 Sonia Narwal
Sonia Narwal Amit Singh
Amit Singh Meghana Tare
Meghana Tare
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 04 January 2024
Sec. Cellular Neuropathology
Volume 17 - 2023 | https://doi.org/10.3389/fncel.2023.1295805
This article is part of the Research Topic Recent Advances in Mitochondrial Dysfunction and Therapeutics for Neurodegeneration and Aging View all 6 articles
One of the hallmarks of Parkinson's Disease (PD) is aggregation of incorrectly folded α-synuclein (SNCA) protein resulting in selective death of dopaminergic neurons. Another form of PD is characterized by the loss-of-function of an E3-ubiquitin ligase, parkin. Mutations in SNCA and parkin result in impaired mitochondrial morphology, causing loss of dopaminergic neurons. Despite extensive research on the individual effects of SNCA and parkin, their interactions in dopaminergic neurons remain understudied. Here we employ Drosophila model to study the effect of collective overexpression of SNCA along with the downregulation of parkin in the dopaminergic neurons of the posterior brain. We found that overexpression of SNCA along with downregulation of parkin causes a reduction in the number of dopaminergic neuronal clusters in the posterior region of the adult brain, which is manifested as progressive locomotor dysfunction. Overexpression of SNCA and downregulation of parkin collectively results in altered mitochondrial morphology in a cluster-specific manner, only in a subset of dopaminergic neurons of the brain. Further, we found that SNCA overexpression causes transcriptional downregulation of parkin. However, this downregulation is not further enhanced upon collective SNCA overexpression and parkin downregulation. This suggests that the interactions of SNCA and parkin may not be additive. Our study thus provides insights into a potential link between α-synuclein and parkin interactions. These interactions result in altered mitochondrial morphology in a cluster-specific manner for dopaminergic neurons over a time, thus unraveling the molecular interactions involved in the etiology of Parkinson's Disease.
Parkinson's Disease (PD), first characterized by James Parkinson in 1817 (Jost and Reichmann, 2017), is the second most common persistent movement neurodegenerative disorder, after Alzheimer's disease, with no cure to date. PD affects 0.8% of the worldwide population among other neurological disorders (Ray Dorsey et al., 2018). It is characterized by the specific loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) region of the midbrain which leads to motor symptoms including bradykinesia, muscular rigidity, resting tremor, postural instability (Wood-Kaczmar et al., 2006; Chartier and Duyckaerts, 2018). Progressive loss of DA neurons and the presence of intra-neuronal Lewy bodies (mainly aggregation of α-synuclein protein) are majorly considered neuropathological features of PD (Chartier and Duyckaerts, 2018; Shahmoradian et al., 2019).
SNCA and parkin are the two major genes involved in both sporadic as well as genetic forms of PD (Corti et al., 2011). SNCA encodes an α-synuclein protein and mutations in SNCA are associated with the Autosomal-Dominant form of PD (Polymeropoulos et al., 1997; Kachergus et al., 2004; Appel-Cresswell et al., 2013). Different in-vitro and in-vivo studies have shown that misfolded α-synuclein aggregation causes neurotoxicity by influencing the neurotransmission, synaptic vesicle exocytosis, recycling as well as endocytosis in the substantia nigra region. Migration of α-synuclein between neurons in a prion-like manner to propagate the formation of Lewy bodies throughout the substantia nigra has also been suggested (Li et al., 2008; Burré et al., 2010; Olanow and Brundin, 2013). Parkin encodes an E3 ubiquitin ligase and is the second most common cause of autosomal recessive early-onset PD (Kitada et al., 1998). Parkin loss-of-function causes neurodegeneration with or without forming Lewy bodies in PD patients (Yokochi, 1997; Pramstaller et al., 2005; Johansen et al., 2018). Studies have reported that overexpression of parkin results in reduced neurotoxicity caused by α-synuclein in different models (Oluwatosin-Chigbu et al., 2003; Haywood and Staveley, 2004; Khandelwal et al., 2010). Studies have also reported that parkin mutation results in no aggregation of α-synuclein in mice (Goldberg et al., 2003; Van Rompuy et al., 2015). However, very limited in-vivo studies have been done to explore the link between SNCA and parkin.
At the cellular level, mitochondria dysfunction has been considered a major hallmark of PD (Nicoletti et al., 2021). Studies in different model systems have shown that α-synuclein causes mitochondria fragmentation, disturbed membrane potential, complex I deficits, and reduced ATP production. In Drosophila, elongated as well as fragmented mitochondria have been reported due to α-synuclein overexpression (Nakamura, 2013; Ordonez et al., 2018; Krzystek et al., 2021). Loss-of-function mutation in parkin has also shown the mitochondrial pathology demonstrated as mitochondrial elongation, swelling, and cristae disruption in in-vitro and in-vivo models (Greene et al., 2003; Pesah et al., 2004; Deng et al., 2008; Yu et al., 2011; Noda et al., 2020). However, fused and fragmented mitochondria have also been reported in DA neurons of parkin mutant Drosophila (Cackovic et al., 2018). Moreover, it has been found that mitochondrial fragmentation caused by α-synuclein overexpression can be rescued by co-expression of parkin, PINK1, or DJ-1, indicating that α-synuclein and parkin may function in the same pathway (Kamp et al., 2010; reviewed in Jeśko et al., 2019). However, limited in-vivo studies have been done to test the effect of the interaction of SNCA and parkin on mitochondrial morphology in Parkinson's disease.
In this report, we have tested the interaction of parkin and SNCA and their effect on mitochondria in DA neurons using a humanized Drosophila melanogaster model of PD. Drosophila provides a simple, yet powerful in-vivo system to study neurodegenerative diseases including Parkinson's disease. It has a compact genome size (1/30th of the human genome), limited genetic redundancy, and a shorter generation time (12–15 days) and life span (60–80 days) (Bier, 2005). Almost 75% of all human disease genes are well conserved in the Drosophila genome sequence (Dawson et al., 2010; Aryal and Lee, 2019). Although Drosophila does not have an SNCA homolog, it mimics major PD symptoms i.e., locomotor dysfunction, age-dependent DA neuronal loss, and Lewy bodies aggregation upon overexpression of human SNCA using the GAL4/UAS system (Feany and Bender, 2000). Drosophila adult fly brain contains approximately 100 DA neurons, which are grouped into different clusters according to their anatomical position: PAL (paired anterior lateral), PAM (paired anterior medial), PPM1/2 and PPM3 (paired posterior medial), and PPL1 and PPL2 (paired posterior lateral) (Monastirioti, 1999; Mao and Davis, 2009). It has been suggested that each cluster-specific DA neuron projects to distinct functional areas of the brain (White et al., 2010), although the function of each of the DA clusters is not completely explored. PPL1 and PPM3 are more explored clusters in the case of PD using animal models because these are functionally homologous to the mammalian substantia nigra pars compacta region (Strausfeld and Hirth, 2013). It has been shown that PPL1 is associated largely with memory formation (Heisenberg, 2003) whereas, PPM3 is reported to be a center for the control of locomotor behavior (Strauss, 2002). In PD models of Drosophila, different clusters have been reported to be affected in a context-dependent manner.
In this study, we have used GAL4/UAS system specific for DA neurons to understand DA neuronal loss because of accumulation of α- synuclein as well as downregulation of parkin in a time-dependent manner in adult fly brain, mimicking the PD in humans. We found DA neuronal loss upon genetic alterations is cluster-specific, exhibiting altered mitochondrial morphology. Our studies have implications in providing leads for cellular and molecular interactions on DA neuronal degeneration during the onset and progression of PD.
All fly stocks, genetic crosses, and F1 progenies were maintained on standard fly food containing agar, maize powder, yeast, and sugar at 25°C. The GAL4/UAS system was used to obtain the desired genotype for the experiments (Brand and Perrimon, 1993; Tare et al., 2011). Transgenic Drosophila lines: UAS-GFP (a kind gift from S.C Lakhotia's Lab), UAS-Mito-HA-GFP.AP (BL-8442), UAS-Hsap/SNCA.F (BL-8146), UAS-SNCA.J}1/CyO (BL-51375), UAS-ParkRNAi (BL-37509) were used. Driver Gal4 line TH-Gal4 or ple-GAL4 (BL-8848) was used to overexpress or downregulate the responder lines of genes in DA neurons.
To determine locomotor activity, climbing assays were performed (Pendleton et al., 2002). Ten flies per genotype were transferred into a cylindrical glass tube after anesthetization and left for 5–10 min for revival and acclimatization at room temperature. Tubes were marked up to 8 cm above the bottom of the vial. After acclimatization, flies were gently tapped down to the bottom of the vial and the number of flies that crossed the 8 cm mark was recorded after 10 s. Three trials were performed, and numbers were then averaged, and the resulting mean was used as the overall value for each single group of flies. For all genotypes, three replicates were carried out.
Total RNA was extracted from 25 to 30 fly heads using the TRIzol method (Invitrogen) specified in the Jove protocol (URL: https://www.jove.com/video/50245) (Mehta et al., 2019). RNA concentrations were measured with a Nanodrop ND-1000 Spectrophotometer and equal amounts of RNA were reverse transcribed using Verso cDNA Synthesis Kit (AB1453A). qPCR was performed using 5x HOT FIREPol® EvaGreen® qPCR Mix Plus (Solis BioDyne). RP49 was used as the internal control gene. Primer sequences were as follows:
RP49 Forward: 5′-CCAAGGACTTCATCCGCCACC-3′
Reverse: 5′- GCGGGTGCGCTTGTTCGATCC-3′
Parkin Forward: 5′-ATTTGCCGGTAAGGAACTAAG C-3′
Reverse: 5′-AAGTGGCCGACTGGATTTTCT-3′
Brain preparation for confocal microscopy imaging was done as described by Tito et al. (2016). Briefly, Adult brains of the desired genotype were dissected in cold 1X PBS and incubated with fixative solution (4% formaldehyde) in 1XPBST (0.1%TritonX-100) for 20 min at room temperature. After three washes with 1XPBST for 10 min each wash, blocking was done using 1% BSA for 1hr at room temperature. Brains were probed with rabbit anti-TH (#AB152) at 1:1,000, overnight (12–16) at 4°C. Following three washes for 10 min, brains were incubated with Goat anti-Rabbit, Alexa-Fluor Plus 555 (#A32732) (1:4,000) secondary antibodies at room temperature for 2 h. Brains were washed three times for 15 min, then they were mounted between two glass coverslips by using an antifade medium on microscope slides. A confocal microscope was used to acquire z-stacks at 1 μm intervals with 20 × /N.A.0.60 Plan-Apochromat objective. The number of TH-positive neurons was counted manually within each cluster of posterior regions of the brains.
Western blot was performed as previously described (Gogia et al., 2017) with some modifications. F1 progenies of the desired genotype were collected in 1.5 ml Eppendorf tubes and snap-frozen in liquid nitrogen. Drosophila heads (~30–50) were decapitated and homogenized in 100 ul 1x RIPA buffer (Merck, #20–188) containing 1% protease inhibitor (Sigma, P8340) using a sterilized pestle. The homogenates were centrifuged at 10,000 rpm at 4 C for 10 min. The supernatant was collected and assayed for protein concentration. The protein (80 μg) was resolved on 12% SDA-PAGE and then transferred to 0.2 μm nitrocellulose membrane (Bio Rad, #1620112). After blocking the membrane with 3% BSA in TBS-T (0.05%Tween-20), the membrane was incubated overnight at 4 C with primary antibodies. The primary antibodies used were rabbit anti-Drosophila Parkin (Merck, SAB1300355, 1:500), mouse E7 anti-beta tubulin (DSHB-S1-810, 1:200). Following three washes with TBS-T, the membrane was incubated with appropriate HRP-conjugated secondary antibodies: goat anti-mouse (Thermo Scientific # 31430, 1:1,000), mouse anti-Rabbit (GenScript, #A01856, 1:1,000) for 2-h at room temperature and the signal was detected using ECL substrate (Bio Rad #1705061). Image analysis and quantification was done using ImageJ software. Western blot was done on the same membranes after stripping between each application of the antibody.
To assess the mitochondrial morphology in DA neurons, the UAS-MitoGFP fly strain was used to tag the mitochondria, and DA neurons were stained using anti TH antibody. Z-stack of one PPL1 and one PPM3 DA neuronal cluster per brain was imaged using confocal microscope at 63 × /N.A.1.30 oil with 1.5 zoom. A total of four brains per genotype were scanned. We used publicly available ImageJ Mito-Morphology Macro created by Dagda et al. (2009), to quantify the mitochondria. Average area and circularity were calculated representing elongation and fragmentation of mitochondria respectively.
GraphPad Prism 8.0.1 was used for statistical analysis and graphical display of the data. Significance is expressed as p values which were determined with one-way ANOVA and Two-way ANOVA followed by Tukey's multiple comparison tests as indicated in the figure legends.
Each result is representative of at least three biological and three technical repeats.
The α-synuclein-induced neurotoxicity (i.e., survival, locomotor defect, and DA neuronal death) is restored by parkin in in-vitro and in-vivo models (Petrucelli et al., 2002; Oluwatosin-Chigbu et al., 2003; Yang et al., 2003; Khandelwal et al., 2010). Hence, to test whether parkin is involved in α-synuclein mediated PD condition, we created double transgenes with RNAi of parkin (parkinIR here onward) and UAS-SNCA and expressed in DA neurons using TH-Gal4. Using H3C (DSHB-S1-890) antibody we confirmed the expression of α-synuclein in DA neurons of UAS-SNCA (Supplementary Figure 1A) and double transgenes through fluorescence microscopy (Supplementary Figure 1B) and downregulation of parkin through real-time PCR (Figures 3A, B, 7-day and 21-day respectively). We observed that flies that expressed SNCA with parkin knockdown (UAS-parkinIR; UAS-SNCA) displayed loss of climbing ability as compared to control (Figure 1); however, this was to a lesser extent with respect to parkin downregulation alone in age an age-dependent manner. Though, both SNCA overexpression and parkin downregulation independently displayed a loss of climbing ability with age (3–21 days) as compared to control (TH>GFP) (Figure 1), supporting the existing studies (Feany and Bender, 2000; Wang et al., 2007; Paricio and Muñoz-Soriano, 2011; Zhang et al., 2013; Mohite et al., 2018). We also confirmed the phenotype of parkin downregulation by overexpressing wild type-parkin with SNCA (UAS-SNCA;UAS-parkin). Notably, wild-type parkin overexpression with SNCA was able to restore the climbing ability (Supplementary Figure 2). We also confirmed the overexpression of α-synuclein using H3C antibody in UAS-SNCA; UAS-parkin transgene through fluorescence microscopy (Supplementary Figure 1C). These observations suggest that SNCA and parkin alteration together do not worsen the locomotor defects.
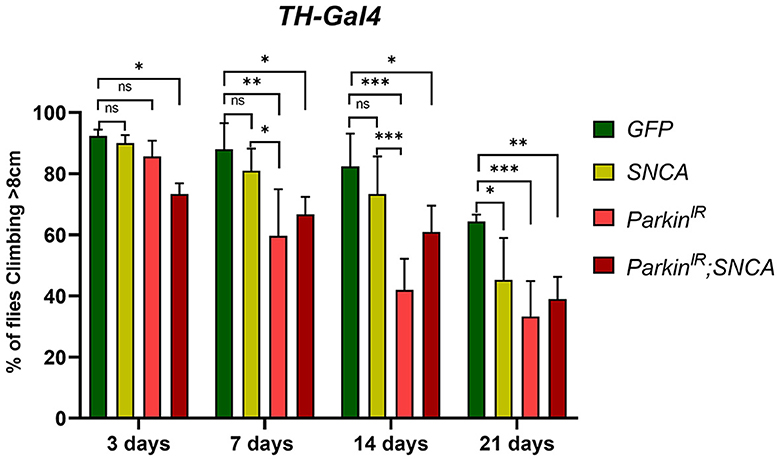
Figure 1. SNCA overexpression and parkin downregulation (parkinIR) independently, and together (parkinIR; SNCA) in dopaminergic neurons (DA), exhibit locomotor dysfunctions: Climbing assay indicates loss of locomotor function with age in flies. A total of 30 (N = 30) flies were used per genotype and 10 flies (n = 10) were used for the climbing assay. Data is represented as mean with SEM. Statistical analysis was performed using Two-way ANOVA followed by Tukey's multiple comparison test. P value: *(0.033), **(0.002), ***(<0.001), ns-not significant (0.12).
In PD, cardinal motor symptoms are caused by the death of DA neurons in the substantia nigra pars compacta (SNpc). Thus, we examined the DA neuronal clusters in adult fly brains using antibodies against tyrosine hydroxylase to investigate whether locomotor defects observed in SNCA and parkin alteration are due to loss of DA neurons. We have explored posterior protocerebrum DA neuronal clusters: PPM1/2, PPM3, PPL1, and PPL2 (Figure 2A). To visualize the DA neurons in the adult brain, we stained the DA neurons of the TH-Gal4 driving GFP flies with TH antibody (Figure 2B), hence confirming the activity of TH-Gal4>UAS-GFP and TH antibody. In our study, only TH-positive neurons were monitored for DA neuron quantification. We have found a substantial reduction in specific DA neuronal clusters in UAS-SNCA, UAS-parkinIR individually, and the UAS-parkinIR; UAS-SNCA transgene adult brain with respect to age-matched controls (Figure 2). In 7-day-old adult fly brains, quantification of PPL1, PPM1&2, PPM3, and PPL2 DA neuron clusters, SNCA, and parkinIR showed significant reduction in number of DA neurons only in PPL1 and PPM1&2 clusters (Figures 2C–E', G). UAS-parkinIR; UAS-SNCA also showed a significant reduction in the number of DA neurons in PPL1 and PPM1&2 as compared to control (Figures 2F, F', G). However, the number of DA neuronal losses in UAS-parkinIR; UAS-SNCA adult fly brains were less pronounced as compared to SNCA and parkinIR alone (Figures 2D–F', G). In 21-day-old fly brains, DA neurons numbers were further decreased in PPL1 and PPM1 & 2 clusters in a similar manner to 7-day-old adult fly brains of SNCA, parkinIR, and in UAS-parkinIR; UAS-SNCA (Figures 2H–K', L). In PPM3 DA neuron clusters, we have observed that the numbers of DA neurons were reduced only in parkinIR adult fly brains (Figures 2G, L). In the PPL2 DA cluster, we have observed no change in the number of DA neurons in UAS-SNCA, UAS-parkinIR, and UAS-parkinIR; UAS-SNCA of 7-day-old and 21-day-old adult fly brains. These observations suggest that SNCA and parkin cause DA clusters specific neuronal loss in the posterior region of adult fly brain.
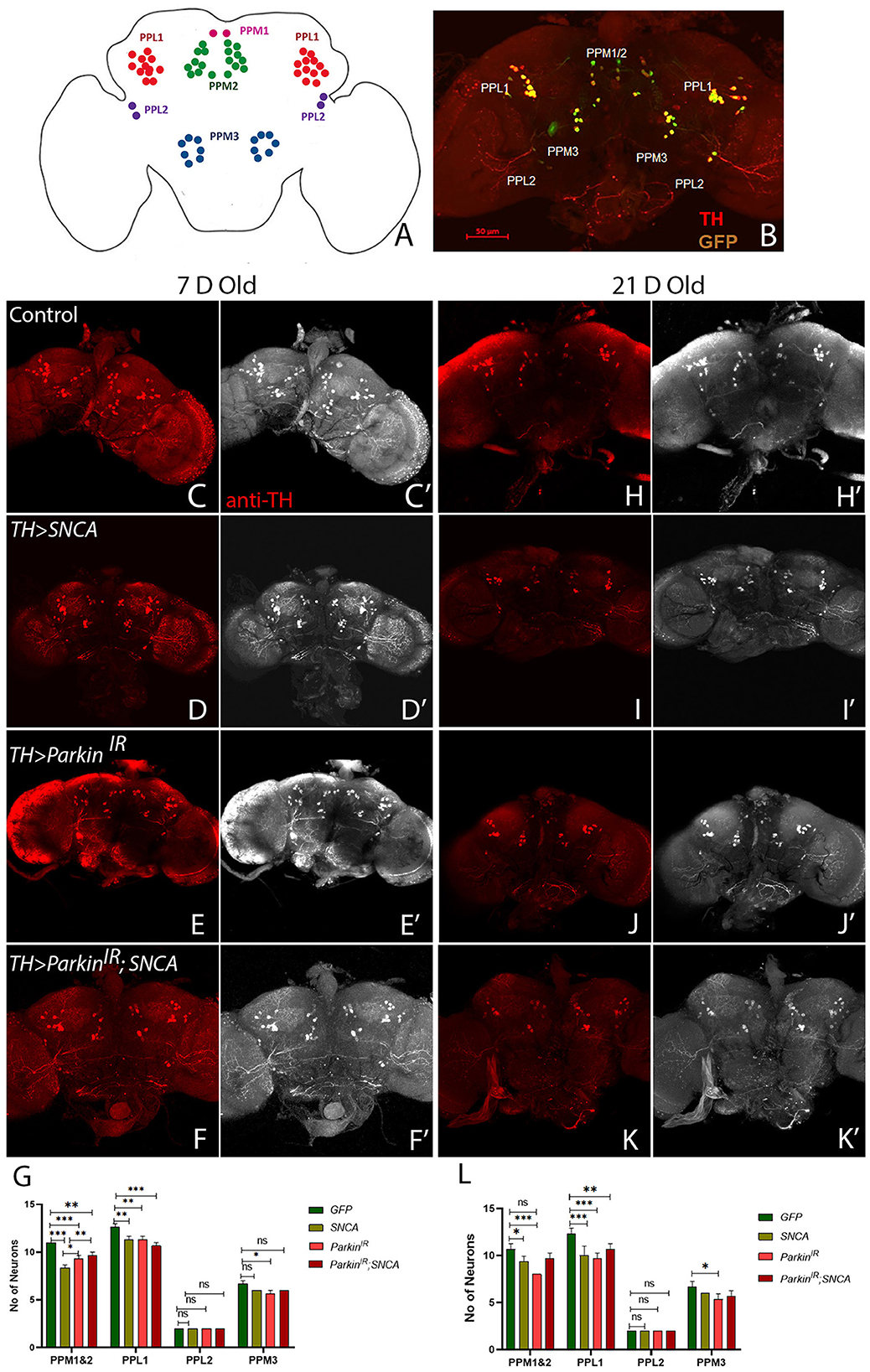
Figure 2. SNCA and parkinIR expression independently and together (parkinIR; SNCA) cause DA cluster specific neuronal loss (A) Schematic representation of DA neuronal clusters (PPL1, PPM1&2, PPM3, PPL2) in the posterior region of adult brain. (B) Representative confocal Maximum Intensity Projection (MIP) of WT adult brain stained with GFP (green) and Tyrosine Hydroxylase (TH) (Red) to reveal DA neurons in the posterior region. An adult brain of the desired genotype was dissected and stained for TH. (C–G) 7-day-old adult fly brains show TH stain. (C, C') Control flies, (D, D') SNCA overexpression, (E, E') ParkinIR expression, (F, F') ParkinIR; SNCA expression, show the TH-stain which is (G) quantified. (H-L) 21-day-old adult fly brains showing TH stain. (H, H') Control flies, (I, I') SNCA overexpression, (J, J') ParkinIR expression, (K, K') ParkinIR; SNCA expression shows the TH-stain and is (L) quantified. Scale bar 50μm. A total of four adult brains were used (n = 4) per genotype. Data is represented as mean with SEM. Statistical analysis was performed using Two-way ANOVA followed by Tukey's multiple comparison test. P value: *(0.033), **(0.002), ***(<0.001), ns-not significant (0.12).
Several in-vitro and in-vivo studies have shown the reduced neurotoxicity caused by SNCA upon parkin overexpression and suggested that parkin plays a vital role in the molecular pathway of PD pathogenesis. It has been reported that mutations in SNCA and parkin affect DA neuronal loss as well as the formation of Lewy bodies (Madsen et al., 2021). However, the effect of wild-type SNCA on parkin is not much explored. Therefore, we tested the parkin mRNA and protein levels of flies with UAS-parkinIR; UAS-SNCA, SNCA overexpression, and parkin downregulation independently. We found decreased parkin mRNA levels in UAS-parkinIR; UAS-SNCA flies as compared to control flies; however, this decreased transcript was at a lesser extent with respect to parkin downregulation and SNCA overexpression independently in 7-day adult fly brains (Figure 3A). In 21-day-old adult fly brains with UAS-parkinIR; UAS-SNCA has not shown a further decrease in parkin mRNA level as compared to control and as well as with SNCA overexpression and parkin downregulation independently (Figure 3B). Wilkaniec et al., 2019 have reported a decreased parkin protein level upon α-synuclein oligomerization which induces cell death in an in-vitro model. In contrast, in our study, we have found no significant change in parkin protein level upon SNCA overexpression in 7 (Figures 3C, E) and 21-day-old (Figures 3D, F) adult fly brains. However, we observed decreased parkin protein levels in flies expressing UAS-parkinIR; UAS-SNCA as compared to control but it was to a lesser extent to parkin downregulation independently (Figures 3E, F). Altogether, these data suggest that SNCA affects the parkin level.
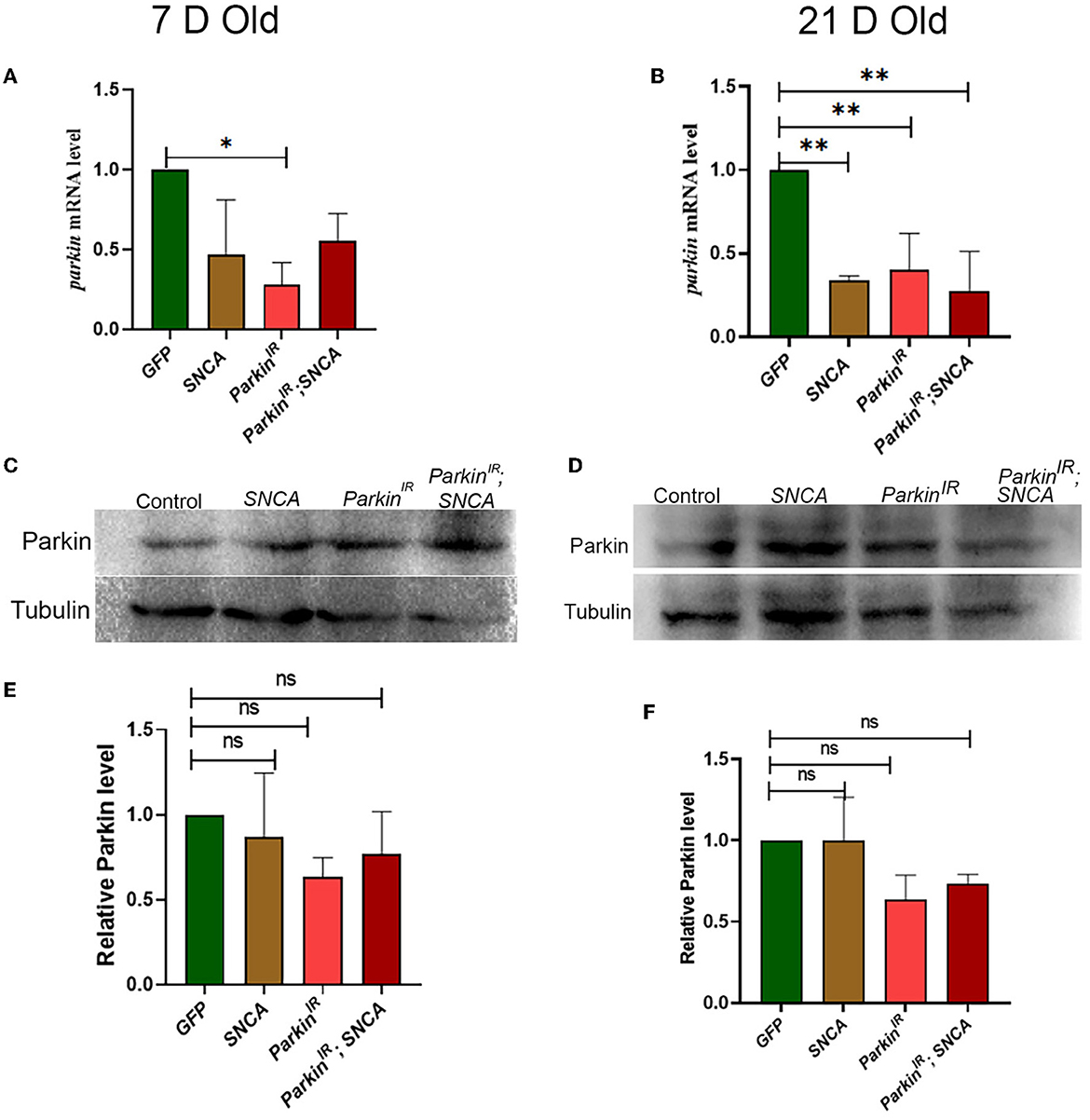
Figure 3. SNCA affects the expression of parkin at the transcriptional level but not the translational level. (A) 7-day old parkin mRNA level (B) 21-day-old parkin mRNA level. (C) Parkin immunoblot and (E) quantification of 7-day-old adult brains normalized to beta-Tubulin. (D) Parkin immunoblot and (F) quantification of 21-day old adult brains normalized to beta-Tubulin. Data is represented as mean with SEM. Statistical analysis was performed using One-way ANOVA followed by Tukey's multiple comparison test. P value: *(0.033), ** (0.002), ns-not significant (0.12).
Mitochondria are highly dynamic organelles and maintenance of mitochondrial morphology is essential for the survival of the neurons. Therefore, we investigated whether α-synuclein and parkin alteration-induced PD phenotypes have any relation with mitochondrial morphology. We have considered only PPL1 and PPM3 DA neuronal clusters for mitochondrial morphology assessment. This is because we have found a decrease in the number of TH-positive DA neurons in PPL1 clusters of UAS-parkinIR; UAS-SNCA (Figure 2), SNCA overexpression, and parkin downregulation independently and in PPM3 due to parkin downregulation only. We assessed the mitochondrial morphology using UAS-mitoGFP in TH-positive neurons. We have found that in PPL1 neuronal clusters, UAS-parkinIR; UAS-SNCA (Figures 4D, D', E; Supplementary Figure 4D) has shown swollen mitochondria as compared to control (TH>mito-GFP) (Figures 4A, A'; Supplementary Figure 4A) in 7-day adult fly brains. Whereas we observed fragmented mitochondria in 21-day-old adult fly brains (Figures 4I, I', J; Supplementary Figure 4H) as compared to control (Figures 4F, F', J; Supplementary Figure 4E). SNCA overexpression (Figures 4B, B', E; Supplementary Figure 4B) and parkin downregulation (Figures 4C, C', E; Supplementary Figure 4C) independently have shown swollen and/or enlarged mitochondria, which degenerate, in 7-day-old adult fly brains, as compared to control (Figures 4A, A', E; Supplementary Figure 4A). SNCA overexpression (Figures 4G, G', J; Supplementary Figure 4F) and parkin downregulation (Figures 4H, H', J; Supplementary Figure 4G) independently have shown further enhanced mitochondrial morphology in 21-day-old adult fly brains as compared to control (Figures 4F, F', J; Supplementary Figure 4E).
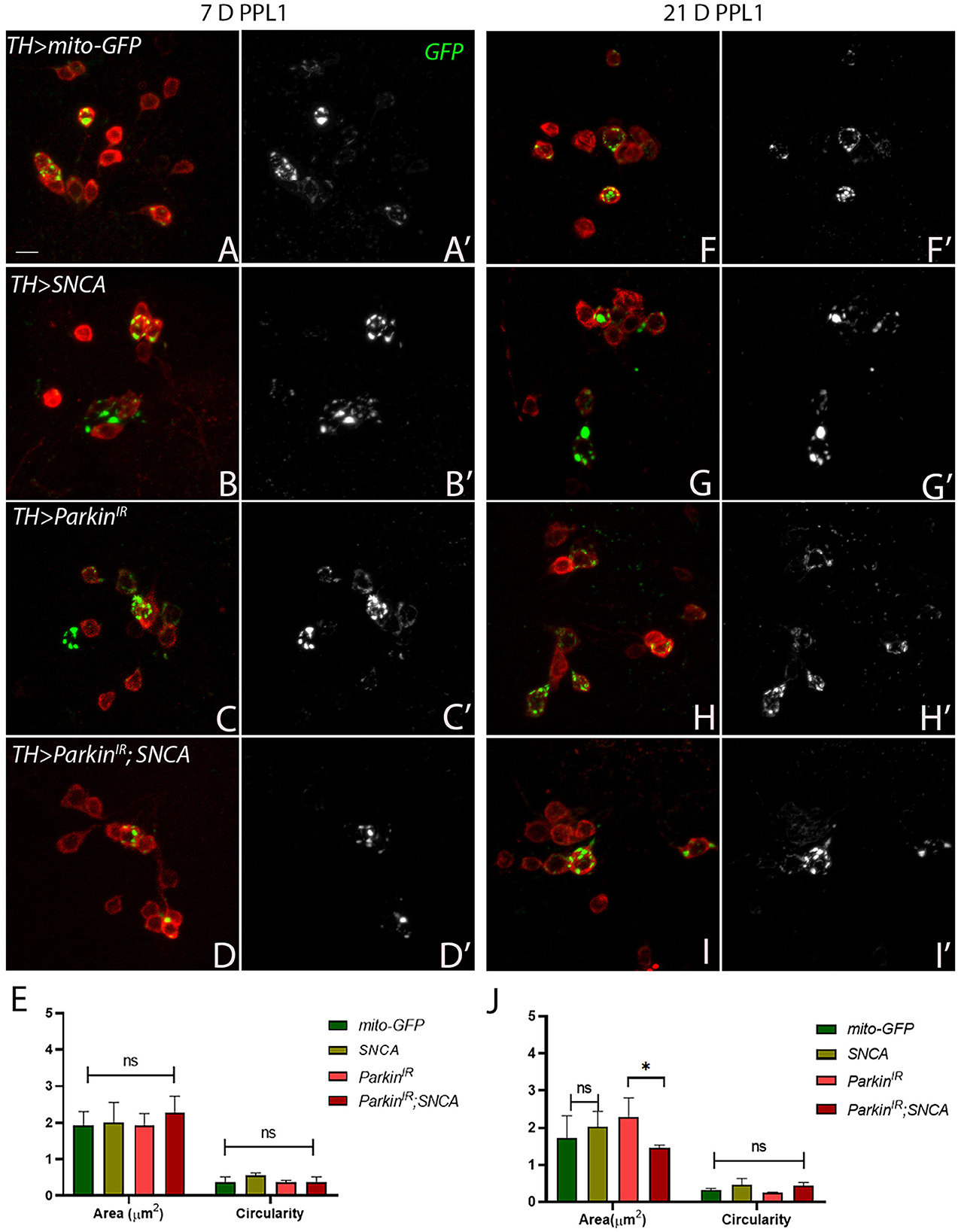
Figure 4. SNCA overexpression results in swollen mitochondria, parkinIR expression has shown elongated whereas together (parkinIR; SNCA) shows fragmented mitochondria in PPL1 DA clusters. Adult brains of the desired genotype expressing the mitochondria-targeted green fluorescent protein (mitoGFP) in TH- TH-positive (red) cells. (A–J) 7-day & 21-day old adult fly brains showing mitoGFP in PPL1 cluster. Control brains showing mitoGFP at (A, A') 7-day and (F, F') 21-day. SNCA overexpressing flies show mitoGFP (B, B') in 7-day and in (G, G') 21-day. ParkinIR-expressing flies show mitoGFP in (C, C') 7-day and in (H, H') 21-day. ParkinIR; SNCA expressing flies show mitoGFP in (D, D') 7-day and further enhanced in (I, I') 21-day. (E) Quantification of mitochondria morphology (area and circularity) was done using ImageJ Mito-Morphology Macro. Scale bar 10μm. A total of four adult brains were used (n = 4) per genotype. Data is represented as mean with SEM. Statistical analysis was performed using Two-way ANOVA followed by Tukey's multiple comparison test. P value: *(0.033), ns-not significant (0.12).
In PPM3 neuronal clusters, UAS-parkinIR; UAS-SNCA (Figures 5D, D', E) and SNCA overexpression (Figures 5B, B', E) have shown fragmented mitochondria in 7-day-old adult fly brains as compared to control (Figures 5A, A', E). In 21-day-old adult brains of UAS-parkinIR; UAS-SNCA (Figures 5I, I', J) and SNCA overexpression (Figures 5G, G', J) have also shown fragmented mitochondria as compared to control (Figures 5F, F', J) which do not degenerate. The parkin downregulation only has shown enlarged and/or swollen mitochondria in PPM3 clusters which degenerate, in 7-day (Figures 5C, C', 5E) as well as in 21-day old adult fly brains (Figures 5H, H', J) as compared to control (Figures 5A, A', E). To confirm the role of parkin in SNCA-induced mitochondrial morphology defects, we performed mitochondrial fractionation. We observed non-statistically different reductions in mitochondrial localization of parkin protein (Supplementary Figure 3). Hence, these data confirm the role of parkin being independent of α-synuclein to cause altered mitochondrial morphology in PD progression.
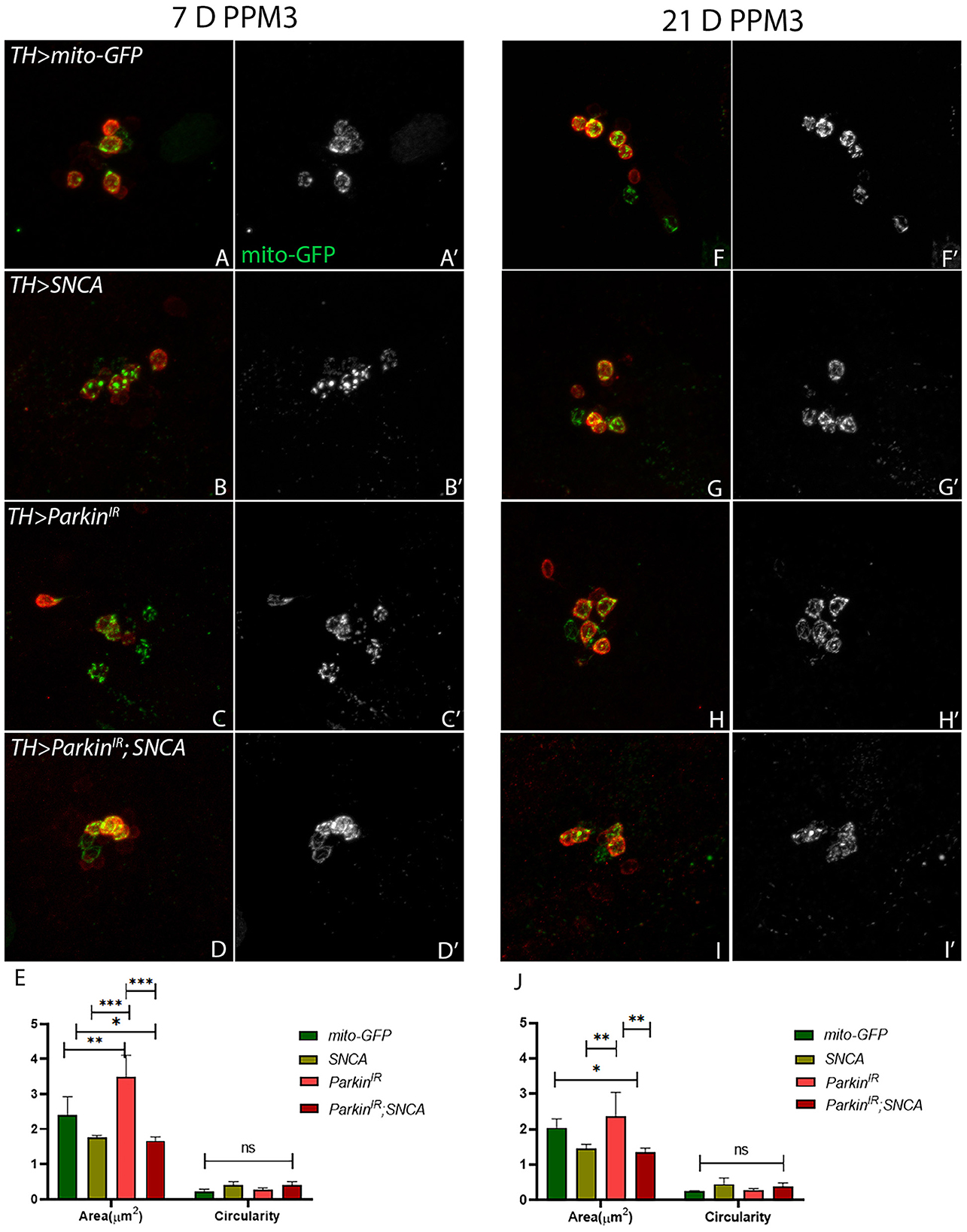
Figure 5. SNCA overexpression and parkinIR; SNCA have shown fragmented mitochondria, whereas parkinIR expression has shown elongated mitochondria in PPM3 DA clusters. Adult brains of the desired genotype expressing the mitochondria-targeted green fluorescent protein (mitoGFP) in TH- TH-positive (red) cells. (A–J) 7-day & 21-day old adult fly brains showing mitoGFP in PPM3 cluster. Control brains showing mitoGFP at (A, A') 7-day and (F, F') 21-day. SNCA overexpressing flies show mitoGFP (B, B') in 7-day and in (G, G') 21-day. ParkinIR expressing flies show mitoGFP in (C, C') 7-day and in (H, H') 21-day. ParkinIR; SNCA expressing flies show mitoGFP in (D, D') 7-day and in (I, I') 21-day also. (E) Quantification of mitochondria morphology (area and circularity) was done using ImageJ Mito-Morphology Macro. Scale bar 10 μm. A total of four adult brains were used (n = 4) per genotype. Data is represented as mean with SEM. Statistical analysis was performed using Two-way ANOVA followed by Tukey's multiple comparison test. P value: *(0.033), **(0.002), ***(<0.001), ns-not significant (0.12).
Here, we characterize the cluster-specific DA neuronal loss associated with the interaction of SNCA and parkin. We have highlighted the effect of SNCA overexpression and parkin knockdown together in terms of DA neuronal loss in PPL1 and PPM3 clusters of adult fly brains over a time period of 21 days.
SNCA and parkin mutations have been found to be involved in motor (postural instability, tremor, bradykinesia) and non-motor symptoms (sleep disorders, depression, anxiety, hallucinations) in PD patients. SNCA overexpression and parkin mutation have also been shown to cause age-dependent locomotor dysfunction and neurodegeneration in in-vivo models (Feany and Bender, 2000; Wang et al., 2007; Ordonez et al., 2018; Yan et al., 2019). In our study, SNCA overexpression and parkin knockdown in DA neurons have also shown progressive locomotor dysfunction. Interaction of SNCA and parkin in PPL1 clusters results in progressive locomotor dysfunctions. However, the effect of genetic alterations is not as pronounced to indicate an additive effect. This is further supported by observed mitochondrial morphology in UAS-parkinIR; UAS-SNCA. With respect to the reports published previously, our work uncovers the relationship between SNCA and parkin in an in-vivo system in a novel way by bringing the gene alterations together. This enables us to understand the relationship, between SNCA and parkin at both transcriptional and translational levels.
Studies have reported that wild-type SNCA causes a reduction in the number of TH-positive neurons in PPM1&2 and PPL1 clusters but not in PPM3 clusters (Trinh et al., 2008, 2010; Barone et al., 2011; Agostini et al., 2023). We have also found that SNCA overexpression has reduced the number of TH-positive neurons in PPM1&2 and PPL1 and not in PPM3, which are consistent with aforementioned reports. Flies with parkin mutation have also been shown to cause progressive loss of DA neurons in the PPL1 cluster but not in PPM3 (Whitworth et al., 2005; Wang et al., 2007) after 20 days post-eclosion. No loss of neurons has been reported in the dorsomedial clusters (DMC) (also known as PPM) in parkin loss of function mutation in the adult fly brain after 21 days post-eclosion (Pesah et al., 2004). However, in our study, parkin knockdown alone shows loss of DA neurons in PPM3 along with PPM1&2 and PPL1 clusters in 7 and 21-day-old adult fly brains. SNCA with parkin knockdown (UAS-parkinIR; UAS-SNCA) showed a decreased number of DA neurons in PPM1&2, PPL1 clusters as compared to control, though at a lesser extent with SNCA and parkin knockdown independently. Numbers of DA neurons in PPL2 clusters were unaltered in SNCA and parkin knockdown independently and together, in 7-day as well as in 21-day-old fly brains. Hence, these observations suggest that DA neuronal loss was correlated with locomotor dysfunctions. Since we did not observe aggravated phenotype in SNCA overexpression and parkin knockdown together (UAS-parkinIR; UAS-SNCA); this may suggest that SNCA doesn't affect parkin directly. Alternatively, this could also mean that parkin downregulation is not the only mechanism involved in SNCA-induced locomotor dysfunction and neurodegeneration.
Neurons have highly dynamic energy requirements, and hence intact mitochondrial morphology is an important aspect to preserve neuronal health. In post-mortem brains of Parkinson's patients, it has been shown that α-synuclein localizes to mitochondria and affects mitochondrial homeostasis (Devi et al., 2008; Nakamura et al., 2011; Wang et al., 2019; Choi et al., 2022). Although, α-synuclein does not have an exact mitochondrial targeting sequence, studies suggest that α-synuclein contains a cryptic mitochondrial targeting sequence in the N-terminus region (Devi et al., 2008). Recently, it has been reported that N-terminus of α-synuclein plays a role in mitochondrial fragmentation via a DRP1-dependent pathway in Drosophila (Krzystek et al., 2021). In Drosophila, C. elegans, dorsal root ganglia of Danio rerio (zebra fish) and in cellular models as well, SNCA overexpression causes mitochondrial fragmentation (Kamp et al., 2010; Butler et al., 2012; O'Donnell et al., 2014). In our study, SNCA overexpression has caused more elongated/or swollen mitochondria in PPL1 clusters (Supplementary Figures 4B, F), while in PPM3 clusters, it results in more fragmented mitochondria in a progressive manner. These results are thus align with the DA neuronal loss in PPL1 clusters but not in PPM3 clusters. This may also be an indication of some other mechanisms involved in rescuing the effect of SNCA overexpression and parkin knockdown together.
Loss-of-function mutations in parkin are the most prevalent cause of recessive form PD (Corti et al., 2011). Upon mitochondria depolarization, parkin is activated by PINK1 and promotes degradation of Mitofusin 1 and 2 (Poole et al., 2008; Gegg et al., 2010; Sarraf et al., 2013) and recruits Drp1 to mitochondria which leads to fission (Buhlman et al., 2014). Parkin is also involved in the selective degradation of damaged mitochondria through the mitophagy process (Pickrell and Youle, 2015). In tissues of parkin-null Drosophila mutants, swollen mitochondria have been observed and this suggests that parkin may either promote fission or inhibit fusion (Greene et al., 2003; Pesah et al., 2004). Conversely, in DA neurons of parkin knockout mice, more fragmented mitochondria have been shown to cause neuronal loss (Noda et al., 2020). The presence of parkin mutation in causing accumulation of dysfunctional mitochondria in PD patients has also been established. In our study, parkin downregulation caused the enlargement of mitochondria in PPL1 (Supplementary Figures 4C, G) and PPM3 clusters in age age-dependent manner. These results were correlating with the DA neuronal loss in PPL1 and PPM3 clusters.
Several studies have reported that overexpression of parkin restores mitochondrial morphology and function caused by SNCA, but it is still not clear whether this is through a direct link between parkin and SNCA, or the neuroprotective role of parkin in maintaining mitochondrial dynamics (Kamp et al., 2010; Lonskaya et al., 2013; Krzystek et al., 2021). In the in-vitro model, exogenous α-synuclein oligomers or fibrils caused a reduction in parkin expression and wild-type parkin overexpression rescues α-synuclein-induced mitochondrial fragmentation (Wilkaniec et al., 2021). However, they have shown that the toxic effects of α-synuclein on mitochondria were higher as compared to parkin silencing-induced mitochondrial dysfunction and suggested that α-synuclein-induced parkin downregulation is not the only mechanism for mitochondrial dysfunction (Wilkaniec et al., 2021). Similarly, in our study, overexpression of SNCA with parkin downregulation (UAS-parkinIR; UAS-SNCA) shows more fragmented mitochondria in PPL1 (Supplementary Figure 4E) as well in PPM3 clusters in age-dependent manner, which is just opposite to SNCA overexpression and parkin downregulation individually. We have found no significant changes in parkin expression at the protein level in SNCA overexpressed flies; however, parkin transcript was significantly reduced. In our fractionation studies as well, we did not see statistically significant differences in the localization of Parkin protein. This warrants that further studies need to be carried out to validate the transcriptional correlation. Also, critical differences in gene expression related to mitochondrial morphology can be explored further. It is important to note that our observations of mitochondrial morphology in specific clusters pertain to the TH-positive neurons only, based on the limited number of trials performed. Thus, further comprehension of mitochondrial morphology within these particular clusters can be attained by exploring other aspects of mitochondrial homeostasis, such as mitochondrial transport and mitophagy, in the progression of Parkinson's Disease.
The current study provides insights into cellular and molecular etiology in the case of PD in a time-dependent manner specifically in DA neurons, using an overexpression system. Since neuronal mitochondria are highly dynamic, depending on ever-changing metabolic requirements, the morphology changes driving degeneration are limited to TH-positive neurons (Figure 6). Using cell isolation techniques from individual DA neuronal clusters of brains will be able to provide more insights at the individual neuronal and organelle level since the number of these neurons is limited. Furthermore, to understand the mechanisms involved in regulation at the organelle level, more research will be required in other animals to understand if these interactions are conserved at the cellular and molecular level affecting the pathogenesis of Parkinson's disease. In addition, it would be crucial to understand if mitochondria are affected by other direct or indirect genetic and molecular factors affecting the progression of Parkinson's disease.
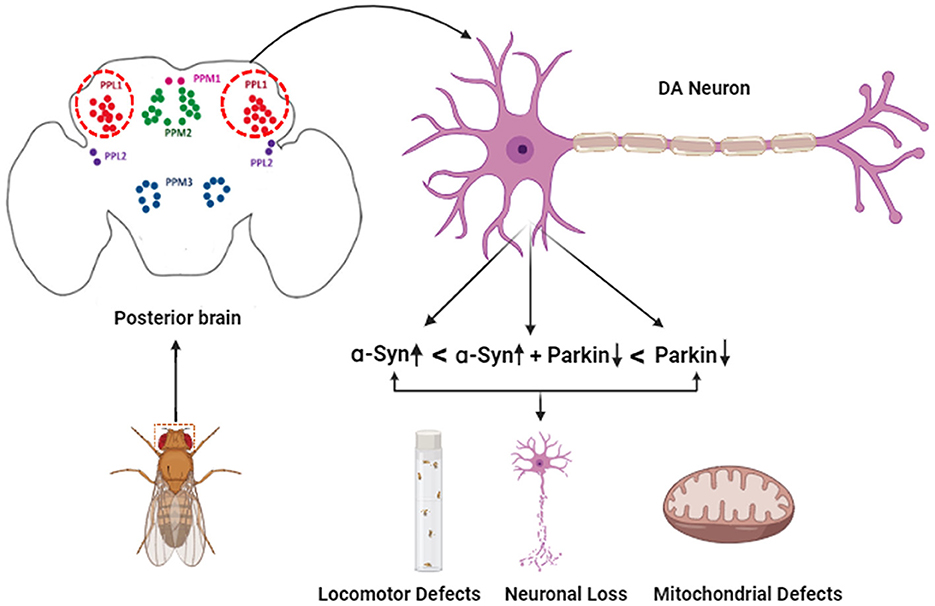
Figure 6. Schematic representation of the effect of α-syn and parkin on specific DA neuronal clusters in the adult fly brain.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
SN: Conceptualization, Data curation, Formal analysis, Investigation, Resources, Visualization, Writing – original draft. AS: Conceptualization, Writing – review & editing. MT: Conceptualization, Writing – review & editing, Project administration, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. SN is currently supported by fellowship from Indian Council of Medical Research 3/1/2/168-Neuro/22-NCD-1. In past, SN had support from Institute fellowship from Birla Institute of Technology and Science, Pilani. MT has research funding from Indian Council of Medical Research, India and Professional Development Funds from Birla Institute of Technology and Science, Pilani, supporting consumables and experimental work for this study.
We thank Prof. Lakhotia, Cytogenetic Lab, BHU, and Prof. Manish Jaiswal, Tata Institute of Fundamental Research, Hyderabad for providing fly reagents. Bloomington Drosophila Stock Centre for procuring fly stocks. BITS-Pilani, Pilani for providing initial funding to SN. The Indian Council of Medical Research (ICMR), New Delhi, India, for providing a Senior Research Fellowship to SN is greatly acknowledged. BITS-Pilani, Pilani, and ICMR for providing financial support for this study to MT.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2023.1295805/full#supplementary-material
Agostini, F., Bubacco, L., Chakrabarti, S., and Bisaglia, M. (2023). α-synuclein toxicity in drosophila melanogaster is enhanced by the presence of iron: implications for Parkinson's disease. Antioxidants 12, 261. doi: 10.3390/antiox12020261
Appel-Cresswell, S., Vilarino-Guell, C., Encarnacion, M., Sherman, H., Yu, I., Shah, B., et al. (2013). Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov. Disord. 28, 811–813. doi: 10.1002/mds.25421
Aryal, B., and Lee, Y. (2019). Disease model organism for Parkinson disease: Drosophila melanogaster. BMB Rep. 52, 250–258. doi: 10.5483/BMBRep.2019.52.4.204
Barone, M. C., Sykiotis, G. P., and Bohmann, D. (2011). Genetic activation of Nrf2 signaling is sufficient to ameliorate neurodegenerative phenotypes in a Drosophila model of Parkinson's disease. DMM Dis. Model. Mech. 4, 701–707. doi: 10.1242/dmm.007575
Bier, E. (2005). Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 6, 9–23. doi: 10.1038/nrg1503
Buhlman, L., Damiano, M., Bertolin, G., Ferrando-Miguel, R., Lombès, A., Brice, A., et al. (2014). Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. Biochim. Biophys. Acta - Mol. Cell Res. 1843, 2012–2026. doi: 10.1016/j.bbamcr.2014.05.012
Burré, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Südhof, T. C. (2010). α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Butler, E. K., Voigt, A., Lutz, A. K., Toegel, J. P., Gerhardt, E., Karsten, P., et al. (2012). The mitochondrial chaperone protein TRAP1 mitigates α-synuclein toxicity. PLoS Genet. 8. doi: 10.1371/journal.pgen.1002488
Cackovic, J., Gutierrez-Luke, S., Call, G. B., Juba, A., O'Brien, S., Jun, C. H., et al. (2018). Vulnerable parkin loss-of-function Drosophila dopaminergic neurons have advanced mitochondrial aging, mitochondrial network loss and transiently reduced autophagosome recruitment. Front. Cell. Neurosci. 12, 1–14. doi: 10.3389/fncel.2018.00039
Chartier, S., and Duyckaerts, C. (2018). Is Lewy pathology in the human nervous system chiefly an indicator of neuronal protection or of toxicity? Cell Tissue Res. 373, 149–160. doi: 10.1007/s00441-018-2854-6
Choi, M. L., Chappard, A., Singh, B. P., Maclachlan, C., Rodrigues, M., Fedotova, E. I., et al. (2022). Pathological structural conversion of α-synuclein at the mitochondria induces neuronal toxicity. Nat. Neurosci. 25, 1134–1148. doi: 10.1038/s41593-022-01140-3
Corti, O., Lesage, S., and Brice, A. (2011). What genetics tells us about the causes and mechanisms of Parkinson's disease. Physiol. Rev. 91, 1161–1218. doi: 10.1152/physrev.00022.2010
Dagda, R. K., Cherra, S. J., Kulich, S. M., Tandon, A., Park, D., and Chu, C. T. (2009). Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 284, 13843–13855. doi: 10.1074/jbc.M808515200
Dawson, T. M., Ko, H. S., and Dawson, V. L. (2010). Genetic animal models of Parkinson's disease. Neuron 66, 646–61. doi: 10.1016/j.neuron.2010.04.034
Deng, H., Dodson, M. W., Huang, H., and Guo, M. (2008). The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl. Acad. Sci. U. S. A. 105, 14503–14508. doi: 10.1073/pnas.0803998105
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson's disease brain. J. Biol. Chem. 283, 9089–9100. doi: 10.1074/jbc.M710012200
Feany, M. B., and Bender, W. W. (2000). A Drosophila model of Parkinson's disease. Nature 404, 394–8. doi: 10.1038/35006074
Gegg, M. E., Cooper, J. M., Chau, K. Y., Rojo, M., Schapira, A. H. V., and Taanman, J. W. (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870. doi: 10.1093/hmg/ddq419
Gogia, N., Sarkar, A., and Singh, A. (2017). An undergraduate cell biology lab: western blotting to detect proteins from Drosophila eye. Drosophila Inform. Serv. 100, 218–225. Available online at: https://www.ou.edu/journals/dis/DIS100/DIS%20100.pdf
Goldberg, M. S., Fleming, S. M., Palacino, J. J., Cepeda, C., Lam, H. A., Bhatnagar, A., et al. (2003). Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 278, 43628–43635. doi: 10.1074/jbc.M308947200
Greene, J. C., Whitworth, A. J., Kuo, I., Andrews, L. A., Feany, M. B., and Pallanck, L. J. (2003). Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. U. S. A. 100, 4078–4083. doi: 10.1073/pnas.0737556100
Haywood, A. F. M., and Staveley, B. E. (2004). Parkin counteracts symptoms in a Drosophila model of Parkinson's disease. BMC Neurosci. 5, 1–12. doi: 10.1186/1471-2202-5-14
Heisenberg, M. (2003). Mushroom body memoir: from maps to models. Nat. Rev. Neurosci. 4, 266–275. doi: 10.1038/nrn1074
Jeśko, H., Lenkiewicz, A. M., Wilkaniec, A., and Adamczyk, A. (2019). The interplay between parkin and alpha-synuclein; possible implications for the pathogenesis of Parkinson's disease. Acta Neurobiol. Exp. (Wars). 79, 279–289. doi: 10.21307/ane-2019-026
Johansen, K. K., Torp, S. H., Farrer, M. J., Gustavsson, E. K., and Aasly, J. O. (2018). A case of Parkinson's disease with no lewy body pathology due to a homozygous exon deletion in parkin. Case Rep. Neurol. Med. 2018, 1–4. doi: 10.1155/2018/6838965
Jost, W. H., and Reichmann, H. (2017). “An essay on the shaking palsy” 200 years old. J. Neural Transm. 124, 899–900. doi: 10.1007/s00702-017-1684-0
Kachergus, J., Roumier, C., Mouroux, V., Douay, X., Lincoln, S., and Levecque, C. (2004). α-synuclein locus duplication as a cause of familial Parkinson's disease. 07, 1167–1169. doi: 10.1016/S0140-6736(04)17103-1
Kamp, F., Exner, N., Lutz, A. K., Wender, N., Hegermann, J., Brunner, B., et al. (2010). Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 29, 3571–3589. doi: 10.1038/emboj.2010.223
Khandelwal, P. J., Dumanis, S. B., Feng, L. R., Maguire-Zeiss, K., Rebeck, G., Lashuel, H. A., et al. (2010). Parkinson-related parkin reduces α-Synuclein phosphorylation in a gene transfer model. Mol. Neurodegener. 5, 47. doi: 10.1186/1750-1326-5-47
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–8. doi: 10.1038/33416
Krzystek, T. J., Banerjee, R., Thurston, L., Huang, J. Q., Swinter, K., Rahman, S. N., et al. (2021). Differential mitochondrial roles for α-synuclein in DRP1-dependent fission and PINK1/Parkin-mediated oxidation. Cell Death Dis. 12, 1–16. doi: 10.1038/s41419-021-04046-3
Li, J. Y., Englund, E., Holton, J. L., Soulet, D., Hagell, P., Lees, A. J., et al. (2008). Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503. doi: 10.1038/nm1746
Lonskaya, I., Desforges, N. M., Hebron, M. L., and Moussa, C. E. H. (2013). Ubiquitination increases parkin activity to promote autophagic a-synuclein clearance. PLoS ONE 8. doi: 10.1371/journal.pone.0083914
Madsen, D. A., Schmidt, S. I., Blaabjerg, M., and Meyer, M. (2021). Interaction between parkin and α-synuclein in park2-mediated Parkinson's disease. Cells 10, 1–30. doi: 10.3390/cells10020283
Mao, Z., and Davis, R. L. (2009). Eight different types of dopaminergic neurons innervate the Drosophila mushroom body neuropil: anatomical and physiological heterogeneity. Front. Neural Circuits 3, 1–17. doi: 10.3389/neuro.04.005.2009
Mehta, A. S., Luz-Madrigal, A., Li, J.-L., Tsonis, P. A., and Singh, A. (2019). Comparative transcriptomic analysis and structure prediction of novel Newt proteins. PLoS ONE 14, e0220416. doi: 10.1371/journal.pone.0220416
Mohite, G. M., Dwivedi, S., Das, S., Kumar, R., Paluri, S., Mehra, S., et al. (2018). Parkinson's disease associated α-synuclein familial mutants promote dopaminergic neuronal death in Drosophila melanogaster. ACS Chem. Neurosci. 9, 2628–2638. doi: 10.1021/acschemneuro.8b00107
Monastirioti, M. (1999). Biogenic amine systems in the fruit fly Drosophila melanogaster. Microsc. Res. Tech. 45, 106–121.
Nakamura, K. (2013). α-Synuclein and mitochondria: partners in crime? Neurotherapeutics 10, 391–9. doi: 10.1007/s13311-013-0182-9
Nakamura, K., Nemani, V. M., Azarbal, F., Skibinski, G., Levy, J. M., Egami, K., et al. (2011). Direct membrane association drives mitochondrial fission by the Parkinson's disease-associated protein α-synuclein. J. Biol. Chem. 286, 20710–26. doi: 10.1074/jbc.M110.213538
Nicoletti, V., Palermo, G., Del Prete, E., Mancuso, M., and Ceravolo, R. (2021). Understanding the multiple role of mitochondria in Parkinson's disease and related disorders: lesson from genetics and protein–interaction network. Front. Cell Dev. Biol. 9, 1–20. doi: 10.3389/fcell.2021.636506
Noda, S., Sato, S., Fukuda, T., Tada, N., Uchiyama, Y., Tanaka, K., et al. (2020). Loss of Parkin contributes to mitochondrial turnover and dopaminergic neuronal loss in aged mice. Neurobiol. Dis. 136, 104717. doi: 10.1016/j.nbd.2019.104717
O'Donnell, K. C., Lulla, A., Stahl, M. C., Wheat, N. D., Bronstein, J. M., and Sagasti, A. (2014). Axon degeneration and PGC-1α-mediated protection in a zebrafish model of α-synuclein toxicity. DMM Dis. Model. Mech. 7, 571–582. doi: 10.1242/dmm.013185
Olanow, C. W., and Brundin, P. (2013). Parkinson's disease and alpha synuclein: is parkinson's disease a prion-like disorder? Mov. Disord. 28, 31–40. doi: 10.1002/mds.25373
Oluwatosin-Chigbu, Y., Robbins, A., Scott, C. W., Arriza, J. L., Reid, J. D., and Zysk, J. R. (2003). Parkin suppresses wild-type α-synuclein-induced toxicity in SHSY-5Y cells. Biochem. Biophys. Res. Commun. 309, 679–684. doi: 10.1016/j.bbrc.2003.08.059
Ordonez, D. G., Lee, M. K., and Feany, M. B. (2018). α-synuclein induces mitochondrial dysfunction through spectrin and the actin cytoskeleton. Neuron 97, 108–124.e6. doi: 10.1016/j.neuron.2017.11.036
Paricio, N., and Muñoz-Soriano, V. (2011). Drosophila models of Parkinson's disease: Discovering relevant pathways and novel therapeutic strategies. Parkinsons. Dis. 2011, 520640. doi: 10.4061/2011/520640
Pendleton, R. G., Parvez, F., Sayed, M., and Hillman, R. (2002). Effects of pharmacological agents upon a transgenic model of Parkinson's disease in Drosophila melanogaster. J. Pharmacol. Exp. Ther. 300, 91–96. doi: 10.1124/jpet.300.1.91
Pesah, Y., Pham, T., Burgess, H., Middlebrooks, B., Verstreken, P., Zhou, Y., et al. (2004). Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 131, 2183–2194. doi: 10.1242/dev.01095
Petrucelli, L., O'Farrell, C., Lockhart, P. J., Baptista, M., Kehoe, K., Vink, L., et al. (2002). Parkin protects against the toxicity associated with mutant α-Synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 36, 1007–1019. doi: 10.1016/S0896-6273(02)01125-X
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in parkinson's disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Poole, A. C., Thomas, R. E., Andrews, L. A., McBride, H. M., Whitworth, A. J., and Pallanck, L. J. (2008). The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. U. S. A. 105, 1638–1643. doi: 10.1073/pnas.0709336105
Pramstaller, P. P., Schlossmacher, M. G., Jacques, T. S., Scaravilli, F., Eskelson, C., Pepivani, I., et al. (2005). Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Ann. Neurol. 58, 411–422. doi: 10.1002/ana.20587
Ray Dorsey, E., Elbaz, A., Nichols, E., Abd-Allah, F., Abdelalim, A., Adsuar, J. C., et al. (2018). Global, regional, and national burden of Parkinson's disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 17, 939–953. doi: 10.1016/S1474-4422(18)30295-3
Sarraf, S. A., Raman, M., Guarani-Pereira, V., Sowa, M. E., Huttlin, E. L., Gygi, S. P., et al. (2013). Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376. doi: 10.1038/nature12043
Shahmoradian, S. H., Lewis, A. J., Genoud, C., Hench, J., Moors, T. E., Navarro, P. P., et al. (2019). Lewy pathology in Parkinson's disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 22, 1099–1109. doi: 10.1038/s41593-019-0423-2
Strausfeld, N. J., and Hirth, F. (2013). Deep homology of arthropod central complex and vertebrate basal ganglia. Science (80-.). 340, 157–161. doi: 10.1126/science.1231828
Strauss, R. (2002). The central complex and the genetic dissection of locomotor behaviour. Curr. Opin. Neurobiol. 12, 633–638. doi: 10.1016/S0959-4388(02)00385-9
Tare, M., Modi, R. M., Nainaparampil, J. J., Puli, O. R., Bedi, S., Fernandez-funez, P., et al. (2011). Activation of JNK signaling mediates amyloid-ß- dependent cell. Death. 6, 1–12. doi: 10.1371/journal.pone.0024361
Tito, A. J., Cheema, S., Jiang, M., and Zhang, S. (2016). A simple one-step dissection protocol for whole-mount preparation of adult drosophila brains. J. Vis. Exp. 2016, 55128. doi: 10.3791/55128-v
Trinh, K., Andrews, L., Krause, J., Hanak, T., Lee, D., Gelb, M., et al. (2010). Decaffeinated coffee and nicotine-free tobacco provide neuroprotection in Drosophila models of Parkinson's disease through an NRF2-dependent mechanism. J. Neurosci. 30, 5525–5532. doi: 10.1523/JNEUROSCI.4777-09.2010
Trinh, K., Moore, K., Wes, P. D., Muchowski, P. J., Dey, J., Andrews, L., et al. (2008). Induction of the phase II detoxification pathway suppresses neuron loss in Drosophila models of Parkinson's disease. J. Neurosci. 28, 465–472. doi: 10.1523/JNEUROSCI.4778-07.2008
Van Rompuy, A. S., Oliveras-Salvá, M., Van Der Perren, A., Corti, O., Van Den Haute, C., and Baekelandt, V. (2015). Nigral overexpression of alpha-synuclein in the absence of parkin enhances alpha-synuclein phosphorylation but does not modulate dopaminergic neurodegeneration. Mol. Neurodegener. 10, 1–14. doi: 10.1186/s13024-015-0017-8
Wang, C., Lu, R., Ouyang, X., Ho, M. W. L., Chia, W., Yu, F., et al. (2007). Drosophila overexpressing parkin R275W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities. J. Neurosci. 27, 8563–8570. doi: 10.1523/JNEUROSCI.0218-07.2007
Wang, X., Becker, K., Levine, N., Zhang, M., Lieberman, A. P., Moore, D. J., et al. (2019). Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 7, 41. doi: 10.1186/s40478-019-0696-4
White, K. E., Humphrey, D. M., and Hirth, F. (2010). The dopaminergic system in the aging brain of Drosophila. Front. Neurosci. 4, 1–12. doi: 10.3389/fnins.2010.00205
Whitworth, A. J., Theodore, D. A., Greene, J. C., Bene,š, H., Wes, P. D., and Pallanck, L. J. (2005). Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc. Natl. Acad. Sci. U. S. A. 102, 8024–8029. doi: 10.1073/pnas.0501078102
Wilkaniec, A., Lenkiewicz, A. M., Babiec, L., Murawska, E., Jeśko, H. M., Cieślik, M., et al. (2021). Exogenous alpha-synuclein evoked parkin downregulation promotes mitochondrial dysfunction in neuronal cells. implications for Parkinson's disease pathology. Front. Aging Neurosci. 13, 1–21. doi: 10.3389/fnagi.2021.591475
Wilkaniec, A., Lenkiewicz, A. M., Czapski, G. A., Jeśko, H. M., Hilgier, W., Brodzik, R., et al. (2019). Extracellular alpha-synuclein oligomers induce parkin s-nitrosylation: relevance to sporadic Parkinson's disease etiopathology. Mol. Neurobiol. 56, 125–140. doi: 10.1007/s12035-018-1082-0
Wood-Kaczmar, A., Gandhi, S., and Wood, N. W. (2006). Understanding the molecular causes of Parkinson's disease. Trends Mol. Med. 12, 521–528. doi: 10.1016/j.molmed.2006.09.007
Yan, C., Liu, J., Gao, J., Sun, Y., Zhang, L., Song, H., et al. (2019). IRE1 promotes neurodegeneration through autophagy-dependent neuron death in the Drosophila model of Parkinson's disease. Cell Death Dis. 10. doi: 10.1038/s41419-019-2039-6
Yang, Y., Nishimura, I., Imai, Y., Takahashi, R., and Lu, B. (2003). Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron 37, 911–924. doi: 10.1016/S0896-6273(03)00143-0
Yu, W., Sun, Y., Guo, S., and Lu, B. (2011). The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum. Mol. Genet. 20, 3227–3240. doi: 10.1093/hmg/ddr235
Keywords: α-synuclein, parkin, mitochondrial morphology, dopaminergic neurons, Parkinson's disease, tyrosine hydroxylase, Drosophila melanogaster
Citation: Narwal S, Singh A and Tare M (2024) Analysis of α-syn and parkin interaction in mediating neuronal death in Drosophila model of Parkinson's disease. Front. Cell. Neurosci. 17:1295805. doi: 10.3389/fncel.2023.1295805
Received: 17 September 2023; Accepted: 01 December 2023;
Published: 04 January 2024.
Edited by:
Neville Ng, University of Wollongong, AustraliaReviewed by:
Venkatachalam Deepa Parvathi, Sri Ramachandra Institute of Higher Education and Research, IndiaCopyright © 2024 Narwal, Singh and Tare. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meghana Tare, bWVnaGFuYS50YXJlQHBpbGFuaS5iaXRzLXBpbGFuaS5hYy5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.