 Edoardo Moretto1,2,3*†
Edoardo Moretto1,2,3*† Skye Stuart2,3†
Skye Stuart2,3† Sunaina Surana2,3,4†
Sunaina Surana2,3,4† Jose Norberto S. Vargas2,3,4†
Jose Norberto S. Vargas2,3,4† Giampietro Schiavo2,3,4*†
Giampietro Schiavo2,3,4*†- 1Institute of Neuroscience, National Research Council, CNR, Milan, Italy
- 2UK Dementia Research Institute, University College London, London, United Kingdom
- 3Department of Neuromuscular Diseases, Queen Square Institute of Neurology, University College London, London, United Kingdom
- 4UCL Queen Square Motor Neuron Disease Centre, University College London, London, United Kingdom
Several neurodegenerative diseases are characterized by the accumulation of aggregated misfolded proteins. These pathological agents have been suggested to propagate in the brain via mechanisms similar to that observed for the prion protein, where a misfolded variant is transferred from an affected brain region to a healthy one, thereby inducing the misfolding and/or aggregation of correctly folded copies. This process has been characterized for several proteins, such as α-synuclein, tau, amyloid beta (Aβ) and less extensively for huntingtin and TDP-43. α-synuclein, tau, TDP-43 and huntingtin are intracellular proteins, and their aggregates are located in the cytosol or nucleus of neurons. They have been shown to spread between cells and this event occurs, at least partially, via secretion of these protein aggregates in the extracellular space followed by re-uptake. Conversely, Aβ aggregates are found mainly extracellularly, and their spreading occurs in the extracellular space between brain regions. Due to the inherent nature of their spreading modalities, these proteins are exposed to components of the extracellular matrix (ECM), including glycans, proteases and core matrix proteins. These ECM components can interact with or process pathological misfolded proteins, potentially changing their properties and thus regulating their spreading capabilities. Here, we present an overview of the documented roles of ECM components in the spreading of pathological protein aggregates in neurodegenerative diseases with the objective of identifying the current gaps in knowledge and stimulating further research in the field. This could potentially lead to the identification of druggable targets to slow down the spreading and/or progression of these pathologies.
Introduction
Most neurodegenerative diseases are characterized by the accumulation of misfolded protein aggregates. These include tau in tauopathies, α-synuclein in synucleinopathies, such as Parkinson’s disease (PD), TDP-43 in frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS), huntingtin in Hungtinton’s disease (HD) and amyloid beta (Aβ) in Alzheimer’s disease (AD) (Ross and Poirier, 2004). These aggregates exert varying degrees of toxicity on neurons and glial cells, ultimately driving their degeneration. Although each of these diseases originates in and affects different regions in the brain, all show some level of spreading of pathology over time. Each disease also exhibits characteristic rates and routes of distribution of pathological protein aggregates to other brain regions (reviewed in Brettschneider et al., 2015). For example, tau aggregation in AD initiates in the locus coeruleus and entorhinal cortex and subsequently spreads to the hippocampus, reaching the neocortex only at later stages of the disease. Aβ plaques instead are first observed in the neocortex, only afterward reaching deeper brain structures in later stages of AD. α-synuclein deposits, on the other hand, are first observed in the olfactory bulb and the dorsal motor nucleus of the vagus nerve, subsequently spreading to the midbrain and later, to the neocortex.
These pathological aggregates have the ability to induce downstream aggregation of natively folded proteins similarly to the prion protein (Vaquer-Alicea and Diamond, 2019). This finding, along with the observation that patterns of diffusion across brain regions are conserved between patients, suggests that some form of seed exist, which have the ability to travel across the brain and spread pathology. This spreading activity is distinguishable in two main classes, one involving intracellular proteins and the other, extracellular aggregates. Tau, α-synuclein, TDP-43 and huntingtin all have intracellular localization, and their aggregates also form intracellularly, whereas Aβ aggregates are found predominantly extracellularly. For intracellular proteins, aggregation is believed to start in a subset of cells from which seeds are then released, either by active secretion or passively due to cell death. Such proteopathic seeds would then be endocytosed by other cells and act as a template for the misfolding of endogenous proteins, thus causing further aggregation. Extracellular aggregation-prone Aβ, on the other hand, could move to different brain regions by simple diffusion in extracellular fluids and nucleate aggregation of locally generated Aβ.
These pathological proteins have been suggested to be present in the extracellular space in free forms, although at least some of them have also been found in extracellular vesicles (e.g., exosomes) or in tunneling nanotubes (Lee et al., 2011). It is thus clear that both classes of proteins will, at some point, be in the extracellular space and therefore enter into contact with components of the extracellular matrix (ECM). The ECM is a ubiquitous and complex protein network present in the space between cells of solid tissues, including the nervous system (Ruoslahti, 1996; Novak and Kaye, 2000; Krishnaswamy et al., 2019). Primarily consisting of laminins, collagens, glycoproteins and proteoglycans, the ECM undergoes regulated remodeling by virtue of extracellular proteases. All of these ECM components are secreted by neurons as well as glia and, in addition to providing physical support for these cells, play pivotal roles in regulating cell division, differentiation and migration, among other functions (Dityatev and Schachner, 2003; Dityatev et al., 2010). Since ECM components form an extracellular meshwork, they regulate the diffusion of molecules in the brain. Thus, rather unsurprisingly, components of the ECM can modulate the properties and spreading of these proteopathic seeds in neurodegenerative diseases. Interestingly, a growing body of evidence suggests that the levels of ECM components are severely affected in several neurodegenerative diseases, such as AD, PD, and ALS (Wong and Venkatachalam, 2019; NeuroLINCS Consortium et al., 2021; Downs et al., 2022; Johnson et al., 2022).
In this review, we summarize the known roles of ECM components in the spreading process, with the largest body of evidence existing for proteoglycans and extracellular proteases. Proteoglycans, which are either found in secreted form or bound to the plasma membrane, are glycosylated proteins which are post-translationally modified by the addition of glycosaminoglycans. The most abundant proteoglycans are heparan sulfate proteoglycans (HSPGs) and chondroitin sulfate proteoglycans (CSPGs), both of which are involved in different steps of the spreading process, including endocytosis of seeds, promoting aggregation and protecting aggregates from degradation (Holmes et al., 2013; Maïza et al., 2018). In addition to typical ECM proteases such as zinc-dependent matrix metalloproteinases (MMPs), additional proteases have been found in the extracellular space and altogether regulate ECM functions (Wang et al., 2008; Krishnaswamy et al., 2019). As detailed below, many of these enzymes cleave proteins that aggregate in neurodegenerative diseases, generating fragments with reported pro- and anti-aggregating effects. Although some of these proteases, such as calpains and cathepsins, mainly localize intracellularly and their activity on proteopathic proteins has been investigated in this context, their presence has also been reported in the extracellular space. Here, we discuss the possibility that cathepsins and calpains retain their activity on protein aggregates in the extracellular milieu. Emerging evidence suggests that this area of research requires further attention, especially as understanding the regulatory roles of ECM components on spreading may identify potential therapeutic targets that could reduce the progression of these devastating diseases.
Tau
Tau is a highly expressed neuronal protein that, through its microtubule binding region (MTBR), binds to and stabilizes axonal microtubules. The MTBR tends to form β-sheets which drive protein aggregation (Wang and Mandelkow, 2016). In pathological conditions, collectively named tauopathies, tau loses its affinity for microtubules, becomes hyper-phosphorylated, and aggregates into oligomers, fibrils and neurofibrillary tangles (NFT) (Braak et al., 1994). Pathological tau aggregation and propagation follows a characteristic spatiotemporal sequence between functionally connected brain regions (Braak and Braak, 1991). This has led to the hypothesis that pathological tau is secreted by a “donor” neuron into the extracellular space before being internalized by “acceptor” neurons (Holmes and Diamond, 2014). This hypothesis implies that tau is exposed to the extracellular environment where different proteases and ECM components could affect its ability to propagate pathology.
Proteoglycans
Heparan sulfate proteoglycans play an important role in the spreading of tau pathology (Figures 1Ai,Bi). HSPGs are proteoglycans characterized by one or more heparan sulfate (HS) groups linked to a protein core. HS is composed of disaccharide chains consisting mainly of glucuronic acid and N-acetyl-D-glucosamine (Sarrazin et al., 2011). HSPGs exist in different classes: transmembrane HSPGs, including glypicans and syndecans; serglycins, found in extracellular vesicles; and secreted HSPGs, such as perlecans and agrins (Sarrazin et al., 2011; Condomitti and de Wit, 2018). HSPGs bind to tau and have been shown to act at all stages of its spreading: secretion into the extracellular space (Zehe et al., 2006; Katsinelos et al., 2018), cellular uptake (Holmes et al., 2013; De La-Rocque et al., 2021) and self-assembly into higher-order states (Zhao et al., 2021). Tau binds to heparin, a densely sulfated form of HSPG, through both the N-terminus and the MTBR (Goedert et al., 1996). The sulfate moieties on HSPGs appear to be crucial for tau binding, requiring both 3-O- and 6-O-sulfation (Zhao et al., 2017, 2020; Stopschinski et al., 2018). Heparin induces a conformational change in the MTBR and its flanking region that exposes previously masked tau phosphorylation sites and can induce oligomerization (Paudel and Li, 1999; Sibille et al., 2006; Elbaum-Garfinkle and Rhoades, 2012). However, heparin and related molecules inhibit tau uptake (Zhao et al., 2017; Weisová et al., 2019; Puangmalai et al., 2020), thus preventing tau fibrils from driving intracellular aggregation (Holmes et al., 2013), as shown by the heparin mimetic F6 (Holmes et al., 2013). These results suggest that an excess of extracellular HSPGs could have similar beneficial effects. However, a synthetic heparinoid with nanomolar affinity for tau failed to show any effect on tau pathology after chronic administration in vivo (Stopschinski et al., 2020). It is important to note that most studies have concluded that cell surface-bound HSPGs are involved in tau transfer without assessing the possible contribution of extracellular HSPGs.
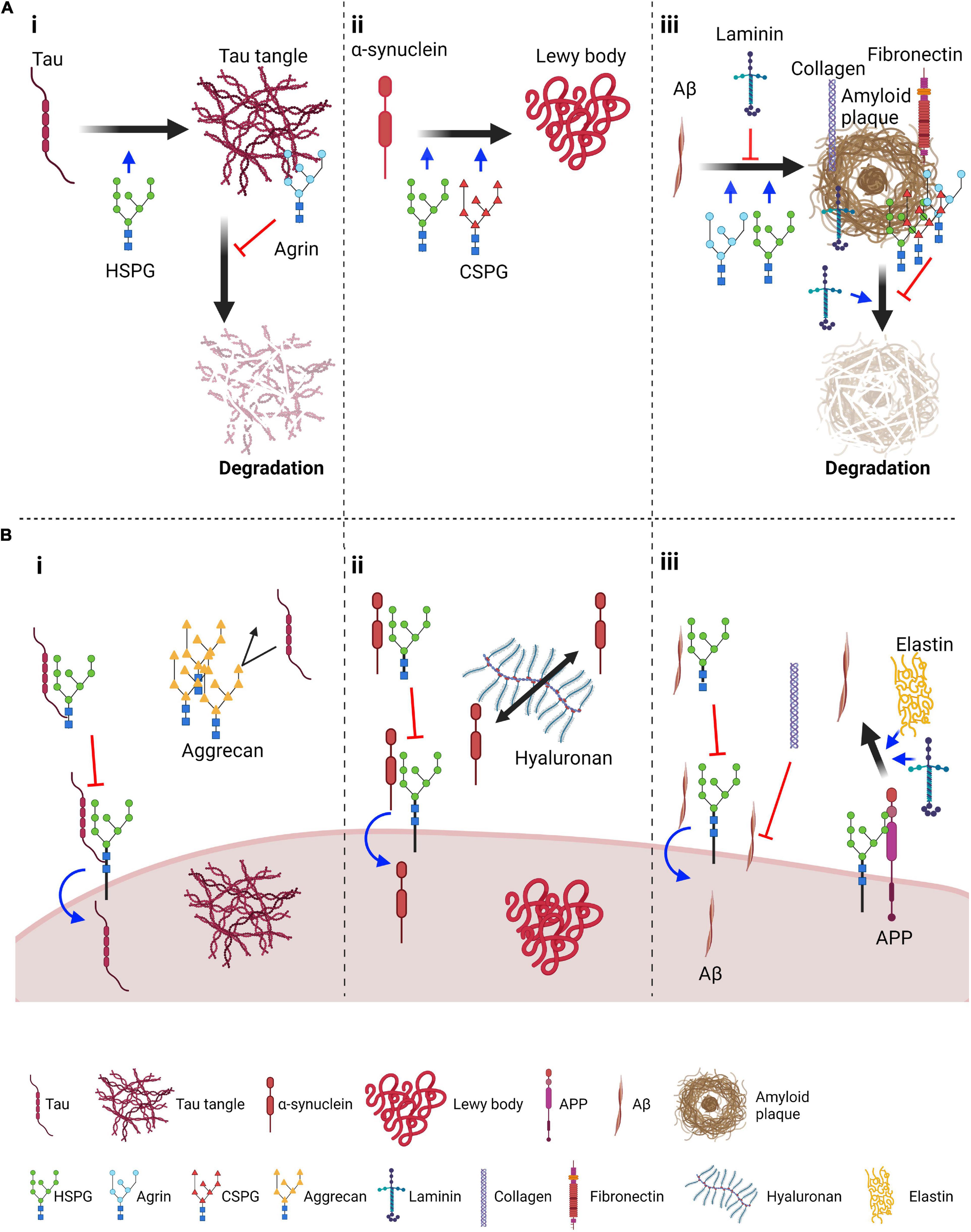
Figure 1. Roles of proteoglycans and ECM components in the aggregation and spreading of tau, α-synuclein and amyloid β. (A) Suggested roles of proteoglycans and core ECM components in aggregation and degradation of pathological protein aggregates: (i) Tau panel: HSPGs promote tau aggregation; Agrin, an extracellular HSPG, localizes on neurofibrillary tangles and prevents their degradation; (ii) α-synuclein panel: HSPGs and CSPGs promote aggregation of α-synuclein; (iii) Aβ panel: HSPGs and agrin can promote Aβ aggregation whereas laminin has been suggested to impair this process. Several proteoglycans, including HSPGs, agrin and CSPGs, and ECM components such as laminin, collagen and fibronectin associate with amyloid plaques. Of these, laminin enhances plaque degradation whereas proteoglycans inhibit this process. (B) Suggested role of proteoglycans and ECM components in endocytosis and spreading: (i) Tau panel: HSPGs on the plasma membrane favor tau seed endocytosis. In contrast, if tau is bound to extracellular HSPGs, its interaction with surface HSPGs might be inhibited, thus reducing tau endocytosis. Aggrecan perineuronal nets have been shown to reduce propagation of tau pathology, possibly by acting as a barrier; (ii) α-synuclein endocytosis is also promoted by HSPGs and similarly extracellular HSPGs can inhibit endocytosis. In the extracellular space, hyaluronan was found to promote spreading of α-synuclein pathology; (iii) Aβ panel: as for tau and α-synuclein, transmembrane HSPGs can promote internalization of Aβ, and this might be inhibited by extracellular interactions. Collagen has been found to reduce the interaction of Aβ peptides with the cell surface. HSPGs have also been found to promote Aβ production from APP, a process that is also promoted by ECM components such as elastin and laminin. Figure was prepared with Biorender (Biorender.com).
Agrin, a major extracellular HSPG, accumulates in AD brains and more specifically, in NFTs (Verbeek et al., 1999; Kroger and Schroder, 2002; Smith and Hilgenberg, 2002; Del Campo Milan et al., 2015). Since agrin contains protease-inhibiting domains, it has been suggested to protect protein aggregates against extracellular proteolysis, leading to the accumulation of these deposits (Verbeek et al., 1999). In addition, agrin may participate in tangle formation because sulfated glycosaminoglycans have been shown to stimulate tau phosphorylation, thus promoting the formation of paired helical filaments (PHFs) (Goedert et al., 1996; Wang et al., 1996; Hasegawa et al., 1997). However, whether or not agrin displays these activities and how it interacts with tau tangles remains to be elucidated.
The CSPG aggrecan forms perineuronal nets predominantly ensheathing fast spiking interneurons (Morawski et al., 2012; Suttkus et al., 2014; van’t Spijker and Kwok, 2017). These nets were proposed to be neuroprotective against pathological tau, since areas with high densities of perineuronal nets in post-mortem AD brains were largely spared of tangles, even at late disease stages (Brückner et al., 1999, 2008; Diamandis et al., 2000; Morawski et al., 2010a,b). Follow-up studies confirmed that the protective action of perineuronal nets was, in fact, mainly mediated by aggrecan. Most interestingly, aggrecan was shown to generate an external barrier restricting internalization and distribution of exogenous tau in organotypic slices (Nowicka et al., 2009; Suttkus et al., 2014, 2016). Indeed, aggrecan knock-out mice presented elevated tau uptake (Suttkus et al., 2016). Based on these results, it was hypothesized that these CSPGs may act by inhibiting the interaction of tau with HSPGs. However, in a recent study, mice expressing human tau carrying the FTD-linked mutation P301L crossed with heterozygous aggrecan KO mice, displayed changes in the expression and phosphorylation of tau but unaltered distribution of tau aggregates (Schmidt et al., 2021).
Proteases
Several proteases have been shown to cleave tau (Table 1). Multiple studies have shown elevated levels of several MMPs in tauopathies as well as demonstrated functional links between different MMPs and tau. For example, upregulated MMP-3 levels were found in the cortex of AD-like amyloidosis transgenic rat models (Pentz et al., 2021), and the concentration of MMP-3 and levels of total and phosphorylated tau positively correlate in the cerebrospinal fluid (CSF) of AD patients (Stomrud et al., 2010; Hanzel et al., 2014). Although MMP-9 is enriched in AD patients’ brains and co-localizes with tau, it is sparsely present in extracellular NFTs (Hernandes-Alejandro et al., 2020). Nübling et al. (2012) tested the effect of MMP-3 and MMP-9 on tau oligomer formation and aggregation. While MMP-3 mildly reduced tau aggregation, MMP-9 processing promoted oligomerization (Nübling et al., 2012; Wang et al., 2014). Multiple potential MMP-3 cleavage sites were identified within the MTBR of tau (Nübling et al., 2012), suggesting this protease could inhibit tau aggregation by degrading regions crucial for oligomer formation. In contrast, MMP-9 cleavage sites were mainly located either in the N-terminal region or close to the C-terminus (Nübling et al., 2012), thus sparing the MTBR and facilitating the generation of tau oligomers. More recently, docking simulations have predicted that a high-affinity complex can be formed between MMP-9 and full-length tau (Hernandes-Alejandro et al., 2020). This binding involves the catalytic domain of MMP-9, suggesting that this interaction could be initiated when MMP-9 is active. Interestingly, MMP-9 can be directly activated by MMP-3 (Ogata et al., 1992; Okada et al., 1992; Shapiro et al., 1995), implying that elevated MMP-3 levels might result in increased MMP-9 activity, indirectly facilitating tau aggregation. MMP-2 also has the capacity to cleave recombinant tau in vitro but fails to process its hyper-phosphorylated forms in NFTs (Terni and Ferrer, 2015). It is thus possible that the accumulation of MMP-2 around NFTs found in the entorhinal cortex at the early stages of AD (Terni and Ferrer, 2015) is a potential response aimed at eliminating the production of toxic fragments in AD brains. The cleavage sites of MMP-2 on tau, however, are yet to be determined. In conclusion, MMP-2, MMP-3 and MMP-9 show differential actions on tau aggregating behavior, suggesting their differential contribution to tau pathology.
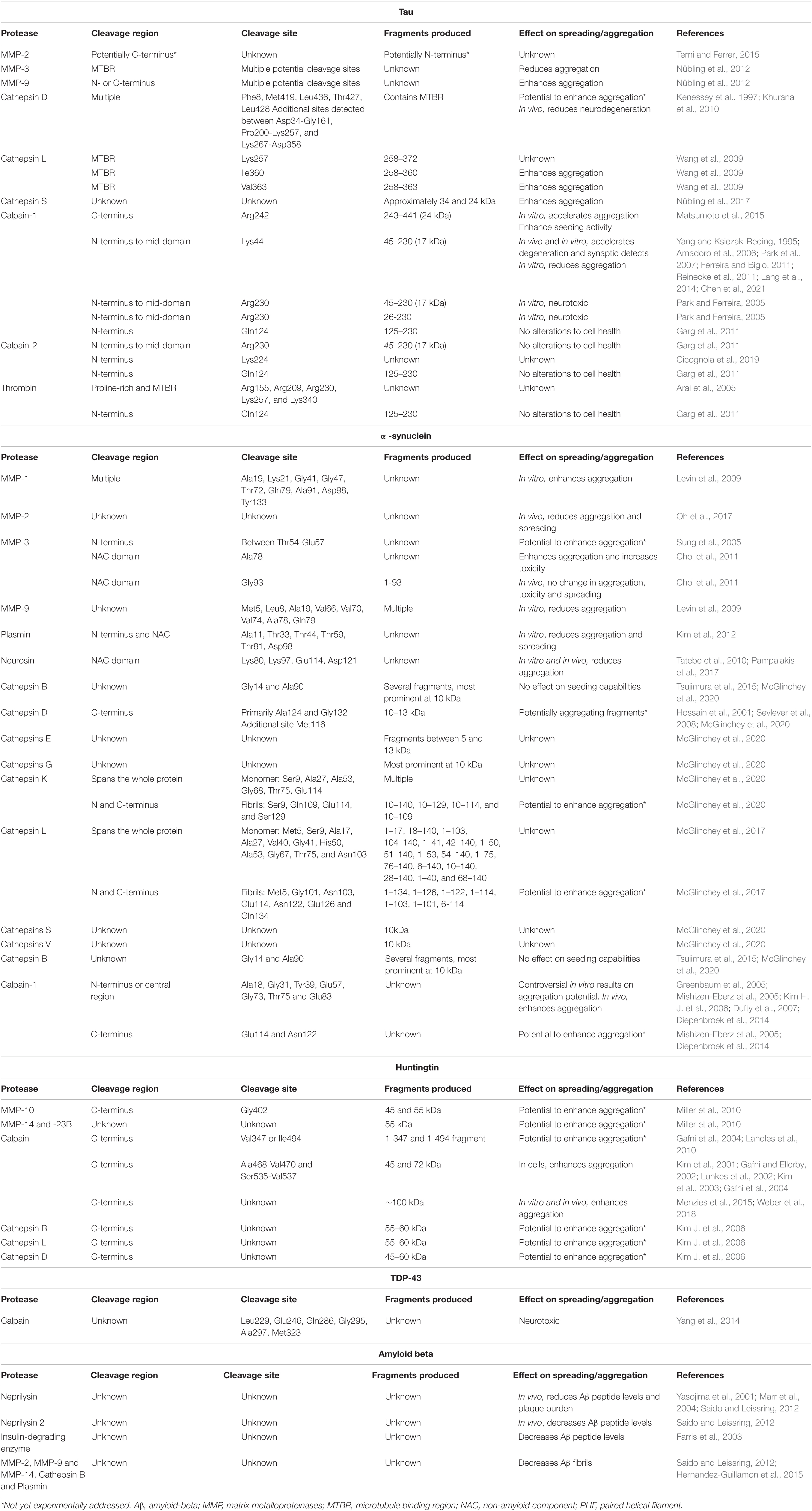
Table 1. This table summarizes the known extracellular proteases of prion-like proteins, their region and site of cleavage, the resulting fragment and the effect of their activity on spreading and aggregation of the pathological proteins.
Thrombin was also found to cleave tau at multiple arginine and lysine residues in the MTBR and proline-rich domains (Arai et al., 2005; Quinn et al., 2018; Zhang et al., 2021). In AD brains, thrombin was found to be upregulated and co-localized with amyloid plaques, microglia and NFTs (Arai et al., 2006). However, PHFs of tau extracted from AD brains were more resistant to thrombin cleavage compared to dephosphorylated PHFs (Arai et al., 2005). The seeding potential of tau fragments produced by thrombin is largely unknown, but tau 125–230 generated by thrombin-mediated cleavage at Gln124-Ala125 was shown to be non-toxic (Garg et al., 2011). This site is also cleaved by calpain-1 and calpain-2 (Garg et al., 2011). However, it is important to note that thrombin-cleaved tau fragments are yet to be identified in tauopathy brains, thereby questioning the pathophysiological relevance of this process.
Several cathepsins have been shown to interact with and modulate tau spreading. As previously mentioned, these proteases are mainly localized to lysosomes, but they can also be secreted in the extracellular space (Boonen et al., 2016; Vidak et al., 2019; Niemeyer et al., 2020). Whilst the majority of data on the action of cathepsins on tau mostly reflects their activity in lysosomes, independent experiments have been conducted either in vitro using recombinant proteins or using cathepsin knockout animals. Therefore, a role for extracellular cathepsins cannot be ruled out. Cathepsins B, D, L, and S have all been proposed to cleave tau. Although cathepsin B accumulates in close proximity to NFTs and amyloid plaques (Ii et al., 1993), there is no direct evidence that this protease can cleave tau. In contrast, cathepsin D cleaves recombinant tau at several sites, mostly sparing the MTBR (Kenessey et al., 1997), and as such these fragments could retain the ability to generate higher-order PHFs. At least one of these fragments was produced at neutral pH in vitro, suggesting that this cleavage could also occur in the extracellular space (Kenessey et al., 1997). Furthermore, cathepsin D upregulation was detected in an aging Drosophila model of tauopathy. The same group observed enhanced tau-induced neurodegeneration in cathepsin D-deficient flies which suggests that this protease may have a neuroprotective effect (Khurana et al., 2010). Cathepsin L has also been found to cleave tau at neutral pH. This activity targets sites within the MTBR, generating several fragments, such as tau 258–360 and tau 258–363, which enhance the aggregation of the full-length protein (García-Sierra et al., 2008; Wang et al., 2009; Zhang et al., 2021). Cathepsin S has been seen to associate with NFTs and its levels are elevated in the brain of AD patients (Lemere et al., 1995; Munger et al., 1995; Nübling et al., 2017). Treatment with cathepsin S in vitro yields a distinct cleavage pattern of tau, in which the MTBR seems to remain intact, allowing its association to NFTs. However, this study did not specify whether cleavage occurred at an acidic or neutral pH (Nübling et al., 2017). It is important to note that cathepsin-cleaved tau fragments have yet to be identified in AD brains, highlighting the need for further investigations to unravel the pathophysiological relevance of this protease in AD.
Calpains are proteases mainly found in the cell cytosol (Goll et al., 2003), albeit secretion of calpain has also been observed (Frangié et al., 2006; Letavernier et al., 2012; McDougall et al., 2021), including from isolated rat brain synaptosomes (Pestereva et al., 2021). Calpain has been detected in the CSF, though it remains unclear whether its presence is due to leakage from dying cells (Laske et al., 2015). This dual localization makes investigations on the role of calpain in the context of tau spreading and seeding challenging and as such, the action of extracellular calpains has not been directly investigated as yet. Both calpain-1 and -2 cleave tau (Chesser et al., 2013), and play opposing roles in inducing neurodegeneration (Baudry and Bi, 2016). Calpain-1 activity is significantly upregulated in AD cortical brain tissue from Braak stage III (Kurbatskaya et al., 2016). Much of the interest surrounding the role of calpain in tau pathology has centered on the 17 kDa N-terminal to mid-domain tau fragment (45–230), which is generated through cleavage by calpain-1 at Lys44-Glu45 (Yang and Ksiezak-Reding, 1995) and by calpain-1 (Park and Ferreira, 2005) or -2 (Garg et al., 2011) at Arg230-Thr231. These events have been observed in vitro, suggesting calpain-1 and -2 could also exert their activity in the extracellular space, although direct evidence is still lacking. This fragment has been observed in patients affected by AD and other tauopathies, such as progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) (Ferreira and Bigio, 2011; Garg et al., 2011). However, the specific effect of this tau fragment remains controversial as it was found to induce apoptosis (Park and Ferreira, 2005; Amadoro et al., 2006; Park et al., 2007; Reinecke et al., 2011; Lang et al., 2014) or morphological changes in cell lines and primary neurons (Chen et al., 2021), whereas other groups have observed no such alterations (Garg et al., 2011). Nonetheless, both Drosophila and mouse models overexpressing tau 45–230 showed accelerated hippocampal degeneration and synaptic defects (Reinecke et al., 2011; Lang et al., 2014). However, when this 17 kDa fragment was incubated with full-length tau, it significantly lowered aggregate formation, suggesting a possible protective effect (Ferreira and Bigio, 2011). Fascinatingly, aggregation and phosphorylation of full-length tau protected it from calpain cleavage (Ferreira and Bigio, 2011), which is in line with previous data indicating aggregated tau is less susceptible to protease cleavage (Yang and Ksiezak-Reding, 1995; Yang et al., 1997). In a recent study investigating stroke biomarkers, this 17 kDa tau fragment accumulated in primary neurons and their media upon hypoxic treatment, and was decreased by calpain inhibitors (Chen et al., 2021). These findings suggest that calpain-mediated cleavage is specific and can be initiated under different pathological conditions. Similarly, the AD-relevant fragment, tau 26–230, is generated by calpain-1 (Park and Ferreira, 2005) and calpain-2 (Garg et al., 2011) cleavage at Arg230-Thr231. In contrast, Matsumoto and colleagues (Matsumoto et al., 2015) have demonstrated that tau is cleaved in vitro by calpain-1 at Arg242-Lys243 producing a 24 kDa C-terminal fragment lacking the N-terminal projection domain (aa 243–441; CTF24). This truncated tau accelerated heparin-induced aggregation and was unable to support microtubule assembly. Furthermore, CTF24 efficiently propagated to other tau-expressing cells, where it displayed higher aggregation and seeding activity than full-length tau (Matsumoto et al., 2015). Interestingly, active calpain-2 co-localizes with tau filaments in AD, Down syndrome and FTD brains (Adamec et al., 2002a,b). A recent study by Cicognola et al. (2020) identified that calpain-2, but not calpain-1, cleaves tau at Lys224, generating an N-terminal fragment previously found to be enriched in CSF in tauopathies (Cicognola et al., 2019). Knockdown of the calpain-2 catalytic subunit gene caused a significant reduction of this N-244 tau fragment in cell-conditioned media (Cicognola et al., 2019). Overall, calpain cleavage appears to promote tau aggregation, thus potentially enhancing its spreading.
α-Synuclein
α-Synuclein is a small cytosolic protein highly abundant in neurons, and is predominately present as a soluble monomer in physiological conditions (Lee and Trojanowski, 2006). It is composed of 140 amino acids forming three main regions: the N-terminus (aa 1–60) which contains apolipoprotein binding motifs, the central non-amyloid component (NAC) (aa 61–95), which has the propensity to fold into beta sheets, and a negatively charged mostly unstructured C-terminus (aa 96–140) (Stefanis, 2012). The function of α-synuclein remains elusive, although reports on its presynaptic localization and ability to interact with lipids suggest it might have a role in neurotransmitter release (Lee and Trojanowski, 2006; Bernal-Conde et al., 2019). The presence of misfolded, aggregated α-synuclein in Lewy bodies is the molecular hallmark of PD and other neurological conditions termed synucleinopathies, and mutations in its coding gene, SNCA, are causative of PD (Stefanis, 2012). α-synuclein was the first pathological protein shown to behave in a manner similar to the prion protein (Karpowicz et al., 2019) and is found extracellularly in the brain and in extracellular fluids, such as CSF (El-Agnaf et al., 2003; Lee et al., 2005). Injection of anti-α-synuclein antibodies in the mouse brain parenchyma halts propagation of α-synuclein pathology (Tran et al., 2014), supporting the possibility of its interneuronal transfer as a free protein. Similar to tau, α-synuclein endocytosis has been shown to occur through interaction with HSPGs and CSPGs (Figures 1Aii,Bii), and it is similarly susceptible to cleavage by extracellular proteases (Table 1).
Proteoglycans
In C17.2 mouse-derived neural stem cells, internalized α-synuclein fibrils colocalize with HSPGs (Holmes et al., 2013). Similar to tau, α-synuclein endocytosis in cell lines is inhibited by co-application of heparin in a dose-dependent manner (Holmes et al., 2013; Ihse et al., 2017). Heparin acts competitively by binding α-synuclein at sites responsible for its interaction with HSPGs, thus blocking its internalization. However, heparin has been found to be less efficient in blocking internalization of monomeric and oligomeric forms of α-synuclein (Ihse et al., 2017). A neuroblastoma cell line exposed to heparin lyases I, II, and III showed reduced endocytosis of α-synuclein fibrils and similarly, cells lacking enzymes responsible for HPSGs biogenesis fail to internalize α-synuclein (Ihse et al., 2017). Additionally, HSPG sulfation was found to be important, as treatment with chlorate, which inhibits sulfation, reduces α-synuclein fibril internalization (Holmes et al., 2013; Hudák et al., 2019). Further analyses on the sulfation requirements of HSPGs was carried out using differentially sulfated heparin. When applied to C17.2 cells, 2-O, 6-O and N-desulphated heparin showed lower efficiency in inhibiting α-synuclein uptake and seeded aggregation compared to standard heparin. Shorter heparin chains are also less efficient in inhibiting α-synuclein uptake and seeding. Accordingly, a CRISPR/Cas9 genetic screen in HEK293 cells identified EXT1, 2, and 3, which mediate the initiation and elongation of the glycosaminoglycan chain in HSPGs, to be involved in α-synuclein uptake and seeding. Similarly, NDTS1, which encodes an enzyme responsible for N-deacetylation and N-sulfation of HSPGs, also appeared to play a role, as its knockout reduced uptake and seeding of α-synuclein. Although the ablation of HS6ST2, an enzyme involved in the 2-O sulfation of HSPGs, did not show overt effects on α-synuclein fibril uptake, its overexpression decreased α-synuclein internalization but increased seeding. This suggests a more complex role of 2-O sulfation on α-synuclein spreading (Stopschinski et al., 2018). Zhang et al. (2020) recently analyzed the interaction between α-synuclein and HSPGs by molecular modeling, suggesting that α-synuclein fibrils display more stable binding to HSPGs compared to its monomeric or dimeric forms, possibly explaining the stronger inhibitory effect of heparin on fibril internalization (Zhang et al., 2020).
In a follow up study, treatment of neuroblastoma cells or primary neurons with heparinase failed to inhibit internalization of N-terminal acetylated α-synuclein monomers or fibrils. Since this modification is present at high levels in vivo (Burré et al., 2013), this result questions the physiological importance of HSPGs in this process. In contrast, peptide-N-glycosidase F (PNGase F), which cleaves complex N-linked glycans from glycoproteins, reduced endocytosis of both fibrillar and monomeric N-acetylated α-synuclein but did not affect non-acetylated α-synuclein. Interestingly, acetylated α-synuclein was found to interact with glycans in the absence of their protein core (Birol et al., 2019). Of note, surface membrane proteins as well as secreted proteins and ECM components are heavily glycosylated (Scott and Panin, 2014), creating an ideal multivalent binding environment for pathological α-synuclein. These data indicate that HSPGs are fundamental players in the internalization of α-synuclein fibrils.
CSPGs have also been implicated in α-synuclein aggregation and spreading (Lehri-Boufala et al., 2015; Mehra et al., 2018). Accordingly, incubation of α-synuclein with chondroitin sulfate A and B in vitro enhances the formation of aggregates able to enter SH-SY5Y cells (Mehra et al., 2018). Interestingly, chondroitin sulfate, and glycosaminoglycans in general, inhibit cathepsin D, suggesting that the endocytosis of an α-synuclein-chondroitin sulfate complex could induce higher seeding effects due to lysosomal inhibition (Lehri-Boufala et al., 2015). However, co-injection of α-synuclein aggregates with chondroitinase in mice did not change the spread of pathology. On the other hand, degradation of hyaluronan, another major component of the ECM, reduced α-synuclein pathology in the same model, although the mechanism at the basis of this effect is currently unclear (Soria et al., 2020).
Proteases
It is generally assumed that fragments of α-synuclein must contain the NAC region to propagate pathology. In vitro studies using recombinant α-synuclein found that negative charges at the α-synuclein C-terminus counteracts the aggregation propensity of the NAC domain (Izawa et al., 2012). As such, cleavage of the C-terminus by proteases increases aggregation and seeding (Sorrentino et al., 2018, 2020; Chakroun et al., 2020; Sorrentino and Giasson, 2020). Interestingly, α-synuclein carrying PD-linked mutations is more efficiently cleaved at the C-terminus than the wildtype protein (Li et al., 2005).
Conversely, the effect of N-terminal truncation is less clear. Most studies report no change (Luk et al., 2009; Volpicelli-Daley et al., 2011) or a decrease (Luk et al., 2016) in seeded aggregation, even though removal of the first two apolipoprotein binding motifs can lead to an increase (Kessler et al., 2003; Sorrentino and Giasson, 2020). Increased propagation of α-synuclein pathology after brain injection of recombinant α-synuclein lacking the first 10 or 30 residues, compared to full length fibrils has been observed (Terada et al., 2018), suggesting that the N-terminus of α-synuclein can also modulate aggregation. Therefore, the action of extracellular proteases has the potential to both negatively and positively modulate α-synuclein propagation.
Different MMPs such as MMP-1, -2, -3, -9, and -14 cleave α-synuclein, with MMP-3 proven to be the most effective (Levin et al., 2009). In vitro tests have shown cleavage at multiple sites at the N-terminus of the NAC (Sung et al., 2005). These cleavage products were observed in the extracellular media of neuroblastoma SK-N-BE cells overexpressing human α-synuclein. MMP-3-cleaved α-synuclein had an elevated propensity to aggregate in vitro and displayed increased toxicity compared to full length α-synuclein when applied to SK-N-BE cells (Sung et al., 2005). Interestingly, MMP-3 is elevated in rat brains exposed to 6-hydroxydopamine or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, two classical models of parkinsonism (Sung et al., 2005; Leem et al., 2020). Accordingly, MMP-3 localization in Lewy bodies was detected in the substantia nigra of PD brains (Choi et al., 2011). Choi et al. (2011) investigated the MMP-3-dependent cleavage of α-synuclein harboring mutations associated with PD, and observed that in vitro, the A53T mutant is processed more efficiently than the wildtype and A30P mutant proteins, especially at sites 78–79 and 91–93. In contrast, the overexpression of MMP-3 and α-synuclein in COS cells produced lower levels of insoluble α-synuclein aggregates. However, overexpression of the A53T-containing 1–93 α-synuclein fragment in mice induced increased toxicity and formation of Lewy body-like structures in dopaminergic neurons of the substantia nigra, both at the site of injection of the α-synuclein-encoding adeno-associated virus and contralateral side (Choi et al., 2011). These results imply that the 1–93 fragment, which contains the full NAC domain and lacks the C-terminus, is prone to aggregation and spreading. Nevertheless, this is not the only fragment produced by MMP-3 cleavage and the overall effect of MMP-3-cleaved α-synuclein has yet to be conclusively evaluated in vivo.
MMP-2 has also been suggested to degrade α-synuclein fibrils. Injection of MMP-2 into animals inoculated with α-synuclein in the cortex and striatum led to reduced levels of insoluble and oligomeric α-synuclein. In addition, human α-synuclein positivity was limited to the injection site in MMP-2 treated animals, suggesting reduced spreading (Oh et al., 2017).
Plasmin is a serine protease that degrades fibrin blood clots, and is also involved in inflammation, collagenase activation and synaptic plasticity (Pang et al., 2004). Application of recombinant plasmin to α-synuclein monomers, oligomers or fibrils resulted in their degradation, including when they harbored several PD-linked mutations, including A53T. Interestingly, α-synuclein found in the media of SH-SY5Y cells was also cleavable by plasmin at different sites spanning the N-terminal domain and the NAC. Using a propagation model utilizing SH-SY5Y cells expressing α-synuclein, co-cultured with the microglia-like cell line BV2 lacking α-synuclein, Kim et al. (2012) showed that exogenous plasmin added to the media could reduce spreading between these two cell types. However, further studies are necessary to confirm the importance of plasmin cleavage on α-synuclein aggregation and spreading in vivo.
α-Synuclein has been found to be degraded by neurosin, also called kallikrein 6. Similar to other members of the kallikrein family, neurosin is a secreted trypsin-like serine protease that is activated extracellularly by sequential cleavage (Yoon et al., 2008) and can be found in human CSF (Diamandis et al., 2000). In vitro treatment of recombinant α-synuclein with neurosin generates several fragments by cleavage within and in the proximity of the NAC domain. Neurosin digestion was found to inhibit α-synuclein aggregation in vitro (Iwata et al., 2003), and treatment of α-synuclein oligomers with neurosin induced their almost complete degradation (Spencer et al., 2013). Pro-aggregation variants such as α-synuclein phosphorylated at Ser129 (Kasai et al., 2008) or carrying the PD-linked mutations A30P, A53T or E46K showed reduced cleavage by neurosin (Iwata et al., 2003; Kasai et al., 2008; Spencer et al., 2013). Of note, neurosin levels are lower in brains of patients affected by dementia with Lewy bodies and in α-synuclein transgenic mouse models (Spencer et al., 2013). Furthermore, overexpression of neurosin in HEK293 cells or in cortical neurons induced degradation of α-synuclein in the culture media, whereas primary neurons from neurosin knockout mice showed increased α-synuclein internalization and aggregation (Tatebe et al., 2010; Pampalakis et al., 2017). In addition, neurosin can also activate proMMP-2 (Pampalakis et al., 2017) and an unidentified extracellular protease (Ximerakis et al., 2014), further promoting α-synuclein degradation. Although it is unclear whether neurosin acts directly or through activation of a downstream protease, its activity appears to strongly reduce both aggregated and monomeric forms of α-synuclein.
Cathepsins B, D, E, G, K, L, S and V have been shown to cleave α-synuclein, both in its monomeric and fibrillar forms. In particular, both cathepsins L and K were shown to completely ablate α-synuclein fibrils, whereas cathepsins B, D, E, G, S and V generate small α-synuclein fragments (McGlinchey and Lee, 2015; McGlinchey et al., 2017, 2020). Importantly, degradation of α-synuclein by cathepsins L and K was achieved upon long incubation (16 h) and at an acidic pH, which would be typically found in lysosomes. Shorter incubation times or treatment at a neutral pH mimicking that of the extracellular space, generated fragments truncated at the N- and C-termini (McGlinchey et al., 2017, 2020). As discussed above, these fragments display an increased aggregation propensity, albeit this was not tested experimentally. Cathepsin B was found to cleave both α-synuclein monomers and fibrils in vitro upon incubation at low pH. It is thus unclear whether this activity could be retained at neutral pH in the extracellular space (McGlinchey and Lee, 2015; Tsujimura et al., 2015). C-terminal cleaved fragments were also observed upon addition of recombinant cathepsin B to lysates of 3D5 cells expressing human α-synuclein, or to mouse and human brain extracts (Sevlever et al., 2008; Tsujimura et al., 2015). Cathepsin D was also found to cleave recombinant α-synuclein in vitro at residues 116, 124 and 132, thus potentially generating aggregating fragments (Hossain et al., 2001; Sevlever et al., 2008). Since the proteolytic activity of both cathepsin B and D on α-synuclein was only observed at acidic pH, it is questionable whether these events occur in the extracellular space. However, cathepsin D knockout mice display insoluble α-synuclein in brain extracts, even in the absence of overexpression. A similar result was observed in human post-mortem brains affected by mutations in the CTSD gene (Cullen et al., 2009). Accordingly, knockdown of the C. elegans Ctsd ortholog caused increased aggregation of overexpressed human α-synuclein, whereas overexpression of Ctsd in α-synuclein expressing worms increased survival of DA neurons, an effect that was not present upon expression of cathepsins B and L (Qiao et al., 2008).
The effects of calpain-1 on α-synuclein have been studied in detail. However, similar to the action of calpain on tau, its activity on α-synuclein has been assumed to occur intracellularly. Calpain cleaves monomeric α-synuclein mostly within the N-terminal or central region (Mishizen-Eberz et al., 2003; Greenbaum et al., 2005; Kim H. J. et al., 2006; Diepenbroek et al., 2014). The aggregation potential of monomeric α-synuclein treated with calpain is still controversial (Mishizen-Eberz et al., 2005; Dufty et al., 2007). In vitro, calpain-1 cleaves both wildtype and PD mutant forms of fibrillar α-synuclein within the C-terminus (Mishizen-Eberz et al., 2003; Diepenbroek et al., 2014), which may increase the aggregating potential of α-synuclein. Diepenbroek and colleagues evaluated the role of calpain in vivo by crossing mice expressing human α-synuclein carrying the A30P mutation with a calpastatin knockout or overexpressing mouse model. Overexpression of calpastatin, which acts as a calpain inhibitor, reduced α-synuclein aggregates in the brain, whereas calpastatin knockout mice showed the opposite effect (Diepenbroek et al., 2014). Similar findings were obtained by treating mice expressing human wildtype α-synuclein with the calpain inhibitors gabadur and neurodur. This treatment decreased insoluble α-synuclein aggregates with a concomitant reduction in gliosis, neurodegeneration and hyperactivity (Hassen et al., 2018). Therefore, calpain processing of α-synuclein appears to increase its aggregation potential.
Huntingtin
Huntingtin is a large 384 kDa protein that contains an N-terminal region, which functions as a nuclear export signal, followed by a stretch of glutamines that contains between 9 and 35 residues in healthy subjects. Higher numbers of CAG repeats, which encode for these glutamines, in the huntingtin gene are associated with aggregation and HD. The remaining portion of this protein is far less characterized, although it presents several HEAT repeats that are important for protein–protein interactions. Huntingtin is involved in several functions from vesicular trafficking to translation and autophagy (Saudou and Humbert, 2016). The neuronal spread of huntingtin pathology has been described; however, the evidence is less definitive compared to other proteins discussed in this review (Pecho-Vrieseling et al., 2014). Aggregates of recombinant polyQ peptides are internalized and induce further aggregation in HEK293 cells (Ren et al., 2009). Moreover, huntingtin aggregates extracted from the brain of R6/2 mice, which overexpress human huntingtin exon1 carrying ∼115 CAG repeats, were able to increase aggregation of overexpressed huntingtin in Neuro2a cells (Nekooki-Machida et al., 2009). However, in HD patients that received fetal neural allografts, aggregated huntingtin was found in the graft region in the extracellular space but not in neurons, arguing against the ability of aggregated huntingtin to spread between cells in vivo (Cicchetti et al., 2014). In this light, further research is required to confirm the relevance of huntingtin spreading in the progression of HD.
Aggregation of huntingtin is driven by polyQ expansion and, as such, this region needs to be retained for huntingtin to act as a seed. In addition, N-terminal fragments of huntingtin have been shown to be more toxic than the full-length protein. For example, shorter versions of huntingtin terminating at residue 145 or 650 are more aggregation-prone in a polyQ length-dependent manner and induce increased sensitivity to oxidative stress in primary neurons compared to the full-length protein. Interestingly, these fragments recruit endogenous huntingtin into aggregates (Martindale et al., 1998). Further studies confirmed that the shorter the huntingtin fragment, the more aggregation prone it is, as long as the polyQ region is retained (Hackam et al., 1998). This observation therefore suggests that extracellular proteolytic cleavage could modulate disease progression (Graham et al., 2006; Ratovitski et al., 2011; O’Brien et al., 2015). C-terminal fragments have been less extensively studied. Work from El-Daher et al. (2015) found that the 587–3144 fragment could cause toxicity on its own, but had a protective effect against fragment 1–167–mediated toxicity, whereas it potentiates 1–586 toxicity in both striatal cells and Drosophila.
Proteases
Truncated huntingtin species are generated through cleavage by different proteases that can be found in the extracellular space such as MMPs, calpains and cathepsins (Table 1). MMP-10, -14 and -23B were identified in an shRNA-based knockdown screen in HEK293 cells looking for a reduction in huntingtin fragments. Knockdown or pharmacological inhibition of these MMPs reduced toxicity in a striatal cell line expressing huntingtin with 111 glutamines, as well as in Drosophila lines expressing the first 336 amino acids of the human huntingtin protein with 128 glutamines. MMP-10 was confirmed to cleave recombinant huntingtin both in vitro and in cell lysates, mostly producing a 45 kDa fragment which could also be observed in brain extracts from HD patients. MMP-10 and -14 activity was increased in a striatal cell line expressing huntingtin with 111 glutamines compared to a non-pathogenic variant with only 7 (Miller et al., 2010). Furthermore, increased MMP-2, -3 and -10 activity was observed in neural stem cells from HD patients compared with their isogenic lines, where the polyQ expansion was corrected. In contrast to this, the activities of MMP-9 and -14 were found to be reduced in HD (Naphade et al., 2017).
Calpain also mediates proteolytic processing of huntingtin. Application of recombinant calpain-1 to recombinant huntingtin or to mouse brain extracts caused the appearance of fragments with molecular weights between 45 and 72 kDa, which could also be observed in post-mortem brain samples of both healthy and HD-affected subjects (Kim et al., 2001, 2003; Gafni and Ellerby, 2002; Lunkes et al., 2002). However, high-polyQ huntingtin appeared to be more resistant to calpain cleavage compared to lower repeat mutants in vitro (Gafni and Ellerby, 2002). Calpain-resistant variants showed reduced aggregation and toxicity in HEK293T cells compared to calpain-cleavable huntingtin (Gafni et al., 2004). In addition, calpain-derived fragments have been observed both in mouse models expressing human huntingtin with 150 polyQ (Gafni et al., 2004; Landles et al., 2010), and in post-mortem caudate samples from HD patients which also presented increased calpain levels (Gafni and Ellerby, 2002; Gafni et al., 2004). These results were confirmed in Drosophila, where knocking out calpain reverted the toxic effects of huntingtin overexpression, although this effect appeared to be due to the intracellular activity of calpain, since a double knockout for calpain and the autophagic protein Atg8 failed to show the same rescue effects (Menzies et al., 2015). Furthermore, crossing calpastatin knockout mice with HD mice with 111 polyQ showed increased production of huntingtin N-terminal fragments and subsequent aggregation (Weber et al., 2018). Similar to tau and α-synuclein, a direct demonstration of calpain-mediated cleavage of huntingtin in the extracellular space is still lacking.
Cathepsins also cleave huntingtin. In particular, cathepsins B, D and L were found to generate two 55–60 kDa fragments when incubated with striatal cell lysates. Interestingly, cathepsin D only generated an N-terminal fragment from wildtype huntingtin, whereas up to four different fragments were detected when huntingtin containing 100 polyQ was exposed to cathepsin D. This cleavage was carried out at a neutral pH, suggesting that it could occur in the extracellular space (Kim J. et al., 2006).
TDP-43
Transactive response (TAR) DNA binding protein 43 (TDP-43) is a protein involved in transcriptional regulation and processing of thousands of different RNAs (Prasad et al., 2019). Under physiological conditions, it is localized in the nucleus but redistributes to the cytosol in several neuropathologies. TDP-43 aggregation is a hallmark of FTD and amyotrophic lateral sclerosis (ALS) and mutations in its coding gene are causative for these diseases (Chhangani et al., 2021). Evidence of its prion-like activity has also been described. For example, injection of brain extracts from FTD patients induced formation of aggregates in neuronal cell lines (Nonaka et al., 2013) and mouse models (Porta et al., 2018; Peng et al., 2020). TDP-43 seeds were also able to transfer between cultured neurons grown in microfluidic devices both as a free protein and inside exosomes (Feiler et al., 2015).
TDP-43 is composed of an N-terminal domain, two RNA recognition motifs and a C-terminal domain. Cryo-electron microscopy studies have identified the core of TDP-43 aggregates to be formed of residues 282–360 (Cao et al., 2019; Arseni et al., 2021). Several TDP-43 fragments have been observed in human samples from ALS-FTD patients (Smethurst et al., 2016) and C-terminal fragments appear to retain aggregation properties (Igaz et al., 2009; Nonaka et al., 2009; Furukawa et al., 2011; Shimonaka et al., 2016). Several studies suggest that a 25 kDa C-terminal fragment, possibly generated by cleavage at residue 216, (Caccamo et al., 2015) promotes seeded aggregation. This fragment is more abundant in the brain than in spinal cord samples isolated from the same FTD patient and, in accordance, brain extracts showed increased seeding capabilities compared to spinal cord extracts (Smethurst et al., 2016). However, other studies have found that expression of this C-terminal fragment in mice does not cause aggregate formation and drives limited behavioral defects (Caccamo et al., 2012, 2015; Akamatsu et al., 2013; Dayton et al., 2013; Walker et al., 2015; Porta et al., 2018). Other C-terminal fragments have also been identified (CT35kDa and CT27kDa), but their aggregation and seeding properties are still controversial (Zhang et al., 2009; Suzuki et al., 2011). In contrast, a recent study showed that injection of aggregates of an N-terminal 1–265 fragment was sufficient to induce formation of TDP-43-positive cytosolic stress granules and cause neuronal death (Pirie et al., 2021).
To the best of our knowledge, there is no evidence directly linking ECM core components with TDP-43 spreading. However, calpains have been found to process TDP-43, and as mentioned for other prion-like aggregates, it is possible that this protease could modify the seeding properties of TDP-43 in the extracellular milieu. Calpains cleave TDP-43 in vitro at multiple sites in mouse and rat brain extracts (Table 1). These fragments retained toxicity when applied to cultured primary neurons. Similar fragments were observed in mouse models of traumatic brain injury (TBI) and in the CSF of TBI patients (Yang et al., 2014). Interestingly, phosphorylated TDP-43 is resistant to calpain cleavage in vitro (Yamashita et al., 2016), whereas the A315T and M337V TDP-43 mutants showed increased processing (Yamashita et al., 2012). TDP-43 fragments corresponding to calpain cleavage events have been observed in extracts from the motor cortex and spinal cord of FTD-ALS patients with a concomitant increase in activated calpain-1 and -2. Interestingly, both fragments and activated calpain were higher in spinal cord samples compared to the motor cortex (Yamashita et al., 2012).
Amyloid Beta
Amyloid precursor protein (APP) is an integral transmembrane protein consisting of a large extracellular domain, a transmembrane domain and a short intracellular tail (Chen et al., 2017). APP is widely expressed in neuronal and non-neuronal cells, and its physiological function is still unclear, although it is believed to function as a cell adhesion molecule (Müller et al., 2017). APP is sequentially cleaved by β- and γ-secretase to produce Aβ peptides (Aβ40 and Aβ42), which form the core of amyloid plaques found in the brain of AD patients (Hardy and Selkoe, 2002; Selkoe, 2002; Haass et al., 2012). In neuronal cells, APP is predominantly found in the secretory pathway, from where it is trafficked to the axon termini and dendrites. Once APP reaches the plasma membrane, it is quickly internalized, leading to only a small fraction being present on the neuronal surface (Haass et al., 2012). Accordingly, Aβ production is likely to occur in the endocytic pathway, since inhibiting the internalization of cell surface APP leads to a significant decrease in Aβ production (Koo and Squazzo, 1994; Koo et al., 1996). The formation of extracellular Aβ aggregates is discussed below, since the involvement of ECM components in this process has been more extensively studied.
Very little is known about the precise molecular mechanisms triggering the conversion of soluble Aβ peptides to oligomers, protofibrils, amyloid fibrils and plaques in the brain (Friesen and Meyer-Luehmann, 2019). Intraneuronal Aβ42 accumulation precedes the deposition of extracellular plaques and has been observed in multivesicular bodies, endosomes and along microtubules (Takahashi et al., 2002, 2004). These intraneuronal Aβ42 clusters are enriched in pyramidal neurons of the hippocampus and entorhinal cortex, both of which are particularly vulnerable to pathology (Gouras et al., 2000). Most Aβ peptides are released from distal axons and synapses, and spread to synaptically connected regions (Friesen and Meyer-Luehmann, 2019); however, no in vivo study has demonstrated the transport and spread of Aβ and the roles that ECM might play. Domert et al. (2014) have shown that oligomeric Aβ is transferred between connecting, differentiated SH-SY5Y cells, and have postulated that this is due to impaired degradation of these. In addition, the cellular mechanism(s) leading to plaque spreading is also not fully elucidated. Thus, many fundamental questions remain about Aβ dynamics in vivo and its deposition in plaques in the AD brain.
Proteoglycans
A class of extracellular proteins which is found to play extensive roles in Aβ uptake, aggregation and deposition is the HSPG family (Figures 1Aiii,Biii). Amyloid plaques were first found to contain HSPGs by Snow and colleagues (Snow et al., 1988), followed by the observation that there is a direct interaction between HSPGs and both APP and Aβ (Narindrasorasak et al., 1991; Brunden et al., 1993). Cell lines deficient in HS biosynthesis, or treated with heparin, show a decrease in the binding of Aβ peptides to the cell membrane and subsequent internalization (Kanekiyo et al., 2011). An AD mouse model with a neuron-specific impairment in HS biosynthesis exhibited decreased plaque burden and Aβ oligomerization (Liu et al., 2016). Interestingly, the low-density lipoprotein-related receptor (LRP1)-dependent endocytosis of Aβ42 relies on HSPGs, since the addition of heparin abrogated the increase in Aβ42 uptake observed by the overexpression of an LRP1 mini-receptor (Kanekiyo et al., 2011). In conjunction with this, the Bu group has observed that ApoE-immunoisolated extracellular vesicles, derived from astrocytes, suppress Aβ binding and uptake in mouse cortical neurons, an effect abrogated by heparin (Fu et al., 2016). These findings suggest that HSPGs are involved in Aβ42 internalization. Although mostly localized at the plasma membrane, syndecans and glypican-1 show extensive co-localization with extracellular amyloid plaques in the brain, with the neuron-specific syndecan-3 being most effective at increasing Aβ42 uptake (van Horssen et al., 2002). By virtue of their HS chains, syndecan-3 and -4 also trigger formation of Aβ42 fibrillar assemblies, underscoring their relevance in plaque formation (Letoha et al., 2019). Glypican-1 interacts with higher-order Aβ structures and reduces SH-SY5Y viability when overexpressing APP (Watanabe et al., 2004). However, its function in plaque seeding or aggregation is currently not known.
Apart from their roles in mediating cell surface binding and endocytosis of APP/Aβ, HSPGs, along with CSPGs, also play a role in clearance and degradation of Aβ peptides (Gupta-Bansal et al., 1995). While post-mortem AD brain samples contain higher than normal levels of HSPGs, clearance of soluble Aβ in the mouse hippocampus is increased upon HSPG depletion (Liu et al., 2016). It is currently postulated that HSPGs function to promote Aβ fibrillation or act as protective chaperones, thus inhibiting Aβ degradation (van Horssen et al., 2003). Both agrin and perlecan, like other HSPGs, bind to Aβ40 fibrils through glycosaminoglycan side chains, protecting them from degradation and accelerating the fibrillation of monomeric Aβ40 (Castillo et al., 1997; Cotman et al., 2000). This role of agrin was recapitulated in an AD mouse model crossed with a conditional Agrn KO line where deletion was driven by an endothelial cell-specific promoter. These mice exhibited increased Aβ generation and deposition (Rauch et al., 2011). However, this effect was not observed when neuronal agrin was deleted, pointing to a non-cell autonomous effect on plaque formation.
Proteases
Proteolytic degradation contributes to the regulation of extracellular Aβ levels and deposition in amyloid plaques. Several extracellular proteases have been implicated in the degradation of Aβ40/42, including neprilysin (NEP) and the insulin-degrading enzyme (IDE) (Table 1; Saido and Leissring, 2012).
Neprilysin is a zinc metalloprotease with broad substrate specificity and cell surface localization. Demonstrating an inverse correlation with age, it is reported to cleave only Aβ monomers along the axon and at synaptic sites, and has negligible enzymatic activity for higher-order Aβ structures (Fukami et al., 2002; Saido and Leissring, 2012). Consistent with its role in the reduction of Aβ42 levels, brain tissues of AD patients exhibit a decrease in NEP expression in AD-vulnerable areas, including the hippocampus, cortex and temporal gyrus (Yasojima et al., 2001). The genetic deletion of NEP or its pharmacological inhibition results in a two-fold increase in Aβ levels in the brain as well as increased hippocampal plaque burden (Marr et al., 2004; Farris et al., 2007). Conversely, overexpression of neuronal NEP results in reduced Aβ levels and deposition, along with a reduction in associated pathology (Leissring et al., 2003). Additionally, its homolog NEP2 also contributes to decreasing Aβ levels (Saido and Leissring, 2012).
The zinc metalloprotease IDE also displays a broad distribution in the extracellular space, cytosol and mitochondria. Similar to NEP, IDE catalyzes the degradation of monomeric Aβ in the brain and its ablation causes an increase in Aβ levels in primary neurons as well as in vivo (Farris et al., 2003). Other extracellular proteases processing Aβ include MMP-2, MMP-9, and MMP-14, cathepsin B and plasmin (Saido and Leissring, 2012; Hernandez-Guillamon et al., 2015). Interestingly, these enzymes are capable of catabolizing Aβ fibrils, in contrast to NEP and IDE (Saido and Leissring, 2012). Their investigation in vivo remains limited, though cathepsin B has been observed in amyloid plaques (Mueller-Steiner et al., 2006).
Other Extracellular Matrix Components
In contrast to HSPGs and extracellular proteases, the role of ECM core proteins in Aβ production, seeding or plaque deposition is less clear. Several ECM proteins, including laminin, collagens and fibronectin co-localize with senile plaques in AD brains (Perlmutter et al., 1991). Proteomic profiling of AD hippocampi has revealed an upregulation of several ECM proteins during all disease stages (Hondius et al., 2016). However, whether these observations are causative, or correlative of amyloid formation, is currently unknown.
Laminin1 binds to soluble Aβ40 through its α-chain and inhibits its fibrillogenesis in a time- and dose-dependent manner, thus influencing the survival of cortical neurons (Drouet et al., 1999). While it increases the amyloidogenic fragment production, laminin1 also promotes Aβ40 depolymerization when incubated with pre-formed fibrils in vitro (Bronfman et al., 1996; Castillo et al., 2000).
Total collagen levels are upregulated in AD; however, the roles of individual isoforms are currently unclear. The Mucke group has shown that synthetic Aβ42 oligomers induce the transcription of the Col6a1 gene in hippocampal and cortical neurons and that collagen VI decreases the interaction of Aβ42 oligomers with the neuronal cell surface, ultimately leading to a reduction in Aβ42 neurotoxicity (Cheng et al., 2009).
In addition, when the 7PA2 cell model of AD is treated with peptides derived from elastin, it shows an elevation in Aβ40 and Aβ42 production. This effect was dependent on the length of the individual peptides, since longer peptides were more potent in Aβ generation and led to AD-related behavior in mice. Significantly, these changes were pinned down to an increased expression of β- and γ-secretase mRNAs (Ma et al., 2019).
Concluding Remarks
In this review we have presented evidence supporting the role of different ECM components in modulating the spread and aggregation of pathological, misfolded proteins. Although the available evidence remains limited, predominately due to the complexity of the ECM and its partial reconstitution observed in cell cultures, available data provide the rationale for further exploration of this area of research. In addition to the proteins discussed in this review, other proteins have been shown to possess the ability of transcellular spread, such as superoxide dismutase 1 (SOD1) (Ayers et al., 2014) and C9orf72 (Westergard et al., 2016) in ALS-FTD. However, to the best of our knowledge, we could not identify studies exploring the contribution of ECM components in mutant SOD1 and C9orf72 spreading.
The findings presented here highlight a high level of overlap between pathways modulating aggregation properties and/or propensity of spreading that affect most of these protein aggregates. In particular, proteoglycans and proteases are shared players in several of these diseases. HSPGs appear to participate in several steps of the spreading process of tau, α-synuclein and Aβ (Figures 1A,B), as well as the prion protein (Horonchik et al., 2005). Furthermore, direct binding to HSPGs has been shown for all three proteins. As mentioned above, HSPGs represent a heterogenous class of molecules. The presence of HSPGs on both the plasma membrane and in the extracellular space further complicates the experimental dissection of the underlying mechanism. Plasma membrane-exposed HSPGs have been clearly linked to internalization of aggregation-prone forms of tau and α-synuclein, a property shared with the prion protein (Horonchik et al., 2005). This suggests that HSPGs represent a common node for the endocytosis of pathological protein aggregates. As discussed above, the interaction appears to be mediated by the glycan groups, rather than the protein core, the contribution of which remains unclear. As such, it is likely that glycans may have some predisposition for binding to aggregated proteins, although the biochemical determinants of these interactions need to be further elucidated. In addition, heparin is routinely used to induce aggregation of recombinant tau and α-synuclein in vitro (Goedert et al., 1996; Cohlberg et al., 2002). This points to a potential role of extracellular HSPGs in inducing aggregation of these proteins, thus facilitating their internalization and as such, favoring the spread of pathology.
However, exogenous application of heparin can inhibit internalization of protein aggregates. This suggests that extracellular HSPGs could also act in a similar manner. However, the only study that has directly addressed extracellular HSPGs in this context has demonstrated that agrin promotes aggregation of Aβ and protects aggregates from degradation (Cotman et al., 2000). A similar function has also been proposed for agrin on tau, given the presence of agrin in NFTs (Verbeek et al., 1999). This multi-pronged action of HSPGs on several steps of the spreading process complicates predictions as to whether HSPGs could be targeted to slow down disease progression. Of note, chronic application of heparin mimetics failed to show significant effects on tau pathology in mice (Stopschinski et al., 2020). Interestingly, a role of HSPGs in the internalization of huntingtin aggregates was excluded (Holmes et al., 2013), suggesting a certain level of specificity. An in-depth characterization of this process is therefore required to understand the role of each HSPG species on the different steps of spreading, and to identify the biochemical determinants driving each of these effects. This could potentially lead to the identification of an endogenous HSPG, the levels of which could be modulated to reduce spreading. Alternatively, synthetic molecules that mimic HSPG protective effects could also be designed. Other ECM components, such as CSPGs, laminin, collagen and elastin, have been found to participate in the spreading of protein aggregates (Figure 1), but their role remains less characterized.
Proteases have emerged as major regulators of prion-like pathological spreading. In addition to a general clearing effect on aggregates, specific proteases can process monomers or oligomers of pathological proteins, generating fragments which display altered aggregation propensity (Table 1). Less is known about how these fragments behave with regard to their internalization properties. This area should be further investigated to conclusively establish the contribution of these proteases to the spreading of protein aggregates. MMPs are active against tau, α-synuclein, huntingtin and Aβ, with each MMP performing distinctive roles. MMP-3, for example, has been shown to reduce the propensity of tau to aggregate, whereas it was suggested to increase α-synuclein aggregation. However, MMP-3 is elevated in AD and PD models (Sung et al., 2005; Leem et al., 2020; Pentz et al., 2021) and in patients (Stomrud et al., 2010; Choi et al., 2011; Hanzel et al., 2014). This suggests that MMP-3 expression might be increased in an attempt to reduce pathology, however with opposite outcomes on tau and α-synuclein. Similarly, MMP-9 levels are increased in AD brains (Hernandes-Alejandro et al., 2020), again as a potential compensatory measure to reduce both Aβ and tau pathology. However, not only does MMP-9 degrade Aβ plaques (Hernandez-Guillamon et al., 2015), but it also induces the production of tau fragments with enhanced aggregation properties (Nübling et al., 2012). Conversely, MMP-2 reduces α-synuclein pathology and Aβ plaque burden in vivo, but was suggested to increase tau aggregation. Similar to other MMPs, its levels are increased in AD (Terni and Ferrer, 2015). In contrast, MMP-2 is reduced in synucleinopathies (Lorenzl et al., 2002), suggesting its causal role in these diseases. With this exception, these observations indicate that MMPs are upregulated in response to pathology, possibly in an attempt to degrade protein aggregates. However, in several cases, protease activity has proven detrimental, potentiating the aggregation of seeds.
Cathepsins and calpains are key intracellular proteases, the roles of which have been poorly characterized in the extracellular space. This makes the interpretation of the data difficult, as most studies do not specifically investigate whether their action is solely exerted intracellularly, or also extracellularly. Therefore, future experiments should be carried out to clarify whether these proteases would retain activity against proteopathic proteins in the extracellular compartment. Despite this, cathepsins seem to have a protective role by reducing the aggregation of tau, Aβ and α-synuclein. As described above, several cathepsins appear to be upregulated in AD suggesting that lysosomal activity is increased in an attempt to reduce the aggregate load. Whether this reflects an increase of extracellular cathepsin is still unclear. In contrast, risk variants in cathepsin B and D genes were identified in synucleinopathies (Robak et al., 2017; Blauwendraat et al., 2020), which support the involvement of these proteases in the mechanism of disease. The role of these proteases in HD and TDP-43 pathology has not yet been investigated in sufficient detail. The well-defined role of cathepsins in lysosomal degradation, in addition to their presence in the extracellular space, makes them an interesting pharmacological target to modulate protein aggregation and spreading in a two-pronged manner. Conversely, calpain cleavage was suggested to promote aggregation of α-synuclein, tau, TDP-43 and huntingtin, with increased levels of calpain detected in related pathologies. This strongly suggests that increased calpain activation could be a common pathological mechanism in neurodegenerative disorders, thus making it a promising therapeutic target. Further research should therefore be carried out to characterize the extracellular proteases involved in the proteolytic processing of prion-like proteins during the prodromic and early symptomatic phases of disease.
The findings presented in this review support the need to clarify the contributions of ECM components in neurodegenerative diseases. To do so, research should include in vivo experiments in animal models of neurodegeneration, which retain the complex ECM web in its native state, and using culture systems better recapitulating this aspect. For example, three-dimensional (3D) organoids generated from human induced pluripotent stem cells (hIPSCs) may allow easier access to study the ECM compared to animal models, albeit maintaining a greater complexity than 2D cultures (Cho et al., 2021). This would not only help the discovery of novel roles for ECM components but could also drive the development of new therapeutic strategies for neurodegenerative diseases. Although these therapies would not directly target initial pathological events, such as the generation of misfolded proteins, they could halt their spreading between brain regions, therefore slowing down disease progression. In addition, given their extracellular localization, these proteins have the advantage of being more easily targetable and their modulation might have reduced side effects as they would not directly affect neuronal and glial cell function. As such, increased efforts in exploring the role of ECM components in modulating the spreading of pathological protein aggregates should be strongly encouraged.
Author Contributions
EM and GS conceptualized the manuscript. EM, SSt, and SSu wrote the manuscript. EM, SSt, SSu, JV, and GS reviewed and edited the manuscript. GS, JV, and EM acquired funding. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Pat Harris Ph.D. studentship in neurodegenerative diseases (SSt); the Human Frontier Science Program long-term fellowship LT000220/2017-L (SSu); the Wellcome Senior Investigator Awards 107116/Z/15/Z and 223022/Z/21/Z (GS), the UK Dementia Research Institute Foundation award UKDRI-1005 (EM and GS), and the UK DRI pilot award PILOT2020-09 (EM and JV).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We kindly thank Ludovica Zaccagnini (UK Dementia Research Institute, UCL) for critical reading of the manuscript.
References
Adamec, E., Murrell, J. R., Takao, M., Hobbs, W., Nixon, R. A., Ghetti, B., et al. (2002a). P301L tauopathy: confocal immunofluorescence study of perinuclear aggregation of the mutated protein. J. Neurol. Sci. 200, 85–93. doi: 10.1016/s0022-510x(02)00150-8
Adamec, E., Mohan, P., Vonsattel, J. P., and Nixon, R. A. (2002b). Calpain activation in neurodegenerative diseases: confocal immunofluorescence study with antibodies specifically recognizing the active form of calpain 2. Acta Neuropathol. 104, 92–104. doi: 10.1007/s00401-002-0528-6
Akamatsu, M., Takuma, H., Yamashita, T., Okada, T., Keino-Masu, K., Ishii, K., et al. (2013). A unique mouse model for investigating the properties of amyotrophic lateral sclerosis-associated protein TDP-43, by in utero electroporation. Neurosci. Res. 77, 234–241. doi: 10.1016/j.neures.2013.09.009
Amadoro, G., Ciotti, M. T., Costanzi, M., Cestari, V., Calissano, P., and Canu, N. (2006). NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc. Natl. Acad. Sci. U.S.A. 103, 2892–2897. doi: 10.1073/pnas.0511065103
Arai, T., Guo, J.-P., and McGeer, P. L. (2005). Proteolysis of non-phosphorylated and phosphorylated tau by thrombin. J. Biol. Chem. 280, 5145–5153. doi: 10.1074/jbc.M409234200
Arai, T., Miklossy, J., Klegeris, A., Guo, J.-P., and McGeer, P. L. (2006). Thrombin and prothrombin are expressed by neurons and glial cells and accumulate in neurofibrillary tangles in Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 65, 19–25. doi: 10.1097/01.jnen.0000196133.74087.cb
Arseni, D., Hasegawa, M., Murzin, A. G., Kametani, F., Arai, M., Yoshida, M., et al. (2021). Structure of pathological TDP-43 filaments from ALS with FTLD. Nature 601, 139–143. doi: 10.1038/s41586-021-04199-3
Ayers, J. I., Fromholt, S., Koch, M., DeBosier, A., McMahon, B., Xu, G., et al. (2014). Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol. 128, 791–803. doi: 10.1007/s00401-014-1342-7
Baudry, M., and Bi, X. (2016). Calpain-1 and Calpain-2: the Yin and Yang of Synaptic Plasticity and Neurodegeneration. Trends Neurosci. 39, 235–245. doi: 10.1016/j.tins.2016.01.007
Bernal-Conde, L. D., Ramos-Acevedo, R., Reyes-Hernández, M. A., Balbuena-Olvera, A. J., Morales-Moreno, I. D., Argüero-Sánchez, R., et al. (2019). Alpha-Synuclein physiology and pathology: a perspective on cellular structures and organelles. Front. Neurosci. 13:1399. doi: 10.3389/fnins.2019.01399
Birol, M., Wojcik, S. P., Miranker, A. D., and Rhoades, E. (2019). Identification of N-linked glycans as specific mediators of neuronal uptake of acetylated α-Synuclein. PLoS Biol. 17:e3000318. doi: 10.1371/journal.pbio.3000318
Blauwendraat, C., Reed, X., Krohn, L., Heilbron, K., Bandres-Ciga, S., Tan, M., et al. (2020). Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 143, 234–248. doi: 10.1093/brain/awz350
Boonen, M., Staudt, C., Gilis, F., Oorschot, V., Klumperman, J., and Jadot, M. (2016). Cathepsin D and its newly identified transport receptor SEZ6L2 can modulate neurite outgrowth. J. Cell Sci. 129, 557–568. doi: 10.1242/jcs.179374
Braak, E., Braak, H., and Mandelkow, E. M. (1994). A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 87, 554–567. doi: 10.1007/BF00293315
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Brettschneider, J., Del Tredici, K., Lee, V. M.-Y., and Trojanowski, J. Q. (2015). Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. 16, 109–120. doi: 10.1038/nrn3887
Bronfman, F. C., Soto, C., Tapia, L., Tapia, V., and Inestrosa, N. C. (1996). Extracellular matrix regulates the amount of the beta-amyloid precursor protein and its amyloidogenic fragments. J. Cell. Physiol. 166, 360–369. doi: 10.1002/(SICI)1097-4652(199602)166:2<360::AID-JCP14>3.0.CO;2-F
Brückner, G., Hausen, D., Härtig, W., Drlicek, M., Arendt, T., and Brauer, K. (1999). Cortical areas abundant in extracellular matrix chondroitin sulphate proteoglycans are less affected by cytoskeletal changes in Alzheimer’s disease. Neuroscience 92, 791–805. doi: 10.1016/s0306-4522(99)00071-8
Brückner, G., Morawski, M., and Arendt, T. (2008). Aggrecan-based extracellular matrix is an integral part of the human basal ganglia circuit. Neuroscience 151, 489–504. doi: 10.1016/j.neuroscience.2007.10.033
Brunden, K. R., Richter-Cook, N. J., Chaturvedi, N., and Frederickson, R. C. (1993). pH-dependent binding of synthetic beta-amyloid peptides to glycosaminoglycans. J. Neurochem. 61, 2147–2154. doi: 10.1111/j.1471-4159.1993.tb07453.x
Burré, J., Vivona, S., Diao, J., Sharma, M., Brunger, A. T., and Südhof, T. C. (2013). Properties of native brain α-synuclein. Nature 498, E4–E6.
Caccamo, A., Majumder, S., and Oddo, S. (2012). Cognitive decline typical of frontotemporal lobar degeneration in transgenic mice expressing the 25-kDa C-terminal fragment of TDP-43. Am. J. Pathol. 180, 293–302. doi: 10.1016/j.ajpath.2011.09.022
Caccamo, A., Shaw, D. M., Guarino, F., Messina, A., Walker, A. W., and Oddo, S. (2015). Reduced protein turnover mediates functional deficits in transgenic mice expressing the 25 kDa C-terminal fragment of TDP-43. Hum. Mol. Genet. 24, 4625–4635. doi: 10.1093/hmg/ddv193
Cao, Q., Boyer, D. R., Sawaya, M. R., Ge, P., and Eisenberg, D. S. (2019). Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nat. Struct. Mol. Biol. 26, 619–627. doi: 10.1038/s41594-019-0248-4
Castillo, G. M., Lukito, W., Peskind, E., Raskind, M., Kirschner, D. A., Yee, A. G., et al. (2000). Laminin inhibition of beta-amyloid protein (Abeta) fibrillogenesis and identification of an Abeta binding site localized to the globular domain repeats on the laminin a chain. J. Neurosci. Res. 62, 451–462. doi: 10.1002/1097-4547(20001101)62:3<451::AID-JNR15>3.0.CO;2-F
Castillo, G. M., Ngo, C., Cummings, J., Wight, T. N., and Snow, A. D. (1997). Perlecan binds to the beta-amyloid proteins (A beta) of Alzheimer’s disease, accelerates A beta fibril formation, and maintains A beta fibril stability. J. Neurochem. 69, 2452–2465. doi: 10.1046/j.1471-4159.1997.69062452.x
Chakroun, T., Evsyukov, V., Nykänen, N.-P., Höllerhage, M., Schmidt, A., Kamp, F., et al. (2020). Alpha-synuclein fragments trigger distinct aggregation pathways. Cell Death Dis. 11:84. doi: 10.1038/s41419-020-2285-7
Chen, G.-F., Xu, T.-H., Yan, Y., Zhou, Y.-R., Jiang, Y., Melcher, K., et al. (2017). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235. doi: 10.1038/aps.2017.28
Chen, Y.-D., Huang, P.-Y., Chiang, C.-S., Huang, Y.-S., and Tang, S.-C. (2021). Generation and Role of Calpain-Cleaved 17-kDa Tau Fragment in Acute Ischemic Stroke. Mol. Neurobiol. 58, 5814–5825. doi: 10.1007/s12035-021-02519-2
Cheng, J. S., Dubal, D. B., Kim, D. H., Legleiter, J., Cheng, I. H., Yu, G.-Q., et al. (2009). Collagen VI protects neurons against Abeta toxicity. Nat. Neurosci. 12, 119–121. doi: 10.1038/nn.2240
Chesser, A. S., Pritchard, S. M., and Johnson, G. V. W. (2013). Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front. Neurol. 4:122. doi: 10.3389/fneur.2013.00122
Chhangani, D., Martín-Peña, A., and Rincon-Limas, D. E. (2021). Molecular, functional, and pathological aspects of TDP-43 fragmentation. iScience 24:102459. doi: 10.1016/j.isci.2021.102459
Cho, A.-N., Jin, Y., An, Y., Kim, J., Choi, Y. S., Lee, J. S., et al. (2021). Microfluidic device with brain extracellular matrix promotes structural and functional maturation of human brain organoids. Nat. Commun. 12:4730. doi: 10.1038/s41467-021-24775-5
Choi, D.-H., Kim, Y.-J., Kim, Y.-G., Joh, T. H., Beal, M. F., and Kim, Y.-S. (2011). Role of matrix metalloproteinase 3-mediated alpha-synuclein cleavage in dopaminergic cell death. J. Biol. Chem. 286, 14168–14177. doi: 10.1074/jbc.M111.222430
Cicchetti, F., Lacroix, S., Cisbani, G., Vallières, N., Saint-Pierre, M., St-Amour, I., et al. (2014). Mutant huntingtin is present in neuronal grafts in Huntington disease patients. Ann. Neurol. 76, 31–42. doi: 10.1002/ana.24174
Cicognola, C., Brinkmalm, G., Wahlgren, J., Portelius, E., Gobom, J., Cullen, N. C., et al. (2019). Novel tau fragments in cerebrospinal fluid: relation to tangle pathology and cognitive decline in Alzheimer’s disease. Acta Neuropathol. 137, 279–296. doi: 10.1007/s00401-018-1948-2
Cicognola, C., Satir, T. M., Brinkmalm, G., Matečko-Burmann, I., Agholme, L., Bergström, P., et al. (2020). Tauopathy-Associated Tau Fragment Ending at Amino Acid 224 Is Generated by Calpain-2 Cleavage. J. Alzheimers Dis. 74, 1143–1156. doi: 10.3233/JAD-191130
Cohlberg, J. A., Li, J., Uversky, V. N., and Fink, A. L. (2002). Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry 41, 1502–1511. doi: 10.1021/bi011711s
Condomitti, G., and de Wit, J. (2018). Heparan sulfate proteoglycans as emerging players in synaptic specificity. Front. Mol. Neurosci. 11:14. doi: 10.3389/fnmol.2018.00014
Cotman, S. L., Halfter, W., and Cole, G. J. (2000). Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer’s disease brain. Mol. Cell. Neurosci. 15, 183–198. doi: 10.1006/mcne.1999.0816
Cullen, V., Lindfors, M., Ng, J., Paetau, A., Swinton, E., Kolodziej, P., et al. (2009). Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol. Brain 2:5. doi: 10.1186/1756-6606-2-5
Dayton, R. D., Gitcho, M. A., Orchard, E. A., Wilson, J. D., Wang, D. B., Cain, C. D., et al. (2013). Selective Forelimb Impairment in Rats Expressing a Pathological TDP-43 25 kDa C-terminal Fragment to Mimic Amyotrophic Lateral Sclerosis. Mol. Ther. 21, 1324–1334. doi: 10.1038/mt.2013.88
De La-Rocque, S., Moretto, E., Butnaru, I., and Schiavo, G. (2021). Knockin’ on heaven’s door: Molecular mechanisms of neuronal tau uptake. J. Neurochem. 156, 563–588. doi: 10.1111/jnc.15144
Del Campo Milan, M., Zuroff, L., Jimenez, C. R., Scheltens, P., and Teunissen, C. E. (2015). Can agrin cerebrospinal fluid concentration be used as an early biomarker for Alzheimer’s disease? Alzheimers Dement. 1, 75–80. doi: 10.1016/j.dadm.2014.11.008
Diamandis, E. P., Yousef, G. M., Soosaipillai, A. R., Grass, L., Porter, A., Little, S., et al. (2000). Immunofluorometric assay of human kallikrein 6 (zyme/protease M/neurosin) and preliminary clinical applications. Clin. Biochem. 33, 369–375. doi: 10.1016/s0009-9120(00)00145-4
Diepenbroek, M., Casadei, N., Esmer, H., Saido, T. C., Takano, J., Kahle, P. J., et al. (2014). Overexpression of the calpain-specific inhibitor calpastatin reduces human alpha-Synuclein processing, aggregation and synaptic impairment in [A30P]αSyn transgenic mice. Hum. Mol. Genet. 23, 3975–3989.
Dityatev, A., and Schachner, M. (2003). Extracellular matrix molecules and synaptic plasticity. Nat. Rev. Neurosci. 4, 456–468.
Dityatev, A., Schachner, M., and Sonderegger, P. (2010). The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat. Rev. Neurosci. 11, 735–746. doi: 10.1038/nrn2898
Domert, J., Rao, S. B., Agholme, L., Brorsson, A.-C., Marcusson, J., Hallbeck, M., et al. (2014). Spreading of amyloid-β peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol. Dis. 65, 82–92. doi: 10.1016/j.nbd.2013.12.019
Downs, M., Sethi, M. K., Raghunathan, R., Layne, M. D., and Zaia, J. (2022). Matrisome changes in Parkinson’s disease. Anal. Bioanal. Chem. doi: 10.1007/s00216-022-03929-4
Drouet, B., Pinçon-Raymond, M., Chambaz, J., and Pillot, T. (1999). Laminin 1 attenuates beta-amyloid peptide Abeta(1-40) neurotoxicity of cultured fetal rat cortical neurons. J. Neurochem. 73, 742–749. doi: 10.1046/j.1471-4159.1999.0730742.x
Dufty, B. M., Warner, L. R., Hou, S. T., Jiang, S. X., Gomez-Isla, T., Leenhouts, K. M., et al. (2007). Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am. J. Pathol. 170, 1725–1738. doi: 10.2353/ajpath.2007.061232
El-Agnaf, O. M. A., Salem, S. A., Paleologou, K. E., Cooper, L. J., Fullwood, N. J., Gibson, M. J., et al. (2003). Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 17, 1945–1947. doi: 10.1096/fj.03-0098fje
Elbaum-Garfinkle, S., and Rhoades, E. (2012). Identification of an aggregation-prone structure of tau. J. Am. Chem. Soc. 134, 16607–16613. doi: 10.1021/ja305206m
El-Daher, M.-T., Hangen, E., Bruyère, J., Poizat, G., Al-Ramahi, I., Pardo, R., et al. (2015). Huntingtin proteolysis releases non-polyQ fragments that cause toxicity through dynamin 1 dysregulation. EMBO J. 34, 2255–2271. doi: 10.15252/embj.201490808
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 4162–4167. doi: 10.1073/pnas.0230450100
Farris, W., Schütz, S. G., Cirrito, J. R., Shankar, G. M., Sun, X., George, A., et al. (2007). Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am. J. Pathol. 171, 241–251. doi: 10.2353/ajpath.2007.070105
Feiler, M. S., Strobel, B., Freischmidt, A., Helferich, A. M., Kappel, J., Brewer, B. M., et al. (2015). TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 211, 897–911. doi: 10.1083/jcb.201504057
Ferreira, A., and Bigio, E. H. (2011). Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Mol. Med. 17, 676–685. doi: 10.2119/molmed.2010.00220
Frangié, C., Zhang, W., Perez, J., Dubois, Y.-C. X., Haymann, J.-P., and Baud, L. (2006). Extracellular calpains increase tubular epithelial cell mobility. Implications for kidney repair after ischemia. J. Biol. Chem. 281, 26624–26632. doi: 10.1074/jbc.M603007200
Friesen, M., and Meyer-Luehmann, M. (2019). Aβ seeding as a tool to study cerebral amyloidosis and associated pathology. Front. Mol. Neurosci. 12:233. doi: 10.3389/fnmol.2019.00233
Fu, Y., Zhao, J., Atagi, Y., Nielsen, H. M., Liu, C.-C., Zheng, H., et al. (2016). Apolipoprotein E lipoprotein particles inhibit amyloid-β uptake through cell surface heparan sulphate proteoglycan. Mol. Neurodegener. 11:37. doi: 10.1186/s13024-016-0099-y
Fukami, S., Watanabe, K., Iwata, N., Haraoka, J., Lu, B., Gerard, N. P., et al. (2002). Abeta-degrading endopeptidase, neprilysin, in mouse brain: synaptic and axonal localization inversely correlating with Abeta pathology. Neurosci. Res. 43, 39–56. doi: 10.1016/s0168-0102(02)00015-9
Furukawa, Y., Kaneko, K., Watanabe, S., Yamanaka, K., and Nukina, N. (2011). A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J. Biol. Chem. 286, 18664–18672. doi: 10.1074/jbc.M111.231209
Gafni, J., and Ellerby, L. M. (2002). Calpain activation in Huntington’s disease. J. Neurosci. 22, 4842–4849.
Gafni, J., Hermel, E., Young, J. E., Wellington, C. L., Hayden, M. R., and Ellerby, L. M. (2004). Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J. Biol. Chem. 279, 20211–20220. doi: 10.1074/jbc.M401267200
García-Sierra, F., Mondragón-Rodríguez, S., and Basurto-Islas, G. (2008). Truncation of tau protein and its pathological significance in Alzheimer’s disease. J. Alzheimers Dis. 14, 401–409. doi: 10.3233/jad-2008-14407
Garg, S., Timm, T., Mandelkow, E.-M., Mandelkow, E., and Wang, Y. (2011). Cleavage of Tau by calpain in Alzheimer’s disease: the quest for the toxic 17 kD fragment. Neurobiol. Aging 32, 1–14. doi: 10.1016/j.neurobiolaging.2010.09.008
Goedert, M., Jakes, R., Spillantini, M. G., Hasegawa, M., Smith, M. J., and Crowther, R. A. (1996). Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383, 550–553. doi: 10.1038/383550a0
Goll, D. E., Thompson, V. F., Li, H., Wei, W., and Cong, J. (2003). The calpain system. Physiol. Rev. 83, 731–801.
Gouras, G. K., Tsai, J., Naslund, J., Vincent, B., Edgar, M., Checler, F., et al. (2000). Intraneuronal Abeta42 accumulation in human brain. Am. J. Pathol. 156, 15–20. doi: 10.1016/s0002-9440(10)64700-1
Graham, R. K., Deng, Y., Slow, E. J., Haigh, B., Bissada, N., Lu, G., et al. (2006). Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 125, 1179–1191. doi: 10.1016/j.cell.2006.04.026
Greenbaum, E. A., Graves, C. L., Mishizen-Eberz, A. J., Lupoli, M. A., Lynch, D. R., Englander, S. W., et al. (2005). The E46K mutation in alpha-synuclein increases amyloid fibril formation. J. Biol. Chem. 280, 7800–7807. doi: 10.1074/jbc.M411638200
Gupta-Bansal, R., Frederickson, R. C., and Brunden, K. R. (1995). Proteoglycan-mediated inhibition of A beta proteolysis. A potential cause of senile plaque accumulation. J. Biol. Chem. 270, 18666–18671. doi: 10.1074/jbc.270.31.18666
Haass, C., Kaether, C., Thinakaran, G., and Sisodia, S. (2012). Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2:a006270.
Hackam, A. S., Singaraja, R., Wellington, C. L., Metzler, M., McCutcheon, K., Zhang, T., et al. (1998). The influence of huntingtin protein size on nuclear localization and cellular toxicity. J. Cell Biol. 141, 1097–1105. doi: 10.1083/jcb.141.5.1097
Hanzel, C. E., Iulita, M. F., Eyjolfsdottir, H., Hjorth, E., Schultzberg, M., Eriksdotter, M., et al. (2014). Analysis of matrix metallo-proteases and the plasminogen system in mild cognitive impairment and Alzheimer’s disease cerebrospinal fluid. J. Alzheimers Dis. 40, 667–678. doi: 10.3233/JAD-132282
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hasegawa, M., Crowther, R. A., Jakes, R., and Goedert, M. (1997). Alzheimer-like changes in microtubule-associated protein Tau induced by sulfated glycosaminoglycans. Inhibition of microtubule binding, stimulation of phosphorylation, and filament assembly depend on the degree of sulfation. J. Biol. Chem. 272, 33118–33124. doi: 10.1074/jbc.272.52.33118
Hassen, G. W., Kesner, L., Stracher, A., Shulman, A., Rockenstein, E., Mante, M., et al. (2018). Effects of Novel Calpain inhibitors in transgenic animal model of Parkinson’s disease/dementia with Lewy bodies. Sci. Rep. 8:18083. doi: 10.1038/s41598-018-35729-1
Hernandes-Alejandro, M., Montaño, S., Harrington, C. R., Wischik, C. M., Salas-Casas, A., Cortes-Reynosa, P., et al. (2020). Analysis of the Relationship between Metalloprotease-9 and Tau Protein in Alzheimer’s Disease. J. Alzheimers Dis. 76, 553–569. doi: 10.3233/JAD-200146
Hernandez-Guillamon, M., Mawhirt, S., Blais, S., Montaner, J., Neubert, T. A., Rostagno, A., et al. (2015). Sequential Amyloid-β Degradation by the Matrix Metalloproteases MMP-2 and MMP-9. J. Biol. Chem. 290, 15078–15091. doi: 10.1074/jbc.M114.610931
Holmes, B. B., DeVos, S. L., Kfoury, N., Li, M., Jacks, R., Yanamandra, K., et al. (2013). Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U.S.A. 110, E3138–E3147. doi: 10.1073/pnas.1301440110
Holmes, B. B., and Diamond, M. I. (2014). Prion-like properties of Tau protein: the importance of extracellular Tau as a therapeutic target. J. Biol. Chem. 289, 19855–19861. doi: 10.1074/jbc.R114.549295
Hondius, D. C., van Nierop, P., Li, K. W., Hoozemans, J. J. M., van der Schors, R. C., van Haastert, E. S., et al. (2016). Profiling the human hippocampal proteome at all pathologic stages of Alzheimer’s disease. Alzheimers Dement. 12, 654–668. doi: 10.1016/j.jalz.2015.11.002
Horonchik, L., Tzaban, S., Ben-Zaken, O., Yedidia, Y., Rouvinski, A., Papy-Garcia, D., et al. (2005). Heparan sulfate is a cellular receptor for purified infectious prions. J. Biol. Chem. 280, 17062–17067. doi: 10.1074/jbc.M500122200
Hossain, S., Alim, A., Takeda, K., Kaji, H., Shinoda, T., and Uéda, K. (2001). Limited proteolysis of NACP/alpha-synuclein. J. Alzheimers Dis. 3, 577–584. doi: 10.3233/jad-2001-3608
Hudák, A., Kusz, E., Domonkos, I., Jósvay, K., Kodamullil, A. T., Szilák, L., et al. (2019). Contribution of syndecans to cellular uptake and fibrillation of α-synuclein and tau. Sci. Rep. 9:16543. doi: 10.1038/s41598-019-53038-z
Igaz, L. M., Kwong, L. K., Chen-Plotkin, A., Winton, M. J., Unger, T. L., Xu, Y., et al. (2009). Expression of TDP-43 C-terminal Fragments in vitro recapitulates pathological features of TDP-43 Proteinopathies. J. Biol. Chem. 284, 8516–8524.
Ihse, E., Yamakado, H., van Wijk, X. M., Lawrence, R., Esko, J. D., and Masliah, E. (2017). Cellular internalization of alpha-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci. Rep. 7:9008. doi: 10.1038/s41598-017-08720-5
Ii, K., Ito, H., Kominami, E., and Hirano, A. (1993). Abnormal distribution of cathepsin proteinases and endogenous inhibitors (cystatins) in the hippocampus of patients with Alzheimer’s disease, parkinsonism-dementia complex on Guam, and senile dementia and in the aged. Virchows Arch. A Pathol. Anat. Histopathol. 423, 185–194. doi: 10.1007/BF01614769
Iwata, A., Maruyama, M., Akagi, T., Hashikawa, T., Kanazawa, I., Tsuji, S., et al. (2003). Alpha-synuclein degradation by serine protease neurosin: implication for pathogenesis of synucleinopathies. Hum. Mol. Genet. 12, 2625–2635. doi: 10.1093/hmg/ddg283
Izawa, Y., Tateno, H., Kameda, H., Hirakawa, K., Hato, K., Yagi, H., et al. (2012). Role of C-terminal negative charges and tyrosine residues in fibril formation of α-synuclein. Brain Behav. 2, 595–605. doi: 10.1002/brb3.86
Johnson, E. C. B., Carter, E. K., Dammer, E. B., Duong, D. M., Gerasimov, E. S., Liu, Y., et al. (2022). Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat. Neurosci. 25, 213–225. doi: 10.1038/s41593-021-00999-y
Kanekiyo, T., Zhang, J., Liu, Q., Liu, C.-C., Zhang, L., and Bu, G. (2011). Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J. Neurosci. 31, 1644–1651. doi: 10.1523/JNEUROSCI.5491-10.2011
Karpowicz, R. J., Trojanowski, J. Q., and Lee, V. M.-Y. (2019). Transmission of α-synuclein seeds in neurodegenerative disease: recent developments. Lab. Invest. 99, 971–981. doi: 10.1038/s41374-019-0195-z
Kasai, T., Tokuda, T., Yamaguchi, N., Watanabe, Y., Kametani, F., Nakagawa, M., et al. (2008). Cleavage of normal and pathological forms of alpha-synuclein by neurosin in vitro. Neurosci. Lett. 436, 52–56. doi: 10.1016/j.neulet.2008.02.057
Katsinelos, T., Zeitler, M., Dimou, E., Karakatsani, A., Müller, H.-M., Nachman, E., et al. (2018). Unconventional secretion mediates the trans-cellular spreading of Tau. Cell Rep. 23, 2039–2055. doi: 10.1016/j.celrep.2018.04.056
Kenessey, A., Nacharaju, P., Ko, L. W., and Yen, S. H. (1997). Degradation of tau by lysosomal enzyme cathepsin D: implication for Alzheimer neurofibrillary degeneration. J. Neurochem. 69, 2026–2038. doi: 10.1046/j.1471-4159.1997.69052026.x
Kessler, J. C., Rochet, J.-C., and Lansbury, P. T. (2003). The N-terminal repeat domain of alpha-synuclein inhibits beta-sheet and amyloid fibril formation. Biochemistry 42, 672–678. doi: 10.1021/bi020429y
Khurana, V., Elson-Schwab, I., Fulga, T. A., Sharp, K. A., Loewen, C. A., Mulkearns, E., et al. (2010). Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet. 6:e1001026. doi: 10.1371/journal.pgen.1001026
Kim, H. J., Lee, D., Lee, C.-H., Chung, K. C., Kim, J., and Paik, S. R. (2006). Calpain-resistant fragment(s) of alpha-synuclein regulates the synuclein-cleaving activity of 20S proteasome. Arch. Biochem. Biophys. 455, 40–47. doi: 10.1016/j.abb.2006.08.019
Kim, J., Sapp, E., Cuiffo, B. G., Sobin, L., Yoder, J., Kegel, K. B., et al. (2006). Lysosomal proteases are involved in generation of N-terminal huntingtin fragments. Neurobiol. Dis. 22, 346–356. doi: 10.1016/j.nbd.2005.11.012
Kim, K. S., Choi, Y. R., Park, J.-Y., Lee, J.-H., Kim, D. K., Lee, S.-J., et al. (2012). Proteolytic cleavage of extracellular α-synuclein by plasmin: implications for Parkinson disease. J. Biol. Chem. 287, 24862–24872. doi: 10.1074/jbc.M112.348128
Kim, M., Roh, J.-K., Yoon, B. W., Kang, L., Kim, Y. J., Aronin, N., et al. (2003). Huntingtin is degraded to small fragments by calpain after ischemic injury. Exp. Neurol. 183, 109–115. doi: 10.1016/s0014-4886(03)00132-8
Kim, Y. J., Yi, Y., Sapp, E., Wang, Y., Cuiffo, B., Kegel, K. B., et al. (2001). Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc. Natl. Acad. Sci. U.S.A. 98, 12784–12789. doi: 10.1073/pnas.221451398
Koo, E. H., and Squazzo, S. L. (1994). Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 269, 17386–17389.
Koo, E. H., Squazzo, S. L., Selkoe, D. J., and Koo, C. H. (1996). Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. J. Cell Sci. 109(Pt 5), 991–998. doi: 10.1242/jcs.109.5.991
Krishnaswamy, V. R., Benbenishty, A., Blinder, P., and Sagi, I. (2019). Demystifying the extracellular matrix and its proteolytic remodeling in the brain: structural and functional insights. Cell. Mol. Life Sci. 76, 3229–3248. doi: 10.1007/s00018-019-03182-6
Kroger, S., and Schroder, J. E. (2002). Agrin in the developing CNS: new roles for a synapse organizer. Physiology 17, 207–212. doi: 10.1152/nips.01390.2002
Kurbatskaya, K., Phillips, E. C., Croft, C. L., Dentoni, G., Hughes, M. M., Wade, M. A., et al. (2016). Upregulation of calpain activity precedes tau phosphorylation and loss of synaptic proteins in Alzheimer’s disease brain. Acta Neuropathol. Commun. 4:34. doi: 10.1186/s40478-016-0299-2
Landles, C., Sathasivam, K., Weiss, A., Woodman, B., Moffitt, H., Finkbeiner, S., et al. (2010). Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J. Biol. Chem. 285, 8808–8823. doi: 10.1074/jbc.M109.075028
Lang, A. E., Riherd Methner, D. N., and Ferreira, A. (2014). Neuronal degeneration, synaptic defect−, and behavioral abnormalities in tau45−230 transgenic mice. Neuroscience 275, 322–339. doi: 10.1016/j.neuroscience.2014.06.017
Laske, C., Stellos, K., Kempter, I., Stransky, E., Maetzler, W., Fleming, I., et al. (2015). Increased cerebrospinal fluid calpain activity and microparticle levels in Alzheimer’s disease. Alzheimers Dement. 11, 465–474. doi: 10.1016/j.jalz.2014.06.003
Lee, H.-J., Patel, S., and Lee, S.-J. (2005). Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 25, 6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005
Lee, S.-J., Lim, H.-S., Masliah, E., and Lee, H.-J. (2011). Protein aggregate spreading in neurodegenerative diseases: problems and perspectives. Neurosci. Res. 70, 339–348. doi: 10.1016/j.neures.2011.05.008
Lee, V. M.-Y., and Trojanowski, J. Q. (2006). Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 52, 33–38. doi: 10.1016/j.neuron.2006.09.026
Leem, Y.-H., Park, J.-S., Park, J.-E., Kim, D.-Y., Kang, J. L., and Kim, H.-S. (2020). Papaverine inhibits α-synuclein aggregation by modulating neuroinflammation and matrix metalloproteinase-3 expression in the subacute MPTP/P mouse model of Parkinson’s disease. Biomed. Pharmacother. 130:110576. doi: 10.1016/j.biopha.2020.110576
Lehri-Boufala, S., Ouidja, M.-O., Barbier-Chassefière, V., Hénault, E., Raisman-Vozari, R., Garrigue-Antar, L., et al. (2015). New roles of glycosaminoglycans in α-synuclein aggregation in a cellular model of Parkinson disease. PLoS One 10:e0116641. doi: 10.1371/journal.pone.0116641
Leissring, M. A., Farris, W., Chang, A. Y., Walsh, D. M., Wu, X., Sun, X., et al. (2003). Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40, 1087–1093. doi: 10.1016/s0896-6273(03)00787-6
Lemere, C. A., Munger, J. S., Shi, G. P., Natkin, L., Haass, C., Chapman, H. A., et al. (1995). The lysosomal cysteine protease, cathepsin S, is increased in Alzheimer’s disease and Down syndrome brain. An immunocytochemical study. Am. J. Pathol. 146, 848–860.
Letavernier, E., Zafrani, L., Perez, J., Letavernier, B., Haymann, J.-P., and Baud, L. (2012). The role of calpains in myocardial remodelling and heart failure. Cardiovasc. Res. 96, 38–45. doi: 10.1093/cvr/cvs099
Letoha, T., Hudák, A., Kusz, E., Pettkó-Szandtner, A., Domonkos, I., Jósvay, K., et al. (2019). Contribution of syndecans to cellular internalization and fibrillation of amyloid-β(1-42). Sci. Rep. 9:1393.
Levin, J., Giese, A., Boetzel, K., Israel, L., Högen, T., Nübling, G., et al. (2009). Increased alpha-synuclein aggregation following limited cleavage by certain matrix metalloproteinases. Exp. Neurol. 215, 201–208. doi: 10.1016/j.expneurol.2008.10.010
Li, W., West, N., Colla, E., Pletnikova, O., Troncoso, J. C., Marsh, L., et al. (2005). Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc. Natl. Acad. Sci. U.S.A. 102, 2162–2167. doi: 10.1073/pnas.0406976102
Liu, C.-C., Zhao, N., Yamaguchi, Y., Cirrito, J. R., Kanekiyo, T., Holtzman, D. M., et al. (2016). Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-β clearance and aggregation in Alzheimer’s disease. Sci. Transl. Med. 8:332ra44. doi: 10.1126/scitranslmed.aad3650
Lorenzl, S., Albers, D. S., Narr, S., Chirichigno, J., and Beal, M. F. (2002). Expression of MMP-2, MMP-9, and MMP-1 and their endogenous counterregulators TIMP-1 and TIMP-2 in postmortem brain tissue of Parkinson’s disease. Exp. Neurol. 178, 13–20. doi: 10.1006/exnr.2002.8019
Luk, K. C., Covell, D. J., Kehm, V. M., Zhang, B., Song, I. Y., Byrne, M. D., et al. (2016). Molecular and biological compatibility with Host Alpha-Synuclein influences fibril pathogenicity. Cell Rep. 16, 3373–3387. doi: 10.1016/j.celrep.2016.08.053
Luk, K. C., Song, C., O’Brien, P., Stieber, A., Branch, J. R., Brunden, K. R., et al. (2009). Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. U.S.A. 106, 20051–20056. doi: 10.1073/pnas.0908005106
Lunkes, A., Lindenberg, K. S., Ben-Haïem, L., Weber, C., Devys, D., Landwehrmeyer, G. B., et al. (2002). Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell 10, 259–269. doi: 10.1016/s1097-2765(02)00602-0
Ma, C., Su, J., Sun, Y., Feng, Y., Shen, N., Li, B., et al. (2019). Significant upregulation of Alzheimer’s β-amyloid levels in a living system induced by Extracellular Elastin Polypeptides. Angew. Chem. Int. Ed. Engl. 58, 18703–18709. doi: 10.1002/anie.201912399
Maïza, A., Chantepie, S., Vera, C., Fifre, A., Huynh, M. B., Stettler, O., et al. (2018). The role of heparan sulfates in protein aggregation and their potential impact on neurodegeneration. FEBS Lett. 592, 3806–3818. doi: 10.1002/1873-3468.13082
Marr, R. A., Guan, H., Rockenstein, E., Kindy, M., Gage, F. H., Verma, I., et al. (2004). Neprilysin regulates amyloid Beta peptide levels. J. Mol. Neurosci. 22, 5–11. doi: 10.1385/JMN:22:1-2:5
Martindale, D., Hackam, A., Wieczorek, A., Ellerby, L., Wellington, C., McCutcheon, K., et al. (1998). Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genet. 18, 150–154. doi: 10.1038/ng0298-150
Matsumoto, S.-E., Motoi, Y., Ishiguro, K., Tabira, T., Kametani, F., Hasegawa, M., et al. (2015). The twenty-four KDa C-terminal tau fragment increases with aging in tauopathy mice: implications of prion-like properties. Hum. Mol. Genet. 24, 6403–6416. doi: 10.1093/hmg/ddv351
McDougall, J. J., McConnell, M., and Reid, A. R. (2021). Intracellular versus extracellular inhibition of calpain I causes differential effects on pain in a rat model of joint inflammation. Mol. Pain 17:17448069211016141. doi: 10.1177/17448069211016141
McGlinchey, R. P., Dominah, G. A., and Lee, J. C. (2017). Taking a bite out of amyloid: mechanistic insights into α-Synuclein Degradation by Cathepsin L. Biochemistry 56, 3881–3884. doi: 10.1021/acs.biochem.7b00360
McGlinchey, R. P., Lacy, S. M., Walker, R. L., and Lee, J. C. (2020). Cathepsin K is a potent disaggregase of α-synuclein fibrils. Biochem. Biophys. Res. Commun. 529, 1106–1111. doi: 10.1016/j.bbrc.2020.06.155
McGlinchey, R. P., and Lee, J. C. (2015). Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 112, 9322–9327. doi: 10.1073/pnas.1500937112
Mehra, S., Ghosh, D., Kumar, R., Mondal, M., Gadhe, L. G., Das, S., et al. (2018). Glycosaminoglycans have variable effects on α-synuclein aggregation and differentially affect the activities of the resulting amyloid fibrils. J. Biol. Chem. 293, 12975–12991. doi: 10.1074/jbc.RA118.004267
Menzies, F. M., Garcia-Arencibia, M., Imarisio, S., O’Sullivan, N. C., Ricketts, T., Kent, B. A., et al. (2015). Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ. 22, 433–444. doi: 10.1038/cdd.2014.151
Miller, J. P., Holcomb, J., Al-Ramahi, I., de Haro, M., Gafni, J., Zhang, N., et al. (2010). Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington’s disease. Neuron 67, 199–212. doi: 10.1016/j.neuron.2010.06.021
Mishizen-Eberz, A. J., Guttmann, R. P., Giasson, B. I., Day, G. A., Hodara, R., Ischiropoulos, H., et al. (2003). Distinct cleavage patterns of normal and pathologic forms of alpha-synuclein by calpain I in vitro. J. Neurochem. 86, 836–847. doi: 10.1046/j.1471-4159.2003.01878.x
Mishizen-Eberz, A. J., Norris, E. H., Giasson, B. I., Hodara, R., Ischiropoulos, H., Lee, V. M.-Y., et al. (2005). Cleavage of alpha-synuclein by calpain: potential role in degradation of fibrillized and nitrated species of alpha-synuclein. Biochemistry 44, 7818–7829. doi: 10.1021/bi047846q
Morawski, M., Brückner, G., Arendt, T., and Matthews, R. T. (2012). Aggrecan: beyond cartilage and into the brain. Int. J. Biochem. Cell Biol. 44, 690–693. doi: 10.1016/j.biocel.2012.01.010
Morawski, M., Pavlica, S., Seeger, G., Grosche, J., Kouznetsova, E., Schliebs, R., et al. (2010a). Perineuronal nets are largely unaffected in Alzheimer model Tg2576 mice. Neurobiol. Aging 31, 1254–1256. doi: 10.1016/j.neurobiolaging.2008.07.023
Morawski, M., Brückner, G., Jäger, C., Seeger, G., and Arendt, T. (2010b). Neurons associated with aggrecan-based perineuronal nets are protected against tau pathology in subcortical regions in Alzheimer’s disease. Neuroscience 169, 1347–1363. doi: 10.1016/j.neuroscience.2010.05.022
Mueller-Steiner, S., Zhou, Y., Arai, H., Roberson, E. D., Sun, B., Chen, J., et al. (2006). Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron 51, 703–714. doi: 10.1016/j.neuron.2006.07.027
Müller, U. C., Deller, T., and Korte, M. (2017). Not just amyloid: physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 18, 281–298. doi: 10.1038/nrn.2017.29
Munger, J. S., Haass, C., Lemere, C. A., Shi, G. P., Wong, W. S., Teplow, D. B., et al. (1995). Lysosomal processing of amyloid precursor protein to A beta peptides: a distinct role for cathepsin S. Biochem. J. 311(Pt 1), 299–305. doi: 10.1042/bj3110299
Naphade, S., Embusch, A., Madushani, K. L., Ring, K. L., and Ellerby, L. M. (2017). Altered expression of matrix metalloproteinases and their endogenous inhibitors in a human isogenic stem cell model of huntington’s disease. Front. Neurosci. 11:736. doi: 10.3389/fnins.2017.00736
Narindrasorasak, S., Lowery, D., Gonzalez-DeWhitt, P., Poorman, R. A., Greenberg, B., and Kisilevsky, R. (1991). High affinity interactions between the Alzheimer’s beta-amyloid precursor proteins and the basement membrane form of heparan sulfate proteoglycan. J. Biol. Chem. 266, 12878–12883.
Nekooki-Machida, Y., Kurosawa, M., Nukina, N., Ito, K., Oda, T., and Tanaka, M. (2009). Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 9679–9684.
NeuroLINCS Consortium, J., Lim, R. G., Kaye, J. A., Dardov, V., Coyne, A. N., et al. (2021). An integrated multi-omic analysis of iPSC-derived motor neurons from C9ORF72 ALS patients. iScience 24:103221. doi: 10.1016/j.isci.2021.103221
Niemeyer, C., Matosin, N., Kaul, D., Philipsen, A., and Gassen, N. C. (2020). The role of cathepsins in memory functions and the pathophysiology of psychiatric disorders. Front. Psychiatry 11:718. doi: 10.3389/fpsyt.2020.00718
Nonaka, T., Kametani, F., Arai, T., Akiyama, H., and Hasegawa, M. (2009). Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet. 18, 3353–3364. doi: 10.1093/hmg/ddp275
Nonaka, T., Masuda-Suzukake, M., Arai, T., Hasegawa, Y., Akatsu, H., Obi, T., et al. (2013). Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 4, 124–134. doi: 10.1016/j.celrep.2013.06.007
Novak, U., and Kaye, A. H. (2000). Extracellular matrix and the brain: components and function. J. Clin. Neurosci. 7, 280–290. doi: 10.1054/jocn.1999.0212
Nowicka, D., Soulsby, S., Skangiel-Kramska, J., and Glazewski, S. (2009). Parvalbumin-containing neurons, perineuronal nets and experience-dependent plasticity in murine barrel cortex. Eur. J. Neurosci. 30, 2053–2063. doi: 10.1111/j.1460-9568.2009.06996.x
Nübling, G., Levin, J., Bader, B., Israel, L., Bötzel, K., Lorenzl, S., et al. (2012). Limited cleavage of tau with matrix-metalloproteinase MMP-9, but not MMP-3, enhances tau oligomer formation. Exp. Neurol. 237, 470–476. doi: 10.1016/j.expneurol.2012.07.018
Nübling, G., Schuberth, M., Feldmer, K., Giese, A., Holdt, L. M., Teupser, D., et al. (2017). Cathepsin S increases tau oligomer formation through limited cleavage, but only IL-6, not cathespin S serum levels correlate with disease severity in the neurodegenerative tauopathy progressive supranuclear palsy. Exp. Brain Res. 235, 2407–2412. doi: 10.1007/s00221-017-4978-4
O’Brien, R., DeGiacomo, F., Holcomb, J., Bonner, A., Ring, K. L., Zhang, N., et al. (2015). Integration-independent transgenic Huntington disease fragment mouse models reveal distinct phenotypes and life span in Vivo. J. Biol. Chem. 290, 19287–19306. doi: 10.1074/jbc.M114.623561
Ogata, Y., Enghild, J. J., and Nagase, H. (1992). Matrix metalloproteinase 3 (stromelysin) activates the precursor for the human matrix metalloproteinase 9. J. Biol. Chem. 267, 3581–3584.
Oh, S. H., Kim, H. N., Park, H. J., Shin, J. Y., Kim, D. Y., and Lee, P. H. (2017). The Cleavage Effect of Mesenchymal Stem Cell and Its Derived Matrix Metalloproteinase-2 on Extracellular α-Synuclein Aggregates in Parkinsonian Models. Stem Cells Transl. Med. 6, 949–961. doi: 10.5966/sctm.2016-0111
Okada, Y., Gonoji, Y., Naka, K., Tomita, K., Nakanishi, I., Iwata, K., et al. (1992). Matrix metalloproteinase 9 (92-kDa gelatinase/type IV collagenase) from HT 1080 human fibrosarcoma cells. Purification and activation of the precursor and enzymic properties. J. Biol. Chem. 267, 21712–21719.
Pampalakis, G., Sykioti, V.-S., Ximerakis, M., Stefanakou-Kalakou, I., Melki, R., Vekrellis, K., et al. (2017). KLK6 proteolysis is implicated in the turnover and uptake of extracellular alpha-synuclein species. Oncotarget 8, 14502–14515. doi: 10.18632/oncotarget.13264
Pang, P. T., Teng, H. K., Zaitsev, E., Woo, N. T., Sakata, K., Zhen, S., et al. (2004). Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 306, 487–491. doi: 10.1126/science.1100135
Park, S.-Y., and Ferreira, A. (2005). The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J. Neurosci. 25, 5365–5375. doi: 10.1523/JNEUROSCI.1125-05.2005
Park, S. Y., Tournell, C., Sinjoanu, R. C., and Ferreira, A. (2007). Caspase-3- and calpain-mediated tau cleavage are differentially prevented by estrogen and testosterone in beta-amyloid-treated hippocampal neurons. Neuroscience 144, 119–127. doi: 10.1016/j.neuroscience.2006.09.012
Paudel, H. K., and Li, W. (1999). Heparin-induced conformational change in microtubule-associated protein Tau as detected by chemical cross-linking and phosphopeptide mapping. J. Biol. Chem. 274, 8029–8038. doi: 10.1074/jbc.274.12.8029
Pecho-Vrieseling, E., Rieker, C., Fuchs, S., Bleckmann, D., Esposito, M. S., Botta, P., et al. (2014). Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat. Neurosci. 17, 1064–1072.
Peng, C., Trojanowski, J. Q., and Lee, V. M.-Y. (2020). Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 16, 199–212.
Pentz, R., Iulita, M. F., Mikutra-Cencora, M., Ducatenzeiler, A., Bennett, D. A., and Cuello, A. C. (2021). A new role for matrix metalloproteinase-3 in the NGF metabolic pathway: proteolysis of mature NGF and sex-specific differences in the continuum of Alzheimer’s pathology. Neurobiol. Dis. 148:105150. doi: 10.1016/j.nbd.2020.105150
Perlmutter, L. S., Barrón, E., Saperia, D., and Chui, H. C. (1991). Association between vascular basement membrane components and the lesions of Alzheimer’s disease. J. Neurosci. Res. 30, 673–681. doi: 10.1002/jnr.490300411
Pestereva, N., Ivleva, I., Zubov, A., Tikhomirova, M., and Karpenko, M. (2021). m-Calpain is released from striatal synaptosomes. Int. J. Neurosci. [Epub ahead of print]. doi: 10.1080/00207454.2021.1901697
Pirie, E., Oh, C.-K., Zhang, X., Han, X., Cieplak, P., Scott, H. R., et al. (2021). S-nitrosylated TDP-43 triggers aggregation, cell-to-cell spread, and neurotoxicity in hiPSCs and in vivo models of ALS/FTD. Proc. Natl. Acad. Sci. U.S.A. 118:e2021368118. doi: 10.1073/pnas.2021368118
Porta, S., Xu, Y., Restrepo, C. R., Kwong, L. K., Zhang, B., Brown, H. J., et al. (2018). Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 9:4220. doi: 10.1038/s41467-018-06548-9
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019). Molecular mechanisms of TDP-43 Misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 12:25. I doi: 10.3389/fnmol.2019.00025
Puangmalai, N., Bhatt, N., Montalbano, M., Sengupta, U., Gaikwad, S., Ventura, F., et al. (2020). Internalization mechanisms of brain-derived tau oligomers from patients with Alzheimer’s disease, progressive supranuclear palsy and dementia with Lewy bodies. Cell Death Dis. 11:314. doi: 10.1038/s41419-020-2503-3
Qiao, L., Hamamichi, S., Caldwell, K. A., Caldwell, G. A., Yacoubian, T. A., Wilson, S., et al. (2008). Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol. Brain 1:17. doi: 10.1186/1756-6606-1-17
Quinn, J. P., Corbett, N. J., Kellett, K. A. B., and Hooper, N. M. (2018). Tau proteolysis in the pathogenesis of tauopathies: neurotoxic fragments and novel biomarkers. J. Alzheimers Dis. 63, 13–33. doi: 10.3233/JAD-170959
Ratovitski, T., Chighladze, E., Waldron, E., Hirschhorn, R. R., and Ross, C. A. (2011). Cysteine proteases bleomycin hydrolase and cathepsin Z mediate N-terminal proteolysis and toxicity of mutant huntingtin. J. Biol. Chem. 286, 12578–12589. doi: 10.1074/jbc.M110.185348
Rauch, S. M., Huen, K., Miller, M. C., Chaudry, H., Lau, M., Sanes, J. R., et al. (2011). Changes in brain β-amyloid deposition and aquaporin 4 levels in response to altered agrin expression in mice. J. Neuropathol. Exp. Neurol. 70, 1124–1137. doi: 10.1097/NEN.0b013e31823b0b12
Reinecke, J. B., DeVos, S. L., McGrath, J. P., Shepard, A. M., Goncharoff, D. K., Tait, D. N., et al. (2011). Implicating calpain in tau-mediated toxicity in vivo. PLoS One 6:e23865. doi: 10.1371/journal.pone.0023865
Ren, P.-H., Lauckner, J. E., Kachirskaia, I., Heuser, J. E., Melki, R., and Kopito, R. R. (2009). Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 11, 219–225. doi: 10.1038/ncb1830
Robak, L. A., Jansen, I. E., van Rooij, J., Uitterlinden, A. G., Kraaij, R., Jankovic, J., et al. (2017). Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140, 3191–3203. doi: 10.1093/brain/awx285
Ross, C. A., and Poirier, M. A. (2004). Protein aggregation and neurodegenerative disease. Nat. Med. 10(Suppl.), S10–S17.
Saido, T., and Leissring, M. A. (2012). Proteolytic degradation of amyloid β-protein. Cold Spring Harb. Perspect. Med. 2:a006379.
Sarrazin, S., Lamanna, W. C., and Esko, J. D. (2011). Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 3
Saudou, F., and Humbert, S. (2016). The biology of huntingtin. Neuron 89, 910–926. doi: 10.1016/j.neuron.2016.02.003
Schmidt, S., Stapf, C., Schmutzler, S., Lachmann, I., Arendt, T., Holzer, M., et al. (2021). Aggrecan modulates the expression and phosphorylation of tau in a novel bigenic TauP301L - Acan mouse model. Eur. J. Neurosci. 53, 3889–3904. doi: 10.1111/ejn.14923
Scott, H., and Panin, V. M. (2014). N-glycosylation in regulation of the nervous system. Adv. Neurobiol. 9, 367–394. doi: 10.1007/978-1-4939-1154-7_17
Selkoe, D. J. (2002). Deciphering the genesis and fate of amyloid beta-protein yields novel therapies for Alzheimer disease. J. Clin. Invest. 110, 1375–1381. doi: 10.1172/JCI16783
Sevlever, D., Jiang, P., and Yen, S.-H. C. (2008). Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 47, 9678–9687. doi: 10.1021/bi800699v
Shapiro, S. D., Fliszar, C. J., Broekelmann, T. J., Mecham, R. P., Senior, R. M., and Welgus, H. G. (1995). Activation of the 92-kDa gelatinase by stromelysin and 4-aminophenylmercuric acetate. Differential processing and stabilization of the carboxyl-terminal domain by tissue inhibitor of metalloproteinases (TIMP). J. Biol. Chem. 270, 6351–6356. doi: 10.1074/jbc.270.11.6351
Shimonaka, S., Nonaka, T., Suzuki, G., Hisanaga, S.-I., and Hasegawa, M. (2016). Templated Aggregation of TAR DNA-binding Protein of 43 kDa (TDP-43) by Seeding with TDP-43 Peptide Fibrils. J. Biol. Chem. 291, 8896–8907. doi: 10.1074/jbc.M115.713552
Sibille, N., Sillen, A., Leroy, A., Wieruszeski, J.-M., Mulloy, B., Landrieu, I., et al. (2006). Structural impact of heparin binding to full-length Tau as studied by NMR spectroscopy. Biochemistry 45, 12560–12572. doi: 10.1021/bi060964o
Smethurst, P., Newcombe, J., Troakes, C., Simone, R., Chen, Y.-R., Patani, R., et al. (2016). In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol. Dis. 96, 236–247.
Smith, M. A., and Hilgenberg, L. G. W. (2002). Agrin in the CNS: a protein in search of a function? Neuroreport 13, 1485–1495. doi: 10.1097/00001756-200208270-00001
Snow, A. D., Mar, H., Nochlin, D., Kimata, K., Kato, M., Suzuki, S., et al. (1988). The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer’s disease. Am. J. Pathol. 133, 456–463.
Soria, F. N., Paviolo, C., Doudnikoff, E., Arotcarena, M.-L., Lee, A., Danné, N., et al. (2020). Synucleinopathy alters nanoscale organization and diffusion in the brain extracellular space through hyaluronan remodeling. Nat. Commun. 11:3440. doi: 10.1038/s41467-020-17328-9
Sorrentino, Z. A., and Giasson, B. I. (2020). The emerging role of α-synuclein truncation in aggregation and disease. J. Biol. Chem. 295, 10224–10244. doi: 10.1074/jbc.REV120.011743
Sorrentino, Z. A., Vijayaraghavan, N., Gorion, K.-M., Riffe, C. J., Strang, K. H., Caldwell, J., et al. (2018). Physiological C-terminal truncation of α-synuclein potentiates the prion-like formation of pathological inclusions. J. Biol. Chem. 293, 18914–18932. doi: 10.1074/jbc.RA118.005603
Sorrentino, Z. A., Xia, Y., Gorion, K.-M., Hass, E., and Giasson, B. I. (2020). Carboxy-terminal truncations of mouse α-synuclein alter aggregation and prion-like seeding. FEBS Lett. 594, 1271–1283. doi: 10.1002/1873-3468.13728
Spencer, B., Michael, S., Shen, J., Kosberg, K., Rockenstein, E., Patrick, C., et al. (2013). Lentivirus mediated delivery of neurosin promotes clearance of wild-type α-synuclein and reduces the pathology in an α-synuclein model of LBD. Mol. Ther. 21, 31–41. doi: 10.1038/mt.2012.66
Stefanis, L. (2012). α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a009399.
Stomrud, E., Björkqvist, M., Janciauskiene, S., Minthon, L., and Hansson, O. (2010). Alterations of matrix metalloproteinases in the healthy elderly with increased risk of prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2, 20. doi: 10.1186/alzrt44
Stopschinski, B. E., Holmes, B. B., Miller, G. M., Manon, V. A., Vaquer-Alicea, J., Prueitt, W. L., et al. (2018). Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus α-synuclein and β-amyloid aggregates. J. Biol. Chem. 293, 10826–10840. doi: 10.1074/jbc.RA117.000378
Stopschinski, B. E., Thomas, T. L., Nadji, S., Darvish, E., Fan, L., Holmes, B. B., et al. (2020). A synthetic heparinoid blocks Tau aggregate cell uptake and amplification. J. Biol. Chem. 295, 2974–2983. doi: 10.1074/jbc.RA119.010353
Sung, J. Y., Park, S. M., Lee, C.-H., Um, J. W., Lee, H. J., Kim, J., et al. (2005). Proteolytic cleavage of extracellular secreted {alpha}-synuclein via matrix metalloproteinases. J. Biol. Chem. 280, 25216–25224.
Suttkus, A., Holzer, M., Morawski, M., and Arendt, T. (2016). The neuronal extracellular matrix restricts distribution and internalization of aggregated Tau-protein. Neuroscience 313, 225–235. doi: 10.1016/j.neuroscience.2015.11.040
Suttkus, A., Rohn, S., Weigel, S., Glöckner, P., Arendt, T., and Morawski, M. (2014). Aggrecan, link protein and tenascin-R are essential components of the perineuronal net to protect neurons against iron-induced oxidative stress. Cell Death Dis. 5:e1119. doi: 10.1038/cddis.2014.25
Suzuki, H., Lee, K., and Matsuoka, M. (2011). TDP-43-induced death is associated with altered regulation of BIM and Bcl-xL and attenuated by caspase-mediated TDP-43 cleavage. J. Biol. Chem. 286, 13171–13183. doi: 10.1074/jbc.M110.197483
Takahashi, R. H., Almeida, C. G., Kearney, P. F., Yu, F., Lin, M. T., Milner, T. A., et al. (2004). Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J. Neurosci. 24, 3592–3599. doi: 10.1523/JNEUROSCI.5167-03.2004
Takahashi, R. H., Milner, T. A., Li, F., Nam, E. E., Edgar, M. A., Yamaguchi, H., et al. (2002). Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 161, 1869–1879. doi: 10.1016/s0002-9440(10)64463-x
Tatebe, H., Watanabe, Y., Kasai, T., Mizuno, T., Nakagawa, M., Tanaka, M., et al. (2010). Extracellular neurosin degrades α-synuclein in cultured cells. Neurosci. Res. 67, 341–346. doi: 10.1016/j.neures.2010.04.008
Terada, M., Suzuki, G., Nonaka, T., Kametani, F., Tamaoka, A., and Hasegawa, M. (2018). The effect of truncation on prion-like properties of α-synuclein. J. Biol. Chem. 293, 13910–13920. doi: 10.1074/jbc.RA118.001862
Terni, B., and Ferrer, I. (2015). Abnormal expression and distribution of MMP2 at initial stages of Alzheimer’s disease-related pathology. J. Alzheimers Dis. 46, 461–469. doi: 10.3233/JAD-142460
Tran, H. T., Chung, C. H.-Y., Iba, M., Zhang, B., Trojanowski, J. Q., Luk, K. C., et al. (2014). A -synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep. 7, 2054–2065. doi: 10.1016/j.celrep.2014.05.033
Tsujimura, A., Taguchi, K., Watanabe, Y., Tatebe, H., Tokuda, T., Mizuno, T., et al. (2015). Lysosomal enzyme cathepsin B enhances the aggregate forming activity of exogenous α-synuclein fibrils. Neurobiol. Dis. 73, 244–253. doi: 10.1016/j.nbd.2014.10.011
van Horssen, J., Kleinnijenhuis, J., Maass, C. N., Rensink, A. A. M., Otte-Höller, I., David, G., et al. (2002). Accumulation of heparan sulfate proteoglycans in cerebellar senile plaques. Neurobiol. Aging 23, 537–545. doi: 10.1016/s0197-4580(02)00010-6
van Horssen, J., Wesseling, P., van den Heuvel, L. P. W. J., de Waal, R. M. W., and Verbeek, M. M. (2003). Heparan sulphate proteoglycans in Alzheimer’s disease and amyloid-related disorders. Lancet Neurol. 2, 482–492. doi: 10.1016/s1474-4422(03)00484-8
van’t Spijker, H. M., and Kwok, J. C. F. (2017). A sweet talk: the molecular systems of perineuronal nets in controlling neuronal communication. Front. Integr. Neurosci. 11:33. doi: 10.3389/fnint.2017.00033
Vaquer-Alicea, J., and Diamond, M. I. (2019). Propagation of protein aggregation in neurodegenerative diseases. Annu. Rev. Biochem. 88, 785–810.
Verbeek, M. M., Otte-Höller, I., van den Born, J., van den Heuvel, L. P., David, G., Wesseling, P., et al. (1999). Agrin is a major heparan sulfate proteoglycan accumulating in Alzheimer’s disease brain. Am. J. Pathol. 155, 2115–2125. doi: 10.1016/S0002-9440(10)65529-0
Vidak, E., Javoršek, U., Vizovišek, M., and Turk, B. (2019). Cysteine Cathepsins and their extracellular roles: shaping the microenvironment. Cells 8:264. doi: 10.3390/cells8030264
Volpicelli-Daley, L. A., Luk, K. C., Patel, T. P., Tanik, S. A., Riddle, D. M., Stieber, A., et al. (2011). Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. doi: 10.1016/j.neuron.2011.08.033
Walker, A. K., Tripathy, K., Restrepo, C. R., Ge, G., Xu, Y., Kwong, L. K., et al. (2015). An insoluble frontotemporal lobar degeneration-associated TDP-43 C-terminal fragment causes neurodegeneration and hippocampus pathology in transgenic mice. Hum. Mol. Genet. 24, 7241–7254. doi: 10.1093/hmg/ddv424
Wang, J. Z., Grundke-Iqbal, I., and Iqbal, K. (1996). Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer’s disease. Nat. Med. 2, 871–875. doi: 10.1038/nm0896-871
Wang, X.-X., Tan, M.-S., Yu, J.-T., and Tan, L. (2014). Matrix metalloproteinases and their multiple roles in Alzheimer’s disease. Biomed Res. Int. 2014:908636. doi: 10.1155/2014/908636
Wang, Y., Luo, W., and Reiser, G. (2008). Trypsin and trypsin-like proteases in the brain: proteolysis and cellular functions. Cell. Mol. Life Sci. 65, 237–252. doi: 10.1007/s00018-007-7288-3
Wang, Y., Martinez-Vicente, M., Krüger, U., Kaushik, S., Wong, E., Mandelkow, E.-M., et al. (2009). Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet. 18, 4153–4170. doi: 10.1093/hmg/ddp367
Watanabe, N., Araki, W., Chui, D.-H., Makifuchi, T., Ihara, Y., and Tabira, T. (2004). Glypican-1 as an Abeta binding HSPG in the human brain: its localization in DIG domains and possible roles in the pathogenesis of Alzheimer’s disease. FASEB J. 18, 1013–1015. doi: 10.1096/fj.03-1040fje
Weber, J. J., Kloock, S. J., Nagel, M., Ortiz-Rios, M. M., Hofmann, J., Riess, O., et al. (2018). Calpastatin ablation aggravates the molecular phenotype in cell and animal models of Huntington disease. Neuropharmacology 133, 94–106. doi: 10.1016/j.neuropharm.2018.01.022
Weisová, P., Cehlár, O., Škrabana, R., Žilková, M., Filipčík, P., Kováčech, B., et al. (2019). Therapeutic antibody targeting microtubule-binding domain prevents neuronal internalization of extracellular tau via masking neuron surface proteoglycans. Acta Neuropathol. Commun. 7:129. doi: 10.1186/s40478-019-0770-y
Westergard, T., Jensen, B. K., Wen, X., Cai, J., Kropf, E., Iacovitti, L., et al. (2016). Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep. 17, 645–652. doi: 10.1016/j.celrep.2016.09.032
Wong, C.-O., and Venkatachalam, K. (2019). Motor neurons from ALS patients with mutations in C9ORF72 and SOD1 exhibit distinct transcriptional landscapes. Hum. Mol. Genet. 28, 2799–2810. doi: 10.1093/hmg/ddz104
Ximerakis, M., Pampalakis, G., Roumeliotis, T. I., Sykioti, V.-S., Garbis, S. D., Stefanis, L., et al. (2014). Resistance of naturally secreted α-synuclein to proteolysis. FASEB J. 28, 3146–3158. doi: 10.1096/fj.13-245852
Yamashita, T., Hideyama, T., Hachiga, K., Teramoto, S., Takano, J., Iwata, N., et al. (2012). A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat. Commun. 3:1307. doi: 10.1038/ncomms2303
Yamashita, T., Teramoto, S., and Kwak, S. (2016). Phosphorylated TDP-43 becomes resistant to cleavage by calpain: a regulatory role for phosphorylation in TDP-43 pathology of ALS/FTLD. Neurosci. Res. 107, 63–69. doi: 10.1016/j.neures.2015.12.006
Yang, L. S., Gordon-Krajcer, W., and Ksiezak-Reding, H. (1997). Tau released from paired helical filaments with formic acid or guanidine is susceptible to calpain-mediated proteolysis. J. Neurochem. 69, 1548–1558. doi: 10.1046/j.1471-4159.1997.69041548.x
Yang, L. S., and Ksiezak-Reding, H. (1995). Calpain-induced proteolysis of normal human tau and tau associated with paired helical filaments. Eur. J. Biochem. 233, 9–17. doi: 10.1111/j.1432-1033.1995.009_1.x
Yang, Z., Lin, F., Robertson, C. S., and Wang, K. K. W. (2014). Dual vulnerability of TDP-43 to calpain and caspase-3 proteolysis after neurotoxic conditions and traumatic brain injury. J. Cereb. Blood Flow Metab. 34, 1444–1452. doi: 10.1038/jcbfm.2014.105
Yasojima, K., Akiyama, H., McGeer, E. G., and McGeer, P. L. (2001). Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of beta-amyloid peptide. Neurosci. Lett. 297, 97–100. doi: 10.1016/s0304-3940(00)01675-x
Yoon, H., Blaber, S. I., Evans, D. M., Trim, J., Juliano, M. A., Scarisbrick, I. A., et al. (2008). Activation profiles of human kallikrein-related peptidases by proteases of the thrombostasis axis. Protein Sci. 17, 1998–2007. doi: 10.1110/ps.036715.108
Zehe, C., Engling, A., Wegehingel, S., Schäfer, T., and Nickel, W. (2006). Cell-surface heparan sulfate proteoglycans are essential components of the unconventional export machinery of FGF-2. Proc. Natl. Acad. Sci. U.S.A. 103, 15479–15484. doi: 10.1073/pnas.0605997103
Zhang, H., Cao, Y., Ma, L., Wei, Y., and Li, H. (2021). Possible mechanisms of tau spread and toxicity in Alzheimer’s disease. Front. Cell Dev. Biol. 9:707268. doi: 10.3389/fcell.2021.707268
Zhang, Q., Xu, Y., Lee, J., Jarnik, M., Wu, X., Bonifacino, J. S., et al. (2020). A myosin-7B-dependent endocytosis pathway mediates cellular entry of α-synuclein fibrils and polycation-bearing cargos. Proc. Natl. Acad. Sci. U.S.A. 117, 10865–10875. doi: 10.1073/pnas.1918617117
Zhang, Y.-J., Xu, Y.-F., Cook, C., Gendron, T. F., Roettges, P., Link, C. D., et al. (2009). Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 7607–7612. doi: 10.1073/pnas.0900688106
Zhao, J., Huvent, I., Lippens, G., Eliezer, D., Zhang, A., Li, Q., et al. (2017). Glycan Determinants of Heparin-Tau Interaction. Biophys. J. 112, 921–932. doi: 10.1016/j.bpj.2017.01.024
Zhao, J., Wu, H., and Tang, X.-Q. (2021). Tau internalization: a complex step in tau propagation. Ageing Res. Rev. 67:101272. doi: 10.1016/j.arr.2021.101272
Keywords: tau, alpha synuclein, amyloid beta, TDP-43, huntingtin, extracellular matrix, HSPG, proteases
Citation: Moretto E, Stuart S, Surana S, Vargas JNS and Schiavo G (2022) The Role of Extracellular Matrix Components in the Spreading of Pathological Protein Aggregates. Front. Cell. Neurosci. 16:844211. doi: 10.3389/fncel.2022.844211
Received: 27 December 2021; Accepted: 08 March 2022;
Published: 29 April 2022.
Edited by:
Chandrakanth Reddy Edamakanti, Northwestern Medicine, United StatesReviewed by:
Mahesh Narayan, The University of Texas at El Paso, United StatesBasant K. Patel, Indian Institute of Technology Hyderabad, India
Carmen Nussbaum-Krammer, Heidelberg University, Germany
Copyright © 2022 Moretto, Stuart, Surana, Vargas and Schiavo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edoardo Moretto, RWRvYXJkby5tb3JldHRvQGluLmNuci5pdA==; Giampietro Schiavo, Z2lhbXBpZXRyby5zY2hpYXZvQHVjbC5hYy51aw==
†ORCID: Edoardo Moretto, orcid.org/0000-0002-3546-6797; Skye Stuart, orcid.org/0000-0002-3503-4687; Sunaina Surana, orcid.org/0000-0002-7017-3105; Jose Norberto S. Vargas, orcid.org/0000-0001-5529-0883; Giampietro Schiavo, orcid.org/0000-0002-4319-8745