
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci., 03 May 2023
Sec. Cellular Neuropathology
Volume 17 - 2023 | https://doi.org/10.3389/fncel.2023.1149391
 Anat Mavashov1,2
Anat Mavashov1,2 Marina Brusel2,3
Marina Brusel2,3 Jiaxing Liu4Victoria Woytowicz4Haneui Bae4
Jiaxing Liu4Victoria Woytowicz4Haneui Bae4 Ying-Hsin Chen4Vardhan S. Dani4
Ying-Hsin Chen4Vardhan S. Dani4 Elena Cardenal-Muñoz5Vittoria Spinosa5
Elena Cardenal-Muñoz5Vittoria Spinosa5 José Ángel Aibar5
José Ángel Aibar5 Moran Rubinstein1,2,3*
Moran Rubinstein1,2,3*Dravet syndrome (Dravet) is a severe congenital developmental genetic epilepsy caused by de novo mutations in the SCN1A gene. Nonsense mutations are found in ∼20% of the patients, and the R613X mutation was identified in multiple patients. Here we characterized the epileptic and non-epileptic phenotypes of a novel preclinical Dravet mouse model harboring the R613X nonsense Scn1a mutation. Scn1aWT/R613X mice, on a mixed C57BL/6J:129S1/SvImJ background, exhibited spontaneous seizures, susceptibility to heat-induced seizures, and premature mortality, recapitulating the core epileptic phenotypes of Dravet. In addition, these mice, available as an open-access model, demonstrated increased locomotor activity in the open-field test, modeling some non-epileptic Dravet-associated phenotypes. Conversely, Scn1aWT/R613X mice, on the pure 129S1/SvImJ background, had a normal life span and were easy to breed. Homozygous Scn1aR613X/R613X mice (pure 129S1/SvImJ background) died before P16. Our molecular analyses of hippocampal and cortical expression demonstrated that the premature stop codon induced by the R613X mutation reduced Scn1a mRNA and NaV1.1 protein levels to ∼50% in heterozygous Scn1aWT/R613X mice (on either genetic background), with marginal expression in homozygous Scn1aR613X/R613X mice. Together, we introduce a novel Dravet model carrying the R613X Scn1a nonsense mutation that can be used to study the molecular and neuronal basis of Dravet, as well as the development of new therapies associated with SCN1A nonsense mutations in Dravet.
Dravet Syndrome (Dravet) is a severe childhood-onset developmental and epileptic encephalopathy. Most cases are caused by de novo mutations in the SCN1A gene, which encodes for the voltage-gated sodium channel NaV1.1 (Claes et al., 2003). Dravet patients develop normally in the first months of life. The first symptom is often a febrile generalized tonic-clonic or focal clonic seizure that can be prolonged. Seizures soon progress to spontaneous afebrile seizures, which can evolve into status epilepticus (SE). Drug treatment in Dravet includes valproate and clobazam as initial therapy, with stiripentol, cannabidiol, or fenfluramine as an adjunct therapy. However, even with polytherapy, patients are rarely seizure-free, with a high risk for sudden unexpected death in epilepsy (SUDEP). Additional non-epileptic comorbidities in Dravet include developmental delay and cognitive impairment (Dravet et al., 2011; Gataullina and Dulac, 2017; Cardenal-Muñoz et al., 2022; Strzelczyk and Schubert-Bast, 2022).
Over 1,000 different SCN1A pathological variants have been identified in Dravet patients. Among these, ∼7% are large deletions, ∼39% are missense mutations, and ∼41% are nonsense or frameshift mutations (Claes et al., 2009; Meng et al., 2015; Xu et al., 2015; Liu et al., 2021). Most of these mutations are de novo, non-recurrent mutations. However, intriguingly, the SCN1A R613X nonsense mutation has been reported to occur de novo in ten different patients (Kearney et al., 2006; Margherita Mancardi et al., 2006; Depienne et al., 2009; Rodda et al., 2012; Wang et al., 2012; Gaily et al., 2013; Moehring et al., 2013; Lee et al., 2015). Here we set out to comprehensively characterize a novel open-access mouse model that harbors this nonsense mutation.
Dravet mouse models (DS mice) recapitulate many aspects of the human disease. To date, fifteen different models have been developed based on various Scn1a mutations. Importantly, all the DS mouse models recapture key pathophysiological phenotypes of Dravet, exhibiting spontaneous seizures, premature mortality, and the presentation of Dravet-associated non-epileptic behavioral traits (Yu et al., 2006; Ogiwara et al., 2007, 2013; Martin et al., 2010; Cheah et al., 2012; Miller et al., 2014; Tsai et al., 2015; Kuo et al., 2019; Ricobaraza et al., 2019; Dyment et al., 2020; Jansen et al., 2020; Uchino et al., 2021; Voskobiynyk et al., 2021; Morey et al., 2022; Valassina et al., 2022; Table 1).
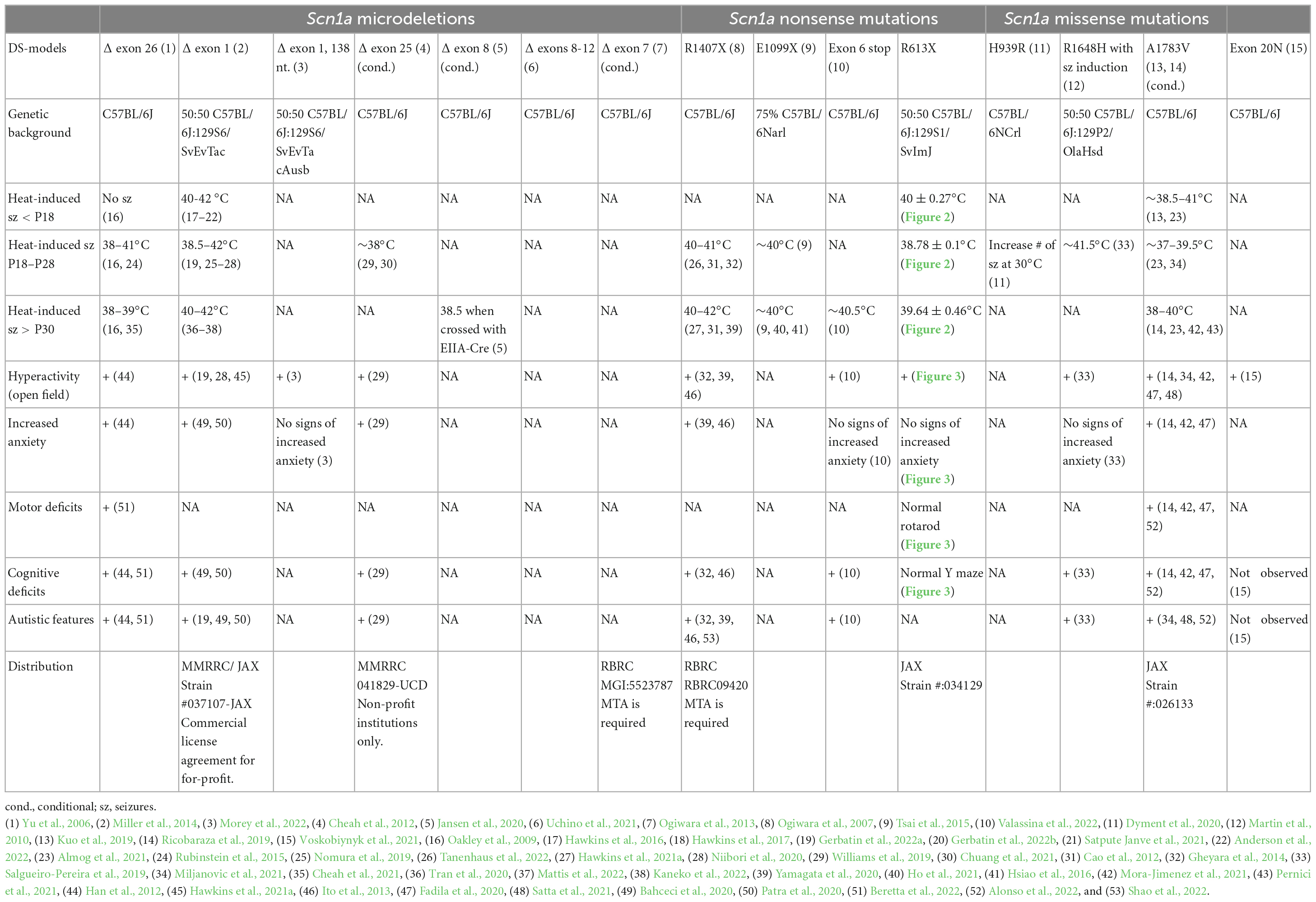
Table 1. Mouse models for DS.
Several DS models are available through international repositories (Table 1), and two lines, developed by the Dravet Syndrome Foundation Spain, are distributed through the Jackson Laboratory for unrestricted use: (i) the conditional DS mice that harbor the Scn1aA1783V missense mutation; and (ii) a new Scn1aR613X line, described here. While the first model has been validated and confirmed to recapitulate Dravet phenotypes (Table 1), the phenotypic and molecular characterization of the new Scn1aR613X model was yet partial (Almog et al., 2022). Here, we show that DS mice carrying the Scn1aR613X nonsense mutation on a mixed C57BL/6J:129S1/SvImJ background recapitulate key phenotypes of Dravet, with heat-induced seizures that occur within the range of physiological fever temperatures at multiple developmental stages, spontaneous convulsive seizures, premature death, and hyperactivity in the open field. Moreover, these mice have reduced levels of Scn1a mRNA and NaV1.1 protein in the hippocampus and the cortex. Together, these data confirm that the Scn1aR613X model is a valid animal model for Dravet research.
All animal experiments were approved by the Animal Care and Use Committee (IACUC) of Tel Aviv University. Mice used in this study were housed in a standard animal facility at the Goldschleger Eye Institute at a constant (22°C) temperature, on a 12-h light/dark cycle, with ad libitum access to food and water.
Mice harboring the global Scn1aR613X mutation were generated by crossing males or females carrying the A-to-T point mutation in nucleotide 1837 (converting arginine 613 to a STOP codon) in addition to a silent C-to-T mutation at position 1833 (129S1/SvImJ-Scn1aem1Dsf/J, The Jackson Laboratory, stock no. 034129) (Figure 1A), with wild-type (WT) mice, females or males (129S1/SvImJ, The Jackson Laboratory, stock no. 002448). Details about allele modification and genotyping are described here: https://www.jax.org/strain/034129. This mouse line was maintained on the pure 129S1/SvImJ genetic background. To produce DS mice (on a mixed background 50:50 C57BL/6J: 129S1/SvImJ), male or female Scn1aWT/R613X mice on the pure 129S1/SvImJ were crossed with WT mice (males or females) on a C57BL/6J background (The Jackson Laboratory, stock no. 000664), generating F1 mice on a 50:50 genetic background. Both male and female offspring were used for experiments. Homozygous mutant mice (Scn1aR613X/R613X) were generated by crossing heterozygous Scn1aWT/R613X mice on the pure 129S1/SvImJ background.
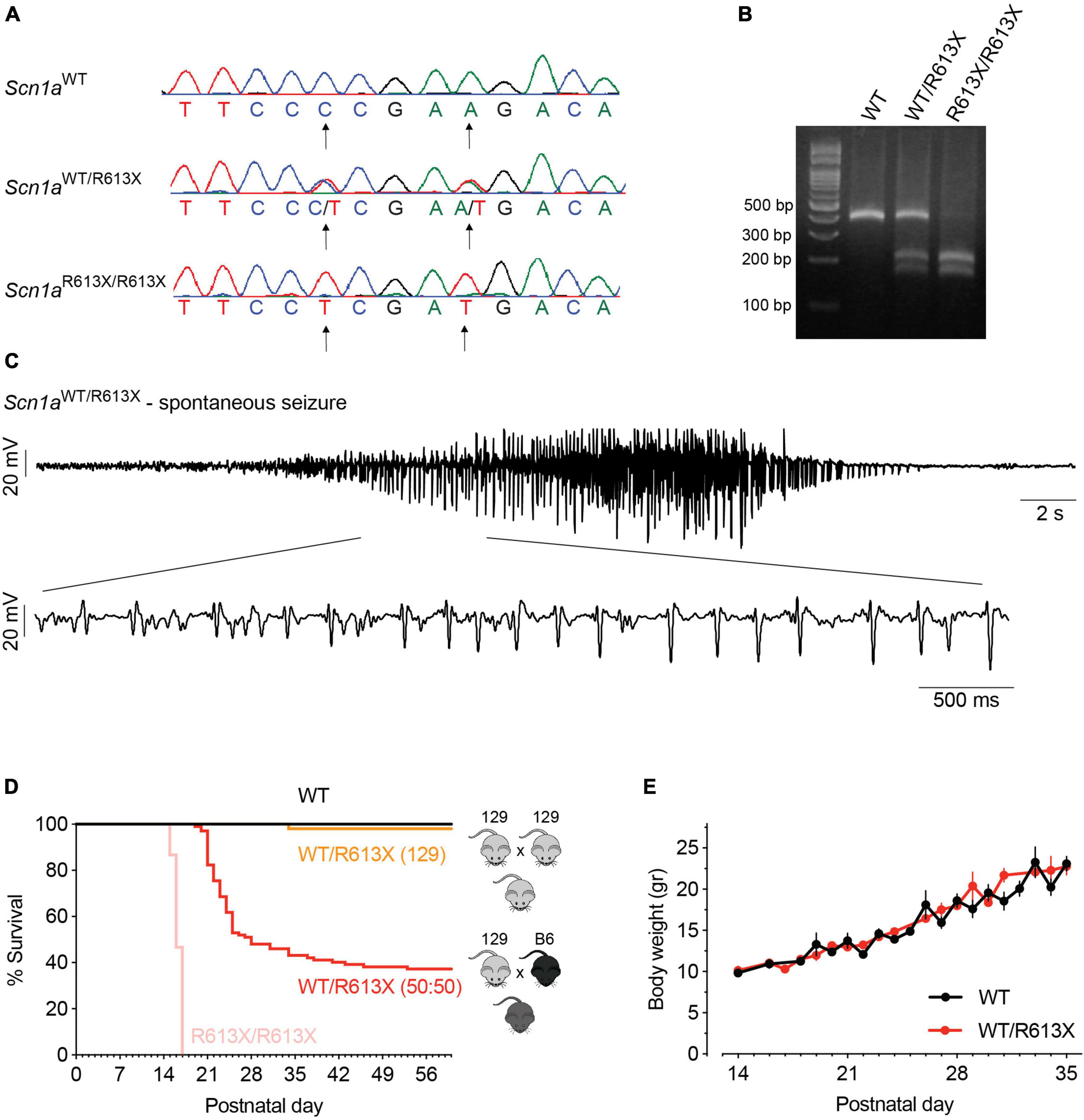
Figure 1. Spontaneous convulsive seizures and premature mortality in DS Scn1aWT/R613X mice. (A) CRISPR/Cas9-generated mutations were introduced to exon 12 of the mouse Scn1a gene: A > T point mutation at nucleotide 1837 (converting Arg613 into a STOP codon) and a silent C > T mutation at position 1833. (B) Genotyping of Scn1aR613X allele using PCR followed by TaqI digestion. (C) A spontaneous seizure captured during an ECoG recording in a Scn1aWT/R613X mouse on a mixed background. (D) Scn1aWT/R613X on a mixed background exhibited premature mortality, in contrast to Scn1aWT/R613X mice on the pure 129S1/SvImJ. Homozygous Scn1aR613X/R613X died prematurely between P14-16. 129 background: WT, n = 24; Scn1aWT/R613X, n = 51; Scn1aR613X/R613X, n = 15. Mixed background: WT, n = 30; Scn1aWT/R613X, n = 102. (E) The growth of Scn1aWT/R613X mice on the mixed background was similar to their WT littermates. WT, n = 10; Scn1aWT/R613X, n = 6.
PCR was performed using the primers and protocol described by the Jackson Laboratory (129S1/SvImJ-Scn1aem1Dsf/J, stock no. 034129). Following the amplification step, 7,000 units of TaqI restriction enzyme (New England Biolabs, Ipswich, MA, USA) were added to 6 μL of the PCR mixture, incubated at 65°C for 15 min, and analyzed on 3% agarose gel (Figure 1B).
Thermal induction of seizures was performed as previously described (Almog et al., 2021). Briefly, the baseline body core temperature was recorded for at least 10 min, allowing the animals to habituate to the recording chamber and rectal probe. Body temperature was then increased by 0.5°C every 2 min with a heat lamp (TCAT-2DF, Physitemp Instruments Inc., Clifton, NJ, USA) until a generalized tonic-clonic seizure was provoked; the temperature was not increased above 42°C. Mice used for thermal induction of seizures were not included in the survival curve.
Electrode implantation was done at P21-P25, as previously described (Fadila et al., 2020). Briefly, the mice were anesthetized with ketamine/xylazine (191/4.25 mg/kg), Carprofen (5 mg/kg) was used as analgesia. A midline incision was made above the skull and five fine silver wire electrodes (130 μm diameter bare; 180 μm diameter coated) were placed at visually identified locations, bilaterally above the somatosensory cortex; a reference electrode was placed on the cerebellum; a ground electrode was placed subcutaneously behind the neck, toward the left shoulder, and an EMG electrode was placed in the neck muscles. The electrodes were connected to a micro-connector system, secured with dental cement, and the skin closed with sutures. Mice were allowed to recover for at least 48 h before recording. For the video-ECoG recordings, the mice were connected to a tethered T8 Headstage (Triangle BioSystems, Durham, NC, USA), and PowerLab 8/35 acquisition hardware with LabChart 8 software (ADInstruments, Sydney, NSW, Australia). Each recording lasted 3–4 h, between 8 am and 6 pm. The electrical signals were recorded and digitized at a sampling rate of 1 KHz with a notch filter at 50 Hz. Seizures were detected by visual inspection of the signal as high amplitude repeated spike-wave events (Figures 1C, 2A).
Behavioral tests were done as described previously (Fadila et al., 2020). For the open-field test, the mice were placed in the center of a square (50 × 50 cm) plexiglas apparatus and their movement was recorded for 10 min. Live tracking was achieved via a monochrome camera (Basler acA1300-60 gm, Basler AG, Ahren, Germany) connected with EthoVision XT 13 software (Noldus Technology, Wageningen, Netherlands). To analyze anxiety-like behavior, the open field arena was digitally divided into 10 × 10 cm squares (25 in total), dividing the arena into “outer” and “inner” zones.
To examine motor functions, the mice were placed on an accelerating rotating rod (acceleration from 3 to 32 RMP, Med Associates, Inc., Fairfax, VT, USA), and the time at which each mouse fell was recorded. The test was repeated five times for each mouse, and the three longest trials were averaged.
To test spatial working memory, a symmetrical Y-maze comprised of three arms (each 35 cm L × 7.6 cm W × 20 cm H) was used. The mouse was placed into one of the Y-maze arms and allowed free exploration for 10 min. Live tracking was achieved via a monochrome camera (Basler acA1300-60 gm, Basler AG, Ahren, Germany) connected with EthoVision XT 13 software (Noldus Technology, Wageningen, Netherlands). Different cohorts of mice were tested at each time point to ensure a reaction to a novel arena. The percentage of spontaneous alternation was calculated as the number of triads divided by the number of possible triads.
Total RNA was isolated using Purelink™ RNA mini kit according to the manufacturer’s instructions (Thermo Fisher Scientific, Life Technologies, Carlsbad, CA, USA). cDNA was synthesized from 500 ng RNA using Maxima H Minus cDNA synthesis kit (Thermo Fisher Scientific, Life Technologies, Carlsbad, CA, USA). Real-time PCR (qPCR) reactions were performed in triplicates in a final volume of 10 μL with 5 ng of RNA as template using the TaqMan Scn1a (Mm00450580_m1) gene expression assay (Applied Biosystems, Thermo Fisher Scientific, Life Technologies, Carlsbad, CA, USA). Two endogenous controls were used: Gusb (Mm00446953_m1) and Tfrc (Mm00441941_m1). Efficiency of 100%, dynamic range, and lack of genomic DNA amplification were verified for all assays.
The hippocampi were extracted and homogenized as previously described (Nissenkorn et al., 2019). Briefly, 0.45–0.7 mg of tissue was homogenized in 0.32 M sucrose supplemented with protease inhibitors (Sigma-Aldrich, St. Louis, MO, USA), 1 mM EDTA and 1 mM PMSF, pH 7.4. Crude membrane preparation was produced by centrifugation at 27,000 × g for 75 min. The pellet was solubilized in 150 mM NaCl, 2% Triton X-100, 25 mM Tris, supplemented with protease inhibitors, 1 mM EDTA and 1 mM PMSF, pH 7.4. 50 μg aliquots of total protein were separated on Tris-acetate gel (6%) and transferred onto PVDF membrane. After overnight blocking in 5% non-fat dry milk in Tris-buffered saline (TBS), the membrane was incubated overnight with anti-NaV1.1 antibody (1:200, Alomone Labs, Jerusalem, Israel; Catalog# ASC-001) or anti-calnexin (1:2,000, Stressgen Biotechnologies, San Diego, CA, USA), followed by 2 h incubation with HRP-conjugated goat anti-rabbit antibody (1:10,000, Sigma-Aldrich, St. Louis, MO, USA). The signal was visualized by chemiluminescent detection using ECL.
Allele-specific digital PCR (dPCR) assays were performed at Tevard Biosciences (Cambridge, MA, USA) on neocortical tissue isolated from WT and Scn1aWT/R613X mice on a mixed 50:50 C57BL/6J:129S1/SvImJ background, generated using a similar breeding strategy as described above. After confirming anesthesia, neocortical tissue was carefully extracted, flash-frozen in liquid nitrogen, and stored at −70°C until processing. Total RNA was extracted from tissues using AllPrep® DNA/RNA/miRNA Universal kit (Qiagen, Hilden, Germany; Catalog# 80224) and reverse transcribed with SuperScript™ IV First-Strand Synthesis System (Thermo Fisher, Waltham, MA, USA; Catalog# 18091050). For allele-specific detection of Scn1a transcripts (WT vs. R613X mRNA), custom-designed Affinity Plus™ qPCR Probes (IDT, Coralville, IA, USA) were used to enable greater SNP target specificity. The primer sequences and probes are as follows (the + sign before a nucleotide indicates locked nucleotides): Forward primer: CACAGCACCTTTGAGGATAAT; Reverse primer: GGTCTGGCTCAGGTTACT; WT probe: TCC + C + CG + A + A + GAC; R613X probe: TCC + T + CG + A + T + GAC + AC. Mouse Gapdh TaqMan probe (Thermo Fisher; Mm99999915_g1) was used as a reference gene. dPCR for absolute quantification of gene targets was performed using naica® System dPCR from Stilla Technologies (Villejuif, France).
Meso scale discovery-electrochemiluminescence assays were performed at Tevard Biosciences (Cambridge, MA, USA). Total protein was isolated from combined neocortex (parietal, temporal, occipital lobes), or dissected cortical lobes (frontal, parietal, temporal, occipital, respectively) and liver. The tissue was homogenized in lysis buffer (1× TBS, 1% TX-100, 0.5% Nonidet P-40, 0.25% Na deoxylate, 1 mM EDTA) supplemented with protease/phosphatase inhibitors (HALT Protease/Phosphatase inhibitor cocktail, Thermo Fisher Catalog # 78440) using beaded tubes (MP Biomedicals, Irvine, CA, USA Catalog # 116913050-CF) and a benchtop homogenizer (MP Biomedicals FastPrep-24 5G) at 4.0 m/s for 5 s. Samples were incubated at 4°C for 15 min with rotation, spun down at 16,000 × g at 4°C for 15 min, and the supernatant containing total protein was collected. Multi-Array 96 Small Spot Plates (Meso Scale Diagnostics, Rockville, MD, USA, Catalog # L45MA) were blocked for 1 h at room temperature with shaking using Blocker B (Meso Scale Diagnostics, Catalog # R93BB). Plates were washed 3 times with TBST, then coated with 5 μg/mL capture antibody for NaV1.1 (UC Davis, Antibodies Inc. Catalog # 75-023) at room temperature for 1 h with shaking. Plates were washed as previously described, and 25 μL of 4 mg/mL standard or protein samples were added to individual wells and incubated overnight at 4°C with shaking. Plates were washed, and the detection antibody for NaV1.1 (Alomone Labs, Catalog # ASC-001-SO) was added at 2.4 μg/mL and incubated at RT for 1 h with shaking. Plates were washed, and secondary detection antibody (SULFO-TAG anti-rabbit antibody; Meso Scale Diagnostics Catalog# R32AB) was added at 2 μg/mL and incubated for 1 h at room temperature with shaking. Plates were washed and read using MSD GOLD Read Buffer B (Meso Scale Diagnostics Catalog # R60AM, 150 μL) on the MSD QuickPlex SQ120 Instrument.
The DS Scn1aR613X mice were generated by the Dravet Syndrome Foundation Spain and deposited for unrestricted distribution by the Jackson Laboratory (stock no. 034129). The Scn1aR613X mutation was generated on the 129S1/SvImJ background (Figures 1A, B). Previous studies demonstrated that mice with Scn1a mutations on the 129 backgrounds have mild epileptic phenotypes, with rare spontaneous seizures and normal lifespan (Yu et al., 2006; Miller et al., 2014; Mistry et al., 2014; Rubinstein et al., 2015; Kang et al., 2018). In agreement, heterozygous Scn1aWT/R613X mice on the pure 129S1/SvImJ background were not witnessed to have spontaneous convulsive seizures or seizures during routine handling, and only one mouse died prematurely (Figure 1D). Conversely, crossing Scn1a mutant mice onto the C57BL/6J background aggravated their epileptic phenotypes with the presentation of spontaneous seizures and premature death (Yu et al., 2006; Miller et al., 2014; Mistry et al., 2014; Rubinstein et al., 2015; Kang et al., 2018). Scn1aWT/R613X on a mixed background (50:50 C57BL/6J:129S1/SvImJ) exhibited spontaneous convulsive seizures, which were often observed unprovoked in their home cage or during routine handling (Figures 1C, D; Supplementary Video 1). Moreover, these mice demonstrated profound premature mortality, with only ∼40% of the mice surviving to P60, and most of the deaths occurring during their fourth week of life (P21-P28, Figure 1D). Scn1aWT/R613X were often found in their cage, with outstretched limbs, suggestive of death during a seizure. Nevertheless, as these deaths were not observed, we cannot exclude the possibility of additional cardiac or respiratory comorbidities (Auerbach et al., 2013; Kalume et al., 2013; Kim et al., 2018; Kuo et al., 2019). Despite poor survival, the growth rate of Scn1aWT/R613X mice on the mixed background was similar to their WT littermates, with no significant differences in body weight (Figure 1E). Homozygous Scn1aR613X/R613X died prematurely between P14-16 (Figure 1D). Together, the Scn1aWT/R613X Dravet mice on a mixed background demonstrated spontaneous seizures and premature mortality, recapitulating Dravet epilepsy and Dravet-associated premature death.
Heat-induced seizures are a hallmark phenotype of SCN1A mutations. We tested the susceptibility of Scn1aWT/R613X mice, on either background, to heat-induced seizures at different ages: the pre-epileptic stage (P14-P16), the severe stage of epilepsy during the fourth week of life (P21-P25), and the stabilization or chronic stage after the fifth week of life (P34-P35) (Fadila et al., 2020; Gerbatin et al., 2022a). All the Scn1a mutant mice exhibited thermally induced seizures below 42°C (Figure 2). The highest susceptibility, demonstrated by the lowest temperature threshold, was observed in Scn1aWT/R613X on the mixed background at their fourth week of life (P21-P25, Figures 2B, C, F). Together, these data confirm that Scn1aWT/R613X mice show a high susceptibility to heat-induced seizures, from the pre-epileptic stage to adulthood, similar to Dravet patients.
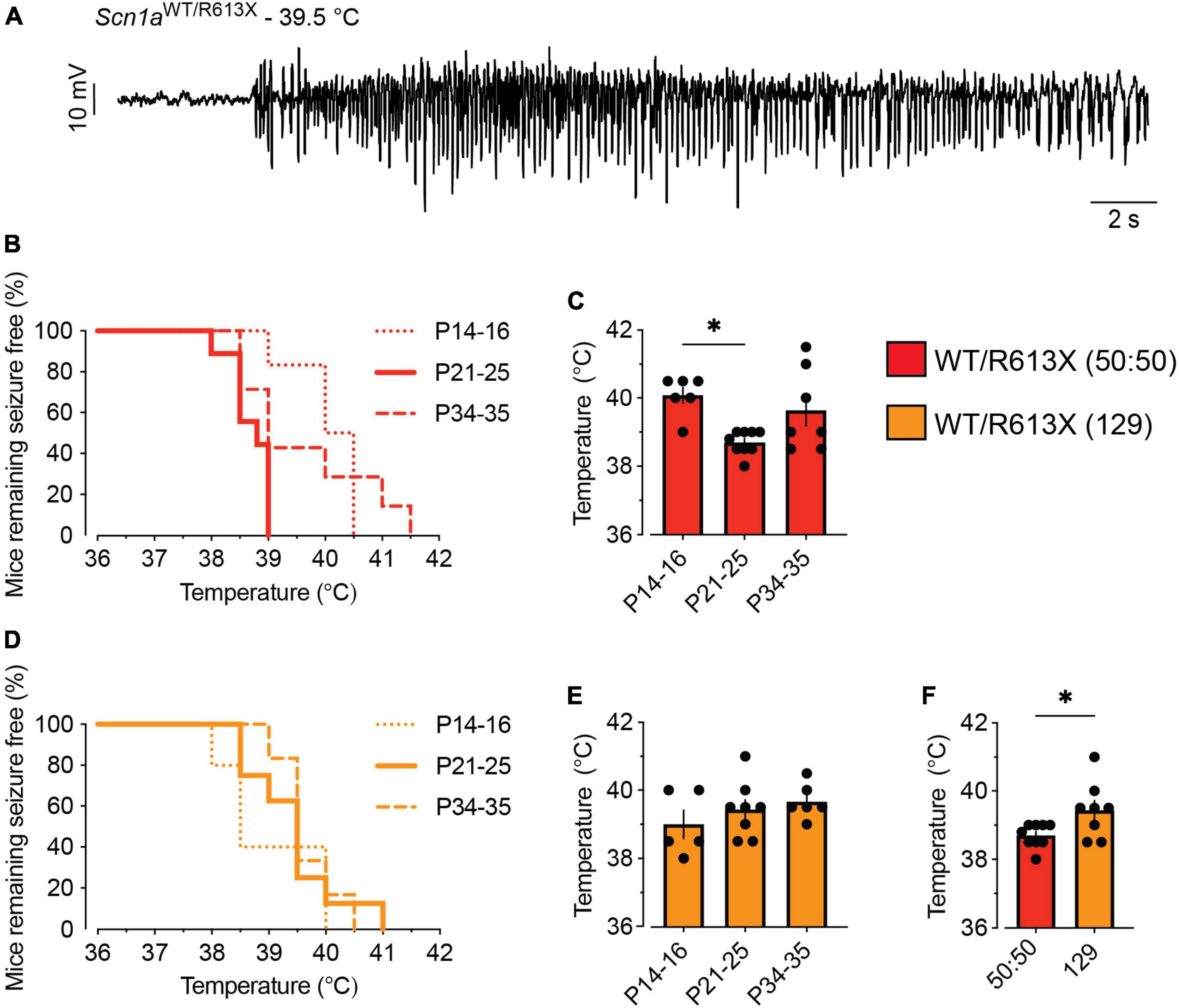
Figure 2. Scn1aWT/R613X are susceptible to heat-induced seizures. (A) An ECoG trace from a Scn1aWT/R613X mouse on a mixed 50:50 background depicting epileptic activity at a temperature of 39.5°C. (B,C) Scn1aWT/R613X mice on a mixed background (50:50), at the indicated ages (P14-16; P21-P25; P34-P35), remaining free of thermally induced seizures (B), and the temperature of seizures (C). P14-16: n = 6; P21-P25: n = 9; P34-P35: n = 7. (D,E) Scn1aWT/R613X mice on the 129S1/SvImJ background, at the indicated ages (P14-16; P21-P25; P34-P35), remaining free of thermally induced seizures (D), and the temperature of seizures (E). P14-16: n = 5; P21-P25: n = 8; P34–P35: n = 6. WT mice did not experience seizures within this temperature range (n = 4–7 at each age group, not shown). (F) At P21-P25, Scn1aWT/R613X mice on a mixed background (50:50) have heat-induced seizures at lower temperatures compared to Scn1aWT/R613X mice on a pure 129 background (129). These are the same data depicted in panels (C,E). These data did not follow a normal distribution. Statistical comparison in panel (C) utilized non-parametric One-Way ANOVA followed by Dunn’s test. Statistical comparison in panel (E) utilized the Mann–Whitney test. *p < 0.05.
To test the presence of non-epileptic phenotypes, in Scn1aWT/R613X mice on the mixed background, we examined hyperactivity in a novel arena, motor functions using the rotarod test, and working memory using the Y-maze spontaneous alternation test. In the open field test, which examines the exploration of a novel arena, heterozygous Scn1aWT/R613X mice displayed increased ambulation and traveled longer distances and at higher velocities compared to their WT littermates (Figures 3A–C). Nevertheless, the time in the center of the arena was similar between Scn1aWT/R613X and WT mice (Figure 3D), indicating that these mice do not exhibit increased anxiety. Next, motor functions were assessed using the rotarod test. As shown in Figure 3E, the latency to fall was comparable between WT and heterozygous Scn1aWT/R613X mice, suggesting normal balance and coordination. Moreover, their performance in the spontaneous alternation Y-maze test, during their fourth week of life, corresponding to the severe stage of Dravet (P21-P25), as well as in a different cohort of mice at their sixth week of life (P36-P43), indicated that these mice do not have a deficit in spatial working memory (Figures 3F, H). Nevertheless, increased locomotor activity was also observed in the Y-maze, further corroborating the hyperactivity observed in the open field. Thus, in addition to susceptibility to thermally induced seizures, spontaneous seizures, and premature death, Scn1aWT/R613X mice on the mixed background also demonstrate hyperactivity.
As Scn1aWT/R613X mice on the pure 129S1/SvImJ background did not exhibit premature mortality (Figure 1) but were susceptible to heat-induced seizures (Figure 2), we wondered if these mice exhibited behavioral deficits. While we did find vast differences in the innate tendency to explore the novel arena (compare Figures 3, 4), the activity of Scn1aWT/R613X mice on the 129S1/SvImJ background was similar to that of their WT littermate in both the open field test and the rotarod test, suggesting that genetic background can also modify Dravet-associated hyperactivity.
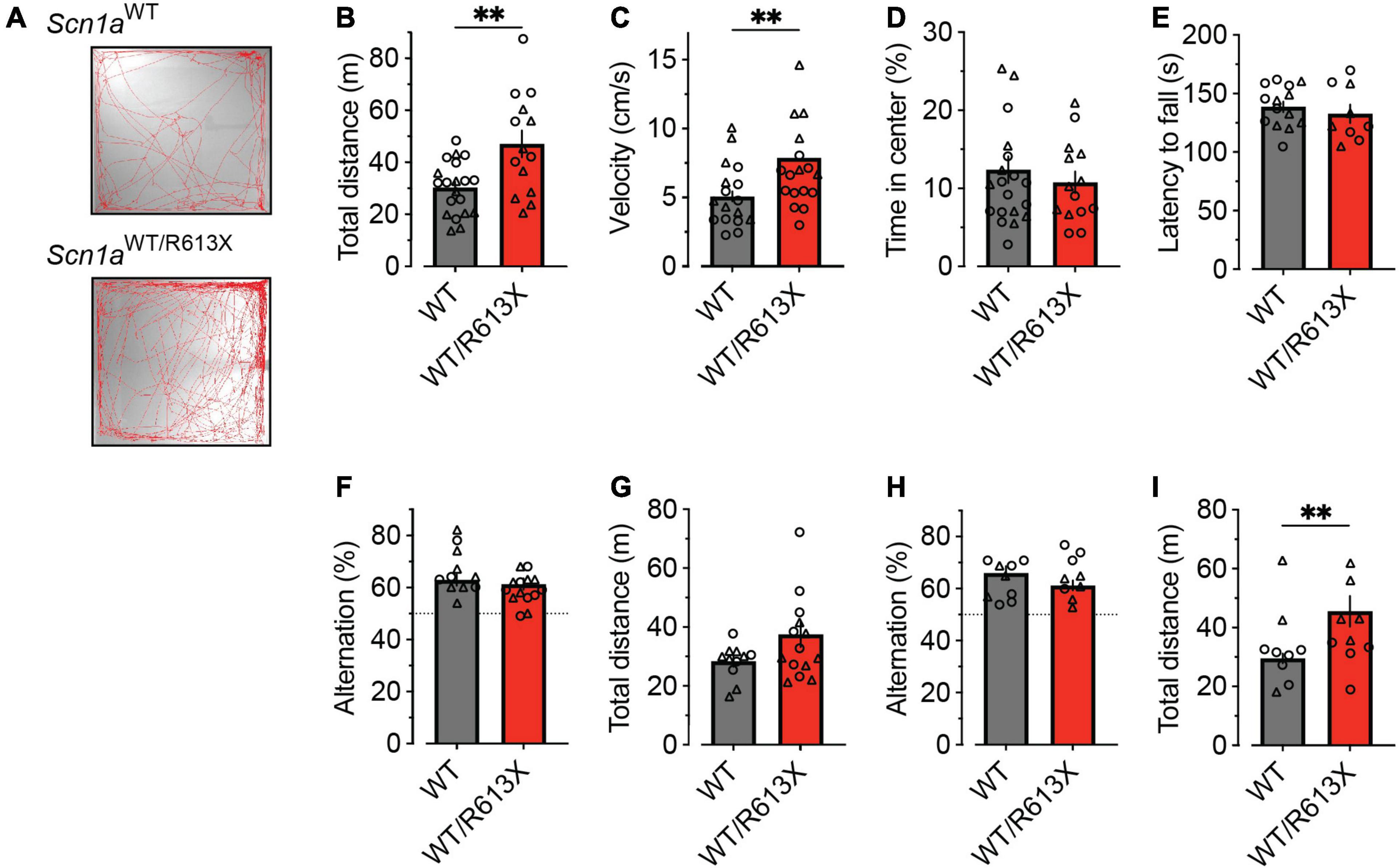
Figure 3. Hyperactivity in Scn1aWT/R613X mice on the mixed background. (A–D) Open field in WT and DS mice at their fourth week of life (P21-P25). (A) Representative examples of exploring the behavior of WT and DS mice during the 10-min test period. (B) Distance moved. (C) Average velocity. (D) Percentage of time spent in the central portion of the arena. WT, n = 20; Scn1aWT/R613X, n = 14. (E) The latency to fall in the rotarod test in WT and DS mice (P34-P42). WT, n = 14; Scn1aWT/R613X, n = 9. (F–I) The Y-maze test in P21-P25 mice (F,G) and in another cohort of older (P36-P43) mice (H,I). The dotted line in panels (F,H) signifies the chance level expected from random alternation. Panels (G,I) depict the total distance moved in the Y-maze arena during exploration. P21-P25: WT, n = 14; Scn1aWT/R613X, n = 11; P36-P43: WT, n = 10; Scn1aWT/R613X, n = 8. In addition to the average and SE, the individual data points from males (triangles) and females (circles) are depicted. As these data were distributed normally, statistical comparison utilized the unpaired t-test. **p < 0.01.
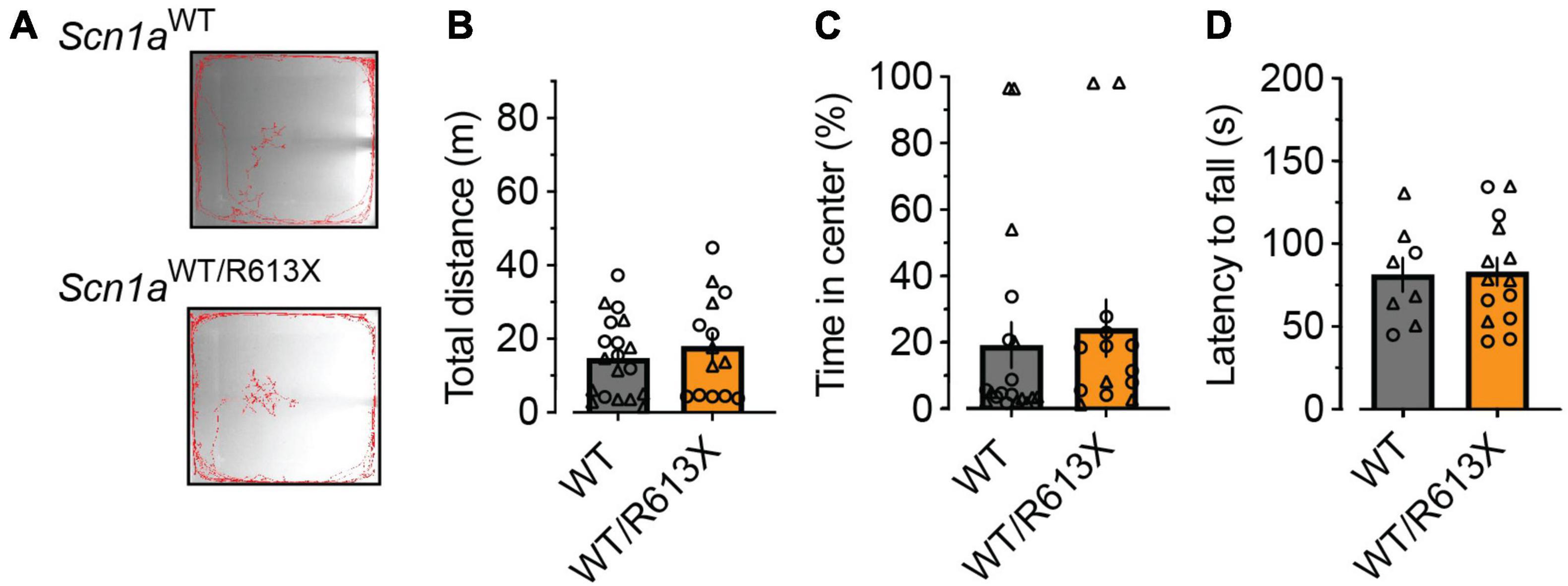
Figure 4. No behavioral deficits in Scn1aWT/R613X mice on the 129S1/SvImJ background. (A) Representative examples of the exploring behavior of WT and Scn1aWT/R613X mice during the 10-min test period. (B) Distance moved in the open field. (C) Percentage of time spent in the central portion of the arena. WT, n = 19; Scn1aWT/R613X, n = 14. (D) The latency to fall in the rotarod test. WT, n = 8; Scn1aWT/R613X, n = 14.
To test the effect of the R613X premature termination at the transcriptional level, we extracted and analyzed mRNA from the hippocampus of P21-P24 WT and Scn1a mutant mice. First, we confirmed that the R613X mutation is also expressed in the hippocampus and translated into mRNA (Figure 5A). Next, quantitative real-time PCR analysis of Scn1a mRNA, using an assay that targets the boundary junction between exons 18–19, demonstrated ∼50% reduction in the expression of the full-length Scn1a mRNA in heterozygous Scn1aWT/R613X mice on either genetic background, and a marginal background expression in homozygous Scn1aR613X/R613X mice (Figures 5B, C).
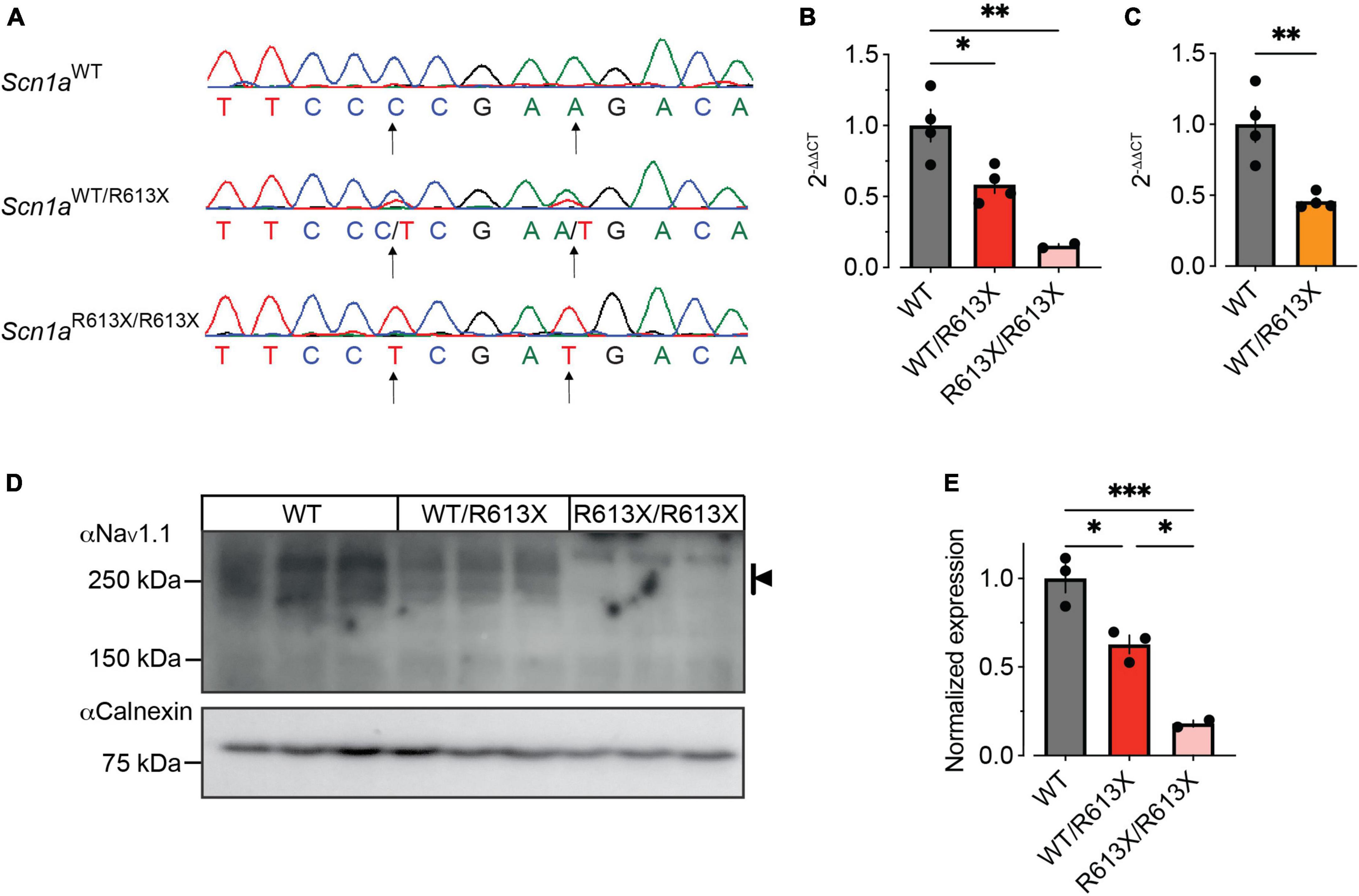
Figure 5. Reduced Scn1a mRNA and NaV1.1 protein expression in the hippocampus of Scn1aWT/R613X mice. (A) Sequencing of hippocampal mRNA confirmed the expression of the Scn1aR613X allele. (B,C) qPCR analysis of Scn1a mRNA expression levels showed a significant reduction to approximately 50% in heterozygous Scn1aWT/R613X on the mixed background (B), and on the pure 129S1/SvImJ background (C). Marginal background expression was found in homozygous Scn1aR613X/R613X mice. Mixed background: WT, n = 4; Scn1aWT/R613X T, n = 4; Scn1aR613X/R613X, n = 2. 129S1/SvImJ background: WT, n = 4; Scn1aWT/R613X, n = 4. (D) Western blot analysis of NaV1.1 protein extracted from the hippocampi of three different mice from each genotype. Kilodaltons = kDa. (E) Quantification of NaV1.1 protein level was based on multiple bands as indicated by the black arrow in panel (D). Only two of the three Scn1aR613X/R613X were included in the quantification due to the dark spot in the middle lane that precluded the inclusion of this sample. Statistical comparison in panels (C,E) utilized One-Way ANOVA followed by Tukey’s multiple comparisons test. Statistical comparison in panel (C) utilized t-test. *p < 0.05, **p < 0.01, ***p < 0.001.
Next, we determine the impact of the R613X mutation on NaV1.1 protein expression levels. In accordance with the decrease observed in Scn1a mRNA, western blot analysis of extracted hippocampi showed that the level of NaV1.1 protein in the hippocampus of heterozygous Scn1aWT/R613X was decreased to ∼50% compared to WT control, with a minimal signal seen in homozygous Scn1aR613X/R613X mice (Figures 5D, E).
To quantitatively assess the level of the R613X transcript in the cortex, we performed an allele-specific dPCR assay, using probes that specifically bind WT Scn1a and R613X sequences (Figure 1A). The R613X allele was not detected in cortical tissue from WT mice, confirming the specificity of the assay (Figure 6A). Conversely, the WT allele was detected at about half the levels in Scn1aWT/R613X tissue compared to WT cortex, as expected for heterozygous animals (Figure 6B). Interestingly, when expressed as the percentage of the WT allele, the steady-state level of the mutant R613X transcript was 8.9 ± 0.9% (Figure 6C), much lower than the expected 50% level, suggesting that transcripts of the mutant allele undergo strong nonsense-mediated decay (NMD) (Jaffrey and Wilkinson, 2018) in neocortical tissue.
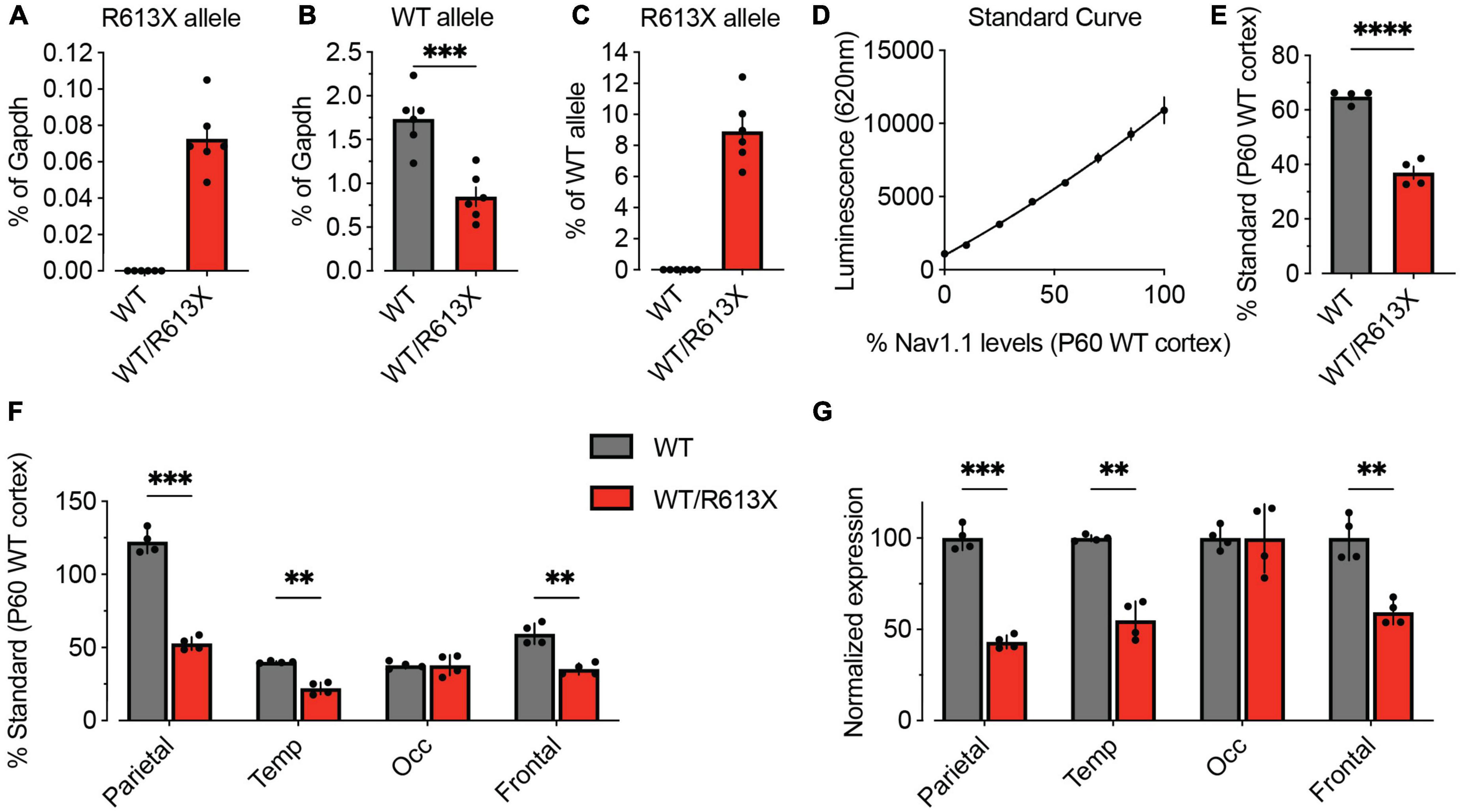
Figure 6. Reduced Scn1a mRNA and NaV1.1 protein expression in the cortex of Scn1aWT/R613X mice. (A–C) Allele-specific quantification of Scn1a transcripts via dPCR. The R613X and WT Scn1a alleles were quantified with Affinity Plus probes on dPCR and normalized to mouse Gapdh levels. (A) Cortical tissue from WT mice showed no detectable levels of the R613X allele transcript, demonstrating the specificity of allelic discrimination using this assay. (B) In cortical tissue from Scn1aWT/R613X mice, the WTScn1a allele is at 48.8% compared to WT cortical tissue. (C) Shown as the ratio of R613X to WT alleles, the mean steady-state level of the Scn1a R613X allele is at 8.9 ± 0.9% of the WT allele in Scn1aWT/R613X animals. WT, n = 6; Scn1aWT/R613X, n = 6. (D–F) Cortical levels of NaV1.1 proteins were quantified using the Meso Scale Discovery Electrochemiluminescence (MSD-ECL) assay. (D) The standard curve was generated by mixing P60 WT cortical and liver proteins at different ratios. The resulting 2nd order polynomial fit, depicted as a solid line (R2 = 0.9924) was used to calculate protein levels relative to P60 WT cortex in panels (E,F). (E) NaV1.1 protein expression in the whole cortex. (F) NaV1.1 protein expression in the parietal, temporal, frontal, and occipital lobes of the neocortex of WT and Scn1aWT/R613X mice. (G) The same data from panel (F), but normalized to NaV1.1 expression in WT mice. WT, n = 4; Scn1aWT/R613X, n = 4. Statistical comparison using mixed model ANOVA followed by Holm–Sidak, to account for analyses of different neocortical sections from each mouse. **p < 0.01, ***p < 0.001, ****p < 0.0001.
For quantitative analysis of NaV1.1 protein levels, we performed the Meso Scale Discovery Electrochemiluminescence (MSD-ECL) assay. MSD-ECL is a highly sensitive ELISA-based assay that enables the detection of small changes in protein levels. Here, we quantified NaV1.1 protein levels in four different cortical lobes (parietal, temporal, occipital, and frontal) on MSD-ECL utilizing two distinct anti-NaV1.1 antibodies (Figures 6D–F). Overall, in the cortex of Scn1aWT/R613X, the level of NaV1.1 was reduced to about 50% compared to WT mice (Figure 6E). However, we found variation in the level of NaV1.1 across the cortex, with the highest NaV1.1 expression in the parietal lobe, and lower levels in other regions (Figure 6F). NaV1.1 expression was reduced to approximately 50% in the parietal, temporal and frontal, but similar to that of WT mice in the occipital lobe (Figure 6G). Together, these data confirm reduced Scn1a mRNA and NaV1.1 protein expression in Scn1aWT/R613X mice, in both the hippocampus and the cortex.
Dravet is a severe form of developmental and epileptic encephalopathy with limited treatment options and poor prognosis. To date, 15 different models have been generated based on microdeletions, nonsense, or missense mutations in the Scn1a gene (Table 1). The novel Scn1aWT/R613X model described here demonstrated core Dravet-associated phenotypes, including spontaneous convulsive seizures, high susceptibility to heat-induced seizures, premature mortality, and several non-epileptic behavioral phenotypes. Thus, this model, which is open-access and publicly available, can be used by the Dravet community for preclinical studies of Dravet mechanisms and therapies.
The genetic background was shown to dramatically modulate the effect of Scn1a mutations (Yu et al., 2006; Miller et al., 2014; Mistry et al., 2014; Rubinstein et al., 2015; Kang et al., 2018). DS mice on the pure C57BL/6J background demonstrate the most severe phenotypes, with high mortality of 60–80% and frequent spontaneous seizures. Conversely, mice with the same Scn1a mutation on the pure 129×1/SvJ or 129S6/SvEvTac background rarely experience spontaneous seizures or premature death. This genetic background effect was attributed to several modifier genes, including Gabra2, Hlf, and Cacna1g (Hawkins and Kearney, 2016; Calhoun et al., 2017; Hawkins et al., 2021b). In accordance, Scn1aWT/R613X mice on the pure 129S1/SvImJ background have a normal life span (Figure 1) and unaltered behavior in the open field test (Figure 4). Despite that, we did observe susceptibility to heat-induced seizures (Figure 2), as well as reduced expression of Scn1a mRNA in the hippocampus (Figure 5).
Multiple studies have used DS mice models to examine the therapeutic potential of current and novel drug treatments (Hawkins et al., 2017; Han et al., 2020; Isom and Knupp, 2021; Pernici et al., 2021; Tanenhaus et al., 2022). Our characterization highlights several readouts that may be useful in future studies to examine the therapeutic benefit while using Scn1aWT/R613X mice on a mixed C57BL/6J:129S1/SvImJ background. Key Dravet-associated phenotypes in these mice include: (i) spontaneous convulsive seizures (Figure 1C; Supplementary Video 1); (ii) profound premature mortality (Figure 1D) with overall survival of less than 50%; (iii) high susceptibility to thermally-induced seizures (Figure 2) with heat-induced seizures at multiple developmental stages occurring within the range of physiological fever temperatures. Heat-induced seizures at relatively low temperatures provide an advantage with a wide measurement range to quantify the effect of current and novel treatments, with the highest sensitivity around P21 (Figure 2C); (iv) developmental changes in the severity of the epileptic phenotypes. Specifically, the pre-epileptic stage in Scn1aWT/R613X DS mice was characterized by susceptibility to heat-induced seizures that preceded the onset of premature mortality (P14-P16); profound mortality during the fourth week of life, with 78% of the deaths occurring between P21 and P28, corresponding to the severe or worsening stage of Dravet, and some stabilization with a reduced rate of premature death in mice that survive beyond P30 (Figure 1D). Thus, relatively restricted age groups should be considered for analysis; (v) the presentation of non-epileptic phenotypes demonstrated here as motor hyperactivity when introduced to a novel arena (Figure 3), modeling Dravet-associated hyperactivity. Of note, we also observed hyperactivity in the Y-maze test (Figure 3), indicating that this is a robust and reliable behavioral readout; (vi) reduced Scn1a mRNA and NaV1.1 protein levels (Figures 5, 6); (vii) impaired firing of inhibitory neurons, a typical neuronal deficit in DS mice, was also observed previously in Scn1aWT/R613X mice (Almog et al., 2022). Importantly, these robust phenotypes highlight this model as an open-access pre-clinical platform to study Dravet therapies.
Nevertheless, some of the non-epileptic phenotypes observed in other DS models were not detected here (Table 1), possibly due to the effect of genetic background on the presentation of Dravet-associated behavioral non-epileptic phenotypes. Specifically, while Scn1aWT/R613X mice demonstrated motor hyperactivity when introduced to a novel arena (Figures 3A–C), no motor deficits or altered spatial working memory were observed (Figure 3). However, these data are in accordance with other DS models that were reported to have normal rotarod performance, including models with deletions in the Scn1a gene (Niibori et al., 2020; Patra et al., 2020; Valassina et al., 2022) or mice harboring the Scn1a R1407X nonsense mutation (Ito et al., 2013). Similarly, normal spontaneous alternations were reported in DS mice with deletion of the 1st exon or mice with knock-in of Scn1a poison exon (Voskobiynyk et al., 2021; Gerbatin et al., 2022a). Conversely, impaired rotarod activity and spontaneous alternations were reported in DS mice with deletion of the last Scn1a exon, as well as in DS mice carrying the Scn1a A1783V missense mutations; both on the pure C57BL/6J background (Fadila et al., 2020; Beretta et al., 2022; Table 1).
In conclusion, the Scn1aWT/R613X DS model, harboring the recurrent nonsense Scn1a mutation and available in open access distribution, including to for-profit organizations through The Jackson Laboratory (Strain no. 034129), demonstrates multiple Dravet-associated phenotypes with robust and specific deficits that can be used as a preclinical model for drug development. Thus, as current treatment options in Dravet are limited and considerable efforts are being made to produce novel and more effective anti-seizure small molecule drugs, as well as disease-modifying genetic treatments (Isom and Knupp, 2021), including those specifically targeting nonsense mutations, we propose that this model can provide a useful and powerful pre-clinical platform for the Dravet research scientific community.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
This animal study was reviewed and approved by the Tel Aviv University, Tevard Biosciences.
AM, EC-M, JA, and MR conceived the project. AM, MB, VW, HB, Y-HC, VD, and MR designed, carried out the experiments and analyzed the data. All authors contributed to the article and approved the submitted version.
This work was supported by the Dravet Syndrome Foundation Spain, the Israel Science Foundation (grant 1454/17, MR), the National Institute of Psychobiology in Israel (MR), the Stolz Foundation Sackler Faculty of Medicine, Tel Aviv University (MR), and Tevard Biosciences.
We thank all the members of the Rubinstein Lab for their constructive criticism and technical support, as well as Natasha Stark (Tevard Biosciences) for technical support. This work was performed in partial fulfillment of the requirements for a Ph.D. degree of AM.
JL, VW, HB, Y-HC, and VD are current or former employees of Tevard Biosciences. EC-M and VS were employed by the Dravet Syndrome Foundation Spain. JA was president of the Dravet Syndrome Foundation Spain, and his role in the Foundation is voluntary and altruistic. The authors declare that this study received funding from Tevard Biosciences. The funder had the following involvement in the study: The data presented in Figure 6 were performed at Tevard Biosciences by its employees, and Tevard Biosciences employees wrote the text related to this Figure. In addition, the rest of the data and the text were discussed with Tevard Biosciences employees.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2023.1149391/full#supplementary-material
Almog, Y., Fadila, S., Brusel, M., Mavashov, A., Anderson, K., and Rubinstein, M. (2021). Developmental alterations in firing properties of hippocampal CA1 inhibitory and excitatory neurons in a mouse model of Dravet syndrome. Neurobiol. Dis. 148:105209. doi: 10.1016/j.nbd.2020.105209
Almog, Y., Mavashov, A., Brusel, M., and Rubinstein, M. (2022). Functional investigation of a neuronal microcircuit in the CA1 area of the hippocampus reveals synaptic dysfunction in Dravet syndrome mice. Front. Mol. Neurosci. 15:823640. doi: 10.3389/fnmol.2022.823640
Alonso, C., Satta, V., Díez-Gutiérrez, P., Fernández-Ruiz, J., and Sagredo, O. (2022). Preclinical investigation of β-caryophyllene as a therapeutic agent in an experimental murine model of Dravet syndrome. Neuropharmacology 205:108914. doi: 10.1016/J.NEUROPHARM.2021.108914
Anderson, L. L., Doohan, P. T., Hawkins, N. A., Bahceci, D., Garai, S., Thakur, G. A., et al. (2022). The endocannabinoid system impacts seizures in a mouse model of Dravet syndrome. Neuropharmacology 205:108897. doi: 10.1016/J.NEUROPHARM.2021.108897
Auerbach, D. S., Jones, J., Clawson, B. C., Offord, J., Lenk, G. M., Ogiwara, I., et al. (2013). Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS One 8:e77843. doi: 10.1371/JOURNAL.PONE.0077843
Bahceci, D., Anderson, L. L., Occelli Hanbury Brown, C. V., Zhou, C., and Arnold, J. C. (2020). Adolescent behavioral abnormalities in a Scn1a+/– mouse model of Dravet syndrome. Epilepsy Behav. 103:106842. doi: 10.1016/J.YEBEH.2019.106842
Beretta, S., Gritti, L., Ponzoni, L., Scalmani, P., Mantegazza, M., Sala, M., et al. (2022). Rescuing epileptic and behavioral alterations in a Dravet syndrome mouse model by inhibiting eukaryotic elongation factor 2 kinase (eEF2K). Mol. Autism 13, 1–22. doi: 10.1186/S13229-021-00484-0/FIGURES/7
Calhoun, J. D., Hawkins, N. A., Zachwieja, N. J., and Kearney, J. A. (2017). Cacna1g is a genetic modifier of epilepsy in a mouse model of Dravet syndrome. Epilepsia 58, e111–e115. doi: 10.1111/EPI.13811
Cao, D., Ohtani, H., Ogiwara, I., Ohtani, S., Takahashi, Y., Yamakawa, K., et al. (2012). Efficacy of stiripentol in hyperthermia-induced seizures in a mouse model of Dravet syndrome. Epilepsia 53, 1140–1145. doi: 10.1111/j.1528-1167.2012.03497.x
Cardenal-Muñoz, E., Auvin, S., Villanueva, V., Cross, J. H., Zuberi, S. M., Lagae, L., et al. (2022). Guidance on Dravet syndrome from infant to adult care: road map for treatment planning in Europe. Epilepsia Open 7, 11–26. doi: 10.1002/epi4.12569
Cheah, C. S., Beckman, M. A., Catterall, W. A., and Oakley, J. C. (2021). Sharp-wave ripple frequency and interictal epileptic discharges increase in tandem during thermal induction of seizures in a mouse model of genetic epilepsy. Front. Cell. Neurosci. 15:751762. doi: 10.3389/FNCEL.2021.751762
Cheah, C. S., Yu, F. H., Westenbroek, R. E., Kalume, F. K., Oakley, J. C., Potter, G. B., et al. (2012). Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. 109, 14646–14651. doi: 10.1073/pnas.1211591109
Chuang, S. H., Westenbroek, R. E., Stella, N., and Catterall, W. A. (2021). Combined antiseizure efficacy of cannabidiol and clonazepam in a conditional mouse model of dravet syndrome. J. Exp. Neurol. 2, 81–85. doi: 10.33696/NEUROL.2.040
Claes, L., Ceulemans, B., Audenaert, D., Smets, K., Löfgren, A., Del-Favero, J., et al. (2003). De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum. Mutat. 21, 615–621. doi: 10.1002/humu.10217
Claes, L. R. F., Deprez, L., Suls, A., Baets, J., Smets, K., Van Dyck, T., et al. (2009). The SCN1A variant database: a novel research and diagnostic tool. Hum. Mutat. 30, E904–E920. doi: 10.1002/humu.21083
Depienne, C., Trouillard, O., Saint-Martin, C., Gourfinkel-An, I., Bouteiller, D., Carpentier, W., et al. (2009). Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J. Med. Genet. 46, 183–191. doi: 10.1136/JMG.2008.062323
Dravet, C., Bureau, M., Bernardina, B. D., and Guerrini, R. (2011). Severe myoclonic epilepsy in infancy (Dravet syndrome) 30 years later. Epilepsia 52, 1–2. doi: 10.1111/j.1528-1167.2011.02993.x
Dyment, D. A., Schock, S. C., Deloughery, K., Tran, M. H., Ure, K., Nutter, L. M. J., et al. (2020). Electrophysiological alterations of pyramidal cells and interneurons of the CA1 region of the hippocampus in a novel mouse model of dravet syndrome. Genetics 215, 1055–1066. doi: 10.1534/genetics.120.303399
Fadila, S., Quinn, S., Turchetti Maia, A., Yakubovich, D., Ovadia, M., Anderson, K. L., et al. (2020). Convulsive seizures and some behavioral comorbidities are uncoupled in the Scn1aA1783V Dravet syndrome mouse model. Epilepsia 61, 2289–2300. doi: 10.1111/epi.16662
Gaily, E., Anttonen, A. K., Valanne, L., Liukkonen, E., Träskelin, A. L., Polvi, A., et al. (2013). Dravet syndrome: new potential genetic modifiers, imaging abnormalities, and ictal findings. Epilepsia 54, 1577–1585. doi: 10.1111/EPI.12256
Gataullina, S., and Dulac, O. (2017). From genotype to phenotype in Dravet disease. Seizure 44, 58–64. doi: 10.1016/j.seizure.2016.10.014
Gerbatin, R. R., Augusto, J., Boutouil, H., Reschke, C. R., and Henshall, D. C. (2022a). Life-span characterization of epilepsy and comorbidities in Dravet syndrome mice carrying a targeted deletion of exon 1 of the Scn1a gene. Exp. Neurol. 354:114090. doi: 10.1016/J.EXPNEUROL.2022.114090
Gerbatin, R. R., Augusto, J., Morris, G., Campbell, A., Worm, J., Langa, E., et al. (2022b). Investigation of MicroRNA-134 as a target against seizures and SUDEP in a mouse model of Dravet syndrome. eNeuro 9:ENEURO.0112-22.2022. doi: 10.1523/eneuro.0112-22.2022
Gheyara, A. L., Ponnusamy, R., Djukic, B., Craft, R. J., Ho, K., Guo, W., et al. (2014). Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol. 76, 443–456. doi: 10.1002/ana.24230
Han, S., Tai, C., Westenbroek, R. E., Yu, F. H., Cheah, C. S., Potter, G. B., et al. (2012). Autistic-like behaviour in Scn1a+/– mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390. doi: 10.1038/nature11356
Han, Z., Chen, C., Christiansen, A., Ji, S., Lin, Q., Anumonwo, C., et al. (2020). Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med. 12:eaaz6100. doi: 10.1126/SCITRANSLMED.AAZ6100
Hawkins, N. A., Anderson, L. L., Gertler, T. S., Laux, L., George, A. L., and Kearney, J. A. (2017). Screening of conventional anticonvulsants in a genetic mouse model of epilepsy. Ann. Clin. Transl. Neurol. 4, 326–339. doi: 10.1002/acn3.413
Hawkins, N. A., Jurado, M., Thaxton, T. T., Duarte, S. E., Barse, L., Tatsukawa, T., et al. (2021a). Soticlestat, a novel cholesterol 24-hydroxylase inhibitor, reduces seizures and premature death in Dravet syndrome mice. Epilepsia 62, 2845–2857. doi: 10.1111/EPI.17062
Hawkins, N. A., Nomura, T., Duarte, S., Barse, L., Williams, R. W., Homanics, G. E., et al. (2021b). Gabra2 is a genetic modifier of Dravet syndrome in mice. Mamm. Genome 32, 350–363. doi: 10.1007/s00335-021-09877-1
Hawkins, N. A., and Kearney, J. A. (2016). Hlf is a genetic modifier of epilepsy caused by voltage-gated sodium channel mutations. Epilepsy Res. 119, 20–23. doi: 10.1016/J.EPLEPSYRES.2015.11.016
Hawkins, N. A., Zachwieja, N. J., Miller, A. R., Anderson, L. L., and Kearney, J. A. (2016). Fine mapping of a Dravet syndrome modifier locus on mouse chromosome 5 and candidate gene analysis by RNA-Seq. PLoS Genet. 12:e1006398. doi: 10.1371/journal.pgen.1006398
Ho, S. Y., Lin, L., Chen, I. C., Tsai, C. W., Chang, F. C., and Liou, H. H. (2021). Perampanel reduces hyperthermia-induced seizures in Dravet syndrome mouse model. Front. Pharmacol. 12:682767. doi: 10.3389/fphar.2021.682767
Hsiao, J., Yuan, T. Y., Tsai, M. S., Lu, C. Y., Lin, Y. C., Lee, M. L., et al. (2016). Upregulation of haploinsufficient gene expression in the brain by targeting a long non-coding RNA improves seizure phenotype in a model of Dravet syndrome. EBioMedicine 9, 257–277. doi: 10.1016/j.ebiom.2016.05.011
Isom, L. L., and Knupp, K. G. (2021). Dravet syndrome: novel approaches for the most common genetic epilepsy. Neurotherapeutics 18, 1524–1534. doi: 10.1007/s13311-021-01095-6
Ito, S., Ogiwara, I., Yamada, K., Miyamoto, H., Hensch, T. K., Osawa, M., et al. (2013). Mouse with NaV1.1 haploinsufficiency, a model for Dravet syndrome, exhibits lowered sociability and learning impairment. Neurobiol. Dis. 49, 29–40. doi: 10.1016/j.nbd.2012.08.003
Jaffrey, S. R., and Wilkinson, M. F. (2018). Nonsense-mediated RNA decay in the brain: emerging modulator of neural development and disease. Nat. Rev. Neurosci. 19, 715–728. doi: 10.1038/s41583-018-0079-z
Jansen, N. A., Dehghani, A., Breukel, C., Tolner, E. A., and van den Maagdenberg, A. M. J. M. (2020). Focal and generalized seizure activity after local hippocampal or cortical ablation of NaV1.1 channels in mice. Epilepsia 61, e30–e36. doi: 10.1111/epi.16482
Kalume, F., Westenbroek, R. E., Cheah, C. S., Yu, F. H., Oakley, J. C., Scheuer, T., et al. (2013). Sudden unexpected death in a mouse model of Dravet syndrome. J. Clin. Invest. 123, 1798–1808. doi: 10.1172/JCI66220
Kang, S. K., Hawkins, N. A., and Kearney, J. A. (2018). C57BL/6J and C57BL/6N substrains differentially influence phenotype severity in the Scn1a+/– mouse model of Dravet syndrome. Epilepsia Open 4, 164–169. doi: 10.1002/epi4.12287
Kaneko, K., Currin, C. B., Goff, K. M., Wengert, E. R., Somarowthu, A., Vogels, T. P. (2022). Developmentally regulated impairment of parvalbumin interneuron synaptic transmission in an experimental model of Dravet syndrome. Cell Rep. 38:110580. doi: 10.1016/j.celrep.2022.110580
Kearney, J. A., Wiste, A. K., Stephani, U., Trudeau, M. M., Siegel, A., Ramachandrannair, R., et al. (2006). Recurrent de novo mutations of SCN1A in severe myoclonic epilepsy of infancy. Pediatr. Neurol. 34, 116–120. doi: 10.1016/J.PEDIATRNEUROL.2005.07.009
Kim, Y., Bravo, E., Thirnbeck, C. K., Smith-Mellecker, L. A., Kim, S. H., Gehlbach, B. K., et al. (2018). Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J. Clin. Invest. 128, 1141–1153. doi: 10.1172/JCI94999
Kuo, F.-S., Cleary, C. M., LoTurco, J. J. L., Chen, X., and Mulkey, D. K. (2019). Disordered breathing in a mouse model of Dravet syndrome. Elife 8:e43387. doi: 10.7554/eLife.43387
Lee, H. F., Chi, C. S., Tsai, C. R., Chen, C. H., and Wang, C. C. (2015). Electroencephalographic features of patients with SCN1A-positive Dravet syndrome. Brain Dev. 37, 599–611. doi: 10.1016/J.BRAINDEV.2014.10.003
Liu, Y. H., Cheng, Y. T., Tsai, M. H., Chou, I. J., Hung, P. C., Hsieh, M. Y., et al. (2021). Genetics and clinical correlation of Dravet syndrome and its mimics – experience of a tertiary center in Taiwan. Pediatr. Neonatol. 62, 550–558. doi: 10.1016/j.pedneo.2021.05.022
Margherita Mancardi, M., Striano, P., Gennaro, E., Madia, F., Paravidino, R., Scapolan, S., et al. (2006). Familial occurrence of febrile seizures and epilepsy in severe myoclonic epilepsy of infancy (SMEI) patients with SCN1A mutations. Epilepsia 47, 1629–1635. doi: 10.1111/J.1528-1167.2006.00641.X
Martin, M. S., Dutt, K., Papale, L. A., Dubé, C. M., Dutton, S. B., De Haan, G., et al. (2010). Altered function of the SCN1A voltage-gated sodium channel leads to γ-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J. Biol. Chem. 285, 9823–9834. doi: 10.1074/jbc.M109.078568
Mattis, J., Somarowthu, A., Goff, K. M., Jiang, E., Yom, J., Sotuyo, N., et al. (2022). Corticohippocampal circuit dysfunction in a mouse model of Dravet syndrome. Elife 11:e69293. doi: 10.7554/ELIFE.69293
Meng, H., Xu, H. Q., Yu, L., Lin, G. W., He, N., Su, T., et al. (2015). The SCN1A mutation database: updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum. Mutat. 36, 573–580. doi: 10.1002/humu.22782
Miljanovic, N., Hauck, S. M., van Dijk, R. M., Di Liberto, V., Rezaei, A., and Potschka, H. (2021). Proteomic signature of the Dravet syndrome in the genetic Scn1a-A1783V mouse model. Neurobiol. Dis. 157:105423. doi: 10.1016/j.nbd.2021.105423
Miller, A. R., Hawkins, N. A., McCollom, C. E., and Kearney, J. A. (2014). Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav. 13, 163–172. doi: 10.1111/gbb.12099
Mistry, A. M., Thompson, C. H., Miller, A. R., Vanoye, C. G., George, A. L., and Kearney, J. A. (2014). Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol. Dis. 65, 1–11. doi: 10.1016/j.nbd.2014.01.006
Moehring, J., Von Spiczak, S., Moeller, F., Helbig, I., Wolff, S., Jansen, O., et al. (2013). Variability of EEG-fMRI findings in patients with SCN1A-positive Dravet syndrome. Epilepsia 54, 918–926. doi: 10.1111/epi.12119
Mora-Jimenez, L., Valencia, M., Sanchez-Carpintero, R., Tønnesen, J., Fadila, S., Rubinstein, M., et al. (2021). Transfer of SCN1A to the brain of adolescent mouse model of Dravet syndrome improves epileptic, motor, and behavioral manifestations. Mol. Ther. Nucleic Acids 25, 585–602. doi: 10.1016/j.omtn.2021.08.003
Morey, N., Przybyla, M., van der Hoven, J., Ke, Y. D., Delerue, F., van Eersel, J., et al. (2022). Treatment of epilepsy using a targeted p38γ kinase gene therapy. Sci. Adv. 8:eadd2577.
Niibori, Y., Lee, S. J., Minassian, B. A., and Hampson, D. R. (2020). Sexually divergent mortality and partial phenotypic rescue after gene therapy in a mouse model of Dravet syndrome. Hum. Gene Ther. 31, 339–351. doi: 10.1089/hum.2019.225
Nissenkorn, A., Almog, Y., Adler, I., Safrin, M., Brusel, M., Marom, M., et al. (2019). In vivo, in vitro and in silico correlations of four de novo SCN1A missense mutations. PLoS One 14:e0211901. doi: 10.1371/journal.pone.0211901
Nomura, T., Hawkins, N. A., Kearney, J. A., George, A. L., and Contractor, A. (2019). Potentiating α2 subunit containing perisomatic GABAA receptors protects against seizures in a mouse model of Dravet syndrome. J. Physiol. 597, 4293–4307. doi: 10.1113/JP277651
Oakley, J. C., Kalume, F., Yu, F. H., Scheuer, T., and Catterall, W. A. (2009). Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc. Natl. Acad. Sci. 106, 3994–3999. doi: 10.1073/pnas.0813330106
Ogiwara, I., Iwasato, T., Miyamoto, H., Iwata, R., Yamagata, T., Mazaki, E., et al. (2013). NaV1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of dravet syndrome. Hum. Mol. Genet. 22, 4784–4804. doi: 10.1093/hmg/ddt331
Ogiwara, I., Miyamoto, H., Morita, N., Atapour, N., Mazaki, E., Inoue, I., et al. (2007). NaV1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 27, 5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007
Patra, P. H., Serafeimidou-Pouliou, E., Bazelot, M., Whalley, B. J., Williams, C. M., McNeish, A., et al. (2020). Cannabidiol improves survival and behavioural co-morbidities of Dravet syndrome in mice. Br. J. Pharmacol. 177, 2779–2792. doi: 10.1111/bph.15003
Pernici, C. D., Mensah, J. A., Dahle, E. J., Johnson, K. J., Handy, L., Buxton, L., et al. (2021). Development of an antiseizure drug screening platform for Dravet syndrome at the NINDS contract site for the epilepsy therapy screening program. Epilepsia 62, 1665–1676. doi: 10.1111/epi.16925
Ricobaraza, A., Mora-Jimenez, L., Puerta, E., Sanchez-Carpintero, R., Mingorance, A., Artieda, J., et al. (2019). Epilepsy and neuropsychiatric comorbidities in mice carrying a recurrent Dravet syndrome SCN1A missense mutation. Sci. Rep. 9:14172. doi: 10.1038/s41598-019-50627-w
Rodda, J. M., Scheffer, I. E., McMahon, J. M., Berkovic, S. F., and Graham, H. K. (2012). Progressive gait deterioration in adolescents with Dravet syndrome. Arch. Neurol. 69, 873–878. doi: 10.1001/ARCHNEUROL.2011.3275
Rubinstein, M., Westenbroek, R. E., Yu, F. H., Jones, C. J., Scheuer, T., and Catterall, W. A. (2015). Genetic background modulates impaired excitability of inhibitory neurons in a mouse model of Dravet syndrome. Neurobiol. Dis. 73, 106–117. doi: 10.1016/j.nbd.2014.09.017
Salgueiro-Pereira, A. R., Duprat, F., Pousinha, P. A., Loucif, A., Douchamps, V., Regondi, C., et al. (2019). A two-hit story: seizures and genetic mutation interaction sets phenotype severity in SCN1A epilepsies. Neurobiol. Dis. 125, 31–44. doi: 10.1016/J.NBD.2019.01.006
Satpute Janve, V., Anderson, L. L., Bahceci, D., Hawkins, N. A., Kearney, J. A., and Arnold, J. C. (2021). The heat sensing Trpv1 receptor is not a viable anticonvulsant drug target in the Scn1a+/– mouse model of Dravet syndrome. Front. Pharmacol. 12:1141. doi: 10.3389/FPHAR.2021.675128/BIBTEX
Satta, V., Alonso, C., Díez, P., Martín-Suárez, S., Rubio, M., Encinas, J. M., et al. (2021). Neuropathological characterization of a Dravet syndrome knock-in mouse model useful for investigating cannabinoid treatments. Front. Mol. Neurosci. 13:602801. doi: 10.3389/fnmol.2020.602801
Shao, E., Chang, C. W., Li, Z., Yu, X., Ho, K., Zhang, M., et al. (2022). TAU ablation in excitatory neurons and postnatal TAU knockdown reduce epilepsy, SUDEP, and autism behaviors in a Dravet syndrome model. Sci. Transl. Med. 14:5527. doi: 10.1126/scitranslmed.abm5527
Strzelczyk, A., and Schubert-Bast, S. (2022). A practical guide to the treatment of Dravet syndrome with anti-seizure medication. CNS Drugs 36, 217–237. doi: 10.1007/s40263-022-00898-1
Tanenhaus, A., Stowe, T., Young, A., McLaughlin, J., Aeran, R., Lin, I. W., et al. (2022). Cell-selective adeno-associated virus-mediated SCN1A gene regulation therapy rescues mortality and seizure phenotypes in a Dravet syndrome mouse model and is well tolerated in nonhuman primates. Hum. Gene Ther. 33, 579–597. doi: 10.1089/hum.2022.037
Tran, C. H., Vaiana, M., Nakuci, J., Somarowthu, A., Goff, K. M., Goldstein, N., et al. (2020). Interneuron desynchronization precedes seizures in a mouse model of Dravet syndrome. J. Neurosci. 40, 2764–2775. doi: 10.1523/JNEUROSCI.2370-19.2020
Tsai, M. S., Lee, M. L., Chang, C. Y., Fan, H. H., Yu, I. S., Chen, Y. T., et al. (2015). Functional and structural deficits of the dentate gyrus network coincide with emerging spontaneous seizures in an Scn1a mutant dravet syndrome model during development. Neurobiol. Dis. 77, 35–48. doi: 10.1016/j.nbd.2015.02.010
Uchino, K., Kawano, H., Tanaka, Y., Adaniya, Y., Asahara, A., Deshimaru, M., et al. (2021). Inhibitory synaptic transmission is impaired at higher extracellular Ca2+ concentrations in Scn1a+/– mouse model of Dravet syndrome. Sci. Rep. 11:10634. doi: 10.1038/s41598-021-90224-4
Valassina, N., Brusco, S., Salamone, A., Serra, L., Luoni, M., Giannelli, S., et al. (2022). Scn1a gene reactivation after symptom onset rescues pathological phenotypes in a mouse model of Dravet syndrome. Nat. Commun. 13:161. doi: 10.1038/s41467-021-27837-w
Voskobiynyk, Y., Battu, G., Felker, S. A., Cochran, J. N., Newton, M. P., Lambert, L. J., et al. (2021). Aberrant regulation of a poison exon caused by a non-coding variant in a mouse model of Scn1a-associated epileptic encephalopathy. PLoS Genet. 17:e1009195. doi: 10.1371/JOURNAL.PGEN.1009195
Wang, J. W., Shi, X. Y., Kurahashi, H., Hwang, S. K., Ishii, A., Higurashi, N., et al. (2012). Prevalence of SCN1A mutations in children with suspected Dravet syndrome and intractable childhood epilepsy. Epilepsy Res. 102, 195–200. doi: 10.1016/J.EPLEPSYRES.2012.06.006
Williams, A. D., Kalume, F., Westenbroek, R. E., and Catterall, W. A. (2019). A more efficient conditional mouse model of Dravet syndrome: implications for epigenetic selection and sex-dependent behaviors. J. Neurosci. Methods 325:108315. doi: 10.1016/J.JNEUMETH.2019.108315
Xu, X., Yang, X., Wu, Q., Liu, A., Yang, X., Ye, A. Y., et al. (2015). Amplicon resequencing identified parental mosaicism for approximately 10% of “de novo” SCN1A mutations in children with Dravet syndrome. Hum. Mutat. 36, 861–872. doi: 10.1002/HUMU.22819
Yamagata, T., Raveau, M., Kobayashi, K., Miyamoto, H., Tatsukawa, T., Ogiwara, I., et al. (2020). CRISPR/dCas9-based Scn1a gene activation in inhibitory neurons ameliorates epileptic and behavioral phenotypes of Dravet syndrome model mice. Neurobiol. Dis. 141:104954. doi: 10.1016/j.nbd.2020.104954
Keywords: Dravet syndrome, mouse model, seizures, hyperactivity, open access
Citation: Mavashov A, Brusel M, Liu J, Woytowicz V, Bae H, Chen Y-H, Dani VS, Cardenal-Muñoz E, Spinosa V, Aibar JÁ and Rubinstein M (2023) Heat-induced seizures, premature mortality, and hyperactivity in a novel Scn1a nonsense model for Dravet syndrome. Front. Cell. Neurosci. 17:1149391. doi: 10.3389/fncel.2023.1149391
Received: 21 January 2023; Accepted: 05 April 2023;
Published: 03 May 2023.
Edited by:
Claudio Rivera, Aix-Marseille University, FranceReviewed by:
Isabella Ferando, University of Miami, United StatesCopyright © 2023 Mavashov, Brusel, Liu, Woytowicz, Bae, Chen, Dani, Cardenal-Muñoz, Spinosa, Aibar and Rubinstein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Moran Rubinstein, bW9yYW5ydWJAdGF1ZXgudGF1LmFjLmls
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.