneurosci.0325-09.2009
Scherer, S. S., Xu, Y. T., Messing, A., Willecke, K., Fischbeck, K. H., and Jeng, L. J. (2005). Transgenic expression of human connexin32 in myelinating Schwann cells prevents demyelination in connexin32-null mice. J. Neurosci. 25, 1550–1559. doi: 10.1523/JNEUROSCI.3082-04.2005
Scherer, S. S., Xu, Y. T., Nelles, E., Fischbeck, K., Willecke, K., and Bone, L. J. (1998). Connexin32-null mice develop demyelinating peripheral neuropathy. Glia 24, 8–20. doi: 10.1002/(sici)1098-1136(199809)24:1<8:aid-glia2<3.0.co;2-3
Schiza, N., Sargiannidou, I., Kagiava, A., Karaiskos, C., Nearchou, M., and Kleopa, K. A. (2015). Transgenic replacement of Cx32 in gap junction-deficient oligodendrocytes rescues the phenotype of a hypomyelinating leukodystrophy model. Hum. Mol. Genet. 24, 2049–2064. doi: 10.1093/hmg/ddu725
Schwämmle, T., and Schulz, E. G. (2023). Regulatory principles and mechanisms governing the onset of random X-chromosome inactivation. Curr. Opin. Genet. Dev. 81:102063. doi: 10.1016/j.gde.2023.102063
Shy, M. E., Siskind, C., Swan, E. R., Krajewski, K. M., Doherty, T., Fuerst, D. R., et al. (2007). CMT1X phenotypes represent loss of GJB1 gene function. Neurology 68, 849–855. doi: 10.1212/01.wnl.0000256709.08271.4d
van de Wiel, J., Meigh, L., Bhandare, A., Cook, J., Nijjar, S., Huckstepp, R., et al. (2020). Connexin26 mediates CO2-dependent regulation of breathing via glial cells of the medulla oblongata. Commun. Biol. 3:521. doi: 10.1038/s42003-020-01248-x
Wang, H. L., Chang, W. T., Yeh, T. H., Wu, T., Chen, M. S., and Wu, C. Y. (2004). Functional analysis of connexin-32 mutants associated with X-linked dominant Charcot-Marie-tooth disease. Neurobiol. Dis. 15, 361–370. doi: 10.1016/j.nbd.2003.11.005
Wang, R., He, J., Li, J. J., Ni, W., Wu, Z. Y., Chen, W. J., et al. (2015). Clinical and genetic spectra in a series of Chinese patients with Charcot-Marie-tooth disease. Clin. Chim. Acta 451(Pt B), 263–270. doi: 10.1016/j.cca.2015.10.007
Weissman, T. A., Riquelme, P. A., Ivic, L., Flint, A. C., and Kriegstein, A. R. (2004). Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron 43, 647–661. doi: 10.1016/j.neuron.2004.08.015
Keywords: connexin, Schwann cell, myelin, CMTX, neuropathy, hemichannel
Citation: Butler J and Dale N (2023) X-linked Charcot Marie Tooth mutations alter CO2 sensitivity of connexin32 hemichannels. Front. Cell. Neurosci. 17:1330983. doi: 10.3389/fncel.2023.1330983
Received: 31 October 2023; Accepted: 07 December 2023;
Published: 21 December 2023.
Edited by:
Zhuofan Lei, University of Maryland, United StatesReviewed by:
Shweta Pradip Jadhav, Consultant, Carlsbad, CA, United StatesCopyright © 2023 Butler and Dale. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas Dale, bi5lLmRhbGVAd2Fyd2ljay5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.