
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 25 July 2022
Sec. Cellular Neuropathology
Volume 16 - 2022 | https://doi.org/10.3389/fncel.2022.953534
This article is part of the Research Topic Modulating Microglia to Enhance Neuroplasticity for Restoring Brain Function After Stroke View all 8 articles
 Congqin Li1†
Congqin Li1† Yong Wang2†
Yong Wang2† Ying Xing1
Ying Xing1 Jing Han3
Jing Han3 Yuqian Zhang1
Yuqian Zhang1 Anjing Zhang1
Anjing Zhang1 Jian Hu1
Jian Hu1 Yan Hua1
Yan Hua1 Yulong Bai1,4,5*
Yulong Bai1,4,5*Microglia are considered the main phagocytic cells in the central nervous system, remodeling neural circuits by pruning synapses during development. Microglial phagocytosis is also a crucial process in maintaining adult brain homeostasis and clearing potential toxic factors, which are recognized to be associated with neurodegenerative and neuroinflammatory disorders. For example, microglia can engulf amyloid-β plaques, myelin debris, apoptotic cells, and extracellular harmful substances by expressing a variety of specific receptors on the cell surface or by reprogramming intracellular glucose and lipid metabolism processes. Furthermore, physical exercise has been implicated to be one of the non-pharmaceutical treatments for various nervous system diseases, which is closely related to neuroplasticity and microglia functions including proliferation, activation, and phagocytosis. This review focuses on the central regulatory mechanisms related to microglia phagocytosis and the potential role of exercise training in this process.
Microglia are the innate immune cells of the central nervous system (CNS) and have a long life span, accounting for 5–20% of glial cells in the adult brain (Mecca et al., 2018). They are malleable in response to different signals (Wendeln et al., 2018) and can undergo extensive morphological, phenotypic, and functional reprogramming to adapt to the changing needs of the developing brain. Microglia play a variety of roles in the brain such as immune monitoring, secretion of cytokines and neurotrophic factors, regulation of inflammation, phagocytosis of cell debris, synaptic connection and pruning, and neural circuit remodeling under physiological and pathological conditions (Kettenmann et al., 2011; Hayashi and Nakanishi, 2013). In addition, microglia are considered crucial regulators of CNS development and homeostasis through the crosstalk with other glia and neurons (Colonna and Butovsky, 2017; Hagemeyer et al., 2017).
In a physiological state, microglia show a highly branched state with a small cell body and long branches, which is called “resting microglia.” They continuously extend and retract through the parenchyma (Nimmerjahn et al., 2005), dynamically surveying the brain parenchyma. In addition, the assembly of neuronal circuits requires a delicate balance between synaptic formation and elimination. Microglia play a defined role in this process by eliminating excess synapses (Ferro et al., 2018). For instance, during brain development, microglia contribute to developmental synaptic remodeling via engulfing synapses from less active neurons (Wu et al., 2015), and thereby, promoting formation of precise neural circuitry (Li et al., 2020). In parallel, microglia dynamically investigate synapses in the adult brain in an activity-dependent manner and play a vital role in neuronal homeostasis and cognitive repair (Bartels et al., 2020). Dysphagocytosis of microglia may lead to synaptic over-pruning or inadequate pruning, which may be a direct culprit in neurodevelopmental and neuropsychiatric disorders such as autism and schizophrenia (Edmonson et al., 2016; Druart and Le Magueresse, 2019). Furthermore, microglia can also normalize dendritic spine increment in an experience-dependent manner (Tuan et al., 2021).
In pathological conditions such as infection, trauma, or cerebral ischemia, microglia rapidly transform from the resting state to the activated state, presenting a typical “amoeboid,” which is characterized by an enlarged cell body, shortened and thickened branches (Wolf et al., 2017), followed by migrating to the inflammatory site and secreting cytokines, chemokines, and neurotoxins, such as reactive oxygen species (ROS), thereby survey surrounding axons and invading pathogens.
Microglia dynamically switch between two polarized states, namely the M1 phenotype (pro-inflammatory) and the M2 phenotype (anti-inflammatory) and are involved in different CNS diseases such as cerebral ischemia, traumatic brain injury, and neurodegenerative diseases such as Alzheimer's disease (AD) and Parkinson's disease (Colonna and Butovsky, 2017; Li and Barres, 2018; Prinz et al., 2019). Microglia can recognize pathogens and other external stimuli on account of a rich array of receptors they express, including toll-like receptors (TLRs) (Li Y. et al., 2021), Fc receptors (Fuller et al., 2014), Ig-superfamily receptors such as triggering receptors expressed on myeloid cells 2 (TREM2) (Zhao et al., 2018; Andreone et al., 2020), scavenger receptors (Wilkinson and El Khoury, 2012), complement receptors (Werneburg et al., 2020), and fractalkine receptors (Gunner et al., 2019).
Compared to brain surgery and medication, exercise therapy is largely non-invasive and free from side effects. A large number of investigations have reported that exercise can induce structural and functional plasticity of the injured CNS, such as promoting the repair of injured neurons and the reconstruction of compensatory neural circuits (Mahalakshmi et al., 2020). This is especially one of the widely used neurorehabilitation methods after stroke (Billinger et al., 2014). Moreover, exercise can regulate brain function by acting on functional changes in microglia. Multiple animal studies have identified the regulation of exercise on the quantity, morphology, phenotype cytokine, and neurotrophic factor expression of microglia in several neurological disorders (Lu et al., 2017; Pignataro et al., 2021; Zhang et al., 2022). Physical exercise also mediates the crosstalk between microglia and astrocytes/neurons to enhance neuroplasticity. For example, the CX3CL1-CX3CL1R axis and the CD200-CD200R axis were more responsive to the same stimulus in the exercised animals than in the sedentary animals (Sung et al., 2012; Fleshner et al., 2014). The molecular mechanisms involved in the crosstalk between astrocytes/neurons and microglia have been elaborated on in recent reviews (Li F. et al., 2021). Mee-Inta et al. (2019) also provided a detailed review of the effect of exercise on microglia activation and its possible regulatory mechanisms. Moreover, Consorti et al. (2021) have reviewed the peripheral mechanisms that drive physical activity to influence the brain, in particular, the physiological processes that transform sensory input in muscles and joints into the activation of brain molecular pathways associated with neuroplasticity.
However, a few reports further summarize microglia functions, such as phagocytosis and environmental surveillance, and the effect of physical exercise on them. As an example, recent studies have shown that exercise reverses behavioral abnormalities in mouse models of autism spectrum disorder by stimulating microglia to engulf excess synapses (Andoh et al., 2019). Treadmill exercise preconditioning for 6 weeks promoted recovery of intracerebral hemorrhage mice with neurological deficits, reduced lesion size, and increased microglial phagocytosis (Kinoshita et al., 2021). This study discusses the possible mechanisms of microglia phagocytosis regulated by exercise. How signals generated by muscle movement are transmitted from the periphery to the center is not explained here, even though it is a crucial and complex process.
Here, we summarize recent knowledge on factors regulating microglial phagocytosis, including TREM2, lipid metabolism, metabolic reprogramming, complement, chemokine receptor, noradrenaline (NA), interleukin (IL)-33/ST2, and TAM system (Table 1). Pharmacological or exercise interventions through some of these factors may contribute to preventing neurodevelopmental dysfunction mediated by microglial phagocytosis of excess living neurons and synapses or neurodegeneration and neuropsychiatric disorders induced by microglia phagocytic dysfunction. Physical exercise has been shown to reverse microglial phagocytosis by modulating several factors. Through this review, we aim to make an overall overview of the molecular mechanism controlling microglia phagocytosis and provide new targets and ideas for physical exercise to promote neuroplasticity from the perspective of microglial phagocytosis.
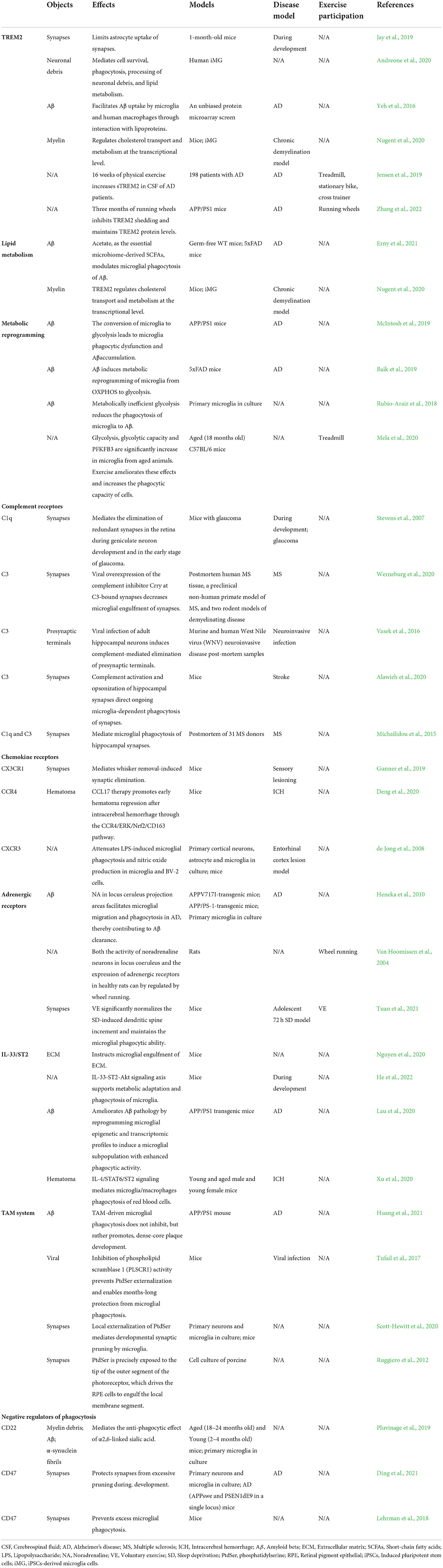
Table 1. Regulation mechanism and exercise participation of microglia phagocytosis in different disease models.
TREM2, an immunoglobulin superfamily receptor, was initially identified as a gene closely associated with the risk of AD in the Genome-wide association (GWAS) study (Jansen et al., 2019). TREM2 is expressed predominantly by microglia in the CNS (Colonna and Wang, 2016). TREM2 enhances microglia phagocytosis of Aβ plaques and ameliorates AD symptoms. During development, microglia rely on TREM2 to restrict the uptake of synapses by astrocytes while there are additional mechanisms that restrict glial cells from improperly eliminating synapses in adulthood. But a high-fat diet is sufficient to restart synaptic loss regulation dependent on TREM2. Defects in TREM2 affect the number of synapses and glial phagocytosis of synaptic buttons in multiple brain regions (Jay et al., 2019). Mechanically, TREM2 binds to its associated immune ligands and signals through tyrosine-based activation motifs (ITAM), such as the adapter protein DNAX activation proteins 12 (DAP12) that recruits downstream tyrosine kinase SYK, ultimately promoting chemotaxis, phagocytosis, and proliferation of microglia (Ulland and Colonna, 2018). The TREM2-DAP12 interaction also activates PI3K/AKT, followed by blocking the MAPK cascade. These signaling pathways ultimately inhibit TLR4-driven inflammatory responses in microglia (Mecca et al., 2018). In good correlation to these findings, Andreone et al. recently confirmed that PLCγ2 regulates microglial survival, myelin phagocytosis, neuronal debris processing, and lipid metabolism as a downstream molecule in TREM2. PLCγ2 also signals TLRs independently of TREM2, mediating an inflammatory response (Andreone et al., 2020). TREM2 has been reported to drive microglia to gather around amyloid-β (Aβ) plaques and engulf them (Yeh et al., 2016). Deletions or mutations in TREM2 exacerbate Aβ toxicity and increase the risk of AD. In the AD model, mice with TREM2 R47H mutations had reduced microglia proliferation, activation, and recruitment to Aβ plaques (Hall-Roberts et al., 2020).
In addition, TREM2 also indirectly affects phagocytic function by regulating microglia metabolism. It has been reported that TREM2 senses lipids and mediates myelin phagocytosis. TREM2 has been identified as a key transcriptional regulator of lipid transport and metabolism. Ligands of TREM2 include lipoprotein particles, such as LDL as well as apolipoproteins (Yeh et al., 2016). Both Apolipoprotein E (APOE) and Aβ oligomers are able to bind to and activate TREM2 (Gratuze et al., 2018; Zhao et al., 2018). Yeh et al. (2016) found that both TREM2 deficiency and APOE deficiency inhibited plaque-associated microglia proliferation in a mouse model of AD. Recent studies have also reported that microglia with TREM2 mutations or lack of TREM2 can engulf myelin fragments but fail to clear cholesteryl ester due to impaired cholesterol transport while accumulation of cholesteryl ester was also observed in APOE-deficient glial cells (Nugent et al., 2020). Overall, TREM2 plays a key role in microglia recruitment, phagocytosis of Aβ plaques, lipid transport, and metabolism. All of these studies suggest that increasing TREM2 expression is an effective treatment to delay the onset of AD (Zhong et al., 2017) and demyelinating disease. Intriguingly, interactions among microglia, neurons, and oligodendrocytes contribute to the selective phagocytosis of myelin sheaths, that is, neuronal activity competitively attracts microglia to the neuronal cell body, thus occupying the microglia used for myelin phagocytosis, while reduced neuronal activity promotes myelin phagocytosis (Hughes and Appel, 2020). Whether this neuronal activity-dependent myelin remodeling cooperates with TREM2 needs to be further verified.
Levels of soluble TREM2 in the cerebrospinal fluid (CSF) were significantly increased in AD patients with long-term physical exercise (Jensen et al., 2019). Similarly, a recent study reported that long-term voluntary running exercise ameliorated cognitive impairment in APP/PS1 mice by inhibiting TREM2 abscission and maintaining TREM2 protein levels while promoting microglial glucose metabolism and hippocampal morphological plasticity in AD mice (Zhang et al., 2022). In brief, exercise regulates microglial phagocytosis by targeting TREM2 directly or indirectly.
In mice and human brains, lipid droplets accumulate significantly in microglia with age. These cells produce an abundance of ROS (Yousef et al., 2019; Marschallinger et al., 2020), secrete pro-inflammatory cytokines, and are deficient in phagocytosis, known as lipid droplet-accumulating microglia (LDAM). More than half of the microglia were found in the LDAM state in the aged hippocampus (Marschallinger et al., 2020).
In activated microglia, metabolic pathways are significantly altered. Microglia activation, phagocytosis, migration, and release of inflammatory factors may be controlled by microglia lipid metabolism (Chausse et al., 2021). In vitro studies indicated that inhibition of lipid droplet formation significantly increased the phagocytosis of BV2 cells, implying that lipid droplets had an adverse effect on phagocytosis. Intriguingly, LDAM contain a large number of lysosomes, which accumulate near the lipid droplets. Therefore, it is hypothesized that in LDAM, lysosomes are more inclined to degrade lipid droplets than the substances being swallowed, resulting in impaired phagocytosis. In addition, there is evidence of lysosome dysfunction in senescent microglia (Mosher and Wyss-Coray, 2014). Therefore, in elderly LDAM, lysosome dysfunction may lead to both phagocytosis impairment and lipid droplet accumulation, which further damages the phagocytosis.
Consistent with this, phagocytosis in macrophages depends on accessible free fatty acids, which are released when the lipid droplets degrade (Chandak et al., 2010). The phagocytic activity of lipid droplet-enriched foam macrophages was lower than that of lipid droplet-free macrophages in atherosclerotic lesions (Chinetti-Gbaguidi et al., 2011). In parallel, recent studies have suggested that acetate, a critical microbiome-derived short-chain fatty acid, regulates disease progression in the mouse model of AD by inhibiting microglial phagocytosis of amyloid-beta (Erny et al., 2021). This suggests that the normal phagocytosis of microglia requires the assistance of increased lipid metabolism (Loving and Bruce, 2020).
At the transcriptional level, genes such as APOE, TREM2, and Lipoprotein Lipase (LPL are increased in microglia both during development and in disease states (Loving and Bruce, 2020). Regulation of lipid metabolism in TREM2 has been described above. In parallel, Nugent et al. reported that TREM2 upregulates APOE and other microglial genes associated with chronic demyelinating injury (Nugent et al., 2020). On the whole, there are some interactions among various lipid metabolism-related genes in microglia, which jointly regulate phagocytosis in different pathological conditions. The specific mechanism by which lipid droplets affect phagocytosis remains to be studied. However, whether exercise training may affect microglial phagocytosis by regulating lipid metabolism remains unclear.
Metabolic reprogramming is a process in which cells exhibit different metabolic characteristics in response to changes in the microenvironment in order to provide energy and biological matter (Mo et al., 2019). Immune cells have a variety of functions, and they would adjust their alternative metabolic pathways in real-time to meet their energy needs based on cell phenotypes. There is increasing evidence that metabolic reprogramming is a key energy basis for the immune response of microglia. Microglia homeostasis depends on oxidative metabolism, while the metabolism of microglia shifts to glycolysis under pro-inflammatory stimulation (Lynch, 2020). The transition from oxidative phosphorylation (OXPHOS) to glycolysis interferes with its phagocytosis. An in vitro study showed that the combination of interferon -γ (IFN-γ) and Aβ can induce glycolysis in microglia and its ability for phagocytosis and chemotaxis is decreased (McIntosh et al., 2019). Besides, TREM2, as mentioned above, is thought to play a key role in regulating microglial metabolic patterns.
Although glycolysis is not as efficient at producing ATP as OXPHOS, glycolysis has a glucose metabolism rate 10–100 times faster than OXPHOS, which enables microglia to complete energy-intensive processes such as phagocytosis. However, only 2 molecules of ATP are produced per glucose due to inefficient glycolysis metabolism, while oxidative metabolism produces about 36 molecules of ATP, so cellular fatigue may lead to the occurrence of functional deficits. Baik et al. have reported acute administration of Aβ-activated microglia, along with metabolic processes from OXPHOS to glycolysis. This process relies on the mTOR-HIF-1a pathway. However, the physiological processes of microglia such as cytokine secretion and phagocytosis are defective in the long run. IFN-γ administration, as a potential treatment, can restore microglia glycolytic metabolism and immune function (Baik et al., 2019). In short, regulating microglia metabolism may be a potential therapeutic strategy.
In contrast, an in vitro study showed that anti-TLR2 antibodies increased the phagocytosis of Aβ by LPS-stimulated microglia, which is associated with inhibition of the activity of the key glycolysis enzyme fructose-6-phospho-2-kinase/fructose-2, 6-bisphosphatase (PFKFB3), and the metabolic pattern of microglia shifted from glycolysis to oxidative metabolism (Rubio-Araiz et al., 2018). The glycolysis, glycolytic capacity, and PFKFB3 were significantly increased in elderly microglia, and exercise could alleviate the above effects and increase the phagocytosis ability (Mela et al., 2020). In brief, exercise reduces the dependence of microglia on metabolically inefficient glycolysis.
C1q and C3, the components of the classical complement cascade, bind to the surface of invading pathogens, which are then cleared by phagocytes expressing corresponding complement receptors in the peripheral immune system (Morgan and Kavanagh, 2018; Reis et al., 2019). Similarly, in the CNS, microglia rely on classical complement cascades to mediate phagocytic signaling, removing excess synapses. Mice with C3, C1q, or complement receptor 3 (CR3) deficiency had persistent synaptic pruning defects, and about 50% of microglial synaptic phagocytosis was blocked in CR3-deficient mice (Schafer et al., 2012; Hong et al., 2016). In parallel, C1q and C3 are also localized in the visual thalamus of developing rodents (Stevens et al., 2007). Microglia expressing CR3 engulf these complement-associated synapses. On the contrary, viral overexpression of the complement inhibitor Crry protects visual function by decreasing microglial engulfment of synapses (Werneburg et al., 2020).
Similar molecular mechanisms also modulate early synaptic loss in neurodegenerative model mice. For example, in demyelinating disease, microglia eliminate synapses through selective complement cascades (Vasek et al., 2016). In a mice model of AD, aggregation of Aβ can activate complement, which influences microglial phagocytosis through complex crosstalk with TLR and inflammasome signaling pathways, thereby, regulating synaptic pruning and loss (Yang et al., 2020). After both reperfused and non-reperfused stroke, complement activation directs ongoing microglia-dependent phagocytosis of synapses for at least 30 d after stroke, leading to a loss of synaptic density that is associated with cognitive decline (Alawieh et al., 2020). Another postmortem report of a multiple sclerosis (MS) patient also showed that C1q and C3 co-located with synaptic proteins in the hippocampus (Michailidou et al., 2015).
The specific mechanism that transforms the combination of complement and complement receptor into the phagocytosis of the synaptic remains unclear. Some studies have suggested that this is associated with Ca2+ influx into boutons in activated neurons, followed by the activation of local Ca2+-dependent scramblases, which lead to the externalization of phosphatidylserine (PtdSer). The externalized PtdSer eventually binds to C1q to trigger synaptic pruning (Païdassi et al., 2008; Lemke, 2017). It is not clear whether the binding of C3 to its receptor requires PtdSer, but Allendorf et al. have suggested that increased sialidase activity in activated microglia leads to desialylation in the cell surface, which stimulates microglia to phagocytose neurons in a CR3-dependent manner (Allendorf et al., 2020).
It has been reported that chronic aerobic exercise can maintain the integrity and function of neurovascular units in aging mice by reducing C1q+ microglia and increasing neuroplasticity (Soto et al., 2015). Furthermore, physical exercise may promote cellular crosstalk between microglia and neurons through complement molecules (Li F. et al., 2021).
Most of these studies have been conducted in the mouse model. However, it has been suggested that mouse models cannot fully solve this problem because human microglia are markedly different from rodent microglia, with higher complement components and multiple important gene expressions during brain development (Gosselin et al., 2017). There is no doubt that further research is needed to clarify this point.
CX3CL1 is one of the critical chemokines and is mainly expressed in neurons in a soluble or membrane-bound state. CX3CR1, a G protein-coupled chemokine receptor, which is highly expressed in microglia (Wolf et al., 2013), keeps microglia homeostasis by binding to CX3CL1 (Madrigal et al., 2017). The CX3CR1/CX3CL1 axis plays an important role in maintaining CNS homeostasis under physiological conditions.
In the developing thalamic cortex, microglia accumulate at synaptic concentration sites and control synaptic maturation through the CX3CR1/CX3CL1 signaling pathway. CX3CR1-deficient mice showed delayed functional maturation of postsynaptic glutamate receptors (Paolicelli et al., 2011; Hoshiko et al., 2012). In addition, artificial removal of whisker induces microglial synaptic phagocytosis and synaptic elimination during development through the CX3CR1-CX3CL1 rather than the CR3 signaling pathway. Gunner et al. showed that Adam10, a gene that encodes a metalloproteinase, plays an important role in cleaving CX3CL1 into a secretory phenotype, and the cleaved CX3CL1 then binds to CX3CR1 and initiates phagocytosis and remodeling of synapses by microglia (Gunner et al., 2019).
Tau and CX3CL1 interact competitively with CX3CR1, and the expression level of CX3CL1 is reduced in the AD brain, so CX3CR1 directly binds to Tau and promotes its internalization and uptake (Chidambaram et al., 2020). In intracerebral hemorrhage model mice, microglial phagocytosis is an important channel to promote hematoma regression. It has been reported that treatment with CCL17, a specific ligand of CCR4, promotes hematoma resolution through the CCR4/ERK/Nrf2/CD163 pathway, thereby, improving neurological function, which is associated with microglia-mediated erythrocyte phagocytosis and clearance of tissue debris (Deng et al., 2020). Additionally, CXCR3b, another splicing variant of CXCR3 expressed in human microvascular endothelial cells, has been shown to bind to CXCL4, which is mainly derived from microglia under neurodegenerative conditions in vitro and in vivo. CXCL4 reduces LPS-induced phagocytosis of microglia and BV-2 cells and the production of nitric oxide (de Jong et al., 2008). Overall, microglia in various disease models appear to activate different chemokine receptors to demonstrate phagocytosis.
Physical exercise is reported to ameliorate cognitive function and neuroplasticity in depressed mice through microglia-mediated CX3CL1-CX3CR1 signaling (Eyre et al., 2013). Similarly, it has been suggested that physical exercise could accelerate the response of the CX3CL1-CX3CR1 axis to stressors and rapidly quieten the activated microglia, thus avoiding the negative cognition-related effects of stress (Fleshner et al., 2014). However, whether exercise training may affect microglial phagocytosis by regulating CX3CL1-CX3CR1 needs further study.
NA in the cortex was derived only from noradrenergic neurons in the locus coeruleus (Berridge and Waterhouse, 2003). Although adrenergic receptors are widely expressed in the neurons and glia (O'Donnell et al., 2012), microglia are the major sites of NA signaling in the cortex. Within the cortex, there is considerable evidence that β1-adrenergic receptor (β1-AR) and β2-adrenergic receptor (β2-AR) are the only functionally significant adrenergic receptors in microglia (Heneka et al., 2010). Moreover, in the non-injured brain, microglia are highly enriched with β2-AR relative to other CNS cell types (Zhang et al., 2014). NA acts as a powerful regulator of microglia function in physiological or pathological settings, including microglial processes motility, arborization, and contact with dendrites (Liu et al., 2019; Stowell et al., 2019). Previous studies have also shown that microglia β2-AR are essential for microglia locomotion, migration, phagocytosis, proliferation, and inflammatory responses (O'Donnell et al., 2012).
In models of AD, there is evidence that the non-selective β-adrenergic receptor agonist isoproterenol indirectly enhances the phagocytosis of Aβ plaques (Heneka et al., 2010) in addition to promoting microglia migration toward Aβ plaques in culture (Kettenmann et al., 2011). Degeneration of the locus coeruleus may contribute to the pathogenesis of AD (Kalinin et al., 2007).
Both the activity of norepinephrine neurons in locus coeruleus and the expression of adrenergic receptors in healthy rats can be regulated by wheel running (Van Hoomissen et al., 2004). Furthermore, voluntary exercise is beneficial to the normalization of dendritic spine's increment and the maintenance of microglial phagocytosis in sleep-deprived mice. The modification of norepinephrine signaling in the CNS may be responsible for the preventive effect of voluntary exercise on microglia and neuron dysfunction induced by sleep deprivation (Tuan et al., 2021). Yet, the impact of physical exercise on the norepinephrine system during brain development remains elusive.
IL-33 is normally inactive in the nucleus and acts as a transcriptional regulator. When cells are subjected to mechanical stress, inflammatory cytokines stimulation, or necrosis, IL-33 is released into the extracellular space and hydrolyzed. Subsequently, the hydrolyzed IL-33 signals adjacent immune cells expressing ST2 receptors in an autocrine/paracrine manner (Altara et al., 2018). During the early stages of synaptic maturation after birth, IL-33 expression is increased in astrocytes in the gray matter of the spinal cord and thalamus, where most synapses are concentrated. In this process, IL-33/ST2 is primarily responsible for driving microglial synapse engulfment and restricted excitatory synapse numbers (Sun et al., 2021). On the other hand, IL-33 is widely expressed in adult brain regions such as the corpus callosum, hippocampus, thalamus, and the granular layer and white matter of the cerebellum. It is also primarily located in astrocytes in the mouse brain and spinal cord (Pichery et al., 2012).
Interestingly, a recent study has shown that most IL-33-expressing cells in the adult hippocampus are neurons and are primarily responsible for instructing microglial engulfment of the extracellular matrix (ECM) and reshaping synapses, which are required for memory consolidation. IL-33 deficiency leads to impaired phagocytosis of ECM by microglia, along with the accumulation of ECM proteins, especially around the synapses and dendritic spines. Treatment with exogenous ECM enzymes contributes to restoring the number of dendritic spines in IL-33-deficient mice (Nguyen et al., 2020). Neuronal IL-33 expression levels are progressively decreased with age, which is consistent with deficits in memory accuracy and the accumulation of ECM around synapses (Végh et al., 2014b). Notably, elimination of ECM or administration of exogenous recombinant IL-33 reverses cognitive decline and memory deficits in the mouse model of AD (Végh et al., 2014a; Fu et al., 2016). Besides, He et al. demonstrated the functional combination of mitochondrial bioenergetics and the phagocytic activity of microglia. They propose a microglia-astrocyte signaling pathway, the IL-33-ST2-Akt signaling axis, which is a crucial pathway for maintaining metabolic adaptation and phagocytic function of microglia during early development and is associated with neurodevelopment and neuropsychiatric disorders. Mechanistically, the IL-33-ST2 axis promoted microglia phagocytosis and energetic metabolism in an AKT-dependent manner. PTEN or SHIP-1 negatively regulates microglial phagocytosis and energy metabolism by inhibiting AKT activation. Inhibition of mitochondria-dependent energy metabolism reduces the ability of microglia to phagocytic synaptosomes (He et al., 2022).
IL-33 and ST2 can also act on their downstream molecules alone and indirectly affect microglial phagocytosis. For example, studies have found that injection of IL-33 in AD transgenic mice enhanced Aβ clearance by reprogramming microglia epigenetics and transcriptome profiling, thereby alleviating AD pathology (Lau et al., 2020). Similarly, in mouse models, microglia promote hematoma regression and functional recovery after intracerebral hemorrhage through the IL-4/STAT6/ST2 signaling pathway. Both in vivo and in vitro experiments showed that STAT6-KO impaired the ability of microglia to phagocytose red blood cells (Xu et al., 2020).
In general, IL-33/ST2 is closely related to microglial energy metabolism, phagocytosis, epigenetics, and transcriptome profiles. A further understanding of how these mechanisms translate between physiological states and different pathologies requires a better understanding of the temporal process of IL-33 expression, defining the dynamics of IL-33 expression at a more precise time and individual neuron level, and mastering the regularity of IL-33 protein release, which are important areas for future research. So far, no studies have examined the impact of exercise training on ST2 in microglia.
Studies have shown that the TAM system is an important mediator for microglia to recognize and phagocytose apoptotic cells (ACs) (Lu and Lemke, 2001) and amyloid plaques (Huang et al., 2021). TAM proteins include cell surface receptor tyrosine kinases (RTKs)- Tyro3, Axl, and Mer (encoded by Mertk) (Lemke, 2013). Human and mouse microglia normally express high levels of Mer and low levels of Axl (Fourgeaud et al., 2016; The ImmGen Consortium, 2016).
PtdSer is present in many different membranes in every cell of the body, including the endoplasmic reticulum, mitochondria, Golgi apparatus, and plasma membrane. In the plasma membrane of normal cells, PtdSer is almost entirely confined to the inner leaflet of the lipid bilayer (Leventis and Grinstein, 2010; van Meer, 2011). When programmed death is initiated, or in some pathological cases, local externalization of eat-me signals such as PtdSer in specific areas of the cell surface allows microglia to recognize and phagocytize pruning of these “locally dead” domains or cells with metabolic damage that are still alive (Lemke, 2019). Inhibition of phospholipid scramblase 1 (PLSCR1) activity avoided intracellular calcium imbalance, prevented extravasation of PtdSer, and protected target cells from microglial phagocytosis for several months (Tufail et al., 2017).
However, PtdSer activates TAM receptors through bridging molecules such as growth arrest-specific (GAS6) and protein S (PROS1) rather than binding directly to TAM proteins (Dransfield et al., 2015). Therefore, GAS6 and PROS1 may be regarded as co-receptors. Specifically, the carboxy-terminal SHBG domain of these ligands binds to Axl and Mer on microglia, whereas the gamma- carboxyglutamic acid (GLA) domain binds to PtdSer (Lemke, 2017).
During synaptic development, microglia engulf PtdSer-labeled hippocampal and retinogeniculate synapses. Blockade of the PtdSer-exposed pathway with Annexin V partially prevents synaptic elimination (Scott-Hewitt et al., 2020). In the APP/PS1 mouse model of AD, gene ablation of Axl and Mer causes microglia to fail to detect, respond to, organize, or engulf amyloid -β plaques properly (Huang et al., 2021). Furthermore, PtdSer and GAS6 were detected on all Aβ plaques surfaces (Huang et al., 2021). After Axl is activated, metalloproteinases cleft the extracellular domain of Axl exposed on the cell surface (Zagórska et al., 2014). Increased levels of the soluble outfield (sAxl) in CSF, combined with GAS6 complex, could predict disease progression and progression in human AD (Mattsson et al., 2013; Sainaghi et al., 2017). Interestingly, the Aβ that was swallowed by microglia mediated by TAM was not eliminated but deposited in the cells. These Aβ fibrils entering the lysosome are compacted into indigestible dense nuclear plaques (Huang et al., 2021). Similarly, the elimination of apoptotic neurons requires Mer and CR3, while Axl is negligible in this process (Anderson et al., 2022). During ACs phagocytosis, GAS6 and/or PROS1 act as a bridge between the externalized PtdSer on ACs and TAM receptors on microglia (Lemke, 2019).
It is worth noting that microglia can selectively phagocytose parts of the cell, instead of the whole cell. For instance, PtdSer is precisely exposed to the tip of the outer segment of the photoreceptor, which drives the retinal pigment epithelial (RPE) cells to engulf the local membrane segment (Ruggiero et al., 2012), while how PtdSer externalization is limited to the small membrane domain of cells remains unknown. It has been suggested that this is related to the local aggregation of Ca2+ channels in the plasma membrane, which locally activates Ca2+-dependent scramblases, thereby promoting the local cleavage of caspase 3 and Caspase 7 (Lemke, 2019). Further research is needed to provide evidence of those processes. Whether physical exercise would drive the externalization of PtdSer or the change of TAM protein expression needs further study.
“Don't-eat-me” is a signal that inhibits the phagocytosis of immune cells. By combining the CRISPR–Cas9 knockout screens with RNA sequencing analysis, Pluvinage et al. screened out CD22, a typical B cell receptor, as a “Don't-eat-me” signal protein, which was upregulated in aging microglia. The anti-phagocytosis of α2, 6-linked sialic acid depends on CD22, which may cooperate with clearance pathways including the perivascular glymphatic (Kress et al., 2014) and meningeal lymphatic systems (Da Mesquita et al., 2018) to control the accumulation of debris and local cytokine concentrations in the aging brain. Inhibition of CD22 by antibody blocking or gene knockout promotes clearance of myelin fragments, Aβ, and α -synuclein fibrils by microglia in vivo (Pluvinage et al., 2019). CD47, a transmembrane protein, is another negative regulator of microglia phagocytosis. It is expressed in most mammalian cells, including neurons, and inhibits phagocytosis in neurodegenerative diseases through signal-regulatory protein alpha (SIRP α) on microglia (Ding et al., 2021). CD47 is also expressed at developing synapses and inhibits microglia-mediated synaptic clearance (Lehrman et al., 2018).
Together, microglial phagocytosis is particularly important in developing synaptic pruning and amyloid clearance in AD mice while evidence for microglial phagocytosis in other disease models is lacking. Many scholars have conducted extensive research on the mechanism of microglial metabolic reprogramming, lipid metabolism, ligand, and receptor binding related to phagocytosis, which provides new targets and new ideas for the treatment of synaptic pruning deficiency and Aβ clearance disorder and also is one of the ways for some drugs and physical exercise to exert neural remodeling effect. TREM2 not only plays a role through its downstream tyrosine kinase SYK, but also indirectly regulates microglia lipid metabolism and glucose metabolism, which may be a vital intervention target for drugs or physical therapy. Based on the previous research, exercise training has been shown to influence microglial phagocytosis by modulating the glycolysis process and expression of TREM2, complement and adrenergic receptors. However, the specific cellular and molecular mechanisms of the effect of exercise training on microglia phagocytosis are still lacking solid evidence and in-depth discussion [Figure 1, created with Adobe Illustrator CC 2018 (USA) and Figdraw].
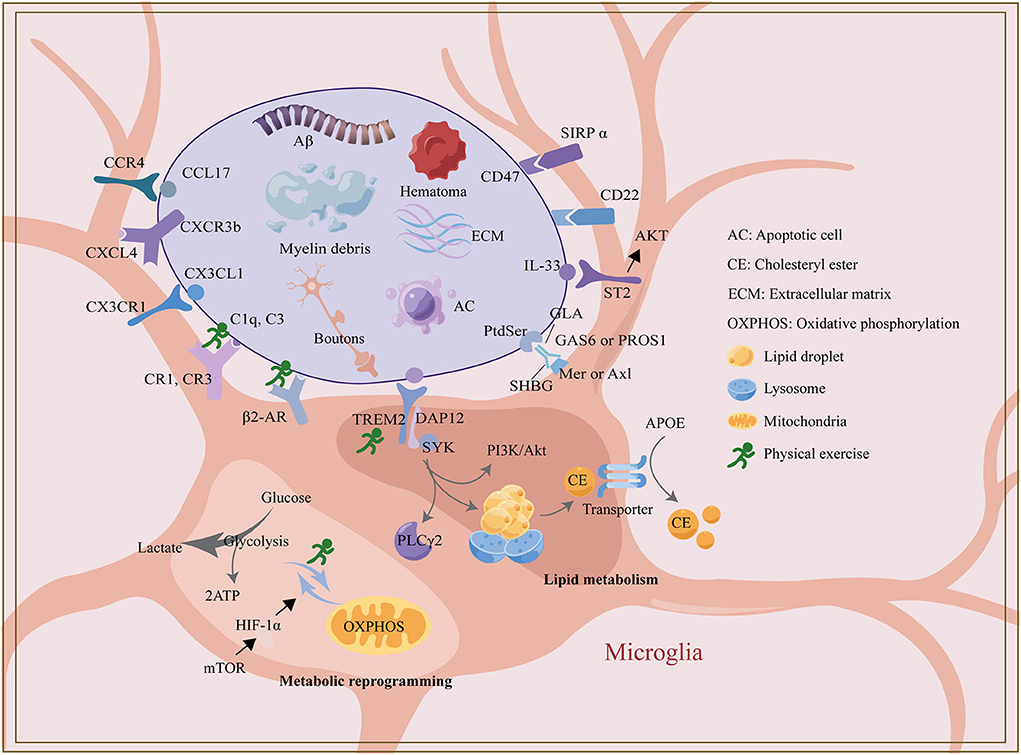
Figure 1. The regulatory mechanism of microglia phagocytosis and exercise.
In summary, the phagocytosis levels of different microglia may be heterogeneous due to the expression levels of immune receptors on the cell surface and their metabolic status. Further research is needed to determine how microglia respond to the regulation of different immune pathways. Deciphering microglia heterogeneity may depend on a combination of single-cell sequencing techniques and targeted mouse models in further study. Furthermore, although microglia are the primary phagocytes scavenging cell debris in the CNS, a growing number of studies have reported astrocyte phagocytosis in microglia-deficient or dysfunctional mice. It has been suggested that astrocytes are on standby when microglia are damaged (Konishi et al., 2020). This compensatory mechanism may be conducive to maintaining or prolonging a healthy CNS, but the extent and duration of this compensation are unclear. However, what kind of “eat-me” signal drives astrocyte phagocytosis remains unclear, which may be a direction for further research.
CL and YB had the initial idea for this article. CL and YW prepared figures and drafted the manuscript. YX and JHan performed the literature search. YB approved the final version of the manuscript and provided the funds. All authors critically revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (grant number 82072540).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alawieh, A. M., Langley, E. F., Feng, W., Spiotta, A. M., and Tomlinson, S. (2020). Complement-dependent synaptic uptake and cognitive decline after stroke and reperfusion therapy. J. Neurosci. 40, 4042–4058. doi: 10.1523/JNEUROSCI.2462-19.2020
Allendorf, D. H., Puigdellívol, M., and Brown, G. C. (2020). Activated microglia desialylate their surface, stimulating complement receptor 3-mediated phagocytosis of neurons. Glia 68, 989–998. doi: 10.1002/glia.23757
Altara, R., Ghali, R., Mallat, Z., Cataliotti, A., Booz, G. W., and Zouein, F. A. (2018). Conflicting vascular and metabolic impact of the IL-33/sST2 axis. Cardiovasc. Res. 114, 1578–1594. doi: 10.1093/cvr/cvy166
Anderson, S. R., Roberts, J. M., Ghena, N., Irvin, E. A., Schwakopf, J., Cooperstein, I. B., et al. (2022). Neuronal apoptosis drives remodeling states of microglia and shifts in survival pathway dependence. Elife 11, e76564. doi: 10.7554/eLife.76564
Andoh, M., Shibata, K., Okamoto, K., Onodera, J., Morishita, K., Miura, Y., et al. (2019). Exercise reverses behavioral and synaptic abnormalities after maternal inflammation. Cell Rep. 27. 2817–2825.e5. doi: 10.1016/j.celrep.2019.05.015
Andreone, B. J., Przybyla, L., Llapashtica, C., Rana, A., Davis, S. S., van Lengerich, B., et al. (2020). Alzheimer's-associated PLCγ2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat. Neurosci. 23, 927–938. doi: 10.1038/s41593-020-0650-6
Baik, S. H., Kang, S., Lee, W., Choi, H., Chung, S., Kim, J. I., et al. (2019). A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer's disease. Cell Metab. 30, 493–507.e6. doi: 10.1016/j.cmet.2019.06.005
Bartels, T., De Schepper, S., and Hong, S. (2020). Microglia modulate neurodegeneration in Alzheimer's and Parkinson's diseases. Science 370, 66–69. doi: 10.1126/science.abb8587
Berridge, C. W., and Waterhouse, B. D. (2003). The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res. Brain Res. Rev. 42, 33–84. doi: 10.1016/S0165-0173(03)00143-7
Billinger, S. A., Arena, R., Bernhardt, J., Eng, J. J., Franklin, B. A., Johnson, C. M., et al. (2014). Physical activity and exercise recommendations for stroke survivors: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 45, 2532–2553. doi: 10.1161/STR.0000000000000022
Chandak, P. G., Radovic, B., Aflaki, E., Kolb, D., Buchebner, M., Fröhlich, E., et al. (2010). Efficient phagocytosis requires triacylglycerol hydrolysis by adipose triglyceride lipase. J. Biol. Chem. 285, 20192–20201. doi: 10.1074/jbc.M110.107854
Chausse, B., Kakimoto, P. A., and Kann, O. (2021). Microglia and lipids: how metabolism controls brain innate immunity. Semin. Cell Dev. Biol. 112, 137–144. doi: 10.1016/j.semcdb.2020.08.001
Chidambaram, H., Das, R., and Chinnathambi, S. (2020). Interaction of Tau with the chemokine receptor, CX3CR1 and its effect on microglial activation, migration and proliferation. Cell Biosci. 10, 109. doi: 10.1186/s13578-020-00474-4
Chinetti-Gbaguidi, G., Baron, M., Bouhlel, M. A., Vanhoutte, J., Copin, C., Sebti, Y., et al. (2011). Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARγ and LXRα pathways. Circ. Res. 108, 985–995. doi: 10.1161/CIRCRESAHA.110.233775
Colonna, M., and Butovsky, O. (2017). microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 35, 441–468. doi: 10.1146/annurev-immunol-051116-052358
Colonna, M., and Wang, Y. (2016). TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 17, 201–207. doi: 10.1038/nrn.2016.7
Consorti, A., Di Marco, I., and Sansevero, G. (2021). Physical exercise modulates brain physiology through a network of long- and short-range cellular interactions. Front. Mol. Neurosci. 14, 710303. doi: 10.3389/fnmol.2021.710303
Da Mesquita, S., Louveau, A., Vaccari, A., Smirnov, I., Cornelison, R. C., Kingsmore, K. M., et al. (2018). Functional aspects of meningeal lymphatics in ageing and Alzheimer's disease. Nature 560, 185–191. doi: 10.1038/s41586-018-0368-8
de Jong, E. K., de Haas, A. H., Brouwer, N., van Weering, H. R., Hensens, M., Bechmann, I., et al. (2008). Expression of CXCL4 in microglia in vitro and in vivo and its possible signaling through CXCR3. J. Neurochem. 105, 1726–1736. doi: 10.1111/j.1471-4159.2008.05267.x
Deng, S., Sherchan, P., Jin, P., Huang, L., Travis, Z., Zhang, J. H., et al. (2020). Recombinant CCL17 enhances hematoma resolution and activation of CCR4/ERK/Nrf2/CD163 signaling pathway after intracerebral hemorrhage in mice. Neurotherapeutics 17, 1940–1953. doi: 10.1007/s13311-020-00908-4
Ding, X., Wang, J., Huang, M., Chen, Z., Liu, J., Zhang, Q., et al. (2021). Loss of microglial SIRPα promotes synaptic pruning in preclinical models of neurodegeneration. Nat. Commun. 12, 2030. doi: 10.1038/s41467-021-22301-1
Dransfield, I., Zagórska, A., Lew, E. D., Michail, K., and Lemke, G. (2015). Mer receptor tyrosine kinase mediates both tethering and phagocytosis of apoptotic cells. Cell Death Dis. 6, e1646. doi: 10.1038/cddis.2015.18
Druart, M., and Le Magueresse, C. (2019). Emerging roles of complement in psychiatric disorders. Front Psychiatry 10, 573. doi: 10.3389/fpsyt.2019.00573
Edmonson, C. A., Ziats, M. N., and Rennert, O. M. (2016). A non-inflammatory role for microglia in autism spectrum disorders. Front. Neurol. 7, 9. doi: 10.3389/fneur.2016.00009
Erny, D., Dokalis, N., Mezö, C., Castoldi, A., Mossad, O., Staszewski, O., et al. (2021). Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 33, 2260𠄃2276.e7. doi: 10.1016/j.cmet.2021.10.010
Eyre, H. A., Papps, E., and Baune, B. T. (2013). Treating depression and depression-like behavior with physical activity: an immune perspective. Front Psychiatry 4, 3. doi: 10.3389/fpsyt.2013.00003
Ferro, A., Qu, W., Lukowicz, A., Svedberg, D., Johnson, A., and Cvetanovic, M. (2018). Inhibition of NF-κB signaling in IKKβF/F;LysM Cre mice causes motor deficits but does not alter pathogenesis of Spinocerebellar ataxia type 1. PLoS ONE 13, e0200013. doi: 10.1371/journal.pone.0200013
Fleshner, M., Greenwood, B. N., and Yirmiya, R. (2014). Neuronal-glial mechanisms of exercise-evoked stress robustness. Curr. Top. Behav. Neurosci. 18, 1–12. doi: 10.1007/7854_2014_277
Fourgeaud, L., Través, P. G., Tufail, Y., Leal-Bailey, H., Lew, E. D., Burrola, P. G., et al. (2016). TAM receptors regulate multiple features of microglial physiology. Nature 532, 240–244. doi: 10.1038/nature17630
Fu, A. K., Hung, K. W., Yuen, M. Y., Zhou, X., Mak, D. S., Chan, I. C., et al. (2016). IL-33 ameliorates Alzheimer's disease-like pathology and cognitive decline. Proc. Natl. Acad. Sci. U. S. A. 113, E2705–E2713. doi: 10.1073/pnas.1604032113
Fuller, J. P., Stavenhagen, J. B., and Teeling, J. L. (2014). New roles for Fc receptors in neurodegeneration-the impact on Immunotherapy for Alzheimer's Disease. Front. Neurosci. 8, 235. doi: 10.3389/fnins.2014.00235
Gosselin, D., Skola, D., Coufal, N. G., Holtman, I. R., Schlachetzki, J. C. M., Sajti, E., et al. (2017). An environment-dependent transcriptional network specifies human microglia identity. Science 356, eaal3222. doi: 10.1126/science.aal3222
Gratuze, M., Leyns, C. E. G., and Holtzman, D. M. (2018). New insights into the role of TREM2 in Alzheimer's disease. Mol. Neurodegener. 13, 66. doi: 10.1186/s13024-018-0298-9
Gunner, G., Cheadle, L., Johnson, K. M., Ayata, P., Badimon, A., Mondo, E., et al. (2019). Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat. Neurosci. 22, 1075–1088. doi: 10.1038/s41593-019-0419-y
Hagemeyer, N., Hanft, K. M., Akriditou, M. A., Unger, N., Park, E. S., Stanley, E. R., et al. (2017). Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 134, 441–458. doi: 10.1007/s00401-017-1747-1
Hall-Roberts, H., Agarwal, D., Obst, J., Smith, T. B., Monzón-Sandoval, J., Di Daniel, E., et al. (2020). TREM2 Alzheimer's variant R47H causes similar transcriptional dysregulation to knockout, yet only subtle functional phenotypes in human iPSC-derived macrophages. Alzheimers. Res. Ther. 12, 151. doi: 10.1186/s13195-020-00709-z
Hayashi, Y., and Nakanishi, H. (2013). [Synaptic plasticity and synaptic reorganization regulated by microglia]. Nihon Shinkei Seishin Yakurigaku Zasshi 33, 211–216.
He, D., Xu, H., Zhang, H., Tang, R., Lan, Y., Xing, R., et al. (2022). Disruption of the IL-33-ST2-AKT signaling axis impairs neurodevelopment by inhibiting microglial metabolic adaptation and phagocytic function. Immunity 55, 159–173.e9. doi: 10.1016/j.immuni.2021.12.001
Heneka, M. T., Nadrigny, F., Regen, T., Martinez-Hernandez, A., Dumitrescu-Ozimek, L., Terwel, D., et al. (2010). Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc. Natl. Acad. Sci. U. S. A. 107, 6058–6063. doi: 10.1073/pnas.0909586107
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hoshiko, M., Arnoux, I., Avignone, E., Yamamoto, N., and Audinat, E. (2012). Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J. Neurosci. 32, 15106–15111. doi: 10.1523/JNEUROSCI.1167-12.2012
Huang, Y., Happonen, K. E., Burrola, P. G., O'Connor, C., Hah, N., Huang, L., et al. (2021). Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 22, 586–594. doi: 10.1038/s41590-021-00913-5
Hughes, A. N., and Appel, B. (2020). Microglia phagocytose myelin sheaths to modify developmental myelination. Nat. Neurosci. 23, 1055–1066. doi: 10.1038/s41593-020-0654-2
Jansen, I. E., Savage, J. E., Watanabe, K., Bryois, J., Williams, D. M., Steinberg, S., et al. (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat. Genet. 51, 404–413. doi: 10.1038/s41588-018-0311-9
Jay, T. R., von Saucken, V. E., Muñoz, B., Codocedo, J. F., Atwood, B. K., Lamb, B. T., et al. (2019). TREM2 is required for microglial instruction of astrocytic synaptic engulfment in neurodevelopment. Glia 67, 1873–1892. doi: 10.1002/glia.23664
Jensen, C. S., Bahl, J. M., Østergaard, L. B., Høgh P, Wermuth, L., Heslegrave, A., Zetterberg, H., et al. (2019). Exercise as a potential modulator of inflammation in patients with Alzheimer's disease measured in cerebrospinal fluid and plasma. Exp. Gerontol. 121, 91–98. doi: 10.1016/j.exger.2019.04.003
Kalinin, S., Gavrilyuk, V., Polak, P. E., Vasser, R., Zhao, J., Heneka, M. T., et al. (2007). Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer's disease. Neurobiol. Aging 28, 1206–1214. doi: 10.1016/j.neurobiolaging.2006.06.003
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Kinoshita, K., Hamanaka, G., Ohtomo, R., Takase, H., Chung, K. K., Lok, J., et al. (2021). Mature adult mice with exercise-preconditioning show better recovery after intracerebral hemorrhage. Stroke 52, 1861–1865. doi: 10.1161/STROKEAHA.120.032201
Konishi, H., Okamoto, T., Hara, Y., Komine, O., Tamada, H., Maeda, M., et al. (2020). Astrocytic phagocytosis is a compensatory mechanism for microglial dysfunction. EMBO J. 39, e104464. doi: 10.15252/embj.2020104464
Kress, B. T., Iliff, J. J., Xia, M., Wang, M., Wei, H. S., Zeppenfeld, D., et al. (2014). Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 76, 845–861. doi: 10.1002/ana.24271
Lau, S. F., Chen, C., Fu, W. Y., Qu, J. Y., Cheung, T. H., Fu, A. K. Y., et al. (2020). IL-33-PU.1 transcriptome reprogramming drives functional state transition and clearance activity of microglia in Alzheimer's disease. Cell Rep. 31, 107530. doi: 10.1016/j.celrep.2020.107530
Lehrman, E. K., Wilton, D. K., Litvina, E. Y., Welsh, C. A., Chang, S. T., Frouin, A., et al. (2018). CD47 protects synapses from excess microglia-mediated pruning during development. Neuron 100, 120–134.e6. doi: 10.1016/j.neuron.2018.09.017
Lemke, G.. (2013). Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 5, a009076. doi: 10.1101/cshperspect.a009076
Lemke, G.. (2017). Phosphatidylserine is the signal for TAM receptors and their ligands. Trends Biochem. Sci. 42, 738–748. doi: 10.1016/j.tibs.2017.06.004
Lemke, G.. (2019). How macrophages deal with death. Nat. Rev. Immunol. 19, 539–549. doi: 10.1038/s41577-019-0167-y
Leventis, P. A., and Grinstein, S. (2010). The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 39, 407–427. doi: 10.1146/annurev.biophys.093008.131234
Li, F., Geng, X., Yun, H. J., Haddad, Y., Chen, Y., and Ding, Y. (2021). Neuroplastic effect of exercise through astrocytes activation and cellular crosstalk. Aging Dis. 12, 1644–1657. doi: 10.14336/AD.2021.0325
Li, Q., and Barres, B. A. (2018). Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225–242. doi: 10.1038/nri.2017.125
Li, T., Chiou, B., Gilman, C. K., Luo, R., Koshi, T., Yu, D., et al. (2020). A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. EMBO J. 39, e104136. doi: 10.15252/embj.2019104136
Li, Y., Xia, Y., Yin, S., Wan, F., Hu, J., Kou, L., et al. (2021). Targeting microglial α-synuclein/TLRs/NF-kappaB/NLRP3 inflammasome axis in Parkinson's disease. Front. Immunol. 12, 719807. doi: 10.3389/fimmu.2021.719807
Liu, Y. U., Ying, Y., Li, Y., Eyo, U. B., Chen, T., Zheng, J., et al. (2019). Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat. Neurosci. 22, 1771–1781. doi: 10.1038/s41593-019-0511-3
Loving, B. A., and Bruce, K. D. (2020). Lipid and lipoprotein metabolism in microglia. Front. Physiol. 11, 393. doi: 10.3389/fphys.2020.00393
Lu, Q., and Lemke, G. (2001). Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 293, 306–311. doi: 10.1126/science.1061663
Lu, Y., Dong, Y., Tucker, D., Wang, R., Ahmed, M. E., Brann, D., et al. (2017). Treadmill exercise exerts neuroprotection and regulates microglial polarization and oxidative stress in a streptozotocin-induced rat model of sporadic Alzheimer's disease. J. Alzheimers. Dis. 56, 1469–1484. doi: 10.3233/JAD-160869
Lynch, M. A.. (2020). Can the emerging field of immunometabolism provide insights into neuroinflammation? Prog. Neurobiol. 184, 101719. doi: 10.1016/j.pneurobio.2019.101719
Madrigal, J. L., Caso, J. R., García-Bueno, B., Gutiérrez, I. L., and Leza, J. C. (2017). Noradrenaline induces CX3CL1 production and release by neurons. Neuropharmacology 114, 146–155. doi: 10.1016/j.neuropharm.2016.12.001
Mahalakshmi, B., Maurya, N., Lee, S. D., and Bharath Kumar, V. (2020). Possible neuroprotective mechanisms of physical exercise in neurodegeneration. Int. J. Mol. Sci. 21, 5895. doi: 10.3390/ijms21165895
Marschallinger, J., Iram, T., Zardeneta, M., Lee, S. E., Lehallier, B., Haney, M. S., et al. (2020). Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 23, 194–208. doi: 10.1038/s41593-019-0566-1
Mattsson, N., Insel, P., Nosheny, R., Zetterberg, H., Trojanowski, J. Q., Shaw, L. M., et al. (2013). CSF protein biomarkers predicting longitudinal reduction of CSF β-amyloid42 in cognitively healthy elders. Transl. Psychiatry 3, e293. doi: 10.1038/tp.2013.69
McIntosh, A., Mela, V., Harty, C., Minogue, A. M., Costello, D. A., Kerskens, C., et al. (2019). Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol. 29, 606–621. doi: 10.1111/bpa.12704
Mecca, C., Giambanco, I., Donato, R., and Arcuri, C. (2018). Microglia and aging: the role of the TREM2-DAP12 and CX3CL1-CX3CR1 axes. Int. J. Mol. Sci. 19, 318. doi: 10.3390/ijms19010318
Mee-Inta, O., Zhao, Z. W., and Kuo, Y. M. (2019). Physical exercise inhibits inflammation and microglial activation. Cells 8, 691. doi: 10.3390/cells8070691
Mela, V., Mota, B. C., Milner, M., McGinley, A., Mills, K. H. G., Kelly, Á. M., et al. (2020). Exercise-induced re-programming of age-related metabolic changes in microglia is accompanied by a reduction in senescent cells. Brain Behav. Immun. 87, 413–428. doi: 10.1016/j.bbi.2020.01.012
Michailidou, I., Willems, J. G., Kooi, E. J., van Eden, C., Gold, S. M., Geurts, J. J., et al. (2015). Complement C1q-C3-associated synaptic changes in multiple sclerosis hippocampus. Ann. Neurol. 77, 1007–1026. doi: 10.1002/ana.24398
Mo, Y., Wang, Y., Zhang, L., Yang, L., Zhou, M., Li, X., et al. (2019). The role of Wnt signaling pathway in tumor metabolic reprogramming. J. Cancer 10, 3789–3797. doi: 10.7150/jca.31166
Morgan, B. P., and Kavanagh, D. (2018). Introduction to complement in health and disease: novel aspects and insights. Semin. Immunopathol. 40, 1–2. doi: 10.1007/s00281-017-0664-7
Mosher, K. I., and Wyss-Coray, T. (2014). Microglial dysfunction in brain aging and Alzheimer's disease. Biochem. Pharmacol. 88, 594–604. doi: 10.1016/j.bcp.2014.01.008
Nguyen, P. T., Dorman, L. C., Pan, S., Vainchtein, I. D., Han, R. T., Nakao-Inoue, H., et al. (2020). Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell 182, 388–403.e15. doi: 10.1016/j.cell.2020.05.050
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Nugent, A. A., Lin, K., van Lengerich, B., Lianoglou, S., Przybyla, L., Davis, S. S., et al. (2020). TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105, 837–854.e9. doi: 10.1016/j.neuron.2019.12.007
O'Donnell, J., Zeppenfeld, D., McConnell, E., Pena, S., and Nedergaard, M. (2012). Norepinephrine: a neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochem. Res. 37, 2496–2512. doi: 10.1007/s11064-012-0818-x
Païdassi, H., Tacnet-Delorme, P., Garlatti, V., Darnault, C., Ghebrehiwet, B., Gaboriaud, C., et al. (2008). C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J. Immunol. 180, 2329–2338. doi: 10.4049/jimmunol.180.4.2329
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Pichery, M., Mirey, E., Mercier, P., Lefrancais, E., Dujardin, A., Ortega, N., et al. (2012). Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J. Immunol. 188, 3488–3495. doi: 10.4049/jimmunol.1101977
Pignataro, P., Dicarlo, M., Zerlotin, R., Zecca, C., Dell'Abate, M. T., Buccoliero, C., et al. (2021). FNDC5/Irisin system in neuroinflammation and neurodegenerative diseases: update and novel perspective. Int. J. Mol. Sci. 22, 1605. doi: 10.3390/ijms22041605
Pluvinage, J. V., Haney, M. S., Smith, B. A. H., Sun, J., Iram, T., Bonanno, L., et al. (2019). CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 568, 187–192. doi: 10.1038/s41586-019-1088-4
Prinz, M., Jung, S., and Priller, J. (2019). Microglia biology: one century of evolving concepts. Cell 179, 292–311. doi: 10.1016/j.cell.2019.08.053
Reis, E. S., Mastellos, D. C., Hajishengallis, G., and Lambris, J. D. (2019). New insights into the immune functions of complement. Nat. Rev. Immunol. 19, 503–516. doi: 10.1038/s41577-019-0168-x
Rubio-Araiz, A., Finucane, O. M., Keogh, S., and Lynch, M. A. (2018). Anti-TLR2 antibody triggers oxidative phosphorylation in microglia and increases phagocytosis of β-amyloid. J. Neuroinflammation 15, 247. doi: 10.1186/s12974-018-1281-7
Ruggiero, L., Connor, M. P., Chen, J., Langen, R., and Finnemann, S. C. (2012). Diurnal, localized exposure of phosphatidylserine by rod outer segment tips in wild-type but not Itgb5-/- or Mfge8-/- mouse retina. Proc. Natl. Acad. Sci. U. S. A. 109, 8145–8148. doi: 10.1073/pnas.1121101109
Sainaghi, P. P., Bellan, M., Lombino, F., Alciato, F., Carecchio, M., Galimberti, D., et al. (2017). Growth arrest specific 6 concentration is increased in the cerebrospinal fluid of patients with Alzheimer's disease. J. Alzheimers. Dis. 55, 59–65. doi: 10.3233/JAD-160599
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Scott-Hewitt, N., Perrucci, F., Morini, R., Erreni, M., Mahoney, M., Witkowska, A., et al. (2020). Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J. 39, e105380. doi: 10.15252/embj.2020105380
Soto, I., Graham, L. C., Richter, H. J., Simeone, S. N., Radell, J. E., Grabowska, W., et al. (2015). APOE stabilization by exercise prevents aging neurovascular dysfunction and complement induction. PLoS Biol. 13, e1002279. doi: 10.1371/journal.pbio.1002279
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Stowell, R. D., Sipe, G. O., Dawes, R. P., Batchelor, H. N., Lordy, K. A., Whitelaw, B. S., et al. (2019). Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nat. Neurosci. 22, 1782–1792. doi: 10.1038/s41593-019-0514-0
Sun, Y., Wen, Y., Wang, L., Wen, L., You, W., Wei, S., et al. (2021). Therapeutic opportunities of interleukin-33 in the central nervous system. Front. Immunol. 12, 654626. doi: 10.3389/fimmu.2021.654626
Sung, Y. H., Kim, S. C., Hong, H. P., Park, C. Y., Shin, M. S., Kim, C. J., et al. (2012). Treadmill exercise ameliorates dopaminergic neuronal loss through suppressing microglial activation in Parkinson's disease mice. Life Sci. 91, 1309–1316. doi: 10.1016/j.lfs.2012.10.003
The ImmGen Consortium (2016). Open-source ImmGen: mononuclear phagocytes. Nat. Immunol. 17, 741. doi: 10.1038/ni.3478
Tuan, L. H., Tsao, C. Y., Lee, L. J., and Lee, L. J. (2021). Voluntary exercise ameliorates synaptic pruning deficits in sleep-deprived adolescent mice. Brain Behav. Immun. 93, 96–110. doi: 10.1016/j.bbi.2020.12.017
Tufail, Y., Cook, D., Fourgeaud, L., Powers, C. J., Merten, K., Clark, C. L., et al. (2017). phosphatidylserine exposure controls viral innate immune responses by microglia. Neuron 93, 574–586.e8. doi: 10.1016/j.neuron.2016.12.021
Ulland, T. K., and Colonna, M. (2018). TREM2 - a key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 14, 667–675. doi: 10.1038/s41582-018-0072-1
Van Hoomissen, J. D., Holmes, P. V., Zellner, A. S., Poudevigne, A., and Dishman, R. K. (2004). Effects of beta-adrenoreceptor blockade during chronic exercise on contextual fear conditioning and mRNA for galanin and brain-derived neurotrophic factor. Behav. Neurosci. 118, 1378–1390. doi: 10.1037/0735-7044.118.6.1378
van Meer, G.. (2011). Dynamic transbilayer lipid asymmetry. Cold Spring Harb. Perspect. Biol. 3, a004671. doi: 10.1101/cshperspect.a004671
Vasek, M. J., Garber, C., Dorsey, D., Durrant, D. M., Bollman, B., Soung, A., et al. (2016). A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534, 538–543. doi: 10.1038/nature18283
Végh, M. J., Heldring, C. M., Kamphuis, W., Hijazi, S., Timmerman, A. J., Li, K. W., et al. (2014a). Reducing hippocampal extracellular matrix reverses early memory deficits in a mouse model of Alzheimer's disease. Acta Neuropathol Commun 2, 76. doi: 10.1186/s40478-014-0076-z
Végh, M. J., Rausell, A., Loos, M., Heldring, C. M., Jurkowski, W., van Nierop, P., et al. (2014b). Hippocampal extracellular matrix levels and stochasticity in synaptic protein expression increase with age and are associated with age-dependent cognitive decline. Mol. Cell. Proteomics 13, 2975–2985. doi: 10.1074/mcp.M113.032086
Wendeln, A. C., Degenhardt, K., Kaurani, L., Gertig, M., Ulas, T., Jain, G., et al. (2018). Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338. doi: 10.1038/s41586-018-0023-4
Werneburg, S., Jung, J., Kunjamma, R. B., Ha, S. K., Luciano, N. J., Willis, C. M., et al. (2020). Targeted complement inhibition at synapses prevents microglial synaptic engulfment and synapse loss in demyelinating disease. Immunity 52, 167–182.e7. doi: 10.1016/j.immuni.2019.12.004
Wilkinson, K., and El Khoury, J. (2012). Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer's disease. Int. J. Alzheimers. Dis. 2012, 489456. doi: 10.1155/2012/489456
Wolf, S. A., Boddeke, H. W., and Kettenmann, H. (2017). Microglia in physiology and disease. Annu. Rev. Physiol. 79, 619–643. doi: 10.1146/annurev-physiol-022516-034406
Wolf, Y., Yona, S., Kim, K. W., and Jung, S. (2013). Microglia, seen from the CX3CR1 angle. Front. Cell. Neurosci. 7, 26. doi: 10.3389/fncel.2013.00026
Wu, Y., Dissing-Olesen, L., MacVicar, B. A., and Stevens, B. (2015). Microglia: dynamic mediators of synapse development and plasticity. Trends Immunol. 36, 605–613. doi: 10.1016/j.it.2015.08.008
Xu, J., Chen, Z., Yu, F., Liu, H., Ma, C., Xie, D., et al. (2020). IL-4/STAT6 signaling facilitates innate hematoma resolution and neurological recovery after hemorrhagic stroke in mice. Proc. Natl. Acad. Sci. U. S. A. 117, 32679–32690. doi: 10.1073/pnas.2018497117
Yang, J., Wise, L., and Fukuchi, K. I. (2020). TLR4 cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer's disease. Front. Immunol. 11, 724. doi: 10.3389/fimmu.2020.00724
Yeh, F. L., Wang, Y., Tom, I., Gonzalez, L. C., and Sheng, M. (2016). TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91, 328–340. doi: 10.1016/j.neuron.2016.06.015
Yousef, H., Czupalla, C. J., Lee, D., Chen, M. B., Burke, A. N., Zera, K. A., et al. (2019). Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat. Med. 25, 988–1000. doi: 10.1038/s41591-019-0440-4
Zagórska, A., Través, P. G., Lew, E. D., Dransfield, I., and Lemke, G. (2014). Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 15, 920–928. doi: 10.1038/ni.2986
Zhang, S. S., Zhu, L., Peng, Y., Zhang, L., Chao, F. L., Jiang, L., et al. (2022). Long-term running exercise improves cognitive function and promotes microglial glucose metabolism and morphological plasticity in the hippocampus of APP/PS1 mice. J. Neuroinflamm. 19, 34. doi: 10.1186/s12974-022-02401-5
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O'Keeffe, S., et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014
Zhao, Y., Wu, X., Li, X., Jiang, L. L., Gui, X., Liu, Y., et al. (2018). TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron 97, 1023–1031.e7. doi: 10.1016/j.neuron.2018.01.031
Keywords: microglia, phagocytosis, exercise, TREM2, synapsis
Citation: Li C, Wang Y, Xing Y, Han J, Zhang Y, Zhang A, Hu J, Hua Y and Bai Y (2022) Regulation of microglia phagocytosis and potential involvement of exercise. Front. Cell. Neurosci. 16:953534. doi: 10.3389/fncel.2022.953534
Received: 26 May 2022; Accepted: 29 June 2022;
Published: 25 July 2022.
Edited by:
Zongjian Liu, Capital Medical University, ChinaReviewed by:
Wen Wu, Southern Medical University, ChinaCopyright © 2022 Li, Wang, Xing, Han, Zhang, Zhang, Hu, Hua and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yulong Bai, ZHJfYmFpeWxAZnVkYW4uZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.