
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci., 02 March 2023
Sec. Cellular Neurophysiology
Volume 17 - 2023 | https://doi.org/10.3389/fncel.2023.1112405
This article is part of the Research TopicRising Stars in Cellular Neurophysiology 2022View all 5 articles
 Brett N. Adey1,2
Brett N. Adey1,2 Johnathan Cooper-Knock3
Johnathan Cooper-Knock3 Ahmad Al Khleifat4
Ahmad Al Khleifat4 Isabella Fogh4
Isabella Fogh4 Philip van Damme5,6,7
Philip van Damme5,6,7 Philippe Corcia8,9
Philippe Corcia8,9 Philippe Couratier10,11Orla Hardiman12Russell McLaughlin13
Philippe Couratier10,11Orla Hardiman12Russell McLaughlin13 Marc Gotkine14,15Vivian Drory16,17
Marc Gotkine14,15Vivian Drory16,17 Vincenzo Silani18,19
Vincenzo Silani18,19 Nicola Ticozzi18,19
Nicola Ticozzi18,19 Jan H. Veldink20
Jan H. Veldink20 Leonard H. van den Berg20
Leonard H. van den Berg20 Mamede de Carvalho21
Mamede de Carvalho21 Susana Pinto21
Susana Pinto21 Jesus S. Mora Pardina22
Jesus S. Mora Pardina22 Mónica Povedano Panades23Peter M. Andersen24
Mónica Povedano Panades23Peter M. Andersen24 Markus Weber25
Markus Weber25 Nazli A. Başak26Christopher E. Shaw4
Nazli A. Başak26Christopher E. Shaw4 Pamela J. Shaw3Karen E. Morrison27John E. Landers28
Pamela J. Shaw3Karen E. Morrison27John E. Landers28 Jonathan D. Glass29Patrick Vourc’h7,30
Jonathan D. Glass29Patrick Vourc’h7,30 Richard J. B. Dobson2,31,32,33Gerome Breen1
Richard J. B. Dobson2,31,32,33Gerome Breen1 Ammar Al-Chalabi4,34Ashley R. Jones4†
Ammar Al-Chalabi4,34Ashley R. Jones4† Alfredo Iacoangeli2,4,31*†
Alfredo Iacoangeli2,4,31*†Introduction: Caveolin-1 and Caveolin-2 (CAV1 and CAV2) are proteins associated with intercellular neurotrophic signalling. There is converging evidence that CAV1 and CAV2 (CAV1/2) genes have a role in amyotrophic lateral sclerosis (ALS). Disease-associated variants have been identified within CAV1/2 enhancers, which reduce gene expression and lead to disruption of membrane lipid rafts.
Methods: Using large ALS whole-genome sequencing and post-mortem RNA sequencing datasets (5,987 and 365 tissue samples, respectively), and iPSC-derived motor neurons from 55 individuals, we investigated the role of CAV1/2 expression and enhancer variants in the ALS phenotype.
Results: We report a differential expression analysis between ALS cases and controls for CAV1 and CAV2 genes across various post-mortem brain tissues and three independent datasets. CAV1 and CAV2 expression was consistently higher in ALS patients compared to controls, with significant results across the primary motor cortex, lateral motor cortex, and cerebellum. We also identify increased survival among carriers of CAV1/2 enhancer mutations compared to non-carriers within Project MinE and slower progression as measured by the ALSFRS. Carriers showed a median increase in survival of 345 days.
Discussion: These results add to an increasing body of evidence linking CAV1 and CAV2 genes to ALS. We propose that carriers of CAV1/2 enhancer mutations may be conceptualised as an ALS subtype who present a less severe ALS phenotype with a longer survival duration and slower progression. Upregulation of CAV1/2 genes in ALS cases may indicate a causal pathway or a compensatory mechanism. Given prior research supporting the beneficial role of CAV1/2 expression in ALS patients, we consider a compensatory mechanism to better fit the available evidence, although further investigation into the biological pathways associated with CAV1/2 is needed to support this conclusion.
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease affecting upper and lower motor neurons. It is characterised by the progressive loss of motor function, leading to muscle weakness, difficulty breathing and swallowing, and paralysis. There is currently no treatment, with a mean life expectancy of 3 years (Al-Chalabi and Hardiman, 2013). ALS is comorbid with fronto-temporal dementia (FTD), with an estimated 50% of ALS patients experiencing impaired executive function (Lomen-Hoerth et al., 2003; Strong et al., 2017). These diseases are often conceptualised as two ends of a disease spectrum with a shared pathogenesis and clinical overlap (Phukan et al., 2007; Conlon et al., 2018).
Individuals who have a first-degree relative with ALS are twice as likely than average to develop ALS (Al-Chalabi et al., 2010), and patients with a family history (familial ALS) make up approximately 5%–10% of cases (Zou et al., 2017). A pathogenic variant for familial patients can be identified in over 50% of cases (Turner et al., 2017). However, most cases have no family history (sporadic ALS), and the majority have no identified genetic aetiology. A recent genome-wide association study (GWAS) estimates the narrow-sense heritability of ALS due to SNPs at 8.5% (Van Rheenen et al., 2016). This represents a minimum heritability value based upon the variation of SNPs included in sequencing arrays. Broad sense heritability estimations for ALS vary between 43% and 53% (Ryan et al., 2019; Trabjerg et al., 2020). Of all currently known pathogenic variants, the most common is a hexanucleotide repeat expansion within the C9orf72 gene, which accounts for 30%–40% of familial cases and 5%–10% of sporadic cases (Brown and Al-Chalabi, 2015; Braems et al., 2020). Individuals with this mutation display an earlier age of onset and faster disease progression (Iacoangeli et al., 2019). A further 5% of sporadic cases are attributable to mutations in SOD1, FUS, and TARDBP genes (Jones et al., 2021).
Despite these known genetic variants, a large proportion of ALS heritability remains unaccounted for. Most ALS genetic studies focus on the study of rare single nucleotide variants (SNVs) and small insertions and deletions (indels) in the coding regions of the genome, or on common single nucleotide polymorphisms (SNPs). As a consequence, structural and rare variants in non-coding regions of the genome are largely under-investigated and could represent a potential source of the missing heritability (Young, 2019; Cooper-Knock et al., 2020; Theunissen et al., 2020).
Caveolin-1 and Caveolin-2 (CAV1 and CAV2, or CAV1/2) genes code for proteins that are associated with the function of membrane lipid rafts. These are regions of low fluidity within the cellular membrane, which act as anchoring points for intercellular signalling (Igarashi et al., 2020). Converging evidence links CAV1 and CAV2 genes to ALS pathology; CAV1 is associated with neuronal survival and is upregulated during induced ischemia in mice, aiding the uptake of extracellular vesicles and reducing apoptosis (Yue et al., 2019). CAV1 may also play a role in the cognitive decline associated with ALS/FTD (Tang et al., 2021), with overexpression increasing neuroplasticity, pro-growth signalling, learning, and memory in mice (Head et al., 2011; Mandyam et al., 2017). Additional evidence using male SOD1 mice showed that the promotion of neuron-specific CAV1 expression increases body weight and improves longevity and motor function (Sawada et al., 2019). In a subsequent mouse study, subpial administration of synapsin-promoted CAV1 also increased survival, although saw no changes to body weight or motor function (Ichinomiya et al., 2021). Conversely, increased neurodegeneration and synaptic reduction were observed in CAV1 knock-out mice (Head et al., 2010).
In humans, CAV1 coding regions are enriched for ALS-associated variants and CAV1 and CAV2 enhancer mutations are significantly associated with an increased risk of ALS (Cooper-Knock et al., 2020). An expression analysis revealed that two mutations within CAV1 and CAV2 enhancer regions reduced CAV1/2 expression in patient-derived non-neuronal cells, which was supported by CRISPR-Cas9 editing in neuronal cells (Cooper-Knock et al., 2020). Together, evidence from human and mouse studies indicate that CAV1/2 is neuroprotective, and CAV1/2 mutations are a risk factor for ALS pathology, likely as a consequence of reduced gene expression.
In this study, we aim to investigate whether these mutations lead to differences in disease-related phenotypes, as well as changes in ALS risk, and explore whether CAV1/2 expression plays a role in the disease beyond enhancer mutations. In the first set of analyses, we used an RNA-sequencing pipeline to perform expression analysis of the CAV1 and CAV2 genes. The results supported our hypothesis that CAV1 and CAV2 genes would be differentially expressed between ALS cases and controls, with patients showing increased expression. In the second set of analyses, we investigated differences in survival duration and age of onset between ALS patients with and without CAV1/2 enhancer mutations. Considering the evidence that CAV1/2 enhancer mutations reduce CAV1/2 expression and that CAV1/2 expression is beneficial to ALS phenotypes, we hypothesised a reduced survival duration and earlier age of onset in ALS patients who have CAV1 or CAV2 enhancer mutations. Results were opposite to our expectation, showing increased survival duration among carriers of CAV1/2 enhancer mutations. No difference in age of onset was observed between groups.
To confirm whether differential expression of CAV1/2 occurred in neurons specifically, we ran an RNA-seq expression analysis in iPSC-derived motor neurons (MNs) from ALS patients and neurologically normal controls. Additionally, we examined the presence of a correlation between the expression of CAV1/2 in the iPSC-derived MNs and survival, age of onset, and disease progression as measured by the ALSFRS.
RNA-seq datasets for the differential expression analyses were obtained from TargetALS at the New York Genome Centre (NYGC; NCBI GEO ID: GSE116622 and GSE124439), the Florida Mayo Clinic (NCBI GEO ID: GSE67196), and the King’s College London and MRC London Neurodegenerative Diseases Brain Bank (Smith et al., 2015; Iacoangeli et al., 2021; Jones et al., 2021).
Sample collection and data generation were previously described (Jones et al., 2021). Briefly, frozen human post-mortem samples were used in all cases, and tissue was taken across multiple brain areas. The KCL MRC Brain Bank samples were taken from the primary motor cortex. The Mayo Clinic samples were obtained from the lateral hemisphere of the cerebellum, Brodmann areas 9 and 44 (prefrontal cortex) and Brodmann area 4 (primary motor cortex). The Target alS (NYGC) samples were obtained from the cerebellum, the lateral and medial motor cortex, and various locations within the frontal cortex.
Whole genome sequencing and clinical data of ALS cases from Project MinE (data freeze 2) were used for the survival and age of onset analyses (Zhang et al., 2022). Samples were filtered to remove common variants (MAF > 0.01) in the enhancer regions of CAV1 and CAV2 genes, which are defined in Cooper-Knock et al. (2020). Individuals with missing data for sex, survival, and age of onset for the corresponding analysis, or those that failed quality controls (Project MinE ALS Sequencing Consortium, 2018) were removed. This retained 5,987 cases for analysis, including 44 individuals with at least one CAV1 or CAV2 enhancer mutation (individual variants and their frequencies can be found in Supplementary Material). Data generation and whole-genome sequencing quality controls, including principal component analysis, were previously described (Project MinE ALS Sequencing Consortium, 2018; Van Rheenen et al., 2021; Zhang et al., 2022).
Total RNA-seq gene expression profiling of iPSC-derived MNs and phenotype data were obtained for 55 ALS patients and 15 controls from AnswerALS (Baxi et al., 2022). Gene expression was normalized for gene length and then sequencing depth to produce transcripts per kilobase million (TPM). Age of onset and disease status were available for all individuals and these parameters were used to check for the correlation between the expression of top-ranked RefMap ALS genes and age at disease onset.
An RNA-seq based differential expression analysis was performed for CAV1 and CAV2 genes on samples across three datasets. A detailed protocol of library preparation is described by Tam et al. (2019) for TargetALS samples, Prudencio et al. (2015) for Mayo Clinic samples, Prudencio et al. (2015) and Jones et al. (2021) for the KCL MRC Brain Bank samples. Figure 1 illustrates the stages performed in the RNA-seq analysis.
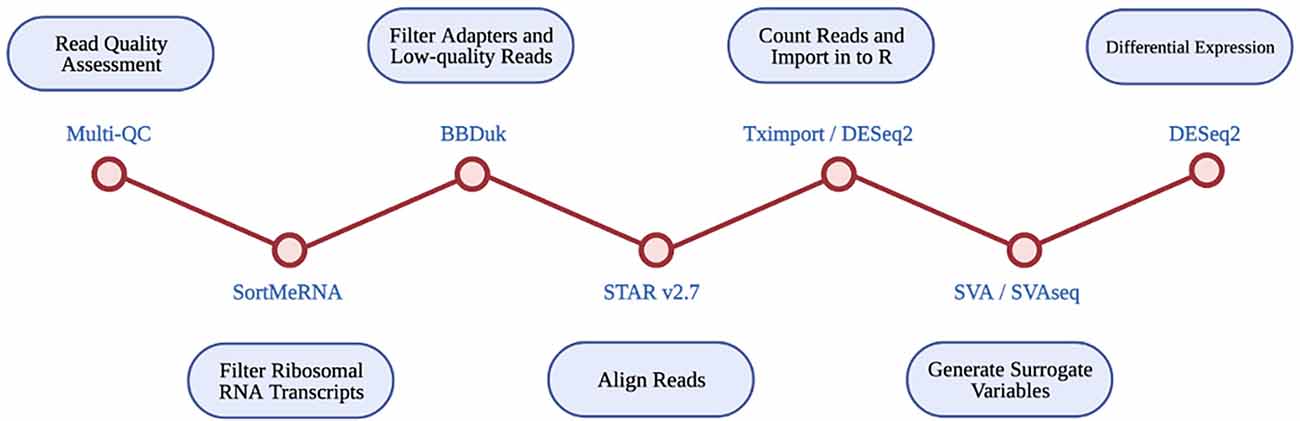
Figure 1. Diagrammatic representation of RNA-seq differential expression pipeline. Each RNA-seq step is shown in the blue circles, with the tool used at each step given beside each red circle.
Multi-Q23 was used for all datasets to assess read quality pre- and post- alignment. The removal of ribosomal RNA transcripts was achieved by filtering with SortMeRNA, using rRNA databases. BBDuk was used to filter adapters and low-quality reads. RNA-seq reads were aligned with STAR v2.7 using the GRCh37.89 reference genome.
Read counts were imported into R using Tximport and DESeq2. Only transcripts with at least 10 reads were retained for analysis. Available data for disease status, gender, quintiles of age, quintiles of PMI, RIN, and flow-cell were imported into R. SVA and SVAseq were used to generate surrogate variables for each sample, which estimate expression heterogeneity. These were included as covariates in subsequent analyses to control for unaccounted confounding factors such as cell heterogeneity and extraneous variation.
Raw read counts were supplied to DESeq2, which was used to perform a differential expression analysis across ALS cases and controls. Differential expression was estimated using log2 fold-change, a wald test, and FDR p-value correction. Analyses were run using covariates of age, gender, post-mortem delay, RIN, and surrogate variables, where data was available.
The final differential expression results were meta-analysed for each brain tissue type using the Stouffer method (Stouffer et al., 1949). This uses the p-value, sample size, and log2 fold-change from each dataset to produce meta-analysed test statistics, and considers the direction of effect.
Multiple cox proportional hazard survival analyses were run and visualised in R using the survival and survminer packages. These analyses were to assess whether the presence of CAV1/2 enhancer mutations impacts patient survival. Analyses were run with sex at birth and age of onset as covariates, using individuals with no CAV1/2 mutations together with: CAV1 mutations only, CAV2 mutations only, and individuals with mutations in either gene.
C9-related ALS is characterised by different clinical presentations (Al-Chalabi et al., 2016, 2017), earlier age of onset, and faster disease progression compared to non-C9 ALS, suggesting a separate disease mechanism (Iacoangeli et al., 2019). Analyses were therefore run with and without individuals carrying a pathogenic repeat expansion of the C9orf72 gene (Iacoangeli et al., 2019) to assess whether increasing sample homogeneity would reveal a stronger effect of CAV1/2 mutations on survival. Analyses were additionally run excluding samples from patients with other well-known ALS mutations (SOD1, FUS, TARDBP), and matching samples based on nationality. Finally, survival analyses were run when stratifying samples by type of CAV enhancer mutation (CAV1 or CAV2).
A second set of analyses were run to determine whether CAV1/2 status affected age of onset, using sex at birth as a covariate. These were linear regression and cox proportional hazard models, run in R using the survival package. Analyses were run with and without carriers of a pathogenic C9orf72 repeat expansion. They compared samples with no CAV1/2 mutation to: (1) samples with CAV1 enhancer mutations; (2) samples with CAV2 enhancer mutations; and (3) samples with either mutation.
Samples were matched across disease status by age and sex within each dataset, where data was permitted. Cases were comprised of samples from sporadic and familial ALS patients, including C9orf72- and SOD1-associated ALS. Control samples were obtained from individuals with non-neurological or non-ALS disease. An outline of each dataset is provided in Figure 2.
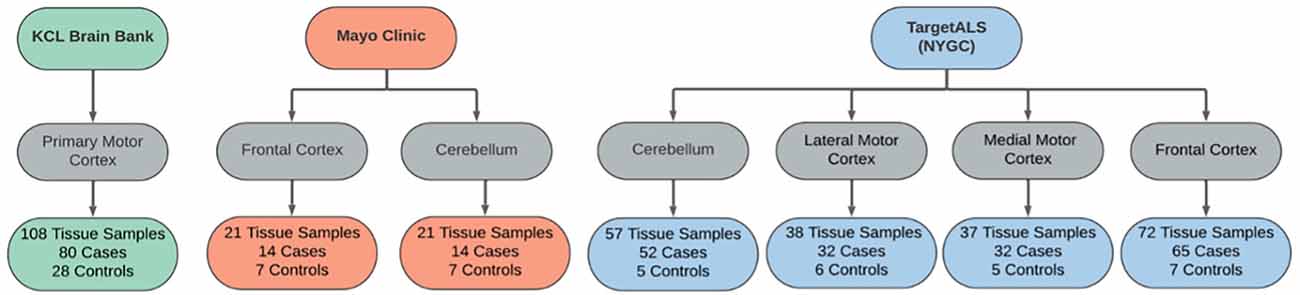
Figure 2. Sample overview across the RNAseq datasets used in the differential expression analyses. Datasets were obtained from the KCL Brain Bank (green), Mayo Clinic (orange), and TargetALS (NYGC; blue).
CAV1/2 enhancer variants of MAF > 0.01 in gnomAD were removed prior to analysis. 5,987 samples passed the quality controls and were used for analysis. Of these, 356 were carriers of the C9orf72 repeat expansion. In total, 44 patients had at least one CAV1/2 enhancer mutation, of which, 34 were carriers of CAV1 mutations, and 10 were carriers of CAV2 mutations. Figure 3 shows sample sizes for the four primary Project MinE survival analyses.
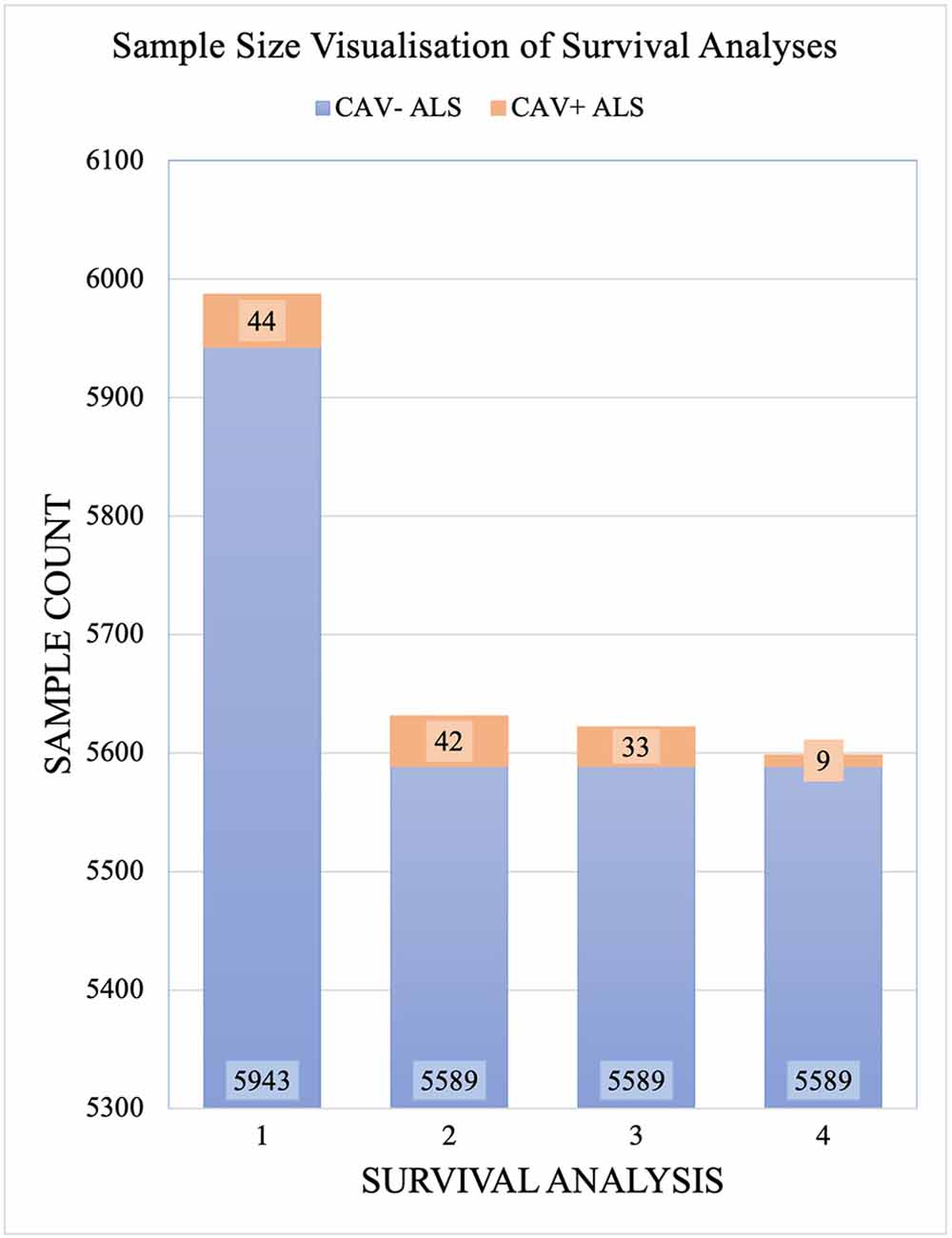
Figure 3. Sample sizes for each Project MinE survival analysis. Samples are divided by those with CAV1/2 enhancer mutations (orange) and without (blue). Analyses are: Full dataset including all rare mutations; excluding C9orf72 samples; and with CAV1 and CAV- samples only with CAV2 and CAV- samples only. CAV- refers to patients who do not carry CAV1/2 enhancer mutations and CAV+ refers to those who do.
Considering converging evidence that CAV1/2 genes are neuroprotective and the previous association between ALS disease status and CAV1/2 enhancer regions, we hypothesised that CAV1 and CAV2 genes would be differentially expressed between ALS patients and controls within brain tissue. Results from the differential expression analysis for CAV1 and CAV2 are outlined in Table 1 and shown in violin plots in Figure 4. CAV1 showed statistically significant differential gene expression within the KCL primary motor cortex (Log2FC = 0.396, p = 0.04) and the NYGC cerebellum (Log2FC = 0.751, p = 0.02). CAV2 was differentially expressed in the primary motor cortex within the KCL BrainBank sample (Log2FC = 0.183, p = 0.01), in addition to the cerebellum (Log2FC = 0.669, p = 0.004) and lateral motor cortex (Log2FC = 0.691, p = 0.029) within Target alS (NYGC) samples. Dataset-tissues almost universally showed a positive log2 fold-change (with the exception of the NYGC frontal cortex), suggesting that CAV1/2 is consistently upregulated among ALS cases. This direction of effect is contrary to previous evidence if we conclude that a higher expression level in cases corresponds to gene expression increasing ALS risk. However, this aligns with a compensatory model, in which expression of CAV1/2 genes is increased to mitigate ALS-related pathology.
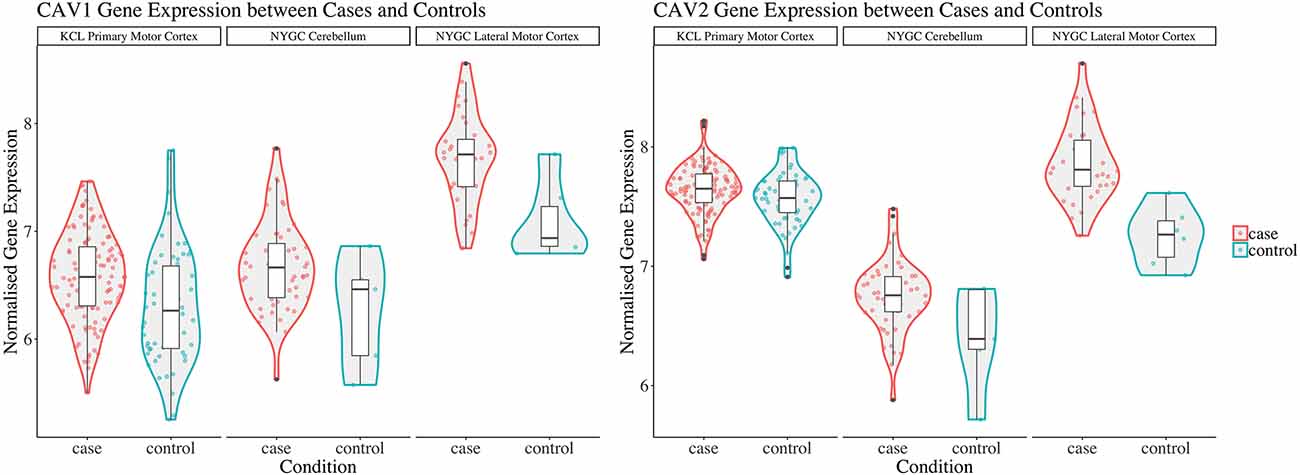
Figure 4. This figure shows violin plots of significant gene expression for CAV1 and CAV2 between cases and controls. The X-axis indicates tissue/dataset combination and case/control status. The Y-axis is normalised gene expression. Coloured dots inside violin plots are jittered gene expressions for each sample. Boxplots inside each violin plot show gene expression for each category. Violin plot colour: Condition (case: red; control: blue). Note that CAV1 differential expression in the lateral motor cortex is significant only to p < 0.1. Violin plots for all analyses are available in Supplementary Figure 1.
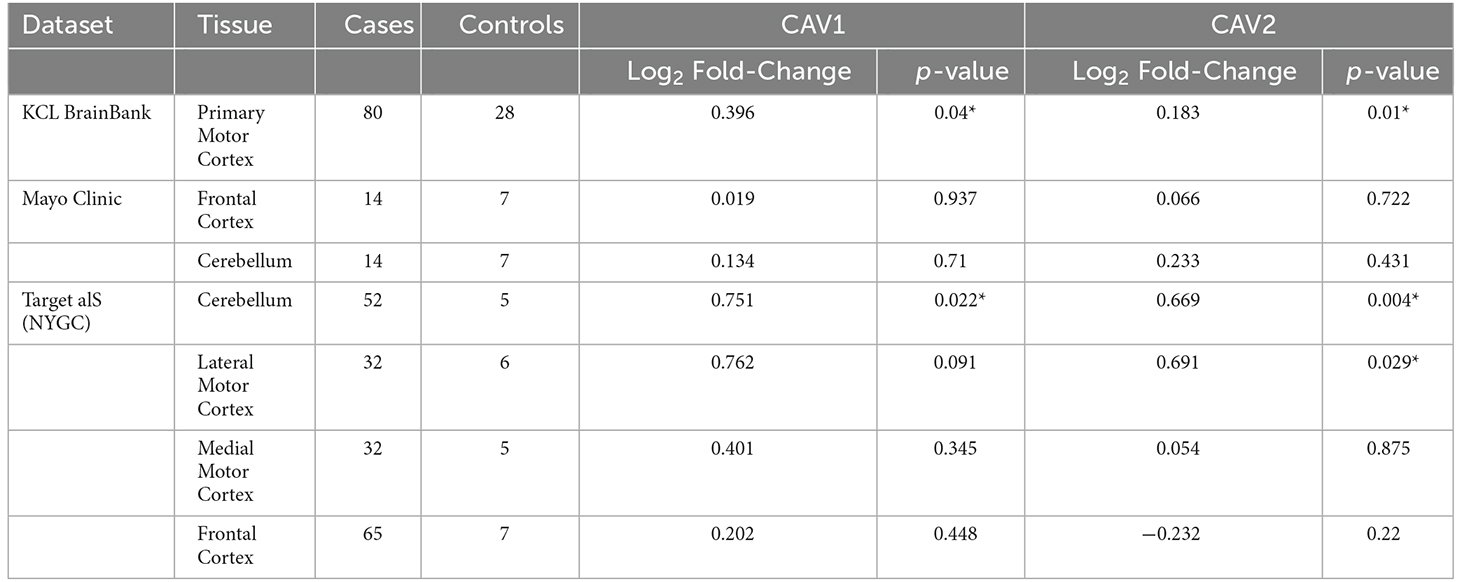
Table 1. The table outlines the differential expression (Log2-fold change) for CAV1 (blue) and CAV2 (green) across brain tissues and datasets, *p < 0.05.
Log2 fold-change for CAV1 and CAV2 were in a consistent direction across all datasets and tissues except for the CAV2 NYGC frontal cortex. For this reason, a Stouffer meta-analysis was run for the motor cortex, frontal cortex, and cerebellum, the results of which are shown in Table 2. Two TargetALS NYGC tissue regions were available within the motor cortex, the lateral and medial motor cortex, of which only the lateral motor cortex reached statistical significance (Log2FC = 0.691, p = 0.029). These datasets were separately meta-analysed with the KCL Brainbank dataset. These analyses were statistically significant for both CAV1 and CAV2 genes, and all showed a large log2 fold-change over 2.
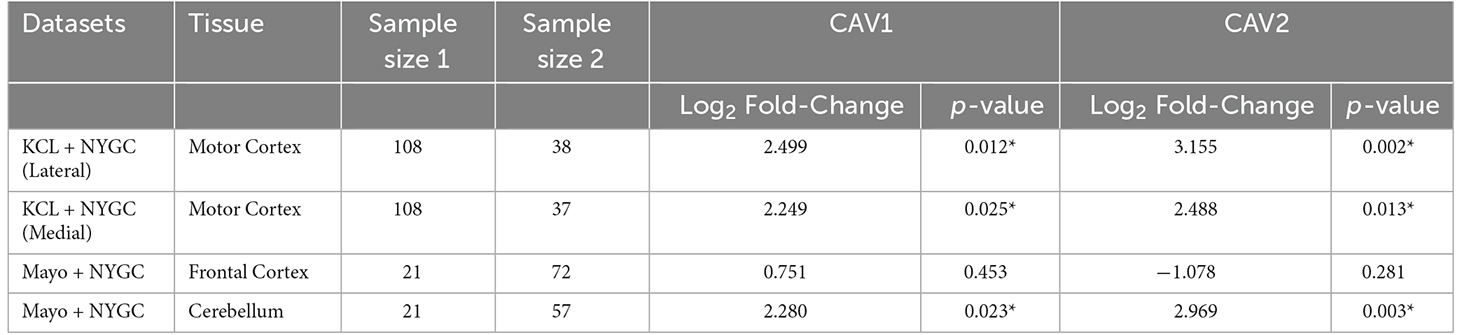
Table 2. Stouffer meta-analysis of differential expression data, *p < 0.05.
Bulk RNA-seq in post-mortem brain tissue has shown that expression of both CAV1 and CAV2 genes is higher in ALS patients compared to controls. Enhanced CAV1 expression has previously been associated with neuroprotection (Sawada et al., 2019) and reduced CAV1 expression has been associated with risk for ALS (Cooper-Knock et al., 2020). Therefore, the observed higher expression of CAV1 and CAV2 might represent a compensatory reaction to neurotoxicity. However, the bulk RNA-seq analysis does not allow us to determine which cell types are responsible for observed changes in CAV1/2 expression. To address this, we analysed gene expression in iPSC-derived MNs from ALS patients (n = 551) and neurologically normal controls (n = 15). Mean expression of both genes was higher in ALS patients compared to controls although this difference was not statistically significant (CAV1: mean ALS = 1.46 TPM, mean control = 1.3 TPM, t = 0.48, Log2FC = 0.1575, p = 0.31. CAV2: mean ALS = 1.67 TPM, mean control = 1.39 TPM, t = 1.43, Log2FC = 0.2647, p = 0.08).
Using RNAseq from iPSC-derived MN, we examined the association between CAV1/2 expression and phenotypic measures. An outline of these results is shown in Table 3. Age of onset was quantified in days; there was no significant correlation between CAV1/2 expression and age of onset (Pearson correlation p > 0.05). Survival was measured in days from the date of onset to death and censored samples were not included because of the lack of longitudinal data; the date of death was available for 27 ALS patients. Cox proportional hazards model was used to determine whether survival was significantly correlated with CAV1/2 expression. The first 10 principal components were used as covariates to control for population structure. Neither CAV1 (p = 0.96) nor CAV2 (p = 0.70) were significantly associated with survival in this cohort.
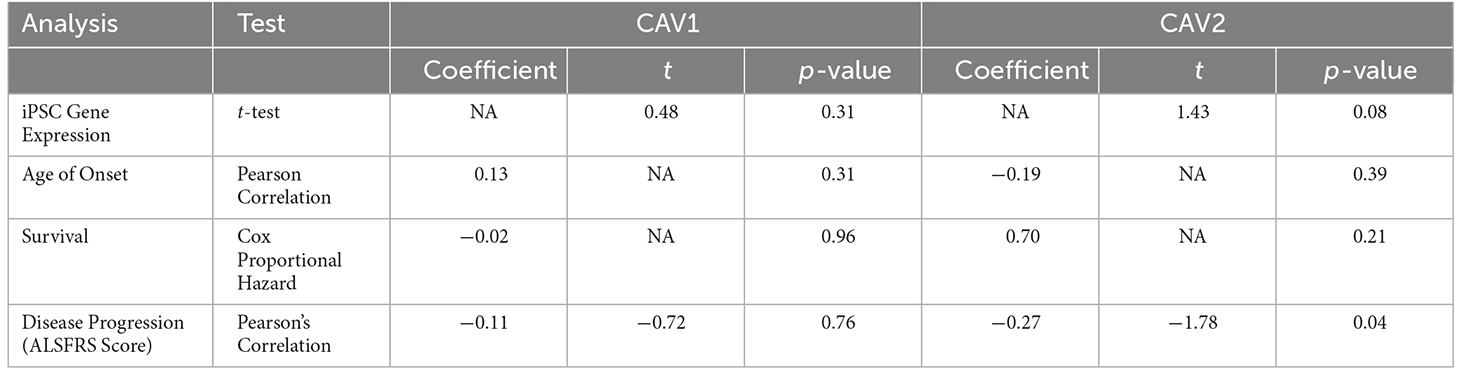
Table 3. Results of AnswerALS RNA-seq expression and phenotypic correlation analyses for CAV1 (blue) and CAV2 (green).
Next, we tested whether CAV1/2 expression was correlated with the rate of change in ALSFRS, which is a measure of the rate of disease progression. The ALSFRS was measured longitudinally between 2 and 10 times (with a median of four measurements). The delta-ALSFRS was calculated using linear regression based upon patient visit time and was available for 43 ALS patients. CAV2 expression but not CAV1 expression was negatively correlated with the rate of change of ALSFRS score (Figure 5); iPSC-derived MN with higher CAV2 expression were derived from patients with a faster rate of decline in the ALSFRS (Pearson correlation p = 0.04, t = −1.78, r = −0.27). In view of our previous data, this could suggest that a compensatory increase in CAV2 expression is highest in patients with more rapid disease progression. It is interesting that CAV1 has been previously associated with neuroprotection but was not significant in this test which may indicate opposing forces of compensatory upregulation with more aggressive disease and a therapeutic effect slowing disease progression.
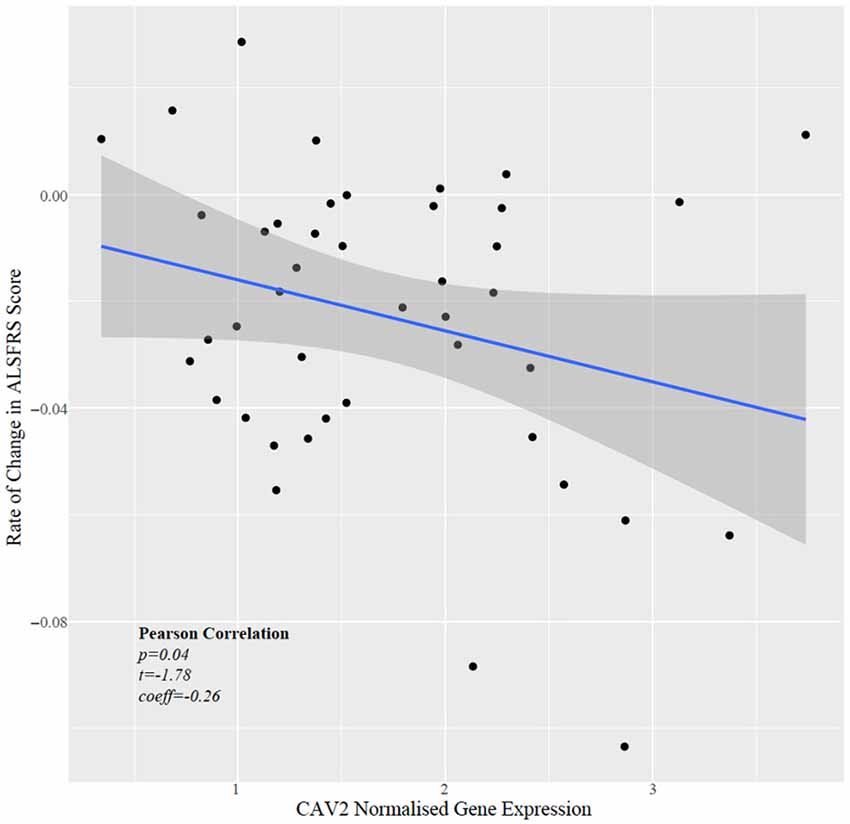
Figure 5. Scatter plot showing normalised CAV2 gene expression against rate of change in the ALSFRS.
Table 4 outlines the results from four of these survival analyses. In the first set of analyses (1–2), we tested the difference in survival of the patients carrying a mutation in the enhancer of either gene (CAV1/2) against non-carriers (Figure 6). The decision was made to combine CAV1 and CAV2 enhancer mutations due to their related biological function, co-expression, overlapping enhancers, and to maximise the statistical power. CAV1/2 mutations were significantly associated with longer survival (HR = 0.694, p = 0.043; HR = 0.674, p = 0.034). This was the case irrespective of whether C9orf72 samples were included or removed.
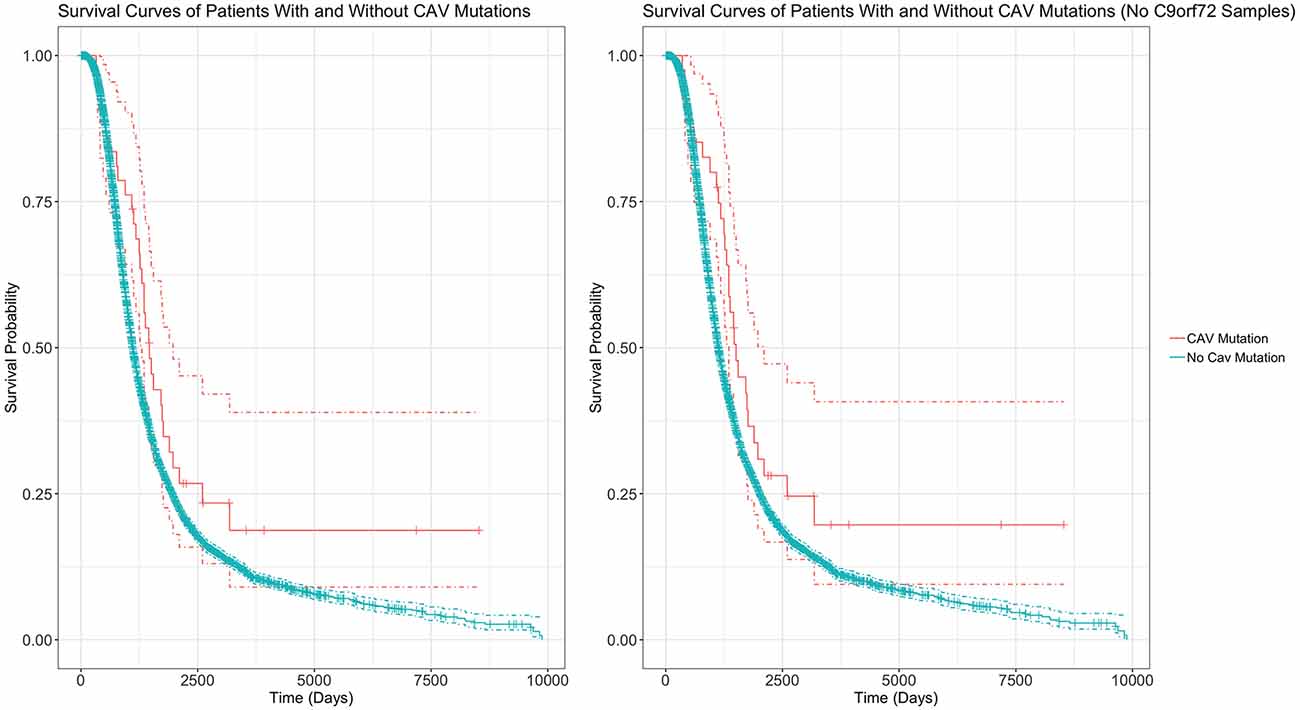
Figure 6. Survival Curves comparing survival of patients with vs without any CAV1/2 mutation. The left graph is based upon data from Analysis 1, inclusive of patients with C9orf72 repeat expansion. The right graph is from Analysis 2, with C9orf72 samples removed. Patients with CAV1/2 mutations have a longer survival time (C9orf72-inclusive analysis: median survival difference of 345 days. See Table 5 for a full descriptive summary). Y-axis is the fraction of surviving sample. X-axis is time in days. Dashed lines indicate 95% confidence intervals. Orthogonal lines indicate death or censoring event. Graphs exclude 22 samples from patients surviving over 10,000 days to improve scaling. Complete graphs are available in Supplementary Figure 2.
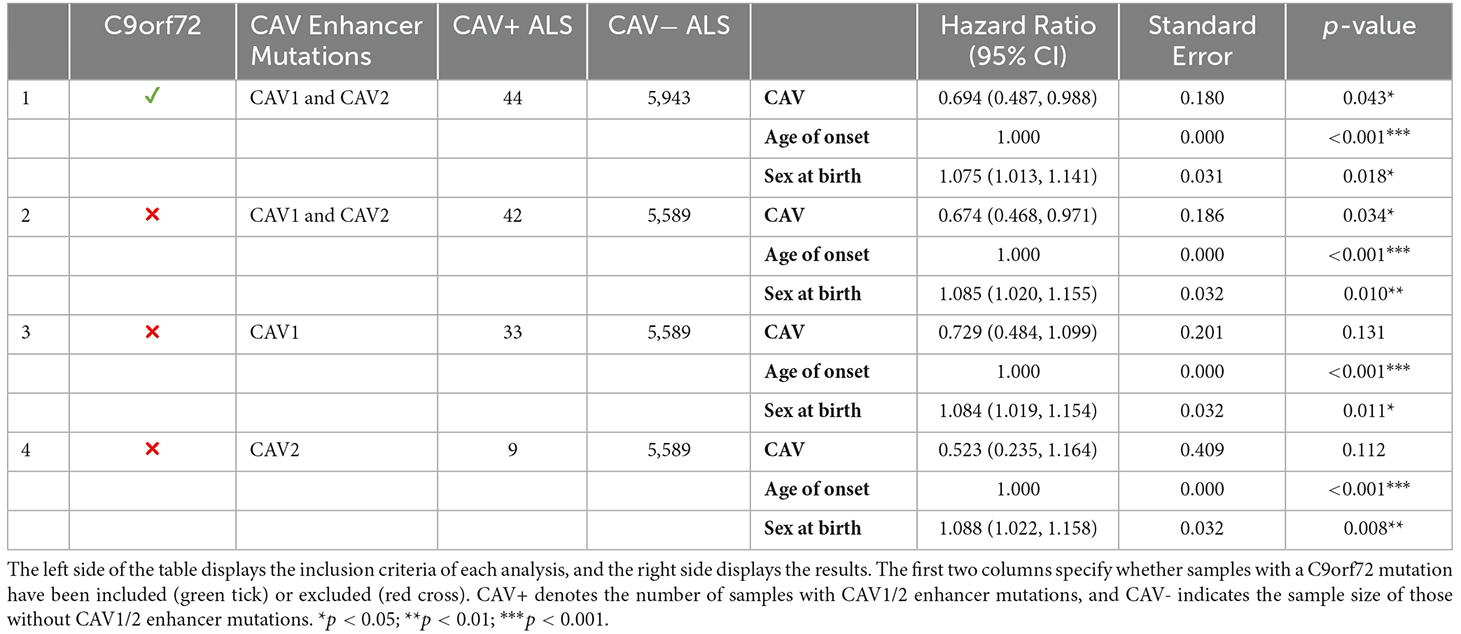
Table 4. Breakdown of results across four survival analyses.
The following analyses were then stratified by the presence of CAV1 or CAV2 enhancer mutations. These analyses excluded C9orf72 samples. Although not significant, the effects on survival of CAV1 and CAV2 enhancer mutations were similar and consistent with the analyses 1–2. This supports our initial choice to aggregate them to increase statistical power based on the hypothesis that mutations in the enhancers of both genes have a similar role in ALS. Descriptive statistics for these analyses are available in Table 5.
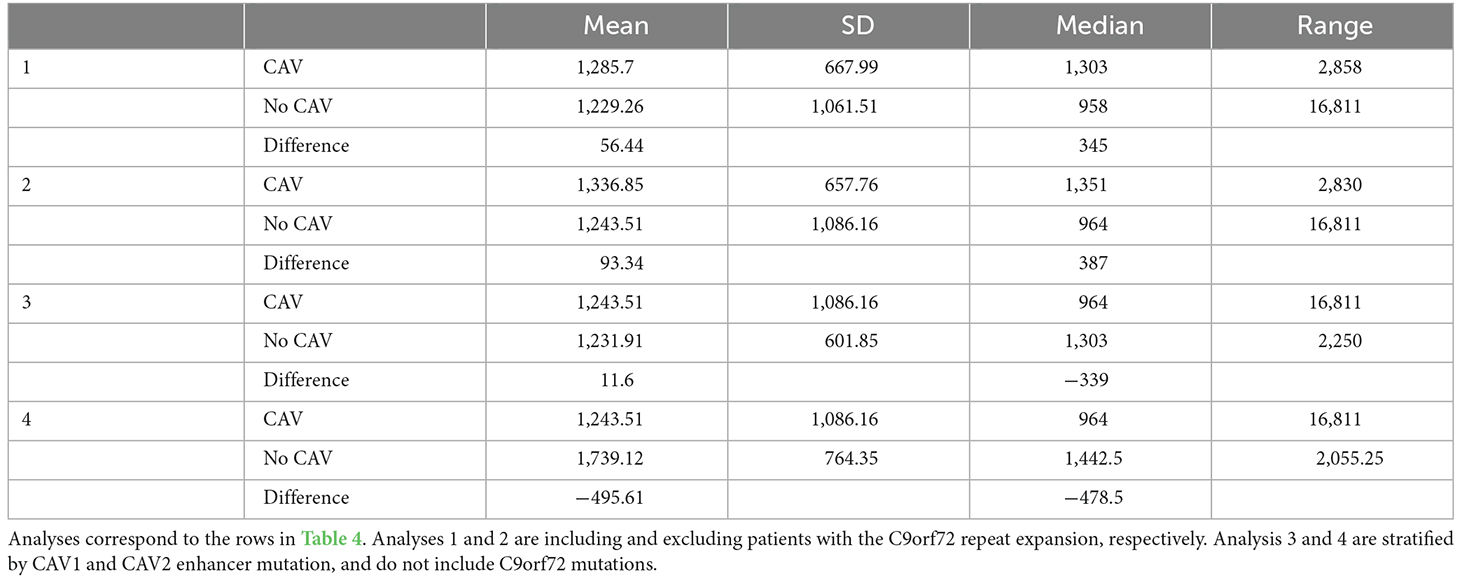
Table 5. This table shows the mean, standard deviation, median, and range of survival in days for individuals with uncensored data.
Similarly to the survival analyses, each age of onset analysis was performed using differing inclusion criteria. Table 6 displays the results for all age of onset analyses and their inclusion criteria. Cox Proportional hazards model was used for each analysis, setting the event status indicator to 1 (the event has occurred) for each sample. In parallel, a linear regression was performed using the same inclusion criteria as analysis 1. No analysis found any effect of CAV1/2 mutation on the age of onset.
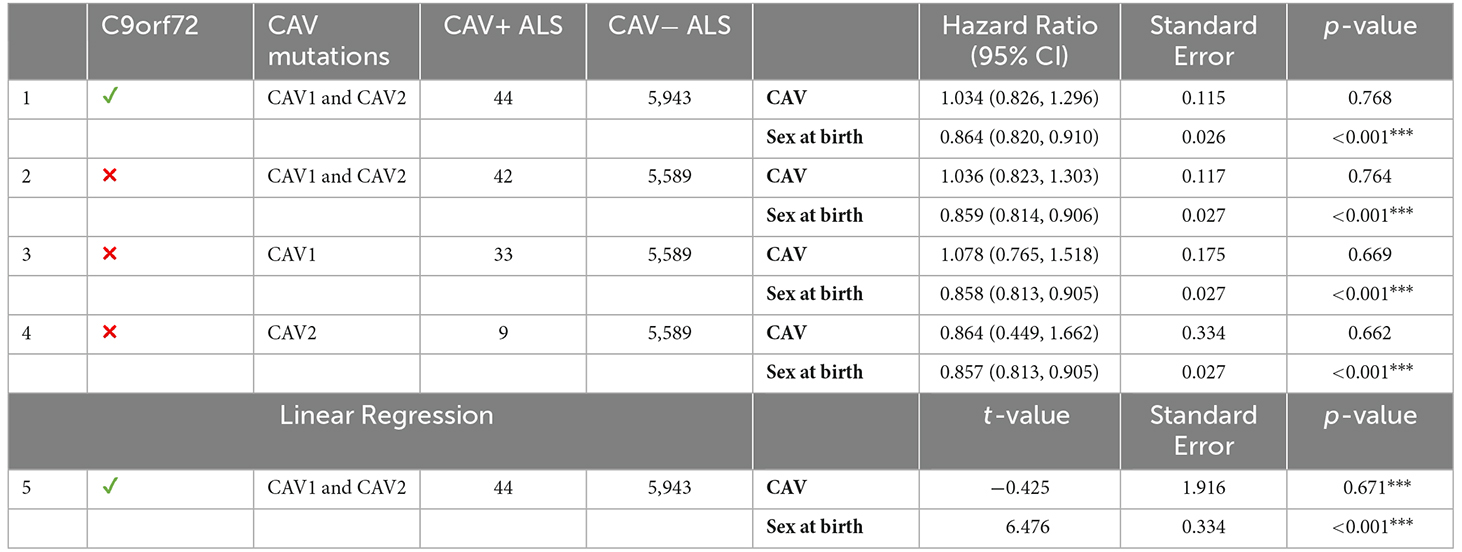
Table 6. The left side of the table describes the inclusion criteria and CAV+/CAV− sample size; the right size shows results from age of onset analyses using Cox proportional hazards and linear regression models; ***p < 0.001.
We report increased expression of CAV1 and CAV2 in ALS cases when compared to controls using bulk RNA sequencing from post-mortem brain tissue samples. Statistically significant differential expression was found in the KCL Brainbank and Target alS (NYGC) samples, but not in Mayo Clinic samples, although the direction of effect was consistent. Non-significant results may be due to a lack of power, as the sample size was substantially smaller in the Mayo Clinic samples than the other datasets. Additionally, meta-analyses revealed significant differences within the cerebellum and motor cortex for both CAV1 and CAV2 expression, but not the frontal cortex. One possible interpretation is that overexpression of CAV1/2 genes increases ALS risk. However, this is inconsistent with evidence that CAV1/2 expression is protective in ALS (Head et al., 2011; Cooper-Knock et al., 2020) and more generally promotes neuronal growth and improves motor function (Egawa et al., 2017, 2018). An alternative interpretation consistent with previous literature is that the gene upregulation is indicative of a compensatory mechanism; CAV1/2 expression is increased as a response to ALS pathology, which affords greater protection.
Survival analyses showed that among ALS patients, carriers of CAV1/2 enhancer mutations had longer survival compared to non-carriers, with a median survival difference of 345 days in the Project MinE dataset. No correlation was demonstrated between gene expression and survival in the AnswerALS iPSC-derived MNs, although this analysis was limited by the small sample size. We observed a negative correlation between CAV2 expression and the rate of change in the ALSFRS in the iPSC-derived MNs. Given the seemingly protective role of CAV1/2, it was expected that mutations in CAV1/2 enhancers, which purportedly decrease CAV1/2 expression, would in turn reduce survival. We consider two possible explanations for observing the opposite outcome. CAV1/2 enhancer mutations exist in non-coding regions and have an unknown impact on gene expression. Cooper-Knock and colleagues (Cooper-Knock et al., 2020) ran an expression analysis using a single CAV1/2 enhancer mutation (chr7:116222625:T > C), finding an association with reduced CAV1/2 expression in patient-derived neuronal cells. However, this is not sufficient evidence to conclude the global effect of CAV1/2 mutations on expression, as enhancer mutations may also increase gene expression (Corradin and Scacheri, 2014; Sur and Taipale, 2016). The effects of other variants on gene expression may account for the increased survival duration that we observed. Further investigation into the of CAV1/2 enhancer mutations on gene expression would be beneficial to build evidence for or against this interpretation.
An alternative hypothesis is that patients with CAV1/2 mutations represent a subset of ALS patients with a less aggressive phenotype. In this framework, CAV1/2 enhancer mutations reduce CAV1/2 expression, leading to dysfunctional neuronal signalling and accelerated neurodegeneration. However, the dysfunction associated with CAV1/2 is on average less severe than non-CAV-related ALS phenotypes, leading to the longer survival time found in our analyses. It is more likely that rare variants occurring within enhancer regions are deleterious, leading to reduced function of the enhancer and therefore reduced expression than to improve function and increase CAV1/2 expression. This prior expectation makes this interpretation more biologically plausible.
Whether or not CAV1/2 enhancer mutations increase or decrease CAV1/2 gene expression, both align with the “compensatory model” of CAV1/2 overexpression in ALS patients. If CAV1/2 are neuroprotective and are upregulated to compensate for ALS pathology, CAV1/2 enhancer mutations which increase expression simply boost this effect, leading to increased survival. If these mutations decrease expression and subsequently increase neurodegeneration, the “increased survival” we observe among patients with CAV1/2 enhancer mutations may be explained by CAV-mediated ALS being on average less severe than non-CAV ALS.
Individuals with CAV1/2 mutations represent a small but relevant proportion of ALS patients (0.7%). Our results add to an increasing body of evidence linking CAV1 and CAV2 genes to ALS, help to elucidate the role of their enhancer mutations and gene expression in ALS, and support the positioning of CAV1/2 genes as potential targets for the development of treatment. However, further research into the functional effect of CAV1/2 mutations is needed to clarify their role in the pathogenesis of ALS.
Publicly available datasets were analyzed in this study. This data can be found here: GSE116622: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE116622 GSE124439: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE124439 GSE67196: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67196 Project MinE: https://www.projectmine.com/research/data-sharing/ AnswerALS: https://dataportal.answerals.org/data-search.
The studies involving human participants were reviewed and approved by the Institute of Psychiatry, Psychology & Neuroscience, King’s College London, and the MRC London Neurodegenerative Diseases Brain Bank, in addition to the following ethical committees: The Netherlands: University Medical Center Utrecht Medical Ethics Committee, Utrecht, Netherlands. UK MNDA Biobank: Trent University Medical Ethics Committee. UK (Sheffield): Yorkshire and the Humber - Sheffield Research Ethics Committee. Turkey: Ethics Committee on Research with Human Participants (INAREK) at Bogazici University, Istanbul, Turkey. Belgium: Ethical Committee of University Hospital Leuven. Ireland: Beaumont Hospital Research & Ethics Committee. Spain (Madrid): Comité de ética de la Investigación del Hospital Carlos III. Spain (Barcelona): Bellvitge University Hospital Ethics Committee, Barcelona, Spain. United States: Committee for the Protection of Human Subjects in Research of the University of Massachusetts Medical School, Worcester, USA. France (Paris): Medical Research Ethics Committee of “Assistance Publique-Hôpitaux de Paris”. France (Tours): The ethics committee of Tours Hospital, France. France (Limoges): Ethics committee of Limoges University Hospital, France. Sweden: Regional Ethical Review Board in Umeå. Israel (Tel-aviv): The Institutional Review Board of Tel Aviv Sourasky Medical Center, Israel. Israel (Jerusalem): Hadassah University Hospital IRB board. Portugal: The Local Research Ethics Committee at the Faculty of Medicine, University of Lisbon, Lisbon, Portugal. Italy: Ethical Committee of Città della Salute Hospital, Torino, Italy. Switzerland: Kantonale Ethikkomission des Kantons St. Gallen, Switzerland. Australia: Sydney South West Area Health Service Human Research Ethics Committee; HREC at the different sites: University of Sydney, Western Sydney Local Health District, Royal Brisbane and Women Hospital Metro North, South Metropolitan Health Service, Macquarie University, QIMR Berghofer Medical Research Institute, University of New South Wales and the University of Melbourne. The patients/participants provided their written informed consent to participate in this study.
BA, JC-K, AJ, and AI contributed to concept, design of the study, running the analyses, and drafted the manuscript. All authors contributed to the article and approved the submitted version.
We would like to acknowledge funding from the following funders: UK Research and Innovation; Medical Research Council; South London and Maudsley NHS Foundation Trust; MND Scotland; Motor Neurone Disease Association; National Institute for Health Research; Spastic Paraplegia Foundation; Rosetrees Trust; Darby Rimmer MND Foundation. Funding for open access charge: UKRI. BA acknowledges funding from an NIHR pre-doctoral fellowship (NIHR301067). AI is funded by the Motor Neurone Disease Association and South London and Maudsley NHS Foundation Trust. JC-K is supported by a Wellcome Trust fellowship (216596/Z/19/Z). AAK is funded by ALS Association Milton Safenowitz Research Fellowship (grant number 22-PDF-609. DOI: 10.52546/pc.gr.150909.), The Motor Neurone Disease Association (MNDA) Fellowship (AAK/Oct21/975-799), The Darby Rimmer Foundation, and The NIHR Maudsley Biomedical Research Centre. This is an EU Joint Programme-Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organizations under the aegis of JPND: http://www.neurodegenerationresearch.eu/ [United Kingdom, Medical Research Council MR/L501529/1 to AA-C, principal investigator (PI) and MR/R024804/1 to AA-C, PI; Economic and Social Research Council ES/L008238/1 to AA-C (co-PI)] and through the Motor Neurone Disease Association. This study represents independent research partly funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The work leading up to this publication was funded by the European Community’s Horizon 2020 Programme (H2020-PHC-2014-two-stage; grant 633413). We acknowledge use of the research computing facility at King’s College London, Rosalind (https://rosalind.kcl.ac.uk), which is delivered in partnership with the National Institute for Health Research (NIHR) Biomedical Research Centres at South London and Maudsley and Guy’s and St. Thomas’ NHS Foundation Trusts and part-funded by capital equipment grants from the Maudsley Charity (award 980) and Guy’s and St Thomas’ Charity (TR130505). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, King’s College London, or the Department of Health and Social Care.
JV reports to have sponsored research agreements with Biogen and Astra Zeneca.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2023.1112405/full#supplementary-material.
Supplementary Figure 1 Violin plots of gene expression for CAV1 and CAV2 between cases and controls. The X-axis indicates tissue/dataset combination and case/control status. The Y-axis is normalised gene expression. Coloured dots inside violin plots are jittered gene expressions for each sample. Boxplots inside each violin plot show gene expression for each category. Violin plot colour: Condition (case: red; control: blue).
Supplementary Figure 2 Uncropped survival curves comparing survival of patients with vs without any CAV1/2 mutation. The top graph is based upon data from Analysis 1, inclusive of patients with C9orf72 repeat expansion. The bottom graph is from Analysis 2, with C9orf72 samples removed. Patients with CAV1/2 mutations have a longer survival time (C9orf72-inclusive analysis: median survival difference of 345 days. See Table 5 for a full descriptive summary). Y-axis is the fraction of surviving sample. X-axis is time in days. Dashed lines indicate 95% confidence intervals. Orthogonal lines indicate death or censoring event.
Supplementary Table 1 List of CAV1/2 enhancer variants and their frequency within the Project MinE dataset.
Al-Chalabi, A., Fang, F., Hanby, M. F., Leigh, P. N., Shaw, C. E., Ye, W., et al. (2010). An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 81, 1324–1326. doi: 10.1136/jnnp.2010.207464
Al-Chalabi, A., and Hardiman, O. (2013). The epidemiology of ALS: a conspiracy of genes, environment and time. Nat. Rev. Neurol. 9, 617–628. doi: 10.1038/nrneurol.2013.203
Al-Chalabi, A., Hardiman, O., Kiernan, M. C., Chiò, A., Rix-Brooks, B., and van den Berg, L. H. (2016). Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. 15, 1182–1194. doi: 10.1016/S1474-4422(16)30199-5
Al-Chalabi, A., Van Den Berg, L. H., and Veldink, J. (2017). Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat. Rev. Neurol. 13, 96–104. doi: 10.1038/nrneurol.2016.182
Baxi, E. G., Thompson, T., Li, J., Kaye, J. A., Lim, R. G., Wu, J., et al. (2022). Answer ALS, a large-scale resource for sporadic and familial ALS combining clinical and multi-omics data from induced pluripotent cell lines. Nat. Neurosci. 25, 226–237. doi: 10.1038/s41593-021-01006-0
Braems, E., Swinnen, B., and Van Den Bosch, L. (2020). C9orf72 loss-of-function: a trivial, stand-alone or additive mechanism in C9 ALS/FTD? Acta Neuropathol. 140, 625–643. doi: 10.1007/s00401-020-02214-x
Brown, R. H., Jr., and Al-Chalabi, A. (2015). Endogenous retroviruses in ALS: a reawakening? Sci. Transl. Med. 7:307fs40. doi: 10.1126/scitranslmed.aad3533
Conlon, E. G., Fagegaltier, D., Agius, P., Davis-Porada, J., Gregory, J., Hubbard, I., et al. (2018). Unexpected similarities between C9ORF72 and sporadic forms of ALS/FTD suggest a common disease mechanism. eLife 7:e37754. doi: 10.7554/eLife.37754
Cooper-Knock, J., Zhang, S., Kenna, K. P., Moll, T., Franklin, J. P., Allen, S., et al. (2020). Rare variant burden analysis within enhancers identifies CAV1 as an ALS risk gene. Cell Rep. 33:108456. doi: 10.1016/j.celrep.2020.108456
Corradin, O., and Scacheri, P. C. (2014). Enhancer variants: evaluating functions in common disease. Genome Med. 6:85. doi: 10.1186/s13073-014-0085-3
Egawa, J., Schilling, J. M., Cui, W., Posadas, E., Sawada, A., Alas, B., et al. (2017). Neuron-specific caveolin-1 overexpression improves motor function and preserves memory in mice subjected to brain trauma. FASEB J. 31, 3403–3411. doi: 10.1096/fj.201601288RRR
Egawa, J., Zemljic-Harpf, A., Mandyam, C. D., Niesman, I. R., Lysenko, L. V., Kleschevnikov, A. M., et al. (2018). Neuron-targeted caveolin-1 promotes ultrastructural and functional hippocampal synaptic plasticity. Cereb. Cortex 28, 3255–3266. doi: 10.1093/cercor/bhx196
Head, B. P., Hu, Y., Finley, J. C., Saldana, M. D., Bonds, J. A., Miyanohara, A., et al. (2011). Neuron-targeted caveolin-1 protein enhances signaling and promotes arborization of primary neurons. J. Biol. Chem. 286, 33310–33321. doi: 10.1074/jbc.M111.255976
Head, B. P., Peart, J. N., Panneerselvam, M., Yokoyama, T., Pearn, M. L., Niesman, I. R., et al. (2010). Loss of caveolin-1 accelerates neurodegeneration and aging. PLoS One 5:e15697. doi: 10.1371/journal.pone.0015697
Iacoangeli, A., Al Khleifat, A., Jones, A. R., Sproviero, W., Shatunov, A., Opie-Martin, S., et al. (2019). C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol. Commun. 7:115. doi: 10.1186/s40478-019-0724-4
Iacoangeli, A., Fogh, I., Selvackadunco, S., Topp, S. D., Shatunov, A., van Rheenen, W., et al. (2021). SCFD1 expression quantitative trait loci in amyotrophic lateral sclerosis are differentially expressed. Brain Commun. 3:fcab236. doi: 10.1093/braincomms/fcab236
Ichinomiya, T., Wang, S., Patel, H., Tadokoro, T., Marsala, M., and Head, B. (2021). Subpial gene delivery of synapsin-promoted Caveolin-1 prolongs Survival in hSODG93A mice Model of ALS. FASEB J. 35. doi: 10.1096/fasebj.2021.35.S1.02086
Igarashi, M., Honda, A., Kawasaki, A., and Nozumi, M. (2020). Neuronal signaling involved in neuronal polarization and growth: lipid rafts and phosphorylation. Front. Mol. Neurosci. 13:150. doi: 10.3389/fnmol.2020.00150
Jones, A., Iacoangeli, A., Adey, B., Bowles, H., Shatunov, A., Troakes, C., et al. (2021). A HML6 endogenous retrovirus on chromosome 3 is upregulated in amyotrophic lateral sclerosis motor cortex. Sci. Rep. 11:14283. doi: 10.1038/s41598-021-93742-3
Lomen-Hoerth, C., Murphy, J., Langmore, S., Kramer, J. H., Olney, R. K., and Miller, B. (2003). Are amyotrophic lateral sclerosis patients cognitively normal? Neurology 60, 1094–1097. doi: 10.1212/01.wnl.0000055861.95202.8d
Mandyam, C. D., Schilling, J. M., Cui, W., Egawa, J., Niesman, I. R., Kellerhals, S. E., et al. (2017). Neuron-targeted caveolin-1 improves molecular signaling, plasticity and behavior dependent on the hippocampus in adult and aged mice. Biol. Psychiatry 81, 101–110. doi: 10.1016/j.biopsych.2015.09.020
Phukan, J., Pender, N. P., and Hardiman, O. (2007). Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 6, 994–1003. doi: 10.1016/S1474-4422(07)70265-X
Project MinE ALS Sequencing Consortium (2018). Project MinE: study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur. J. Hum. Genet. 26, 1537–1546. doi: 10.1038/s41431-018-0177-4
Prudencio, M., Belzil, V. V., Batra, R., Ross, C. A., Gendron, T. F., Pregent, L. J., et al. (2015). Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 18, 1175–1182. doi: 10.1038/nn.4065
Ryan, M., Heverin, M., McLaughlin, R. L., and Hardiman, O. (2019). Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol. 76, 1367–1374. doi: 10.1001/jamaneurol.2019.2044
Sawada, A., Wang, S., Jian, M., Leem, J., Wackerbarth, J., Egawa, J., et al. (2019). Neuron—targeted caveolin—1 improves neuromuscular function and extends survival in SOD1G93A mice. FASEB J. 33, 7545–7554. doi: 10.1096/fj.201802652RR
Smith, L., Cupid, B. C., Dickie, B. G. M., Al-Chalabi, A., Morrison, K. E., Shaw, C. E., et al. (2015). Establishing the UK DNA Bank for motor neuron disease (MND). BMC Genet. 16:84. doi: 10.1186/s12863-015-0236-6
Stouffer, S. A., Suchman, E. A., DeVinney, L. C., Star, S. A., Williams, R. M., Jr. (1949). The American Soldier: Adjustment During Army Life. (Studies In Social Psychology In World War II) Vol. 1. Princeton, NJ: Princeton University Press.
Strong, M. J., Abrahams, S., Goldstein, L. H., Woolley, S., Mclaughlin, P., Snowden, J., et al. (2017). Amyotrophic lateral sclerosis-frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph. Lateral Scler. Frontotemporal Degener. 18, 153–174. doi: 10.1080/21678421.2016.1267768
Sur, I., and Taipale, J. (2016). The role of enhancers in cancer. Nat. Rev. Cancer 16, 483–493. doi: 10.1038/nrc.2016.62
Tam, O. H., Rozhkov, N. V., Shaw, R., Kim, D., Hubbard, I., Fennessey, S., et al. (2019). Postmortem cortex samples identify distinct molecular subtypes of ALS: retrotransposon activation, oxidative stress and activated glia. Cell Rep. 29, 1164–1177.e5. doi: 10.1016/j.celrep.2019.09.066
Tang, W., Li, Y., Li, Y., and Wang, Q. (2021). Caveolin-1, a novel player in cognitive decline. Neurosci. Biobehav. Rev. 129, 95–106. doi: 10.1016/j.neubiorev.2021.06.044
Theunissen, F., Flynn, L. L., Anderton, R. S., Mastaglia, F., Pytte, J., Jiang, L., et al. (2020). Structural variants may be a source of missing heritability in sALS. Front. Neurosci. 14:47. doi: 10.3389/fnins.2020.00047
Trabjerg, B. B., Garton, F. C., van Rheenen, W., Fang, F., Henderson, R. D., Mortensen, P. B., et al. (2020). ALS in danish registries: heritability and links to psychiatric and cardiovascular disorders. Neurol. Genet. 6:e398. doi: 10.1212/NXG.0000000000000398
Turner, M. R., Al-Chalabi, A., Chio, A., Hardiman, O., Kiernan, M. C., Rohrer, J. D., et al. (2017). Genetic screening in sporadic ALS and FTD. J. Neurol. Neurosurg. Psychiatry 88, 1042–1044. doi: 10.1136/jnnp-2017-315995
Van Rheenen, W., Shatunov, A., Dekker, A. M., McLaughlin, R. L., Diekstra, F. P., Pulit, S. L., et al. (2016). Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048. doi: 10.1038/ng.3622
Van Rheenen, W., Van Der Spek, R. A. A., Bakker, M. K., Van Vugt, J. J. F. A., Hop, P. J., Zwamborn, R. A. J., et al. (2021). Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 53, 1636–1648. doi: 10.1038/s41588-021-00973-1
Young, A. I. (2019). Solving the missing heritability problem. PLoS Genet. 15:e1008222. doi: 10.1371/journal.pgen.1008222
Yue, K.-Y., Zhang, P.-R., Zheng, M.-H., Cao, X.-L., Cao, Y., Zhang, Y.-Z., et al. (2019). Neurons can upregulate Cav-1 to increase intake of endothelial cells-derived extracellular vesicles that attenuate apoptosis via miR-1290. Cell Death Dis. 10:869. doi: 10.1038/s41419-019-2100-5
Zhang, S., Cooper-Knock, J., Weimer, A. K., Shi, M., Moll, T., Marshall, J. N. G., et al. (2022). Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis. Neuron 110, 992–1008.e11. doi: 10.1016/j.neuron.2021.12.019
Keywords: ALS (Amyotrophic lateral sclerosis), neurodegeneration, differential expression analysis (DEA), survival analysis, caveolin, Cav, CAV1 and CAV2, enhancer variant
Citation: Adey BN, Cooper-Knock J, Al Khleifat A, Fogh I, van Damme P, Corcia P, Couratier P, Hardiman O, McLaughlin R, Gotkine M, Drory V, Silani V, Ticozzi N, Veldink JH, van den Berg LH, de Carvalho M, Pinto S, Mora Pardina JS, Povedano Panades M, Andersen PM, Weber M, Başak NA, Shaw CE, Shaw PJ, Morrison KE, Landers JE, Glass JD, Vourc’h P, Dobson RJB, Breen G, Al-Chalabi A, Jones AR and Iacoangeli A (2023) Large-scale analyses of CAV1 and CAV2 suggest their expression is higher in post-mortem ALS brain tissue and affects survival. Front. Cell. Neurosci. 17:1112405. doi: 10.3389/fncel.2023.1112405
Received: 30 November 2022; Accepted: 06 February 2023;
Published: 02 March 2023.
Edited by:
Tomomi Ichinose, Wayne State University, United StatesReviewed by:
Sokol V. Todi, Wayne State University, United StatesCopyright © 2023 Adey, Cooper-Knock, Al Khleifat, Fogh, van Damme, Corcia, Couratier, Hardiman, McLaughlin, Gotkine, Drory, Silani, Ticozzi, Veldink, van den Berg, de Carvalho, Pinto, Mora Pardina, Povedano Panades, Andersen, Weber, Başak, Shaw, Shaw, Morrison, Landers, Glass, Vourc’h, Dobson, Breen, Al-Chalabi, Jones and Iacoangeli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alfredo Iacoangeli, YWxmcmVkby5pYWNvYW5nZWxpQGtjbC5hYy51aw==
† These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.