 Toni Christoph Berger
Toni Christoph Berger Erik Taubøll
Erik Taubøll Kjell Heuser
Kjell Heuser- 1Department of Neurology, Oslo University Hospital, Oslo, Norway
- 2Faculty of Medicine, University of Oslo, Oslo, Norway
Pharmacological therapy of epilepsy has so far been limited to symptomatic treatment aimed at neuronal targets, with the result of an unchanged high proportion of patients lacking seizure control. The dissection of the intricate pathological mechanisms that transform normal brain matter to a focus for epileptic seizures—the process of epileptogenesis—could yield targets for novel treatment strategies preventing the development or progression of epilepsy. While many pathological features of epileptogenesis have been identified, obvious shortcomings in drug development are now believed to be based on the lack of knowledge of molecular upstream mechanisms, such as DNA methylation (DNAm), and as well as a failure to recognize glial cell involvement in epileptogenesis. This article highlights the potential role of DNAm and related gene expression (GE) as a treatment target in epileptogenesis.
Introduction
Epileptogenesis is the transformation of a physiologically functioning brain into an epileptic one, and the progression of manifested epilepsy (Pitkanen and Engel, 2014) (Figure 1). Although often initially not detectable clinically, this process is assumed to have a temporal and spatial starting point in the brain, involving an initial incident, such as trauma, hypoxia, infection, or complex febrile seizures. According to the current practical definition, this is followed by a latent phase devoid of clinical seizures, evolving to the chronic state that features the occurrence of spontaneous, and often progressive, epileptic seizures (Boison et al., 2013; Pitkanen and Engel, 2014). A prerequisite for the development of true anti-epileptogenic drugs preventing the emergence of clinical seizures, or stopping the worsening of chronic epilepsy, is to improve our understanding of the underlying pathological processes of epileptogenesis (Aronica and Gorter, 2007; Pitkanen and Lukasiuk, 2011; Loscher et al., 2013; Pitkänen et al., 2013; Löscher, 2020). Meanwhile, several pathological hallmarks of epileptogenesis are known. These are, for example, neuronal death, reactive gliosis, blood-brain barrier (BBB) disruption, axonal damage and sprouting, network reorganization, alteration of the extracellular matrix (ECM), and astrocyte uncoupling, as well as substantial changes of the molecular architecture of both neurons and glial cells (Tauck and Nadler, 1985; de Lanerolle et al., 1989, 2012; Houser, 1990; Houser et al., 1990; Mathern et al., 1995; Eid et al., 2005, 2019; Vezzani and Granata, 2005; Dityatev, 2010; Blumcke et al., 2013; Bedner et al., 2015; Patel et al., 2019; Bruxel et al., 2021). What is more delicate is the lack of strategies for how to prevent the formation of these pathological features. Obvious shortcomings of novel preventive drug development are based on the lack of knowledge of molecular upstream mechanisms, such as DNA methylation (DNAm) or gene expression (GE) (Perucca et al., 2020), and also the neglect of the importance of glial cell involvement in the inflammatory processes accompanying epileptogenesis (Kalozoumi et al., 2018; Patel et al., 2019). The goal of this review is to discuss the significance of DNAm as a potential target for anti-epileptogenic treatment in the scope of novel knowledge of glial involvement in this process.
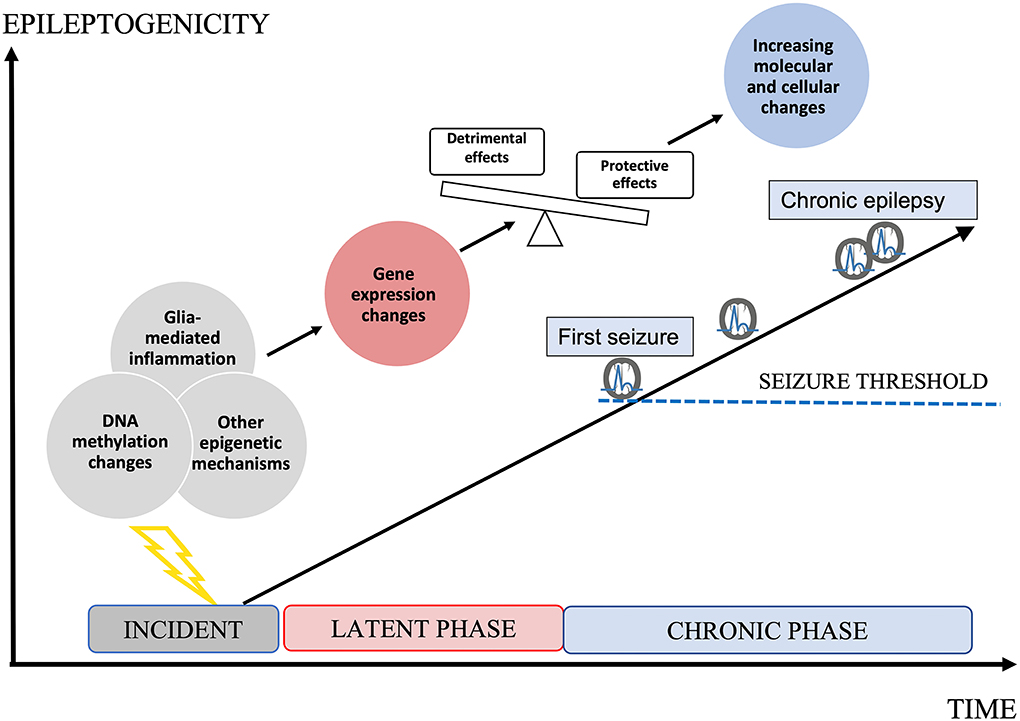
Figure 1. Epileptogenesis. Epileptogenesis describes the development of a brain becoming prone to generate epileptic seizures and further progression. An initial incident occurs to the brain, initiating the latent phase in which molecular and cellular changes increasingly develop. Epigenetic mechanisms, such as DNA methylation (DNAm) changes may determine changes in gene expression (GE) and by this cellular responses in both neurons and glial cells. Both protective and detrimental changes occur. Imbalance in favor to detrimental effects leads to increasing epileptogenicity up to the threshold of clinical seizures. During the chronic phase, epileptogenicity increases leading to the progression of clinical epilepsy, accompanied by increasing molecular and cellular changes. Both the latent phase and the chronic phase could be relevant to the development of anti-epileptogenic medication.
DNAm: Function and potential therapeutical target
After the human genome was deciphered in 2003, there were high expectations that pathological phenotypes would be linked to genomic variants (Collins et al., 2003; Green et al., 2015). However, the expectations did not come to fruition, and in the field of epilepsy, only a few genomic loci could be linked to epileptic conditions (International League Against Epilepsy Consortium on Complex Epilepsies, 2014; Abou-Khalil et al., 2018). The field of “epigenomics” arose with the hope to decipher the missing link between genotype and phenotype. Today, epigenetics can be defined as mechanisms that largely determine the “transcriptome,” meaning which genes are “used” to define a certain phenotype (Feinberg, 2007; McClung and Nestler, 2008; Gräff et al., 2011).
At the molecular level, epigenetics includes several mechanisms: DNAm, histone modifications, and non-coding RNAs are the most integral parts of this machinery (Gräff et al., 2011; Cavalli and Heard, 2019). These mechanisms are heavily interacted (Li et al., 2008; Cedar and Bergman, 2009; Wang et al., 2015). They determine chromatin accessibility, quality, and quantity of GE, and by this, tissue development in normal as well as pathological states. They can be altered by environmental factors (Danchin et al., 2011) and are mitotically inheritable (Cavalli and Heard, 2019). As such, they are potential upstream mechanisms for various molecular pathways, including those prompting pathological features of epileptogenesis. In the following, we will focus on DNA methylation in epileptogenesis.
DNA methylation is so far the most comprehensively studied epigenetic mechanism (Dor and Cedar, 2018) and is defined as the methylation (adding of a -CH3 group) of the DNA-base cytosine in a CpG (Cytosine – phosphate – Guanine) dinucleotide (CpG) (Luo et al., 2018). Oxidated variants of 5-methylcytosine (5-mC—in this review simply referred to as DNAm) include 5-hydroxymethylcytosine [5-hmC, relevant in post-mitotic neurons (Mellén et al., 2017)], 5-formylcytosine (5-fC), and 5-carboxylcytosine (5-caC), all of which potentially exert distinct effects on the GE and chromatin accessibility (Song and He, 2013). Other types of methylation include: non-CpG DNAm in, e.g., neurons (Kozlenkov et al., 2014), RNA methylation (Widagdo and Anggono, 2018), and mitochondrial DNAm (Cavalcante et al., 2020). In the central nervous system (CNS), DNAm has a major role in brain development, cell differentiation, and disease (Lister et al., 2013; Smith and Meissner, 2013; Sanosaka et al., 2017; Greenberg and Bourc'his, 2019). DNAm has been shown to influence the GE in a tissue-, context-, and cell-dependent manner (Smith and Meissner, 2013; Greenberg and Bourc'his, 2019), usually in a close interaction with transcription factors (Kribelbauer et al., 2019). How far DNAm states correlate with GE is still a matter of debate (Luo et al., 2018). GE is, on the other hand, regarded as an indicator of protein abundance and biological function, and provides a relevant estimate of downstream effects (Wang et al., 2009; Liu Y. et al., 2016; Silva and Vogel, 2016). Thus, DNAm might influence downstream molecular and cellular processes via GE regulation. DNAm is modifiable by environmental factors (Martin and Fry, 2018; Cavalli and Heard, 2019), hormones (Kovács et al., 2020), and even by neuronal activity (Guo et al., 2011). Recently, the site-specific modification of DNAm was shown to specifically alter GE (Liu X. S. et al., 2016; Liu and Jaenisch, 2019), which is especially interesting when it comes to novel therapeutic concepts. Further, disease-specific blood-DNAm pattern alterations have been reported in several pathological conditions (Fransquet et al., 2018; Agha et al., 2019; Henderson-Smith et al., 2019; Somineni et al., 2019) and proposed as possible biomarkers, with adjunct therapeutical implications (Kim et al., 2018; Berdasco and Esteller, 2019).
DNAm in epilepsy
Epilepsy-related alterations in DNAm have been shown in previous studies in both animal models (Miller-Delaney et al., 2012; Kobow et al., 2013; Machnes et al., 2013; Ryley Parrish et al., 2013; Williams-Karnesky et al., 2013; Li et al., 2015; Lusardi et al., 2015; Debski et al., 2016; Liu X. et al., 2016; Zybura-Broda et al., 2016) and in humans (Zhu et al., 2012; Miller-Delaney et al., 2015; Liu X. et al., 2016; Zhang et al., 2021) (Table 1). The most consistent finding is a state of DNA hypermethylation occurring in chronic-epilepsy states in both animal models (Kobow et al., 2013) and human hippocampal tissues (Miller-Delaney et al., 2015). Although some studies associate alterations in DNAm with GE changes (Kobow et al., 2009; Li et al., 2015; Debski et al., 2016), a recent study was more hesitant to reach this conclusion (Lipponen et al., 2018).

Table 1. Relevant studies assessing DNA methylation (DNAm) in epileptogenesis.
At the early stages of epileptogenesis, no changes (Ryley Parrish et al., 2013), or a slight tendency toward general DNA hypomethylation (Miller-Delaney et al., 2012), have been found. At single genes, DNAm changes have been recorded as soon as 1 h after status epilepticus (SE) initiation by intra-peritoneal (i.p.) kainate treatment in rats (Ryley Parrish et al., 2013). At chronic time points, a general tendency toward hypermethylation was found, and potential associations between DNAm and GE at specific genomic loci identified (Kobow et al., 2013). A follow-up study reanalyzed these results and compared them to two other murine models of focal epilepsy, traumatic brain injury (TBI) and amygdala stimulation, at chronic time points. This study found both a general tendency toward CpG hypomethylation (amygdala stimulation) and hypermethylation (pilocarpine, TBI) methylation and little overlap regarding DNAm between these models (Debski et al., 2016).
Some studies investigated both DNAm and GE changes to identify potential correlations. Although no general correlations between DNAm and GE were found, coincidental occurrences of these two phenomena were detected at some genomic loci (Kobow et al., 2013; Debski et al., 2016). Other studies have used more of a “black box” approach, investigating general methylation levels and testing interventions by means of DNA-methyl-transferase inhibitors—targeting the “hypermethylated” state in chronic epilepsy and potentially attenuating epileptogenesis. In one study (rat model, i.p. kainate) the application of a DNMT inhibitor did not significantly alter the disease course (some/despite of later onset of SE), but it did reduce the general hypermethylation state and also DNAm at one of the investigated genes (Ryley Parrish et al., 2013). Another group reported reversed hypermethylation, attenuated seizure severity, and later onset of epileptogenesis (kindling model in mice and rats using pentylenetetrazol) associated with both the application of a DNMT inhibitor and adenosine (intracranial implants) (Williams-Karnesky et al., 2013). In our previous studies employing the intracortical kainate mouse model of mesial temporal lobe epilepsy, we observed vast and mainly cell-specific changes in DNAm in both the ipsilateral (Berger et al., 2019) and contralateral (Berger et al., 2020) hippocampi, with hypermethylation generally outweighing hypomethylation in both hippocampi. DNAm alterations near epilepsy-relevant genes and genes within epilepsy relevant Gene Ontology/Kyoto Encyclopedia of Genes and Genomes (GO/KEGG) pathways (Berger et al., 2019, 2020). However, we did not detect general correlations between DNAm and GE on genomic features (Berger et al., 2019, 2020). Nevertheless, at several potentially epileptogenesis-related genes, statistically significant alterations in DNAm and GE coincided (Berger et al., 2019). In summary, in this specific model, we were not able to conclude that DNAm facilitates a direct regulation of GE in early epileptogenesis. This conclusion concurs with results from work on the potential role of DNAm for GE in epileptogenesis (Lipponen et al., 2018) and the theory that DNAm mostly represents a secondary molecular marker of long-term gene silencing (Dor and Cedar, 2018; Luo et al., 2018; Greenberg and Bourc'his, 2019). In addition, prior studies investigating general methylation trends or associations have not demonstrated mechanistic correlations between DNAm and GE/protein alterations/epileptogenesis, but rather have shown co-incidences. Nevertheless, many previous studies and our data suggest wide-spread changes in DNAm in epileptogenesis. As the different epigenetic mechanisms (histone modifications and microRNA [miRNA]) are inter-linked (Li et al., 2008; Cedar and Bergman, 2009; Wang et al., 2015), DNAm may still affect downstream effects, not necessarily directly, but indirectly.
Increasing detection sensitivity by cell-specific approaches including focus on glial cells
Several recent studies have revealed a possible involvement of DNAm in both epileptogenesis and GE (Table 1). However, reports are not consistent and possible shortcomings may be associated with the widely used simultaneous bulk analysis of several different CNS cell types. The analysis of DNAm and GE in neurons and glia cells individually may rectify a crucial shortcoming of previous studies and be one prudent step toward deciphering the “DNA methylome.” Neurons and glia facilitate mostly complementary tasks in physiological and pathological CNS states, such as epileptogenesis (Cahoy et al., 2008; Doyle et al., 2008; Zamanian et al., 2012; Patel et al., 2019), and exhibit different DNAm methylation profiles (Kozlenkov et al., 2014; Sanosaka et al., 2017). DNAm both in ways of 5hmC and non-CpG methylation are of a much higher significance in neurons than in glial cells (Kozlenkov et al., 2014; Mellén et al., 2017). Analyzing molecular mechanisms in these individual cell types separately enhances resolution, and information can be obtained about the cellular origin of detected effects (e.g., changes in DNAm and GE, as well as their possible correlation). Further, the methylation of several genomic regions, such as regulatory elements exerts cell-specific effects (Li et al., 2021).
We recently introduced a more cell-specific methodology in 2 separate publications using cell sorting of brain tissue into neuronal and non-neuronal (glial) cells prior to DNAm and GE analysis (Berger et al., 2019, 2020). We found considerable changes in both DNAm and GE at 24 h post-initiation of SE in a mouse model of mesial temporal lobe epilepsy with hippocampal sclerosis (Berger et al., 2019, 2020). Most of these molecular alterations were specific to either neurons or glia. Further, GE changes uncovered a substantial involvement of glia cells in processes crucial to epileptogenesis, such as inflammation, neuronal death, neurogenesis, and Ca2+ signaling. Furthermore, we found a substantial overlap of genomic dysregulation with other epilepsy models, and even with other neurodegenerative diseases, such as Parkinson's disease or multiple sclerosis (Berger et al., 2019, 2020). These studies underscored our postulation that a cell-specific analysis of DNAm and GE in epileptogenesis provides deeper knowledge about the cellular origin of molecular mechanisms and by this, it provides a clearer view on putative upstream targets for drug development. This makes expressly sense in the scope of novel knowledge on glial cells as important orchestrators of brain inflammation and epileptogenesis. In the following, we focus on differentially regulated glial genes and their probable contribution to epileptogenesis.
Glial contribution to epileptogenesis
With our novel understanding of glial cells as central organizers of homeostatic functions and as major contributors to inflammation and brain excitability, we observe a paradigm shift where glial cells are included in the equation of epilepsy pathogenesis. Through this, we expect to approach novel curative treatment strategies for epilepsy (Heuser et al., 2021).
In the following, the most prominent hallmarks of epileptogenesis and specifically their glial contribution are discussed in detail, as alteration of glia-mediated downstream effects may represent novel treatment targets for anti-epileptogenic intervention.
Differentially regulated glial genes and their involvement in neuronal death
Death of pyramidal neurons in CA3/1 is a major hallmark of human mTLE-HS (Blumcke et al., 2013) and is reproduced in many animal models, including the intracortical kainic acid mouse model of mTLE-HS (Bedner et al., 2015). In this now widely used and well-characterized model, apoptosis is first detectable after 6 h post-injection (hpi)—but not at 4 h—in CA1 neurons (Bedner et al., 2015). At 24 hpi, both CA1 and CA3 exhibit apoptotic pyramidal neurons, and, from then on, a progressive neurodegeneration leads eventually to 90% neuronal death in CA1 and CA3 at 28 days post-injection (dpi), and a complete absence of hippocampal neurons at 9 months post-injection (mpi) (Bedner et al., 2015). Further, the majority of GABAergic interneurons in CA1 and the dentate gyrus responsible for tonic inhibition are diminished substantially already at 5 dpi (Müller et al., 2020). At 24 hpi, we detected that 8 genes related to the regulation of neuronal death were upregulated in glia, while 2 genes in this category were downregulated in neurons (Berger et al., 2019). Many of these glial genes are associated with tumor necrosis factor alpha (TNF-α), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), and interleukin 1 beta (IL-1β) related pathways (Berger et al., 2019) and have previously been shown to be upregulated to some extent in microglia, but mostly astrocytes, in reactive states (Schlomann et al., 2000; Almeida et al., 2005; Koyama and Ikegaya, 2005; Saha et al., 2006; Groves et al., 2018; Iughetti et al., 2018). Although these results do not necessarily imply a causal role, they at least indicate an important involvement of glia in general, and astrocytes in particular, for neuronal death and already in the early latent phase of epileptogenesis.
Differentially regulated glial genes and their involvement in reactive astrogliosis
Reactive astrogliosis is a common feature of various neurodegenerative states (Zamanian et al., 2012) and pathognomonic for several epileptic conditions, such as mesial temporal lobe epilepsy with hippocampal sclerosis (mTLE-HS) in humans, successfully mimicked in the i.c. mouse model of mTLE-HS (Blumcke et al., 2013; Bedner et al., 2015). In general, this phenomenon comprises morphological and molecular reshaping of astrocytes in response to an external stimulus, and it is associated with astrocyte proliferation, immune-cell recruitment, and scar formation (Escartin et al., 2021). These responses are not necessarily dichotomous as previously proposed (good vs. bad astrocytes) (Zamanian et al., 2012), but represent a continuum of various molecular responses, the cumulative consequences of which are at best unknown (Escartin et al., 2021).
In mTLE-HS, astrocytes undergo functional and morphological changes already in the early stages of epileptogenesis. As shown in the i.c. mouse model, already at 4 hpi, cell death (primarily via necroptosis and autophagy) is detectable in astrocytes and the number of astrocytes in the CA1 region is reduced (Wu et al., 2021). These structural alterations are accompanied by decreased coupling and impaired capability of K+ buffering (Bedner et al., 2015; Wu et al., 2021), which in turn are associated with increased extracellular glutamate levels and epileptic seizures (Pannasch et al., 2011). Over the course of epileptogenesis, astrocytes proliferate, express more GFAP, and at 9 mpi, a time point relevant to findings in humans with mTLE-HS, clear structural integration into the surrounding tissue is absent (Bedner et al., 2015). Our data at 24 hpi in the ipsilateral hippocampus (Berger et al., 2019), reveal that Serpina3n is the most significantly differentially expressed gene in glia. This astrocytic gene is associated with inflammation (Takamiya et al., 2002) and neuronal damage (Gesase and Kiyama, 2007) and has previously been identified as one of the major molecular hallmarks of reactive astrogliosis (Zamanian et al., 2012). Other interesting genes upregulated at 24 hpi in the ipsilateral hippocampus are Cox2 (Ptgs2) and Cxcl10 (Berger et al., 2020), both of which are pro-epileptic and potentially pro-epileptogenic inflammatory agents (Nelson and Gruol, 2004; Sui et al., 2006; Rojas et al., 2014) that are dysregulated in reactive astrogliosis (Zamanian et al., 2012). Other potential pathways involved in functional astrocytic alterations, such as astrocyte uncoupling, are the upregulation of mitogen-activated protein kinase (MAPK) pathways observed at 24 hpi mainly in glia (Berger et al., 2019). Early astrocytic uncoupling has been linked to altered phosphorylation of Cx43 via MAPK (Deshpande et al., 2017). This potentially involves TNF-α and IL-1β (Retamal et al., 2007) [mostly increased in glia (Berger et al., 2019)], which have been previously shown to have a negative effect on astrocyte coupling in the i.c. mouse model of mTLE-HS (Bedner et al., 2015).
Differentially regulated glial genes and their involvement in brain inflammation
Inflammation is closely associated with epileptogenesis (Vezzani et al., 2011). Both astrocytes and microglia can initiate and modulate inflammatory responses (Aronica et al., 2012; Devinsky et al., 2013; Eyo et al., 2017; Liddelow et al., 2017) and orchestrate downstream alterations, such as neuronal death or reactive gliosis (Jha et al., 2019; Patel et al., 2019). Apart from the activation of astrocytes at early time points of epileptogenesis in the i.c. mouse model of mTLE-HS, microglia are both prone to necroptosis already at 4 hpi and activated (elevated Iba1) at 14 dpi (Deshpande et al., 2020; Wu et al., 2021). Elevations in the levels of both TNF-α and IL-1β have also been measured at the early stages of epileptogenesis in this model. At 24 hpi, we observed increased TNF-α and IL-1β pathways, mainly in glia (Berger et al., 2019). Both these cytokines effect glutamate uptake and release from astrocytes (Hu et al., 2000; Bezzi et al., 2001; Santello et al., 2011). They also affect astrocyte coupling and neuronal death. Inflammation is also connected to the disruption of the BBB, angiogenesis, alterations of the ECM, and aberrant neurogenesis, all important elements of epileptogenesis (Pitkanen and Lukasiuk, 2011; Patel et al., 2019). In the i.c. kainic acid model of mTLE-HS, albumin extravasation is detected at 5 dpi and throughout epileptogenesis (3 and 9 mpi) (Deshpande et al., 2017). At 28 dpi, CD31 as a marker of endothelial cells, and as such of angiogenesis, is increased in the ipsilateral hippocampus 3-fold in both CA1 and DG (Deshpande et al., 2017). This is in line with our findings in the same model, where we at 24 hpi, detected a total of 19 glial genes and 11 neuronal genes involved in angiogenesis (Berger et al., 2019). Albumin extravasation is known to induce the activation of transforming growth factor beta (TGF-β) in astrocytes (Heinemann et al., 2012), which, in turn induces MAPKs potentially leading to Cx43 phosphorylation and uncoupling of astrocytes, a mechanism believed to be important in epileptogenesis (Deshpande et al., 2017). TGF-β further induces increased neuronal excitability via astrocyte-mediated reduction of Kir4.1, AQP channels, and glutamate transporters (Ivens et al., 2007; Kim et al., 2012). We found TGF-β pathways to be increased in both neurons and glia at 24 hpi (Berger et al., 2019).
Potential strategies and challenges of anti-epileptogenic intervention
Hypermethylation in chronic epilepsy states in both human and murine CNS tissue, represents the most consistent finding of DNAm alterations. Alas, the potential attenuation of epileptogenesis via DNAm inhibition could serve as an anti-epileptic or even anti-epileptogenic target (Ryley Parrish et al., 2013; Williams-Karnesky et al., 2013). Mechanistically, this may be linked to direct anti-epileptic/anti-epileptogenic effects of adenosine, as this molecule is metabolically connected to DNAm (Weltha et al., 2019).
Anti-epileptogenic therapy could either enforce endogenic homeostatic pathways or attenuate detrimental responses. Genes dysregulated in the latent phase but also in the early chronic state of epileptogenesis are potential upstream targets of anti-epileptogenic intervention. In general, all the above-mentioned genes with CpG islands in their promoters (possibly other genomic features also) could be targeted to alter GE via DNAm alteration [as shown feasible in ref. (Liu X. S. et al., 2016)]. Even if DNAm changes do not overlap with GE changes in different epilepsy models and at different specific time points, it may still be possible to alter the expression of crucial genes and pathways by means of epigenetic editing as described elsewhere (Liu X. S. et al., 2016; Holtzman and Gersbach, 2018; Liu and Jaenisch, 2019). To achieve this goal, an epigenetic tool, such as a modified CRISPR system with either a DNMT (to facilitate hypermethylation and potentially gene silencing) or a TET oxidase (to gain hypomethylation and gene activation), applicable both to the anatomical region and cell type of choice, would be necessary.
Further, specific post-transcriptional (Desi and Tay, 2019) or post-translational modifications (Wang et al., 2014) could be employed to intervene between gene transcription and protein synthesis/downstream effects of the given target. As proposed by previous studies and supported by our findings (Berger et al., 2019, 2020), glia plays an important role in inflammatory pathways (Devinsky et al., 2013; Patel et al., 2019). Thus, the development of glia-specific drugs may lead the way to the next generation of anti-epileptogenic treatment. Nevertheless, a long way is ahead to reach these goals. One way to decipher the “epileptogenic code” would be to analyze all known molecular factors (e.g., DNAm, histone modifications, transcription factor binding, GE, proteomics, EEG, and clinical parameters) simultaneously and using comparable methods, crystalize patterns with the help of artificial intelligence, as has been performed with a fraction of parameters in ref. (Myszczynska et al., 2020). The next pragmatic steps toward this long-term goal include the assessment of cell-specific DNAm and throughout the whole process of epileptogenesis in animal models, with confirmation of results in human studies. Potential outcomes of such efforts include cell-specific molecular treatment targets and biomarkers for epileptogenesis. However, one of the most prominent challenges is the versatility of the epilepsies. Seizures are symptomatic manifestations of cerebral dysfunction and can be caused by a multitude of conditions, ranging from unknown idiopathic to the acquired causes, such as TBI, ischemic cerebral insults, infections, or hypoxia. Moreover, we must rely on simplified models restricted to only mimicking a certain group of the human epilepsies. Further, we must face concerns on comparability, interpretability, and translation of various research results based on diverse analytical tools (e.g., for GE—RNA-Seq or various kits) and bioinformatics processing.
Another important issue worth mentioning is the role of DNAm for diagnostic and prognostic approaches in epilepsy. As an example, the blood of patients with mTLE-HS has shown specific DNAm patterns (Long et al., 2017) and a concordant-twin study detected patterns that enabled distinction between focal and generalized epilepsy (Mohandas et al., 2019). These results suggest that blood-based DNAm could be a potential biomarker for epilepsy and epileptogenesis, paving the way to a more personalized management of people with epilepsy (PWE). Blood-based DNAm as a biomarker could also help to predict the patients risk for developing comorbidities, such as anxiety, depression, or cognitive decline and predict treatment response with existing anti-seizure medication (ASM).
Conclusions
Neurons and glia orchestrate epileptogenesis, with glia playing a crucial part in neuronal death, reactive gliosis, and inflammation in early epileptogenesis. Several, mostly inflammation-related and glia-derived genes may serve as targets for anti-epileptogenic intervention, potentially by means of epigenetic modification. Several recent studies have revealed widespread and cell-specific alterations in DNAm in epileptogenesis. To what extent and how exactly DNAm influences GE (and downstream effects) in epileptogenesis, is challenging to determine, and methodological shortcomings as well as inadequate processing of complex data seem to muddy the waters. We have yet to fathom the full complexity of interplay at various levels of molecular interactions in epileptogenesis. It is possible, or even likely, that not a single gene or pathway determines epileptogenesis, but rather the interaction of genes at a given time, the resulting levels of proteins, and cellular interactions that determine the epileptogenic phenotype.
Author contributions
KH and TB contributed to conception, design, writing, and revision of the article. ET contributed to the writing and revision of the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the European Commission (ERA-NET NEURON, Brain Inflammation, Glia and Epilepsy), the European Union's Horizon 2020 research and innovation program (Marie Sklodowska-Curie grant agreement No. 722053), and the South-Eastern Norway Regional Health Authority (No. 2014018).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abou-Khalil, B., Auce, P., Avbersek, A., Bahlo, M., Balding, D. J., Bast, T., et al. (2018). Genome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nat. Commun. 9, 5269. doi: 10.1038/s41467-018-07524-z
Agha, G., Mendelson, M. M., Ward-Caviness, C. K., Joehanes, R., Huan, T., Gondalia, R., et al. (2019). Blood leukocyte DNA methylation predicts risk of future myocardial infarction and coronary heart disease. Circulation. 140, 645–657. doi: 10.1161/CIRCULATIONAHA.118.039357
Almeida, R. D., Manadas, B. J., Melo, C. V., Gomes, J. R., Mendes, C. S., Grãos, M. M., et al. (2005). Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 12, 1329–1343. doi: 10.1038/sj.cdd.4401662
Aronica, E., and Gorter, J. A. (2007). Gene expression profile in temporal lobe epilepsy. Neuroscientist. 13, 100–108. doi: 10.1177/1073858406295832
Aronica, E., Ravizza, T., Zurolo, E., and Vezzani, A. (2012). Astrocyte immune responses in epilepsy. Glia 60, 1258–1268. doi: 10.1002/glia.22312
Bedner, P., Dupper, A., Huttmann, K., Muller, J., Herde, M. K., Dublin, P., et al. (2015). Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain J. Neurol. 138, 1208–1222. doi: 10.1093/brain/awv067
Berdasco, M., and Esteller, M. (2019). Clinical epigenetics: seizing opportunities for translation. Nat. Rev. Genet. 20, 109–127. doi: 10.1038/s41576-018-0074-2
Berger, T. C., Vigeland, M. D., Hjorthaug, H. S., Etholm, L., Nome, C. G., Tauboll, E., et al. (2019). Neuronal and glial DNA methylation and gene expression changes in early epileptogenesis. PLoS ONE. 14, e0226575. doi: 10.1371/journal.pone.0226575
Berger, T. C., Vigeland, M. D., Hjorthaug, H. S., Nome, C. G., Taubøll, E., Selmer, K. K., et al. (2020). Differential glial activation in early epileptogenesis-insights from cell-specific analysis of DNA methylation and gene expression in the contralateral hippocampus. Front. Neurol. 11, 573575. doi: 10.3389/fneur.2020.573575
Bezzi, P., Domercq, M., Brambilla, L., Galli, R., Schols, D., Clercq, D., et al. (2001). E, et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat. Neurosci. 4, 702–710. doi: 10.1038/89490
Blumcke, I., Thom, M., Aronica, E., Armstrong, D. D., Bartolomei, F., Bernasconi, A., et al. (2013). International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a task force report from the ILAE commission on diagnostic methods. Epilepsia 54, 1315–1329. doi: 10.1111/epi.12220
Boison, D., Sandau, U., Ruskin, D., Kawamura, M., and Masino, S. (2013). Homeostatic control of brain function—new approaches to understand epileptogenesis. Front. Cell. Neurosci. 7, 109. doi: 10.3389/fncel.2013.00109
Bruxel, E. M., Bruno, D. C. F., do Canto, A. M., Geraldis, J. C., Godoi, A. B., Martin, M., et al. (2021). Multi-omics in mesial temporal lobe epilepsy with hippocampal sclerosis: clues into the underlying mechanisms leading to disease. Seizure 90, 34–50. doi: 10.1016/j.seizure.2021.03.002
Cahoy, J. D., Emery, B., Kaushal, A., Foo, L. C., Zamanian, J. L., Christopherson, K. S., et al. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278. doi: 10.1523/JNEUROSCI.4178-07.2008
Cavalcante, G. C., Magalhães, L., Ribeiro-Dos-Santos, Â., and Vidal, A. F. (2020). Mitochondrial epigenetics: non-coding RNAs as a novel layer of complexity. Int. J. Mol. Sci. 21, 1838. doi: 10.3390/ijms21051838
Cavalli, G., and Heard, E. (2019). Advances in epigenetics link genetics to the environment and disease. Nature 571, 489–499. doi: 10.1038/s41586-019-1411-0
Cedar, H., and Bergman, Y. (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10, 295–304. doi: 10.1038/nrg2540
Collins, F. S., Morgan, M., and Patrinos, A. (2003). The human genome project: lessons from large-scale biology. Science 300, 286–290. doi: 10.1126/science.1084564
Danchin, É., Charmantier, A., Champagne, F. A., Mesoudi, A., Pujol, B., Blanchet, S., et al. (2011). Beyond DNA: integrating inclusive inheritance into an extended theory of evolution. Nat. Rev. Genet. 12, 475–486. doi: 10.1038/nrg3028
de Lanerolle, N. C., Kim, J. H., Robbins, R. J., and Spencer, D. D. (1989). Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 495, 387–395. doi: 10.1016/0006-8993(89)90234-5
de Lanerolle, N. C., Lee, T. S., and Spencer, D. D. (2012). “Histopathology of human epilepsy,” in Jasper's Basic Mechanisms of the Epilepsies, eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen and A. V. Delgado-Escueta (Bethesda, MD: National Center for Biotechnology Information, US) Copyright ©. Michael A Rogawski, Antonio V Delgado-Escueta, Jeffrey L Noebels, Massimo Avoli and Richard W Olsen.
Debski, K. J., Pitkanen, A., Puhakka, N., Bot, A. M., Khurana, I., Harikrishnan, K. N., et al. (2016). Etiology matters - genomic DNA methylation patterns in three rat models of acquired epilepsy. Sci. Rep. 6, 25668. doi: 10.1038/srep25668
Deshpande, T., Li, T., Henning, L., Wu, Z., Müller, J., Seifert, G., et al. (2020). Constitutive deletion of astrocytic connexins aggravates kainate-induced epilepsy. Glia 68, 2136–2147. doi: 10.1002/glia.23832
Deshpande, T., Li, T., Herde, M. K., Becker, A., Vatter, H., Schwarz, M. K., et al. (2017). Subcellular reorganization and altered phosphorylation of the astrocytic gap junction protein connexin43 in human and experimental temporal lobe epilepsy. Glia 65, 1809–1820. doi: 10.1002/glia.23196
Desi, N., and Tay, Y. (2019). The butterfly effect of RNA alterations on transcriptomic equilibrium. Cells 8, 1634. doi: 10.3390/cells8121634
Devinsky, O., Vezzani, A., Najjar, S., De Lanerolle, N. C., and Rogawski, M. A. (2013). Glia and epilepsy: excitability and inflammation. Trends Neurosci. 36, 174–184. doi: 10.1016/j.tins.2012.11.008
Dityatev, A. (2010). Remodeling of extracellular matrix and epileptogenesis. Epilepsia. 51, 61–65. doi: 10.1111/j.1528-1167.2010.02612.x
Dixit, A. B., Sharma, D., Tripathi, M., Srivastava, A., Paul, D., Prakash, D., et al. (2018). Genome-wide DNA methylation and RNAseq analyses identify aberrant signalling pathways in focal cortical dysplasia (FCD) Type II. Sci. Rep. 8, 17976. doi: 10.1038/s41598-018-35892-5
Dixit, A. B., Srivastava, A., Sharma, D., Tripathi, M., Paul, D., Lalwani, S., et al. (2020). Integrated genome-wide DNA methylation and RNAseq analysis of hippocampal specimens identifies potential candidate genes and aberrant signalling pathways in patients with hippocampal sclerosis. Neurol India 68, 307–313. doi: 10.4103/0028-3886.280649
Dor, Y., and Cedar, H. (2018). Principles of DNA methylation and their implications for biology and medicine. Lancet 392, 777–786. doi: 10.1016/S0140-6736(18)31268-6
Doyle, J. P., Dougherty, J. D., Heiman, M., Schmidt, E. F., Stevens, T. R., Ma, G., et al. (2008). Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135, 749–762. doi: 10.1016/j.cell.2008.10.029
Eid, T., Lee, T. S., Thomas, M. J., Amiry-Moghaddam, M., Bjornsen, L. P., Spencer, D. D., et al. (2005). Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc. Natl. Acad. Sci. U.S.A. 102, 1193–1198. doi: 10.1073/pnas.0409308102
Eid, T., Lee, T. W., Patrylo, P., and Zaveri, H. P. (2019). Astrocytes and glutamine synthetase in epileptogenesis. J. Neurosci. Res. 97, 1345–1362. doi: 10.1002/jnr.24267
Escartin, C., Galea, E., Lakatos, A., O'Callaghan, J. P., Petzold, G. C., Serrano-Pozo, A., et al. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 24, 312–325. doi: 10.1038/s41593-020-00783-4
Eyo, U. B., Murugan, M., and Wu, L. J. (2017). Microglia-neuron communication in epilepsy. Glia 65, 5–18. doi: 10.1002/glia.23006
Feinberg, A. P. (2007). Phenotypic plasticity and the epigenetics of human disease. Nature 447, 433–440. doi: 10.1038/nature05919
Fransquet, P. D., Lacaze, P., Saffery, R., McNeil, J., Woods, R., Ryan, J., et al. (2018). Blood DNA methylation as a potential biomarker of dementia: a systematic review. Alzheimers Dement. 14, 81–103. doi: 10.1016/j.jalz.2017.10.002
Gesase, A. P., and Kiyama, H. (2007). Peripheral nerve injury induced expression of mRNA for serine protease inhibitor 3 in the rat facial and hypoglossal nuclei but not in the spinal cord. Ital. J. Anat. Embryol. 112, 157–168.
Gräff, J., Kim, D., Dobbin, M. M., and Tsai, L-.H. (2011). Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol. Rev. 91, 603–649. doi: 10.1152/physrev.00012.2010
Green, E. D., Watson, J. D., and Collins, F. S. (2015). Human genome project: twenty-five years of big biology. Nature 526, 29–31. doi: 10.1038/526029a
Greenberg, M. V. C., and Bourc'his, D. (2019). The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 20, 590–607. doi: 10.1038/s41580-019-0159-6
Groves, A., Kihara, Y., Jonnalagadda, D., Rivera, R., Kennedy, G., Mayford, M., et al. (2018). A Functionally defined in vivo astrocyte population identified by c-fos activation in a mouse model of multiple sclerosis modulated by S1P signaling: immediate-early astrocytes (ieAstrocytes). eNeuro 5. doi: 10.1523/ENEURO.0239-18.2018
Guo, J. U., Ma, D. K., Mo, H., Ball, M. P., Jang, M. H., Bonaguidi, M. A., et al. (2011). Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 14, 1345–1351. doi: 10.1038/nn.2900
Heinemann, U., Kaufer, D., and Friedman, A. (2012). Blood-brain barrier dysfunction, TGFβ signaling, and astrocyte dysfunction in epilepsy. Glia 60, 1251–1257. doi: 10.1002/glia.22311
Henderson-Smith, A., Fisch, K. M., Hua, J., Liu, G., Ricciardelli, E., Jepsen, K., et al. (2019). DNA methylation changes associated with Parkinson's disease progression: outcomes from the first longitudinal genome-wide methylation analysis in blood. Epigenetics. 14, 365–382. doi: 10.1080/15592294.2019.1588682
Heuser, K., de Curtis, M., and Steinhäuser, C. (2021). Editorial: glial dysfunction in epileptogenesis. Front. Neurol. 12, 716308. doi: 10.3389/fneur.2021.716308
Holtzman, L., and Gersbach, C. A. (2018). Editing the epigenome: reshaping the genomic landscape. Annu. Rev. Genomics Hum. Genet. 19, 43–71. doi: 10.1146/annurev-genom-083117-021632
Houser, C., Miyashiro, J., Swartz, B., Walsh, G., Rich, J., Delgado-Escueta, A., et al. (1990). Altered patterns of dynorphin immunoreactivity suggest mossy fiber reorganization in human hippocampal epilepsy. J. Neurosci. 10, 267–282. doi: 10.1523/JNEUROSCI.10-01-00267.1990
Houser, C. R. (1990). Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res. 535, 195–204. doi: 10.1016/0006-8993(90)91601-C
Hu, S., Sheng, W. S., Ehrlich, L. C., Peterson, P. K., and Chao, C. C. (2000). Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 7, 153–159. doi: 10.1159/000026433
International League Against Epilepsy Consortium on Complex Epilepsies (2014). Electronic address e-auea. Genetic determinants of common epilepsies: a meta-analysis of genome-wide association studies. Lancet Neurol. 13, 893–903. doi: 10.1016/S1474-4422(14)70171-1
Iughetti, L., Lucaccioni, L., Fugetto, F., Predieri, B., Berardi, A., Ferrari, F., et al. (2018). Brain-derived neurotrophic factor and epilepsy: a systematic review. Neuropeptides 72, 23–29. doi: 10.1016/j.npep.2018.09.005
Ivens, S., Kaufer, D., Flores, L. P., Bechmann, I., Zumsteg, D., Tomkins, O., et al. (2007). TGF-β receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 130, 535–547. doi: 10.1093/brain/awl317
Jha, M. K., Jo, M., Kim, J. H., and Suk, K. (2019). Microglia-astrocyte crosstalk: an intimate molecular conversation. Neuroscientist 25, 227–240. doi: 10.1177/1073858418783959
Kalozoumi, G., Kel-Margoulis, O., Vafiadaki, E., Greenberg, D., Bernard, H., Soreq, H., et al. (2018). Glial responses during epileptogenesis in Mus musculus point to potential therapeutic targets. PLoS ONE. 13, e0201742. doi: 10.1371/journal.pone.0201742
Kim, H., Wang, X., and Jin, P. (2018). Developing DNA methylation-based diagnostic biomarkers. J. Genet. Genom. 45, 87–97. doi: 10.1016/j.jgg.2018.02.003
Kim, S. Y., Buckwalter, M., Soreq, H., Vezzani, A., and Kaufer, D. (2012). Blood-brain barrier dysfunction-induced inflammatory signaling in brain pathology and epileptogenesis. Epilepsia 53, 37–44. doi: 10.1111/j.1528-1167.2012.03701.x
Kobow, K., Jeske, I., Hildebrandt, M., Hauke, J., Hahnen, E., Buslei, R., et al. (2009). Increased reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy. J. Neuropathol. Exp. Neurol. 68, 356–364. doi: 10.1097/NEN.0b013e31819ba737
Kobow, K., Kaspi, A., Harikrishnan, K. N., Kiese, K., Ziemann, M., Khurana, I., et al. (2013). Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropathol. 126, 741–756. doi: 10.1007/s00401-013-1168-8
Kobow, K., Ziemann, M., Kaipananickal, H., Khurana, I., Mühlebner, A., Feucht, M., et al. (2019). Genomic DNA methylation distinguishes subtypes of human focal cortical dysplasia. Epilepsia 60, 1091–1103. doi: 10.1111/epi.14934
Kovács, T., Szabó-Meleg, E., and Ábrahám, I. M. (2020). Estradiol-induced epigenetically mediated mechanisms and regulation of gene expression. Int. J. Mol. Sci. 21, 3177. doi: 10.3390/ijms21093177
Koyama, R., and Ikegaya, Y. (2005). To BDNF or not to BDNF: that is the epileptic hippocampus. Neuroscientist 11, 282–287. doi: 10.1177/1073858405278266
Kozlenkov, A., Roussos, P., Timashpolsky, A., Barbu, M., Rudchenko, S., Bibikova, M., et al. (2014). Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 42, 109–127. doi: 10.1093/nar/gkt838
Kribelbauer, J. F., Lu, X. J., Rohs, R., Mann, R. S., and Bussemaker, H. J. (2019). Toward a mechanistic understanding of DNA methylation readout by transcription factors. J. Mol. Biol. 432, 1801–1805. doi: 10.1016/j.jmb.2019.10.021
Li, H. J., Wan, R. P., Tang, L. J., Liu, S. J., Zhao, Q. H., Gao, M. M., et al. (2015). Alteration of Scn3a expression is mediated via CpG methylation and MBD2 in mouse hippocampus during postnatal development and seizure condition. Biochim. Biophys. Acta. 1849, 1–9. doi: 10.1016/j.bbagrm.2014.11.004
Li, X., Wang, X., He, K., Ma, Y., Su, N., He, H., et al. (2008). High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression. Plant Cell 20, 259–276. doi: 10.1105/tpc.107.056879
Li, Y. E., Preissl, S., Hou, X., Zhang, Z., Zhang, K., Qiu, Y., et al. (2021). An atlas of gene regulatory elements in adult mouse cerebrum. Nature 598, 129–136. doi: 10.1038/s41586-021-03604-1
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lipponen, A., El-Osta, A., Kaspi, A., Ziemann, M., Khurana, I., Kn, H., et al. (2018). Transcription factors Tp73, Cebpd, Pax6, and Spi1 rather than DNA methylation regulate chronic transcriptomics changes after experimental traumatic brain injury. Acta Neuropathol. Commun. 6, 17. doi: 10.1186/s40478-018-0519-z
Lister, R., Mukamel, E. A., Nery, J. R., Urich, M., Puddifoot, C. A., Johnson, N. D., et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341. doi: 10.1126/science.1237905
Liu, X., Ou, S., Xu, T., Liu, S., Yuan, J., Huang, H., et al. (2016). New differentially expressed genes and differential DNA methylation underlying refractory epilepsy. Oncotarget. 7, 87402–87416. doi: 10.18632/oncotarget.13642
Liu, X. S., and Jaenisch, R. (2019). Editing the epigenome to tackle brain disorders. Trends Neurosci. 42, 861–870. doi: 10.1016/j.tins.2019.10.003
Liu, X. S., Wu, H., Ji, X., Stelzer, Y., Wu, X., Czauderna, S., et al. (2016). Editing DNA methylation in the mammalian genome. Cell 167, 233–47.e17. doi: 10.1016/j.cell.2016.08.056
Liu, Y., Beyer, A., and Aebersold, R. (2016). On the dependency of cellular protein levels on mRNA abundance. Cell 165, 535–550. doi: 10.1016/j.cell.2016.03.014
Long, H-. Y., Feng, L., Kang, J., Luo, Z-H., Xiao, W-., et al. (2017). Blood DNA methylation pattern is altered in mesial temporal lobe epilepsy. Sci. Rep. 7, 43810. doi: 10.1038/srep43810
Löscher, W. (2020). The holy grail of epilepsy prevention: Preclinical approaches to antiepileptogenic treatments. Neuropharmacology. 167, 107605. doi: 10.1016/j.neuropharm.2019.04.011
Loscher, W., Klitgaard, H., Twyman, R. E., and Schmidt, D. (2013). New avenues for anti-epileptic drug discovery and development. Nat. Rev. Drug Discov. 12, 757–776. doi: 10.1038/nrd4126
Luo, C., Hajkova, P., and Ecker, J. R. (2018). Dynamic DNA methylation: In the right place at the right time. Science 361, 1336–1340. doi: 10.1126/science.aat6806
Lusardi, T. A., Akula, K. K., Coffman, S. Q., Ruskin, D. N., Masino, S. A., Boison, D., et al. (2015). Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology 99, 500–509. doi: 10.1016/j.neuropharm.2015.08.007
Machnes, Z. M., Huang, T. C., Chang, P. K., Gill, R., Reist, N., Dezsi, G., et al. (2013). DNA methylation mediates persistent epileptiform activity in vitro and in vivo. PLoS ONE 8, e76299. doi: 10.1371/journal.pone.0076299
Martin, E. M., and Fry, R. C. (2018). Environmental influences on the epigenome: exposure- associated DNA methylation in human populations. Annu. Rev. Public Health. 39, 309–333. doi: 10.1146/annurev-publhealth-040617-014629
Mathern, G. W., Babb, T. L., Vickrey, B. G., Melendez, M., and Pretorius, J. K. (1995). The clinical-pathogenic mechanisms of hippocampal neuron loss and surgical outcomes in temporal lobe epilepsy. Brain J. Neurol. 118, 105–118. doi: 10.1093/brain/118.1.105
McClung, C. A., and Nestler, E. J. (2008). Neuroplasticity mediated by altered gene expression. Neuropsychopharmacology 33, 3–17. doi: 10.1038/sj.npp.1301544
Mellén, M., Ayata, P., and Heintz, N. (2017). 5-hydroxymethylcytosine accumulation in postmitotic neurons results in functional demethylation of expressed genes. Proc. Nat. Acad. Sci. 114, E7812–E21. doi: 10.1073/pnas.1708044114
Miller-Delaney, S. F., Das, S., Sano, T., Jimenez-Mateos, E. M., Bryan, K., Buckley, P. G., et al. (2012). Differential DNA methylation patterns define status epilepticus and epileptic tolerance. J. Neurosci. 32, 1577–1588. doi: 10.1523/JNEUROSCI.5180-11.2012
Miller-Delaney, S. F. C., Bryan, K., Das, S., McKiernan, R. C., Bray, I. M., Reynolds, J. P., et al. (2015). Differential DNA methylation profiles of coding and non-coding genes define hippocampal sclerosis in human temporal lobe epilepsy. Brain 138, 616–631. doi: 10.1093/brain/awu373
Mohandas, N., Loke, Y. J., Hopkins, S., Mackenzie, L., Bennett, C., Berkovic, S. F., et al. (2019). Evidence for type-specific DNA methylation patterns in epilepsy: a discordant monozygotic twin approach. Epigenomics 11, 951–968. doi: 10.2217/epi-2018-0136
Müller, J., Timmermann, A., Henning, L., Müller, H., Steinhäuser, C., Bedner, P., et al. (2020). Astrocytic GABA accumulation in experimental temporal lobe epilepsy. Front. Neurol. 11, 1762. doi: 10.3389/fneur.2020.614923
Myszczynska, M. A., Ojamies, P. N., Lacoste, A. M. B., Neil, D., Saffari, A., Mead, R., et al. (2020). Applications of machine learning to diagnosis and treatment of neurodegenerative diseases. Nat. Rev. Neurol. 16, 440–456. doi: 10.1038/s41582-020-0377-8
Nelson, T. E., and Gruol, D. L. (2004). The chemokine CXCL10 modulates excitatory activity and intracellular calcium signaling in cultured hippocampal neurons. J. Neuroimmunol. 156, 74–87. doi: 10.1016/j.jneuroim.2004.07.009
Pannasch, U., Vargová, L., Reingruber, J., Ezan, P., Holcman, D., Giaume, C., et al. (2011). Astroglial networks scale synaptic activity and plasticity. Proc. Natl. Acad. Sci. U.S.A. 108, 8467–8472. doi: 10.1073/pnas.1016650108
Patel, D. C., Tewari, B. P., Chaunsali, L., and Sontheimer, H. (2019). Neuron–glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 20, 282–297. doi: 10.1038/s41583-019-0126-4
Perucca, E., Brodie, M. J., Kwan, P., and Tomson, T. (2020). 30 years of second-generation antiseizure medications: impact and future perspectives. Lancet Neurol. 19, 544–556. doi: 10.1016/S1474-4422(20)30035-1
Pitkanen, A., and Engel, J. (2014). Past and present definitions of epileptogenesis and its biomarkers. Neurotherapeutics 11, 231–241. doi: 10.1007/s13311-014-0257-2
Pitkanen, A., and Lukasiuk, K. (2011). Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 10, 173–186. doi: 10.1016/S1474-4422(10)70310-0
Pitkänen, A., Nehlig, A., Brooks-Kayal, A. R., Dudek, F. E., Friedman, D., Galanopoulou, A. S., et al. (2013). Issues related to development of antiepileptogenic therapies. Epilepsia 54, 35–43. doi: 10.1111/epi.12297
Retamal, M. A., Froger, N., Palacios-Prado, N., Ezan, P., Sáez, P. J., Sáez, J. C., et al. (2007). Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 27, 13781–13792. doi: 10.1523/JNEUROSCI.2042-07.2007
Rojas, A., Jiang, J., Ganesh, T., Yang, M. S., Lelutiu, N., Gueorguieva, P., et al. (2014). Cyclooxygenase-2 in epilepsy. Epilepsia 55, 17–25. doi: 10.1111/epi.12461
Ryley Parrish, R., Albertson, A. J., Buckingham, S. C., Hablitz, J. J., Mascia, K. L., Davis Haselden, W., et al. (2013). Status epilepticus triggers early and late alterations in brain-derived neurotrophic factor and NMDA glutamate receptor Grin2b DNA methylation levels in the hippocampus. Neuroscience 248, 602–619. doi: 10.1016/j.neuroscience.2013.06.029
Saha, R. N., Liu, X., and Pahan, K. (2006). Up-regulation of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective role of cytokine. J. Neuroimmune Pharmacol. 1, 212–222. doi: 10.1007/s11481-006-9020-8
Sanosaka, T., Imamura, T., Hamazaki, N., Chai, M., Igarashi, K., Ideta-Otsuka, M., et al. (2017). DNA ethylome analysis identifies transcription factor-based epigenomic signatures of multilineage competence in neural stem/progenitor cells. Cell Rep. 20, 2992–3003. doi: 10.1016/j.celrep.2017.08.086
Santello, M., Bezzi, P., and Volterra, A. (2011). TNFα controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 69, 988–1001. doi: 10.1016/j.neuron.2011.02.003
Schlomann, U., Rathke-Hartlieb, S., Yamamoto, S., Jockusch, H., and Bartsch, J. W. (2000). Tumor necrosis factor α induces a metalloprotease-disintegrin, ADAM8 (CD 156): implications for neuron–glia interactions during neurodegeneration. J. Neurosci. 20, 7964. doi: 10.1523/JNEUROSCI.20-21-07964.2000
Silva, G. M., and Vogel, C. (2016). Quantifying gene expression: the importance of being subtle. Mol. Syst. Biol. 12, 885. doi: 10.15252/msb.20167325
Smith, Z. D., and Meissner, A. D. N. A. (2013). methylation: roles in mammalian development. Nat. Rev. Genet. 14, 204–220. doi: 10.1038/nrg3354
Somineni, H. K., Venkateswaran, S., Kilaru, V., Marigorta, U. M., Mo, A., Okou, D. T., et al. (2019). Blood-derived DNA methylation signatures of crohn's disease and severity of intestinal inflammation. Gastroenterology. 156, 2254–65.e3. doi: 10.1053/j.gastro.2019.01.270
Song, C. X., and He, C. (2013). Potential functional roles of DNA demethylation intermediates. Trends Biochem. Sci. 38, 480–484. doi: 10.1016/j.tibs.2013.07.003
Sui, Y., Stehno-Bittel, L., Li, S., Loganathan, R., Dhillon, N. K., Pinson, D., et al. (2006). CXCL10-induced cell death in neurons: role of calcium dysregulation. Eur. J. Neurosci. 23, 957–964. doi: 10.1111/j.1460-9568.2006.04631.x
Takamiya, A., Takeda, M., Yoshida, A., and Kiyama, H. (2002). Inflammation induces serine protease inhibitor 3 expression in the rat pineal gland. Neuroscience 113, 387–394. doi: 10.1016/S0306-4522(02)00198-7
Tauck, D. L., and Nadler, J. V. (1985). Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J. Neurosci. 5, 1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985
Vezzani, A., French, J., Bartfai, T., and Baram, T. Z. (2011). The role of inflammation in epilepsy. Nat. Rev. Neurol. 7, 31–40. doi: 10.1038/nrneurol.2010.178
Vezzani, A., and Granata, T. (2005). Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia 46, 1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x
Wang, L., Fu, X., Peng, X., Xiao, Z., Li, Z., Chen, G., et al. (2016). DNA methylation profiling reveals correlation of differential methylation patterns with gene expression in human epilepsy. J. Mol. Neurosci. 59, 68–77. doi: 10.1007/s12031-016-0735-6
Wang, X., Zheng, G., and Dong, D. (2015). Coordinated action of histone modification and microRNA regulations in human genome. Gene 570, 277–281. doi: 10.1016/j.gene.2015.06.046
Wang, Y.-C., Peterson, S. E., and Loring, J. F. (2014). Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 24, 143–160. doi: 10.1038/cr.2013.151
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Weltha, L., Reemmer, J., and Boison, D. (2019). The role of adenosine in epilepsy. Brain Res. Bull. 151, 46–54. doi: 10.1016/j.brainresbull.2018.11.008
Widagdo, J., and Anggono, V. (2018). The m6A-epitranscriptomic signature in neurobiology: from neurodevelopment to brain plasticity. J. Neurochem. 147, 137–152. doi: 10.1111/jnc.14481
Williams-Karnesky, R. L., Sandau, U. S., Lusardi, T. A., Lytle, N. K., Farrell, J. M., Pritchard, E. M., et al. (2013). Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J. Clin. Invest. 123, 3552–3563. doi: 10.1172/JCI65636
Wu, Z., Deshpande, T., Henning, L., Bedner, P., Seifert, G., Steinhäuser, C., et al. (2021). Cell death of hippocampal CA1 astrocytes during early epileptogenesis. Epilepsia 62, 1569–1583. doi: 10.1111/epi.16910
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012
Zhang, W., Wang, H., Liu, B., Jiang, M., Gu, Y., Yan, S., et al. (2021). Differential DNA Methylation profiles in patients with temporal lobe epilepsy and hippocampal sclerosis ILAE Type I. J. Mol. Neurosci. 71, 1951–1966. doi: 10.1007/s12031-020-01780-9
Zhu, Q., Wang, L., Zhang, Y., Zhao, F. H., Luo, J., Xiao, Z., et al. (2012). Increased expression of DNA methyltransferase 1 and 3a in human temporal lobe epilepsy. J. Mol. Neurosci. 46, 420–426. doi: 10.1007/s12031-011-9602-7
Keywords: epilepsy, epileptogenesis, DNA methylation, gene expression, therapy, epigenetics, glia, inflammation
Citation: Berger TC, Taubøll E and Heuser K (2022) The potential role of DNA methylation as preventive treatment target of epileptogenesis. Front. Cell. Neurosci. 16:931356. doi: 10.3389/fncel.2022.931356
Received: 28 April 2022; Accepted: 27 June 2022;
Published: 22 July 2022.
Edited by:
Sandra Henriques Vaz, Universidade de Lisboa, PortugalReviewed by:
Marco I. Gonzalez, University of California, Davis, United StatesCopyright © 2022 Berger, Taubøll and Heuser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toni Christoph Berger, dC5jLmJlcmdlckBtZWRpc2luLnVpby5ubw==; Kjell Heuser, ZHIuaGV1c2VyQGdtYWlsLmNvbQ==