 Kazuyuki Fujihara
Kazuyuki Fujihara
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci., 24 April 2023
Sec. Cellular Neuropathology
Volume 17 - 2023 | https://doi.org/10.3389/fncel.2023.1161608
This article is part of the Research TopicMapping the Pathophysiology of Schizophrenia: Interactions Between Multiple Cellular Pathways, Volume IIView all 8 articles
Abnormalities in the γ-aminobutyric acid (GABA) system have been reported in the postmortem brains of individuals with schizophrenia. In particular, the reduction of one of the GABA-synthesizing enzymes, the 67-kDa isoform of glutamate decarboxylase (GAD67), has garnered interest among researchers because of its role in the formation of γ-oscillations and its potential involvement in the cognitive dysfunction observed in schizophrenia. Although several animal models have been generated to simulate the alterations observed in postmortem brain studies, they exhibit inconsistent behavioral phenotypes, leading to conflicting views regarding their contributions to the pathogenesis and manifestation of schizophrenia symptoms. For instance, GAD67 knockout rats (also known as Gad1 knockout rats) exhibit marked impairments in spatial working memory, but other model animals do not. In this review, we summarize the phenotypic attributes of these animal models and contemplate the potential for secondary modifications that may arise from the disruption of the GABAergic nervous system.
Schizophrenia is one of the most common psychiatric disorders with a lifetime morbid risk of approximately 0.72% (Saha et al., 2005). Although there are exceptions, schizophrenia generally develops during adolescence and young adulthood and has a chronic course. The symptoms of schizophrenia are highly varied; however, there exist several characteristic clusters of symptoms (Liddle, 1987). The first cluster is called the positive symptom or reality distortion syndrome. This comprises hallucinations and delusions. The second is negative symptoms or psychomotor poverty syndrome, which includes avolition and poor emotional expression (Nakaya and Ohmori, 2008; Correll and Schooler, 2020). The third is the disorganization syndrome, which represents conceptual disorganization and bizarre behavior. In addition, impairments in cognitive function, such as working memory deficits, are considered important prognostic factors (Keefe and Harvey, 2012). Psychomotor poverty and disorganization syndrome are also associated with cognitive impairment (Altshuler et al., 2004; Liddle, 2019). It is noteworthy that these various symptoms are not unique to schizophrenia but also occur with certain types of encephalitis and substance abuse (e.g., amphetamines and phencyclidine). Therefore, clinicians must rule out organic diseases or drug effects during diagnosis (American Psychiatric Association, 2013). Conversely, this implies that unknown biological factors may be responsible for schizophrenia. Indeed, since Kraepelin’s conceptualization, it has been assumed that there is some biological basis for this disorder (Hoenig, 1983). Therefore, several pathophysiological hypotheses have been proposed.
The classical hypothesis of schizophrenia is the dopamine hypothesis (Howes and Kapur, 2009). This was proposed based on the finding that antipsychotics are dopamine D2 receptor antagonists (Meltzer and Stahl, 1976; Seeman, 2021). The dopamine hypothesis is consistent with the fact that amphetamines, which increase extracellular dopamine levels, produce positive symptom-like symptoms. However, the presence of treatment-resistant cases and the limited efficacy of the drug in treating negative symptoms and cognitive dysfunction make it difficult to explain the mechanisms of schizophrenia through the dopaminergic system alone (Correll and Schooler, 2020). Another promising hypothesis is the glutamate hypothesis (Uno and Coyle, 2019), as the N-methyl-D-aspartate (NMDA)-type glutamate receptor antagonist phencyclidine causes both positive and negative symptoms. In addition, encephalitis, in which autoantibodies to the NMDA receptor are produced, causes psychiatric symptoms similar to schizophrenia (Dalmau et al., 2011). Although various other pathological hypotheses have been proposed, a unified understanding has not yet been reached. The concept of schizophrenia is based on its symptoms and course, and there are currently no biomarkers. It is crucial to keep in mind that there is no evidence that schizophrenia is a single biological disease but only a clinical syndrome.
As mentioned above, dopamine and glutamate hypotheses were proposed based on pharmacological findings. In contrast, the γ-aminobutyric acid (GABA) hypothesis is based on results in postmortem brain studies (Gonzalez-Burgos and Lewis, 2008; Lewis and Sweet, 2009; Dienel et al., 2022). GABA, an inhibitory neurotransmitter, is biosynthesized by decarboxylation of glutamate (Bu et al., 1992; Soghomonian and Martin, 1998; Obata, 2013). This reaction is catalyzed by glutamate decarboxylase (GAD), which is encoded by two different genes, GAD1 and GAD2 in humans (Gad1 and Gad2 in rodents). These genes correspond to the 67-kDa isoform (GAD67) and the 65-kDa isoform (GAD65), respectively. Of the two isoforms, GAD67 has been reported to be decreased in the postmortem brains of individuals with schizophrenia. In addition, other marker molecules associated with GABAergic neurons are also altered in expression. The replicability of GABAergic nervous system disturbances observed in the cerebral cortex and hippocampus has attracted the attention of many researchers over the past two decades. In particular, GABAergic systems are involved in working memory through the formation of γ-oscillations, which have been postulated to be associated with cognitive dysfunction in schizophrenia (Gonzalez-Burgos et al., 2010).
In recent years, the pathologies in the neurotransmitters and neurons as well as glial cells, including microglia (Volk, 2017; Murphy et al., 2021; Parellada and Gassó, 2021; Dienel et al., 2022) and astrocyte (Notter, 2021; de Oliveira Figueiredo et al., 2022), have also been implicated in schizophrenia. For example, the activation of microglia has been reported in schizophrenia (Radewicz et al., 2000; Wierzba-Bobrowicz et al., 2005; Fillman et al., 2013; Bloomfield et al., 2016; Marques et al., 2019). Interestingly, some microglia express GABA receptors and can monitor ambient GABA concentrations (Kuhn et al., 2004). Therefore, some researchers refer to GABA as a “neurotransmitter” that mediates signaling between neurons and microglia (Barragan et al., 2015). Impairment of GABAergic neurotransmission in schizophrenia can affect glial cells.
With these considerations in mind, this mini-review first outlines the GABA hypothesis and the animal models based on this hypothesis. This review will primarily focus on GAD67 because it directly affects GABA synthesis, has been independently reported to be downregulated in different postmortem brain banks, and various types of genetically modified animal models have been generated. Further, we discuss the potential of the neuroimmune system as a critical player that interacts with the GABAergic nervous system to further the pathogenesis of the disorder and discuss directions for future research.
In the 1970s, a reduction in GAD activity and a decrease in GABA content in the nucleus accumbens and thalamus were reported, which marked the inception of the GABA hypothesis (Bird et al., 1977; Perry et al., 1979). Later, brain banks were established, and the mRNA and protein levels of genes related to the GABAergic nervous system were more precisely analyzed. In the prefrontal cortex of schizophrenia, many studies have documented a reduction in GAD67 mRNA (Akbarian et al., 1995; Guidotti et al., 2000; Volk et al., 2000; Hashimoto et al., 2003, 2008b,a; Veldic et al., 2005; Ruzicka et al., 2007; Tsubomoto et al., 2019) or protein levels (Guidotti et al., 2000; Curley et al., 2011, 2013; Fish et al., 2021). The expression of GAD67 has also been reported to be decreased in the temporal cortex (Impagnatiello et al., 1998), the hippocampus (Heckers et al., 2002), and the cingulate cortex (Woo et al., 2007, 2012). Although it has received little attention, a recent report also suggested decreased level of GAD65 in GABAergic terminals (Fish et al., 2021). However, GAD65 is outside the scope of this review. It has also been reported that the impairment of GABAergic neurons is accompanied by decreased input from excitatory neurons (Chung et al., 2016a,b). These findings are illustrated in Figures 1A, B.
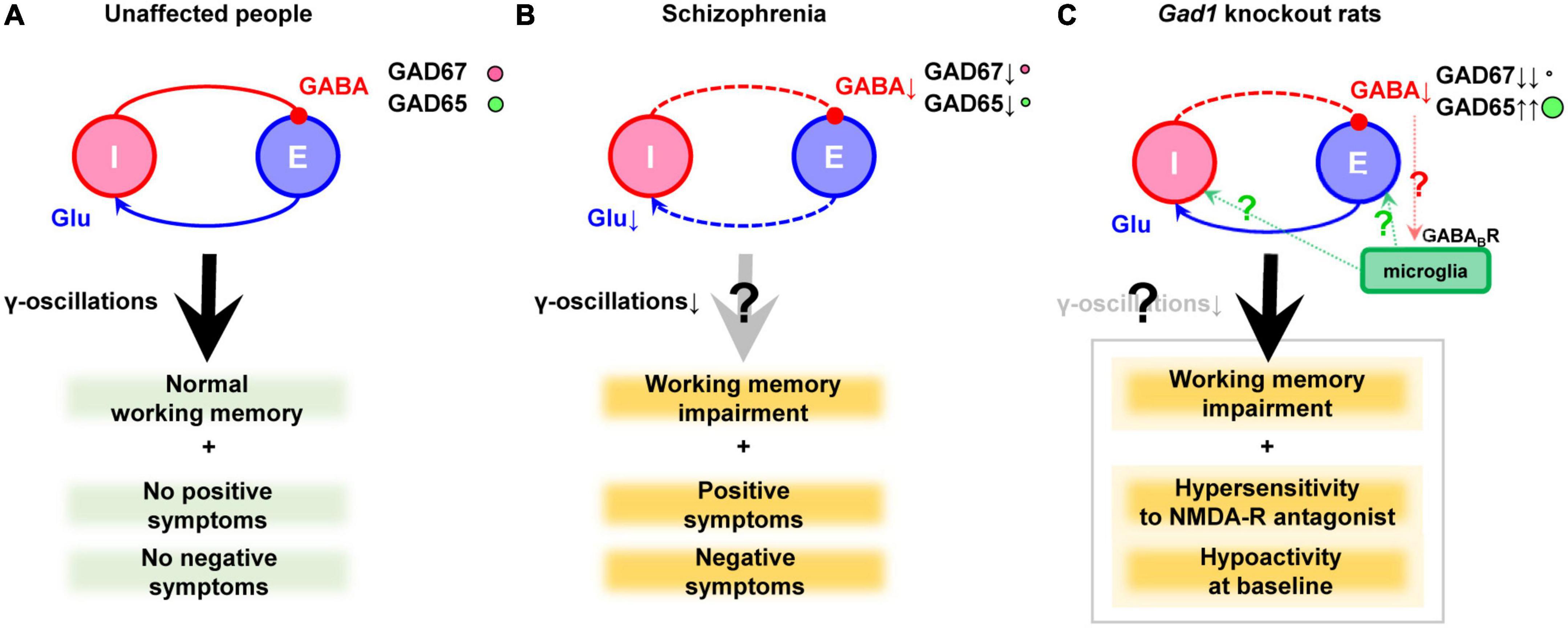
Figure 1. GABAergic dysfunction in the cortex of schizophrenia and Gad1 knockout rats. (A) Balanced neurotransmission of excitatory (E) glutamate (Glu) and inhibitory (I) GABA. (B) Impaired inhibitory transmission caused by GAD67 (plus GAD65) reduction in schizophrenia. Deficient excitatory input probably exists as well. Notably, the cause and result relationship between the impaired transmission and clinical symptoms cannot be demonstrated only postmortem studies alone. (C) Deficient inhibitory transmission caused by complete loss of GAD67 in Gad1 knockout rats. The compensatory increase in GAD65 cannot prevent behavioral abnormalities relevant to schizophrenia. The presence or absence of γ-oscillations abnormalities has not yet been verified. In addition, a hypothetical involvement of microglia is also shown (see the main text). Causal relationships that have not been directly proven are indicated with question marks to denote their hypothetical nature.
Decreased GAD67 expression is particularly prominent in parvalbumin-positive cells (PV neurons) among the inhibitory interneurons (Hashimoto et al., 2003; Curley et al., 2011). PV neurons are major GABAergic neurons, accounting for approximately 25% of all GABAergic neurons in humans and 40% in rodents (Tamamaki et al., 2003; Uematsu et al., 2008). Since PV neurons (especially PV basket cells) play an essential role in synchronizing action potentials within the neuronal population during working memory tasks (γ-oscillations; Gonzalez-Burgos and Lewis, 2008; Gonzalez-Burgos et al., 2010), GAD67 reduction is hypothesized to be the origin of cognitive impairment in schizophrenia. In addition to GAD67, PV expression is reduced in the postmortem brains of patients with schizophrenia (Hashimoto et al., 2003). Other findings include decreased expression of somatostatin, another marker of GABAergic neuron subtypes, reduced expression of the GABA-A receptor α1 subunit, the δ subunit, GABA transporter (GAT-1), neuropeptide Y, and cholecystokinin (Hashimoto et al., 2008a,b). The reduced expression of GAT-1 has been suggested to occur at PV neuron terminals (Bitanihirwe and Woo, 2014), further supporting the involvement of PV neurons in the pathogenesis of schizophrenia.
Moreover, attempts have been made to measure GABA concentrations in the brains of people with schizophrenia in vivo. Nuclear magnetic resonance spectroscopy (MRS) has been used to detect and quantify substances in the human brain. In standard clinical MRS, traces of GABA are buried in the signals of other substances, but a particular sequence called MEGA-PRESS makes it possible to measure it (Rothman et al., 1993). Our group has also used this technique to reveal correlations between impulse control and GABA concentrations in healthy subjects and discussed their relationship with a predisposition to psychiatric disorders (Fujihara et al., 2015b). Several GABA measurements have also been made in schizophrenia using GABA-MRS (Yoon et al., 2010, 2020). A recent meta-analysis supports a decrease in GABA concentrations in the cingulate gyrus in first-episode psychosis (Nakahara et al., 2022). Despite variability across studies (Egerton et al., 2017), the meta-analysis results are consistent with the reduction of GABA synthesis suggested by postmortem brain studies. It has also been reported that GABA concentrations in the dorsolateral prefrontal cortex are positively associated with working memory, although these findings were only found in healthy individuals (Yoon et al., 2016).
A genome-wide association study identified 108 loci associated with schizophrenia (Ripke et al., 2014). The list included genes that could be related to GABAergic neurons but not GAD67 or PV. In contrast, smaller genetic studies have shown that single nucleotide polymorphisms (SNPs) and loss-of-function mutations in the GAD1 gene were involved in GAD67 expression levels and schizophrenia (Addington et al., 2005; Straub et al., 2007; Du et al., 2008; Hyde et al., 2011; Giacopuzzi et al., 2017; Magri et al., 2018). These results suggest that there is no direct GAD1 gene vulnerability in all cases of schizophrenia, but rather that the reduction in GAD67 occurs because of other biological factors. For example, maternal infection, a risk factor for schizophrenia (Brown and Derkits, 2010), can reduce GAD67 levels (Giovanoli et al., 2015; Volk, 2017). Genetic variation in C4, a component of the complement system in the innate immune system, has been associated with an increased risk of developing schizophrenia (Sekar et al., 2016). The observed decrease in GAD67 expression in mice overexpressing C4 protein suggests that changes in the neuroimmune system may influence GAD67 expression (Druart et al., 2021). Other upstream factors, such as decreased input to GABAergic neurons (Akbarian and Huang, 2006), and a reduction in Zif268, a transcription factor that regulates GAD67 expression, in postmortem brains of individuals with schizophrenia have also been reported (Kimoto et al., 2014). Even if upstream biological processes vary, the consistent decrease in GAD67 found in postmortem brain studies may represent a common pathophysiology of the syndrome.
Given that the aforementioned studies were observational in nature, it can be challenging to definitively determine the psychiatric symptoms resulting from decreased GAD67 or GABA levels. However, this can be achieved through the creation of animal models in which GABA synthesis has been experimentally reduced, and its behavior has been examined. This section discusses the phenotypes of rodents with diminished GAD67 expression (Table 1).
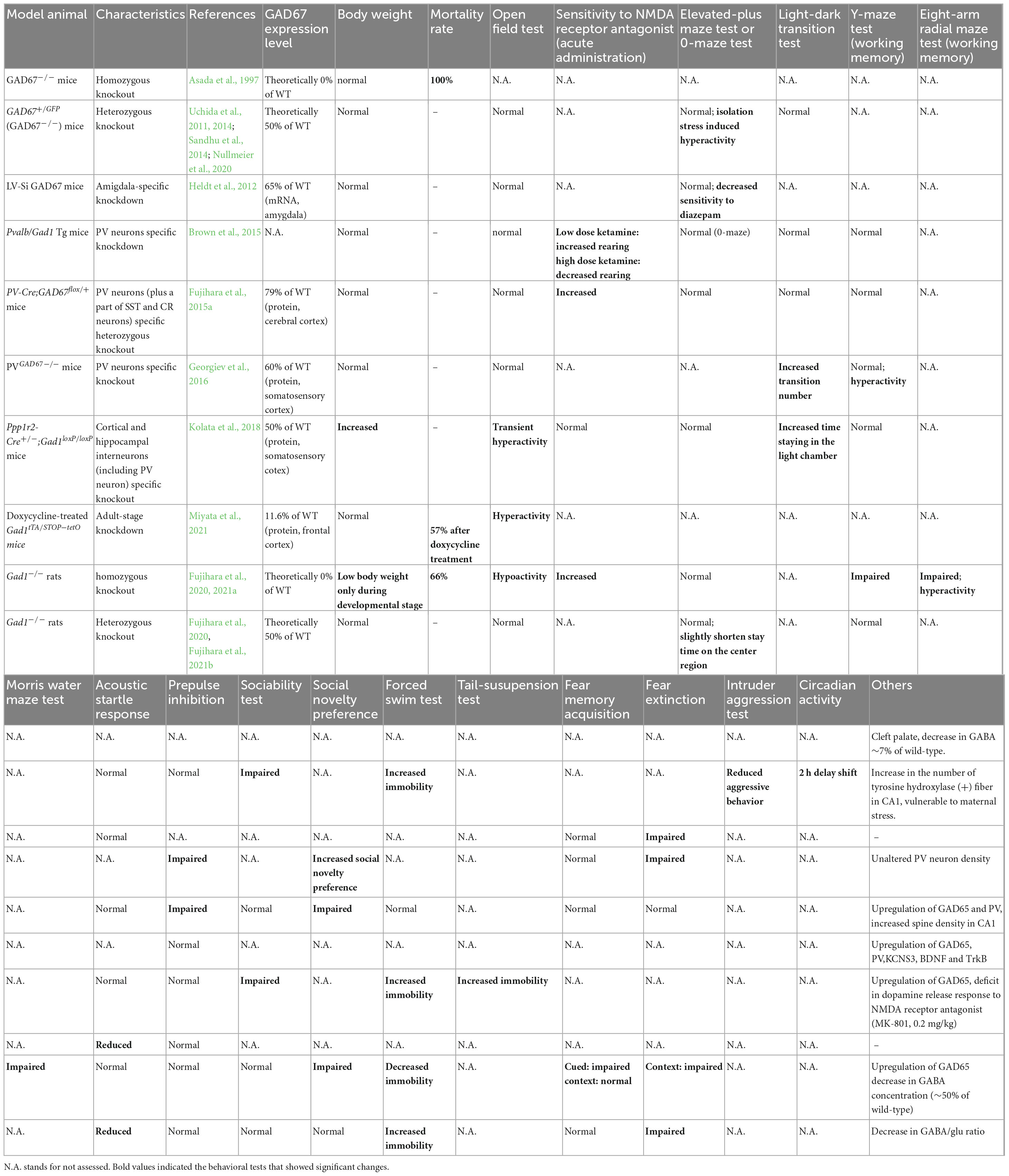
Table 1. Behavioral phenotype comparison of Gad1 (GAD67) knockout/knockdown animals.
The ablation of GAD67 in mice (GAD67 knockout mice, also known as Gad1–/– mice) resulted in a reduction in GABA levels to 7% of that observed in wild-type mice. As embryonic GAD67 serves as the primary GABA-producing enzyme in mice, its complete loss is detrimental to survival, resulting in the manifestation of cleft palate and death shortly after birth (Asada et al., 1997; Kakizaki et al., 2015). Consequently, behavioral analysis of adult animals is infeasible, rendering GAD67 knockout mice unsuitable as a model for investigating psychiatric disorders.
In contrast, GAD67 heterozygous knockout mice (Gad1–/– mice) survive to adulthood. Female mutants demonstrate increased vulnerability to stress during pregnancy, which leads to reduced body weight in their offspring (Uchida et al., 2011). Furthermore, GAD67 heterozygote fetuses exhibit elevated corticosterone levels and lower body weight than wild-type littermates. Examination of the offspring after birth revealed that the number of PV neurons in the prefrontal cortex was reduced in GAD67 heterozygous knockout mice subjected to maternal stress (Uchida et al., 2014). Regrettably, behavioral experiments have yet to be performed on these mice, thus the presence or absence of behavior analogous to schizophrenia remains unknown. GAD67 heterozygous knockout mice exhibit decreased sociability and aggressiveness in the absence of maternal stress (Sandhu et al., 2014), as well as elevated immobility during the forced swim test (Nullmeier et al., 2020). Furthermore, social isolation stress elicits an increase in locomotor activity in GAD67 heterozygous knockout mice, but does not elicit a similar response in their wild-type littermates. Impaired social behavior and stress-induced hyperactivity may reflect negative and positive symptom-like behaviors, respectively. Histological analysis revealed that tyrosine hydroxylase-positive nerve fibers in the hippocampal CA1 region were increased, suggesting dopaminergic neural changes even under maternal stress-free conditions (Nullmeier et al., 2020). In summary, while the phenotype of Gad1–/– mice exhibits some similarities to schizophrenia, the effect it has on working memory remained inconclusive.
As noted earlier, the postmortem brains of patients with schizophrenia show a significant decrease in GAD67 expression, especially in PV neurons. Several groups have created conditional knockout mice that mimic this finding. Two of these mouse models (Pvalb/Gad1-silencing mice and PV-Cre; GAD67+/flox mice) showed altered prepulse inhibition and responsiveness to NMDA receptor antagonists and a partially schizophrenia-like phenotype but no apparent working memory impairment (Brown et al., 2015; Fujihara et al., 2015a). However, because both mouse models were evaluated only with the Y-maze task, a very simple spatial working memory task, more sensitive tasks such as the eight-arm radial maze test will be needed (Olton and Samuelson, 1976; David and Olton, 1979). Another strain of PV neuron-specific GAD67 knockout mice (PVGAD67–/– mice), reported by Georgiev et al. (2016), showed a phenotype different from the first two, as they did not show impaired prepulse inhibition. Moreover, their locomotor activity increased in the light/dark transition and Y-maze tests without affecting working memory. They analyzed the cerebral cortex of mice using a method similar to that used in postmortem brain studies (in situ hybridization). They found increased expression of PV, GAD65, brain-derived neurotrophic factor (BDNF), and TrkB, a profile that differs from the actual postmortem findings in schizophrenia. Among these, the increased expression of PV and GAD65 was consistent with the mouse model of Fujihara et al. (2015a). As well-known, the BDNF-TrkB signaling pathway was attenuated in schizophrenia. From this, they speculated that the reduced expression of GAD67 in PV neurons may be downstream of the disease pathway and not enough to produce schizophrenia-like behaviors and histological characteristics (Georgiev et al., 2016). Another study generated a conditional knockout mouse that covers more widespread GABAergic neurons, including PV neurons (Ppp1r2-Cre+/–; Gad1loxP/loxP; Kolata et al., 2018). This mouse model is inconsistent in its behavioral phenotype with the first two conditional knockout mice, as it shows no impairment in prepulse inhibition and does not respond to NMDA receptor antagonist administration. However, it displayed increased mobility in the forced swim test and reduced sociability in the social interaction test, showing similarity to GAD67 conventional heterozygous knockout mice. In addition, this mouse model had no tail-suspension-induced dopamine release in the anterior cingulate cortex. It has been noted that these findings may be related to negative symptoms of schizophrenia (Kolata et al., 2018).
Site-specific GAD67 knockdown mice also provided insight into the association between GAD67 and psychiatric symptoms. A lentiviral RNA interference strategy was used to perform amygdala-specific knockdown of GAD67 (Heldt et al., 2012). The model mice displayed impaired fear memory elimination, as in Pvalb/Gad1-silencing mice (Table 1). Time-specific GAD67 global knockdown mice have also been developed. Gad1tTA/STOP–tetO mice show a progressive loss of GAD67 expression upon doxycycline treatment at the adult stage (Miyata et al., 2021). This process causes mice to exhibit hyperlocomotion and anxiety-like behaviors. These results suggest that a decrease in GAD67 also results in emotional abnormalities.
Although the model animals described above are mice, recently, GAD67 knockout rats (Gad1–/– rats) have been generated using the CRISPR/Cas9 system. Unlike Gad1–/– mice, they do not exhibit cleft palate, and approximately 33% of individuals can grow into adults (Fujihara et al., 2020; Jiang et al., 2022). The mortality rate of Gad1–/– rats after adulthood is the same as that of wild-type rats, allowing for long-term breeding and behavioral tests. Gad1–/– rats show reduced spontaneous locomotion, hypersensitivity to NMDA receptor antagonists, and impaired social recognition memory, spatial reference memory, spatial working memory, and cued-fear memory (Fujihara et al., 2020, Fujihara et al., 2021b). These symptoms may correspond to negative symptoms, positive symptoms, and cognitive dysfunction. This rat model is the only one that exhibits a distinct working memory deficit among GAD67 knockout/knockdown animals. In contrast, Gad1–/– rats showed only very mild behavioral changes that did not necessarily match the phenotype of Gad1–/– mice (Fujihara et al., 2021a). The authors repeatedly exposed Gad1–/– rats to a low-dose NMDA receptor antagonist during adolescence but did not find as many behavioral abnormalities as Gad1–/–. Of note, Gad1–/– rats also showed an increase in GAD65 levels in the cerebral cortex, similar to the conditional knockout mice (Fujihara et al., 2020). Although this compensatory increase is not seen in schizophrenia, a higher level of GAD65 is not sufficient to prevent behavioral alterations in Gad1–/– rats (Figure 1C).
One major limitation in model animal research is the species difference between humans and rodents. For example, it has been established that humans have specialized GABAergic neurons (Boldog et al., 2018) and different GABAergic neuron markers compared to rodents (Krienen et al., 2020). Although it is unclear how these differences affect behavior, they may contribute to phenotypic differences between human schizophrenia and animal models. In addition, it should be noted that even when the same gene is knocked out, phenotypic differences may exist between mice and rats (Fujihara et al., 2020), highlighting the importance of considering species differences in future research.
Glutamic acid decarboxylase 67 knockout animals show a variety of behavioral changes, some of which may be related to schizophrenia. However, the symptom domains that are affected depend on the animal species, the method used, and the extent of knockout/knockdown. Additionally, certain model animals exhibit phenotypes that are diametrically opposed to one another (e.g., locomotor activity in the open field test; see Table 1). Patients who have schizophrenia may manifest psychomotor agitation in conjunction with positive symptoms or alternatively, they may display a diminished level of activity with negative symptoms predominating. If reduced GAD67 is involved in both positive and negative symptoms in human schizophrenia, it is unclear what would cause such variability. It may be possible to answer this question by analyzing what makes animals with similar genetic vulnerabilities different. For example, the difference between Gad1–/– rats and doxycycline-treated Gad1tTA/STOP–tetO mice is that, in addition to species differences, GAD67 is deficient from the embryo in the former, whereas in the latter, the decrease proceeds as an adult (Fujihara et al., 2020; Miyata et al., 2021). Nevertheless, no clear rule of thumb on the phenotype differences among GAD67 knockout/knockdown animals can be found at this time.
The impairment in fear extinction is the most commonly observed phenotype in the GAD67 knockout/knockdown model animals (Table 1). Individuals with schizophrenia also have impairments in extinction memory recall, which is associated with decreased ventromedial prefrontal activity in patients (Holt et al., 2009, 2012). In schizophrenia, the functional connectivity between amygdala and medial prefrontal cortex is diminished. The amygdala-specific GAD67 knockdown is sufficient to cause this phenotype, suggesting GABAergic dysfunction in amygdala underlies the abnormal fear memory in schizophrenia.
Notably, although working memory deficits are thought to be derived from abnormalities in the GABAergic nervous system, they have only been observed in Gad1–/– rats (Fujihara et al., 2020). Theoretically, the capacity for GABA production in Gad1–/– rats is the lowest among the model animals that can undergo behavioral tests. As the reduction of GAD67 is supposed to be milder in human schizophrenia, we can speculate that another biological factor (s) possibly interacts with GAD67 reduction, resulting in working memory impairment.
Finally, we discuss a perspective that has not yet been evaluated using these animal models. Psychological stress, which is crucial for the development of psychiatric disorders (Norman and Malla, 1993), can induce immune activation (Volk, 2017). If the neuroimmune system is altered by GABAergic dysfunction in schizophrenia, it may also affect the progression of the disorder. Although GABA-A receptor expression in microglia is unknown (Barragan et al., 2015), microglia express GABA-B receptors, which alter K+ permeability upon GABA signaling. Microglia release interleukin-6 (IL-6) in response to stimulation with lipopolysaccharides, but this release is inhibited by the simultaneous activation of GABA-B receptors (Kuhn et al., 2004). Taken together, it is also speculated that microglia are more likely to be activated under low GABA conditions, such as schizophrenia.
Yeung et al. (2018) have already reported the relationship between the GABAergic system and microglia in a model animal of schizophrenia. Mice with knockout of the β2 subunit of the GABA-A receptor (Gabrb2 knockout mice) show an increased number of microglia in the cortex and hippocampus (Yeung et al., 2018), accompanied by increased levels of cytokines such as TNF-α and IL-6. In schizophrenia patients, the serum IL-6 level is also elevated in untreated schizophrenia by approximately 1.5 standard deviation above that in healthy controls (Zhou et al., 2021). At present, there is no evidence that GABA-A receptors, which contain the β2 subunit, exist in microglia. Thus, the mechanism of microglial activation in Gabrb2 knockout mice is different from that discussed in the previous paragraph. Yeung et al. (2018) speculated that microglial activation is caused by an astrocyte-mediated mechanism in Gabrb2 knockout mice.
Recently, microglia with GABA-B receptors have been found to be involved in pruning GABAergic synapses (Favuzzi et al., 2021). Loss of microglia-specific GABA-B receptors significantly impairs GABAergic synaptic pruning. As a result, the number of GABAergic synapses increased at P30 but decreased by P60. The mouse model was hypoactive at P30 and hyperactive at P60. Although Favuzzi et al. (2021) noted an association with attention deficit hyperactivity disorder (ADHD), the transition from hypoactivity to hyperactivity is not identical to the course of ADHD in humans. However, the change in symptoms as the animal reaches adulthood and the marked reduction in the number of GABAergic terminals also reminds us of an association with schizophrenia.
Collectively, these findings suggest that the disruption of inhibitory neurotransmission may create an environment in which microglia are more likely to become activated, further altering the pathology via microglial responses. In Gad1–/– rats, tissue GABA levels were reduced to approximately 50% of those in wild-type rats (Fujihara et al., 2020). Although this reduction may be too extreme compared with schizophrenia patients, Gad1–/– rats are expected to be a good experimental system to investigate the extent to which reduced tissue GABA levels affect microglial properties (Figure 1C). Imposing stress or more direct inflammation in these systems may also be useful in elucidating the mechanisms by which environmental factors and predisposing factors interact to cause disease progression.
There is no guarantee that schizophrenia is a single biological disorder; rather, it is regarded as a syndrome conceptualized based on psychopathological symptoms and course. Therefore, biological studies on schizophrenia should focus on “common pathways” shared by as many patients as possible and on therapeutic interventions to address them rather than on a complete understanding of all cases.
Just as conventional therapies targeting the dopaminergic nervous system are not perfect, it may be too much to hope for the ideal efficacy of new treatment strategies. However, in schizophrenia research, where major breakthroughs have not been achieved since the discovery of antipsychotics, it is expected that as many therapeutic strategies as possible will be added to this mix. The disturbance in GABAergic system remains a promising candidate as a therapeutic target, because of its replicability in the postmortem brain studies.
To produce significant working memory deficits in rodents, complete loss of GAD67 was required. This is a considerably more severe abnormality than that observed in postmortem brain studies. Therefore, in many actual cases of schizophrenia, a milder abnormality in the GABAergic nervous system may combine with other biological factors to form working memory impairment. This could be an abnormality of excitatory input onto GABAergic neurons (Figure 1B), or it could be an interaction between specific environmental factors and GABAergic abnormalities. To test these possibilities, it would be helpful to expose the GAD67 mutants discussed here to various environmental factors, including stress. To date, there have been no reports of stress-induced cognitive dysfunction in GAD67 mutants. However, it can cause activity changes in Gad1–/– rats (Nullmeier et al., 2020). Analysis of the environmental factors that interact with the vulnerability of GABAergic neurons, the timing and mechanism of these interactions, and the resulting phenotypic changes can lead to a better understanding of schizophrenia’s pathophysiology. In doing so, it will be necessary to pay attention to the intervention of microglia and other factors, rather than focusing only on neurons, as has been done previously.
KF reviewed the literature, wrote the manuscript, and approved the submitted version.
This work was supported by JSPS KAKENHI (grant numbers: JP17K17628 and JP21K15742) and the Takeda Science Foundation.
The author thanks Takako Fujihara, Yuchio Yanagawa, and Masato Fukuda for their encouragement.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Addington, A. M., Gornick, M., Duckworth, J., Sporn, A., Gogtay, N., Bobb, A., et al. (2005). GAD1 (2q31.1), which encodes glutamic acid decarboxylase (GAD 67), is associated with childhood-onset schizophrenia and cortical gray matter volume loss. Mol. Psychiatry 10, 581–588. doi: 10.1038/sj.mp.4001599
Akbarian, S., and Huang, H. S. (2006). Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res. Rev. 52, 293–304. doi: 10.1016/j.brainresrev.2006.04.001
Akbarian, S., Kim, J. J., Potkin, S. G., Hagman, J. O., Tafazzoli, A., Bunney, W. E., et al. (1995). Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch. Gen. Psychiatry 52, 258–266.
Altshuler, L. L., Ventura, J., Van Gorp, W. G., Green, M. F., Theberge, D. C., and Mintz, J. (2004). Neurocognitive function in clinically stable men with bipolar I disorder or schizophrenia and normal control subjects. Biol. Psychiatry 56, 560–569. doi: 10.1016/j.biopsych.2004.08.002
American Psychiatric Association (2013). Diagnostic and statistical manual of mental disorders (DSM-5), 5th Edn. Virginia, VA: American Psychiatric Association. doi: 10.1176/appi.books.9780890425787
Asada, H., Kawamura, Y., Maruyama, K., Kume, H., Ding, R.-G., Kanbara, N., et al. (1997). Cleft palate and decreased brain -aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc. Natl. Acad. Sci. U.S.A. 94, 6496–6499. doi: 10.1073/pnas.94.12.6496
Barragan, A., Weidner, J. M., Jin, Z., Korpi, E. R., and Birnir, B. (2015). GABAergic signalling in the immune system. Acta Physiol. 213, 819–827. doi: 10.1111/apha.12467
Bird, E. D., Barnes, J., Iversen, L. L., Spokes, E. G., Mackay, A. V., and Shepherd, M. (1977). Increased brain dopamine and reduced glutamic acid decarboxylase and choline acetyl transferase activities in schizophrenia and related psychoses. Lancet 310, 1157–1159.
Bitanihirwe, B. K. Y., and Woo, T. U. W. (2014). Transcriptional dysregulation of γ-aminobutyric acid transporter in parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. Psychiatry Res. 220, 1155–1159. doi: 10.1016/j.psychres.2014.09.016
Bloomfield, P. S., Selvaraj, S., Veronese, M., Rizzo, G., Bertoldo, A., Owen, D. R., et al. (2016). Microglial activity in people at ultra high risk of psychosis and in schizophrenia: An [11C]PBR28 PET brain imaging study. Am. J. Psychiatry 173, 44–52. doi: 10.1176/appi.ajp.2015.14101358
Boldog, E., Bakken, T. E., Hodge, R. D., Novotny, M., Aevermann, B. D., Baka, J., et al. (2018). Transcriptomic and morphophysiological evidence for a specialized human cortical GABAergic cell type. Nat. Neurosci. 21, 1185–1195. doi: 10.1038/s41593-018-0205-2
Brown, A. S., and Derkits, E. J. (2010). Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am. J. Psychiatry 167, 261–280. doi: 10.1176/appi.ajp.2009.09030361
Brown, J. A., Ramikie, T. S., Schmidt, M. J., Báldi, R., Garbett, K., Everheart, M. G., et al. (2015). Inhibition of parvalbumin-expressing interneurons results in complex behavioral changes. Mol. Psychiatry 20, 1499–1507. doi: 10.1038/mp.2014.192
Bu, D. F., Erlander, M. G., Hitz, B. C., Tillakaratne, N. J., Kaufman, D. L., Wagner-McPherson, C. B., et al. (1992). Two human glutamate decarboxylases, 65-kDa GAD and 67-kDa GAD, are each encoded by a single gene. Proc. Natl. Acad. Sci. U.S.A. 89, 2115–2119.
Chung, D. W., Fish, K. N., and Lewis, D. A. (2016a). Pathological basis for deficient excitatory drive to cortical parvalbumin interneurons in schizophrenia. Am. J. Psychiatry 173, 1131–1139. doi: 10.1176/appi.ajp.2016.16010025
Chung, D. W., Volk, D. W., Arion, D., Zhang, Y., Sampson, A. R., and Lewis, D. A. (2016b). Dysregulated ErbB4 splicing in schizophrenia: Selective effects on parvalbumin expression. Am. J. Psychiatry 173, 60–68. doi: 10.1176/appi.ajp.2015.15020150
Correll, C. U., and Schooler, N. R. (2020). Negative symptoms in schizophrenia: A review and clinical guide for recognition, assessment, and treatment. Neuropsychiatr. Dis. Treat. 16, 519–534. doi: 10.2147/NDT.S225643
Curley, A. A., Arion, D., Volk, D. W., Asafu-adjei, J. K., Sampson, A. R., Fish, K. N., et al. (2011). Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: Clinical, protein, and cell type-specific features. Am. J. Psychiatry 168, 921–929.
Curley, A. A., Eggan, S. M., Lazarus, M. S., Huang, Z. J., Volk, D. W., and Lewis, D. A. (2013). Role of glutamic acid decarboxylase 67 in regulating cortical parvalbumin and GABA membrane transporter 1 expression: Implications for schizophrenia. Neurobiol. Dis. 50, 179–186. doi: 10.1016/j.nbd.2012.10.018
Dalmau, J., Lancaster, E., Martinez-Hernandez, E., Rosenfeld, M. R., and Balice-Gordon, R. (2011). Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 10, 63–74. doi: 10.1016/S1474-4422(10)70253-2
David, S., and Olton, B. C. P. (1979). Spatial memory and hippocampal function. Neuropsychologia 17, 669–682.
de Oliveira Figueiredo, E. C., Calì, C., Petrelli, F., and Bezzi, P. (2022). Emerging evidence for astrocyte dysfunction in schizophrenia. Glia 70, 1585–1604. doi: 10.1002/glia.24221
Dienel, S. J., Schoonover, K. E., and Lewis, D. A. (2022). Cognitive dysfunction and prefrontal cortical circuit alterations in schizophrenia: Developmental trajectories. Biol. Psychiatry 92, 450–459. doi: 10.1016/j.biopsych.2022.03.002
Druart, M., Nosten-Bertrand, M., Poll, S., Crux, S., Nebeling, F., Delhaye, C., et al. (2021). Elevated expression of complement C4 in the mouse prefrontal cortex causes schizophrenia-associated phenotypes. Mol. Psychiatry 26, 3489–3501. doi: 10.1038/s41380-021-01081-6
Du, J., Duan, S., Wang, H., Chen, W., Zhao, X., Zhang, A., et al. (2008). Comprehensive analysis of polymorphisms throughout GAD1 gene: A family-based association study in schizophrenia. J. Neural Transm. 115, 513–519. doi: 10.1007/s00702-007-0844-z
Egerton, A., Modinos, G., Ferrera, D., and McGuire, P. (2017). Neuroimaging studies of GABA in schizophrenia: A systematic review with meta-analysis. Transl. Psychiatry 7:e1147. doi: 10.1038/tp.2017.124
Favuzzi, E., Huang, S., Saldi, G. A., Binan, L., Ibrahim, L. A., Fernández-Otero, M., et al. (2021). GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell 184, 4048.e32–4063.e32. doi: 10.1016/j.cell.2021.06.018
Fillman, S. G., Cloonan, N., Catts, V. S., Miller, L. C., Wong, J., Mccrossin, T., et al. (2013). Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol. Psychiatry 18, 206–214. doi: 10.1038/mp.2012.110
Fish, K. N., Rocco, B. R., DeDionisio, A. M., Dienel, S. J., Sweet, R. A., and Lewis, D. A. (2021). Altered parvalbumin basket cell terminals in the cortical visuospatial working memory network in schizophrenia. Biol. Psychiatry 90, 47–57. doi: 10.1016/j.biopsych.2021.02.009
Fujihara, K., Narita, K., Suzuki, Y., Takei, Y., Suda, M., Tagawa, M., et al. (2015b). Relationship of γ-aminobutyric acid and glutamate+glutamine concentrations in the perigenual anterior cingulate cortex with performance of Cambridge Gambling Task. Neuroimage 109, 102–108. doi: 10.1016/j.neuroimage.2015.01.014
Fujihara, K., Miwa, H., Kakizaki, T., Kaneko, R., Mikuni, M., Tanahira, C., et al. (2015a). Glutamate decarboxylase 67 deficiency in a subset of GABAergic neurons induces schizophrenia-related phenotypes. Neuropsychopharmacology 40, 2475–2486. doi: 10.1038/npp.2015.117
Fujihara, K., Sato, T., Miyasaka, Y., and Mashimo, T. (2021b). Genetic deletion of glutamate decarboxylase 67-kDa isoform alters conditioned fear behavior in rats. FEBS Open Bio 11, 340–353. doi: 10.1002/2211-5463.13065
Fujihara, K., Sato, T., Higeta, K., Miyasaka, Y., Mashimo, T., and Yanagawa, Y. (2021a). Behavioral consequences of a combination of Gad1 haplodeficiency and adolescent exposure to an NMDA receptor antagonist in long-evans rats. Front. Pharmacol. 12:646088. doi: 10.3389/fphar.2021.646088
Fujihara, K., Yamada, K., Ichitani, Y., Kakizaki, T., Jiang, W., Miyata, S., et al. (2020). CRISPR/Cas9-engineered Gad1 elimination in rats leads to complex behavioral changes: Implications for schizophrenia. Transl. Psychiatry 10:426. doi: 10.1038/s41398-020-01108-6
Georgiev, D., Yoshihara, T., Kawabata, R., Matsubara, T., Tsubomoto, M., Minabe, Y., et al. (2016). Cortical gene expression after a conditional knockout of 67 kda glutamic acid decarboxylase in parvalbumin neurons. Schizophr. Bull. 42, 992–1002. doi: 10.1093/schbul/sbw022
Giacopuzzi, E., Gennarelli, M., Minelli, A., Gardella, R., Valsecchi, P., Traversa, M., et al. (2017). Exome sequencing in schizophrenic patients with high levels of homozygosity identifies novel and extremely rare mutations in the GABA/glutamatergic pathways. PLoS One 12:e0182778. doi: 10.1371/journal.pone.0182778
Giovanoli, S., Notter, T., Richetto, J., Labouesse, M. A., Vuillermot, S., Riva, M. A., et al. (2015). Late prenatal immune activation causes hippocampal deficits in the absence of persistent inflammation across aging. J. Neuroinflammation 12:221. doi: 10.1186/s12974-015-0437-y
Gonzalez-Burgos, G., and Lewis, D. A. (2008). GABA neurons and the mechanisms of network oscillations: Implications for understanding cortical dysfunction in schizophrenia. Schizophr. Bull. 34, 944–961. doi: 10.1093/schbul/sbn070
Gonzalez-Burgos, G., Hashimoto, T., and Lewis, D. A. (2010). Alterations of cortical GABA neurons and network oscillations in schizophrenia. Curr. Psychiatry Rep. 12, 335–344. doi: 10.1007/s11920-010-0124-8
Guidotti, A., Auta, J., Davis, J. M., Gerevini, V. D., Dwivedi, Y., Grayson, D. R., et al. (2000). Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: A postmortem brain study. Arch. Gen. Psychiatry 57, 1061–1069. doi: 10.1001/archpsyc.57.11.1061
Hashimoto, T., Arion, D., Unger, T., Maldonado-Avilés, J. G., Morris, H. M., Volk, D. W., et al. (2008a). Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 13, 147–161. doi: 10.1038/sj.mp.4002011
Hashimoto, T., Ph, D., Bazmi, H. H., Mirnics, K., Wu, Q., Ph, D., et al. (2008b). Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am. J. Psychiatry 165, 479–489.
Hashimoto, T., Volk, D. W., Eggan, S. M., Mirnics, K., Pierri, J. N., Sun, Z., et al. (2003). Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J. Neurosci. 23, 6315–6326.
Heckers, S., Stone, D., Walsh, J., Shick, J., Koul, P., and Benes, F. M. (2002). Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch. Gen. Psychiatry 59, 521–529. doi: 10.1001/archpsyc.59.6.521
Heldt, S. A., Mou, L., and Ressler, K. J. (2012). In vivo knockdown of GAD67 in the amygdala disrupts fear extinction and the anxiolytic-like effect of diazepam in mice. Transl. Psychiatry 2:e181. doi: 10.1038/tp.2012.101
Hoenig, J. (1983). The concept of schizophrenia kraepelin-bleuler-schneider. Br. J. Pharmacol. 142, 547–556.
Holt, D. J., Coombs, G., Zeidan, M. A., Goff, D. C., and Milad, M. R. (2012). Failure of neural responses to safety cues in schizophrenia. Arch. Gen. Psychiatry 69, 893–903. doi: 10.1001/archgenpsychiatry.2011.2310
Holt, D. J., Lebron-Milad, K., Milad, M. R., Rauch, S. L., Pitman, R. K., Orr, S. P., et al. (2009). Extinction memory is impaired in schizophrenia. Biol. Psychiatry 65, 455–463. doi: 10.1016/j.biopsych.2008.09.017
Howes, O. D., and Kapur, S. (2009). The dopamine hypothesis of schizophrenia: Version III - The final common pathway. Schizophr. Bull. 35, 549–562. doi: 10.1093/schbul/sbp006
Hyde, T. M., Lipska, B. K., Ali, T., Mathew, S. V., Law, A. J., Metitiri, O. E., et al. (2011). Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J. Neurosci. 31, 11088–11095. doi: 10.1523/jneurosci.1234-11.2011
Impagnatiello, F., Guidotti, A. R., Pesold, C., Dwivedi, Y., Caruncho, H., Pisu, M. G., et al. (1998). A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 95, 15718–15723. doi: 10.1073/pnas.95.26.15718
Jiang, W., Kakizaki, T., Fujihara, K., Miyata, S., Zhang, Y., Suto, T., et al. (2022). Impact of GAD65 and/or GAD67 deficiency on perinatal development in rats. FASEB J. 36, 1–10. doi: 10.1096/fj.202101389R
Kakizaki, T., Oriuchi, N., and Yanagawa, Y. (2015). GAD65/GAD67 double knockout mice exhibit intermediate severity in both cleft palate and omphalocele compared with GAD67 knockout and VGAT knockout mice. Neuroscience 288, 86–93. doi: 10.1016/j.neuroscience.2014.12.030
Keefe, R. S. E., and Harvey, P. D. (2012). “Cognitive impairment in schizophrenia,” in Novel antischizophrenia treatments, (Berlin: Springer), 11–38. doi: 10.1007/978-3-642-25758-2
Kimoto, S., Bazmi, H. H., and Lewis, D. A. (2014). Lower expression of glutamic acid decarboxylase 67 in the prefrontal cortex in schizophrenia: Contribution of altered regulation by Zif268. Am. J. Psychiatry 171, 969–978. doi: 10.1176/appi.ajp.2014.14010004
Kolata, S. M., Nakao, K., Jeevakumar, V., Farmer-Alroth, E. L., Fujita, Y., Bartley, A. F., et al. (2018). Neuropsychiatric phenotypes produced by GABA reduction in mouse cortex and hippocampus. Neuropsychopharmacology 43, 1445–1456. doi: 10.1038/npp.2017.296
Krienen, F. M., Goldman, M., Zhang, Q., C H del Rosario, R., Florio, M., Machold, R., et al. (2020). Innovations present in the primate interneuron repertoire. Nature 586, 262–269. doi: 10.1038/s41586-020-2781-z
Kuhn, S. A., Van Landeghem, F. K. H., Zacharias, R., Färber, K., Rappert, A., Pavlovic, S., et al. (2004). Microglia express GABAB receptors to modulate interleukin release. Mol. Cell. Neurosci. 25, 312–322. doi: 10.1016/j.mcn.2003.10.023
Lewis, D. A., and Sweet, R. A. (2009). Schizophrenia from a neural circuitry perspective: Advancing toward rational pharmacological therapies. J. Clin. Invest. 119, 706–716. doi: 10.1172/JCI37335
Liddle, P. F. (1987). The symptoms of chronic schizophrenia. A re-examination of the positive-negative dichotomy. Br. J. Psychiatry 151, 145–151.
Liddle, P. F. (2019). The core deficit of classical schizophrenia: Implications for predicting the functional outcome of psychotic illness and developing effective treatments. Can. J. Psychiatry 64, 680–685. doi: 10.1177/0706743719870515
Magri, C., Giacopuzzi, E., La Via, L., Bonini, D., Ravasio, V., Elhussiny, M. E. A., et al. (2018). A novel homozygous mutation in GAD1 gene described in a schizophrenic patient impairs activity and dimerization of GAD67 enzyme. Sci. Rep. 8:15470. doi: 10.1038/s41598-018-33924-8
Marques, T. R., Ashok, A. H., Pillinger, T., Veronese, M., Turkheimer, F. E., Dazzan, P., et al. (2019). Neuroinflammation in schizophrenia: Meta-analysis of in vivo microglial imaging studies. Psychol. Med. 49, 2186–2196. doi: 10.1017/S0033291718003057
Meltzer, H. Y., and Stahl, S. M. (1976). The dopamine hypothesis of schizophrenia. Schizophr. Bull. 2, 19–76. doi: 10.1111/j.1744-6163.1990.tb00312.x
Miyata, S., Kakizaki, T., Fujihara, K., Obinata, H., Hirano, T., Nakai, J., et al. (2021). Global knockdown of glutamate decarboxylase 67 elicits emotional abnormality in mice. Mol. Brain 14:5. doi: 10.1186/s13041-020-00713-2
Murphy, C. E., Walker, A. K., and Weickert, C. S. (2021). Neuroinflammation in schizophrenia: The role of nuclear factor kappa B. Transl. Psychiatry 11:528. doi: 10.1038/s41398-021-01607-0
Nakahara, T., Tsugawa, S., Noda, Y., Ueno, F., Honda, S., Kinjo, M., et al. (2022). Glutamatergic and GABAergic metabolite levels in schizophrenia-spectrum disorders: A meta-analysis of 1H-magnetic resonance spectroscopy studies. Mol. Psychiatry 27, 744–757. doi: 10.1038/s41380-021-01297-6
Nakaya, M., and Ohmori, K. (2008). A two-factor structure for the schedule for the deficit syndrome in schizophrenia. Psychiatry Res. 158, 256–259. doi: 10.1016/j.psychres.2007.10.008
Norman, R. M. G., and Malla, A. K. (1993). Stressful life events and schizophrenia. I: A review of the research. Br. J. Psychiatry 162, 161–166. doi: 10.1192/bjp.162.2.161
Notter, T. (2021). Astrocytes in schizophrenia. Brain Neurosci. Adv. 5:23982128211009148. doi: 10.1177/23982128211009148
Nullmeier, S., Elmers, C., D’Hanis, W., Sandhu, K. V. K., Stork, O., Yanagawa, Y., et al. (2020). Glutamic acid decarboxylase 67 haplodeficiency in mice: Consequences of postweaning social isolation on behavior and changes in brain neurochemical systems. Brain Struct. Funct. 225, 1719–1742. doi: 10.1007/s00429-020-02087-6
Obata, K. (2013). Synaptic inhibition and γ-aminobutyric acid in the mammalian central nervous system. Proc. Japan Acad. Ser. B Phys. Biol. Sci. 89, 139–156. doi: 10.2183/pjab.89.139
Olton, D. S., and Samuelson, R. J. (1976). Remembrance of places passed: Spatial memory in rats. J. Exp. Psychol. Anim. Behav. Process. 2, 97–116. doi: 10.1037/0097-7403.2.2.97
Parellada, E., and Gassó, P. (2021). Glutamate and microglia activation as a driver of dendritic apoptosis: A core pathophysiological mechanism to understand schizophrenia. Transl. Psychiatry 11:271. doi: 10.1038/s41398-021-01385-9
Perry, T. L., Buchanan, J., Kish, S. J., and Hansen, S. (1979). γ-Aminobutyric-acid deficiency in brain of schizophrenic patients. Lancet 313, 237–239.
Radewicz, K., Garey, L. J., Gentleman, S. M., and Reynolds, R. (2000). Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. J. Neuropathol. Exp. Neurol. 59, 137–150. doi: 10.1093/jnen/59.2.137
Ripke, S., Neale, B. M., Corvin, A., Walters, J. T. R., Farh, K. H., Holmans, P. A., et al. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Rothman, D. L., Petroff, O. A. C., Behar, K. L., and Mattson, R. H. (1993). Localized 1H NMR measurements of γ-aminobutyric acid in human brain in vivo. Proc. Natl. Acad. Sci. U.S.A. 90, 5662–5666. doi: 10.1073/pnas.90.12.5662
Ruzicka, W. B., Zhubi, A., Veldic, M., Grayson, D. R., Costa, E., and Guidotti, A. (2007). Selective epigenetic alteration of layer I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection. Mol. Psychiatry 12, 385–397. doi: 10.1038/sj.mp.4001954
Saha, S., Chant, D., Welham, J., and McGrath, J. (2005). A systematic review of the prevalence of schizophrenia. PLoS Med. 2:e141. doi: 10.1371/journal.pmed.0020141
Sandhu, K. V., Lang, D., Müller, B., Nullmeier, S., Yanagawa, Y., Schwegler, H., et al. (2014). Glutamic acid decarboxylase 67 haplodeficiency impairs social behavior in mice. Genes Brain Behav. 13, 439–450. doi: 10.1111/gbb.12131
Seeman, M. V. (2021). History of the dopamine hypothesis of antipsychotic action. World J. Psychiatry 11, 355–364. doi: 10.5498/wjp.v11.i7.355
Sekar, A., Bialas, A. R., De Rivera, H., Davis, A., Hammond, T. R., Kamitaki, N., et al. (2016). Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183. doi: 10.1038/nature16549
Soghomonian, J., and Martin, D. L. (1998). Two isoforms of glutamate decarboxylase: Why? Trends Pharmacol. Sci. 19, 500–505.
Straub, R. E., Lipska, B. K., Egan, M. F., Goldberg, T. E., Callicott, J. H., Mayhew, M. B., et al. (2007). Allelic variation in GAD1 (GAD 67) is associated with schizophrenia and influences cortical function and gene expression. Mol. Psychiatry 12, 854–869. doi: 10.1038/sj.mp.4001988
Tamamaki, N., Yanagawa, Y., Tomioka, R., Miyazaki, J. I., Obata, K., and Kaneko, T. (2003). Green Fluorescent Protein Expression and Colocalization with Calretinin, Parvalbumin, and Somatostatin in the GAD67-GFP Knock-In Mouse. J. Comp. Neurol. 467, 60–79. doi: 10.1002/cne.10905
Tsubomoto, M., Kawabata, R., Zhu, X., Minabe, Y., Chen, K., Lewis, D. A., et al. (2019). Expression of transcripts selective for GABA neuron subpopulations across the cortical visuospatial working memory network in the healthy state and schizophrenia. Cereb. Cortex 29, 3540–3550. doi: 10.1093/cercor/bhy227
Uchida, T., Furukawa, T., Iwata, S., Yanagawa, Y., and Fukuda, A. (2014). Selective loss of parvalbumin-positive GABAergic interneurons in the cerebral cortex of maternally stressed Gad1-heterozygous mouse offspring. Transl. Psychiatry 4:e371. doi: 10.1038/tp.2014.13
Uchida, T., Oki, Y., Yanagawa, Y., and Fukuda, A. (2011). A heterozygous deletion in the glutamate decarboxylase 67 gene enhances maternal and fetal stress vulnerability. Neurosci. Res. 69, 276–282. doi: 10.1016/j.neures.2010.12.010
Uematsu, M., Hirai, Y., Karube, F., Ebihara, S., Kato, M., Abe, K., et al. (2008). Quantitative chemical composition of cortical GABAergic neurons revealed in transgenic venus-expressing rats. Cereb. Cortex 18, 315–330. doi: 10.1093/cercor/bhm056
Uno, Y., and Coyle, J. T. (2019). Glutamate hypothesis in schizophrenia. Psychiatry Clin. Neurosci. 73, 204–215. doi: 10.1111/pcn.12823
Veldic, M., Guidotti, A., Maloku, E., Davis, J. M., and Costa, E. (2005). In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proc. Natl. Acad. Sci. U.S.A. 102, 2152–2157. doi: 10.1073/pnas.0409665102
Volk, D. W. (2017). Role of microglia disturbances and immune-related marker abnormalities in cortical circuitry dysfunction in schizophrenia. Neurobiol. Dis. 99, 58–65. doi: 10.1016/j.nbd.2016.12.019
Volk, D. W., Austin, M. C., Pierri, J. N., Sampson, A. R., and Lewis, D. A. (2000). Decreased glutamic acid decarboxylase 67 messenger. Arch. Gen Psychiatry 57, 237–245. doi: 10.1001/archpsyc.57.3.237
Wierzba-Bobrowicz, T., Lewandowska, E., Lechowicz, W., Stêpieñ, T., and Pasennik, E. (2005). Quantitative analysis of activated microglia, ramified and damage of processes in the frontal and temporal lobes of chronic schizophrenics. Folia Neuropathol. 43, 81–89.
Woo, T. U. W., Shrestha, K., Amstrong, C., Minns, M. M., Walsh, J. P., and Benes, F. M. (2007). Differential alterations of kainate receptor subunits in inhibitory interneurons in the anterior cingulate cortex in schizophrenia and bipolar disorder. Schizophr. Res. 96, 46–61. doi: 10.1016/j.schres.2007.06.023
Woo, T. W., Walsh, J. P., and Benes, F. M. (2012). Density of glutamic acid decarboxylase 67 messenger RNA–containing neurons that express the N-methyl-. Arch. Gen. Psychiatry 61, 649–657.
Yeung, R. K., Xiang, Z. H., Tsang, S. Y., Li, R., Ho, T. Y. C., Li, Q., et al. (2018). Gabrb2-knockout mice displayed schizophrenia-like and comorbid phenotypes with interneuron-astrocyte-microglia dysregulation. Transl. Psychiatry 8:128. doi: 10.1038/s41398-018-0176-9
Yoon, J. H., Grandelis, A., and Maddock, R. J. (2016). Dorsolateral prefrontal cortex GABA concentration in humans predicts working memory load processing capacity. J. Neurosci. 36, 11788–11794. doi: 10.1523/jneurosci.1970-16.2016
Yoon, J. H., Maddock, R. J., DongBo Cui, E., Minzenberg, M. J., Niendam, T. A., Lesh, T., et al. (2020). Reduced in vivo visual cortex GABA in schizophrenia, a replication in a recent onset sample. Schizophr. Res. 215, 217–222. doi: 10.1016/j.schres.2019.10.025
Yoon, J. H., Maddock, R. J., Rokem, A., Silver, M. A., Minzenberg, M. J., Ragland, J. D., et al. (2010). GABA concentration is reduced in visual cortex in schizophrenia and correlates with orientation-specific surround suppression. J. Neurosci. 30, 3777–3781. doi: 10.1523/jneurosci.6158-09.2010
Keywords: inhibitory neurons, psychiatric disorders, knockout rats, genome editing, microglia
Citation: Fujihara K (2023) Beyond the γ-aminobutyric acid hypothesis of schizophrenia. Front. Cell. Neurosci. 17:1161608. doi: 10.3389/fncel.2023.1161608
Received: 08 February 2023; Accepted: 04 April 2023;
Published: 24 April 2023.
Edited by:
Chao Deng, University of Wollongong, AustraliaReviewed by:
Byron Bitanihirwe, The University of Manchester, United KingdomCopyright © 2023 Fujihara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuyuki Fujihara, cHN5X2Z1amloYXJhQGd1bm1hLXUuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.