
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 14 December 2023
Sec. Cellular Neuropathology
Volume 17 - 2023 | https://doi.org/10.3389/fncel.2023.1295840
This article is part of the Research Topic Crosstalk between Peripheral and Local Immune Response in the Pathophysiology of Stroke and Neurodegeneration Diseases, Volume II View all 10 articles
 Estrid Thougaard1,2
Estrid Thougaard1,2 Brianna Carney3
Brianna Carney3 Agnieszka Wlodarczyk1,2
Agnieszka Wlodarczyk1,2 Roberta Brambilla1,2,3*†
Roberta Brambilla1,2,3*† Kate Lykke Lambertsen1,2,4*†
Kate Lykke Lambertsen1,2,4*†In central nervous system (CNS) injury and disease, peripherally derived myeloid cells infiltrate the CNS parenchyma and interact with resident cells, propagating the neuroinflammatory response. Because peripheral myeloid populations differ profoundly depending on the type and phase of injury, their crosstalk with CNS resident cells, particularly microglia, will lead to different functional outcomes. Thus, understanding how peripheral myeloid cells affect the phenotype and function of microglia in different disease conditions and phases may lead to a better understanding of disease-specific targetable pathways for neuroprotection and neurorepair. To this end, we set out to develop an in vitro system to investigate the communication between peripheral myeloid cells and microglia, with the goal of uncovering potential differences due to disease type and timing. We isolated peripheral myeloid cells from mice undergoing experimental autoimmune encephalomyelitis (EAE), a model of multiple sclerosis, or acute cerebral ischemia by permanent middle cerebral artery occlusion (pMCAO) at different times after disease and probed their ability to change the phenotype of primary microglia isolated from the brain of adult mice. We identified changes not only dependent on the disease model, but also on the timepoint after disease onset from which the myeloid cells were isolated. Peripheral myeloid cells from acute EAE induced morphological changes in microglia, followed by increases in expression of genes involved in inflammatory signaling. Conversely, it was the peripheral myeloid cells from the chronic phase of pMCAO that induced gene expression changes in genes involved in inflammatory signaling and phagocytosis, which was not followed by a change in morphology. This underscores the importance of understanding the role of infiltrating myeloid cells in different disease contexts and phases. Furthermore, we showed that our assay is a valuable tool for investigating myeloid cell interactions in a range of CNS neuroinflammatory conditions.
Diseases of the central nervous system (CNS) are etiopathologically diverse, ranging from acute disorders such as traumatic brain injury and stroke, to chronic neurodegenerative diseases like multiple sclerosis (MS) and Alzheimer’s. Because an immune-inflammatory response is common to virtually all of them (Lucas et al., 2006), targeting specific immune processes has been pursued for the treatment of neurological diseases (Correale et al., 2017; Lambertsen et al., 2019; Ruiz et al., 2019; Iadecola et al., 2020).
Cells of the myeloid lineage play an important role in the response to CNS injury and disease (Brendecke and Prinz, 2015). Microglia are the CNS-resident myeloid cells and are key regulators of neural homeostasis (Nayak et al., 2014). They are also a crucial component of the response to injury/disease and, in disease states, they undergo profound phenotypic changes signaling their activation. These include morphological changes, such as the transition to an amoeboid shape, and transcriptional changes that lead to increased expression and release of cytokines and chemokines (DiSabato et al., 2016; Becher et al., 2017). The advent of single cell omics has made it clear that microglial responses are highly heterogeneous and vary in time and space depending on the type, severity, and localization of injury/disease within the CNS. This has led to the identification of multiple coexisting microglial phenotypes with distinct and often opposite functions (Zheng et al., 2022; Vainchtein et al., 2023).
In MS, microglia can have both detrimental and protective effects (Rawji and Yong, 2013; Guerrero and Sicotte, 2020; Plastini et al., 2020). They are involved in demyelination (Zrzavy et al., 2017; Savarin et al., 2018), and in the experimental autoimmune encephalomyelitis (EAE) model, the early depletion of microglia or reduction of their activation improves the outcome (Goldmann et al., 2013; Nissen et al., 2018). Microglia can also be protective in EAE through production of anti-inflammatory cytokines and clearance of myelin debris (Kocur et al., 2015; Wlodarczyk et al., 2015). Depletion of microglia at late timepoints after EAE exacerbates inflammation and demyelination (Tanabe et al., 2019), indicating a finely tuned microglial function that differs according to the disease phase.
In acute neurological disease, such as ischemic stroke, microglia are the first cells to be activated and, as in MS, they have dual functions (Ma et al., 2017; Qin et al., 2019). Microglia release pro-inflammatory cytokines after experimental stroke, leading to disruption of the blood-brain-barrier (Clausen et al., 2008; Jolivel et al., 2015), synapse elimination, and neuronal death (Neher et al., 2013; Shi et al., 2021). Reducing the number of activated microglia increases the number of new neurons and improves functional outcome (Liu et al., 2007). Microglia can also limit the ischemic damage by reducing the size of the ischemic lesion, thus improving the outcome (Lalancette-Hébert et al., 2007; Lambertsen et al., 2009; Szalay et al., 2016), and the phagocytic properties of microglia are important in the recovery post-stroke (Kawabori et al., 2015).
Peripheral immune cells of the myeloid lineage are the first to infiltrate the CNS in neurological disease. Neutrophils are present in the inflamed areas shortly after the onset of ischemic stroke and EAE (Lewis et al., 2014; Beuker et al., 2021) and have been proposed as a biomarker for MS (Bisgaard et al., 2017). Macrophages also infiltrate the ischemic brain in large numbers early after stroke (Beuker et al., 2021), and in EAE, where T-cells are required for the induction of disease, monocytes were shown to be necessary for disease progression (Ajami et al., 2011). When participating in the neuroinflammatory response, peripheral myeloid cells carry out many of the same functions as their CNS-resident counterpart, microglia. However, their disease-induced phenotypic changes may occur with different timing and localization, leading to distinct and sometimes opposing functions (Ajami et al., 2011; Giles et al., 2018; Werner et al., 2020; Gao et al., 2017, 2023). Peripherally derived myeloid cells interact with CNS cells driving the acute neuroinflammatory response but can also persist long-term in the CNS parenchyma participating in the chronic neuroinflammatory response (Garcia-Bonilla et al., 2016), where they sustain various processes (Brendecke and Prinz, 2015; Giles et al., 2018; Lambertsen et al., 2019).
Because of the co-existence of microglia and peripherally derived myeloid cells in CNS disease, it is important to understand how their cross-talk might affect the outcome in different CNS pathologies (degenerative vs. traumatic) and disease stages (acute vs. chronic). The complexity and plurality of CNS cellular networks make it challenging to study discrete cell-cell interactions in vivo, and even more so under inflammatory conditions. A further obstacle is the functional and transcriptional similarities of the two populations, making it difficult to distinguish them in vivo. To overcome these challenges, we developed an in vitro system to investigate the communication between peripheral myeloid cells and microglia, with the goal of uncovering potential differences due to disease type and timing.
Adult female C57Bl/6 mice (2 months old) were used for this study. The animals were kept in group housing under controlled temperature and humidity, a 12/12-h light/dark cycle, and with food and water ad libitum. The experiments carried out at the Animal Core Facility of The Miami Project to Cure Paralysis were performed according to protocols and guidelines approved by the Institutional Animal Care and Use Committee of the University of Miami. The experiments conducted at the Animal Facility of the University of Southern Denmark were performed in accordance with approved permits (J. no. 2019-15-0201-01620).
The distal part of the left middle cerebral artery (MCA) was permanently coagulated to induce an experimental stroke, as previously described (Lambertsen et al., 2009; Yli-Karjanmaa et al., 2019). The surgery was performed under anesthesia with a mix of Hypnorm [fentanyl citrate (0.135 mg/mL, VetaPharma) and fluanisone (10 mg/mL, VetaPharma)], Midazolam (5 mg/mL, Hameln), and sterile water in the ratio 1:1:2. When the mouse was fully sedated, the skin was incised from the eye to the ear on the left side, the muscles were dissected to expose the skull, and a small hole was drilled above the MCA. The MCA was coagulated by pinching it with bipolar forceps and administering an electric current, hereby inducing a permanent middle cerebral artery occlusion (pMCAO). Mice were given subcutaneous injections of analgesics [Temgesic (0.001/20 mg body weight buprenorphine; Indivior)] every 8 h for 24 h, starting at the time of surgery, while being kept in a temperature-controlled heating cabinet. Eyes were coated in Viscotears ointment (2 mg/g, Bausch and Lomb) to prevent dehydration. The mice were euthanized at acute disease (1 day post-injury, dpi) or chronic disease (14 dpi) for isolation of peripheral myeloid cells, using an intraperitoneal injection of 0.2 mL pentobarbital (200 mg/mL) containing lidocaine (10 mg/mL) followed by transcardial perfusion with 20 ml phosphate-buffered saline (PBS).
Mice were induced with experimental autoimmune encephalomyelitis (EAE) following a protocol well established in the laboratory (Brambilla et al., 2011; Madsen et al., 2020). Briefly, on day 0 and 2, pertussis toxin (PTX, List Biological Laboratories, #181) diluted in PBS was injected intraperitoneally (500 μg/mouse). On day 1, myelin oligodendrocyte glycoprotein peptide 35–55 (MOG35–55, Bio-Synthesis, #A9349-1) emulsified in complete Freund’s adjuvant was injected subcutaneously (0.33 mg/mouse). Disease development was tracked daily, and locomotor dysfunction was scored on a standard 0–6 scale where 0 = no symptoms; 1 = loss of tail tone; 2 = fully flaccid tail; 3 = complete hind limb paralysis; 4 = complete forelimb paralysis; 5 = moribund; 6 = dead. Mice were euthanized at acute disease (20 days post-induction, dpi) or chronic disease (30 dpi) for isolation of peripheral myeloid cells. Inclusion criterion for the acute timepoint was presence of symptoms for a maximum of 7 days. Inclusion criterion for the chronic timepoint was presence of symptoms for a minimum of 17 days.
Primary microglia cultures were generated from whole brains of naïve adult (2 months old) C57Bl/6 mice. Following transcardial perfusion with PBS, brains were dissected out and enzymatically dissociated into a single-cell suspension using Neural Tissue Dissociation Kit (P) (Miltenyi Biotec, #130-092-628) followed by myelin removal with debris removal solution (Miltenyi Biotec, #130-109-398). Microglia were then isolated by magnetic-activated cell sorting (MACS) with LS columns (Miltenyi Biotec, #130-042-401) after incubation with anti-CD11b conjugated magnetic microbeads (Miltenyi Biotec, #130-093-634) according to the manufacturer’s protocol. Cells were seeded on poly-D-lysine coated plates (Sigma-Aldrich, #P7280) and maintained in complete medium consisting of RPMI 1640 Medium (Gibco, A1049101) supplemented with 20% L929 fibroblast conditioned media, 10% fetal bovine serum (FBS) (GEMINI, #900-108), 50 μM beta-mercaptoethanol, and 1% antibiotic-antimycotic (Gibco, #15240-062). Cells were plated in 24-well plates at a density of 37,000 cells/cm2 for RNA isolation, and in 24-well plates (indirect co-cultures) or 96-well plates (direct co-cultures) at a density of 16,000 cells/cm2 for morphological analysis. Microglia were cultured for 5 days with a partial media change (50%) every other day.
The primary microglia culture was assessed for purity by immunohistochemical staining and cell counting. Paraformaldehyde (PFA) fixed cells were incubated with goat anti-Iba1 (1:200, Novus Biologicals, #NB100-1028), rat anti-glial fibrillary acidic protein (GFAP) (1:500, Invitrogen, #13-0300) and rabbit anti-oligodendrocyte transcription factor 2 (Olig2) (1:200, Sigma-Aldrich, #AB9610) overnight at 4°C, followed by washing and subsequent 1-h incubation with secondary antibodies (1:750, Thermo Fisher, Donkey anti-goat 594 #A-11058; Donkey anti-rat 488 #A-21208; Donkey anti-rabbit 647 #A-31573). The cells were washed, and nuclei stained with 4′,6-diamidino-2-phenylindole (DAPI) (1:6000, Invitrogen, #D1306). Seventeen (17) randomly selected fields of view from 6 individual cell culture wells were imaged at 20X magnification, and the percentage of Iba1+ microglia, GFAP+ astrocytes, Olig2+ oligodendrocytes and Iba1–GFAP–Olig2– cells out of the total number of DAPI+ cells were estimated using ImageJ software.
Since secondary lymphoid organs, such as the spleen, coordinate the immune response in immune-mediated pathologies such as MS and EAE, (Zhu et al., 2007), while in ischemic stroke the direct output of myeloid cells from the bone marrow drives the peripheral innate immune response (Courties et al., 2015), we isolated myeloid cells from the spleen of EAE induced mice and from the bone marrow of pMCAO injured mice at acute and chronic disease phases. After perfusion with PBS, spleen or bone marrow was isolated and manually dissociated into single cell suspensions followed by red blood cell lysis. Myeloid cells were isolated by magnetic bead cell separation with anti-F4/80 MicroBeads (Miltenyi Biotec, #130-110-443) and LS columns (Miltenyi Biotec, #130-042-401) according to the manufacturer’s protocol. For indirect co-cultures, 100,000 myeloid cells in 1 ml media (density 100 cells/μl) were plated in cell culture inserts (cellQART 24-well Cell Culture Insert, Sterlitech, #9320402), which were then placed on top of naïve microglia that were maintained in culture for 5 days. For direct co-cultures, myeloid cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) (2 μM dilution in PBS, BioLegend, #423801) for 10 min at 37°C. Then 50,000 myeloid cells in 500 μl media were plated in the 24-well plates and 10,000 myeloid cells in 100 μl media were plated in the 96-well plates (density 100 cells/μl) directly on top of naïve microglia maintained in culture for 5 days. Naïve unstimulated microglia without myeloid cells were used as controls. Three-five (3–5) biological replicates/condition with 2–6 technical replicates for gene expression analysis and 1–8 technical replicates for morphological analysis were plated.
After 24 h in co-culture, microglia (either alone or mixed with peripheral myeloid cells) were fixed with 4% PFA, washed, and stained with CellMask Deep Red Plasma Membrane Stain (1:5000, Invitrogen, #C10046) and DAPI (1:2000, Invitrogen, #D1306). Cells were imaged with the Opera Phenix Plus High Content Screening System (Perkin Elmer), and micrographs were analyzed with Harmony® software package. Multiple filters were applied to eliminate artifacts using empirically determined cutoffs. For the purpose of this study, we extracted the following parameters: area, perimeter, roundness, length/width ratio, and percentage of elongated cells (with length/width ratio above 3). Within each well, multiple fields of view were automatedly sampled, and the average value per well was calculated for each parameter. For direct co-cultures, the algorithm was instructed not to include CFSE-labeled cells (peripheral myeloid cells) in the analysis. Morphological data were calculated as the mean of 1–8 technical replicates per mouse. To account for variability between cultures, the results were normalized to internal unstimulated microglia controls, before being calculated as fold change of the acute timepoint with direct exposure.
Direct co-cultures underwent fluorescence-activated cell sorting (FACS) to separate microglia from peripheral myeloid cells after 24 h in co-culture. Briefly, cells were lifted using a cell scraper, spun down, and resuspended in DMEM-F12 (Gibco, #11320-033) supplemented with 5% FBS (GEMINI, #900-108). Cell suspensions were passed through a strainer to ensure single cell separation, and DAPI was added to label dead cells (NucBlue Fixed Cell Stain ReadyProbes reagent, Invitrogen, #R37606). Live DAPI–CFSE– microglia were sorted directly into RNA extraction buffer from the Arcturus PicoPure RNA Isolation Kit (Applied Biosystems, #12204-01). Microglia from indirect co-cultures after 24 h in co-culture and unstimulated microglia controls were collected as described above and immediately resuspended in RNA extraction buffer from the Arcturus PicoPure RNA Isolation Kit. In all experiments, 2–6 technical replicates were pooled to obtain 3–5 samples/condition.
Microglial RNA was isolated with the Arcturus PicoPure RNA Isolation Kit (Applied Biosystems, #12204-01). RNA concentration and purity were measured using a 2100 Agilent Bioanalyzer. RNA was reverse-transcribed to cDNA with the Sensiscript RT Kit (Qiagen, #205211) together with random primers (Promega, #C1181). Semiquantitative RT-PCR was performed with PowerUp SYBR Green Master Mix (Applied Biosystems, #A25742) and specific primers for the genes of interest (Table 1), according to the manufacturer’s instructions. cDNA samples from the pMCAO co-cultures underwent pre-amplification (14 cycles at 57°C) due to low concentrations. The PCR data were normalized to Gapdh expression before being calculated as fold change of unstimulated naïve microglia using the deltadeltaCt method.
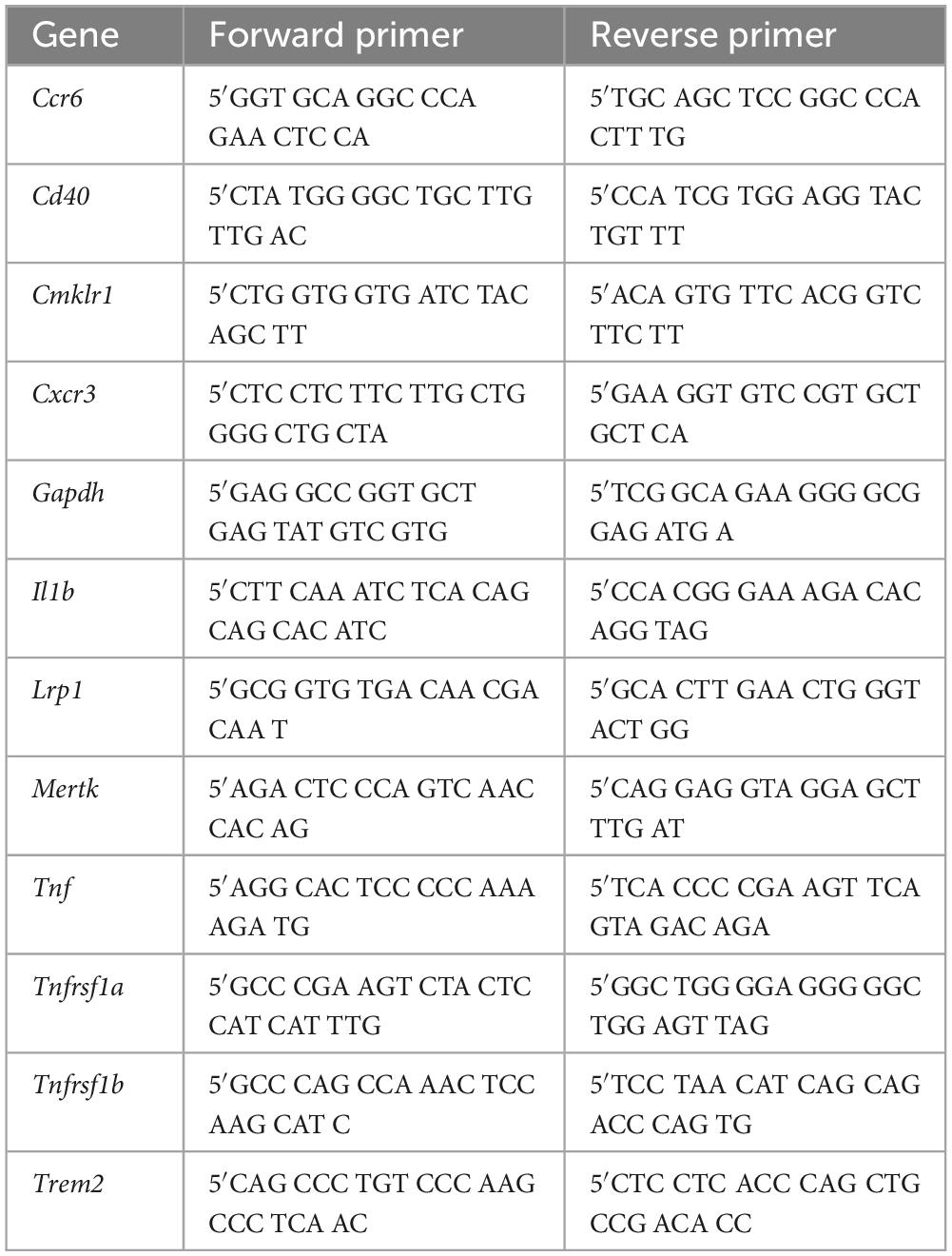
Table 1. Primers used for RT-PCR.
All data are expressed as mean ± SEM of 3–5 biological replicates. Data were analyzed with unpaired two-tailed t-test, and differences between groups were considered statistically significant at p-values ≤ 0.05.
To investigate the cell-cell communication between myeloid cell populations in different disease conditions, we devised an in vitro assay in which peripherally derived myeloid cells isolated from mice undergoing EAE or cerebral ischemia (pMCAO) were co-cultured with microglia. This enabled us to study both direct communication requiring cell-to-cell contact and indirect communication driven by soluble factors between peripherally derived and CNS-resident myeloid cells. Adult microglia were cultured from naïve mice for 5 days and then exposed to peripherally derived myeloid cells (either from pMCAO or EAE mice) that were plated directly over the microglia monolayer or seeded in cell culture inserts (Figure 1A). In EAE, myeloid cells were obtained from the spleen at 20 dpi (the most acute phase of disease) and 30 dpi (during chronic disease) (Figure 1B). In pMCAO, myeloid cells were obtained from the bone marrow at 1 dpi (acute disease) and 14 dpi (chronic disease). The purity of adult primary microglia was confirmed by immunohistochemistry, with 99% of cells being positive for Iba1, and no contamination by GFAP+ astrocytes or Olig2+ oligodendrocytes (Figures 1C, D). In direct co-cultures (Figure 1E), to allow for discrimination of peripherally derived myeloid cells from microglia, myeloid cells were pre-labeled with CFSE and CFSE– microglia were isolated by FACS (Figure 1F). Taken together, these validation experiments confirm that our in vitro assay works as intended and can be used to study different types of interactions between myeloid cell populations.
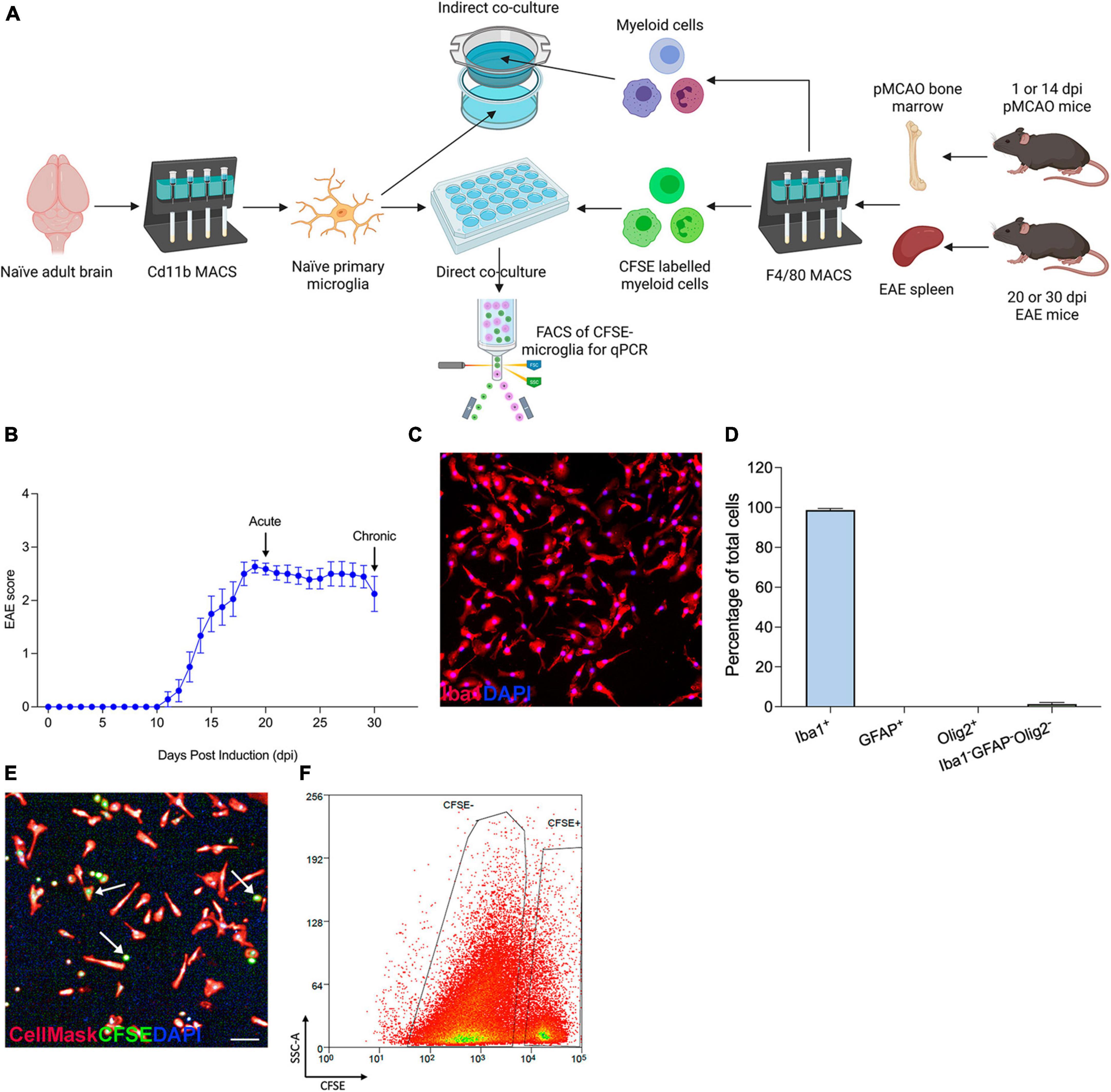
Figure 1. In vitro assay setup and validation. (A) Schematics of co-culture experiments. (B) Daily clinical scores of the EAE mice used in the present study. Data are shown as mean ± SEM, n = 14. (C) Representative 10X image of Iba1+DAPI+ naïve microglia. (D) Assessment of microglial purity by quantification of Iba1+ microglia, GFAP+ astrocytes, Olig2+ oligodendrocytes, and Iba1–GFAP–Olig2– cells out of total DAPI+ cells. Data are expressed as mean ± SEM of 17 randomly selected areas imaged from 6 cell culture wells. (E) Representative image of a direct co-culture, with microglia stained with CellMask (red) and peripherally derived myeloid cells labeled with CFSE (green); scale bar = 100 μm. Arrows point at examples of CFSE-stained myeloid cells. (F) CSFE– microglia isolated by FACS to exclude peripherally derived CSFE+ myeloid cells. Figure created with BioRender.com.
Myeloid cells respond to injury/disease by transitioning to an activated phenotype that is often characterized by upregulation of inflammatory genes such as cytokines and chemokines (Brendecke and Prinz, 2015; Becher et al., 2017). To investigate how peripheral myeloid cells in acute traumatic and chronic neurological disease may differ in their ability to drive microglial responses, we analyzed the expression of key inflammatory genes in microglia from our co-culture systems. Myeloid cells from both pMCAO and EAE mice increased microglial expression of Tnf compared to naïve unstimulated microglia, but no differences were observed between acute and chronic myeloid cell exposure (Figure 2A). Expression of TNF receptors Tnfrsf1a and Tnfrsf1b was also increased compared to baseline levels (Figures 2B, C). Direct contact with pMCAO myeloid cells induced higher expression of Tnfrsf1a and Tnfrsf1b when cells were obtained at the chronic timepoint, whereas direct contact with EAE myeloid cells induced lower expression of Tnfrsf1a and Tnfrsf1b when cells were obtained at the chronic timepoint. The highest expression of Tnfrsf1a was observed in pMCAO (chronic 14 dpi), and the highest expression of Tnfrsf1b was in EAE (acute 20 dpi) (Figures 2B, C). Interestingly, indirect interaction had minimal effect on microglial expression of these Tnf signaling genes, except for Tnfrsf1b which was elevated in both acute and chronic EAE (Figure 2C). For Il1b, direct contact with myeloid cells from acute disease (both pMCAO and EAE) increased microglial expression levels (Figure 2D). Finally, expression of Cd40, a member of the TNF superfamily whose ligand, CD40L, is important for microglial activation in inflammatory conditions (Ponomarev et al., 2006), was elevated by direct contact with chronic EAE myeloid cells compared to acute (Figure 2E). A similar trend (p = 0.065) was observed with pMCAO myeloid cells (Figure 2E).
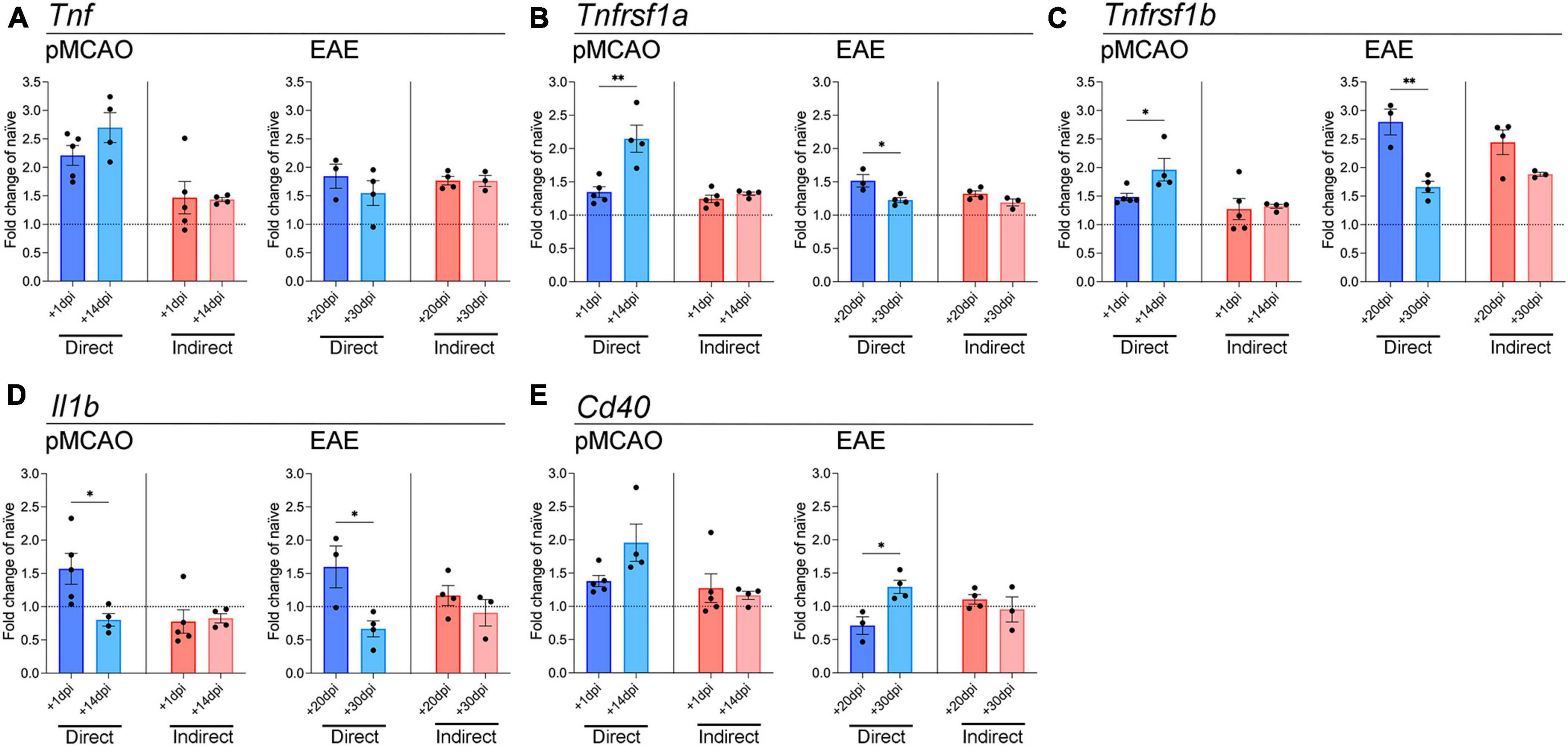
Figure 2. Gene expression analysis of pro-inflammatory cytokines and TNF receptor superfamily members in microglia. qPCR analysis of Tnf (A), Tnfrsf1a (B), Tnfrsf1b (C), Il1b (D), and Cd40 (E) in adult microglia exposed to pMCAO or EAE myeloid cells. Direct: Direct contact and interaction between myeloid cells and microglia. Indirect: Indirect interaction between myeloid cells and microglia, only via soluble factors. Data are expressed as fold change of naïve microglia after normalization to Gapdh gene expression. The horizontal line at y = 1 indicates baseline expression in naïve microglia. Data are presented as mean ± SEM of 3–5 biological replicates. Each biological replicate is derived from 2–6 technical replicates. *p ≤ 0.05, **p ≤ 0.01, unpaired two-tailed t-test.
To look further into microglial inflammatory changes induced by myeloid cells, we examined genes involved in chemokine signaling, which are important for microglial migration toward sites of injury/disease (Becher et al., 2017; Ifergan and Miller, 2020). Cmklr1, the receptor for chemerin, was mildly elevated over baseline levels with EAE-derived cells at both timepoints, and with pMCAO-derived cells, but only with direct cell-cell interaction (Figure 3A). Cxcr3 expression changed primarily with pMCAO myeloid cells in direct contact with microglia, showing a significant increase in chronic cells over acute (Figure 3B). With EAE-derived myeloid cells, Cxcr3 changed minimally, showing a significant increase in acute over chronic cells after indirect cell-cell interaction (Figure 3B). No notable differences were found in Ccr6 in any of the conditions (Figure 3C).
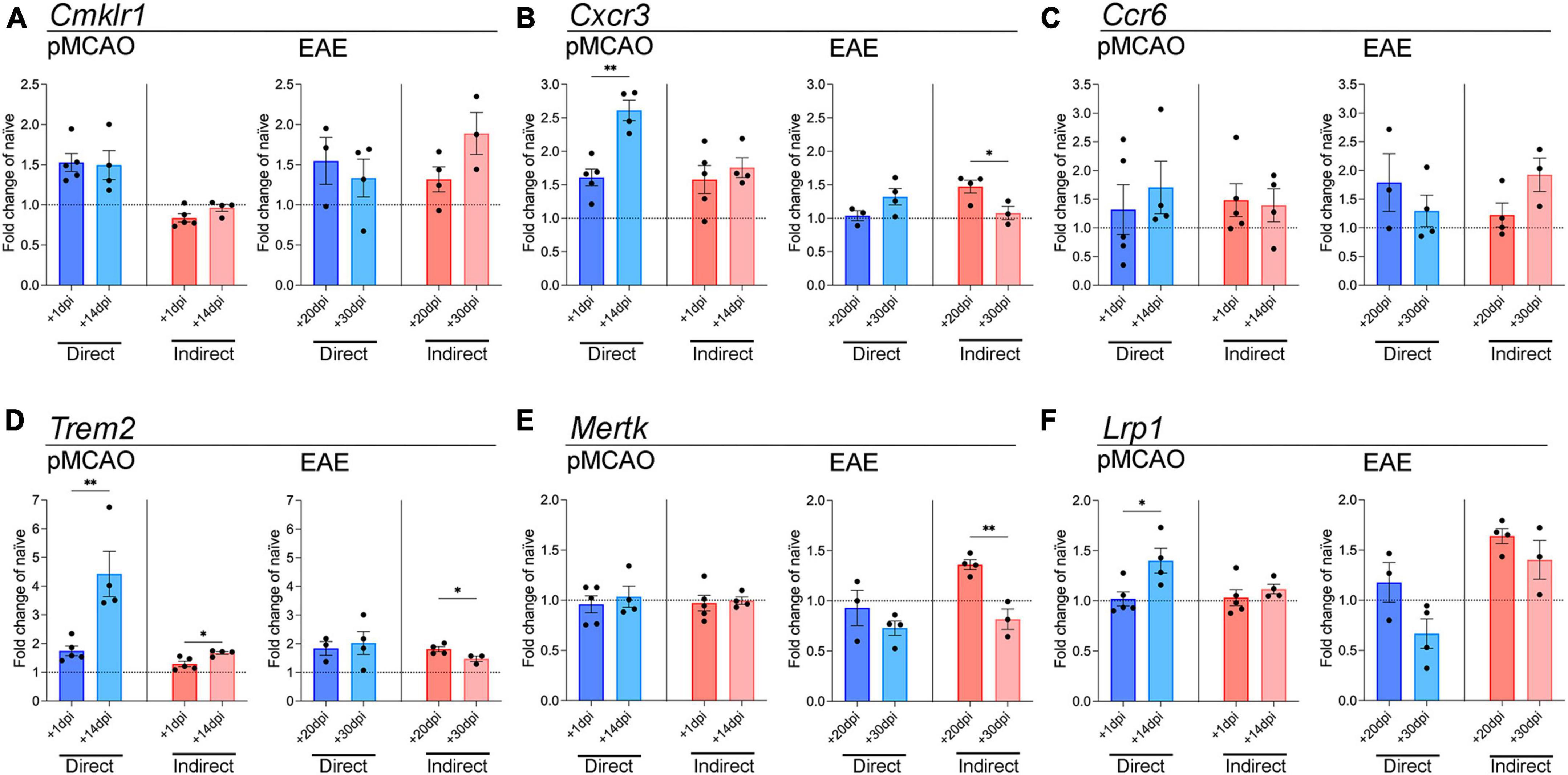
Figure 3. Gene expression analysis of chemokine receptor and phagocytosis genes in microglia. qPCR analysis of Cmklr1 (A), Cxcr3 (B), Ccr6 (C), Trem2 (D), Mertk (E), and Lrp1 (F) in adult microglia exposed to pMCAO or EAE myeloid cells. Direct: Direct contact and interaction between myeloid cells and microglia. Indirect: Indirect interaction between myeloid cells and microglia, only via soluble factors. Data are expressed as fold change of naïve microglia after normalization to Gapdh gene expression. The horizontal line at y = 1 indicates baseline expression in naïve microglia. Data are presented as mean ± SEM of 3–5 biological replicates. Each biological replicate is derived from 2–6 technical replicates. *p ≤ 0.05, **p ≤ 0.01, unpaired two-tailed t-test.
Next, we tested whether myeloid cells might affect the microglial phagocytic function. Phagocytosis is key in maintaining CNS homeostasis, and in disease states it is important to clear toxic cell debris (Fu et al., 2014). We examined the expression of Trem2, Mertk, and Lrp1, which have all been shown to be important for microglia phagocytic properties (Fu et al., 2014; Chuang et al., 2016; Shi et al., 2021). Myeloid cells from the chronic phase of pMCAO induced a higher expression of microglial Trem2 compared to acute pMCAO cells, both with direct and indirect interaction (Figure 3D). Microglial Trem2 expression with direct exposure to chronic pMCAO myeloid cells was the highest compared to naïve microglia (Figure 3D). Interestingly, Trem2 changes with EAE myeloid cells were minimal. Trem2 was mildly elevated over baseline levels in all conditions, and acute EAE myeloid cells induced higher expression than chronic EAE myeloid cells with indirect interaction (Figure 3D). A similar pattern with EAE myeloid cells was observed for Mertk, where expression was higher with acute than chronic cells after indirect interaction (Figure 3E). pMCAO myeloid cells did not induce any changes in Mertk expression (Figure 3E). Similar to Trem2, Lrp1 was higher with chronic pMCAO myeloid cells compared to acute. EAE myeloid cells increased Lrp1 expression after indirect contact, with no difference between acute and chronic (Figure 3F).
Collectively, these data illustrate that peripherally derived myeloid cells can induce transcriptional changes in naïve microglia, dependent on disease type and stage and mode of interaction (direct or indirect).
Microglial activation driven by injury and disease is denoted by a series of phenotypic changes that include morphological adaptations whereby microglia transition from a ramified, branched phenotype to an ameboid phenotype with larger cell body and shorter processes (Woodburn et al., 2021). To further investigate whether myeloid cells changed microglial activation state, we evaluated microglial morphology using high content analysis, focusing on area, roundness, perimeter, length/width ratio, and quantifying the percentage of elongated cells. Overall, the morphological changes were relatively mild in all experimental conditions (Figures 4A–E). When exposed to myeloid cells from pMCAO mice, the most notable change was observed in cell size, where indirect cell interaction led to a reduced cell area (Figure 4A) and perimeter (Figure 4C) with acute myeloid cells, showing a significant difference compared to chronic. However, the microglia exposed to pMCAO myeloid cells appeared to have a similar morphology across conditions (Figure 4F). When exposed to myeloid cells from EAE mice, direct contact most notably affected microglial length/width ratio (Figure 4D) and % of elongated cells (Figure 4E), which decreased with acute interaction compared to chronic. These changes were evident as microglia became visibly more rounded (Figures 4B, G).
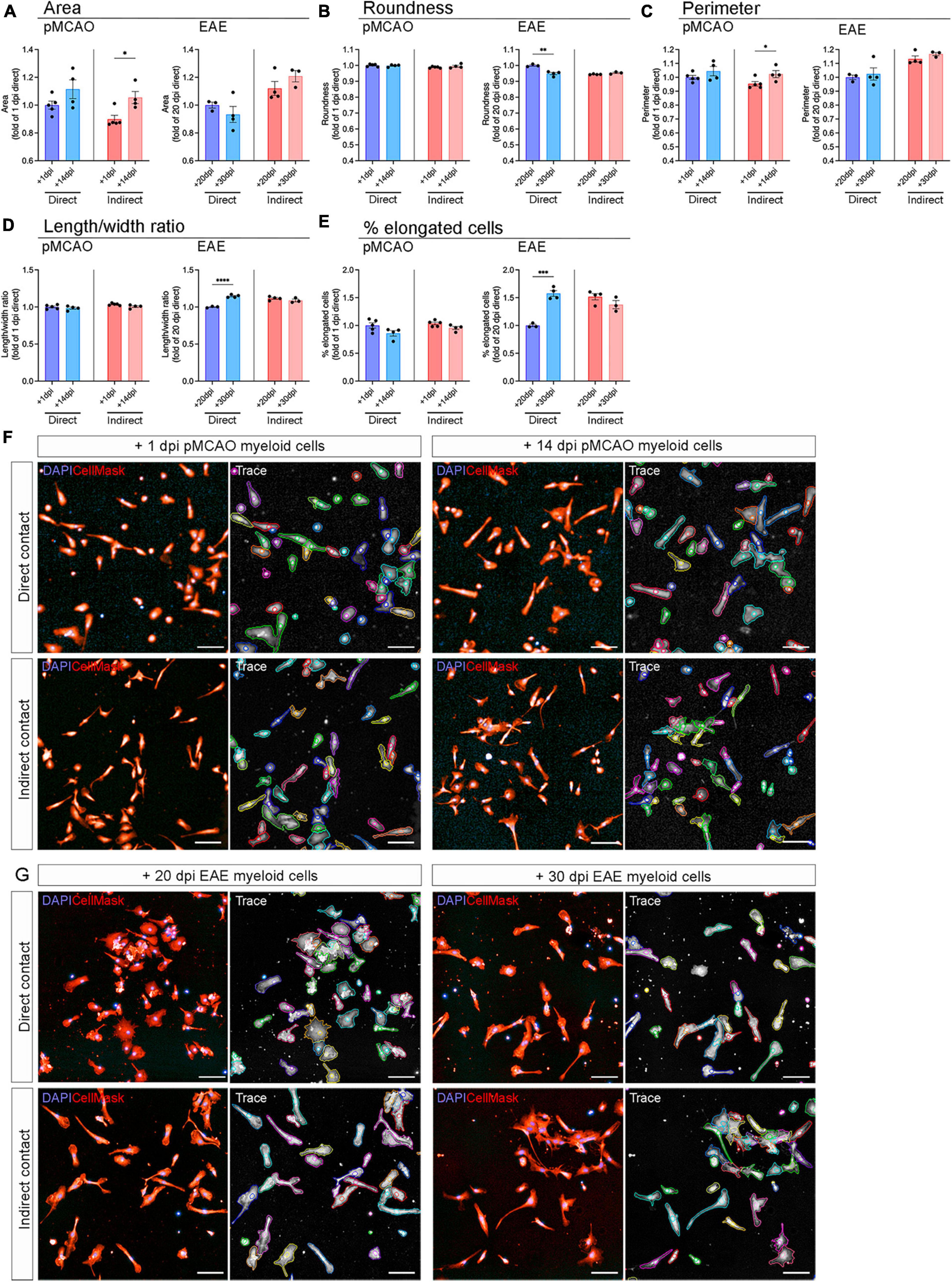
Figure 4. Microglial morphology after exposure to peripheral myeloid cells. Adult naïve microglia exposed to peripheral myeloid cells from pMCAO or EAE mice were evaluated for area (A), roundness (B), perimeter (C), length/width ratio (D), and percentage of elongated cells (E). Data are normalized to naïve controls in each experiment and calculated as fold change of the acute timepoint with direct contact. Direct: Direct contact and interaction between myeloid cells and microglia. Indirect: Indirect interaction between myeloid cells and microglia, only via soluble factors. Data are presented as mean ± SEM of 3–5 biological replicates. Each biological replicate is the average of 1–8 technical replicates. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, unpaired two-tailed t-test. Representative images of microglia exposed to pMCAO (F) and EAE (G) myeloid cells with tracing generated by Harmony® high content analysis software. Scale bars = 100 μm.
Although microglia and infiltrating myeloid cells have previously been thought to act in similar ways in neurological disease, RNA sequencing studies revealed distinct microglia and myeloid cell populations with key differences in disease conditions (Lewis et al., 2014; Yamasaki et al., 2014; Guo et al., 2021; Zheng et al., 2022). Despite the considerable involvement of peripheral myeloid cells in neurological disease, we still lack a full understanding of how they drive different disease processes and affect CNS-resident cells. In the current study, we developed an in vitro assay to identify potential differences between acute traumatic and chronic neurodegenerative disease by investigating how peripheral myeloid cells from pMCAO or EAE mice affect naïve microglia. As the spleen coordinates the immune response in MS and EAE (Zhu et al., 2007) and cells from the bone marrow drive the immune response in ischemic stroke (Courties et al., 2015), we isolated myeloid cells from the spleen of EAE induced mice and from the bone marrow of pMCAO injured mice at acute and chronic disease phases. We found key differences in their ability to modulate gene expression and morphology of microglia, not only between the two disease models, but also through the course of disease. These changes were dependent on whether the cells were in direct contact or had interactions via soluble factors only. Peripheral myeloid cells increased microglial expression of most of the investigated genes compared to the baseline expression. This validates our in vitro system, as an increase in microglial gene expression is expected with exposure to myeloid cells derived from disease conditions.
We tested Il1b expression as it is released from microglia and infiltrating myeloid cells acutely in neuroinflammatory conditions and is involved in EAE progression and ischemic damage (Clausen et al., 2008; Yang et al., 2014; Lévesque et al., 2016). The finding that peripheral myeloid cells from the acute phase of both EAE and pMCAO mice induced higher Il1b expression in microglia compared to chronic phase myeloid cells suggests that activated peripherally derived myeloid cells might activate pathogen recognition receptors (PRRs) and inflammasome assembly acutely (Lee et al., 2013; Yang et al., 2014). Interestingly, even though microglial Tnf expression was increased upon interaction with peripheral myeloid cells from both disease models, no changes were seen between acute and chronic timepoints, indicating that differences between disease phases are not triggered by peripheral myeloid cells. Expression of microglial Tnfrsf1a and Tnfrsf1b was increased with direct exposure to peripheral myeloid cells from both pMCAO and EAE mice. In the acute phase of EAE, peripheral myeloid cells highly express TNF and other cytokines (Renno et al., 1995; Ifergan and Miller, 2020), which may cause the increased expression of microglial Tnfrsf1a and Tnfrsf1b after interaction with acute EAE peripheral myeloid cells in our experimental setting. In stroke, increased TNF receptor expression peak days after pMCAO induction (Lambertsen et al., 2007), supporting our finding that peripherally derived chronic pMCAO myeloid cells increase microglial TNF receptor expression to a larger extent than acute myeloid cells. It is noteworthy that peripherally derived myeloid cells in both disease models induced changes in TNFR1 and TNFR2 simultaneously, as these two receptors have distinct and opposing functions in neurological disease (Dong et al., 2016; Papazian et al., 2021). This suggests that peripheral myeloid cells can induce both detrimental and protective effects in the microglia.
CD40 is upregulated with microglial activation and increases the release of cytokines and chemokines (Benveniste et al., 2004; Plastini et al., 2020). CD40 has a major role in EAE progression (Hart et al., 2005). Indeed, full activation of microglia in EAE depends on CD40, and lack of CD40 leads to lower T-cell proliferation and less severe disease (Ponomarev et al., 2006). The increase in microglial Cd40 expression with exposure to peripheral myeloid cells from chronic EAE indicates that myeloid cells can induce Cd40 expression, which might drive disease progression in the chronic stage through increased microglial activation and possibly T-cell proliferation.
Peripheral myeloid cells might have a more detrimental phenotype at the chronic timepoint following pMCAO, for example by providing “eat-me” signals to microglia, which in turn upregulate the expression of genes involved in phagocytosis. Thus, the increases in Trem2 and Lrp1 in microglia exposed to peripherally derived chronic pMCAO myeloid cells could be protective by eliminating detrimental infiltrating myeloid cells (Neumann et al., 2008). This is supported by several studies reporting an important role of Trem2 and Lrp1 in phagocytosis and neuroprotection in neurological disease (Takahashi et al., 2005; Fernandez-Castaneda et al., 2013; Kawabori et al., 2015; Chuang et al., 2016). Increased phagocytosis may also be detrimental if the targets are viable neurons (Neher et al., 2013; Shi et al., 2021), leading to a worsening of the ischemic damage at the chronic stage. However, phagocytosis of viable neurons depends primarily on Mertk (Neher et al., 2013; Shi et al., 2021), which was not upregulated in microglia following exposure to pMCAO peripheral myeloid cells in our study. Future studies using metabolomics and/or lipidomics, as well as scRNAseq, would help identify and quantify pathways and networks of cellular lipids and metabolites that change in microglia following exposure to myeloid cells at acute and chronic time points after EAE and pMCAO.
RNA sequencing studies reveal that peripheral myeloid cells from EAE mice upregulate multiple soluble cytokines and chemokines, especially in the acute phase of disease (Lewis et al., 2014; Yamasaki et al., 2014; Gao et al., 2023). This may explain the upregulation in microglial Trem2, Mertk, and Cxcr3 that we observed with indirect contact with peripheral myeloid cells from acute EAE. Additionally, several studies demonstrated that infiltrating monocytes have very different transcriptional profiles at onset and peak phases of EAE compared to the recovery phase (Yamasaki et al., 2014; Gao et al., 2023). This fits well with our data, where we found microglia interacting with peripheral myeloid cells from acute EAE to have higher gene expression than with myeloid cells from chronic EAE, with the exception of Cd40. Also it was the exposure to acute EAE peripheral myeloid that altered microglial morphology to become more rounded, which is a hallmark of microglial activation (Stence et al., 2001; Woodburn et al., 2021). Overall, peripherally derived myeloid cells from acute EAE appear to be the most effective at inducing microglia activation, leading to changes in both gene expression and morphology.
This study prioritized investigating changes in gene expression, as a measure of whether peripheral myeloid cells were able to induce changes in naïve microglia using our in vitro assay. Future studies should aim to investigate whether the changes observed in microglial gene expression, persist as changes in protein levels. Moreover, as microglia will be activated in disease conditions (Woodburn et al., 2021), repeating the study with microglia isolated from EAE and pMCAO mice, to study the effect of activated peripheral myeloid cells on activated microglia, should be persued. Using naïve microglia in the present study was prioritized, as it allowed us to study the specific effect of activated peripheral myeloid cells, without involvement from intrinsic microglial activation. Additionally, it would be interesting to reverse the assay, to study the effect of microglia derived from different disease conditions on naïve peripheral myeloid cells.
In conclusion, we developed an in vitro system that allowed us to investigate the communication between peripheral myeloid cells and microglia. We identified peripheral myeloid cell-induced changes in microglia not only dependent on the disease model, but also on the disease phase at which myeloid cells were isolated. This underscores that neuroprotective and neuroreparative therapies must be tailored to each condition, and no myeloid modulating approach fits all. Our assay can be used to identify potential targets in myeloid cells under specific disease conditions, opening up the possibility of modulating these in vivo, either genetically or pharmacologically, in future studies, to assess their specific role in disease development. Furthermore, we showed that our assay is a valuable tool for investigating myeloid cell interactions in a range of CNS neuroinflammatory conditions.
The raw data supporting the conclusions of this article will be made available by the authors upon reasonable request.
The animal study was approved by the Institutional Animal Care and Use Committee of the University of Miami and the Danish Animal Inspectorate (J. no. 2019-15-0201-01620). The study was conducted in accordance with the local legislation and institutional requirements.
ET: Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Writing – original draft, Writing – review and editing. BC: Methodology, Writing – review and editing. AW: Data curation, Methodology, Writing – review and editing. RB: Conceptualization, Formal analysis, Funding acquisition, Resources, Supervision, Validation, Writing – review and editing. KLL: Conceptualization, Formal analysis, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by: Danish Multiple Sclerosis Society (A41489), A.P. Møller Foundation (20-L-0269 and L-2022-00268), Overlaegerådets Forskningsfond, Odense University Hospital (110-A4683), Faculty of Health Sciences at the University of Southern Denmark to ET; NIH NINDS (1RO1NS094522-01 and NS084303-01A1), Italian Multiple Sclerosis Foundation (FISM) (2015/R/7), The Miami Project To Cure Paralysis and the Buoniconti Fund to RB; Independent Research Fund Denmark (0134-00084B), Carlsberg Foundation (CF21-0215), and Danish Alzheimer Association’s Research Prize in Basic Research to KLL.
We thank Dr. Hassan A. Ali and Yan Shi at the High Content Screening (HCS) Core Facility of the University of Miami for their assistance with HCS imaging and analysis. We thank Dr. Oliver Umland at the DRI Flow Cytometry Core Facility of the University of Miami and Lars Vitved from the Flow Cytometry and Confocal Microscopy Core Facility of the University of Southern Denmark for their help with flow cytometric analyses and cell sorting. We thank Eva Juarez (University of Miami) for her assistance with animal care, and Maureen Ascona (University of Miami) for technical support in the Brambilla laboratory. Finally, we also thank Claire Gudex for proof-reading the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ccr, CC chemokine receptor; CD, cluster of differentiation; cDNA, copy deoxyribonucleic acid; CFSE, carboxyfluorescein succinimidyl ester; Cmklr, chemerin chemokine-like receptor; CNS, central nervous system; Cxcr, C-X-C motif chemokine receptor; DAPI, 4′,6-Diamidino-2-Phenylindole; Dpi, days post injury/induction; EAE, experimental autoimmune encephalomyelitis; FACS, fluorescence-activated cell sorting; FBS, fetal bovine serum; Gapdh, Glyceraldehyde-3-Phosphate Dehydrogenase; GFAP, glial fibrillary acidic protein; Iba1, ionized calcium-binding adapter molecule 1; Il1b, interleukin-1 beta; Lrp, low density lipoprotein receptor-related protein; MACS, magnetic-activated cell sorting; MCA, middle cerebral artery; Mertk, MER proto-oncogene tyrosine kinase; MS, multiple sclerosis; Olig2, oligodendrocyte transcription factor 2; PBS, phosphate buffered saline; PFA, paraformaldehyde; pMCAO, permanent middle cerebral artery occlusion; PCR, polymerase chain reaction; PPR, pathogen recognition receptor; RNA, ribonucleic acid; SEM, standard error of the mean; Tnf, tumor necrosis factor; Tnfrsf, tumor necrosis factor receptor; Trem2, triggering receptor expressed on myeloid cells 2.
Ajami, B., Bennett, J. L., Krieger, C., McNagny, K. M., and Rossi, F. M. V. (2011). Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 14, 1142–1150. doi: 10.1038/nn.2887
Becher, B., Spath, S., and Goverman, J. (2017). Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 17, 49–59. doi: 10.1038/nri.2016.123
Benveniste, E. N., Nguyen, V. T., and Wesemann, D. R. (2004). Molecular regulation of CD40 gene expression in macrophages and microglia. Brain Behav. Immun. 18, 7–12. doi: 10.1016/j.bbi.2003.09.001
Beuker, C., Strecker, J. K., Rawal, R., Schmidt-Pogoda, A., Ruck, T., Wiendl, H., et al. (2021). Immune cell infiltration into the brain after ischemic stroke in humans compared to mice and rats: A systematic review and meta-analysis. Transl. Stroke Res. 12, 976–990. doi: 10.1007/s12975-021-00887-4
Bisgaard, A. K., Pihl-Jensen, G., and Frederiksen, J. L. (2017). The neutrophil-to-lymphocyte ratio as disease actvity marker in multiple sclerosis and optic neuritis. Mult. Scler. Relat. Disord. 18, 213–217. doi: 10.1016/j.msard.2017.10.009
Brambilla, R., Ashbaugh, J. J., Magliozzi, R., Dellarole, A., Karmally, S., Szymkowski, D. E., et al. (2011). Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain 134, 2736–2754. doi: 10.1093/brain/awr199
Brendecke, S. M., and Prinz, M. (2015). Do not judge a cell by its cover—diversity of CNS resident, adjoining and infiltrating myeloid cells in inflammation. Semin. Immunopathol. 37, 591–605. doi: 10.1007/s00281-015-0520-6
Chuang, T. Y., Guo, Y., Seki, S. M., Rosen, A. M., Johanson, D. M., Mandell, J. W., et al. (2016). LRP1 expression in microglia is protective during CNS autoimmunity. Acta Neuropathol. Commun. 4:68. doi: 10.1186/s40478-016-0343-2
Clausen, B. H., Lambertsen, K. L., Babcock, A. A., Holm, T. H., Dagnaes-Hansen, F., and Finsen, B. (2008). Interleukin-1 beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J. Neuroinflammat. 5, 1–18. doi: 10.1186/1742-2094-5-46
Correale, J., Gaitán, M. I., Ysrraelit, M. C., and Fiol, M. P. (2017). Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain 140, 527–546. doi: 10.1093/brain/aww258
Courties, G., Herisson, F., Sager, H. B., Heidt, T., Ye, Y., Wei, Y., et al. (2015). Ischemic stroke activates hematopoietic bone marrow stem cells. Circ. Res. 116, 407–417. doi: 10.1161/CIRCRESAHA.116.305207
DiSabato, D. J., Quan, N., and Godbout, J. P. (2016). Neuroinflammation: The devil is in the details. J. Neurochem. 139, 136–153. doi: 10.1111/jnc.13607
Dong, Y., Fischer, R., Naudé, P. J. W., Maier, O., Nyakas, C., Duffey, M., et al. (2016). Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc. Natl. Acad. Sci. U. S. A. 113, 12304–12309. doi: 10.1073/pnas.1605195113
Fernandez-Castaneda, A., Arandjelovic, S., Stiles, T. L., Schlobach, R. K., Mowen, K. A., Gonias, S. L., et al. (2013). Identification of the low density lipoprotein (LDL) receptor-related protein-1 interactome in central nervous system myelin suggests a role in the clearance of necrotic cell debris. J. Biol. Chem. 288, 4538–4548. doi: 10.1074/jbc.M112.384693
Fu, R., Shen, Q., Xu, P., Luo, J. J., and Tang, Y. (2014). Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 49, 1422–1434. doi: 10.1007/s12035-013-8620-6
Gao, H., Danzi, M. C., Choi, C. S., Taherian, M., Dalby-Hansen, C., Ellman, D. G., et al. (2017). Opposing functions of microglial and macrophagic TNFR2 in the pathogenesis of experimental autoimmune encephalomyelitis. Cell Rep. 18, 198–212. doi: 10.1016/j.celrep.2016.11.083
Gao, H., Di, J., Clausen, B. H., Wang, N., Zhu, X., Zhao, T., et al. (2023). Distinct myeloid population phenotypes dependent on TREM2 expression levels shape the pathology of traumatic versus demyelinating CNS disorders. Cell Rep. 42:112629. doi: 10.1016/j.celrep.2023.112629
Garcia-Bonilla, L., Faraco, G., Moore, J., Murphy, M., Racchumi, G., Srinivasan, J., et al. (2016). Spatio-temporal profile, phenotypic diversity, and fate of recruited monocytes into the post-ischemic brain. J. Neuroinflammat. 13, 1–13. doi: 10.1186/s12974-016-0750-0
Giles, D. A., Washnock-Schmid, J. M., Duncker, P. C., Dahlawi, S., Ponath, G., Pitt, D., et al. (2018). Myeloid cell plasticity in the evolution of central nervous system autoimmunity. Ann. Neurol. 83, 131–141. doi: 10.1002/ana.25128
Goldmann, T., Wieghofer, P., Müller, P. F., Wolf, Y., Varol, D., Yona, S., et al. (2013). A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 16, 1618–1626. doi: 10.1038/nn.3531
Guerrero, B. L., and Sicotte, N. L. (2020). Microglia in multiple sclerosis: Friend or foe? Front. Immunol. 11:374. doi: 10.3389/fimmu.2020.00374
Guo, K., Luo, J., Feng, D., Wu, L., Wang, X., Xia, L., et al. (2021). Single-cell RNA sequencing with combined use of bulk RNA sequencing to reveal cell heterogeneity and molecular changes at acute stage of ischemic stroke in mouse cortex penumbra area. Front. Cell Dev. Biol. 9:624711. doi: 10.3389/fcell.2021.624711
Hart, B. A., Blezer, E. L. A., Brok, H. P. M., Boon, L., De Boer, M., Bauer, J., et al. (2005). Treatment with chimeric anti-human CD40 antibody suppresses MRI-detectable inflammation and enlargement of pre-existing brain lesions in common marmosets affected by MOG-induced EAE. J. Neuroimmunol. 163, 31–39. doi: 10.1016/j.jneuroim.2005.02.005
Iadecola, C., Buckwalter, M. S., and Anrather, J. (2020). Immune responses to stroke: Mechanisms, modulation, and therapeutic potential. J. Clin. Invest. 130, 2777–2788. doi: 10.1172/JCI135530
Ifergan, I., and Miller, S. D. (2020). Potential for targeting myeloid cells in controlling CNS inflammation. Front. Immunol. 11:571897. doi: 10.3389/fimmu.2020.571897
Jolivel, V., Bicker, F., Binamé, F., Ploen, R., Keller, S., Gollan, R., et al. (2015). Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 129, 279–295. doi: 10.1007/s00401-014-1372-1
Kawabori, M., Kacimi, R., Kauppinen, T., Calosing, C., Kim, J. Y., Hsieh, C. L., et al. (2015). Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J. Neurosci. 35, 3384–3396. doi: 10.1523/JNEUROSCI.2620-14.2015
Kocur, M., Schneider, R., Pulm, A. K., Bauer, J., Kropp, S., Gliem, M., et al. (2015). IFNβ secreted by microglia mediates clearance of myelin debris in CNS autoimmunity. Acta Neuropathol. Commun. 3:20. doi: 10.1186/s40478-015-0192-4
Lalancette-Hébert, M., Gowing, G., Simard, A., Yuan, C. W., and Kriz, J. (2007). Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 27, 2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007
Lambertsen, K. L., Clausen, B. H., Babcock, A. A., Gregersen, R., Fenger, C., Nielsen, H. H., et al. (2009). Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J. Neurosci. 29, 1319–1330. doi: 10.1523/JNEUROSCI.5505-08.2009
Lambertsen, K. L., Clausen, B. H., Fenger, C., Wulf, H., Owens, T., Dagnaes-Hansen, F., et al. (2007). Microglia and macrophages express tumor necrosis factor receptor p75 following middle cerebral artery occlusion in mice. Neuroscience 144, 934–949. doi: 10.1016/j.neuroscience.2006.10.046
Lambertsen, K. L., Finsen, B., and Clausen, B. H. (2019). Post-stroke inflammation—target or tool for therapy? Acta Neuropathol. 137, 693–714. doi: 10.1007/s00401-018-1930-z
Lee, H. M., Kang, J., Lee, S. J., and Jo, E. K. (2013). Microglial activation of the NLRP3 inflammasome by the priming signals derived from macrophages infected with mycobacteria. Glia 61, 441–452. doi: 10.1002/glia.22448
Lévesque, S. A., Paré, A., Mailhot, B., Bellver-Landete, V., Kébir, H., Lécuyer, M. A., et al. (2016). Myeloid cell transmigration across the CNS vasculature triggers IL-1ß-driven neuroinflammation during autoimmune encephalomyelitis in mice. J. Exp. Med. 213, 929–949. doi: 10.1084/jem.20151437
Lewis, N. D., Hill, J. D., Juchem, K. W., Stefanopoulos, D. E., and Modis, L. K. (2014). RNA sequencing of microglia and monocyte-derived macrophages from mice with experimental autoimmune encephalomyelitis illustrates a changing phenotype with disease course. J. Neuroimmunol. 277, 26–38. doi: 10.1016/j.jneuroim.2014.09.014
Liu, Z., Fan, Y., Won, S. J., Neumann, M., Hu, D., Zhou, L., et al. (2007). Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke 38, 146–152. doi: 10.1161/01.STR.0000251791.64910.cd
Lucas, S. M., Rothwell, N. J., and Gibson, R. M. (2006). The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 147, 232–240. doi: 10.1038/sj.bjp.0706400
Ma, Y., Wang, J., Wang, Y., and Yang, G. Y. (2017). The biphasic function of microglia in ischemic stroke. Prog. Neurobiol. 157, 247–272. doi: 10.1016/j.pneurobio.2016.01.005
Madsen, P. M., Desu, H. L., Vaccari, J. P., de, R., Florimon, Y., Ellman, D. G., et al. (2020). Oligodendrocytes modulate the immune-inflammatory response in EAE via TNFR2 signaling. Brain Behav. Immun. 84, 132–146. doi: 10.1016/j.bbi.2019.11.017
Nayak, D., Roth, T. L., and McGavern, D. B. (2014). Microglia development and function. Annu. Rev. Immunol. 32, 367–402. doi: 10.1146/annurev-immunol-032713-120240
Neher, J. J., Emmrich, J. V., Fricker, M., Mander, P. K., Théry, C., and Brown, G. C. (2013). Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. U. S. A. 110, E4098–E4107. doi: 10.1073/pnas.1308679110
Neumann, J., Sauerzweig, S., Rönicke, R., Gunzer, F., Dinkel, K., Ullrich, O., et al. (2008). Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: A new mechanism of CNS immune privilege. J. Neurosci. 28, 5965–5975. doi: 10.1523/JNEUROSCI.0060-08.2008
Nissen, J. C., Thompson, K. K., West, B. L., and Tsirka, S. E. (2018). Csf1R inhibition attenuates experimental autoimmune encephalomyelitis and promotes recovery. Exp. Neurol. 307, 24–36. doi: 10.1016/j.expneurol.2018.05.021
Papazian, I., Tsoukala, E., Boutou, A., Karamita, M., Kambas, K., Iliopoulou, L., et al. (2021). Fundamentally different roles of neuronal TNF receptors in CNS pathology: TNFR1 and IKKβ promote microglial responses and tissue injury in demyelination while TNFR2 protects against excitotoxicity in mice. J. Neuroinflammat. 18, 1–21. doi: 10.1186/s12974-021-02200-4
Plastini, M. J., Desu, H. L., and Brambilla, R. (2020). Dynamic responses of microglia in animal models of multiple sclerosis. Front. Cell Neurosci. 14:269. doi: 10.3389/fncel.2020.00269
Ponomarev, E. D., Shriver, L. P., and Dittel, B. N. (2006). CD40 expression by microglial cells is required for their completion of a two-step activation process during central nervous system autoimmune inflammation. J. Immunol. 176, 1402–1410.
Qin, C., Zhou, L. Q., Ma, X. T., Hu, Z. W., Yang, S., Chen, M., et al. (2019). Dual functions of microglia in ischemic stroke. Neurosci. Bull. 35, 921–933. doi: 10.1007/s12264-019-00388-3
Rawji, K. S., and Yong, V. W. (2013). The benefits and detriments of macrophages/microglia in models of multiple sclerosis. Clin. Dev. Immunol. 2013:948976. doi: 10.1155/2013/948976
Renno, T., Krakowski, M., Piccirillo, C., Lin, J. Y., and Owens, T. (1995). TNF-alpha expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. Regulation by Th1 cytokines. J. Immunol. 154, 944–953. doi: 10.4049/JIMMUNOL.154.2.944
Ruiz, F., Vigne, S., and Pot, C. (2019). Resolution of inflammation during multiple sclerosis. Semin. Immunopathol. 41, 711–726. doi: 10.1007/s00281-019-00765-0
Savarin, C., Dutta, R., and Bergmann, C. C. (2018). Distinct gene profiles of bone marrow-derived macrophages and microglia during neurotropic coronavirus-induced demyelination. Front. Immunol. 9:1325. doi: 10.3389/fimmu.2018.01325
Shi, X., Luo, L., Wang, J., Shen, H., Li, Y., Mamtilahun, M., et al. (2021). Stroke subtype-dependent synapse elimination by reactive gliosis in mice. Nat. Commun. 12:6943. doi: 10.1038/s41467-021-27248-x
Stence, N., Waite, M., and Dailey, M. E. (2001). Dynamics of microglial activation: A confocal time-lapse analysis in hippocampal slices. Glia 33, 256–266.
Szalay, G., Martinecz, B., Lénárt, N., Környei, Z., Orsolits, B., Judák, L., et al. (2016). Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun 7:11499. doi: 10.1038/ncomms11499
Takahashi, K., Rochford, C. D. P., and Neumann, H. (2005). Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657. doi: 10.1084/jem.20041611
Tanabe, S., Saitoh, S., Miyajima, H., Itokazu, T., and Yamashita, T. (2019). Microglia suppress the secondary progression of autoimmune encephalomyelitis. Glia 67, 1694–1704. doi: 10.1002/glia.23640
Vainchtein, I. D., Alsema, A. M., Dubbelaar, M. L., Grit, C., Vinet, J., van Weering, H. R. J., et al. (2023). Characterizing microglial gene expression in a model of secondary progressive multiple sclerosis. Glia 71, 588–601. doi: 10.1002/glia.24297
Werner, Y., Mass, E., Kumar, P. A., Ulas, T., Händler, K., Horne, A., et al. (2020). Cxcr4 distinguishes HSC-derived monocytes from microglia and reveals monocyte immune responses to experimental stroke. Nat. Neurosci. 23, 351–362. doi: 10.1038/s41593-020-0585-y.Cxcr4
Wlodarczyk, A., Cédile, O., Jensen, K. N., Jasson, A., Mony, J. T., Khorooshi, R., et al. (2015). Pathologic and protective roles for microglial subsets and bone marrow- and blood-derived myeloid cells in central nervous system inflammation. Front. Immunol. 6:463. doi: 10.3389/fimmu.2015.00463
Woodburn, S. C., Bollinger, J. L., and Wohleb, E. S. (2021). The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflammat. 18, 1–16. doi: 10.1186/s12974-021-02309-6
Yamasaki, R., Lu, H., Butovsky, O., Ohno, N., Rietsch, A. M., Cialic, R., et al. (2014). Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 211, 1533–1549. doi: 10.1084/jem.20132477
Yang, F., Wang, Z., Wei, X., Han, H., Meng, X., Zhang, Y., et al. (2014). NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J. Cereb. Blood Flow Metab. 34, 660–667. doi: 10.1038/jcbfm.2013.242
Yli-Karjanmaa, M., Clausen, B. H., Degn, M., Novrup, H. G., Ellman, D. G., Toft-Jensen, P., et al. (2019). Topical administration of a soluble TNF inhibitor reduces infarct volume after focal cerebral ischemia in mice. Front. Neurosci. 13:781. doi: 10.3389/fnins.2019.00781
Zheng, K., Lin, L., Jiang, W., Chen, L., Zhang, X., Zhang, Q., et al. (2022). Single-cell RNA-seq reveals the transcriptional landscape in ischemic stroke. J. Cereb. Blood Flow Metab. 42, 56–73. doi: 10.1177/0271678X211026770
Zhu, B., Bando, Y., Xiao, S., Yang, K., Anderson, A. C., Kuchroo, V. K., et al. (2007). CD11b+Ly-6Chi suppressive monocytes in experimental autoimmune encephalomyelitis. J. Immunol. 179, 5228–5237. doi: 10.4049/jimmunol.179.8.5228
Keywords: myeloid cells, microglia, multiple sclerosis, ischemic stroke, neuroinflammation
Citation: Thougaard E, Carney B, Wlodarczyk A, Brambilla R and Lambertsen KL (2023) Peripherally derived myeloid cells induce disease-dependent phenotypic changes in microglia. Front. Cell. Neurosci. 17:1295840. doi: 10.3389/fncel.2023.1295840
Received: 17 September 2023; Accepted: 23 November 2023;
Published: 14 December 2023.
Edited by:
Anwen Shao, Zhejiang University, ChinaReviewed by:
Ganapathi Kandasamy, University of Miami, United StatesCopyright © 2023 Thougaard, Carney, Wlodarczyk, Brambilla and Lambertsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kate Lykke Lambertsen, a2xhbWJlcnRzZW5AaGVhbHRoLnNkdS5kaw==; Roberta Brambilla, cmJyYW1iaWxsYUBtZWQubWlhbWkuZWR1
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.