 Miren Revuelta
Miren Revuelta Janire Urrutia
Janire Urrutia Alvaro Villarroel
Alvaro Villarroel Oscar Casis
Oscar Casis- 1Department of Physiology, Faculty of Medicine and Nursery, University of the Basque Country (UPV/EHU), Leioa, Spain
- 2Instituto Biofisika, Consejo Superior de Investigaciones Científicas (CSIC)-University of the Basque Country/Euskal Herriko Unibertsitatea (UPV/EHU), Leioa, Spain
- 3Department of Physiology, Faculty of Pharmacy, University of the Basque Country (UPV/EHU), Vitoria-Gasteiz, Spain
Increase of deposits of amyloid β peptides in the extracellular matrix is landmark during Alzheimer’s Disease (AD) due to the imbalance in the production vs. clearance. This accumulation of amyloid β deposits triggers microglial activation. Microglia plays a dual role in AD, a protective role by clearing the deposits of amyloid β peptides increasing the phagocytic response (CD163, IGF-1 or BDNF) and a cytotoxic role, releasing free radicals (ROS or NO) and proinflammatory cytokines (TNF-α, IL-1β) in response to reactive gliosis activated by the amyloid β aggregates. Microglia activation correlated with an increase KV1.3 channels expression, protein levels and current density. Several studies highlight the importance of KV1.3 in the activation of inflammatory response and inhibition of neural progenitor cell proliferation and neuronal differentiation. However, little is known about the pathways of this activation in neural stem cells differentiation and proliferation and the role in amyloid β accumulation. In recent studies using in vitro cells derived from mice models, it has been demonstrated that KV1.3 blockers inhibit microglia-mediated neurotoxicity in culture reducing the expression and production of the pro-inflammatory cytokines IL-1β and TNF-α through the NF-kB and p38MAPK pathway. Overall, we conclude that KV1.3 blockers change the course of AD development, reducing microglial cytotoxic activation and increasing neural stem cell differentiation. However, further investigations are needed to establish the specific pathway and to validate the use of this blocker as therapeutic treatment in Alzheimer patients.
Introduction
Alzheimer’s Disease (AD) is one of the main progressive neurodegenerative disorders and the most common cause of dementia affecting principally the elderly (Bateman et al., 2012). The histopathology is characterized by brain atrophy, deposits of amyloid β (Aβ) peptides in the extracellular matrix, neurofibrillary tangles (mainly tau protein), loss of neurons and synapses and dystrophic neurites (Hansen et al., 2018).
The increased number of Aβ amyloid plaques in the extracellular matrix, due to the imbalance in the production vs. clearance, is believed to be the principal pathogenic mechanism (Selkoe and Hardy, 2016). Familial AD is characterized by excessive production of Aβ, caused by a mutation in amyloid precursor protein (APP) or in the APP processing enzyme. However, Familial AD is extremely rare, while the majority of the AD cases are “sporadic” and occur late in life. Late AD is thought to be a result of genetic and environmental factors, and mainly aging that reduce the brain’s ability to clear Aβ (Mawuenyega et al., 2010; Wildsmith et al., 2013).
Microglial activation was initially thought to be incidental and triggered by the accumulation of amyloid deposits. Interestingly, it has been established recently that many genes found in or near AD risk loci are genes mainly expressed in microglia (Hemonnot et al., 2019). Among these genes, Apoliprotein E (APOE), SP1l, TREM2, or CD33 code for proteins that are expressed principally or exclusively in microglia (Verheijen and Sleegers, 2018).
Microglia, the major inflammatory cells of the brain, play a dual role in AD. On the one hand, they play a protective role by clearing the deposits of Aβ peptides increasing the phagocytic activity (Miners et al., 2011) and on the other hand, they play a cytotoxic role by releasing cytotoxic substances and pro-inflammatory cytokines in response to reactive gliosis activated by the Aβ aggregates (Glass et al., 2010).
Microglial Activation in Alzheimer’s Disease
Neuroinflammation is driven mostly by glial cells such as microglia and astrocytes (Forloni and Balducci, 2018). Microglia, which represent around 10–15% of human brain cells, are immune cells that first respond to nervous system changes (Kwon and Koh, 2020; Liu et al., 2021). Microglia are categorized mainly in two opposite phenotypes depending on their specific markers that define cell type and state; the pro-inflammatory M1 (classical activation) and M2 (alternative activation) phenotypes (Mills et al., 2000; Bi et al., 2021). Depending on the activated phenotype, microglia can produce either cytotoxic or neuroprotective effects. The classical activation is related to pro-inflammatory cytokine production, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and reactive oxygen species (ROS) or nitric oxide (NO) production. Meanwhile, the alternative activation promotes anti-inflammatory response with increased IL-4, IL-10, CD36 and phagocytic response expressing CD163, insulin-like growth factor 1 (IGF-1) and brain derived neurotrophic factor (BDNF) (Le et al., 2016; Chen et al., 2021; Figure 1). The microglial polarization has not been supported by single-cell RNA-seq, while this transcriptomic analysis have been used to distinguish between diseased associated microglia (DAM) or activated response microglia (ARM) (Keren-Shaul et al., 2017; Sala Frigerio et al., 2019). Transcriptome data show that during neurodegenerative diseases both phenotypes, the neurotoxic and the neuroprotective, are expressed (Sarlus and Heneka, 2017; Figure 1).
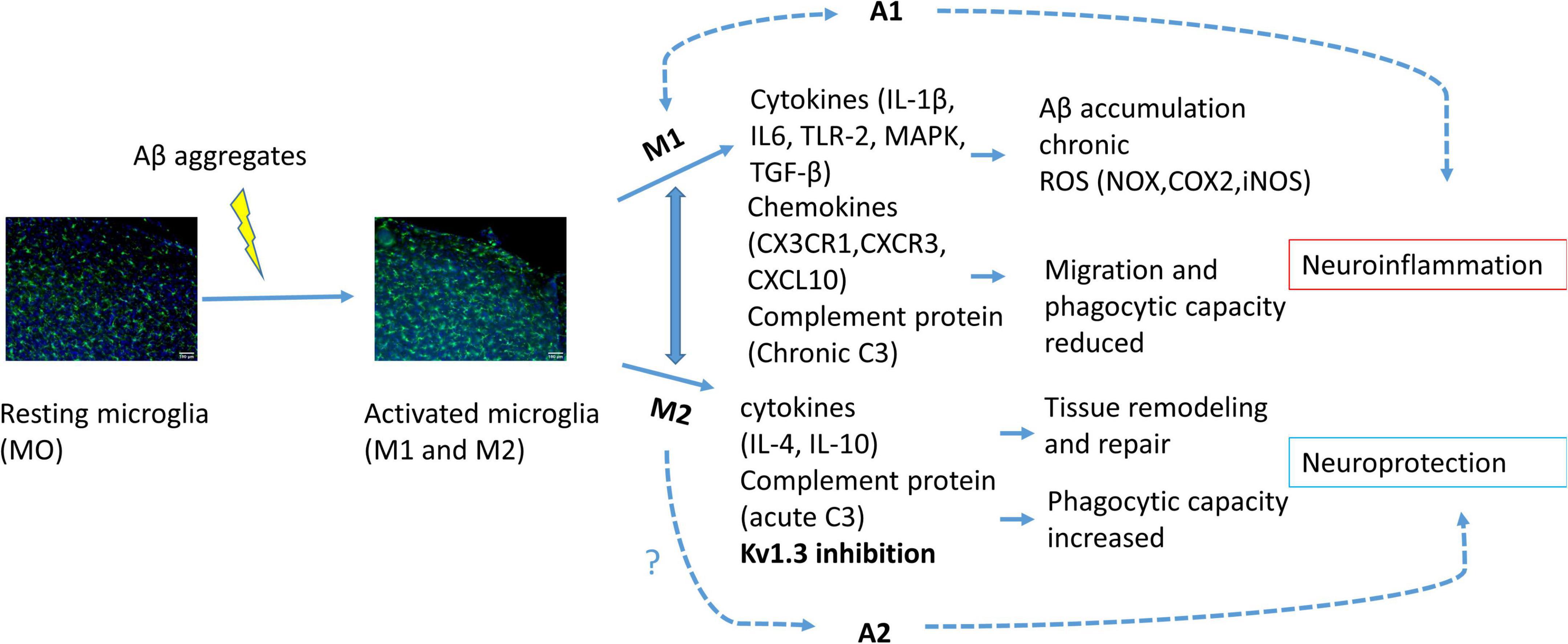
Figure 1. Schematic illustration of microglial activation due to Aβ accumulation. Activated microglia polarized into M1 or M2 phenotype. Activation of M1 phenotype induces pro-inflammatory cytokine, chemokine and complement protein release provoking citotoxicity and consequently astrocyte A1 activation, neuroinflammation and neuronal cell death. M2 phenotype activation induces anti-inflammatory response and consequently A2 activation and neuroprotection. KV1.3 inhibition has been related to M2 phenotype polarization.
During AD, microglial activation is associated with Aβ deposits in human and mice brains (Kamphuis et al., 2012; Olmos-Alonso et al., 2016). Indeed, activated microglia in AD mouse models express increased inflammatory markers CD36, CD14, CD11c, major histocompatibility complex-II (MHC-II), and inducible nitric oxide synthase (iNOS), as well as M1 phenotype markers.
The inflammatory response is typically composed of three main stages. First, toll-like receptor (TLR)-mediated NF-κB formation. This leads to an increase of mainly members of the nod-like receptor (NLR) that assemble the inflammasome that third, activates caspase-1, which cleaves the precursor protein and release IL-1β (Yin et al., 2016).
While anti-inflammatory cytokines may have a deleterious role in AD (Guillot-Sestier et al., 2015), TLR activation and IL-1β secretion may also have protective effects (Shaftel et al., 2007; Richard et al., 2008).
Cytokines
Post-mortem immunohistochemical studies of brain tissues show that among all cytokines that are highly expressed during AD, IL-1β, IL-6, and TNF-α are the most abundant (Bernhardi et al., 2015). Increased levels of these cytokines may play different roles in the context of Aβ deposition. Remarkably, IL-1β, a key cytokine of innate immune response, enhance Aβ and tau pathology (Lee et al., 2013), while increased levels of TNF-α may facilitate the Aβ clearance (Montgomery et al., 2011; Sarlus and Heneka, 2017).
Particularly, IL-1β activates astrocytes that may contribute to plaque formation due to the release of astrocyte-derived proteins, such as IL-6, APOE and some complement proteins. Furthermore, IL-1β induces neurite growth promoting the cytokine S100β. S100β induces the increase of Aβ precursor protein, so it has been linked with the initial deposition of Aβ (Griffin and Mrak, 2002). The abnormal accumulation of Aβ plaques also triggers the excessive release of other anti-inflammatory cytokines, such as IL-4, IL-10, IL-13 that accelerates tissue remodeling, repair and angiogenesis and inhibits the production of other pro-inflammatory cytokines (Stamouli and Politis, 2016; Kaur et al., 2019).
During neuroinflammation in AD there is also an activation of TLR-2. This activation triggers the nuclear translocation of NF-κB and provokes Aβ-induced inflammation and chronicity of AD (Zhao et al., 2013).
Some protein kinases such as mitogen-activated protein kinase (MAPK), cell division cycle 2 kinase (CDC2) and Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathways have been also identified in AD progression (Rather et al., 2021). Activated MAPK and NF-κB increase the production of pro-inflammatory cytokines promoting APP processing, blood-brain barrier (BBB) disintegration and aggravates tau protein phosphorylation. Moreover, the formation of neurofibrillary tangles due to p38-MAPK activation leads to neuronal degeneration and finally neuronal death (Jeong et al., 2014; Rather et al., 2021).
Chemokines
Chemokines, a large family of small (8–14 kDa) basic proteins, are also important inflammatory mediators overexpressed during inflammatory events in the CNS. During AD, several chemokines have been associated with microglia activation due to Aβ depositions. For example, CCR2 (C-C motif chemokine receptor type 2) is a chemokine expressed on microglia that accumulates mononuclear phagocytes in inflammatory sites. Studies show that lack of CCR2 decreases microglial accumulation and results in an increased Aβ deposition, indicating that CCR2 may play a protective role in AD promoting Aβ clearance (El Khoury et al., 2007).
Moreover the lack of CCR2 stimulates the expression of TGF-β and CX3CR1 (CX3C chemokine receptor 1) in microglia (Guedes et al., 2018). Interestingly, several murine AD mice models revealed that genetic elimination of CX3CR1, a chemokine receptor predominantly found in microglia, resulted in a decrease of amyloid plaques due to the increase of phagocytic capacity in the activated microglia (Guedes et al., 2018).
Complement Proteins
The complement system, composed of about 30 proteins, plays an important role in host defense and in the inflammatory regulation (Crehan et al., 2012). The accumulation of Aβ plaques and increased neurofibrillary tangles activate the classical complement pathway in microglia within the collagen-like domain of C1q (Shen et al., 2001). Nevertheless, complement’s role needs to be further studied (Rasmussen et al., 2018), as C3, a central component in the activation of the complement system, provokes different responses to microglial phagocytosis.
The complement-dependent mechanism can also mediate synapse loss by swallowing this synapse. During AD, this synapse loss involves a pathway in which the complement clears pathogens and apoptotic cells after binding of complement protein C1q. Thus, blocking microglial activation or the activation of complement mechanism may have beneficial effects in AD reducing synapse and neuronal loss (Hansen et al., 2018).
Free Radicals
Some authors described that the abnormal accumulation of Aβ and the deposition of neurofibrillary tangles extend oxidative damage, impair Ca2+ homeostasis and produce mitochondrial dysfunction during AD (Bello-Medina et al., 2021). Nevertheless, other studies claim that during AD the increased ROS production and altered Ca2+ homeostasis precede Aβ accumulation and is due to mitochondrial dysfunction (Yoo et al., 2020). Anyway, increased microglial ROS production contributes to oxidative stress resulting in neuronal dysfunction and neurotoxicity. Moreover, microglia respond to damage-associated molecular patterns (DAMPs) released from damaged cells, activating NADPH oxidase (NOX). In fact, the activation of the phagocyte NOX2 in microglia seems to play an important role in neuroinflammation and in neuronal death (Qin et al., 2013; Jiang et al., 2015).
Microglia produce pattern recognition receptors (PRR) also in response to DAMPs stimuli, such as Complement receptor 3 (CR3) or TLR. These PRR mediate activation of pro-inflammatory signaling traducers NLRP3 inflammasome, NF-κB and MAPKs (Simpson and Oliver, 2020).
Microglial Ion Channels and Alzheimer’s Disease
In healthy brains, microglia regulate the correct development and function of synapses and synaptic plasticity. Microglia-synapse disruption may contribute to synapse loss, dysfunction and, consequently, disease (Hong et al., 2016). There are many studies concerning the effect of changes in cytokines, chemokines or ROS production in microglial activation. However, little is known about the effects of changes in the intracellular ionic homeostasis and microglial activation (Izquierdo et al., 2019). Ion channels are involved in many microglial functions, such as cytokine production, migration, production or proliferation, among others.
Microglia membrane express different ion channels, such as Ca2+-, K+-, Na+-, H+- and Cl–-channels, in order to face all physiological functions. For instance, Ca2+-channels are important for intracellular Ca2+ homeostasis in microglia. Store-operated Ca2+-channels, voltage-gated Ca2+-channels and transient receptor potential channels control Ca2+ signaling for microglial activation (Luo et al., 2021). Inward rectifier channels and voltage-gated K+-channels as well as Cl–-channels (volume regulated Cl–-channels and chloride intracellular channels), are not only necessary for cell hyperpolarization, but for cell activation and proliferation. This is because they supply the driving force that allow an increase intracellular Ca2+ concentration via Ca2+-channels (Nguyen et al., 2017).
Voltage-gated and acid-sensing Na+-channels are also relevant in these non-excitable cells to regulate cell migration, phagocytosis, and secretion of cytokines (Pappalardo et al., 2016). Finally, voltage-gated H+-channels are also important to regulate cells’ pH.
Glial cells express the voltage-gated K+-channels KV1.3 and KV1.5. These channels activity changes in microglial activation by modifying their relative expression. In fact, one characteristic of activated inflammatory cells is an increased expression and function of the KV1.3 channels (Pérez-Verdaguer et al., 2016). In quiescent cells KV1.5 regulates the proliferation rate (Pannasch et al., 2006; Gubiè et al., 2021) while KV1.3 is dominant in activated microglial cells (Gubiè et al., 2021). Besides, KV1.5 seems to be essential for NO production (Pannasch et al., 2006), but causes cell cycle arrest. On the other hand, KV1.3 participates in microglial proliferation and migration, as well as in the cytokine release (Charolidi et al., 2015; Stebbing et al., 2015). LPS-mediated microglial activation induces cytokine release, but decreases proliferation (Pannasch et al., 2006). In response to Aβ accumulation KV1.3, KV1.5 and calcium-activated K+-channels (KCa3.1, KCa2.3, or BK channels) increase voltage-dependent Ca2+ entry provoking a disruption in Ca2+ homeostasis and consequently neurodegeneration (Dolga et al., 2012; Kumar et al., 2016; Huang et al., 2021). Among all these channels, the most studied is KV1.3.
KV1.3 in Microglia During Alzheimer’s Disease
Within all type of microglial channels, KV1.3 has a fundamental role in the activation of these cells, since it contributes to maintaining the negative membrane potential. This channel is a Shaker-type voltage-gated K+-channel with six transmembrane domains (Wulff and Zhorov, 2008) and it is widely distributed throughout the whole body, being highly expressed in both nervous and immune systems. First described in T cells (DeCoursey et al., 1984), it has been related with autoimmune diseases mostly as it plays an important role in immune cell activation by modulating Ca2+ signaling (Wulff et al., 2007; Feske et al., 2015).
Microglia activation provokes an overexpression of KV1.3 mRNA and protein levels, which lead to increased current densities (Nguyen et al., 2017). KV1.3 is required for microglial pro-inflammatory activation and neurotoxicity (Figure 2A) and is highly expressed by microglia in human AD brains and AD mice models (Rangaraju et al., 2015).
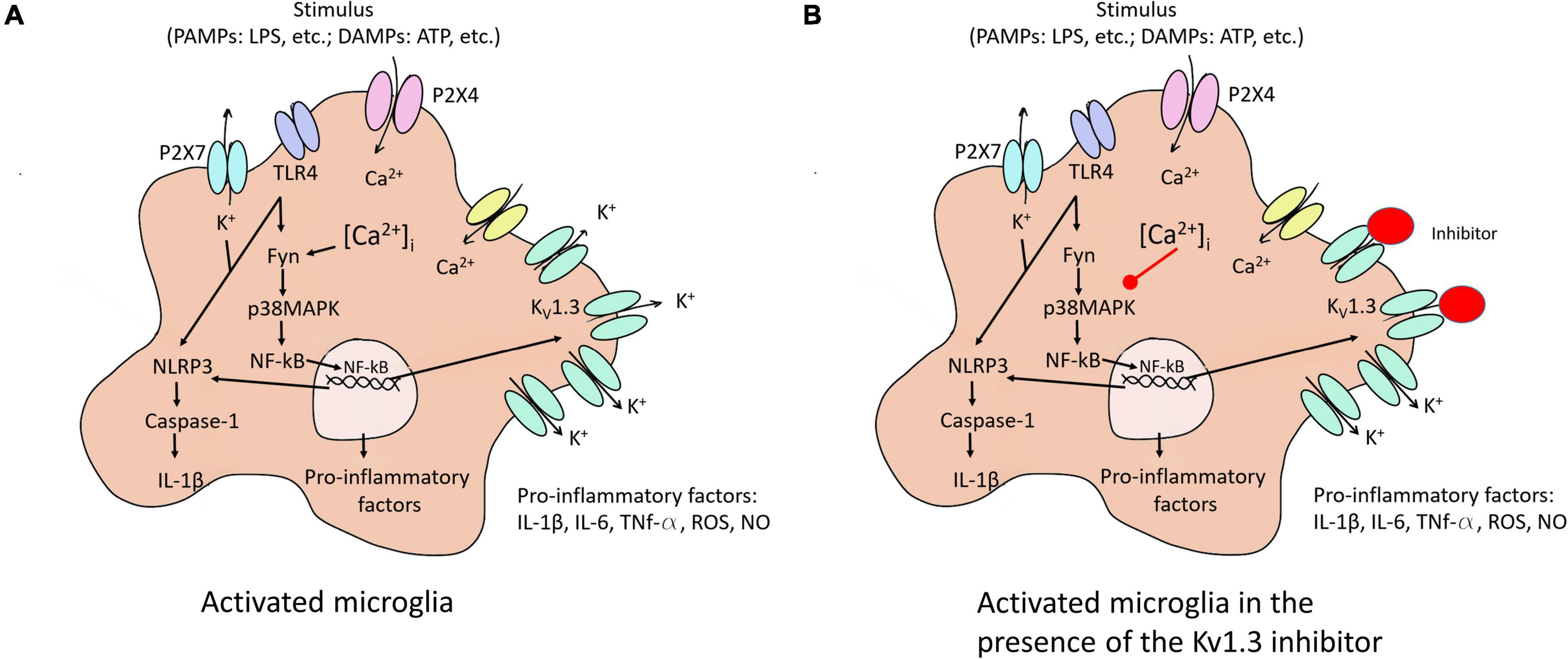
Figure 2. Microglia activation to M1 and the KV1.3 blockade effect on this activation. (A) Different stimuli activate microglia to pro-inflammatory state (M1). The activation of the NF-κB pathway induced an increase of KV1.3 protein in the membrane, among others effects. (B) KV1.3 inhibitor provokes a smaller Ca2+ entry reducing secretion of pro-inflammatory factors due to the decrease in the activation of NF-κB pathway. P2 × 7 and P2 × 4, purinergic receptors; TLR4, toll like receptor 4; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; PAMPs, exposure to pathogen-associated molecular patterns; DAMPs, endogenous damage-associated molecular patterns; ROS, reactive oxygen species; Fyn, non-receptor tyrosine-protein kinase; IL, interleukin; TNF, tumor necrosis factor; iNOS, nitric oxide synthase, NO, nitric oxide.
Overexpression of KV1.3 channels in a microglial cell line increases the expression and secretion of different interleukins (Sarkar et al., 2020). Mice exposed to experimental stroke (Chen et al., 2016) as well as treatment with lipopolysaccharide (LPS) or a combination of LPS and IFN-γ in the microglia of mouse models (Nguyen et al., 2017; Di Lucente et al., 2018) exhibit increased KV1.3 current in affected areas. LPS-mediated microglial activation induces cytokine release, but decreases proliferation (Pannasch et al., 2006). In addition, stimulus such LPS or ATP activates the NF-κB pathway via different membrane receptors [purinergic receptor (P2 × 4 and P2 × 7) and TLR4] leading to an overexpression of KV1.3 and pro-inflammatory factors provoking neuroinflammation (Figure 2A). Concerning this, Di Lucente et al. (2018) demonstrated that blockade KV1.3 blockade after LPS treatment induces M2 microglia polarization reducing pro-inflammatory markers.
ATP and the increased K+ efflux augment Ca2+ entry, which raises the inflammatory state of the cell through the activation of NF-κB pathway via p38MAPK phosphorylation. At the same time, NF-κB interacts with their binding sites in the KV1.3 promotor. All these changes cause NLRP3 inflammasome activation conducting IL-1β secretion (Figure 2A). Besides, inhibition of NF-κB or up-regulation of KV1.3 provoked by αSynAgg stimulation indicates that both p38MAPK and NF-κB pathways intervene in the transcriptional regulation of the channel (Sarkar et al., 2020).
Effect of Microglia on Neural Stem Cell Differentiation in Alzheimer’s Disease
There is a controversy about human neurogenesis. Some authors conclude that hippocampal neurogenesis is extremely rare in the adult brain, as they did not detect new neurons in the dental gyrus (Sorrells et al., 2018). However, other authors observed immature neurons, neuroblast and neural progenitor cells in aged human hippocampus (Boldrini et al., 2018; Tobin et al., 2019).
In the adult mammalian brain, neural stem cells (NSCs) are localized in two major neurological niches, the subgranular zone of the hippocampus (SGZ) and the subventricular zone of the lateral ventricle (SVZ). These cells retain the ability to proliferate and differentiate into neurons and glial cells (Moreno-Cugnon et al., 2019).
Some studies report that microglia in the hippocampus are more active than in other brain regions, playing an important role in refining neuronal circuits (Rao et al., 2022). Furthermore, microglia release several cytokines that promote microglial migration, neuroblast generation and neurogenesis and is considered a crucial component for determine NSC fate (Shigemoto-Mogami et al., 2014; Geribaldi-Doldán et al., 2021).
The hippocampus is one of the most affected brain regions in AD with altered dentate granule cells. There have been several studies using NSC/induced pluripotent stem cells (iPSCs) derived from Alzheimer patients with the objective of promoting neurogenesis and ameliorating the progression of the disease (Wu et al., 2021). In one of these studies, for example, they demonstrate that the release of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IGF-1 by microglia enhance the dopaminergic differentiation of neural stem cells and promote neurogenesis (Boyd et al., 2021; Schmidt et al., 2021).
As mentioned, impaired CX3CR1 has been described in AD (Guedes et al., 2018). In the hippocampus, this impairment has been linked to adult hippocampal neurogenesis disruption, with spatial and fear-memory and motor learning loss due mainly to the increase of IL-1β by microglial activation (Parkitny and Maletic-Savatic, 2021). Remarkably, the importance of IL-1β in adult hippocampal neurogenesis was recognized over a decade ago when it was associated with anti-proliferative and anti-neurogenic effects (Crampton et al., 2012). Moreover, PRR seem to be implicated in the modulation of adult neurogenesis as they are expressed also in neural progenitor cells (NPCs), providing communication pathways from apoptotic or injured cells (Parkitny and Maletic-Savatic, 2021).
Role of KV1.3 in Neural Stem Cell Differentiation
Recent studies support the hypothesis that cell proliferation and division depend on K+-channels activity (Gallo et al., 1996; Jäger and Grissmer, 2004). KV1.3 channels control action potential firing of hippocampal and OB neurons, representing around 60–80% of all K+-channels in these areas (Martínez-Mármol et al., 2016). This channel has also been found in NPC. Moreover, Liebau et al., 2006 demonstrated that blockade of KV1.3 by Psora-4 [5-(4-Phenylbutoxy) psoralen] increases the number of NPC in vitro. In 2010, Wang et al. (2010) also exposed the importance of KV1.3 in the activation of inflammatory response and inhibition of NPC proliferation and neuronal differentiation. However, little is known about the pathways of this activation in NSC differentiation and proliferation.
KV1.3 as a Potential Therapeutic Target in Alzheimer’s Disease
To date, there is no treatment to cure or prevent AD. Current treatments are only useful in slowing down the progression of the disease and in managing some behavioral and cognitive symptoms of AD patients. Because of the complex pathophysiology, sometimes the treatment needs to be a combination of therapies. Currently, only six treatments are approved in the US. These include three cholinesterase inhibitors (donepezil, galantamine, and rivastigmine), one N-methyl-D-aspartate receptor antagonist (memantine) (Cummings et al., 2019), a fixed-dose combination with donepezil and memantine and finally, the recently approved aducanumab, a human monoclonal antibody that targets, and reduces Aβ accumulations in the brain.
As mentioned, during AD there is a release of cytotoxic substances and pro-inflammatory cytokines by the M1 activation provoking neuronal damage and aggravating AD pathology (Heneka et al., 2015). In this M1 activated state, KV1.3 channels are upregulated. Little is known about the mechanism in which these channels are activated and the consequences in AD. Recent studies have analyzed the effect of KV1.3 blockers on microglial profiles in AD models and confirm that pro-inflammatory and neurotoxic microglia functions are reduced with different KV1.3 inhibitors (Figure 2B). KV1.3 blockade decreases cerebral amyloid load, enhances hippocampal neuronal plasticity, and improves behavioral deficits by a reduction of microglia activation and inflammatory cytokines levels in transgenic AD mouse models (Chen et al., 2018; Maezawa et al., 2018; Ma et al., 2020). Accordingly, KV1.3 blockers inhibit microglia-mediated neurotoxicity in culture (Fordyce et al., 2005) and protect mice from microglia-mediated radiation-induced brain injury in vivo (Peng et al., 2014).
Classification of KV1.3 channel blockers depends on their selectivity and blockade potency. The most effective inhibitors are the natural peptides such as the sea anemone Stichodactyla helianthus toxin ShK and scorpion toxins HsTx, OSK1 and Vm24. These molecules present high affinity for different channels. An analog of Shk, the ShK-223, diminished the activity of the pro-inflammatory microglia and elevated Aβ clearance in AD models (Rangaraju et al., 2018; Ramesha et al., 2021). HsTX1 [R14A] mutant, is a potent, selective and highly stable peptide inhibitor. It has been shown that this inhibitor reduces the release of TNF-α and IL-6 by LPS-mediated BV-2 microglia activation improving neuroinflammation (Nicolazzo et al., 2022).
Beside these peptides, there are different synthetic organic small-molecules inhibitors such as, PAP−1 [5-(4-phenoxybutoxy) psoralen], Psora−4, dihydroquinoline, benzamides, clofazimine, furoquinoline, acridinone, furochromene−7−thione, diphenoxylate, and several analogs are used. All these inhibitors differ in the potency to block KV1.3 channel and the selectivity for the target (Gubiè et al., 2021).
In rodents, PAP-1 and ShK-223 reduce the expression and production of several cytokines (IL-1β, IL-4, IL-5, IL-10, IL-12, IFNγ and TNF-α) (Nguyen et al., 2017; Zayas-Arrabal et al., 2021) and decreased Aβ plaque burden in the 5xFAD mice brain. Furthermore, these molecules increased Aβ phagocytosis by microglia and not blood derived monocytes due to the KV1.3 channel blockade (Maezawa et al., 2018; Ramesha et al., 2021). The pharmacological blockade of the channel in AD mice model promotes synaptogenesis and polarizes microglial phenotype toward M2 (Ramesha et al., 2021).
However, the use of pharmacological KV1.3 blockers may have several adverse effects. ShK and HsTX1 blockers, for instance, have difficulties to penetrating the intestinal mucosa so they can’t be taken orally and they neither can cross the BBB. Therefore, there is a need for cell penetrating peptides to assist the passage of the drug across the cell membrane (Wang et al., 2020). Other problem relates to the selective of KV1.3 blockade. It is important to mention that KV1.3 is also expressed in mitochondria. This channel controls cell-proliferation and has an important role in cellular respiration (Styles et al., 2021). Thus, the use of some small-molecule KV1.3 blockers as therapy could also block mitochondrial channels, inducing apoptosis in cancer cells (Teisseyre et al., 2019). According to recent studies, the most potent and selective small-molecule KV1.3 inhibitor available is PAP-1 (Peruzzo et al., 2020).
However, PAP-1, PSORA-4 and some derivatives produce apoptosis in cancer cell lines. These blockers inhibit KV1.3 channels both at the plasma and mitochondrial membranes, causing an increased ROS production and, finally, apoptosis. The activation of the apoptotic pathway by these inhibitors is mainly due to cancer cells’ massive ROS release (Checchetto et al., 2019). Therefore, the therapeutic use of these inhibitors in AD may induce microglial apoptosis by further increasing the cell’s basal level of ROS. Additional studies need to be performed in order to determine the cytotoxicity of this KV1.3 channel blocker in AD.
In summary, KV1.3 plays important roles in regulating membrane potential, preventing depolarization and controlling Ca2+ signaling events reducing microglia activation. However, further investigation is needed to achieve a deeper understanding of the role of KV1.3 in the microglial immune response and to identify specific pathways for enhancement of Aβ plaque formation or NPC differentiation. Moreover, the potential pharmacological use of drugs targeting KV1.3 channels requires further characterization.
Author Contributions
MR and JU wrote the article. AV and OC reviewed. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the MICINN (PID2020-118814RB-I00), the Government of the Autonomous Community of the Basque Country (IT1165-19 and KK-2020/00110), and the Spanish Ministry of Science and Innovation (RTI2018-097839-B-100 to AV).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bateman, R. J., Xiong, C., Benzinger, T. L. S., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Bello-Medina, P. C., González-Franco, D. A., Vargas-Rodríguez, I., and Díaz-Cintra, S. (2021). Oxidative stress, the immune response, synaptic plasticity, and cognition in transgenic models of Alzheimer disease. Neurología doi: 10.1016/j.nrleng.2019.06.008 [Epub Online ahead of print]
Bernhardi, Rv, Cornejo, F., Parada, G. E., and Eugenín, J. (2015). Role of TGFβ signaling in the pathogenesis of Alzheimer’s disease. Front. Cell. Neurosci. 9:426. doi: 10.3389/fncel.2015.00426
Bi, R., Fang, Z., You, M., He, Q., and Hu, B. (2021). Microglia Phenotype and Intracerebral Hemorrhage: A Balance of Yin and Yang. Front. Cell. Neurosci. 15:765205. doi: 10.3389/fncel.2021.765205
Boldrini, M., Fulmore, C. A., Tartt, A. N., Simeon, L. R., Pavlova, I., Poposka, V., et al. (2018). Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell 22, 589–599.e5. doi: S1934-5909(18)30121-8
Boyd, A., Byrne, S., Middleton, R. J., Banati, R. B., and Liu, G. (2021). Control of Neuroinflammation through Radiation-Induced Microglial Changes. Cells 10:2381. doi: 10.3390/cells10092381
Charolidi, N., Schilling, T., and Eder, C. (2015). Microglial Kv1.3 Channels and P2Y12 Receptors Differentially Regulate Cytokine and Chemokine Release from Brain Slices of Young Adult and Aged Mice. PLoS One 10:e0128463. doi: 10.1371/journal.pone.0128463
Checchetto V., Prosdocimi E., and Leanza L. (2019). Mitochondrial Kv1.3: a New Target in Cancer Biology? Cell Physiol. Biochem. 53, 52–62 doi: 10.33594/000000195
Chen, M., Lai, X., Wang, X., Ying, J., Zhang, L., Zhou, B., et al. (2021). Long Non-coding RNAs and Circular RNAs: Insights Into Microglia and Astrocyte Mediated Neurological Diseases. Front. Mol. Neurosci. 14:745066. doi: 10.3389/fnmol.2021.745066
Chen, Y., Nguyen, H. M., Maezawa, I., Grössinger, E. M., Garing, A. L., Köhler, R., et al. (2016). The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J. Cereb. Blood Flow Metabol. 36, 2146–2161. doi: 10.1177/0271678X15611434
Chen, Y., Nguyen, H. M., Maezawa, I., Jin, L., and Wulff, H. (2018). Inhibition of the potassium channel Kv1.3 reduces infarction and inflammation in ischemic stroke. Ann. Clin. Translat. Neurol. 5, 147–161. doi: 10.1002/acn3.513
Crampton, S. J., Collins, L. M., Toulouse, A., Nolan, Y. M., and O’Keeffe, G. W. (2012). Exposure of foetal neural progenitor cells to IL-1β impairs their proliferation and alters their differentiation - a role for maternal inflammation? J. Neurochem. 120, 964–973. doi: 10.1111/j.1471-4159.2011.07634.x
Crehan, H., Holton, P., Wray, S., Pocock, J., Guerreiro, R., and Hardy, J. (2012). Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology 217, 244–250. doi: 10.1016/j.imbio.2011.07.017
Cummings, J. L., Tong, G., and Ballard, C. (2019). Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimer’s Dis. 67, 779–794. doi: 10.3233/JAD-180766
DeCoursey, T. E., Chandy, K. G., Gupta, S., and Cahalan, M. D. (1984). Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature 307, 465–468. doi: 10.1038/307465a0
Di Lucente, J., Nguyen, H. M., Wulff, H., Jin, L. W., and Maezawa, I. (2018). The voltage-gated potassium channel Kv1.3 is required for microglial pro-inflammatory activation in vivo. Glia 66, 1881–1895. doi: 10.1002/glia.23457
Dolga, A. M., Letsche, T., Gold, M., Doti, N., Bacher, M., Chiamvimonvat, N., et al. (2012). Activation of KCNN3/SK3/K(Ca)2.3 channels attenuates enhanced calcium influx and inflammatory cytokine production in activated microglia. Glia 60, 2050–2064. doi: 10.1002/glia.22419
El Khoury, J., Toft, M., Hickman, S. E., Means, T. K., Terada, K., Geula, C., et al. (2007). Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 13, 432–438. doi: 10.1038/nm1555
Feske, S., Wulff, H., and Skolnik, E. Y. (2015). Ion Channels in Innate and Adaptive Immunity. Annu. Rev. Immunol. 33, 291–353. doi: 10.1146/annurev-immunol-032414-112212
Fordyce, C. B., Jagasia, R., Zhu, X., and Schlichter, L. C. (2005). Microglia Kv1.3 Channels Contribute to Their Ability to Kill Neurons. J. Neurosci. 25, 7139–7149. doi: 10.1523/JNEUROSCI.1251-05.2005
Forloni, G., and Balducci, C. (2018). Alzheimer’s Disease, Oligomers, and Inflammation. J. Alzheimer’s Dis. 62, 1261–1276. doi: 10.3233/JAD-170819
Gallo, V., Zhou, J., McBain, C., Wright, P., Knutson, P., and Armstrong, R. (1996). Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. J. Neurosci. 16, 2659–2670. doi: 10.1523/JNEUROSCI.16-08-02659.1996
Geribaldi-Doldán, N., Fernandez-Ponce, C., Quiroz, R. N., Sanchez-Gomar, I., Escorcia, L. G., Velasquez, E. P., et al. (2021). The Role of Microglia in Glioblastoma. Front. Oncol. 10:603495. doi: 10.3389/fonc.2020.603495
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms Underlying Inflammation in Neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Griffin, W. S. T., and Mrak, R. E. (2002). Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer’s disease. J. Leukoc Biol. 72, 233–238.
Gubiè, S., Hendrickx, L. A., Toplak, Z., Sterle, M., Peigneur, S., Tomasic, T., et al. (2021). Discovery of KV 1.3 ion channel inhibitors: medicinal chemistry approaches and challenges. Med. Res. Rev. 41, 2423–2473. doi: 10.1002/med.21800
Guedes, J. R., Lao, T., Cardoso, A. L., and Khoury, J. E. (2018). Roles of Microglial and Monocyte Chemokines and Their Receptors in Regulating Alzheimer’s Disease-Associated Amyloid-β and Tau Pathologies. Front. Neurol. 9:549. doi: 10.3389/fneur.2018.00549
Guillot-Sestier, M. V., Doty, K. R., and Town, T. (2015). Innate Immunity Fights Alzheimer’s Disease. Trends Neurosci. 38, 674–681. doi: 10.1016/j.tins.2015.08.008
Hansen, D. V., Hanson, J. E., and Sheng, M. (2018). Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459–472. doi: 10.1083/jcb.201709069
Hemonnot, A., Hua, J., Ulmann, L., and Hirbec, H. (2019). Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front. Aging Neurosci. 11:233. doi: 10.3389/fnagi.2019.00233
Heneka, M. T., Carson, M. J., Khoury, J. E., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Hong, S., Dissing-Olesen, L., and Stevens, B. (2016). New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 36, 128–134. doi: 10.1016/j.conb.2015.12.004
Huang, S., Chen, T., Suo, Q., Shi, R., Khan, H., Ma, Y., et al. (2021). BK Channel-Mediated Microglial Phagocytosis Alleviates Neurological Deficit After Ischemic Stroke. Front. Cell Neurosci. 15:683769. doi: 10.3389/fncel.2021.683769
Izquierdo, P., Attwell, D., and Madry, C. (2019). Ion Channels and Receptors as Determinants of Microglial Function. Trends Neurosci. 42, 278–292. doi: 10.1016/j.tins.2018.12.007
Jäger, H., and Grissmer, S. (2004). Characterization of the outer pore region of the apamin-sensitive Ca2+-activated K+ channel rSK2. Toxicon 43, 951–960. doi: 10.1016/j.toxicon.2004.03.025
Jeong, S., Lei, B., Wang, H., Dawson, H. N., and James, M. L. (2014). Intravenous immunoglobulin G improves neurobehavioral and histological outcomes after traumatic brain injury in mice. J. Neuroimmunol. 276, 112–118. doi: 10.1016/j.jneuroim.2014.08.626
Jiang, L., Chen, S., Chu, C., Wang, S., Oyarzabal, E., Wilson, B., et al. (2015). A novel role of microglial NADPH oxidase in mediating extra-synaptic function of norepinephrine in regulating brain immune homeostasis. Glia 63, 1057–1072. doi: 10.1002/glia.22801
Kamphuis, W., Mamber, C., Moeton, M., Kooijman, L., Sluijs, J. A., Jansen, A. H. P., et al. (2012). GFAP Isoforms in Adult Mouse Brain with a Focus on Neurogenic Astrocytes and Reactive Astrogliosis in Mouse Models of Alzheimer Disease. PLoS One 7:e42823. doi: 10.1371/journal.pone.0042823
Kaur, D., Sharma, V., and Deshmukh, R. (2019). Activation of microglia and astrocytes: a roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 27, 663–677. doi: 10.1007/s10787-019-00580-x
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. doi: 10.1016/j.cell.2017.05.018
Kumar, P., Kumar, D., Jha, S. K., Jha, N. K., and Ambasta, R. K. (2016). Ion Channels in Neurological Disorders. Adv. Protein Chem. Struct. Biol. 103, 97–136. doi: 10.1016/bs.apcsb.2015.10.006
Kwon, H. S., and Koh, S. (2020). Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Translat. Neurodegen. 9:42. doi: 10.1186/s40035-020-00221-2
Le, W., Wu, J., and Tang, Y. (2016). Protective Microglia and Their Regulation in Parkinson’s Disease. Front. Mol. Neurosci. 9:89. doi: 10.3389/fnmol.2016.00089
Lee, D. C., Rizer, J., Hunt, J. B., Selenica, M.-B., Gordon, M. N., and Morgan, D. (2013). Review: experimental manipulations of microglia in mouse models of Alzheimer’s pathology: activation reduces amyloid but hastens tau pathology. Neuropathol. Appl. Neurobiol. 39, 69–85. doi: 10.1111/nan.12002
Liebau, S., Pröpper, C., Böckers, T., Lehmann-Horn, F., Storch, A., Grissmer, S., et al. (2006). Selective blockage of K v 1.3 and K v 3.1 channels increases neural progenitor cell proliferation. J. Neurochem. 99, 426–437. doi: 10.1111/j.1471-4159.2006.03967.x
Liu, J., Liu, L., Wang, X., Jiang, R., Bai, Q., and Wang, G. (2021). Microglia: A Double-Edged Sword in Intracerebral Hemorrhage From Basic Mechanisms to Clinical Research. Front. Immunol. 12:675660. doi: 10.3389/fimmu.2021.675660
Luo, L., Song, S., Ezenwukwa, C. C., Jalali, S., Sun, B., and Sun, D. (2021). Ion channels and transporters in microglial function in physiology and brain diseases. Neurochem. Int. 142:104925. doi: 10.1016/j.neuint.2020.104925
Ma, D., Zhang, N., Zhang, Y., and Chen, H. (2020). Kv1.3 channel blockade alleviates cerebral ischemia/reperfusion injury by reshaping M1/M2 phenotypes and compromising the activation of NLRP3 inflammasome in microglia. Exp. Neurol. 332:113399. doi: 10.1016/j.expneurol.2020.113399
Maezawa, I., Nguyen, H. M., Lucente, J. D., Jenkins, D. P., Singh, V., Hilt, S., et al. (2018). Kv1.3 inhibition as a potential microglia-targeted therapy for Alzheimer’s disease: preclinical proof of concept. Brain 141, 596–612. doi: 10.1093/brain/awx346
Martínez-Mármol, R., Comes, N., Styrczewska, K., Pérez-Verdaguer, M., Vicente, R., Pujadas, L., et al. (2016). Unconventional EGF-induced ERK1/2-mediated Kv1.3 endocytosis. Cell. Mol. Life Sci. 73, 1515–1528. doi: 10.1007/s00018-015-2082-0
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased Clearance of CNS β-Amyloid in Alzheimer’s Disease. Science 330:1774. doi: 10.1126/science.1197623
Mills, C. D., Kincaid, K., Alt, J. M., Heilman, M. J., and Hill, A. M. M. - (2000). 1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 164, 6166–6173. doi: 10.4049/jimmunol.164.12.6166
Miners, J. S., Barua, N., Kehoe, P. G., Gill, S., and Love, S. (2011). Aβ-Degrading Enzymes: Potential for Treatment of Alzheimer Disease. J. Neuropathol. Exp. Neurol. 70, 944–959. doi: 10.1097/NEN.0b013e3182345e46
Montgomery, S. L., Mastrangelo, M. A., Habib, D., Narrow, W. C., Knowlden, S. A., Wright, T. W., et al. (2011). Ablation of TNF-RI/RII Expression in Alzheimer’s Disease Mice Leads to an Unexpected Enhancement of Pathology. Am. J. Pathol. 179, 2053–2070. doi: 10.1016/j.ajpath.2011.07.001
Moreno-Cugnon, L., Revuelta, M., Arrizabalaga, O., Colie, S., Moreno-Valladares, M., Jimenez-Blasco, D., et al. (2019). Neuronal p38α mediates age-associated neural stem cell exhaustion and cognitive decline. Aging Cell 18:e13044. doi: 10.1111/acel.13044
Nguyen, H. M., Grössinger, E. M., Horiuchi, M., Davis, K. W., Jin, L., Maezawa, I., et al. (2017). Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 65, 106–121. doi: 10.1002/glia.23078
Nicolazzo, J. A., Pan, Y., Di Stefano, I., Choy, K. H. C., Reddiar, S. B., Low, Y. L., et al. (2022). Blockade of Microglial Kv1.3 Potassium Channels by the Peptide HsTX1[R14A] Attenuates Lipopolysaccharide-mediated Neuroinflammation. J. Pharm. Sci. 111, 638–647. doi: 10.1016/j.xphs.2021.11.003
Olmos-Alonso, A., Schetters, S. T. T., Sri, S., Askew, K., Mancuso, R., Vargas-Caballero, M., et al. (2016). Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 139, 891–907. doi: 10.1093/brain/awv379
Pannasch, U., Farber, K., Nolte, C., Blonski, M., Yan Chiu, S., Messing, A., et al. (2006). The potassium channels Kv1.5 and Kv1.3 modulate distinct functions of microglia. Mol. Cell Neurosci. 33, 401–411. doi: 10.1016/j.mcn.2006.08.009
Pappalardo, L. W., Black, J. A., and Waxman, S. G. (2016). Sodium channels in astroglia and microglia. Glia 64, 1628–1645. doi: 10.1002/glia.22967
Parkitny, L., and Maletic-Savatic, M. (2021). Glial PAMPering and DAMPening of Adult Hippocampal Neurogenesis. Brain Sci. 11:1299. doi: 10.3390/brainsci11101299
Peng, Y., Lu, K., Li, Z., Zhao, Y., Wang, Y., Hu, B., et al. (2014). Blockade of Kv1.3 channels ameliorates radiation-induced brain injury. Neuro-oncology 16, 528–539. doi: 10.1093/neuonc/not221
Pérez-Verdaguer, M., Capera, J., Serrano-Novillo, C., Estadella, I., Sastre, D., and Felipe, A. (2016). The voltage-gated potassium channel Kv1.3 is a promising multitherapeutic target against human pathologies. Expert Opin. Ther. Targets 20, 577–591. doi: 10.1517/14728222.2016.1112792
Peruzzo, R., Mattarei, A., Azzolini, M., Becker-Flegler, K. A., Romio, M., Rigoni, G., et al. (2020). Insight into the mechanism of cytotoxicity of membrane-permeant psoralenic Kv1.3 channel inhibitors by chemical dissection of a novel member of the family. Redox Biol. 37, 101705. doi: 10.1016/j.redox.2020.101705
Qin, L., Liu, Y., Hong, J., and Crews, F. T. (2013). NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia 61, 855–868. doi: 10.1002/glia.22479
Ramesha, S., Rayaprolu, S., Bowen, C. A., Giver, C. R., Bitarafan, S., Nguyen, H. M., et al. (2021). Unique molecular characteristics and microglial origin of Kv1.3 channel-positive brain myeloid cells in Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 118:e2013545118. doi: 10.1073/pnas.2013545118
Rangaraju, S., Dammer, E. B., Raza, S. A., Rathakrishnan, P., Xiao, H., Gao, T., et al. (2018). Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegen. 13:24. doi: 10.1186/s13024-018-0254-8
Rangaraju, S., Gearing, M., Jin, L., and Levey, A. (2015). Potassium Channel Kv1.3 Is Highly Expressed by Microglia in Human Alzheimer’s Disease. J. Alzheimer’s Dis. 44, 797–808. doi: 10.3233/JAD-141704
Rao, Y. L., Ganaraja, B., Murlimanju, B. V., Joy, T., Krishnamurthy, A., and Agrawal, A. (2022). Hippocampus and its involvement in Alzheimer’s disease: a review. 3 Biotech 12:55. doi: 10.1007/s13205-022-03123-4
Rasmussen, K. L., Nordestgaard, B. G., Frikke-Schmidt, R., and Nielsen, SF. (2018). An updated Alzheimer hypothesis: complement C3 and risk of Alzheimer‘s disease—A cohort study of 95,442 individuals. Alzheimer’s & Dementia 14, 1589–1601. doi: 10.1016/j.jalz.2018.07.223
Rather, M. A., Khan, A., Alshahrani, S., Rashid, H., Qadri, M., Rashid, S., et al. (2021). Inflammation and Alzheimer’s Disease: Mechanisms and Therapeutic Implications by Natural Products. Mediators Inflamm. 2021:9982954. doi: 10.1155/2021/9982954
Richard, K. L., Filali, M., Prefontaine, P., and Rivest, S. (2008). Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1-42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. J. Neurosci. 28, 5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008
Sala Frigerio, C., Wolfs, L., Fattorelli, N., Thrupp, N., Voytyuk, I., Schmidt, I., et al. (2019). The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Abeta Plaques. Cell Rep. 27, 1293–1306.e6. doi: 10.1016/j.celrep.2019.03.099
Sarkar, S., Nguyen, H. M., Malovic, E., Luo, J., Langley, M., Palanisamy, B. N., et al. (2020). Kv1.3 modulates neuroinflammation and neurodegeneration in Parkinson’s disease. J. Clin. Invest. 130, 4195–4212. doi: 10.1172/JCI136174
Sarlus, H., and Heneka, M. T. (2017). Microglia in Alzheimer’s disease. J. Clin. Invest. 127, 3240–3249. doi: 10.1172/JCI90606
Schmidt, S. I., Bogetofte, H., Ritter, L., Agergaard, J. B., Hammerich, D., Kabiljagic, A. A., et al. (2021). Microglia-Secreted Factors Enhance Dopaminergic Differentiation of Tissue- and iPSC-Derived Human Neural Stem Cells. Stem Cell Rep. 16, 281–294. doi: 10.1016/j.stemcr.2020.12.011
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Shaftel, S. S., Kyrkanides, S., Olschowka, J. A., Miller, J. N., Johnson, R. E., and O’Banion, M. K. (2007). Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Invest. 117, 1595–1604. doi: 10.1172/JCI31450
Shen, Y., Lue, L., Yang, L., Roher, A., Kuo, Y., Strohmeyer, R., et al. (2001). Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neurosci. Lett. 305, 165–168. doi: 10.1016/S0304-3940(01)01842-0
Shigemoto-Mogami, Y., Hoshikawa, K., Goldman, J. E., Sekino, Y., and Sato, K. (2014). Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. J. Neurosci. 34, 2231–2243. doi: 10.1523/JNEUROSCI.1619-13.2014
Simpson, D. S. A., and Oliver, P. L. (2020). ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 9:743. doi: 10.3390/antiox9080743
Sorrells, S. F., Paredes, M. F., Cebrian-Silla, A., Sandoval, K., Qi, D., Kelley, K. W., et al. (2018). Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 555, 377–381. doi: 10.1038/nature25975
Stamouli, E. C., and Politis, A. M. (2016). Pro-inflammatory cytokines in Alzheimer’s disease. Psychiatriki 27, 264–275. doi: 10.22365/jpsych.2016.274.264
Stebbing, M. J., Cottee, J. M., and Rana, I. (2015). The Role of Ion Channels in Microglial Activation and Proliferation - A Complex Interplay between Ligand-Gated Ion Channels, K(+) Channels, and Intracellular Ca(2.). Front. Immunol. 6:497. doi: 10.3389/fimmu.2015.00497
Styles, F. L., Al-Owais, M. M., Scragg, J. L., Chuntharpursat-Bon, E., Hettiarachchi, N. T., Lippiat, J. D., et al. (2021). Kv1.3 voltage-gated potassium channels link cellular respiration to proliferation through a non-conducting mechanism. Cell Death Dis. 12:372. doi: 10.1038/s41419-021-03627-6
Teisseyre, A., Palko-Labuz, A., Sroda-Pomianek, K., and Michalak, K. (2019). Voltage-Gated Potassium Channel Kv1.3 as a Target in Therapy of Cancer. Front. Oncol. 9:933. doi: 10.3389/fonc.2019.00933
Tobin, M. K., Musaraca, K., Disouky, A., Shetti, A., Bheri, A., Honer, W. G., et al. (2019). Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell Stem Cell 24, 974–982.e3. doi: 10.1016/j.stem.2019.05.003
Verheijen, J., and Sleegers, K. (2018). Understanding Alzheimer Disease at the Interface between Genetics and Transcriptomics. Trends Gen. 34, 434–447. doi: 10.1016/j.tig.2018.02.007
Wang, T., Lee, M. H., Johnson, T., Allie, R., Hu, L., Calabresi, P. A., et al. (2010). Activated T-Cells Inhibit Neurogenesis by Releasing Granzyme B: Rescue by Kv1.3 Blockers. J. Neurosci. 30, 5020–5027. doi: 10.1523/JNEUROSCI.0311-10.2010
Wang, X., Li, G., Guo, J., Zhang, Z., Zhang, S., Zhu, Y., et al. (2020). Kv1.3 Channel as a Key Therapeutic Target for Neuroinflammatory Diseases: State of the Art and Beyond. Front. Neurosci. 13:1393. doi: 10.3389/fnins.2019.01393
Wildsmith, K. R., Holley, M., Savage, J. C., Skerrett, R., and Landreth, G. E. (2013). Evidence for impaired amyloid β clearance in Alzheimer’s disease. Alzheimer’s Res. Ther. 5:33. doi: 10.1186/alzrt187
Wu, X., Shen, Q., Zhang, Z., Zhang, D., Gu, Y., and Xing, D. (2021). Photoactivation of TGFβ/SMAD signaling pathway ameliorates adult hippocampal neurogenesis in Alzheimer’s disease model. Stem Cell Res. Ther. 12:345. doi: 10.1186/s13287-021-02399-2
Wulff, H., Kolski-Andreaco, A., Sankaranarayanan, A., Sabatier, J., and Shakkottai, V. (2007). Modulators of Small- and Intermediate-Conductance Calcium-Activated Potassium Channels and their Therapeutic Indications. Curr. Med. Chem. 14, 1437–1457. doi: 10.2174/092986707780831186
Wulff, H., and Zhorov, B. S. (2008). K + Channel Modulators for the Treatment of Neurological Disorders and Autoimmune Diseases. Chem. Rev. 108, 1744–1773. doi: 10.1021/cr078234p
Yin, F., Sancheti, H., Patil, I., and Cadenas, E. (2016). Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radical Biol. Med. 100, 108–122. doi: 10.1016/j.freeradbiomed.2016.04.200
Yoo, S., Park, J., Kim, S., and Jung, Y. (2020). Emerging perspectives on mitochondrial dysfunction and inflammation in Alzheimer’s disease. BMB Rep. 53, 35–46. doi: 10.5483/BMBRep.2020.53.1.274
Zayas-Arrabal, J., Alquiza, A., Rodríguez-de-Yurre, A., Echeazarra, L., Fernández-López, V., Gallego, M., et al. (2021). Kv1.3 Channel Blockade Improves Inflammatory Profile, Reduces Cardiac Electrical Remodeling, and Prevents Arrhythmia in Type 2 Diabetic Rats. Cardiovascul. Drugs Ther. doi: 10.1007/s10557-021-07264-1 [Epub Online ahead of print]
Keywords: Alzheimer’s disease, microglia, KV1.3, inflammation, neurodegenaration, neural stem cell (NSC), therapeutic targets
Citation: Revuelta M, Urrutia J, Villarroel A and Casis O (2022) Microglia-Mediated Inflammation and Neural Stem Cell Differentiation in Alzheimer’s Disease: Possible Therapeutic Role of KV1.3 Channel Blockade. Front. Cell. Neurosci. 16:868842. doi: 10.3389/fncel.2022.868842
Received: 03 February 2022; Accepted: 31 March 2022;
Published: 21 April 2022.
Edited by:
Pedro M. Pimentel-Coelho, Federal University of Rio de Janeiro, BrazilReviewed by:
Marilia Zaluar P. Guimaraes, Federal University of Rio de Janeiro, BrazilCopyright © 2022 Revuelta, Urrutia, Villarroel and Casis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miren Revuelta, bWlyZW4ucmV2dWVsdGFAZWh1LmV1cw==; Janire Urrutia, amFuaXJlLnVycnV0aWFAZWh1LmV1cw==
†These authors have contributed equally to this work and share first authorship