 Georgios Michalettos
Georgios Michalettos Karsten Ruscher
Karsten Ruscher- 1Laboratory for Experimental Brain Research, Division of Neurosurgery, Department of Clinical Sciences, Wallenberg Neuroscience Center, Lund University, Lund, Sweden
- 2LUBIN Lab—Lunds Laboratorium för Neurokirurgisk Hjärnskadeforskning, Division of Neurosurgery, Department of Clinical Sciences, Lund University, Lund, Sweden
Adaptive plasticity processes are required involving neurons as well as non-neuronal cells to recover lost brain functions after an ischemic stroke. Recent studies show that gamma-Aminobutyric acid (GABA) has profound effects on glial and immune cell functions in addition to its inhibitory actions on neuronal circuits in the post-ischemic brain. Here, we provide an overview of how GABAergic neurotransmission changes during the first weeks after stroke and how GABA affects functions of astroglial and microglial cells as well as peripheral immune cell populations accumulating in the ischemic territory and brain regions remote to the lesion. Moreover, we will summarize recent studies providing data on the immunomodulatory actions of GABA of relevance for stroke recovery. Interestingly, the activation of GABA receptors on immune cells exerts a downregulation of detrimental anti-inflammatory cascades. Conversely, we will discuss studies addressing how specific inflammatory cascades affect GABAergic neurotransmission on the level of GABA receptor composition, GABA synthesis, and release. In particular, the chemokines CXCR4 and CX3CR1 pathways have been demonstrated to modulate receptor composition and synthesis. Together, the actual view on the interactions between GABAergic neurotransmission and inflammatory cascades points towards a specific crosstalk in the post-ischemic brain. Similar to what has been shown in experimental models, specific therapeutic modulation of GABAergic neurotransmission and inflammatory pathways may synergistically promote neuronal plasticity to enhance stroke recovery.
Introduction
Stroke is among the most common and important causes of disability and death worldwide. Annually, approximately 12 million people suffer a stroke worldwide; thereof 6 million patients do not survive the insult. Approximately 5 million stroke victims acquire life-long disabilities and will need support for daily living by next of kin or at institutions (Feigin et al., 2021). The societal cost associated with stroke is huge, amounting to 60 billion euros in the EU in 2019 (Luengo-Fernandez et al., 2020), alarming figures we expect to grow with an increasing aging population and health care costs. Stroke causes loss of neurons and of neurological function due to cell loss predominantly in the affected neuronal tracts and circuits. In addition, neurological deficits are also due to dysfunction of remaining neurons in the vicinity to or in areas remote from the infarct connected through brain-wide neural networks (Carter et al., 2010). The affected neuronal networks, therefore, are considered as neuronal substrates for recovery-promoting therapies modulating mechanisms of brain plasticity, i.e., the innate ability of the brain to remodel neural network connections.
Brain plasticity comprises the ability of the brain to reorganize its cellular structures and its function in response to intrinsic and extrinsic stimuli (Wieloch and Nikolich, 2006; Cramer et al., 2011). Brain plasticity can be stimulated in stroke patients by multimodal rehabilitation (Bunketorp-Kall et al., 2017). For instance, various advanced training paradigms, assisted by virtual reality, computer gaming, are thought to stimulate brain plasticity, and have shown promise in supporting rehabilitation (Hatem et al., 2016). In the experimental setting, this is accomplished by an enriched environment (EE), comprising large cages with toys, tubes, ladders, and larger groups of animals with the opportunity for more complex social interaction that activates various neural networks of the brain. Furthermore, transcranial magnetic stimulation (TMS) or direct current stimulation (tDCS) enhances recovery even when treatment is instituted months after the stroke. However, pharmacological interventions are still limited to support rehabilitation after stroke.
Appropriate neuronal function depends on interconnected and well-organized circuits of inhibitory interneurons and excitatory projection of cortical pyramidal neurons. Any changes in the synaptic stability and organization of inhibitory neurons may impair the regulation of excitatory circuits. GABAergic neurotransmission, therefore, is a key element regulating the excitation/inhibition balance, and brain connectivity.
Background—Elements of GABAergic Neurotransmission
γ-aminobutyric acid (GABA) is the main inhibitory neurotransmitter in the adult brain. Together with the excitatory neurotransmitter glutamate, GABA neurotransmission regulates the inhibitory-excitatory balance necessary for adequate brain function. In the adult brain, GABA is responsible for the hyperpolarization of the cell, preventing the conveyance of neuronal action potentials. The main functions of GABA in mediating inhibitory neuronal activity have been extensively studied. In addition, research is focused on unraveling new roles of the amino acid in non-neuronal cell functions and pathologies. There are two main types of GABA receptors ionotropic GABAA(GABAARs) receptors and metabotropic GABAB receptors (GABABRs; Wu and Sun, 2015).
Glutamic Acid Decarboxylase Isoforms
Synthesis of GABA in neurons is catalyzed by two isoforms of the glutamicacid decarboxylase, GAD65, and GAD67 (Pinal and Tobin, 1998). These key enzymes convert glutamate to GABA. Different functions of each isoform appeared mainly due to studies from knock-out mice (Asada et al., 1997; Condie et al., 1997; Kash et al., 1997; Tian et al., 1999). GAD67 has been found to be constitutively active and provide the majority of the cytosolic GABA, while GAD65 is mainly responsible for synaptic GABA production released from synaptic vesicles during neurotransmission (Pinal and Tobin, 1998; Battaglioli et al., 2003). Together, both isoforms exhibit different cellular distribution and structural properties (Dupuy and Houser, 1996; Jin et al., 2003). Their function strongly depends on post-translational modifications including phosphorylation, palmitoylation, and cleavage (Lee et al., 2019).
While GAD67 is a hydrophilic cytoplasmic protein, GAD65 undergoes post-translational hydrophobic modifications, which enable it to be anchored on the membrane compartments of the synaptic vesicles (Kanaani et al., 1999, 2002). Further studies have confirmed that the two isoforms can interact with each other, creating heterodimers in the membrane of the synaptic vesicles (Kanaani et al., 2004, 2010). This indicates that GAD67 is not only involved in maintaining metabolic levels of cellular GABA but may also contribute to the rapid vesicular accumulation of GABA in the presynaptic terminal for covering the incremental needs of synaptic neurotransmission by vesicular release into the synaptic cleft (Kanaani et al., 2010).
GABAA Receptors
GABAA receptors belong to the family of Cys-loop ligand-gated ion channels and are responsible for mediating most of the fast inhibitory neurotransmission in the central nervous system (CNS). When GABA binds to these receptors at post-synaptic sites, the ion channel opens, enabling the influx of chloride (Cl−) ions into the cell along a concentration gradient resulting in a change in the membrane potential. Together with other factors such as the maturation status of cells, either de- or hyperpolarization of the post-synaptic mature neuron occurs (Fritschy and Panzanelli, 2014). GABAA receptors are pentameric channels which are composed of a variety of subunits. Nineteen genes have been identified encoding GABAA receptor subunits (α1–α6, β1–β3, γ1–γ3, δ, ε, π, θ, ρ1–ρ3) in mammals, demonstrating that there is high heterogeneity assembling the receptor (Barnard et al., 1998; Bonnert et al., 1999). Experimental evidence suggests that the GABAA receptor assembled of different subunit compositions provide variable functions and pharmacological properties (Gingrich et al., 1995; Browne et al., 2001; Dixon et al., 2014). The second type of GABAA receptor previously described as the GABAC receptor, is a homopentameric ion channel solely comprised of ρ1-ρ3 subunits with distinct distribution in the CNS (Wegelius et al., 1998).
Depending on their cellular localization, GABAA receptors have been categorized into synaptic and extra-synaptic, mediating synaptic (phasic), and extra-synaptic (tonic) inhibition, respectively (Farrant and Nusser, 2005; Glykys and Mody, 2007; Brickley and Mody, 2012). The most common stoichiometry, accounting for the majority of the overall distribution of GABAA receptors in the brain, is the heteropentameric form consisting of two α-subunits, two β- and one γ-subunit, with the α1α1β2β2γ2 combination (Sieghart and Sperk, 2002; Goetz et al., 2007). Initially, segregation was made between synaptic and extrasynaptic receptors with synaptic (phasic) GABAA receptors being composed primarily of α1–3, β1–3, and γ1–3 subunits and extrasynaptic GABAA receptors consisting of α4, α5, α6, or δ-subunits. However, electrophysiological studies combined with pharmacological application of GABAA receptor positive allosteric modulators have revealed that subunits, previously defined as “synaptic” are also found in somatic membranes of neurons (Lindquist and Birnir, 2006; Stojanovic et al., 2016). In specific, zolpidem, a positive allosteric compound of α1-, α2-, α3-, and γ2- containing GABAA receptors, activated such receptors in granule neurons of the dentate gyrus, however, to a lesser extent compared to compounds that target α4-, α6-, and δ- containing receptors (Lindquist and Birnir, 2006). These results indicate that a higher receptor subunit diversity of extrasynaptic receptors.
Depending on the neuronal cell type, GABAA receptors can be localized in different subcellular regions such as in somata, dendrites, synapses, and in the non-synaptic membrane (Somogyi et al., 1989). Furthermore, the δ-subunit is assembled in the place of the γ2 subunit and is typically associated with the α4 and α6-subunit isoforms (Clarkson, 2012; Fritschy and Panzanelli, 2014). Several studies have revealed that the β2, β3, γ2 variants are the most abundant isoforms participating in the assembly of the majority of GABAA receptors subtypes, with α4βδ complexes being mostly located in the cortex, hippocampus, and thalamus and α6βδ complexes being located in the cerebellum (Fritschy and Panzanelli, 2014; Nguyen and Nicoll, 2018).
Similar distribution patterns of GABAA receptor subunits have been found in rodents and immunohistochemical and in situ hybridization studies on post-mortem human tissue (Waldvogel et al., 2010). Most of the αsubunits exhibit laminar expression in the cerebral cortex similar to that of rodents (Akbarian et al., 1995; Lewohl et al., 1997; Waldvogel et al., 2010; Stojanovic et al., 2016). In addition, structures such as the hippocampus, the basal ganglia, and the thalamus in the human brain exhibit a high degree of similarity to rodents regarding the expression profile of GABAA receptor subunit subtypes and their subcellular localization (Houser et al., 1988; Waldvogel et al., 1999; Loup et al., 2000; Popken et al., 2002; Stojanovic et al., 2016). However, it should be noted that differences have been observed, mostly on the expression level of individual subunit subtypes (Waldvogel et al., 2010). Furthermore, due to the difficulty of conducting relevant experiments in humans, there is limited data on the dynamics of receptor composition in the human brain, an aspect taken into consideration when translating preclinical results to the human brain.
Topological Dynamics of GABAA Receptor Subunits
The localization of GABAA receptors is a dynamic process, which involves the trafficking of the ion-channel receptors along the surface of the neurons (Choii and Ko, 2015; Lorenz-Guertin and Jacob, 2018). When focusing on synaptic inhibition, GABAA receptors, either through lateral diffusion or exocytosis, accumulate in the membrane of the post-synaptic neuron. Synaptic localization of the receptor is determined by its interaction with a “local” anchoring complex, consisting of gephyrin molecules (Craig et al., 1996; Studler et al., 2005; Goetz et al., 2007).
Several studies have focused on the importance of the γ2-subunit maintaining the ability to cluster on the inhibitory synapses (Kittler and Moss, 2003; Kittler et al., 2004). Lack of the γ2-subunit and replacement by δ-containing GABAA receptors, along with their respective α4, α5, and α6 variants, prevents localization at the synaptic cleft and such GABAA receptors are found almost exclusively extrasynaptically, mediating tonic inhibition through ambient GABA levels (Nusser and Mody, 2002; Stell et al., 2003; Zheleznova et al., 2009). However, this model seems not rigid, since GABAA receptors containing ofα1β2γ2 or α3β3γ2 subunits can also be found in extrasynaptic membranes, although less frequently, thus determining a dynamic system regarding the localization of GABAA receptors (Nusser et al., 1998; Mortensen et al., 2012).
The above-mentioned distribution pattern of extrasynaptic receptors is found in cerebellar granular cells, the dentate gyrus, the thalamus, in the granular cortical layers as well as in the hippocampus (Nusser and Mody, 2002; Sieghart and Sperk, 2002; Zheleznova et al., 2009). The α1-α3 subunits exhibit unique distribution patterns and partial overlap, with the α1 subunit being the most prevalent throughout the adult mouse forebrain (Sieghart and Sperk, 2002; Fritschy and Panzanelli, 2014).
Spatiotemporal and Age-Related Alterations in GABAA Receptor Subunits Expression
The GABAergic neurotransmitter system is highly modular and dynamic. Variables such as age, sex, environmental factors, dietary habits, circadian cycle, contribute to the complexity of successfully translating preclinical findings into precise personalized clinical care. For example, spectroscopy studies on humans regarding the response of GABA to motor learning revealed a decrease of the inhibitory neurotransmitters in the motor cortex (M1) in intervals of neuronal plasticity changes (Sampaio-Baptista et al., 2015; Kolasinski et al., 2019). Furthermore, the progression of the ovarian cycle alters the number of δ-containing GABAARs in the hippocampus, ultimately affecting the electrophysiological properties of the local circuitry (Barth et al., 2014). In parallel, GABAAR subunit expression is modulated by estrogens, however, in-depth studies are warranted if these changes depend on the brain region (Herbison and Fénelon, 1995). Subunit expression does not differ between males and females in the temporal cortex, although differences in other brain regions cannot be excluded (Pandya et al., 2019). Furthermore, alcohol dependence studies focused on post-mortem human brains have revealed spatial alterations in the expression of certain GABAA subunits, indicating possible changes in receptor composition that could be indicative of the behavioral pathology in dependence (Jin et al., 2012; Bhandage et al., 2014).
Interestingly, subunits such as the ε and ρ2 subtypes which are less frequently studied were not only confirmed on transcript level in the human brain but were also demonstrated to undergo regulation (Bhandage et al., 2014). Nevertheless, the extent to which functional subunits are integrated into receptors and are modulated by the same post-translational modifications observed in rodents remains to be elucidated in humans.
In the context of aging, evidence shows that the GABAergic system might be affected in older age, unable to exert its fine-tuned inhibitory efficacy on the neural circuits of the brain, ultimately affecting plasticity and adaptiveness to brain injury. In specific, electrophysiological studies conducted on transgenic APPSwe mice, a rodent model of Alzheimer’s disease (AD), revealed that GABA-induced currents in the dentate gyrus (DG) of the hippocampus are not altered in this area by age alone but in combination with the presence of Alzheimer’s disease pathology (Hammoud et al., 2021). In accordance with this observation, electrophysiological recordings on human temporal cortices with AD demonstrated impaired GABAergic signaling (Limon et al., 2012). This effect was attributed to possible alterations in the composition of GABAA receptors, overall affecting the brain’s response to GABA (Limon et al., 2012). On the other hand, quantitative studies on human cortical samples revealed that the expression of most of the elements comprising the GABAergic system remains robust throughout age with the exception of certain subunits and GAT-1 (Pandya et al., 2019). Additionally, alterations in subunit expression differ throughout aging between males and females (Ethiraj et al., 2021).
GABAB Receptors
GABAB receptors belong to the family of G protein-coupled receptors (GPCR). They are responsible for the later and slower component of inhibitory transmission and are found both on the pre- and post-synaptic membrane (Huang et al., 1995; Sakaba and Neher, 2003; Ulrich and Bettler, 2007). GABAB receptors are comprised of two subunits, R1 and R2. Their regulation varies on the transcriptional and post-translational levels dependent on the physiological or pathological condition (Benke et al., 1999; Billinton et al., 2000; Terunuma et al., 2010; Kantamneni et al., 2014). The majority of GABAB receptors inhibit isoforms of the adenyl cyclase (AC) through the Gαi/o subunits (Wojcik and Neff, 1984; Xu and Wojcik, 1986; Terunuma, 2018). In parallel, different isoforms of AC have been demonstrated to undergo stimulation as a result of ligand binding due to activation from Gβγ subunits of GPCR receptors (Tang and Gilman, 1991; Terunuma, 2018), indicating a multi-functional role of these receptors in modulating intracellular signaling pathways and neuronal activity.
Activation of GABAB receptors is also coupled to K+ and/or Ca2+ channels via G-protein mediated pathways (Gähwiler and Brown, 1985; Huang et al., 1995; Misgeld et al., 1995; Sodickson and Bean, 1996; Filippov et al., 2000). Both channels are either activated or inhibited by β and γ subunits of the G protein. Upon opening of G protein-gated inward rectifying potassium (GIRK) channels, membrane potential changes, reducing the excitability of neurons in their resting phase. In addition, GABAB receptors inhibit voltage-sensitive calcium (Ca2+) channels (VSCC) controlling the rate of neuronal firing as well as neuronal processes dependent on the dynamics of intracellular calcium (Mintz and Bean, 1993; Pfrieger et al., 1994; Lambert and Wilson, 1996; Limon et al., 2012; Bhandage et al., 2014; Hammoud et al., 2021). As such, both channels have been demonstrated to be determinant elements in defining synaptic transmission and plasticity in neuronal networks under physiological conditions as well as brain pathologies (Chung et al., 2009; Frank, 2014; Marron Fernandez de Velasco et al., 2015; Sánchez-Rodríguez et al., 2017; Nanou and Catterall, 2018).
In this review, we will focus on the relevance of the aforementioned elements of the GABAergic system in the context of brain injury, specifically ischemia and stroke, and their possible interactions with the inflammatory and immune response found in the post-ischemic brain during the recovery phase of stroke. We will focus on the involvement of GABAA receptors as mediators of neuronal activity and synaptic plasticity and the perspective of pharmacological intervention, contributing to neuroprotection and the recruitment of cellular repair mechanisms necessary to facilitate stroke outcome.
GABAergic Neurotransmission After Stroke
An initial increase in the release of GABA is found in the ischemic brain following global ischemia (Globus et al., 1991) and focal ischemia induced by permanent occlusion of the middle cerebral artery (Ruan et al., 2017). Interestingly, to counteract the excitotoxic insult various studies demonstrate that an enhanced GABAergic tonus i.e., by administration of GABAergic agents prior to ischemia or shortly after the ischemic episode provides neuro protection in preclinical stroke models (Corbett et al., 2008). In addition, the identification of molecules and cascades that enhance GABAergic neurotransmission during the acute phase after stroke has been a promising field defining neuroprotective compounds. For example, peptide hormones, such as oxytocin and insulin, have been characterized for their in vitro neuroprotective role through mechanisms of GABAA receptor subunit upregulation and increased post-ischemic cell-surface receptor stability, respectively (Mielke and Wang, 2005; Kaneko et al., 2016). In addition, erythropoietin, a glycoprotein whose application has been extensively investigated in stroke for its protective effect, has been demonstrated to enhance GABAergic activity (Ruscher et al., 2002; Gonzalez et al., 2007; Juenemann et al., 2020; Roseti et al., 2020). While levels of GABA increased, the immediate intrinsic reaction of insulted cells appears to downregulate and internalize cell-surface GABAA receptors after the insult (Kittler et al., 2005; Mielke and Wang, 2005; Mele et al., 2014; Costa et al., 2016). Decreased density of plasma-membrane receptors has been associated with truncation of anchoring structures as well as post-translation modifications on regulatory residues of receptor subunits that promote clathrin-dependent endocytosis (Kittler et al., 2005; Mielke and Wang, 2005; Mele et al., 2014; Costa et al., 2016). Despite promising preclinical results, early administration of benzodiazepines did not favor outcomes in patients and even increased post-stroke mortality at 90 days (Lodder et al., 2006; Colin et al., 2019). The discussion of unsuccessful translation is beyond the scope of this review and will need to involve all aspects of translation of preclinical data into clinical development and practice.
Modulation of processes that contribute to recovery beyond the time window of neuroprotection represents a paradigm shift aiming at enhancing brain plasticity mechanisms following stroke. Results from preclinical studies and clinical observations provide evidence of an increased inhibition of neuronal function that impedes the recovery of lost neuronal function, importantly independent of the lesion size (Hagemann et al., 1998; Bütefisch et al., 2003; Xie et al., 2014). This elevated inhibitory tonus is mediated by the GABAergic system and prevents the restoration of impaired neuronal function in the area of the lesion, hindering the effectiveness of repair mechanisms such as axonal regrowth, synapse formation, and cytoskeletal rearrangement (Paik and Yang, 2014; Joy and Carmichael, 2021).
After the acute phase, layer 2/3 pyramidal neurons are exposed to a high degree of GABAergic tonic inhibition mediated by extrasynaptic receptors and attributed to GABA transporter (GAT-3/4) dysfunction (Clarkson et al., 2010). This effect lasts for up to 2 weeks after focal permanent ischemia induced by photothrombosis (Clarkson et al., 2010). Specific pharmacological inverse antagonism on the α5 subunit of GABAA receptors improved functional outcomes (Clarkson et al., 2010; Wang et al., 2018). At the same time, the brain may intrinsically lower tonic inhibition. This mechanism, however, is associated with an increased frequency of epileptic discharges (Jaenisch et al., 2016).
In addition, enhanced phasic GABAergic signaling has been found in cortical layer 5 of the peri-infarct area during the first and second week of the recovery phase of stroke (Hiu et al., 2016; Feng et al., 2020). This effect seems to be mediated by α1-containing GABAA receptors (Hiu et al., 2016; Neumann et al., 2019). It is of note that an increase of α1-containing GABAA receptors in the synapses of lower cortical areas adjacent to the ischemic core might be highly region-specific. Interestingly, transcript levels of the α1 subunit are decreased in the proximal peri-infarct cortical area 7 days after photothrombosis (Kharlamov et al., 2008). This indicates the regulation on both the level of gene transcription as well as post-translational modifications, which may affect recycling/trafficking of receptors (Han et al., 2021). An overview on changes in the expression of GABAA receptor subunits in different rodent stroke models over time is summarized in Table 1.
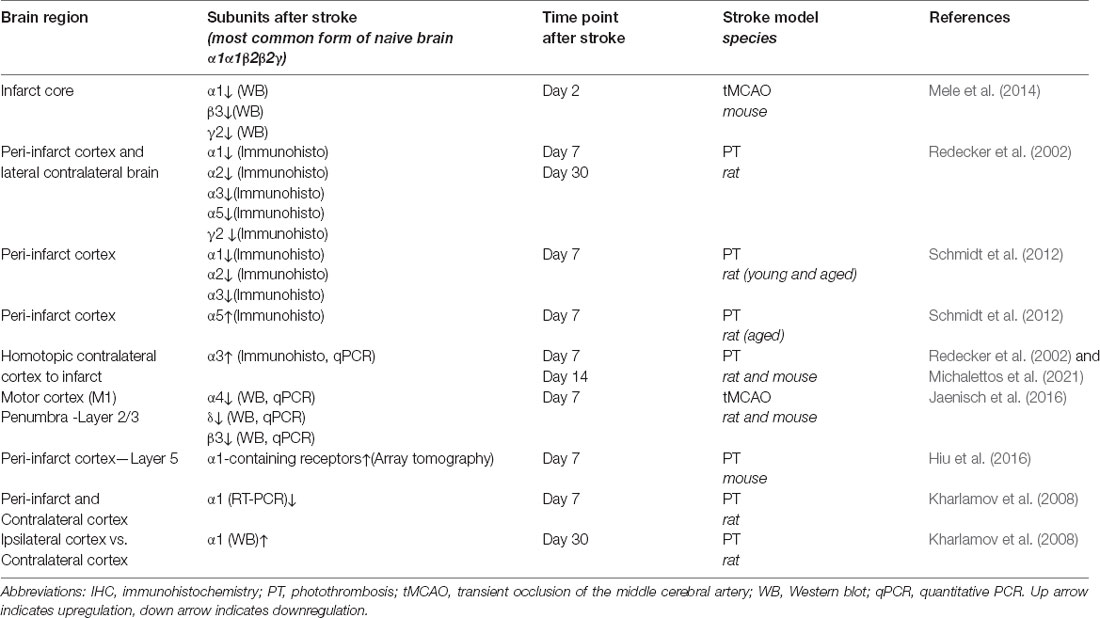
Table 1. Changes of GABAA receptor subunita expression in rodent models of stroke.
Furthermore, adaptive plasticity processes in the lesioned hemisphere function together with remote neuronal networks, i.e., in the homotopic regions of non-lesioned hemisphere (Cramer, 2008; Carmichael, 2012; Boddington and Reynolds, 2017; Hakon et al., 2018). Interestingly, synaptic α3 subunits are upregulated in the contralateral motor cortex of rats subjected to photothrombosis (Redecker et al., 2002). We recently also found an interhemispheric asymmetry of the α3 subunit on transcript level during the recovery phase of stroke (Michalettos et al., 2021). This might be interpreted as intrinsic action to avert a preponderance of neuronal activity on the contralateral side. In addition, α3-containing GABAA receptors may possess distinct kinetic and functional properties, regarding to their response to GABA activation (Gingrich et al., 1995; Browne et al., 2001). However, despite these studies, the exact role and regulation of synaptic GABAergic neurotransmission in the the modulation of neuronal function and plasticity remains to be studied in detail.
Data from these studies are obtained from young male rodents. Further studies will be required to understand how age and sex contribute to GABAergic neurotransmission following stroke. From a GABAergic perspective, adaptive plasticity mechanisms may be impaired in the aged brain due to a prevalenceof GABAA receptors showing different pharmacological kinetics and response to GABA compared to young animals. In addition, evaluating inhibitory tonus as a measure of repressed plasticity in the post-injured brain, concomitant changes in the excitatory input may occur, making it difficult to estimate changes in the overall excitatory/inhibitory balance of the neural circuits undergoing rearrangement. In specific, both GABAA and GABAB receptors have been demonstrated to interact with α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptors in a positive feedback manner, both on the level of neurotransmitter release and excitatory and inhibitory post-synaptic currents (Ben-Ari et al., 1997; Chen et al., 2000; Fiszman et al., 2005; Kantamneni, 2015; Schulz et al., 2018).
Despite GABAA receptors being the receptor involved in the majority of cascades, activation of GABAB receptors has also been proposed to participate in cellular responses mediating neuronal survival (Costa et al., 2004), further contributing to the role of GABA as an all-around protecting-mediator of neuronal injury.
Immune/Inflammatory Response in Stroke
Inflammation is one of the core processes involved in the pathophysiology of stroke (Iadecola and Anrather, 2011). Following neuronal cell death and the initiation of the ischemic cascade, several immunological cascades take place in order to isolate and restore function to the lesioned area (Lakhan et al., 2009). However, depending on the severity of the damage and the magnitude of the immune response, secondary inflammation can further contribute to the collateral damage of the injured area, prevailing for weeks after the end of the acute ischemic phase (Rayasam et al., 2018). Processes such as microglial activation and migration, upregulation of pro-inflammatory cytokines, sealing of the injured area through glial scar formation, leucocyte chemotaxis, and infiltration, increase in blood-brain barrier (BBB) permeability, and recruitment of adaptive immunity mechanisms characterize the early environment of the brain in the acute and sub-acute phase of stroke (Morioka et al., 1993; Vila et al., 2000; Gelderblom et al., 2009; Lakhan et al., 2013; Pawluk et al., 2020). However, it is still unclear which components of the inflammatory response, either innate or adaptive, are responsible for providing a neuro-protective role and which contribute to further induced collateral damage to the lesioned ischemic tissue.
Modulation of Glial Function by GABA—Relevance for Post-stroke Inflammation
Upon stroke, microglia, pericytes, and astrocytes, begin to seal the injured area through a process termed reactive gliosis (Burda and Sofroniew, 2014; Sims and Yew, 2017). The glial scar separates the necrotic area of the infarct core from the adjacent tissue and potentially viable neurons capable of retaining their cellular physiology. The glial scar not only provides a physical barrier but also represents a rather dynamic microenvironment regulating biochemical, intracellular, and extracellular functions in the vicinity of the injured area (Sofroniew, 2005; Becerra-Calixto and Cardona-Gómez, 2017; Sims and Yew, 2017). The physiological role of astrocytes in processes of homeostasis of neurotransmitters, transport of water, ion buffering, metabolic surveillance, and immunomodulation, has been well-established (Sofroniew, 2005; Becerra-Calixto and Cardona-Gómez, 2017; Sims and Yew, 2017). In addition, microglia, the resident immune cell of the brain, play a pivotal role in mediating inflammation and neuronal plasticity after CNS injury (Lull and Block, 2010; Anttila et al., 2017).
Interestingly, astrocytes possess components of the molecular machinery to synthesize, metabolize and store GABA, 4-aminobutyrate transaminase (GABA-T), GABA transporters as well as ionotropic and metabotropic receptors (Lee et al., 2011; Höft et al., 2014). The repertoire of GABAAR subunits also varies between astrocytes dependent on their localization (Riquelme et al., 2002; Höft et al., 2014). For instance, electrophysiological studies on spinal cord astrocytes showed opposite effects of inverse benzodiazepine agonism in fibrous and protoplasmic types suggesting alterations of receptor composition while undergoing morphological transitions (Rosewater and Sontheimer, 1994). In addition, astrocytes mediate neurotransmitter homeostasis through non-synaptic interactions such as uptake via GAT2 and GAT3 and metabolic conversion (Schousboe et al., 2013). Furthermore, with the exception of GAD, microglia also express ionotropic and metabotropic receptors as well as GABA-T (Kuhn et al., 2004; Lee et al., 2011; Nieman et al., 2020). However, the function of these receptors and transporters is poorly understood.
In contrast to astrocytes, microglia express the γ2-subunit (Höft et al., 2014; Nieman et al., 2020). It has been demonstrated that microglia actively survey and interact with synapses, extending their processes over the bulbous area of the synaptic buttons (Wake et al., 2009). The dynamics of these interactions are altered following transient middle cerebral artery occlusion (tMCAO) in mice (Wake et al., 2009). As such, microglial GABAA receptors may directly associate with gephyrin complexes in the synaptic clusters, possibly affecting the trafficking of receptors (Schousboe et al., 2013). This type of direct interaction has not been verified for GABAA receptors in spinal cord microglia, but rather exclusively for glycinergic receptors (Cantaut-Belarif et al., 2017). Further studies are required to evaluate similar mechanisms in brain resident microglia.
The effect of GABA on astrocytes and microglia reducing the activation of inflammatory mediators NF-κB and p38 and the release of TNFα and Il-6 after stimulation with lipopolysaccharide (LPS) and interferon-γ in vitro has been previously reported (Kuhn et al., 2004; Lee et al., 2011). NF-κB has been proposed as a detrimental inflammatory mediator in stroke, contributing to collateral neuronal damage (Schneider et al., 1999; Crack et al., 2006; Inta et al., 2006; Saggu et al., 2016). The Delta subunit-selective compound DS2, a positive allosteric modulator of extrasynaptic δ-containing GABAA receptors (Jensen et al., 2013), has demonstrated neuroprotective properties after photothrombosis and, to some extent, the treatment exerted functional recovery following stroke by attenuation of the NF-κB response (Neumann et al., 2019). However, due to low BBB penetration, it has been speculated that the mechanism of action of DS2 is not associated with modulatory effects on brain resident glial cells but rather through functional changes in peripheral immune cells (Jin et al., 2013; Neumann et al., 2019). These findings indicate that GABA could potentially act specifically on astrocytes of the glial scar, preventing an excessive activation of NF-κB. Likewise, the reduced transcriptional activity of NF-κB in reactive astrocytes may facilitate axon regeneration and thus neural repair mechanisms (Saggu et al., 2016; Becerra-Calixto and Cardona-Gómez, 2017).
Effects of GABA on Immune Cell Function in the Post-ischemic Brain
Different populations of immune cells accumulate in the ischemic territory (Gelderblom et al., 2009), integrated in an inflammatory/immune response (Iadecola et al., 2020). Functional GABAA receptors have been found on microglia, dendritic cells, T cells, natural killer (NK) cells, monocytes/macrophages, B cells, and neutrophils, respectively, all cells have been reported to be involved in post-stroke inflammation. However, for most of immune cell populations, an exact link between GABA and functional changes in immune cells has not been provided following stroke. The following review of studies, therefore, summarizes potential mechanisms how GABA may regulate immune cell functions in the post-ischemic brain (Figure 1).
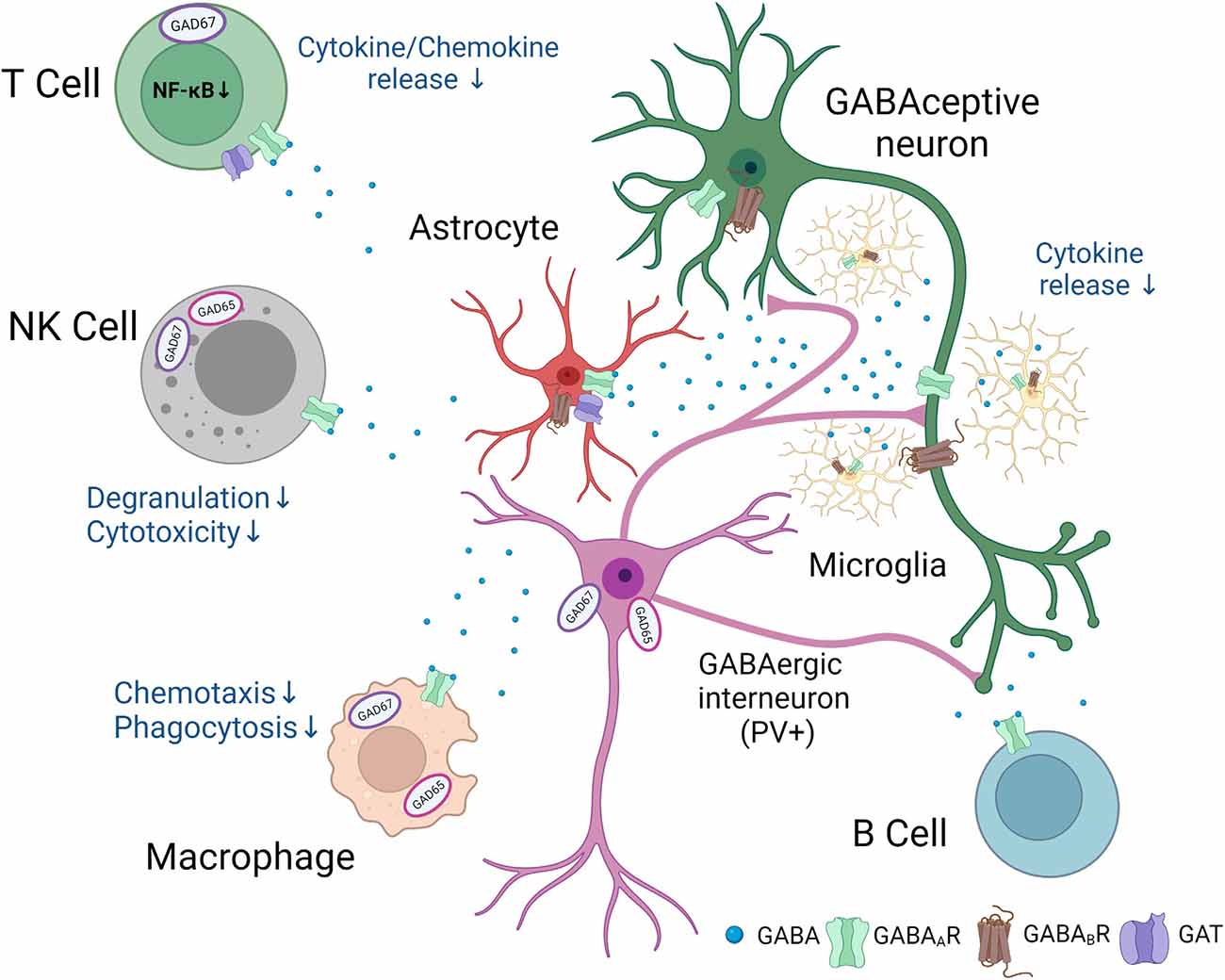
Figure 1. Putative involvement of GABAergic signaling in neuro-immunological crosstalk after stroke.Following stroke, neurons, glial, and immune cells synthesize and release GABA. In addition, GABAA and GABAB receptors and specific GABA transporters are found in a number of immune and glial cells. GABA exerts anti-inflammatory effects and changes in the function of GABAceptive neurons indicated in the figure. The figure was created with Biorender.com.
Microglia/Dendritic Cells
Microglia express both GABAA and GABAB receptors (Kuhn et al., 2004; Cheung et al., 2009). Activation of mainly the GABAB type receptors attenuated the release of lipopolysaccharide-induced IL-6 and IL-12p40, the latter acting as a chemo attractant for macrophages and promoting the migration of dendritic cells (Cooper and Khader, 2007). In addition, GABAergic signaling has been directly linked to an increased migratory activity of dendritic cells infected with Toxoplasma gondii (Fuks et al., 2012).
Moreover, stimulation of microglia with either GABA or muscimol, a selective GABAA receptor agonist, resulted in different levels of radical species production in cultured microglia indicating differently composed GABAA receptors and intracellular cascades involved in mediating GABAergic signals (Mead et al., 2012). In addition, administration of muscimol (Lee et al., 2011) to microglia stimulated with lipopolysaccharide (LPS)/interferon-γ, the latter typically shows elevated levels following stroke (Kuric and Ruscher, 2014) significantly reduced the level of pro-inflammatory cytokines interleukin 6 (IL-6) and tumor necrosis factor alpha (TNFα). Together, results point towards anti-inflammatory actions of GABA on microglial cells. Further studies will be required to exactly determine the composition as well as functionality of different GABAA and GABAB type receptors in the post-ischemic brain.
Monocytes/Macrophages
Likewise to microglia, GABAA receptors, as well as GABA synthesizing enzymes, are found in cells of the monocytic lineages (Wheeler et al., 2011). Interestingly, treatment with GABA downregulates phagocytosis and motility of macrophages and monocytic cells (Wheeler et al., 2011). Thus, an increased GABAergic tonus that develops during the first days after stroke onset might be beneficial to counteract the excessive phagocytic activity of phagocytes. In addition, GABAA receptor signaling restrains “M1” activation but fosters “M2” polarization in pulmonary macrophages (Januzi et al., 2018). This is of importance since increased levels of GABA in the ischemic territory may restrain the release of cytokines that otherwise would perpetuate detrimental actions pro-inflammatory actions.
T Cell Populations
Similar to other immune cell populations, GABA exerts immunomodulatory actions on T cells (Bjurstöm et al., 2008; Dionisio et al., 2011). Effects of an increased GABAergic tonus on the number of different T cells subpopulations after stroke have not been determined. Hence, GABA contributes to TCR-mediated T cell cycle progression silencing CD4+ T cells in the G0/G1 phase consisting of a higher portion of CD3+/CD28+ cells without affecting their viability (Tian et al., 2004). In addition, it has been demonstrated that administration of GABA or homotaurine, a GABAA receptor-specific agonist, is involved in increasing the number of regulatory T cells in EAE as well as type1 Diabetes models (Tian et al., 2018). Specifically, in the experimental autoimmune encephalitis (EAE) model, treatment enhanced the number of CD8+CD122+PD-1+ and CD4+Foxp3+ Treg cells. Regulatory CD19+IL-10+ B cells were not affected. Interestingly, IL-10+B-cell treated mice show an increased number of IL-10+CD8+CD122+ Treg population. Generation of these cells has been associated with spleen preservation and reduced CNS inflammation after tMCAO (Bodhankar et al., 2015). The role of CD4+Foxp3+ Treg cells in stroke recovery, however, remains divergent in might be dependent on the stroke model, different microenvironments in the post-ischemic brain as well as the time intervals after the insult (Liesz and Kleinschnitz, 2016). In addition, homotaurine inhibits autoreactive Th17 and Th1 responses as well as relevance for stroke recovery processes. Stimulation of T cells with GABA also has been shown to inhibit the production of pro-inflammatory cytokines and therefore it attenuates the T cell response in inflammatory disease models such as EAE but also in Diabetes models (Soltani et al., 2011; Prud’homme et al., 2015). In addition to GABAA receptor activation-mediated effects on T cells, a reasonable number of publications demonstrate some of the GABAergic effects are at least partially mediated by activating the GABA transporter type 1 (GAT-1; Wang et al., 2008). It is exclusively expressed on activated T cells primed with antigens. Increased influx of GABA via the transporter downregulates proliferation of the CD4+ T cells (Wang et al., 2008).
Natural Killer Cells
NK cells represent a population of innate immune cells accounting for about 5%–20% of human blood (Perera Molligoda Arachchige, 2021). Recently, this population has been defined as GABAergic cells due to the release of the neurotransmitter upon stimulation/exposure to pathogens and/or inflammatory stimuli (Bhandage et al., 2021). This would predestine NK cells’ interaction with other immune cells and brain resident GABAceptive cells. One possible GABA-driven interaction comes from experiments performed in NK cells and dendritic cells infected with Toxoplasma gondii (Bhandage et al., 2021). Here, stimulation with exogenous GABA reduced degranulation and cytotoxicity of NK cells. Conversely, NK cells conditioned medium containing GABA enhanced migration of parasitized dendritic cells. Such interactions might be of relevance in the post-ischemic brain since both cell types significantly contribute to post-stroke inflammatory cascades (Gan et al., 2014).
Impact of Chemokine Pathways on GABAergic Neurotransmission
Chemokines and respective receptor driven inflammatory cascades have been identified as an essential component in stroke recovery mechanism and may offer a promising field identifying novel targets to improve functional outcome.
The absence of the C-C chemokine receptor type 5 (CCR5) receptor, for example, results in worse outcomes in mice after stroke exhibiting bigger infarct sizes, sustained invasion of neutrophils during the first 7 days, and reduced brain plasticity in the chronic recovery phase (Sorce et al., 2010; Ping et al., 2021). In contrast, conditional knockdown of neuronal CCR5 prior to stroke or pharmacological antagonism of CCR5 1 day after the stroke incident were shown to be beneficial for the neurological outcome and enhanced brain plasticity (Joy et al., 2019). Similarly, the C-X3-C Motif Chemokine Ligand 1/C-X3-C Motif Chemokine Receptor 1 (CX3CL1/CX3CR1) pathway, which is unique in the CNS, has been implied to possess divergent functions. While CX3CL1 or CX3CR1 deficiency points towards a neuroprotective role (Soriano et al., 2002; Dénes et al., 2008), administration of CXC3L1 to wild-type mice or cx3cl1−/− mice showed reduced ischemic lesions in wildtype animals while an increase in lesion sizes was found in knockout littermates, respectively (Cipriani et al., 2011). We have previously reported that CX3CR1 deficiency does not affect infarct size and outcome, but causes alterations in the morphology of microglia populating the peri-infarct area (van der Maten et al., 2017).
Pharmacological antagonism of C-X-C Motif Chemokine Receptor 1/C-X-C Motif Chemokine Receptor 2 (CXCR1/CXCR2) by the C-X-C Motif Chemokine Ligand 8 (CXCL8) receptor blocker reparixin aiming at attenuating leukocyte infiltration promoted functional outcome and reduced infarct size (Villa et al., 2007). Likewise, C-C Motif Chemokine Receptor 2CCR2−/− knockout mice exhibited beneficial outcome after tMCAO, with reduced monocyte and macrophage infiltration as well as reduced BBB permeability (Dimitrijevic et al., 2007). We have previously shown that pharmacological antagonism of C-X-C Motif Chemokine Receptor 4 (CXCR4) with AMD3100 attenuates the accumulation of CX3CR1-positive microglia and contributes to enhanced recovery of lost neurological function (Walter et al., 2015). Likewise, conditional knockout of the CXCR4 gene in hematopoietic stem cells (HSCs) and their derivatives, such as circulating monocytes and monocyte-derived macrophages, results in a reduced population of immune cells in the ischemic territory, after both PT and tMCAO (Werner et al., 2020).
Besides their function on inflammatory cells, experimental evidence emerge revealing a role of chemokine-driven cascades in neurons and neuronal function in the post-ischemic brain (de Haas et al., 2007). Following a stroke, an upregulation of CCR5 transcripts has been detected in neuronal cells, which was not detectable before the insult and in naïve mice (Joy et al., 2019). To which extent transcripts are translated into functional proteins remains to be elucidated. We previously found that NeuN+ neurons express CXCR4 in the peri-infarct area, as a target for AMD3100 treatment to specifically modulate this pathway (Ruscher et al., 2013). Similarly, CCL2 and CX3CR1 are upregulated in neurons following hypoxia or ischemia (Andres et al., 2011; Wang et al., 2018). CCR2 has also been demonstrated to affect GABA-induced currents in spinal neurons, indicating mechanistic interactions between chemokine receptors and GABAA receptors (Gosselin et al., 2005). GABA-induced currents are also affected by cytokines (Giacco et al., 2019). Susceptibility of neurons to chemokines and cytokines potentially modulates mechanisms of synaptic plasticity, neurotransmitter receptor expression and neurotransmitter-producing enzymes. Therefore, we will summarize the current evidence on how chemokine pathways interact with the GABAergic neurotransmission following stroke.
Cross Talk Between the CX3CL1/CX3CR1 Pathway and GABAergic Neurotransmission
It has been previously described that CX3CL1, as a membrane-bound protein found both in neurons and glial cells, undergoes cleavage after excitotoxic conditioning. Shedding of the protein is characteristic of the ischemia onset (Chapman et al., 2000; Meucci et al., 2000; Wang et al., 2018). As such, the soluble form of the chemokine acts in a paracrine fashion on microglia and neurons, as well as a chemotactic agent for infiltrating immune cells (Imai et al., 1997; Dichmann et al., 2001; Tarozzo et al., 2002; Wang et al., 2018). Electrophysiological studies on hippocampal neurons and serotonin neurons of the dorsal raphe nucleus have demonstrated that the application of CX3CL1 enhances inhibitory post-synaptic currents through GABAA receptors but depresses excitatory inputs from AMPA receptors through post-translational modifications (Ragozzino et al., 2006; Heinisch and Kirby, 2009). From a GABAergic perspective, the neuroprotective role of CX3CL1 could be attributed in part to enhanced inhibitory currents and suppressed AMPA receptor function during the acute excitotoxic phase of ischemia (Cipriani et al., 2011).
Studies on both physiological and pathological conditions, such as epilepsy, demonstrate that the CX3CR1 pathway may regulate the number of post-synaptic GABAA receptors or their sensitivity to GABA and thus their subunit composition (Heinisch and Kirby, 2009; Roseti et al., 2013). We have shown that CX3CR1 deficiency modulates the expression of GABAA receptor subunits in the recovery phase of stroke both in the ischemic and contralateral to the lesion hemisphere (Michalettos et al., 2021). This indicates that plastic procedures occurring in the homotopic contralateral motor region may be partially mediated by a CX3CR1-dependent mechanism. However, it needs to be further elucidated whether this effect is the result of neuronal receptor absence or an altered microglia-synapse interaction (Wake et al., 2009).
Involvement of CXCR4/CXCL12 in Neuromodulation After Stroke
The upregulation of the CXCR4/CXCL12 pathway in the ischemic hemisphere has been extensively described (Stumm et al., 2002; Schönemeier et al., 2008; Wang et al., 2012; Ruscher et al., 2013). However, there is limited data on the aspect of how the CXCR4/CXCR7/CXCL12 axis modulates neuronal function following stroke. It is well established that post-natal neurons express functional CXCR4 receptors, including GABAergic interneurons (Trecki et al., 2010; Wu et al., 2017). Electrophysiological studies point towards a pre-synaptic mode of action of the receptor, mediating Ca2+-dependent release of GABA (Guyon et al., 2006; Heinisch and Kirby, 2010), and a post-synaptic mode of action, possibly involving direct interactions of the receptor with GABAB receptors (Guyon et al., 2006). In addition, it has been demonstrated that CXCL12 modulates CX3CL1 homeostasis by regulating CX3CL1 expression as well as CX3CL1 cleavage rate under physiological conditions (Cook et al., 2010). We have confirmed this interaction in vivo, in the recovery phase of mice subjected to PT, in which pharmacological antagonism with AMD3100 resulted in reduced CX3CL1 levels, both membrane-bound and soluble (Walter et al., 2015). Therefore, low levels of CX3CL1 may shift the inhibitory-excitatory balance towards an excitatory tissue environment, allowing for beneficial plastic procedures to take place, a process which might be age-dependent.
As such, the potential effects of AMD3100 on the inhibitory-excitatory balance of the post-ischemic brain involve two distinct levels of interactions, including modulation of inflammatory response and regulation of neuronal function. Increased levels of GAD enzymes and GAD67-positive neurons have been observed in the ipsilateral striatum 1 to 2 days after transient forebrain ischemia (Li et al., 2010). An increase of inhibitory neurons in the vicinity of the ischemic tissue was attributed to resident somatostatin-expressing neurons shifting to a GABAergic phenotype and not the maturation of migrating neural precursor cells (Li et al., 2010). In vitro studies, on the other hand, showed that the CXCR4 pathway is related to the maturation process of embryonic hippocampal neurons through the induction of GAD67 expression (Luo et al., 2008). We have demonstrated that administration of AMD3100 for 2 weeks resulted in decreased expression of GAD67 and GABAA subunits in the peri-infarct area (Michalettos et al., 2021). This might be related to a reduced formation of GAD67-positive interneurons or direct regulation of GAD67 expression downstream of neuronal CXCR4 receptors. The downregulation of GABAA receptors by the treatment was not subunit-specific. Therefore, it is likely that inflammatory mediators are responsible for the synthesis/turnover of several types of GABAA receptors. Further research is required to delineate the exact mechanism of action of CXCR4 regarding the regulation of GABAA receptors in healthy and injured neurons undergoing adaptive plastic procedures.
Conclusions
Based on solid preclinical studies, post-stroke GABAergic neurotransmission and detrimental inflammatory cascades have been targeted in clinical trials. However, studies did not meet primary endpoints for several reasons. From the preclinical point of view, we are beginning to understand the complex interaction between inflammatory cascades and neuronal functions. Only with full comprehension about pathophysiology of adaptive neuronal plasticity and definitions in conjunction with well-designed clinical trials will allow for implementation of new adjuvant treatments to enhance neurological functions after ischemic stroke.
Author Contributions
Both authors contributed equally to the first draft and revision of the mansucript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Swedish Brain Fund (Hjärnfonden, grant no. FO2020-0138), the Hans-Gabriel and Alice Trolle Wachtmeister Foundation, a regional grant via ALF Skåne and Sparbankstiftelsen Färs & Frosta.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AD, Alzheimer’s disease; AMD3100, 1,1′-[1,4-phenylenebis(methylene)]bis[1,4,8,11-tetraazacyclotetradecane]; CCR2, C-C chemokine receptor type 2; CCR5, C-C chemokine receptor type 5; CX3CL1, C-X3-C Motif Chemokine Ligand 1; CX3CR1, C-X3-C Motif Chemokine Receptor 1; CXCL8, C-X-C Motif Chemokine Ligand 8; CXCR4, C-X-C Motif Chemokine Receptor 4; CNS, central nervous system; EAE, experimental autoimmune encephalitis; GABA, γ-aminobutyric acid; GABAAR, γ-aminobutyric acid type A receptor; GABABR, γ-aminobutyric acid type B receptor; GAD65, glutamic acid decarboxylase 65; GAD67, glutamic acid decarboxylase 67; GAT, γ-aminobutyric acid transporter; GIRK, G protein-gated inward rectifying potassium; NK, natural killer; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PT, photothrombosis; TCR, T-cell receptor; tMCAO, transient middle cerebral artery occlusion; TNFα, Tumor necrosis factor alpha; VSCC, voltage-sensitive calcium channel.
References
Akbarian, S., Huntsman, M. M., Kim, J. J., Tafazzoli, A., Potkin, S. G., Bunney, W. E. J., et al. (1995). GABAA receptor subunit gene expression in human prefrontal cortex: comparison of schizophrenics and controls. Cereb. Cortex 5, 550–560. doi: 10.1093/cercor/5.6.550
Andres, R. H., Choi, R., Pendharkar, A. V., Gaeta, X., Wang, N., Nathan, J. K., et al. (2011). The CCR2/CCL2 interaction mediates the transendothelial recruitment of intravascularly delivered neural stem cells to the ischemic brain. Stroke 42, 2923–2931. doi: 10.1161/STROKEAHA.110.606368
Anttila, J. E., Whitaker, K. W., Wires, E. S., Harvey, B. K., and Airavaara, M. (2017). Role of microglia in ischemic focal stroke and recovery: focus on Toll-like receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry 79, 3–14. doi: 10.1016/j.pnpbp.2016.07.003
Asada, H., Kawamura, Y., Maruyama, K., Kume, H., Ding, R. G., Kanbara, N., et al. (1997). Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc. Natl. Acad. Sci. U S A 94, 6496–6499. doi: 10.1073/pnas.94.12.6496
Barnard, E. A., Skolnick, P., Olsen, R. W., Mohler, H., Sieghart, W., Biggio, G., et al. (1998). International union of pharmacology. XV. subtypes of gamma-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function. Pharmacol. Rev. 50, 291–313.
Barth, A. M. I., Ferando, I., and Mody, I. (2014). Ovarian cycle-linked plasticity of δ-GABAA receptor subunits in hippocampal interneurons affects γ oscillations in vivo. Front. Cell. Neurosci. 8:222. doi: 10.3389/fncel.2014.00222
Battaglioli, G., Liu, H., and Martin, D. L. (2003). Kinetic differences between the isoforms of glutamate decarboxylase: implications for the regulation of GABA synthesis. J. Neurochem. 86, 879–887. doi: 10.1046/j.1471-4159.2003.01910.x
Becerra-Calixto, A., and Cardona-Gómez, G. P. (2017). The role of astrocytes in neuroprotection after brain stroke: potential in cell therapy. Front. Mol. Neurosci. 10:88. doi: 10.3389/fnmol.2017.00088
Ben-Ari, Y., Khazipov, R., Leinekugel, X., Caillard, O., and Gaiarsa, J. L. (1997). GABAA, NMDA and AMPA receptors: a developmentally regulated “ménage à trois”. Trends Neurosci. 20, 523–529. doi: 10.1016/s0166-2236(97)01147-8
Benke, D., Honer, M., Michel, C., Bettler, B., and Mohler, H. (1999). gamma-aminobutyric acid type B receptor splice variant proteins GBR1a and GBR1b are both associated with GBR2 in situ and display differential regional and subcellular distribution. J. Biol. Chem. 274, 27323–27330. doi: 10.1074/jbc.274.38.27323
Bhandage, A. K., Friedrich, L. M., Kanatani, S., Jakobsson-Björkén, S., Escrig-Larena, J. I., Wagner, A. K., et al. (2021). GABAergic signaling in human and murine NK cells upon challenge with Toxoplasma gondii. J. Leukoc. Biol. 110, 617–628. doi: 10.1002/JLB.3HI0720-431R
Bhandage, A. K., Jin, Z., Bazov, I., Kononenko, O., Bakalkin, G., Korpi, E. R., et al. (2014). GABA-A and NMDA receptor subunit mRNA expression is altered in the caudate but not the putamen of the postmortem brains of alcoholics. Front. Cell. Neurosci. 8:415. doi: 10.3389/fncel.2014.00415
Billinton, A., Stean, T. O., Bowery, N. G., and Upton, N. (2000). GABAB(1) splice variant mRNAs are differentially affected by electroshock induced seizure in rats. Neuroreport 11, 3817–3822. doi: 10.1097/00001756-200011270-00044
Bjurstöm, H., Wang, J., Ericsson, I., Bengtsson, M., Liu, Y., Kumar-Mendu, S., et al. (2008). GABA, a natural immunomodulator of T lymphocytes. J. Neuroimmunol. 205, 44–50. doi: 10.1016/j.jneuroim.2008.08.017
Boddington, L. J., and Reynolds, J. N. J. (2017). Targeting interhemispheric inhibition with neuromodulation to enhance stroke rehabilitation. Brain Stimul. 10, 214–222. doi: 10.1016/j.brs.2017.01.006
Bodhankar, S., Chen, Y., Lapato, A., Vandenbark, A. A., Murphy, S. J., Saugstad, J. A., et al. (2015). Regulatory CD8+CD122+ T-cells predominate in CNS after treatment of experimental stroke in male mice with IL-10-secreting B-cells. Metab. Brain Dis. 30, 911–924. doi: 10.1007/s11011-014-9639-8
Bonnert, T. P., McKernan, R. M., Farrar, S., le Bourdellès, B., Heavens, R. P., Smith, D. W., et al. (1999). θ, a novel γ-aminobutyric acid type A receptor subunit. Proc. Natl. Acad. Sci. U S A 96, 9891–9896. doi: 10.1073/pnas.96.17.9891
Brickley, S. G., and Mody, I. (2012). Extrasynaptic GABAA receptors: their function in the CNS and implications for disease. Neuron 73, 23–34. doi: 10.1016/j.neuron.2011.12.012
Browne, S. H., Kang, J., Akk, G., Chiang, L. W., Schulman, H., Huguenard, J. R., et al. (2001). Kinetic and pharmacological properties of GABAA receptors in single thalamic neurons and GABAA subunit expression. J. Neurophysiol. 86, 2312–2322. doi: 10.1152/jn.2001.86.5.2312
Bunketorp-Kall, L., Lundgren-Nilsson, A., Samuelsson, H., Pekny, T., Blomve, K., Pekna, M., et al. (2017). Long-term improvements after multimodal rehabilitation in late phase after stroke: a randomized controlled trial. Stroke 48, 1916–1924. doi: 10.1161/STROKEAHA.116.016433
Burda, J. E., and Sofroniew, M. V. (2014). Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248. doi: 10.1016/j.neuron.2013.12.034
Bütefisch, C. M., Netz, J., Wessling, M., Seitz, R. J., and Hömberg, V. (2003). Remote changes in cortical excitability after stroke. Brain 126, 470–481. doi: 10.1093/brain/awg044
Cantaut-Belarif, Y., Antri, M., Pizzarelli, R., Colasse, S., Vaccari, I., Soares, S., et al. (2017). Microglia control the glycinergic but not the GABAergic synapses via prostaglandin E2 in the spinal cord. J. Cell Biol. 216, 2979–2989. doi: 10.1083/jcb.201607048
Carmichael, S. T. (2012). Brain excitability in stroke: the yin and yang of stroke progression. Arch. Neurol. 69, 161–167. doi: 10.1001/archneurol.2011.1175
Carter, A. R., Astafiev, S. V., Lang, C. E., Connor, L. T., Rengachary, J., Strube, M. J., et al. (2010). Resting interhemispheric functional magnetic resonance imaging connectivity predicts performance after stroke. Ann. Neurol. 67, 365–375. doi: 10.1002/ana.21905
Chapman, G. A., Moores, K., Harrison, D., Campbell, C. A., Stewart, B. R., and Strijbos, P. J. (2000). Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J. Neurosci. 20:RC87. doi: 10.1523/JNEUROSCI.20-15-j0004.2000
Chen, W. R., Xiong, W., and Shepherd, G. M. (2000). Analysis of relations between NMDA receptors and GABA release at olfactory bulb reciprocal synapses. Neuron 25, 625–633. doi: 10.1016/s0896-6273(00)81065-x
Cheung, G., Kann, O., Kohsaka, S., Făerber, K., and Kettenmann, H. (2009). GABAergic activities enhance macrophage inflammatory protein-1α release from microglia (brain macrophages) in postnatal mouse brain. J. Physiol. 587, 753–768. doi: 10.1113/jphysiol.2008.163923
Choii, G., and Ko, J. (2015). Gephyrin: a central GABAergic synapse organizer. Exp. Mol. Med. 47:e158. doi: 10.1038/emm.2015.5
Chung, H. J., Ge, W.-P., Qian, X., Wiser, O., Jan, Y. N., and Jan, L. Y. (2009). G protein-activated inwardly rectifying potassium channels mediate depotentiation of long-term potentiation. Proc. Natl. Acad. Sci. U S A 106, 635–640. doi: 10.1073/pnas.0811685106
Cipriani, R., Villa, P., Chece, G., Lauro, C., Paladini, A., Micotti, E., et al. (2011). CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J. Neurosci. 31, 16327–16335. doi: 10.1523/JNEUROSCI.3611-11.2011
Clarkson, A. N. (2012). Perisynaptic GABA receptors the overzealous protector. Adv. Pharmacol. Sci. 2012:708428. doi: 10.1155/2012/708428
Clarkson, A. N., Huang, B. S., Macisaac, S. E., Mody, I., and Carmichael, S. T. (2010). Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 468, 305–309. doi: 10.1038/nature09511
Colin, O., Labreuche, J., Deguil, J., Mendyk, A.-M., Deken, V., Cordonnier, C., et al. (2019). Preadmission use of benzodiazepines and stroke outcomes: the Biostroke prospective cohort study. BMJ Open 9:e022720. doi: 10.1136/bmjopen-2018-022720
Condie, B. G., Bain, G., Gottlieb, D. I., and Capecchi, M. R. (1997). Cleft palate in mice with a targeted mutation in the gamma-aminobutyric acid-producing enzyme glutamic acid decarboxylase 67. Proc. Natl. Acad. Sci. U S A 94, 11451–11455. doi: 10.1073/pnas.94.21.11451
Cook, A., Hippensteel, R., Shimizu, S., Nicolai, J., Fatatis, A., and Meucci, O. (2010). Interactions between chemokines: regulation of fractalkine/CX3CL1 homeostasis by SDF/CXCL12 in cortical neurons. J. Biol. Chem. 285, 10563–10571. doi: 10.1074/jbc.M109.035477
Cooper, A. M., and Khader, S. A. (2007). IL-12p40: an inherently agonistic cytokine. Trends Immunol. 28, 33–38. doi: 10.1016/j.it.2006.11.002
Corbett, D., Larsen, J., and Langdon, K. D. (2008). Diazepam delays the death of hippocampal CA1 neurons following global ischemia. Exp. Neurol. 214, 309–314. doi: 10.1016/j.expneurol.2008.08.018
Costa, C., Leone, G., Saulle, E., Pisani, F., Bernardi, G., and Calabresi, P. (2004). Coactivation of GABAA and GABAB receptor results in neuroprotection during in vitro ischemia. Stroke 35, 596–600. doi: 10.1161/01.STR.0000113691.32026.06
Costa, J. T., Mele, M., Baptista, M. S., Gomes, J. R., Ruscher, K., Nobre, R. J., et al. (2016). Gephyrin cleavage in in vitro brain ischemia decreases GABAA receptor clustering and contributes to neuronal death. Mol. Neurobiol. 53, 3513–3527. doi: 10.1007/s12035-015-9283-2
Crack, P. J., Taylor, J. M., Ali, U., Mansell, A., and Hertzog, P. J. (2006). Potential contribution of NF-κB in neuronal cell death in the glutathione peroxidase-1 knockout mouse in response to ischemia-reperfusion injury. Stroke 37, 1533–1538. doi: 10.1161/01.STR.0000221708.17159.64
Craig, A. M., Banker, G., Chang, W., McGrath, M. E., and Serpinskaya, A. S. (1996). Clustering of gephyrin at GABAergic but not glutamatergic synapses in cultured rat hippocampal neurons. J. Neurosci. 16, 3166–3177. doi: 10.1523/JNEUROSCI.16-10-03166.1996
Cramer, S. C. (2008). Repairing the human brain after stroke: I. mechanisms of spontaneous recovery. Ann. Neurol. 63, 272–287. doi: 10.1002/ana.21393
Cramer, S. C., Sur, M., Dobkin, B. H., O’Brien, C., Sanger, T. D., Trojanowski, J. Q., et al. (2011). Harnessing neuroplasticity for clinical applications. Brain 134, 1591–1609. doi: 10.1093/brain/awr039
Dénes, A., Ferenczi, S., Halász, J., Környei, Z., and Kovács, K. J. (2008). Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 28, 1707–1721. doi: 10.1038/jcbfm.2008.64
de Haas, A. H., van Weering, H. R. J., de Jong, E. K., Boddeke, H. W. G. M., and Biber, K. P. H. (2007). Neuronal chemokines: versatile messengers in central nervous system cell interaction. Mol. Neurobiol. 36, 137–151. doi: 10.1007/s12035-007-0036-8
Dichmann, S., Herouy, Y., Purlis, D., Rheinen, H., Gebicke-Härter, P., and Norgauer, J. (2001). Fractalkine induces chemotaxis and actin polymerization in human dendritic cells. Inflamm. Res. 50, 529–533. doi: 10.1007/PL00000230
Dimitrijevic, O. B., Stamatovic, S. M., Keep, R. F., and Andjelkovic, A. V. (2007). Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 38, 1345–1353. doi: 10.1161/01.STR.0000259709.16654.8f
Dionisio, L., José De Rosa, M., Bouzat, C., and Esandi, M. D. C. (2011). An intrinsic GABAergic system in human lymphocytes. Neuropharmacology 60, 513–519. doi: 10.1016/j.neuropharm.2010.11.007
Dixon, C., Sah, P., Lynch, J. W., and Keramidas, A. (2014). GABAA receptor α and γ subunits shape synaptic currents via different mechanisms. J. Biol. Chem. 289, 5399–5411. doi: 10.1074/jbc.M113.514695
Dupuy, S. T., and Houser, C. R. (1996). Prominent expression of two forms of glutamate decarboxylase in the embryonic and early postnatal rat hippocampal formation. J. Neurosci. 16, 6919–6932. doi: 10.1523/JNEUROSCI.16-21-06919.1996
Ethiraj, J., Palpagama, T. H., Turner, C., van der Werf, B., Waldvogel, H. J., Faull, R. L. M., et al. (2021). The effect of age and sex on the expression of GABA signaling components in the human hippocampus and entorhinal cortex. Sci. Rep. 11:21470. doi: 10.1038/s41598-021-00792-8
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat. Rev. Neurosci. 6, 215–229. doi: 10.1038/nrn1625
Feigin, V. L., Stark, B. A., Johnson, C. O., Roth, G. A., Bisignano, C., Abady, G. G., et al. (2021). Global, regional and national burden of stroke and its risk factors, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 20, 795–820. doi: 10.1016/S1474-4422(21)00252-0
Feng, Y.-W., Huang, Y.-Q., Yan, Y., Li, G., He, X.-F., Liang, F.-Y., et al. (2020). Phasic GABA signaling mediates the protective effects of cTBS against cerebral ischemia in mice. Neurosci. Lett. 715:134611. doi: 10.1016/j.neulet.2019.134611
Filippov, A. K., Couve, A., Pangalos, M. N., Walsh, F. S., Brown, D. A., and Moss, S. J. (2000). Heteromeric assembly of GABABR1 and GABABR2 receptor subunits inhibits Ca2+ current in sympathetic neurons. J. Neurosci. 20, 2867–2874. doi: 10.1523/JNEUROSCI.20-08-02867.2000
Fiszman, M. L., Barberis, A., Lu, C., Fu, Z., Erdélyi, F., Szabó, G., et al. (2005). NMDA receptors increase the size of GABAergic terminals and enhance GABA release. J. Neurosci. 25, 2024–2031. doi: 10.1523/JNEUROSCI.4980-04.2005
Frank, C. A. (2014). How voltage-gated calcium channels gate forms of homeostatic synaptic plasticity. Front. Cell. Neurosci. 8:40. doi: 10.3389/fncel.2014.00040
Fritschy, J.-M., and Panzanelli, P. (2014). GABAA receptors and plasticity of inhibitory neurotransmission in the central nervous system. Eur. J. Neurosci. 39, 1845–1865. doi: 10.1111/ejn.12534
Fuks, J. M., Arrighi, R. B. G., Weidner, J. M., Kumar Mendu, S., Jin, Z., Wallin, R. P. A., et al. (2012). GABAergic signaling is linked to a hypermigratory phenotype in dendritic cells infected by Toxoplasma gondii. PLoS Pathog. 8:e1003051. doi: 10.1371/journal.ppat.1003051
Gähwiler, B. H., and Brown, D. A. (1985). GABAB-receptor-activated K+ current in voltage-clamped CA3 pyramidal cells in hippocampal cultures. Proc. Natl. Acad. Sci. U S A 82, 1558–1562. doi: 10.1073/pnas.82.5.1558
Gan, Y., Liu, Q., Wu, W., Yin, J.-X., Bai, X.-F., Shen, R., et al. (2014). Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc. Natl. Acad. Sci. U S A 111, 2704–2709. doi: 10.1073/pnas.1315943111
Gelderblom, M., Leypoldt, F., Steinbach, K., Behrens, D., Choe, C.-U., Siler, D. A., et al. (2009). Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 40, 1849–1857. doi: 10.1161/STROKEAHA.108.534503
Giacco, V., Panattoni, G., Medelin, M., Bonechi, E., Aldinucci, A., Ballerini, C., et al. (2019). Cytokine inflammatory threat, but not LPS one, shortens GABAergic synaptic currents in the mouse spinal cord organotypic cultures. J. Neuroinflammation 16:127. doi: 10.1186/s12974-019-1519-z
Gingrich, K. J., Roberts, W. A., and Kass, R. S. (1995). Dependence of the GABAA receptor gating kinetics on the alpha-subunit isoform: implications for structure-function relations and synaptic transmission. J. Physiol. 489, 529–543. doi: 10.1113/jphysiol.1995.sp021070
Globus, M. Y., Busto, R., Martinez, E., Valdés, I., Dietrich, W. D., and Ginsberg, M. D. (1991). Comparative effect of transient global ischemia on extracellular levels of glutamate, glycine and gamma-aminobutyric acid in vulnerable and nonvulnerable brain regions in the rat. J. Neurochem. 57, 470–478. doi: 10.1111/j.1471-4159.1991.tb03775.x
Glykys, J., and Mody, I. (2007). Activation of GABAA receptors: views from outside the synaptic cleft. Neuron 56, 763–770. doi: 10.1016/j.neuron.2007.11.002
Goetz, T., Arslan, A., Wisden, W., and Wulff, P. (2007). GABAA receptors: structure and function in the basal ganglia. Prog. Brain Res. 160, 21–41. doi: 10.1016/S0079-6123(06)60003-4
Gonzalez, F. F., McQuillen, P., Mu, D., Chang, Y., Wendland, M., Vexler, Z., et al. (2007). Erythropoietin enhances long-term neuroprotection and neurogenesis in neonatal stroke. Dev. Neurosci. 29, 321–330. doi: 10.1159/000105473
Gosselin, R. D., Varela, C., Banisadr, G., Mechighel, P., Rostene, W., Kitabgi, P., et al. (2005). Constitutive expression of CCR2 chemokine receptor and inhibition by MCP-1/CCL2 of GABA-induced currents in spinal cord neurones. J. Neurochem. 95, 1023–1034. doi: 10.1111/j.1471-4159.2005.03431.x
Guyon, A., Skrzydelsi, D., Rovère, C., Rostène, W., Parsadaniantz, S. M., and Nahon, J. L. (2006). Stromal cell-derived factor-1α modulation of the excitability of rat substantia nigra dopaminergic neurones: presynaptic mechanisms. J. Neurochem. 96, 1540–1550. doi: 10.1111/j.1471-4159.2006.03659.x
Hagemann, G., Redecker, C., Neumann-Haefelin, T., Freund, H. J., and Witte, O. W. (1998). Increased long-term potentiation in the surround of experimentally induced focal cortical infarction. Ann. Neurol. 44, 255–258. doi: 10.1002/ana.410440217
Hakon, J., Quattromani, M. J., Sjölund, C., Tomasevic, G., Carey, L., Lee, J.-M., et al. (2018). Multisensory stimulation improves functional recovery and resting-state functional connectivity in the mouse brain after stroke. Neuroimage Clin. 17, 717–730. doi: 10.1016/j.nicl.2017.11.022
Hammoud, H., Netsyk, O., Tafreshiha, A. S., Korol, S. V., Jin, Z., Li, J.-P., et al. (2021). Insulin differentially modulates GABA signalling in hippocampal neurons and, in an age-dependent manner, normalizes GABA-activated currents in the tg-APPSwe mouse model of Alzheimer’s disease. Acta Physiol. (Oxf) 232:e13623. doi: 10.1111/apha.13623
Han, W., Shepard, R. D., and Lu, W. (2021). Regulation of GABAARs by transmembrane accessory proteins. Trends Neurosci. 44, 152–165. doi: 10.1016/j.tins.2020.10.011
Hatem, S. M., Saussez, G., Della Faille, M., Prist, V., Zhang, X., Dispa, D., et al. (2016). Rehabilitation of motor function after stroke: a multiple systematic review focused on techniques to stimulate upper extremity recovery. Front. Hum. Neurosci. 10:442. doi: 10.3389/fnhum.2016.00442
Heinisch, S., and Kirby, L. G. (2009). Fractalkine/CX3CL1 enhances GABA synaptic activity at serotonin neurons in the rat dorsal raphe nucleus. Neuroscience 164, 1210–1223. doi: 10.1016/j.neuroscience.2009.08.075
Heinisch, S., and Kirby, L. G. (2010). SDF-1alpha/CXCL12 enhances GABA and glutamate synaptic activity at serotonin neurons in the rat dorsal raphe nucleus. Neuropharmacology 58, 501–514. doi: 10.1016/j.neuropharm.2009.08.022
Herbison, A. E., and Fénelon, V. S. (1995). Estrogen regulation of GABAA receptor subunit mRNA expression in preoptic area and bed nucleus of the stria terminalis of female rat brain. J. Neurosci. 15, 2328–2337. doi: 10.1523/JNEUROSCI.15-03-02328.1995
Hiu, T., Farzampour, Z., Paz, J. T., Wang, E. H. J., Badgely, C., Olson, A., et al. (2016). Enhanced phasic GABA inhibition during the repair phase of stroke: a novel therapeutic target. Brain 139, 468–480. doi: 10.1093/brain/awv360
Höft, S., Griemsmann, S., Seifert, G., and Steinhäuser, C. (2014). Heterogeneity in expression of functional ionotropic glutamate and GABA receptors in astrocytes across brain regions: insights from the thalamus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130602. doi: 10.1098/rstb.2013.0602
Houser, C. R., Olsen, R. W., Richards, J. G., and Möhler, H. (1988). Immunohistochemical localization of benzodiazepine/GABAA receptors in the human hippocampal formation. J. Neurosci. 8, 1370–1383. doi: 10.1523/JNEUROSCI.08-04-01370.1988
Huang, C. L., Slesinger, P. A., Casey, P. J., Jan, Y. N., and Jan, L. Y. (1995). Evidence that direct binding of Gβγ to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron 15, 1133–1143. doi: 10.1016/0896-6273(95)90101-9
Iadecola, C., and Anrather, J. (2011). The immunology of stroke: from mechanisms to translation. Nat. Med. 17, 796–808. doi: 10.1038/nm.2399
Iadecola, C., Buckwalter, M. S., and Anrather, J. (2020). Immune responses to stroke: mechanisms, modulation and therapeutic potential. J. Clin. Invest. 130, 2777–2788. doi: 10.1172/JCI135530
Imai, T., Hieshima, K., Haskell, C., Baba, M., Nagira, M., Nishimura, M., et al. (1997). Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 91, 521–530. doi: 10.1016/s0092-8674(00)80438-9
Inta, I., Paxian, S., Maegele, I., Zhang, W., Pizzi, M., Spano, P., et al. (2006). Bim and Noxa are candidates to mediate the deleterious effect of the NF-κB subunit RelA in cerebral ischemia. J. Neurosci. 26, 12896–12903. doi: 10.1523/JNEUROSCI.3670-06.2006
Jaenisch, N., Liebmann, L., Guenther, M., Hübner, C. A., Frahm, C., and Witte, O. W. (2016). Reduced tonic inhibition after stroke promotes motor performance and epileptic seizures. Sci. Rep. 6:26173. doi: 10.1038/srep26173
Januzi, L., Poirier, J. W., Maksoud, M. J. E., Xiang, Y.-Y., Veldhuizen, R. A. W., Gill, S. E., et al. (2018). Autocrine GABA signaling distinctively regulates phenotypic activation of mouse pulmonary macrophages. Cell Immunol. 332, 7–23. doi: 10.1016/j.cellimm.2018.07.001
Jensen, M. L., Wafford, K. A., Brown, A. R., Belelli, D., Lambert, J. J., and Mirza, N. R. (2013). A study of subunit selectivity, mechanism and site of action of the delta selective compound 2 (DS2) at human recombinant and rodent native GABAA receptors. Br. J. Pharmacol. 168, 1118–1132. doi: 10.1111/bph.12001
Jin, Z., Bazov, I., Kononenko, O., Korpi, E. R., Bakalkin, G., and Birnir, B. (2012). Selective changes of GABAA channel subunit mRNAs in the Hippocampus and orbitofrontal cortex but not in prefrontal cortex of human alcoholics. Front. Cell. Neurosci. 5:30. doi: 10.3389/fncel.2011.00030
Jin, Z., Mendu, S. K., and Birnir, B. (2013). GABA is an effective immunomodulatory molecule. Amino Acids 45, 87–94. doi: 10.1007/s00726-011-1193-7
Jin, H., Wu, H., Osterhaus, G., Wei, J., Davis, K., Sha, D., et al. (2003). Demonstration of functional coupling between γ-aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc. Natl. Acad. Sci. U S A 100, 4293–4298. doi: 10.1073/pnas.0730698100
Joy, M. T., Ben Assayag, E., Shabashov-Stone, D., Liraz-Zaltsman, S., Mazzitelli, J., Arenas, M., et al. (2019). CCR5 is a therapeutic target for recovery after stroke and traumatic brain injury. Cell 176, 1143–1157.e13. doi: 10.1016/j.cell.2019.01.044
Joy, M. T., and Carmichael, S. T. (2021). Encouraging an excitable brain state: mechanisms of brain repair in stroke. Nat. Rev. Neurosci. 22, 38–53. doi: 10.1038/s41583-020-00396-7
Juenemann, M., Braun, T., Schleicher, N., Yeniguen, M., Schramm, P., Gerriets, T., et al. (2020). Neuroprotective mechanisms of erythropoietin in a rat stroke model. Transl Neurosci. 11, 48–59. doi: 10.1515/tnsci-2020-0008
Kanaani, J., Diacovo, M. J., El-Husseini, A. E.-D., Bredt, D. S., and Baekkeskov, S. (2004). Palmitoylation controls trafficking of GAD65 from Golgi membranes to axon-specific endosomes and a Rab5a-dependent pathway to presynaptic clusters. J. Cell Sci. 117, 2001–2013. doi: 10.1242/jcs.01030
Kanaani, J., el-Husseini, A. e.-D., Aguilera-Moreno, A., Diacovo, J. M., Bredt, D. S., and Baekkeskov, S. (2002). A combination of three distinct trafficking signals mediates axonal targeting and presynaptic clustering of GAD65. J. Cell Biol. 158, 1229–1238. doi: 10.1083/jcb.200205053
Kanaani, J., Kolibachuk, J., Martinez, H., and Baekkeskov, S. (2010). Two distinct mechanisms target GAD67 to vesicular pathways and presynaptic clusters. J. Cell Biol. 190, 911–925. doi: 10.1083/jcb.200912101
Kanaani, J., Lissin, D., Kash, S. F., and Baekkeskov, S. (1999). The hydrophilic isoform of glutamate decarboxylase, GAD67, is targeted to membranes and nerve terminals independent of dimerization with the hydrophobic membrane-anchored isoform, GAD65. J. Biol. Chem. 274, 37200–37209. doi: 10.1074/jbc.274.52.37200
Kaneko, Y., Pappas, C., Tajiri, N., and Borlongan, C. V. (2016). Oxytocin modulates GABAAR subunits to confer neuroprotection in stroke in vitro. Sci. Rep. 6:35659. doi: 10.1038/srep35659
Kantamneni, S. (2015). Cross-talk and regulation between glutamate and GABAB receptors. Front. Cell. Neurosci. 9:135. doi: 10.3389/fncel.2015.00135
Kantamneni, S., Gonzàlez-Gonzàlez, I. M., Luo, J., Cimarosti, H., Jacobs, S. C., Jaafari, N., et al. (2014). Differential regulation of GABAB receptor trafficking by different modes of N-methyl-D-aspartate (NMDA) receptor signaling. J. Biol. Chem. 289, 6681–6694. doi: 10.1074/jbc.M113.487348
Kash, S. F., Johnson, R. S., Tecott, L. H., Noebels, J. L., Mayfield, R. D., Hanahan, D., et al. (1997). Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc. Natl. Acad. Sci. U S A 94, 14060–14065. doi: 10.1073/pnas.94.25.14060
Kharlamov, E. A., Downey, K. L., Jukkola, P. I., Grayson, D. R., and Kelly, K. M. (2008). Expression of GABAA receptor α1 subunit mRNA and protein in rat neocortex following photothrombotic infarction. Brain Res. 1210, 29–38. doi: 10.1016/j.brainres.2008.02.070
Kittler, J. T., Chen, G., Honing, S., Bogdanov, Y., McAinsh, K., Arancibia-Carcamo, I. L., et al. (2005). Phospho-dependent binding of the clathrin AP2 adaptor complex to GABAA receptors regulates the efficacy of inhibitory synaptic transmission. Proc. Natl. Acad. Sci. U S A. 102, 14871–14876. doi: 10.1073/pnas.0506653102
Kittler, J. T., and Moss, S. J. (2003). Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr. Opin. Neurobiol. 13, 341–347. doi: 10.1016/s0959-4388(03)00064-3
Kittler, J. T., Thomas, P., Tretter, V., Bogdanov, Y. D., Haucke, V., Smart, T. G., et al. (2004). Huntingtin-associated protein 1 regulates inhibitory synaptic transmission by modulating gamma-aminobutyric acid type A receptor membrane trafficking. Proc. Natl. Acad. Sci. U S A 101, 12736–12741. doi: 10.1073/pnas.0401860101
Kolasinski, J., Hinson, E. L., Divanbeighi Zand, A. P., Rizov, A., Emir, U. E., and Stagg, C. J. (2019). The dynamics of cortical GABA in human motor learning. J. Physiol. 597, 271–282. doi: 10.1113/JP276626
Kuhn, S. A., van Landeghem, F. K. H., Zacharias, R., Färber, K., Rappert, A., Pavlovic, S., et al. (2004). Microglia express GABAB receptors to modulate interleukin release. Mol. Cell Neurosci. 25, 312–322. doi: 10.1016/j.mcn.2003.10.023
Kuric, E., and Ruscher, K. (2014). Reduction of rat brain CD8+ T-cells by levodopa/benserazide treatment after experimental stroke. Eur. J. Neurosci. 40, 2463–2470. doi: 10.1111/ejn.12598
Lakhan, S. E., Kirchgessner, A., and Hofer, M. (2009). Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J. Transl. Med. 7:97. doi: 10.1186/1479-5876-7-97
Lakhan, S. E., Kirchgessner, A., Tepper, D., and Leonard, A. (2013). Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front. Neurol. 4:32. doi: 10.3389/fneur.2013.00032
Lambert, N. A., and Wilson, W. A. (1996). High-threshold Ca2+ currents in rat hippocampal interneurones and their selective inhibition by activation of GABA(B) receptors. J. Physiol. 492, 115–127. doi: 10.1113/jphysiol.1996.sp021294
Lee, S.-E., Lee, Y., and Lee, G. H. (2019). The regulation of glutamic acid decarboxylases in GABA neurotransmission in the brain. Arch. Pharm. Res. 42, 1031–1039. doi: 10.1007/s12272-019-01196-z
Lee, M., Schwab, C., and McGeer, P. L. (2011). Astrocytes are GABAergic cells that modulate microglial activity. Glia 59, 152–165. doi: 10.1002/glia.21087
Lewohl, J. M., Crane, D. I., and Dodd, P. R. (1997). Expression of the α1, α2 and α3 isoforms of the GABAA receptor in human alcoholic brain. Brain Res. 751, 102–112. doi: 10.1016/s0006-8993(96)01396-0
Li, Y., Blanco, G. D., Lei, Z., and Xu, Z. C. (2010). Increased GAD expression in the striatum after transient cerebral ischemia. Mol. Cell Neurosci. 45, 370–377. doi: 10.1016/j.mcn.2010.07.010
Liesz, A., and Kleinschnitz, C. (2016). Regulatory T cells in post-stroke immune homeostasis. Transl. Stroke Res. 7, 313–321. doi: 10.1007/s12975-016-0465-7
Limon, A., Reyes-Ruiz, J. M., and Miledi, R. (2012). Loss of functional GABAA receptors in the Alzheimer diseased brain. Proc. Natl. Acad. Sci. U S A 109, 10071–10076. doi: 10.1073/pnas.1204606109
Lindquist, C. E. L., and Birnir, B. (2006). Graded response to GABA by native extrasynaptic GABA receptors. J. Neurochem. 97, 1349–1356. doi: 10.1111/j.1471-4159.2006.03811.x
Lodder, J., van Raak, L., Hilton, A., Hardy, E., and Kessels, A. (2006). Diazepam to improve acute stroke outcome: results of the early GABA-Ergic activation study in stroke trial. a randomized double-blind placebo-controlled trial. Cerebrovasc. Dis. 21, 120–127. doi: 10.1159/000090210
Lorenz-Guertin, J. M., and Jacob, T. C. (2018). GABA type a receptor trafficking and the architecture of synaptic inhibition. Dev. Neurobiol. 78, 238–270. doi: 10.1002/dneu.22536
Loup, F., Wieser, H. G., Yonekawa, Y., Aguzzi, A., and Fritschy, J. M. (2000). Selective alterations in GABAA receptor subtypes in human temporal lobe epilepsy. J. Neurosci. 20, 5401–5419. doi: 10.1523/JNEUROSCI.20-14-05401.2000
Luengo-Fernandez, R., Violato, M., Candio, P., and Leal, J. (2020). Economic burden of stroke across Europe: a population-based cost analysis. Eur. Stroke J. 5, 17–25. doi: 10.1177/2396987319883160
Lull, M. E., and Block, M. L. (2010). Microglial activation and chronic neurodegeneration. Neurotherapeutics 7, 354–365. doi: 10.1016/j.nurt.2010.05.014
Luo, Y., Lathia, J., Mughal, M., and Mattson, M. P. (2008). SDF1α/CXCR4 signaling, via ERKs and the transcription factor Egr1, induces expression of a 67-kDa form of glutamic acid decarboxylase in embryonic hippocampal neurons. J. Biol. Chem. 283, 24789–24800. doi: 10.1074/jbc.M800649200
Marron Fernandez de Velasco, E., McCall, N., and Wickman, K. (2015). GIRK channel plasticity and implications for drug addiction. Int. Rev. Neurobiol. 123, 201–238. doi: 10.1016/bs.irn.2015.05.011
Mead, E. L., Mosley, A., Eaton, S., Dobson, L., Heales, S. J., and Pocock, J. M. (2012). Microglial neurotransmitter receptors trigger superoxide production in microglia; consequences for microglial-neuronal interactions. J. Neurochem. 121, 287–301. doi: 10.1111/j.1471-4159.2012.07659.x
Mele, M., Ribeiro, L., Inácio, A. R., Wieloch, T., and Duarte, C. B. (2014). GABAA receptor dephosphorylation followed by internalization is coupled to neuronal death in in vitro ischemia. Neurobiol. Dis. 65, 220–232. doi: 10.1016/j.nbd.2014.01.019
Meucci, O., Fatatis, A., Simen, A. A., and Miller, R. J. (2000). Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc. Natl. Acad. Sci. U S A 97, 8075–8080. doi: 10.1073/pnas.090017497
Michalettos, G., Walter, H. L., Antunes, A. R. P., Wieloch, T., Talhada, D., and Ruscher, K. (2021). Effect of anti-inflammatory treatment with AMD3100 and CX3CR1 deficiency on GABAA receptor subunit and expression of glutamate decarboxylase isoforms after stroke. Mol. Neurobiol. 58, 5876–5889. doi: 10.1007/s12035-021-02510-x
Mielke, J. G., and Wang, Y. T. (2005). Insulin exerts neuroprotection by counteracting the decrease in cell-surface GABA receptors following oxygen-glucose deprivation in cultured cortical neurons. J. Neurochem. 92, 103–113. doi: 10.1111/j.1471-4159.2004.02841.x
Mintz, I. M., and Bean, B. P. (1993). GABAB receptor inhibition of P-type Ca2+ channels in central neurons. Neuron. 10, 889–898. doi: 10.1016/0896-6273(93)90204-5
Misgeld, U., Bijak, M., and Jarolimek, W. (1995). A physiological role for GABAB receptors and the effects of baclofen in the mammalian central nervous system. Prog. Neurobiol. 46, 423–462. doi: 10.1016/0301-0082(95)00012-k
Morioka, T., Kalehua, A. N., and Streit, W. J. (1993). Characterization of microglial reaction after middle cerebral artery occlusion in rat brain. J. Comp. Neurol. 327, 123–132. doi: 10.1002/cne.903270110
Mortensen, M., Patel, B., and Smart, T. G. (2012). GABA potency at GABAA receptors found in synaptic and extrasynaptic zones. Front Cell Neurosci. 6:1. doi: 10.3389/fncel.2012.00001
Nanou, E., and Catterall, W. A. (2018). Calcium channels, synaptic plasticity and neuropsychiatric disease. Neuron. 98, 466–481. doi: 10.1016/j.neuron.2018.03.017
Neumann, S., Boothman-Burrell, L., Gowing, E. K., Jacobsen, T. A., Ahring, P. K., Young, S. L., et al. (2019). The delta-subunit selective GABAA receptor modulator, DS2, improves stroke recovery via an anti-inflammatory mechanism. Front. Neurosci. 13:1133. doi: 10.3389/fnins.2019.01133
Neumann, E., Rudolph, U., Knutson, D. E., Li, G., Cook, J. M., Hentschke, H., et al. (2019). Zolpidem activation of alpha 1-containing GABAA receptors selectively inhibits high frequency action potential firing of cortical neurons. Front. Pharmacol. 9:1523. doi: 10.3389/fphar.2018.01523
Nguyen, Q.-A., and Nicoll, R. A. (2018). The GABAA receptor β subunit is required for inhibitory transmission. Neuron. 98, 718–725.e3. doi: 10.1016/j.neuron.2018.03.046
Nieman, A. N., Li, G., Zahn, N. M., Mian, M. Y., Mikulsky, B. N., Hoffman, D. A., et al. (2020). Targeting nitric oxide production in microglia with novel imidazodiazepines for nonsedative pain treatment. ACS Chem. Neurosci. 11, 2019–2030. doi: 10.1021/acschemneuro.0c00324
Nusser, Z., and Mody, I. (2002). Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J. Neurophysiol. 87, 2624–2628. doi: 10.1152/jn.2002.87.5.2624
Nusser, Z., Sieghart, W., and Somogyi, P. (1998). Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J. Neurosci. 18, 1693–1703. doi: 10.1523/JNEUROSCI.18-05-01693.1998
Paik, N.-J., and Yang, E. (2014). Role of GABA plasticity in stroke recovery. Neural Regen. Res. 9, 2026–2028. doi: 10.4103/1673-5374.147920
Pandya, M., Palpagama, T. H., Turner, C., Waldvogel, H. J., Faull, R. L., and Kwakowsky, A. (2019). Sex- and age-related changes in GABA signaling components in the human cortex. Biol. Sex Differ. 10:5. doi: 10.1186/s13293-018-0214-6
Pawluk, H., Woźniak, A., Grześk, G., Kołodziejska, R., Kozakiewicz, M., Kopkowska, E., et al. (2020). The role of selected pro-inflammatory cytokines in pathogenesis of ischemic stroke. Clin. Interv. Aging 15, 469–484. doi: 10.2147/CIA.S233909
Perera Molligoda Arachchige, A. S. (2021). Human NK cells: from development to effector functions. Innate Immun. 27, 212–229. doi: 10.1177/17534259211001512
Pfrieger, F. W., Gottmann, K., and Lux, H. D. (1994). Kinetics of GABAB receptor-mediated inhibition of calcium currents and excitatory synaptic transmission in hippocampal neurons in vitro. Neuron 12, 97–107. doi: 10.1016/0896-6273(94)90155-4
Pinal, C. S., and Tobin, A. J. (1998). Uniqueness and redundancy in GABA production. Perspect. Dev. Neurobiol. 5, 109–118.
Ping, S., Qiu, X., Kyle, M., and Zhao, L.-R. (2021). Brain-derived CCR5 contributes to neuroprotection and brain repair after experimental stroke. Aging Dis. 12, 72–92. doi: 10.14336/AD.2020.0406
Popken, G. J., Leggio, M. G., Bunney, W. E., and Jones, E. G. (2002). Expression of mRNAs related to the GABAergic and glutamatergic neurotransmitter systems in the human thalamus: normal and schizophrenic. Thalamus Relat. Syst. 1, 349–369. doi: 10.1016/S1472-9288(02)00008-0
Prud’homme, G. J., Glinka, Y., and Wang, Q. (2015). Immunological GABAergic interactions and therapeutic applications in autoimmune diseases. Autoimmun. Rev. 14, 1048–1056. doi: 10.1016/j.aquatox.2021.105860
Ragozzino, D., Di Angelantonio, S., Trettel, F., Bertollini, C., Maggi, L., Gross, C., et al. (2006). Chemokine fractalkine/CX3CL1 negatively modulates active glutamatergic synapses in rat hippocampal neurons. J. Neurosci. 26, 10488–10498. doi: 10.1523/JNEUROSCI.3192-06.2006
Rayasam, A., Hsu, M., Kijak, J. A., Kissel, L., Hernandez, G., Sandor, M., et al. (2018). Immune responses in stroke: how the immune system contributes to damage and healing after stroke and how this knowledge could be translated to better cures? Immunology 154, 363–376. doi: 10.1111/imm.12918
Redecker, C., Wang, W., Fritschy, J.-M., and Witte, O. W. (2002). Widespread and long-lasting alterations in GABAA-receptor subtypes after focal cortical infarcts in rats: mediation by NMDA-dependent processes. J. Cereb. Blood Flow Metab. 22, 1463–1475. doi: 10.1097/01.WCB.0000034149.72481.BD
Riquelme, R., Miralles, C. P., and De Blas, A. L. (2002). Bergmann glia GABAA receptors concentrate on the glial processes that wrap inhibitory synapses. J. Neurosci. 22, 10720–10730. doi: 10.1523/JNEUROSCI.22-24-10720.2002
Roseti, C., Cifelli, P., Ruffolo, G., Barbieri, E., Guescini, M., Esposito, V., et al. (2020). Erythropoietin increases GABAA currents in human cortex from TLE patients. Neuroscience. 439, 153–162. doi: 10.1016/j.neuroscience.2019.04.013
Roseti, C., Fucile, S., Lauro, C., Martinello, K., Bertollini, C., Esposito, V., et al. (2013). Fractalkine/CX3CL1 modulates GABAA currents in human temporal lobe epilepsy. Epilepsia 54, 1834–1844. doi: 10.1111/epi.12354
Rosewater, K., and Sontheimer, H. (1994). Fibrous and protoplasmic astrocytes express GABAA receptors that differ in benzodiazepine pharmacology. Brain Res. 636, 73–80. doi: 10.1016/0006-8993(94)90177-5
Ruan, L., Wang, Y., Chen, S.-C., Zhao, T., Huang, Q., Hu, Z.-L., et al. (2017). Metabolite changes in the ipsilateral and contralateral cerebral hemispheres in rats with middle cerebral artery occlusion. Neural Regen. Res. 12, 931–937. doi: 10.4103/1673-5374.208575
Ruscher, K., Freyer, D., Karsch, M., Isaev, N., Megow, D., Sawitzki, B., et al. (2002). Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J. Neurosci. 22, 10291–10301. doi: 10.1523/JNEUROSCI.22-23-10291.2002
Ruscher, K., Kuric, E., Liu, Y., Walter, H. L., Issazadeh-Navikas, S., Englund, E., et al. (2013). Inhibition of CXCL12 signaling attenuates the postischemic immune response and improves functional recovery after stroke. J. Cereb. Blood Flow Metab. 33, 1225–1234. doi: 10.1038/jcbfm.2013.71
Saggu, R., Schumacher, T., Gerich, F., Rakers, C., Tai, K., Delekate, A., et al. (2016). Astroglial NF-kB contributes to white matter damage and cognitive impairment in a mouse model of vascular dementia. Acta Neuropathol. Commun. 4:76. doi: 10.1186/s40478-016-0350-3
Sakaba, T., and Neher, E. (2003). Direct modulation of synaptic vesicle priming by GABAB receptor activation at a glutamatergic synapse. Nature 424, 775–778. doi: 10.1038/nature01859
Sampaio-Baptista, C., Filippini, N., Stagg, C. J., Near, J., Scholz, J., and Johansen-Berg, H. (2015). Changes in functional connectivity and GABA levels with long-term motor learning. Neuroimage 106, 15–20. doi: 10.1016/j.neuroimage.2014.11.032
Sánchez-Rodríguez, I., Temprano-Carazo, S., Nájera, A., Djebari, S., Yajeya, J., Gruart, A., et al. (2017). Activation of G-protein-gated inwardly rectifying potassium (Kir3/GirK) channels rescues hippocampal functions in a mouse model of early amyloid-β pathology. Sci. Rep. 7:14658. doi: 10.1038/s41598-017-15306-8
Schmidt, S., Bruehl, C., Frahm, C., Redecker, C., and Witte, O. W. (2012). Age dependence of excitatory-inhibitory balance following stroke. Neurobiol. Aging 33, 1356–1363. doi: 10.1016/j.neurobiolaging.2010.11.019
Schneider, A., Martin-Villalba, A., Weih, F., Vogel, J., Wirth, T., and Schwaninger, M. (1999). NF-κB is activated and promotes cell death in focal cerebral ischemia. Nat. Med. 5, 554–559. doi: 10.1038/8432
Schousboe, A., Bak, L. K., and Waagepetersen, H. S. (2013). Astrocytic control of biosynthesis and turnover of the neurotransmitters glutamate and GABA. Front. Endocrinol. (Lausanne) 4:102. doi: 10.3389/fendo.2013.00102
Schönemeier, B., Schulz, S., Hoellt, V., and Stumm, R. (2008). Enhanced expression of the CXCl12/SDF-1 chemokine receptor CXCR7 after cerebral ischemia in the rat brain. J. Neuroimmunol. 198, 39–45. doi: 10.1016/j.jneuroim.2008.04.010
Schulz, J. M., Knoflach, F., Hernandez, M.-C., and Bischofberger, J. (2018). Dendrite-targeting interneurons control synaptic NMDA-receptor activation via nonlinear α5-GABAA receptors. Nat. Commun. 9:3576. doi: 10.1038/s41467-018-06004-8
Sieghart, W., and Sperk, G. (2002). Subunit composition, distribution and function of GABA-A receptor subtypes. Curr. Top. Med. Chem. 2, 795–816. doi: 10.2174/1568026023393507
Sims, N. R., and Yew, W. P. (2017). Reactive astrogliosis in stroke: contributions of astrocytes to recovery of neurological function. Neurochem. Int. 107, 88–103. doi: 10.1016/j.neuint.2016.12.016
Sodickson, D. L., and Bean, B. P. (1996). GABAB receptor-activated inwardly rectifying potassium current in dissociated hippocampal CA3 neurons. J. Neurosci. 16, 6374–6385. doi: 10.1523/JNEUROSCI.16-20-06374.1996
Sofroniew, M. V. (2005). Reactive astrocytes in neural repair and protection. Neuroscientist 11, 400–407. doi: 10.1177/1073858405278321
Soltani, N., Qiu, H., Aleksic, M., Glinka, Y., Zhao, F., Liu, R., et al. (2011). GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc. Natl. Acad. Sci. U S A 108, 11692–11697. doi: 10.1073/pnas.1102715108
Somogyi, P., Takagi, H., Richards, J. G., and Mohler, H. (1989). Subcellular localization of benzodiazepine/GABAA receptors in the cerebellum of rat, cat and monkey using monoclonal antibodies. J. Neurosci. 9, 2197–2209. doi: 10.1523/JNEUROSCI.09-06-02197.1989
Sorce, S., Bonnefont, J., Julien, S., Marq-Lin, N., Rodriguez, I., Dubois-Dauphin, M., et al. (2010). Increased brain damage after ischaemic stroke in mice lacking the chemokine receptor CCR5. Br. J. Pharmacol. 160, 311–321. doi: 10.1111/j.1476-5381.2010.00697.x
Soriano, S. G., Amaravadi, L. S., Wang, Y. F., Zhou, H., Yu, G. X., Tonra, J. R., et al. (2002). Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J. Neuroimmunol. 125, 59–65. doi: 10.1016/s0165-5728(02)00033-4
Stell, B. M., Brickley, S. G., Tang, C. Y., Farrant, M., and Mody, I. (2003). Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by δ subunit-containing GABAA receptors. Proc. Natl. Acad. Sci. U S A 100, 14439–14444. doi: 10.1073/pnas.2435457100
Stojanovic, T., Capo, I., Aronica, E., Adle-Biassette, H., Höger, H., Sieghart, W., et al. (2016). The α1, α2, α3 and γ2 subunits of GABAA receptors show characteristic spatial and temporal expression patterns in rhombencephalic structures during normal human brain development. J. Comp. Neurol. 524, 1805–1824. doi: 10.1002/cne.23923
Studler, B., Sidler, C., and Fritschy, J.-M. (2005). Differential regulation of GABAA receptor and gephyrin postsynaptic clustering in immature hippocampal neuronal cultures. J. Comp. Neurol. 484, 344–355. doi: 10.1002/cne.20472
Stumm, R. K., Rummel, J., Junker, V., Culmsee, C., Pfeiffer, M., Krieglstein, J., et al. (2002). A dual role for the SDF-1/CXCR4 chemokine receptor system in adult brain: isoform-selective regulation of SDF-1 expression modulates CXCR4-dependent neuronal plasticity and cerebral leukocyte recruitment after focal ischemia. J. Neurosci. 22, 5865–5878. doi: 10.1523/JNEUROSCI.22-14-05865.2002
Tang, W. J., and Gilman, A. G. (1991). Type-specific regulation of adenylyl cyclase by G protein βγ subunits. Science 254, 1500–1503. doi: 10.1126/science.1962211
Tarozzo, G., Campanella, M., Ghiani, M., Bulfone, A., and Beltramo, M. (2002). Expression of fractalkine and its receptor, CX3CR1, in response to ischaemia-reperfusion brain injury in the rat. Eur. J. Neurosci. 15, 1663–1668. doi: 10.1046/j.1460-9568.2002.02007.x
Terunuma, M. (2018). Diversity of structure and function of GABAB receptors: a complexity of GABAB-mediated signaling. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 94, 390–411. doi: 10.2183/pjab.94.026
Terunuma, M., Pangalos, M. N., and Moss, S. J. (2010). Functional modulation of GABAB receptors by protein kinases and receptor trafficking. Adv. Pharmacol. 58, 113–122. doi: 10.1016/S1054-3589(10)58005-0
Tian, J., Dang, H., Wallner, M., Olsen, R., and Kaufman, D. L. (2018). Homotaurine, a safe blood-brain barrier permeable GABAA-R-specific agonist, ameliorates disease in mouse models of multiple sclerosis. Sci. Rep. 8:16555. doi: 10.1038/s41598-018-32733-3
Tian, J., Lu, Y., Zhang, H., Chau, C. H., Dang, H. N., and Kaufman, D. L. (2004). Gamma-aminobutyric acid inhibits T cell autoimmunity and the development of inflammatory responses in a mouse type 1 diabetes model. J. Immunol. 173, 5298–5304. doi: 10.4049/jimmunol.173.8.5298
Tian, N., Petersen, C., Kash, S., Baekkeskov, S., Copenhagen, D., and Nicoll, R. (1999). The role of the synthetic enzyme GAD65 in the control of neuronal γ-aminobutyric acid release. Proc. Natl. Acad. Sci. U S A 96, 12911–12916. doi: 10.1073/pnas.96.22.12911
Trecki, J., Brailoiu, G. C., and Unterwald, E. M. (2010). Localization of CXCR4 in the forebrain of the adult rat. Brain Res. 1315, 53–62. doi: 10.1016/j.brainres.2009.12.015
Ulrich, D., and Bettler, B. (2007). GABA(B) receptors: synaptic functions and mechanisms of diversity. Curr. Opin. Neurobiol. 17, 298–303. doi: 10.1016/j.conb.2007.04.001
van der Maten, G., Henck, V., Wieloch, T., and Ruscher, K. (2017). CX3C chemokine receptor 1 deficiency modulates microglia morphology but does not affect lesion size and short-term deficits after experimental stroke. BMC Neurosci. 18:11. doi: 10.1186/s12868-016-0325-0
Vila, N., Castillo, J., Dávalos, A., and Chamorro, A. (2000). Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke 31, 2325–2329. doi: 10.1161/01.str.31.10.2325
Villa, P., Triulzi, S., Cavalieri, B., Di Bitondo, R., Bertini, R., Barbera, S., et al. (2007). The interleukin-8 (IL-8/CXCL8) receptor inhibitor reparixin improves neurological deficits and reduces long-term inflammation in permanent and transient cerebral ischemia in rats. Mol. Med. 13, 125–133. doi: 10.2119/2007-00008.Villa
Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S., and Nabekura, J. (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 29, 3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009
Waldvogel, H., Baer, K., and Faull, R. (2010). “Distribution of GABAA receptor subunits in the human brain,” in GABA and Sleep: Molecular, Functional and Clinical Aspects, eds J. Monti, S. Pandi-Perumal, and H. Möhler (Basel: Springer), 73–93. doi: 10.1007/978-3-0346-0226-6_3
Waldvogel, H. J., Kubota, Y., Fritschy, J., Mohler, H., and Faull, R. L. (1999). Regional and cellular localisation of GABAA receptor subunits in the human basal ganglia: an autoradiographic and immunohistochemical study. J. Comp. Neurol. 415, 313–340. doi: 10.1002/(sici)1096-9861(19991220)415:3<313::aid-cne2>3.0.co;2-7
Walter, H. L., van der Maten, G., Antunes, A. R., Wieloch, T., and Ruscher, K. (2015). Treatment with AMD3100 attenuates the microglial response and improves outcome after experimental stroke. J. Neuroinflammation 12:24. doi: 10.1186/s12974-014-0232-1
Wang, Y.-C., Dzyubenko, E., Sanchez-Mendoza, E. H., Sardari, M., Silva de Carvalho, T., Doeppner, T. R., et al. (2018). Postacute delivery of GABAA α5 antagonist promotes postischemic neurological recovery and peri-infarct brain remodeling. Stroke 49, 2495–2503. doi: 10.1161/STROKEAHA.118.021378
Wang, Y., Feng, D., Liu, G., Luo, Q., Xu, Y., Lin, S., et al. (2008). Gamma-aminobutyric acid transporter 1 negatively regulates T cell-mediated immune responses and ameliorates autoimmune inflammation in the CNS. J. Immunol. 181, 8226–8236. doi: 10.4049/jimmunol.181.12.8226
Wang, J., Gan, Y., Han, P., Yin, J., Liu, Q., Ghanian, S., et al. (2018). Ischemia-induced neuronal cell death is mediated by chemokine receptor CX3CR1. Sci. Rep. 8:556. doi: 10.1038/s41598-017-18774-0
Wang, Y., Huang, J., Li, Y., and Yang, G.-Y. (2012). Roles of chemokine CXCL12 and its receptors in ischemic stroke. Curr. Drug Targets 13, 166–172. doi: 10.2174/138945012799201603
Wegelius, K., Pasternack, M., Hiltunen, J. O., Rivera, C., Kaila, K., Saarma, M., et al. (1998). Distribution of GABA receptor rho subunit transcripts in the rat brain. Eur. J. Neurosci. 10, 350–357. doi: 10.1046/j.1460-9568.1998.00023.x
Werner, Y., Mass, E., Ashok Kumar, P., Ulas, T., Händler, K., Horne, A., et al. (2020). Cxcr4 distinguishes HSC-derived monocytes from microglia and reveals monocyte immune responses to experimental stroke. Nat. Neurosci. 23, 351–362. doi: 10.1038/s41593-020-0585-y
Wheeler, D. W., Thompson, A. J., Corletto, F., Reckless, J., Loke, J. C. T., Lapaque, N., et al. (2011). Anaesthetic impairment of immune function is mediated via GABAA receptors. PLoS One 6:e17152. doi: 10.1371/journal.pone.0017152
Wieloch, T., and Nikolich, K. (2006). Mechanisms of neural plasticity following brain injury. Curr. Opin. Neurobiol. 16, 258–264. doi: 10.1016/j.conb.2006.05.011
Wojcik, W. J., and Neff, N. H. (1984). gamma-aminobutyric acid B receptors are negatively coupled to adenylate cyclase in brain and in the cerebellum these receptors may be associated with granule cells. Mol. Pharmacol. 25, 24–28.
Wu, P.-R., Cho, K. K. A., Vogt, D., Sohal, V. S., and Rubenstein, J. L. R. (2017). The cytokine CXCL12 promotes basket interneuron inhibitory synapses in the medial prefrontal cortex. Cereb. Cortex 27, 4303–4313. doi: 10.1093/cercor/bhw230
Wu, C., and Sun, D. (2015). GABA receptors in brain development, function and injury. Metab. Brain Dis. 30, 367–379. doi: 10.1007/s11011-014-9560-1
Xie, Y., Chen, S., Wu, Y., and Murphy, T. H. (2014). Prolonged deficits in parvalbumin neuron stimulation-evoked network activity despite recovery of dendritic structure and excitability in the somatosensory cortex following global ischemia in mice. J. Neurosci. 34, 14890–14900. doi: 10.1523/JNEUROSCI.1775-14.2014
Xu, J., and Wojcik, W. J. (1986). Gamma aminobutyric acid B receptor-mediated inhibition of adenylate cyclase in cultured cerebellar granule cells: blockade by islet-activating protein. J. Pharmacol. Exp. Ther. 239, 568–573.
Keywords: stroke recovery, inflammation, GABA, immune cell, neurotransmission, chemokine, glutamate decarboxylate
Citation: Michalettos G and Ruscher K (2022) Crosstalk Between GABAergic Neurotransmission and Inflammatory Cascades in the Post-ischemic Brain: Relevance for Stroke Recovery. Front. Cell. Neurosci. 16:807911. doi: 10.3389/fncel.2022.807911
Received: 02 November 2021; Accepted: 28 February 2022;
Published: 23 March 2022.
Edited by:
Camila Zaverucha Do Valle, Instituto Nacional de Infectologia Evandro Chagas (INI), BrazilReviewed by:
Bryndis Birnir, Uppsala University, SwedenMiranda Mele, University of Coimbra, Portugal
Copyright © 2022 Michalettos and Ruscher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karsten Ruscher, a2Fyc3Rlbi5ydXNjaGVyQG1lZC5sdS5zZQ==