 Ricardo Martins-Ferreira
Ricardo Martins-Ferreira Bárbara Guerra Leal
Bárbara Guerra Leal Paulo Pinho Costa
Paulo Pinho Costa- 1Epigenetics and Immune Disease Group, Josep Carreras Research Institute (IJC), Barcelona, Spain
- 2Immunogenetics Lab, Molecular Pathology and Immunology, Instituto de Ciências Biomédicas Abel Salazar—Universidade do Porto (ICBAS-UPorto), Porto, Portugal
- 3Autoimmunity and Neuroscience Group, Unit for Multidisciplinary Research in Biomedicine (UMIB), ICBAS-UPorto, Porto, Portugal
- 4Laboratory for Integrative and Translational Research in Population Health (ITR), Porto, Portugal
- 5Instituto Nacional de Saúde Dr. Ricardo Jorge, Department of Human Genetics, Porto, Portugal
Circulating cell-free DNA (cfDNA) are highly degraded DNA fragments shed into the bloodstream. Apoptosis is likely to be the main source of cfDNA due to the matching sizes of cfDNA and apoptotic DNA cleavage fragments. The study of cfDNA in liquid biopsies has served clinical research greatly. Genetic analysis of these circulating fragments has been used in non-invasive prenatal testing, detection of graft rejection in organ transplants, and cancer detection and monitoring. cfDNA sequencing is, however, of limited value in settings in which genetic association is not well-established, such as most neurodegenerative diseases.Recent studies have taken advantage of the cell-type specificity of DNA methylation to determine the tissue of origin, thus detecting ongoing cell death taking place in specific body compartments. Such an approach is yet to be developed in the context of epilepsy research. In this article, we review the different approaches that have been used to monitor cell-type specific death through DNA methylation analysis, and recent data detecting neuronal death in neuropathological settings. We focus on the potential relevance of these tools in focal epilepsies, like Mesial Temporal Lobe Epilepsy with Hippocampal Sclerosis (MTLE-HS), characterized by severe neuronal loss. We speculate on the potential relevance of cfDNA methylation screening for the detection of neuronal cell death in individuals with high risk of epileptogenesis that would benefit from early diagnosis and consequent early treatment.
Introduction
The search for effective biomarkers is one of the most challenging tasks in epilepsy research. Epileptogenesis is now considered a progressive process, with seizure activity contributing to extensive damage and seizure recurrence (Pitkänen et al., 2015). Epilepsy diagnosis must thus be viewed as a race against the clock, aiming for the detection, as early as possible, of individuals with a high risk of epilepsy development, that could be candidates for preventive pharmacological intervention. The search for novel viable biomarkers is considered crucial to improve the current hurdles to early diagnosis and prognostic monitoring in epileptic patients. A major issue continues to be the high refractoriness rates (estimated at 30%) across different types of epilepsy (Pitkänen et al., 2016). Epilepsy treatment is based on the control of ictogenesis. Current anti-seizure drugs target seizure control and are not directed to the whole epileptogenic landscape. Additionally, such biomarkers would facilitate economically feasible clinical trials for the development of new and more effective anti-epileptic therapies by allowing a more precise categorization of individuals with high probability of developing epilepsy (Engel and Pitkänen, 2020).
Nucleic acid-based disease biomarkers have represented one of the most exciting developments in recent years, with non-coding RNAs, specifically microRNAs, gaining center stage (Henshall et al., 2016). In this review, we consider the potential of the still largely unexplored circulating cell-free (cf) DNA. We will focus on cfDNA methylation and its ability to predict cell and tissue of origin in epilepsies with a histopathological phenotype of neuronal damage and death.
Biogenesis and The Apoptotic Cascade
The first description of cfDNA in human plasma dates to 1948 (Mandel and Metais, 1948). Nowadays, it is broadly described as highly degraded DNA fragments circulating freely in the peripheral blood. However, the full spectrum of processes responsible for cfDNA production and release remains elusive. There is a perception that cfDNA may be originated from countless biological features, both physiological and pathological, such as inflammation, aging, exercise, or cancer. The main mechanisms of cfDNA release are considered to be active release or cellular breakdown (Aucamp et al., 2018). The overall conception, on which most of the studies in this field base their hypotheses, is that cfDNA originates predominantly from apoptosis. This relies on the matching sizes between cfDNA and DNA fragments resultant from the apoptotic cascade. Caspase-activated DNase is one of the main effectors of DNA fragmentation during apoptosis. It is a double-stranded endonuclease which lacks exonuclease activity, thus being only capable of fragmenting DNA in inter-nucleosomal linker regions (Heitzer et al., 2020). Human circulating cfDNA presents a predominant and consistent fragment length of approximately 167 bp (Snyder et al., 2016), which corresponds to the length of DNA wrapped around a nucleosome (~147 bp) plus linker fragments (Heitzer et al., 2020).
Genetic Sequencing and Liquid Biopsies
In practice, the study of cfDNA has consisted predominantly of its genetic characterization. Sequencing of cfDNA is nowadays routinely used in some clinic settings. Screening of fetal DNA circulating in the mother’s bloodstream, non-invasive prenatal testing, is used to detect chromosomal abnormalities (Fan et al., 2012). Transplant graft rejection is also monitored by the detection of donor-derived cfDNA sequences (De Vlaminck et al., 2014). Throughout the last two decades, cfDNA sequencing has been increasingly applied to cancer detection (Chabon et al., 2020) and also in post-therapy prognosis (Nabet et al., 2020; Powles et al., 2021). These liquid biopsies provide for non-invasive detection of tumor DNA in biological fluids, also denominated as circulating tumor DNA.
The relevance and potential of cfDNA does not end in its genetics, though. The utility of nucleotide sequencing is narrow in non-mutation rich pathologies, meaning conditions in which a clear association with a mutated genetic profile is not observed. These include a considerable number of neurodegenerative diseases such as, in the interest of this review, different forms of epilepsy.
Deconvoluting Cell-Of-Origin Through DNA Methylation
The strong association between cfDNA and apoptosis suggests the occurrence of cell death somewhere in the organism. However, genetic screening of the circulating DNA is unable to specifically identify tissue or cell of origin. In this context, epigenetics and, specifically, DNA methylation have entered and significantly upgraded cfDNA research. DNA methylation consists of the covalent addition of a methyl group to cytosine residues in the genomic sequence. It occurs predominantly in CpG dinucleotides and is associated with transcriptional regulation. Certain patterns of DNA methylation are highly tissue and cell-specific and conserved in both physiologic and pathologic conditions (Dor and Cedar, 2018; Figure 1).
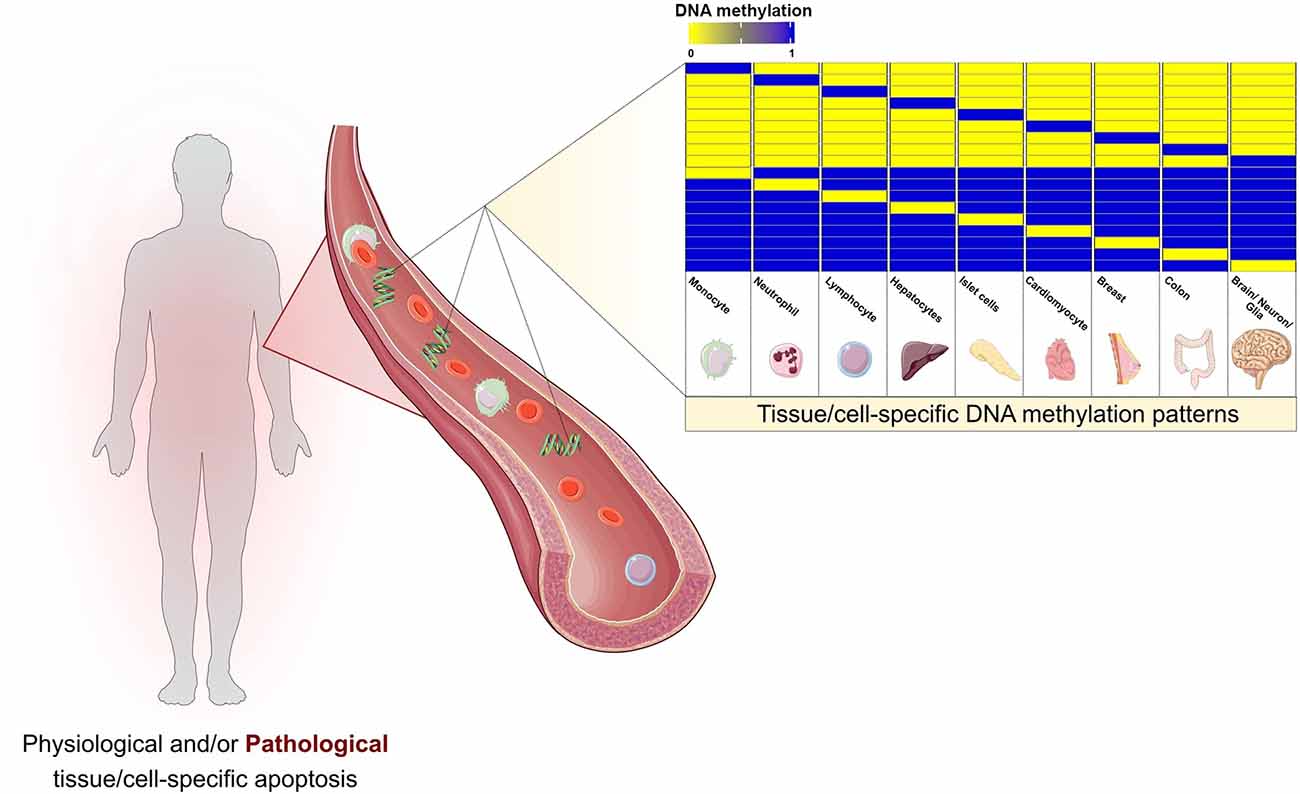
Figure 1. Cell damage and apoptotic events during both physiological and pathological conditions are thought to release cell-free DNA (cfDNA) into the bloodstream. The pool of cfDNA in circulation present specific DNA methylation patterns that would allow the detection and localization of cell death, opening the doors for a wide spectrum of prospective clinical applications. The selected cell and tissue types are intended to be representative. Hematopoietic cells, the main contributors to cfDNA release, are depicted by monocytes, neutrophils, and lymphocytes. Hepatocytes, pancreatic islet cells, cardiomyocytes, breast, and colon are some examples of tissues with previous evidence on the estimation of cell/tissue of origin based on cfDNA methylation. Brain, neurons, and glia represent the potential of applying such pipelines in neurodegenerative diseases.
Alterations of DNA methylation in cfDNA may result from alterations in specific cells or tissue between the pathologic and disease-free settings. Additionally, it can also originate from changes in the composition of cfDNA due to differential cell death rates (Figure 1). The analysis of the cfDNA methylome has been conducted in two main lines of thought. The first opts for directly calculating differential methylation patterns, such as differentially methylated positions (DMPs) and differentially methylated regions (DMRs) between different conditions (Hatt et al., 2015; Jensen et al., 2015; Legendre et al., 2015; Xu et al., 2017; Gallardo-Gómez et al., 2018; Gordevičius et al., 2018). However, the direct usage of cfDNA profiles as predictors may be associated with an increased signal-to-noise ratio, since DMPs and even DMRs can be non-specifical and more difficult to interpret biologically. Cell or tissue-of-origin estimation may be considered as a reduction step, decreasing background noise, and increasing prediction accuracy (Feng et al., 2019). Prediction of cfDNA origin can be approached through two different strategies. The first consists of targeted DNA methylation analysis, that is predicting cell-of-origin by searching for previously identified specific DNA methylation alterations characteristic of a cell type (DMPs and DMRs). This approach has been applied throughout a wide spectrum of settings, including the detection of cfDNA derived from pancreatic β-cell in Type 1 diabetic patients (Olsen et al., 2016a), cardiomyocytes in cardiac infarction and sepsis patients (Zemmour et al., 2018) and hepatocytes in liver transplant recipients, individuals who underwent hepatectomy and sepsis patients (Lehmann-Werman et al., 2018). The second approach consists of genome-wide deconvolution tools that have been developed to simultaneously estimate the percentages of the contribution of multiple tissue and cell types to circulating cfDNA. Deconvolution algorithms have been predominantly used in cancer DNA methylation research (Sun et al., 2015; Guo et al., 2017; Kang et al., 2017; Li et al., 2018). A study by Moss et al. (2018) developed a comprehensive reference-based deconvolution tool accounting for a total of 25 cell/tissue types obtained from 450k and EPIC arrays of primary tissue sources. They validated the previous conception that cfDNA in plasma of healthy individuals as originating predominantly from hematopoietic cells. Furthermore, the authors demonstrated promising results in pathological contexts, by detecting increased plasma cfDNA of pancreatic origin in islet transplant recipients, significant correlation of hepatocyte-derived cfDNA and clinical liver damage markers, and detection of tissue-specific cfDNA in three cancer settings (colon, lung, and breast). Interestingly, cfDNA estimated origin showed good prognostic ability in prostate cancer patients after treatment, and also showed promise in the prediction of tissue of origin in cancer patients with no clear primary (Cancer of Unknown Primary; Moss et al., 2018). An additional interesting aspect of this study is the fact that this deconvolution algorithm accounts for primary cortical neurons, thus allowing its usage in detecting neuronal cell death across neurodegenerative diseases.
Brain-Derived cfDNA in Neurodegeneration
The application of cfDNA methylation-based analysis to neurodegenerative pathologies are still in their infancy. Research has consisted mostly of target-based studies, meaning that the detection of circulating brain-derived DNA relies on precisely measuring small regions, with specifically differential methylation behavior in the CNS in comparison to other tissues, mainly hematopoietic cells. A summary of the current data on CNS-derived cfDNA in a neurodegenerative context is presented in Table 1. The first such study that we are aware of is one by Lehmann-Werman et al. (2016), in which the authors first identified a pattern of clear demethylation in two clusters of CpGs in sorted oligodendrocytes’ methylome (located at the MBP3 gene and in an unannotated region). By bisulfite sequencing such regions, they described increased oligodendrocyte-derived cfDNA in serum or plasma of multiple sclerosis patients. Using the same strategy (known unmethylation of a cluster of nine CpGs of brain tissue), they observed an increase in brain-derived cfDNA in cardiac arrest patients with documented brain damage, and also in traumatic brain injury patients (Lehmann-Werman et al., 2016). In line with this, Olsen et al. (2016b) demonstrated increased demethylation of the MOG gene in sera of active multiple sclerosis patients. In amyotrophic lateral sclerosis patients, plasma cfDNA methylation levels of the RHBDF promoter were increased, relative to healthy controls (Mendioroz et al., 2018). Lastly, Alzheimer’s patients have been shown to have higher levels of LHX2 methylation in plasma-extracted cfDNA (Pai et al., 2019).
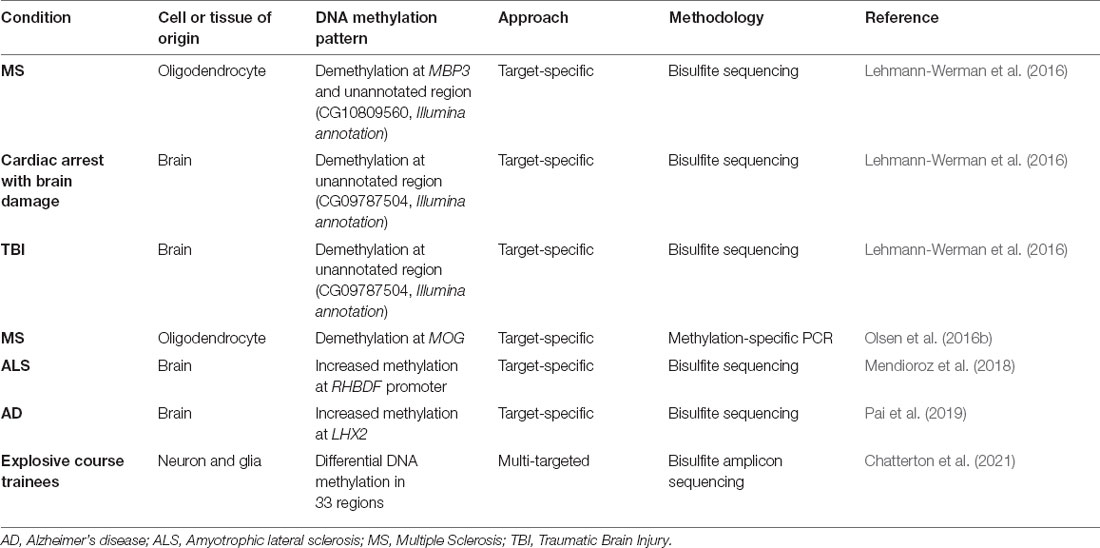
Table 1. Evidence of CNS-derived cfDNA in neurodegenerative settings.
In a more technically evolved and encompassing study, Chatterton et al. (2021) achieved an interesting proof-of-concept in which they were able to detect neuron and glia-derived cfDNA in human plasma samples. They developed a bisulfite amplicon sequencing protocol of a total of 33 regions with neuron and glial-specific DNA methylation patterns identified with an in-house pipeline from 450k microarray profiles. In blood plasma samples of entry personnel during a training course using explosives, the authors report evidence of neuron and glial cfDNA. Furthermore, significantly increased levels of neuron-derived cfDNA were observed on the day of training in which exposure to higher pressures was experienced (Chatterton et al., 2021).
Tissue biopsies are extremely invasive and, in the case of neurologic conditions, particularly inaccessible. Cerebrospinal fluid (CSF) analysis is an option but still causes major discomfort to patients. Along the neurodegeneration spectrum, there are multiple profiles of neurotrauma and damage. One can infer that CNS-derived cfDNA shed into the bloodstream may become a more accessible and interpretable tool for neurodegenerative biomarker research.
Potential of cfDNA Methylome Profiling in Epilepsy
To the best of our knowledge, to date, there are only two studies on cfDNA studies in epilepsy. Both analyzing the concentration of cfDNA. Liimatainen et al. (2013) observed increased sera cfDNA concentration in 167 focal epilepsy patients vs. 250 healthy controls. Alapirtti et al. (2016) on the other hand, failed to verify differences between 52 refractory epilepsy patients (both focal and generalized) and 250 controls. One of the limitations of these studies is the heterogeneity of the studied populations, which include a large spectrum of epileptic syndromes. Given the variability within epilepsies and the increasingly evident differing etiopathogenic fingerprints between syndromes, more narrowly specified epilepsy cohorts would be of more relevance.
As for cfDNA methylation, nothing has, to the best of our knowledge, been reported yet. Nonetheless, altered DNA methylation patterns in brain tissue of epilepsy patients have been consistently described (Miller-Delaney et al., 2012; Liu et al., 2016; Wang et al., 2016; Zhang et al., 2021; Martins-Ferreira et al., 2022). Major efforts have been made for the use of DNA methylation as peripheral biomarkers, namely by demonstrating the correlation of DNA methylation patterns in brain tissue and peripheral tissues, such as blood, saliva, and buccal mucosa (Braun et al., 2019). Reinforcing the aforementioned idea, cfDNA methylation may be more representative of a specific pathological phenotype due to the link between cfDNA methylome, differing of tissue/cell contribution, and pattern of cell death, as described throughout this review.
Mesial Temporal Lobe Epilepsy (MTLE) is one of the most studied epilepsies due to its high incidence (it is the most common focal epilepsy in adults) and its high refractoriness rates. Hippocampal sclerosis is the most prevalent histopathological feature in MTLE. It is characterized by an exacerbated state of gliosis and a severe neuronal death landscape in the mesial temporal regions (Blümcke et al., 2013). A pertinent question regarding MTLE’s etiopathology is the time frame of HS occurrence. It has been considered a consequence of recurrent exposure to seizure activity. However, it can also be speculated that HS may have a role in promoting pro-epileptogenic activity onset and progression. This is, in part, due to the fact that the diagnosis often occurs in the chronic stage, upon the appearance of spontaneous recurrent seizures. The most recently reviewed definition of epileptogenesis describes it as a continuous event. After the initial precipitating injury, the brain undergoes a latent phase in which the ability to produce spontaneous recurrent seizures is acquired. The chronic stage, initiated upon the first unprovoked seizure, is no longer considered stationary, as was previously the case. It is rather a progressive phenomenon, with pathological molecular and cellular alterations extending as the disease progresses (Pitkänen et al., 2016). A recent study by our group has demonstrated major DNA methylation alterations in both hippocampus and anterior temporal neocortex of MTLE-HS patients in comparison to autopsied non-epileptic controls, with enrichment for a wide spectrum of epileptogenesis-related pathways. Additionally, we demonstrate a potential progressive remodeling of DNA methylation in inflammatory genes with increased disease duration (Martins-Ferreira et al., 2022).
The main argument of interest of this review focus on the fact that cell death occurs in the MTLE-HS brain. Despite the time-point at which it may be occurring. Thus, it is plausible that neuron- or glia-derived cfDNA may be released into the circulation during neuro damage. We call for novel cfDNA methylation screening studies in epileptic patients, from targeted approaches like bisulfite sequencing and methylation-specific PCR to genome-wide screenings using microarrays, whole-genome bisulfite sequencing, and reduced representation bisulfite sequencing. Such studies would be potentially helpful in all stages of the epileptogenic landscape. It could promote detection of patients at high risk of epileptogenesis after a brain trauma incident and during latency, facilitate earlier diagnosis and better monitoring, and provide for therapeutic outcome prediction (Figure 2).
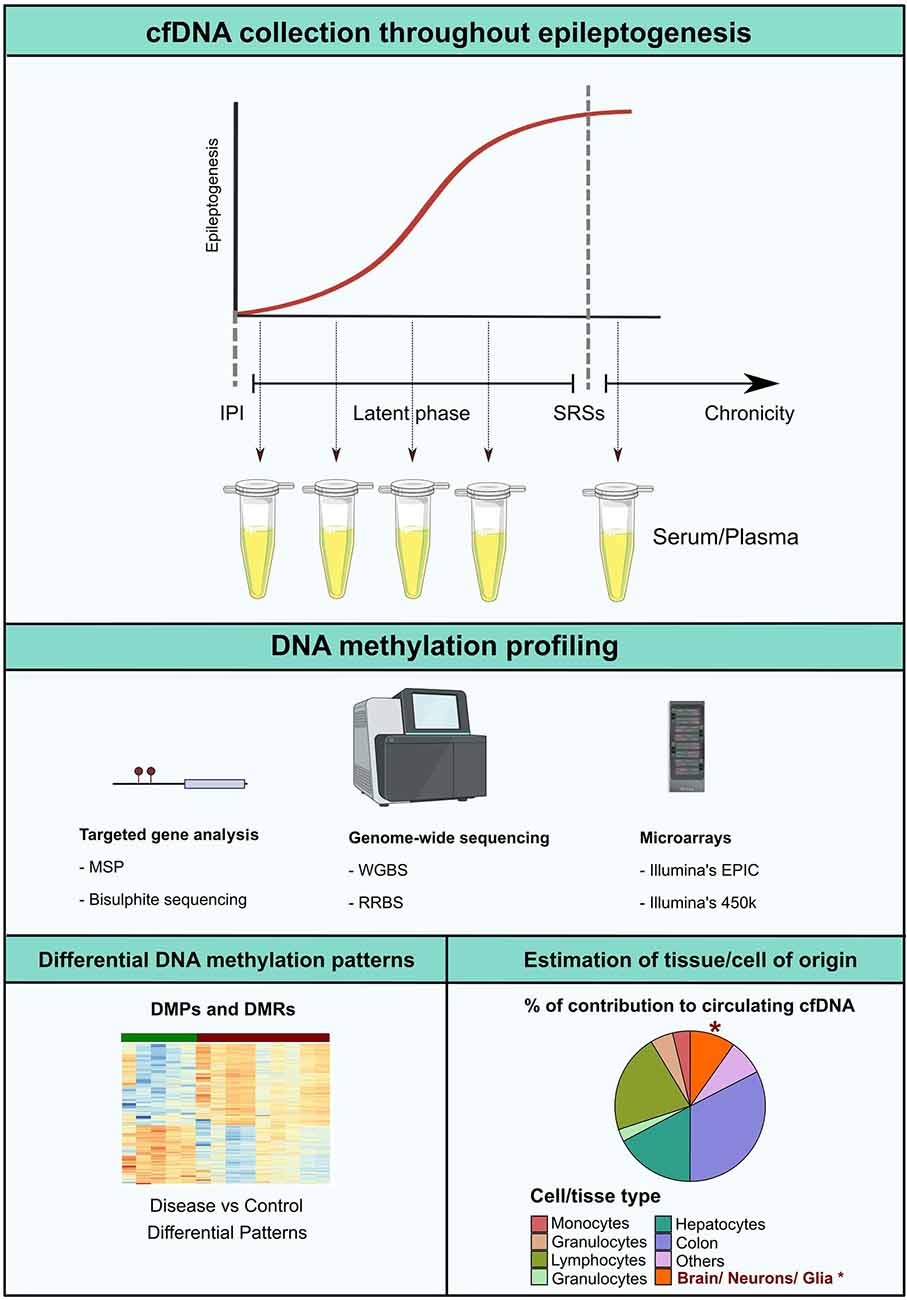
Figure 2. Epileptogenesis is considered to follow a progressive model. After the initial precipitating injury (IPI), the brain parenchyma enters a latent phase in which the ability to generate spontaneous recurrent seizures (SRSs) is established. In the chronic stage, upon initiation of unprovoked seizure activity, epileptogenic molecular and structural remodeling progress with time. DNA methylation profiling along the different timeframes of epileptogenesis would potentially represent a valuable approach for the development of novel non-invasive biomarkers. Possible analytical strategies include detection of altered cfDNA methylation patterns in pathological settings in comparison to controls, or estimation of the percentage of tissue or cell type contribution to the circulating cfDNA pool, which potentially insights on occurring CNS-specific cell death in the early stages of the disease. MSP, Methylation-Specific PCR; RRBS, reduced representation bisulfite sequencing; WBGS, whole genome bisulfite sequencing.
Concluding Remarks
The research on circulating cfDNA has evolved exponentially during the last decades, from the proven clinical utility of genetic characterization to the emerging applications of DNA methylation. Identification of biomarkers is an urgent need across various pathologies, particularly in neurologic diseases in which tissue biopsies are extremely difficult to obtain or simply inaccessible. We propose that cfDNA methylation should be investigated in epileptic patients, as we consider it to be and intriguing and promising beam of light in what has been the gloomy field of epilepsy biomarker research.
Author Contributions
All authors were responsible for the development of the conceptual map. RM-F performed literature search and drafted the manuscript. BL and PC were responsible for the critical revision and final approval of the final version to be published. All authors contributed to the article and approved the submitted version.
Funding
RM-F was funded by an FCT (Fundação para a Ciência e Tecnologia) fellowship (grant number SFRH/BD/137900/2018). Unit for Multidisciplinary Research in Biomedicine (UMIB) was funded by FCT Portugal (grant numbers UIDB/00215/2020 and UIDP/00215/2020), and Laboratory for Integrative and Translational Research in Population Health (ITR) (LA/P/0064/2020).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge Professor Berta Martins da Silva and the remaining members of the Immunogenetics Laboratory of the Molecular Pathology and Immunology Department of ICBAS-UP. We thank the Neurology, Neurophysiology and Neurosurgery Departments of Hospital de Santo António—Centro Hospitalar e Universitário do Porto. We also acknowledge Professor Esteban Ballestar and all members of Epigenetics and Immune Disease of the Josep Carreras Research Institute (IJC).
Abbreviations
cfDNA, cell-free DNA; DMP, differentially methylated position; DMR, differentially methylated region; HS, hippocampal sclerosis; MTLE, mesial temporal lobe epilepsy.
References
Alapirtti, T., Jylhävä, J., Raitanen, J., Mäkinen, R., Peltola, J., Hurme, M. A., et al. (2016). The concentration of cell-free DNA in video-EEG patients is dependent on the epilepsy syndrome and duration of epilepsy. Neurol. Res. 38, 45–50. doi: 10.1080/01616412.2015.1127004
Aucamp, J., Bronkhorst, A. J., Badenhorst, C. P. S., and Pretorius, P. J. (2018). The diverse origins of circulating cell-free DNA in the human body: a critical re-evaluation of the literature. Biol. Rev. Camb. Philos. Soc. 93, 1649–1683. doi: 10.1111/brv.12413
Blümcke, I., Thom, M., Aronica, E., Armstrong, D. D., Bartolomei, F., Bernasconi, A., et al. (2013). International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a task force report from the ILAE commission on diagnostic methods. Epilepsia 54, 1315–1329. doi: 10.1111/epi.12220
Braun, P. R., Han, S., Hing, B., Nagahama, Y., Gaul, L. N., Heinzman, J. T., et al. (2019). Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl. Psychiatry 9:47. doi: 10.1038/s41398-019-0376-y
Chabon, J. J., Hamilton, E. G., Kurtz, D. M., Esfahani, M. S., Moding, E. J., Stehr, H., et al. (2020). Integrating genomic features for non-invasive early lung cancer detection. Nature 580, 245–251. doi: 10.1038/s41586-020-2140-0
Chatterton, Z., Mendelev, N., Chen, S., Carr, W., Kamimori, G. H., Ge, Y., et al. (2021). Bisulfite amplicon sequencing can detect glia and neuron cell-free DNA in blood plasma. Front. Mol. Neurosci. 14:672614. doi: 10.3389/fnmol.2021.672614
Dor, Y., and Cedar, H. (2018). Principles of DNA methylation and their implications for biology and medicine. Lancet 392, 777–786. doi: 10.1016/S0140-6736(18)31268-6
Engel, J, Jr., and Pitkänen, A. (2020). Biomarkers for epileptogenesis and its treatment. Neuropharmacology 167:107735. doi: 10.1016/j.neuropharm.2019.107735
Fan, H. C., Gu, W., Wang, J., Blumenfeld, Y. J., El-Sayed, Y. Y., and Quake, S. R. (2012). Non-invasive prenatal measurement of the fetal genome. Nature 487, 320–324. doi: 10.1038/nature11251
Feng, H., Jin, P., and Wu, H. (2019). Disease prediction by cell-free DNA methylation. Brief Bioinform. 20, 585–597. doi: 10.1093/bib/bby029
Gallardo-Gómez, M., Moran, S., de la Cadena, M. P., Martínez-Zorzano, V. S., Rodríguez-Berrocal, F. J., Rodríguez-Girondo, M., et al. (2018). A new approach to epigenome-wide discovery of non-invasive methylation biomarkers for colorectal cancer screening in circulating cell-free DNA using pooled samples. Clin. Epigenetics 10:53. doi: 10.1186/s13148-018-0487-y
Gordevičius, J., Kriščiūnas, A., Groot, D. E., Yip, S. M., Susic, M., Kwan, A., et al. (2018). Cell-free DNA modification dynamics in abiraterone acetate-treated prostate cancer patients. Clin. Cancer Res. 24, 3317–3324. doi: 10.1158/1078-0432.CCR-18-0101
Guo, S., Diep, D., Plongthongkum, N., Fung, H.-L., Zhang, K., and Zhang, K. (2017). Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat. Genet. 49, 635–642. doi: 10.1038/ng.3805
Hatt, L., Aagaard, M. M., Graakjaer, J., Bach, C., Sommer, S., Agerholm, I. E., et al. (2015). Microarray-based analysis of methylation status of CpGs in placental DNA and maternal blood DNA – potential new epigenetic biomarkers for cell free fetal DNA-based diagnosis. PLoS One 10:e0128918. doi: 10.1371/journal.pone.0128918
Heitzer, E., Auinger, L., and Speicher, M. R. (2020). Cell-free DNA and apoptosis: how dead cells inform about the living. Trends Mol. Med. 26, 519–528. doi: 10.1016/j.molmed.2020.01.012
Henshall, D. C., Hamer, H. M., Pasterkamp, R. J., Goldstein, D. B., Kjems, J., Prehn, J. H. M., et al. (2016). MicroRNAs in epilepsy: pathophysiology and clinical utility. Lancet Neurol. 15, 1368–1376. doi: 10.1016/S1474-4422(16)30246-0
Jensen, T. J., Kim, S. K., Zhu, Z., Chin, C., Gebhard, C., Lu, T., et al. (2015). Whole genome bisulfite sequencing of cell-free DNA and its cellular contributors uncovers placenta hypomethylated domains. Genome Biol. 16:78. doi: 10.1186/s13059-015-0645-x
Kang, S., Li, Q., Chen, Q., Zhou, Y., Park, S., Lee, G., et al. (2017). Cancerlocator: non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA. Genome Biol. 18:53. doi: 10.1186/s13059-017-1191-5
Legendre, C., Gooden, G. C., Johnson, K., Martinez, R. A., Liang, W. S., and Salhia, B. (2015). Whole-genome bisulfite sequencing of cell-free DNA identifies signature associated with metastatic breast cancer. Clin. Epigenetics 7:100. doi: 10.1186/s13148-015-0135-8
Lehmann-Werman, R., Magenheim, J., Moss, J., Neiman, D., Abraham, O., Piyanzin, S., et al. (2018). Monitoring liver damage using hepatocyte-specific methylation markers in cell-free circulating DNA. JCI Insight 3:e120687. doi: 10.1172/jci.insight.120687
Lehmann-Werman, R., Neiman, D., Zemmour, H., Moss, J., Magenheim, J., Vaknin-Dembinsky, A., et al. (2016). Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. U S A 113, E1826–1834. doi: 10.1073/pnas.1519286113
Li, W., Li, Q., Kang, S., Same, M., Zhou, Y., Sun, C., et al. (2018). Cancerdetector: ultrasensitive and non-invasive cancer detection at the resolution of individual reads using cell-free DNA methylation sequencing data. Nucleic Acids Res. 46:e89. doi: 10.1093/nar/gky423
Liimatainen, S. P., Jylhävä, J., Raitanen, J., Peltola, J. T., and Hurme, M. A. (2013). The concentration of cell-free DNA in focal epilepsy. Epilepsy Res. 105, 292–298. doi: 10.1016/j.eplepsyres.2013.03.005
Liu, X., Ou, S., Xu, T., Liu, S., Yuan, J., Huang, H., et al. (2016). New differentially expressed genes and differential DNA methylation underlying refractory epilepsy. Oncotarget 7, 87402–87416. doi: 10.18632/oncotarget.13642
Mandel, P., and Metais, P. (1948). [Nuclear acids in human blood plasma]. Comptes rendus des seances de la Societe de biologie et de ses filiales 142, 241–243.
Martins-Ferreira, R., Leal, B., Chaves, J., Li, T., Ciudad, L., Rangel, R., et al. (2022). Epilepsy progression is associated with cumulative DNA methylation changes in inflammatory genes. Prog. Neurobiol. 209:102207. doi: 10.1016/j.pneurobio.2021.102207
Mendioroz, M., Martínez-Merino, L., Blanco-Luquin, I., Urdánoz, A., Roldán, M., and Jericó, I. (2018). Liquid biopsy: a new source of candidate biomarkers in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 5, 763–768. doi: 10.1002/acn3.565
Miller-Delaney, S. F. C., Das, S., Sano, T., Jimenez-Mateos, E. M., Bryan, K., Buckley, P. G., et al. (2012). Differential DNA methylation patterns define status epilepticus and epileptic tolerance. J. Neurosci. 32, 1577–1588. doi: 10.1523/JNEUROSCI.5180-11.2012
Moss, J., Magenheim, J., Neiman, D., Zemmour, H., Loyfer, N., Korach, A., et al. (2018). Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 9:5068. doi: 10.1038/s41467-018-07466-6
Nabet, B. Y., Esfahani, M. S., Moding, E. J., Hamilton, E. G., Chabon, J. J., Rizvi, H., et al. (2020). Noninvasive early identification of therapeutic benefit from immune checkpoint inhibition. Cell 183, 363–376.e13. doi: 10.1016/j.cell.2020.09.001
Olsen, J. A., Kenna, L. A., Spelios, M. G., Hessner, M. J., and Akirav, E. M. (2016a). Circulating differentially methylated amylin DNA as a biomarker of β-cell loss in type 1 diabetes. PLoS One 11:e0152662. doi: 10.1371/journal.pone.0152662
Olsen, J. A., Kenna, L. A., Tipon, R. C., Spelios, M. G., Stecker, M. M., and Akirav, E. M. (2016b). A minimally-invasive blood-derived biomarker of oligodendrocyte cell-loss in multiple sclerosis. EBioMedicine 10, 227–235. doi: 10.1016/j.ebiom.2016.06.031
Pai, M.-C., Kuo, Y.-M., Wang, I.-F., Chiang, P.-M., and Tsai, K.-J. (2019). The role of methylated circulating nucleic acids as a potential biomarker in Alzheimer’s disease. Mol. Neurobiol. 56, 2440–2449. doi: 10.1007/s12035-018-1229-z
Pitkänen, A., Löscher, W., Vezzani, A., Becker, A. J., Simonato, M., Lukasiuk, K., et al. (2016). Advances in the development of biomarkers for epilepsy. Lancet Neurol. 15, 843–856. doi: 10.1016/S1474-4422(16)00112-5
Pitkänen, A., Lukasiuk, K., Dudek, F. E., and Staley, K. J. (2015). Epileptogenesis. Cold Spring Harb. Perspect. Med. 5:a022822. doi: 10.1101/cshperspect.a022822
Powles, T., Assaf, Z. J., Davarpanah, N., Banchereau, R., Szabados, B. E., Yuen, K. C., et al. (2021). CtDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature 595, 432–437. doi: 10.1038/s41586-021-03642-9
Snyder, M. W., Kircher, M., Hill, A. J., Daza, R. M., and Shendure, J. (2016). Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 164, 57–68. doi: 10.1016/j.cell.2015.11.050
Sun, K., Jiang, P., Allen Chan, K. C., Wong, J., Cheng, Y. K. Y., Liang, R. H. S., et al. (2015). Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer and transplantation assessments. Proc. Natl. Acad. Sci. U S A 112, E5503–5512. doi: 10.1073/pnas.1508736112
De Vlaminck, I., Valantine, H. A., Snyder, T. M., Strehl, C., Cohen, G., Luikart, H., et al. (2014). Circulating cell-free DNA enables noninvasive diagnosis of heart transplant rejection. Sci. Transl. Med. 6:241ra77. doi: 10.1126/scitranslmed.3007803
Wang, L., Fu, X., Peng, X., Xiao, Z., Li, Z., Chen, G., et al. (2016). DNA methylation profiling revealss correlation of differential methylation patterns with gene expression in human epilepsy. J. Mol. Neuroscience. 59, 68–77. doi: 10.1007/s12031-016-0735-6
Xu, R.-H., Wei, W., Krawczyk, M., Wang, W., Luo, H., Flagg, K., et al. (2017). Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat. Mater. 16, 1155–1161. doi: 10.1038/nmat4997
Zemmour, H., Planer, D., Magenheim, J., Moss, J., Neiman, D., Gilon, D., et al. (2018). Non-invasive detection of human cardiomyocyte death using methylation patterns of circulating DNA. Nat. Commun. 9:1443. doi: 10.1038/s41467-018-03961-y
Keywords: cell-free DNA, DNA methylation, epilepsy, epileptogenesis, MTLE-HS, biomarker
Citation: Martins-Ferreira R, Leal BG and Costa PP (2022) The Potential of Circulating Cell-Free DNA Methylation as an Epilepsy Biomarker. Front. Cell. Neurosci. 16:852151. doi: 10.3389/fncel.2022.852151
Received: 10 January 2022; Accepted: 24 February 2022;
Published: 24 March 2022.
Edited by:
Diana Cunha-Reis, University of Lisbon, PortugalReviewed by:
Ana Laura Márquez-Aguirre, CONACYT Centro de Investigación y Asistencia en Tecnolog’i-a y Diseño del Estado de Jalisco (CIATEJ), MexicoCopyright © 2022 Martins-Ferreira, Leal and Costa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bárbara Guerra Leal, bWFpbHRvOmJhZ3VlcnJhbGVhbEBnbWFpbC5jb20=