 Feixia Pan
Feixia Pan Weize Xu
Weize Xu Jieying Ding2
Jieying Ding2 Chencen Wang
Chencen Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 11 January 2023
Sec. Cellular Neuropathology
Volume 16 - 2022 | https://doi.org/10.3389/fncel.2022.1067570
This article is part of the Research Topic Ferroptosis in Stroke, Neurotrauma and Neurodegeneration, Volume II View all 10 articles
Hemorrhagic stroke is a devastating cerebrovascular disease with high morbidity and mortality, for which effective therapies are currently unavailable. Based on different bleeding sites, hemorrhagic stroke can be generally divided into intracerebral hemorrhage (ICH) and subarachnoid hemorrhage (SAH), whose pathogenesis share some similarity. Ferroptosis is a recently defined programmed cell deaths (PCDs), which is a critical supplement to the hypothesis on the mechanism of nervous system injury after hemorrhagic stroke. Ferroptosis is characterized by distinctive morphological changes of mitochondria and iron-dependent accumulation of lipid peroxides. Moreover, scientists have successfully demonstrated the involvement of ferroptosis in animal models of ICH and SAH, indicating that ferroptosis is a promising target for hemorrhagic stroke therapy. However, the studies on ferroptosis still faces a serious of technical and theoretical challenges. This review systematically elaborates the role of ferroptosis in the pathogenesis of hemorrhagic stroke and puts forward some opinions on the dilemma of ferroptosis research.
Hemorrhagic stroke is an acute disease caused by the sudden rupture of a specific blood vessel in the brain. It refers to two main diseases based on bleeding sites: bleeding within brain parenchyma termed intracerebral hemorrhage (ICH) or within the subarachnoid space called subarachnoid hemorrhage (SAH) (Doria and Forgacs, 2019; Fang et al., 2020). Hemorrhagic stroke accounts for 10–20% of all strokes and is responsible for more than 40% of stroke-related deaths caused by severe primary and complicated secondary brain injury (Doria and Forgacs, 2019). Currently, the treatment of hemorrhagic stroke has not been well-established. Apart from the recognized supportive care, the efficacy of antihypertensive therapy, complication prevention, surgery, and hemostatic therapy to reduce primary brain injury remain controversial (Grysiewicz et al., 2008; Xu Y. et al., 2022). Moreover, despite the considerable pre-clinical efficacy of several pharmacological agents targeting pathological events of hemorrhagic stroke, they failed to perform in clinical trials as factors such as the short half-life periods of agents and the role of the blood-brain barrier (BBB) complicated the scenario (Grysiewicz et al., 2008). Further efforts are necessary to develop potential drugs against novel pathological targets to facilitate functional recovery for hemorrhagic stroke patients. Numerous pathological processes during the secondary brain injury lead to the death of neuronal cells after hemorrhagic stroke. In addition to extensively researched apoptosis, many other modes of cell death, including ferroptosis, have been explored in ICH and SAH animal models in recent years (Fang et al., 2020; Shen et al., 2020).
Based on previous studies, Dixon et al. (2012), defined a mode of programmed cell death (PCD) caused by iron overload-dependent accumulation of lethal lipid peroxides as “ferroptosis” (Dixon et al., 2012). Morphologically, ferroptosis is characterized by mitochondrial shrinkage, loss of mitochondrial cristae, and increased mitochondrial membrane density. Biochemically, the ferroptosis-mediated changes include defective glutathione (GSH) activity, excessive accumulation of lethal lipid reactive oxygen species (ROS) on the plasma membrane, the inactivation of glutathione peroxidase 4 (GPX4), and the abnormal activation of some lipid metabolism regulatory molecules (Dixon et al., 2012; Stockwell et al., 2017; Wang et al., 2021). Before the concept was proposed, most of these ferroptosis-associated biochemical changes were detected in animal models of hemorrhagic stroke (Chang et al., 2014; Gao et al., 2020; Cao et al., 2021; Duan et al., 2021; Qu et al., 2021). Several recent studies have systematically examined these markers and monitored key targets regulating ferroptosis, confirming the presence of ferroptosis in hemorrhagic stroke diseases (Alim et al., 2019; Cao et al., 2021). More importantly, many studies demonstrated that inhibition or negative modulation of ferroptosis in neuronal cells was promising as a potential therapy for treating hemorrhagic stroke (Chang et al., 2014; Shao et al., 2019; Qu et al., 2021; Ren et al., 2022). However, limited by the objective reasons of experimental technological challenges, many critical issues still remain to be discussed and resolved. Therefore, in this review, we explored the current status of ferroptosis research advances in animal models of SAH and ICH. We also summarized the findings of the studies on the drugs or inhibitors targeting ferroptosis and further discussed their feasibility as potential therapies for treating hemorrhagic stroke.
Search strategy: A systematic search was implemented on two databases PubMed and Web of Science. The overall retrieval terms are “intracerebral hemorrhage AND ferroptosis,” and “subarachnoid hemorrhage AND ferroptosis.” In the part of Iron overload, retrieval terms “(intracerebral hemorrhage or subarachnoid hemorrhage) AND (iron OR iron disorder OR iron overload)” were used. In the part of Lipid peroxidation, retrieval terms “(intracerebral hemorrhage or subarachnoid hemorrhage) AND (lipid peroxidation OR reactive oxygen species)” were used. In the part of antioxidant system Lipid peroxidation, retrieval terms “(intracerebral hemorrhage or subarachnoid hemorrhage) AND (FSP1/CoQ10 OR GSH/GPX4)” were used. All the references were loaded into Zotero (version 5.0.96) and combined to exclude duplicate and triplicate.
A decade before the term “ferroptosis” was defined, the basic features of ferroptosis were identified in quick succession (Figure 1). The two most used drugs for inducing ferroptosis, erastin, and Ras-selective lethal small molecules 3 (RSL3), were identified by the same research team in 2003 and 2008, respectively (Dolma et al., 2003; Yang and Stockwell, 2008). This group reported that they induce a form of PCD different from apoptosis. It was shown that iron chelator (deferoxamine) and certain antioxidants explicitly inhibit this type of cell death. Based on previous studies, Dixon et al. (2012) named this form of PCD as “ferroptosis” in 2012 and systematically described its biological, biochemical, and genetic characteristics. After about 5 years, studies from several different laboratories comprehensively summarized the morphological features of ferroptosis, which manifest as damaged mitochondria including mitochondrial shrinkage, reduced amounts of cristae, and outer membrane rupturing (Kagan et al., 2017). Mitochondrial structure monitoring by transmission electron microscope (TEM) is crucial for determining the occurrence of ferroptosis in the cells (Dixon et al., 2012). From the biochemical standpoint, ferroptosis involves a complex molecular biological network leading to the overproduction and accumulation of lipid peroxidation products and disrupting the antioxidant systems (Dixon et al., 2012; Stockwell et al., 2017). These include landmarks that are widely recognized by researchers like GPX4, SLC7A11 (xCT), and acyl-CoA synthetase long-chain family member 4 (ACSL4). Moreover, there are also many new targets that have been successively proven to be related to ferroptosis in recent years (Lei et al., 2022). In this review, we attempt to make a succinct summary of all such information below.
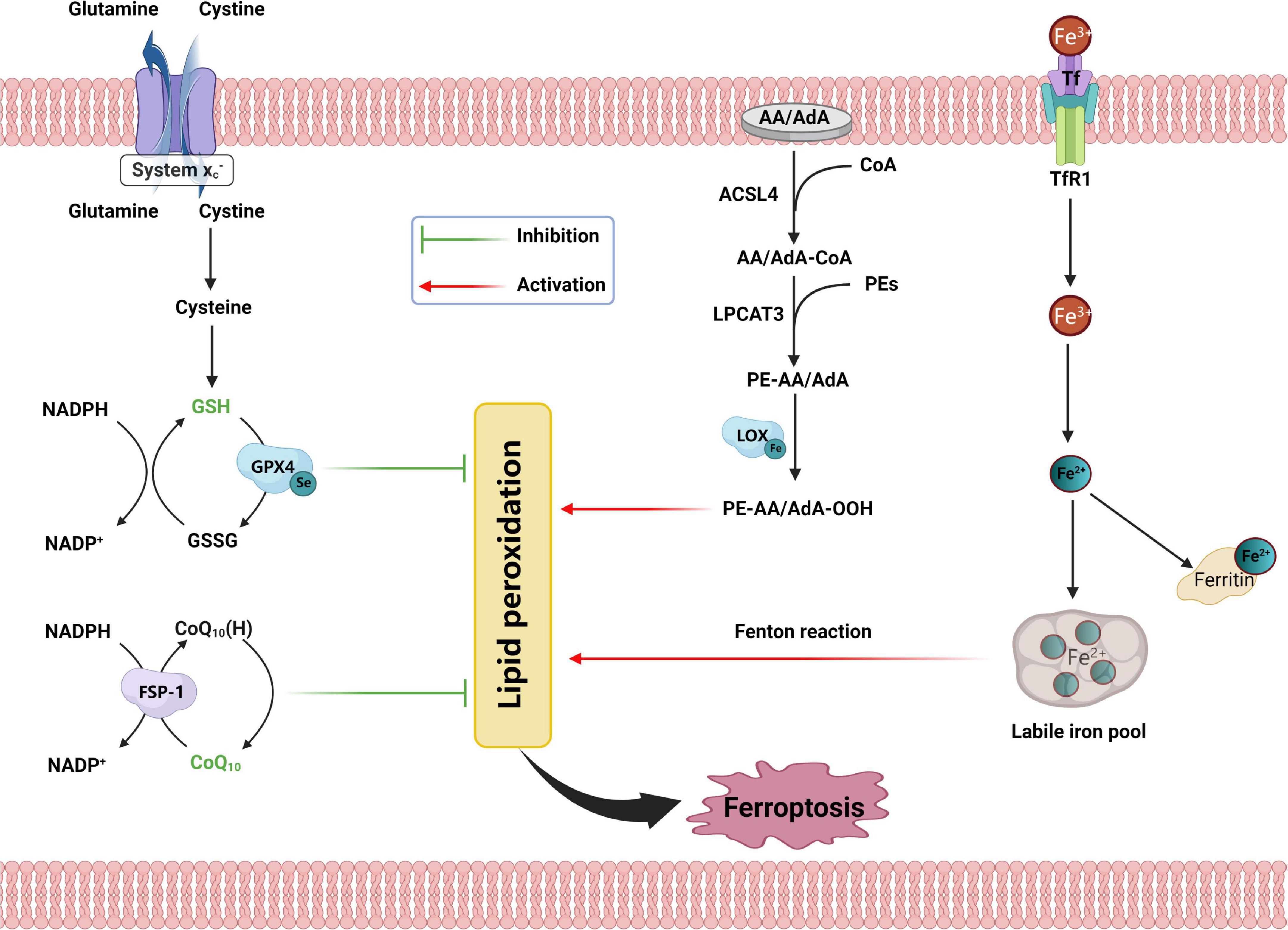
Figure 1. Schematic representation for the mechanism of ferroptosis, including antioxidant systems, iron disorder, and lipid peroxidation. SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; GSH, glutathione; GSSG, oxidized glutathione; GPX4, glutathione peroxidase 4; NADPH, nicotinamide adenine dinucleotide phosphate; GPX4, glutathione peroxidase 4; CoQ10, coenzyme Q10; FSP1, ferroptosis suppressor protein 1; AA, arachidonic acid; AdA, adrenic acid; CoA, coenzyme A; ACSL4, acyl-CoA synthetase long-chain family member 4; AA/AdA-CoA, adrenic acid/arachidonic acid-CoA; PEs, phosphatidylethanolamines; LPCAT3, lysophosphatidylcholine acyltransferase 3; PE-AA/AdA, phosphatidylethanolamine-adrenic acid/arachidonic acid; LOX, lipoxygenase; PE-AA/AdA-OOH, phosphatidylethanolamine- adrenic acid/arachidonic acid bis/trioxide; Tf, transferrin; TFR1, transferrin receptor 1.
Ferroptosis is inhibited by iron chelators, which have led scientists to initially name it a new PCD with a root “ferr-” representing the then-popular belief that iron is indispensable in this process. However, this led to a misconception for future research, whereby people believed that ferroptosis must be accompanied by the disturbance of iron metabolism or even iron overload. Many subsequent landmark studies did not pay attention to iron metabolism-related factors because lethal lipid-related ROS and disruption of the antioxidant system are the key factors mediating ferroptosis (Li and Li, 2020). Moreover, the construction of many in vitro ferroptosis models does not depend on any form of iron (Ding et al., 2020; Liu et al., 2020). Despite no apparent link between iron overload and ferroptosis, substantial attention is warranted to investigate the possibility of ferroptosis during iron overload.
Under normal circumstances, ferrous (Fe2+) ions from physiological or pathological origin get oxidized to ferric (Fe3+) ions. They combine with free transferrin (TF), which is recognized by membrane protein transferrin receptor 1 (TFR1). Then the TF/TFR1 complex will be taken up in a manner of endocytosis. Because of the acidic environment in endosome, Fe3+ will immediately be reduced to Fe2+ by ferric reductase sixtransmembrane epithelial antigen of prostate 3 (STEAP3) after being released from TF (Louandre et al., 2013). After that, divalent metal transporter 1 (DMT1) transports Fe2+ into the labile iron pool in the cytoplasm (Katsarou and Pantopoulos, 2020). It is noteworthy that ferroportin-1 (FPN1) on the cell membrane is currently the only known molecule to actively transport Fe2+ from the intracellular to the extracellular region, maintaining the stability of the intracellular iron pool (Troadec et al., 2010). The change in the function or content of the above-mentioned iron regulatory molecules may cause the iron overload in cells, inducing ferroptosis. A recent study found that activation of nuclear receptor coactivator 4 (NCOA4) destabilizes ferritin in an autophagy-dependent manner, increasing the labile iron level and promoting ferroptosis (Zhang Y. et al., 2021).
Lipid peroxides not removed in time are the most direct executors of ferroptosis. The following three pathways generate the free radical reactions mediating the formation of lethal lipid peroxides: (i) non-enzymatic Fenton reaction; (ii) iron-catalyzed auto-oxidation, and (iii) oxidase- and esterase-mediated peroxidation of polyunsaturated fatty acids (PUFA) (Yang and Stockwell, 2016; Li and Li, 2020). The polyolefinic structure of PUFAs, especially arachidonic acid (AA) and adrenic acid (ADA), makes them susceptible to oxidation to form lipid peroxides. However, studies show that excess peroxidation of PUFAs (AA/ADA-OOH) does not explicitly indicate ferroptosis. AA/ADA must be catalyzed by acyl-CoA synthetase long-chain family member 4 (ACSL4) to form AA/ADA-CoA, followed by esterification into membrane phospholipids before becoming a crucial precursor of lethal lipid-related ROS (Kagan et al., 2017; Wenzel et al., 2017). Two independent groups have implicated using lipid metabolomics screening that the product of phosphatidylethanolamines (PEs)-AA/ADA, namely, PEs-AA/ADA-OOH, oxidized by lipoxygenase, as the critical substance mediating ferroptosis. There are numerous ways of catalyzing PEs-AA/ADA to form peroxides (Kagan et al., 2017; Wenzel et al., 2017). It has been shown that the rate-limiting steps in acetylation and esterification make ACSL4 and lysophosphatidylcholine acyltransferase 3 (LPCAT3) the critical promoters in the regulation of ferroptosis (Feng et al., 2022). Multiple studies have confirmed the roles of these enzymes. Numerous recent studies have indicated how inhibition of ACSL4 and LPCAT3 might be promising therapeutic strategies for treating ferroptosis-related injuries (Cui et al., 2021a; Sha et al., 2021; Reed et al., 2022). However, how PEs-AA/ADA-OOH relay the signals for ferroptosis remains to be interpreted. Plausible underlying mechanisms include binding excess lipid peroxides to the membrane, causing lethal changes in membrane permeability and ion concentrations. It is also believed that the downstream products of these lipid peroxides, like malondialdehyde (MDA) and 4-hydroxy-nonenal (4-HNE), exert ferroptotic damage by degrading proteins or nucleic acids (Liu et al., 2020, 2021). Many studies have implicated MDA and 4-HNE as markers of ferroptosis.
GPX4 is an important peroxide-decomposing enzyme ubiquitously present in the body. GSH can complete the conversion of lipid peroxides to non-toxic lipids (Fraternale et al., 2009). This property protects cell membranes made up of large lipid molecules from peroxides interference and damage. GSH/GPX4 antioxidant system is one of the most crucial endogenous factors against ferroptosis (Li F. et al., 2022). GSH is synthesized by three amino acids: glutamate, cysteine, and glycine. Cysteine, mainly derived from extracellular cystine after entering the cell and being reduced, is present in the lowest concentration in the cell, making it the critical rate-limiting factor for the de novo synthesis of GSH. The cystine/glutamate antiporter (system XC–) on the cell membrane transports the extracellular cysteine into the cell in a 1:1 form while transporting out the intracellular glutamate (Yuan Y. et al., 2022). The system XC– on the cell membrane is a heterodimer consisting of the substrate-specific subunit SLC7A11 (xCT) and the regulatory subunit SLC3A2 (Koppula et al., 2021). GPX4 converts the reduced form of GSH into oxidized glutathione (GSSG) and promotes the reduction reaction from lipid peroxides (L–OOH) to corresponding alcohols (L–OH) (Li F. et al., 2022).
In many pathological conditions or artificial interventions, increased extracellular glutamate and disrupted xCT structure or function lead to intracellular GSH depletion, thereby inducing ferroptosis (Chen et al., 2017; Yadav et al., 2021). Meanwhile, the down-regulation or functional disruption of GPX is also essential for promoting ferroptosis. The most used inducers of ferroptosis, erastin, and RSL3, are inhibitors of system XC– and direct inhibitors of GPX4, respectively (Stockwell et al., 2017). In addition, there are many compounds that can induce ferroptosis by inhibiting GPX4, like DPI7 and DPI10 in a directing binding manner and FIN56 in a form that induces GPX4 degradation (Mou et al., 2019; Sun et al., 2021). On the other hand, selenium promotes GPX4 transcription and prevents it from irreversible degradation, thereby inhibiting ferroptosis (Alim et al., 2019; Yao et al., 2021). Increasing evidence delineate the role of the GSH/GPX4 antioxidant system in the mechanism of ferroptosis.
The ferroptosis suppressor protein 1 (FSP1)/coenzyme Q10 (CoQ10) pathway is another efficient endogenous antioxidant system independent of GPX4/GSH system, that critically regulates cellular sensitivity toward ferroptosis. FSP1, alternatively known as apoptosis-inducing factor mitochondria 2 (AIFM2), has significantly high homology with apoptosis-inducing factor (AIF) (Miriyala et al., 2016). The name and homology details of AIFM2 indicate how it was identified by its function to induce apoptosis by releasing cytochrome c and caspase 9 (Miriyala et al., 2016). However, a recent study found that AIFM2 had a marked inhibitory effect on GPX4 deficiency that leads to ferroptosis, renamed “FSP1” (Doll et al., 2019). The lethal lipid peroxidation event in ferroptosis mainly starts at the plasma membrane, leading to the transfer of FSP1 by myristoylation for subsequent free radical scavenging (Doll et al., 2019). FSP1 uses the reducing equivalents of nicotinamide adenine dinucleotide phosphate [NAD(P)H] to reduce CoQ10 to ubiquinol, which acts as a lipophilic radical-trapping antioxidant (RTA) to remove lipid peroxides from phospholipid bilayers (Bersuker et al., 2019).
NAD(P)H is also indispensable for reducing GSSG to GSH. Therefore, involved in two important antioxidant systems, the level of NAD(P)H have also been indicated by several studies as a predictor of cellular susceptibility to ferroptosis (Bersuker et al., 2019). This molecular crosstalk means that the intracellular defenses against lipid peroxides are not dependent on a single system. FSP1 was also recently reported to inhibit ferroptosis in endosomal required transport (ESCRT)-III-dependent manner (Dai et al., 2020). ESCRT-III is a membrane-associated protein complex enhancing plasma membrane repair by activating subtypes of charged multivesicular body proteins (Dai et al., 2020). In addition, tetrahydrobiopterin (BH4) and its synthetic rate-limiting enzyme Guanosine triphosphate cyclohydrolase 1 (GCH1) are involved in the ferroptosis-resistant oxidative network by regulating CoQ10 synthesis (Mishima and Conrad, 2022). However, more investigation is required to confirm these systems’ effectiveness and underlying mechanisms.
According to the bleeding site, a hemorrhagic stroke can be divided into two subtypes, ICH and SAH (Doria and Forgacs, 2019). Their pathological mechanisms occur in two steps and involve multiple similarities. The first stage involves primary brain injury dominated by mechanical damage caused by the traction of the hematoma, accompanied by increased intracranial pressure and secondary cerebral infarction (Magid-Bernstein et al., 2022). The second stage consists of pathophysiological events caused by blood components and their metabolites, including BBB destruction, neural excitatory events, ion disturbances, oxidative stress, neuroinflammation, cell death, etc (Magid-Bernstein et al., 2022). In SAH, early brain injury (EBI) is crucial, indicating the pathological damage within 3 days after SAH (Kusaka et al., 2004; Fujii et al., 2013). The recovery degree of the pathological damage during EBI will substantially impact the recovery of subsequent neurological functions.
Several animal models were adopted for hemorrhagic stroke research. For ICH, collagenase injection or autologous blood injection at the basal ganglia are two common modeling methods (Krafft et al., 2012; Lei et al., 2014). The former causes cerebral hemorrhage by decomposing collagen on the cell matrix and vascular basement membrane, which has the advantage of high efficiency and convenience. While the latter can better simulate the pathophysiological changes of patients with cerebral hemorrhage. For SAH, the endovascular perforation model is widely used because it can simulate SAH caused by rupture of aneurysm (Du et al., 2016). These models can cause a large amount of instantaneous hemorrhage in specific regions of the brain, and are valuable experimental models of hemorrhagic stroke in preclinical research.
Degradation products of hemoglobin (ferrous iron and heme) play an essential initiating role in the induction of these pathophysiological events. Among them, the abnormal accumulation of iron ions leads to severe tissue damage (Gaasch et al., 2007). It generates a large amount of ROS, providing a solid foundation for ferroptosis to set in Gaasch et al. (2007). This validates the hypothesis that ferroptosis plays a critical pathological role after a hemorrhagic stroke. A series of subsequent studies also confirmed the occurrence of ferroptosis in animal and in vitro models of ICH and SAH (Karuppagounder et al., 2018; Chen et al., 2019; Cao et al., 2021; Qu et al., 2021). Li et al. (2018) first observed specific mitochondrial morphology of ferroptosis in perihematoma neurons in a collagenase-induced mouse ICH model. Then in 2020, ferroptotic mitochondria were first observed in temporal cortical neurons in a mouse model of SAH (Li Y. et al., 2021). The TEM pictures of mitochondria provide strong evidence supporting ferroptosis. In addition, the down-regulation of the antioxidant molecule GPX4 and upregulation of ACSL4 and lipid peroxides were also validated in these two models by various groups (Zhang et al., 2018; Gao et al., 2020; Jin et al., 2021; Qu et al., 2021). These results demonstrate how ferroptosis, an essential complement to other PCDs, plays a vital role in hemorrhagic stroke. Moreover, two critical inhibitors of ferroptosis, Ferrostatin-1 (fer-1) and Liproxstatin-1 (Lip-1), inhibited neuronal ferroptosis and neuroinflammation in animal models of hemorrhagic stroke, reducing neurological impairment (Li et al., 2017a; Zhang et al., 2018; Cao et al., 2021). In addition, various chemical agents and natural medicines improve neurological damage in animals with ICH or SAH by inhibiting ferroptosis, suggesting ferroptosis as a promising potential target for therapeutic strategies for hemorrhagic stroke (Li et al., 2019; Duan et al., 2021; Gao et al., 2022).
However, the knowledge about ferroptosis in hemorrhagic stroke is still minimal. To achieve clinical translation, sufficient preclinical experiments must identify effective therapeutic agents and the exact molecular mechanisms.
In addition to ferroptosis, other PCDs such as apoptosis, pyroptosis, necroptosis, and autophagy are also widely studied in the field of hemorrhagic stroke. There are also various crosstalk between ferroptosis and these PCDs. Apoptosis is the most well-studied forms of PCD in hemorrhagic stroke, which mainly induced by two pathways: extrinsic and intrinsic pathways (Sekerdag et al., 2018). The characteristic features of apoptotic neuron include chromatin condensation, nuclear shrinkage, and DNA fragmentation, which is completely different from ferroptosis (Weiland et al., 2019). Hence, apoptosis, being considered as the main form of neuronal death after hemorrhagic stroke, is mainly studied as an exclusionary form in iron death related studies. A recent study suggests that there may be a certain correlation between ferroptosis and necroptosis through NADPH (Hou et al., 2019). Since autophagy can regulate intracellular iron homeostasis and ROS synthesis, current research indicates that many ferroptosis are autophagic dependent (Ma et al., 2022; Tao et al., 2022). The latest research of Tao et al. (2022) indicates that S100A8 can mediate autophagy-dependent ferroptosis of microglia after SAH. In addition, mitophagy has also been reported to mediate ferroptosis (Li J. et al., 2022), which has not been verified in hemorrhagic stroke, indicating it may be a valuable research direction in the future.
Subarachnoid hemorrhage (SAH) and intracerebral hemorrhage (ICH) share many pathological similarities, and similar hypotheses have been formed to identify the mechanism of ferroptosis in these two hemorrhagic strokes. However, to date, the underlying mechanisms of ferroptosis have not been understood to the same extent in ICH and SAH, triggered by differential research investments. Because the incidence of SAH is much lower than ICH worldwide, the progress of basic research in decoding the mechanisms of ferroptosis is also relatively inadequate in SAH (Doria and Forgacs, 2019). However, the advances in research to decode ferroptosis mechanisms in SAH have not been summarized. Therefore, in this part, we will systematically review the research progress in understanding ferroptosis mechanisms in ICH and SAH.
The direct accumulation of blood components in the injured area is a critical pathogenic symptom of hemorrhagic disease. Therefore, since the discovery of ICH, researchers have long been critically concerned about the disturbance of iron metabolism (Uselis, 1970). As early as 2004, a study identified iron deposition in the basal ganglia in a rat model of ICH (Nakamura et al., 2004). This deposited iron is mainly derived from the lysed red blood cell hemoglobin, which is degraded into heme and free iron. In addition, Nakamura et al. (2005) indicated that holo-transferrin interacts with thrombin to exacerbate brain damage upon the onset of ICH. Iron accumulation and ferritin upregulation also cause long-term neurological impairment in animal models of ICH (Nakamura et al., 2005). Deferoxamine (DFO) is a drug that eliminates iron accumulation and has been validated by multiple research groups in various animal models. Its therapeutic mechanism has been hypothesized to involve DNA oxidative damage repair, neuronal apoptosis inhibition, and brain edema reduction (Farr and Xiong, 2021). In addition, Zhao et al. (2011) reported another potent iron chelator, minocycline, which alleviates neuronal apoptosis after ICH by inhibiting the pathways that upregulate iron handling proteins of the brain. DFO and minocycline have been selected for clinical trials because of their excellent efficacy in animal experiments. Unfortunately, no drug targeting iron chelation has successfully achieved clinical translation (Selim et al., 2011). Moreover, the specific mechanism underlying cellular damage due to ROS generation by iron overload has not been elucidated.
Introducing the concept of ferroptosis provides insights into a new pathogenic mechanism of iron-mediated diseases, encouraging researchers to delve deep into it. Hemorrhage leads to iron deposition and regulates iron’s entry and exit pathways by several vital molecules, including TF, TFR1, DMT1, and FPN1 (Vogt et al., 2021). Hepcidin has received more attention in recent years due to its ability to change intracellular iron content by regulating the expression of TF, TFR1, DMT1, and FPN1 (Camaschella et al., 2020). TF, TFR1, and DMT1 are responsible for iron uptake, while FPN1 is the only regulatory molecule aiding in the transport of iron out of the cells (Vogt et al., 2021). Yang et al. (2021) have demonstrated how ICH mice exhibited less iron overload, brain edema, and neuronal damage after adeno-associated virus-mediated overexpression of hepcidin. Down-regulating TF and TFR1 mainly achieve this effect after ICH. In recent years, many new iron chelators have been tested on animal models of ICH and achieved considerable preclinical efficacy. VK-28, Deferiprone (DFP), and Deferasirox (DFX) are some representative drugs (Wang et al., 2016; Li et al., 2017b; Imai et al., 2021). However, it is worth pointing out that although most studies of ferroptosis in ICH involve some iron deposition, few studies have directly explored the association between iron deposition and ferroptosis. We believe this action stems from the lack of a specific mechanism of iron-mediated ferroptosis.
The STICH II trial showed that withdrawal after open surgery didn’t significantly reduce the mortality of cerebral hemorrhage compared with the initial conservative treatment (Mendelow et al., 2013). However, the subgroup of MISTIE III trial showed that compared with the conservative treatment group, the patients whose blood clot size decreased less than 15 ml after minimally invasive surgery live with better neurological recovery (Hanley et al., 2019). In addition to iron chelating agent drugs, surgical removal of hematoma or endogenous phagocytosis of ectopic erythrocyte from the source to reduce the degradation products of erythrocyte are effective treatments for hemorrhagic stroke (Zheng et al., 2022). The phagocytosis of erythrocytes is mainly completed by microglia and macrophages, which are regulated by several signal pathways. Peroxisome proliferator-activated receptor γ (PPAR-γ) is one of the most widely studied regulatory molecules, and its activation can promote the phagocytosis of microglia on hematoma. Drugs including monascin, simvastatin, wogonin can activate PPAR-γ, thus promoting phagocytosis (Wang et al., 2018; Fu et al., 2020; Zhuang et al., 2021). In addition, the activation of nuclear factor erythroid 2-related factor2 (Nrf2) pathway has also been proved to promote hematoma phagocytosis (Zheng et al., 2022). Xu C. et al. (2022) demonstrated that CD47 also plays an important role in the process of hematoma phagocytosis in the SAH model.
Before the concept of early brain injury was proposed, the academic research on the pathogenesis of SAH mainly focused on secondary ischemia caused by vasospasm (Lin et al., 2004). In 1988, the non-glucocorticoid 21-aminosteroid U74006F, an inhibitor of iron-dependent lipid peroxidation, alleviated the acutely progressive hypoperfusion state in a cat SAH model (Hall and Travis, 1988). In the early 1990s, Utkan et al. (1996) demonstrated for the first time that the iron chelator DFO could prevent SAH-induced vasospasm in a rabbit model (Utkan et al., 1996). They also hypothesized that the underlying mechanism might involve the inhibition of iron-induced ROS and lipid peroxides (Utkan et al., 1996). Subsequent studies have repeatedly demonstrated the reduction of vasospasm by iron chelation (Luo et al., 1995). Smith et al. (1996) primarily focused on iron-induced neuronal damage. They elucidated how a lipid peroxidation inhibitor, tirilazad mesylate (U-74006F), and its metabolites attenuated early BBB damage and attenuated iron-induced neuronal lipid peroxidative damage. This may be the earliest proof of the underlying mechanism of ferroptosis in hemorrhagic stroke.
Similar to studies on ICH, around 2004, researchers systematically studied the metabolism and role of hemoglobin, heme, and iron in SAH and also pointed out the predictive effect of serum ferritin content on the severity of SAH (O’Connell and Watson, 2003; Suzuki et al., 2003). In addition, the up-regulation of TF and TFR1 and ferritin in brain tissue was also verified by Lee et al. (2010) in a rat model of SAH. Their team also showed that iron overload in the acute phase of SAH leads to oxidative stress and neuronal damage, which can be reversed by the application of DFO (Lee et al., 2010). However, since the advent of the concept of ferroptosis, few studies have systematically verified the association between iron overload and cellular ferroptosis in SAH models. Moreover, no study has investigated the neurological rescue effect of various iron chelators achieved by inhibiting ferroptosis. However, many studies have reported a positive correlation between iron accumulation and ferroptosis in ischemic stroke. One study indicated that tau-mediated iron export could reduce ferroptosis after ischemic stroke (Tuo et al., 2017). Mitochondrial ferritin has also been upregulated in the brain of mice with ischemic stroke upon FPN1 upregulation to attenuate ferroptosis and achieve neuroprotective effects (Wang P. et al., 2022). Recently, Zhang H. et al. (2021) pointed out that hepcidin can promote ferroptosis after SAH by regulating iron metabolism, and this effect may be performed by upregulating DMT1. Their research on iron metabolism regulation lacks comprehensiveness, warranting more in-depth studies to explore the correlation between iron overload and ferroptosis.
Oxidative stress is essential in ICH pathogenesis, primarily mediated by excessive accumulation of ROS and reactive nitrogen species (RNS) (Li et al., 2011). ROS is a group of partially reduced oxygen-containing molecules or radicals, and its simple form mainly includes hydrogen peroxide (H2O2) and some radicals such as hydroxyl radical (⋅OH) and superoxide anion (O2–) (Zhu et al., 2021). Lipid peroxidation involves ROS-mediated electron loss, forming various reactive intermediates. Before introducing the concept of ferroptosis, numerous studies demonstrated how ROS and end-products of lipid peroxidation like MDA damage proteins and DNA, resulting in cell injury (Wang et al., 2011). Some classic antioxidant drugs, such as metformin and melatonin, provide neuroprotective effects to ICH mice by reducing ROS and MDA levels (Lekic et al., 2010; Wu et al., 2016). However, the mechanisms underlying the pathogenicity of lipid peroxides remain unclear. The synthesis of lipid peroxides relies on several specific regulatory processes, which are thought to be critical factors regulating the initiation of ICH ferroptosis.
The high level of PUFAs in the brain is beneficial for inducing ferroptosis signaling in ICH. 20-hydroxyeicosatetraenoic acid (20-HETE), a metabolite of ADA, was recently shown to be involved in neuronal ferroptosis upon ICH induction in mice (Han et al., 2021). And reduction of its production with the specific inhibitor HET0016 significantly inhibited lipid peroxidation levels and cell death in an in vitro model of ICH (Han et al., 2021). This significantly contributed to research on lipid peroxidation in ferroptosis after ICH due to limited previous research exploring this direct relationship. Previous studies indicated that N-acetylcysteine could neutralize the peroxidative toxicity of arachidonic acid catalyzed by 5-lipoxygenase to inhibit ferroptosis after ICH (Karuppagounder et al., 2018). Moreover, baicalin, a non-specific inhibitor of 12/15-lipoxygenase, significantly increases the detection indices related to ferroptosis after ICH (Duan et al., 2021), suggesting that 12/15-lipoxygenase may play a critical role in the process of ferroptosis after ICH.
ACSL4 has also been implicated in the process of ferroptosis in ICH by two recent independent studies (Chen et al., 2021; Jin et al., 2021). It is highly expressed in the brain tissue surrounding the hematoma of ICH mice. Moreover, ACSL4 can also be regulated by long non-coding RNAs (LncRNAs). LncRNA H19 positively regulates ACSL4 expression upon ICH onset (Chen et al., 2021). Another study pointed out that LncRNA HOX transcript antisense RNA (HOTAIR) can bind to UPF1 to degrade ACSL4, thereby inhibiting ferroptosis and the impairment of neurological function in ICH mice (Jin et al., 2021).
As early as the 1980s, a team led by Asano explored the roles of lipid peroxides in SAH (Suzuki et al., 1983). They used a combination of high-performance liquid chromatography (HPLC) and gas chromatography-mass spectrometry (GC-MS) techniques to identify hydroperoxy HETEs (HPETEs) and HETEs in the cerebrospinal fluid (CSF) of SAH patients as well as healthy individuals (Suzuki et al., 1983). The results showed that compared with the average population, the content of 5-HETE in the CSF of SAH patients was significantly increased. However, the knowledge about SAH at that time led them to hypothesize that the content of this 5-HETE was related to the degree of vasospasm (Suzuki et al., 1983). Their follow-up studies also showed that 5-HETE levels were also elevated in the walls of blood vessels and the temporal cortex adjacent to SAH blood clots (Sakaki et al., 1986). Later, another group reported the up-regulation of another class of lipid peroxides, phosphatidylcholine hydroperoxide, and cholesteryl ester hydroperoxide, in the CSF of SAH patients (Kamezaki et al., 2002). However, our current knowledge indicates that neither of these two lipid peroxides mediates ferroptosis. After an extended period, there has been a lacuna in studies on lipid peroxidation in patients or animals with SAH. It is worth pointing out that 5-HETE is formed when 5-lipoxygenase catalyzes ADA (Stockwell et al., 2017). Unfortunately, no studies have investigated the mechanism of action and role of 5-lipoxygenase in SAH. In animal models of ICH and ischemic stroke, 5-lipoxygenase has been demonstrated to induce ferroptosis induction critically. 15-lipoxygenase performs a similar function as 5-lipoxygenase, only differing in the site of oxidized PUFAs (Çolakoğlu et al., 2018). It is highly expressed in microglia upon the onset of SAH. Reduction of 15-lipoxygenase level by drug application inhibits microglial ferroptosis (Gao et al., 2022). Moreover, applying its inhibitor baicalin can reduce ferroptosis after SAH and thus alleviate EBI (Zhang et al., 2020). Such lines of research on lipoxygenase may be valuable in depicting SAH pathogenesis.
Qu et al. (2021) verified the essential role of ASCL4 in ferroptosis induction in the SAH mice model for the first time. They found that the down-regulation of ACSL4 significantly inhibits lipid peroxidation and neurological damage in SAH. Two subsequent studies also examined the role of ACSL4 as a protein-level biomarker for ferroptosis (Huang et al., 2022; Yuan B. et al., 2022). The alteration of ASCL4 has higher stability than other marker molecules and has a better significance in detection. This characteristic has been confirmed in various disease models.
Moreover, GSH/GPX antioxidant system received attention simultaneously as more studies on lipid peroxidation were performed. In the ICH disease model, the role of GSH was first revealed by the team led by Dhandapani, who successively reported that GSH depletion leads to cell damage and death in hemin-induced astrocytes and endothelial cells (Laird et al., 2008; Sukumari-Ramesh et al., 2010). However, these forms of cell death do not accord with the features of ferroptosis. Zhang et al. (2018), for the first time, explored the expression changes and effects of GPX4 in a mouse model of ICH and showed how its levels in mouse brain tissue surrounding the hematoma were significantly reduced 2 days after ICH. Moreover, overexpression of GPX4 with adeno-associated virus significantly attenuated neuronal cell death and improved mice behavior (Zhang et al., 2018). Unfortunately, the identification of ferroptosis was not rigorous enough. In 2019, a cellular study found that selenium upregulates GPX4 by activating the transcription factors TFAP2c and Sp1 of GPX4 (Alim et al., 2019). Upregulated GPX4 can protect neurons from ferroptosis. They reported that GPX4 was upregulated in hemin-induced primary neurons, which may be related to the negative feedback regulation after ferroptosis. GSH and GPX have been repeatedly verified as critical negative regulators of oxidative stress in subsequent ICH studies. Most studies demonstrated how GSH content and the expression of GPX4 are downregulated, subject to reversing by regulating antioxidant drugs like dauricine or microRNAs to exert neuroprotective effects (Zhang et al., 2018; Peng et al., 2022).
A recent study showed in the FSP1/CoQ10 antioxidant system that the level of FSP1 was significantly reduced in the brain tissue surrounding hematomas in ICH mice, which could be treated with the drug dexpramipexole to reverse this harmful change (Wang B. et al., 2022). However, the expression pattern and the underlying mechanism with the potential role of CoQ10 have not been adequately investigated.
In SAH, the GSH/GPX4 antioxidant system research has been significantly more robust than in ICH, due to the early successful identification of lipid peroxides in CSF and the correlation between its content and disease prognosis (Suzuki et al., 1983; Sakaki et al., 1986). Lipid peroxidation in CSF also revealed decreased GPX activity and decreased GSH content in CSF of SAH patients (Suzuki et al., 1983). Another animal experiment showed reduced superoxide-dismutase (SOD) and GPX levels could be observed in the hippocampus of the SAH rat model (Sakaki et al., 1986). Based on such observations, the imbalance of this antioxidant system in the temporal cortex of SAH patients have also been elucidated.
Moreover, a multicenter, double-blind clinical trial showed how a seleno-organic compound, ebselen, alleviate neurological deficits in SAH patients through a GPX-like mechanism of action (Handa et al., 2000). Later, more antioxidant drugs, such as melatonin and N-acetylcysteine, upregulated the antioxidant activity of GSH/GPX, causing neuroprotection in SAH experimental animals (Ayer et al., 2008; Lu et al., 2009). Notably, any of these neuroprotective effects involve inhibition of neuronal apoptosis.
After introducing the ferroptosis concept, the main focus of the research was on a subtype of the GPX family, GPX4, which also acts as a biomarker in SAH ferroptosis. Gao et al. (2020) were the first to explore the role of GPX4 in SAH. They showed that SAH significantly reduced GPX4, which is mainly expressed in neurons, and overexpression of GPX4 alleviated lipid peroxidation-mediated cell death. Most drug-related studies also focus on the up-regulation of GPX4 and whether it can be achieved as a criterion for determining whether a drug is effective for ferroptosis treatment (Huang et al., 2022; Yuan B. et al., 2022). SLC7A11, as the critical target regulating GSH synthesis, was also found to be impaired in SAH models (Liu et al., 2022). But in terms of protein expression, the reduction is not as significant as GPX4.
A recent study also validated the inhibition of the FSP1/CoQ10 antioxidant system in vivo and in vitro SAH models. They highlighted how the activation of a well-known epigenetic regulator, Sirtuin 1 (SIRT1), alleviates neuronal ferroptosis in SAH by upregulating the expression of FSP1 and CoQ10B (Yuan B. et al., 2022). Their finding adds critically to the existing knowledge about the antioxidant mechanism underlying ferroptosis after a hemorrhagic stroke and deserves more investment in future research.
Apoptosis, pyroptosis, and autophagy involve hallmark molecules that can execute the cell death program, enabling researchers to detect these PCDs by simple but specific techniques (Fang et al., 2020). In addition, technical means such as TUNNEL staining and annexin v-pi staining help to locate the apoptotic cells (Luchetti et al., 2023). For autophagy, the autophagic-lysosome is its identification marker (Rodgers et al., 2022). However, the currently established markers for ferroptosis, GPX4, SLC7A11, ACSL4, and several iron metabolism-related indicators fail to point to ferroptosis directly. In 2017, two independent groups established that PEs-AA/ADA-OOH are the direct executor of ferroptosis, providing a novel yet convincing biomarker for validating ferroptosis (Kagan et al., 2017; Wenzel et al., 2017). However, separating and purifying this specific phospholipid is challenging as the tedious and elaborate process involves high-performance liquid chromatography and mass spectrometry with high-purity standards for comparison. Very few laboratories worldwide accurately detect PEs-AA/ADA-OOH. Hence minimal testing has been performed to evaluate its content in brain tissue after a hemorrhagic stroke. Moreover, no ferroptosis-specific tissue staining techniques or flow cytometry have been developed. Therefore, the identification of ferroptosis still involves evaluating morphological changes observed by TEM and verifying many biomarkers.
Evidence from most of the current studies suggests that the cell types undergoing ferroptosis after hemorrhagic stroke mainly involve neurons (Zhang et al., 2018; Gao et al., 2020; Huang et al., 2022). The recovery of neural function is largely due to the repair of neurons. Moreover, the primary regulators of ferroptosis, ACSL4, and GPX4, were mainly localized in neurons, further validating the hypothesis that ferroptosis occurs in neurons after an injury. Mitochondrial ferroptotic damage within neurons under TEM observation is critical for identification. However, multiple other cell types in the brain have also been reported to undergo ferroptosis.
Several studies have reported the role of ferroptosis in microglia, which put forward by Kapralov et al.’s (2020) research on macrophage ferroptosis. They found M2-type macrophages to be more sensitive than M1-type macrophages to ferroptosis. Studies have shown that ferroptosis is prevalent in SAH microglia, and the sensitivity of M1 and M2 microglia to ferroptosis is also similar to that of peripheral macrophages (Wu et al., 2020). Two subsequent studies indicated that this sensitivity of microglia to ferroptosis may be regulated by the expression of intracellular NRF2 or iNOS (Cui et al., 2021b; Qu et al., 2022). Nevertheless, the specific role of microglia upon the onset of ferroptosis has not been fully explored.
Li et al. (2018) used TEM to observe mitochondria characteristics of ferroptosis in axons of oligodendrocytes. Subsequently, they further systematically presented ample evidence of ferroptosis in oligodendrocytes after ICH (Shen et al., 2022). Dexpramipexole has also been reported to attenuate white matter damage caused by oligodendrocyte ferroptosis in mice after ICH, thereby improving spatial localization and motor function (Wang B. et al., 2022). Moreover, ferroptosis has also been reported to be involved in white matter damage after spinal cord injury, which can be suppressed by the application of Fer-1 (Chen et al., 2022).
In an Alzheimer’s disease study, ferroptosis was shown to occur in astrocytes and is positively regulated by NADPH oxidase 4-mediated impairment of mitochondrial metabolism (Park et al., 2021). Another study identified ferroptosis in cultured astrocytes. Although the validation evidence for ferroptosis in this in vitro experiment is not comprehensive (Li S. et al., 2021), it still provides an outlook for the existence of ferroptosis in astrocytes.
In addition, some studies or reviews have proposed the feasibility and essential role of ferroptosis in endothelial cells (Qin et al., 2021). However, evidence for it, especially in the central nervous system (CNS), still appears to be lacking. In summary, studying the ferroptosis of cells other than neurons holds great promise in the CNS. The lack of practical means makes this line of research somewhat challenging to identify the specific ferroptotic cell localization. We have presented the current studies for various types of ferroptosis in the CNS in Table 1.

Table 1. Ferroptosis in different cell types in the central nervous system (CNS).
Developing clinically effective drugs against ferroptosis is a crucial goal of our research. The elucidation of the molecular mechanism of ferroptosis after hemorrhagic new stroke will facilitate the development of novel and more efficacious drugs. There is electron microscopic evidence that ferrotropis existed earlier in ICH than necrossis and autophagy (Li et al., 2018). However, a significant challenge in this area of research involves the failure to define the contribution of ferroptosis to neurological impairment after hemorrhagic stroke and the difficulty of determining which specific period after hemorrhagic stroke plays a vital role in the ferroptosis effect. The crucial questions remain: faced with various PCDs after injury, which one aspect of research should we focus on? Is it possible to target in combination some of these forms of death to achieve the highest therapeutic effect? Are the dominant PCDs different across periods after hemorrhagic stroke? In fact, a large amount of basic studies related to ferroptosis are lack of reliable experimental evidence. For example, many articles have not provided evidence of TEM, or arbitrarily concluded that ferroptosis changes only through the changes in the end products of lipid peroxidation. And most of studies have not been repeatedly verified or even can’t be reproduced at all. It has to be admitted that in view of the above problems, there is no treatment based on iron death that can be carried out in a phase I clinical trial. These issues are of paramount importance to be addressed in the clinical translation of preclinical drug research for hemorrhagic stroke. In the face of the above problems, using single cell sequencing technology to classify dying cells in biology may be a direction worth trying. Combining with the pseudotime analysis, we may classify the outcomes of these cells by detecting the marker molecules of PCDs, so as to solve the difficult problems of time point after stroke and proportion of ferroptosis in PCDs.
FP and CW designed and wrote the manuscript. WX and JD contributed to the literature searches and analyses. FP and WX critically revised the manuscript. All authors approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alim, I., Caulfield, J. T., Chen, Y., Swarup, V., Geschwind, D. H., Ivanova, E., et al. (2019). Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177, 1262.e–1279.e. doi: 10.1016/j.cell.2019.03.032
Ayer, R. E., Sugawara, T., Chen, W., Tong, W., and Zhang, J. H. (2008). Melatonin decreases mortality following severe subarachnoid hemorrhage. J. Pineal Res. 44, 197–204. doi: 10.1111/j.1600-079X.2007.00508.x
Bersuker, K., Hendricks, J. M., Li, Z., Magtanong, L., Ford, B., Tang, P. H., et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. doi: 10.1038/s41586-019-1705-2
Camaschella, C., Nai, A., and Silvestri, L. (2020). Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 105, 260–272. doi: 10.3324/haematol.2019.232124
Cao, Y., Li, Y., He, C., Yan, F., Li, J., Xu, H., et al. (2021). Selective ferroptosis inhibitor liproxstatin-1 attenuates neurological deficits and neuroinflammation after subarachnoid hemorrhage. Neurosci. Bull. 37, 535–549. doi: 10.1007/s12264-020-00620-5
Chang, C. F., Cho, S. and Wang, J. (2014). (-)-Epicatechin protects hemorrhagic brain via synergistic Nrf2 pathways. Ann. Clin. Transl. Neurol. 1, 258–271. doi: 10.1002/acn3.54
Chen, B., Chen, Z., Liu, M., Gao, X., Cheng, Y., Wei, Y., et al. (2019). Inhibition of neuronal ferroptosis in the acute phase of intracerebral hemorrhage shows long-term cerebroprotective effects. Brain Res. Bull. 153, 122–132. doi: 10.1016/j.brainresbull.2019.08.013
Chen, B., Wang, H., Lv, C., Mao, C., and Cui, Y. (2021). Long non-coding RNA H19 protects against intracerebral hemorrhage injuries via regulating microRNA-106b-5p/acyl-CoA synthetase long chain family member 4 axis. Bioengineered 12, 4004–4015. doi: 10.1080/21655979.2021.1951070
Chen, D., Fan, Z., Rauh, M., Buchfelder, M., Eyupoglu, I. Y., and Savaskan, N. (2017). ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 36, 5593–5608. doi: 10.1038/onc.2017.146
Chen, Y., Zuliyaer, T., Liu, B., Guo, S., Yang, D., Gao, F., et al. (2022). Sodium selenite promotes neurological function recovery after spinal cord injury by inhibiting ferroptosis. Neural. Regen. Res. 17, 2702–2709. doi: 10.4103/1673-5374.314322
Çolakoğlu, M., Tunçer, S., Banerjee, S. (2018). , Emerging cellular functions of the lipid metabolizing enzyme 15-Lipoxygenase-1. Cell Prolif. 51:e12472. doi: 10.1111/cpr.12472
Cui, Y., Zhang, Y., Zhao, X., Shao, L., Liu, G., Sun, C., et al. (2021a). ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav. Immun. 93, 312–321. doi: 10.1016/j.bbi.2021.01.003
Cui, Y., Zhang, Z., Zhou, X., Zhao, Z., Zhao, R., Xu, X., et al. (2021b). Microglia and macrophage exhibit attenuated inflammatory response and ferroptosis resistance after RSL3 stimulation via increasing Nrf2 expression. J. Neuroinflammation 18:249. doi: 10.1186/s12974-021-02231-x
Dai, E., Zhang, W., Cong, D., Kang, R., Wang, J., Tang, D., et al. (2020). AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem. Biophys. Res. Commun. 523, 966–971. doi: 10.1016/j.bbrc.2020.01.066
Ding, C., Ding, X., Zheng, J., Wang, B., Li, Y., Xiang, H., et al. (2020). miR-182-5p and miR-378a-3p regulate ferroptosis in I/R-induced renal injury. Cell Death Dis. 11:929. doi: 10.1038/s41419-020-03135-z
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Doll, S., Freitas, F. P., Shah, R., Aldrovandi, M., Silva, M. C., Ingold, I., et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. doi: 10.1038/s41586-019-1707-0
Dolma, S., Lessnick, S. L., Hahn, W. C., and Stockwell, B. R. (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296. doi: 10.1016/S1535-6108(03)00050-3
Doria, J. W., and Forgacs, P. B. (2019). Incidence, implications, and management of seizures following ischemic and hemorrhagic stroke. Curr. Neurol. Neurosci. Rep. 19:37. doi: 10.1007/s11910-019-0957-4
Du, G. J., Lu, G., Zheng, Z. Y., Poon, W. S., and Wong, K. C. (2016). Endovascular perforation murine model of subarachnoid hemorrhage. Acta Neurochir. Suppl. 121, 83–88. doi: 10.1007/978-3-319-18497-5_14
Duan, L., Zhang, Y., Yang, Y., Su, S., Zhou, L., Lo, P., et al. (2021). Baicalin inhibits ferroptosis in intracerebral hemorrhage. Front. Pharmacol. 12:629379. doi: 10.3389/fphar.2021.629379
Fang, Y., Gao, S., Wang, X., Cao, Y., Lu, J., Chen, S., et al. (2020). Programmed cell deaths and potential crosstalk with blood-brain barrier dysfunction after hemorrhagic stroke. Front. Cell Neurosci. 14:68. doi: 10.3389/fncel.2020.00068
Farr, A. C., and Xiong, M. P. (2021). Challenges and opportunities of deferoxamine delivery for treatment of Alzheimer’s Disease, Parkinson’s Disease, and intracerebral hemorrhage. Mol. Pharm. 18, 593–609. doi: 10.1021/acs.molpharmaceut.0c00474
Feng, H., Liu, Q., Deng, Z., Li, H., Zhang, H., Song, J., et al. (2022). Human umbilical cord mesenchymal stem cells ameliorate erectile dysfunction in rats with diabetes mellitus through the attenuation of ferroptosis. Stem Cell Res. Ther. 13:450. doi: 10.1186/s13287-022-03147-w
Fraternale, A., Paoletti, M. F., Casabianca, A., Nencioni, L., Garaci, E., Palamara, A. T., et al. (2009). GSH and analogs in antiviral therapy. Mol. Aspects Med. 30, 99–110. doi: 10.1016/j.mam.2008.09.001
Fu, P., Liu, J., Bai, Q., Sun, X., Yao, Z., Liu, L., et al. (2020). Long-term outcomes of monascin - a novel dual peroxisome proliferator-activated receptor γ/nuclear factor-erythroid 2 related factor-2 agonist in experimental intracerebral hemorrhage. Ther. Adv. Neurol. Disord. 13:1756286420921083. doi: 10.1177/1756286420921083
Fujii, M., Yan, J., Rolland, W. B., Soejima, Y., Caner, B., and Zhang, J. H. (2013). Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Transl. Stroke Res. 4, 432–446. doi: 10.1007/s12975-013-0257-2
Gaasch, J. A., Lockman, P. R., Geldenhuys, W. J., Allen, D. D., and Schyf, C. J. (2007). Brain iron toxicity: Differential responses of astrocytes, neurons, and endothelial cells. Neurochem. Res. 32, 1196–1208. doi: 10.1007/s11064-007-9290-4
Gao, S., Liu, J., Han, Y., Deji, Q., Zhaba, W., Deng, H., et al. (2020). Neuroprotective role of glutathione peroxidase 4 in experimental subarachnoid hemorrhage models. Life Sci. 257:118050. doi: 10.1016/j.lfs.2020.118050
Gao, S., Zhou, L., Lu, J., Fang, Y., Wu, H., Xu, W., et al. (2022). Cepharanthine attenuates early brain injury after subarachnoid hemorrhage in mice via inhibiting 15-Lipoxygenase-1-Mediated microglia and endothelial cell ferroptosis. Oxid. Med. Cell Longev. 2022:4295208. doi: 10.1155/2022/4295208
Grysiewicz, R. A., Thomas, K., and Pandey, D. K. (2008). Epidemiology of ischemic and hemorrhagic stroke: Incidence, prevalence, mortality, and risk factors. Neurol. Clin. 26, 871–895. doi: 10.1016/j.ncl.2008.07.003
Hall, E. D., and Travis, M. A. (1988). Effects of the nonglucocorticoid 21-aminosteroid U74006F on acute cerebral hypoperfusion following experimental subarachnoid hemorrhage. Exp. Neurol. 102, 244–248. doi: 10.1016/0014-4886(88)90100-8
Han, R., Wan, J., Han, X., Ren, H., Falck, J. R., Munnuri, S., et al. (2021). 20-HETE participates in intracerebral hemorrhage-induced acute injury by promoting cell ferroptosis. Front. Neurol. 12:763419. doi: 10.3389/fneur.2021.763419
Handa, Y., Kaneko, M., Takeuchi, H., Tsuchida, A., Kobayashi, H., and Kubota, T. (2000). Effect of an antioxidant, ebselen, on development of chronic cerebral vasospasm after subarachnoid hemorrhage in primates. Surg. Neurol. 53, 323–329. doi: 10.1016/S0090-3019(00)00168-3
Hanley, D. F., Thompson, R. E., Rosenblum, M., Yenokyan, G., Lane, K., McBee, N., et al. (2019). Efficacy and safety of minimally invasive surgery with thrombolysis in intracerebral haemorrhage evacuation (MISTIE III): A randomised, controlled, open-label, blinded endpoint phase 3 trial. Lancet 393, 1021–1032. doi: 10.1016/S0140-6736(19)30195-3
Hou, L., Huang, R., Sun, F., Zhang, L., and Wang, Q. (2019). NADPH oxidase regulates paraquat and maneb-induced dopaminergic neurodegeneration through ferroptosis. Toxicology 417, 64–73. doi: 10.1016/j.tox.2019.02.011
Huang, Y., Wu, H., Hu, Y., Zhou, C., Wu, J., Wu, Y., et al. (2022). Puerarin attenuates oxidative stress and ferroptosis via AMPK/PGC1α/Nrf2 pathway after subarachnoid hemorrhage in rats. Antioxidants 11:1259 doi: 10.3390/antiox11071259
Imai, T., Tsuji, S., Matsubara, H., Ohba, T., Sugiyama, T., Nakamura, S., et al. (2021). Deferasirox, a trivalent iron chelator, ameliorates neuronal damage in hemorrhagic stroke models. Naunyn Schmiedebergs Arch. Pharmacol. 394, 73–84. doi: 10.1007/s00210-020-01963-6
Jhelum, P., Santos-Nogueira, E., Teo, W., Haumont, A., Lenoël, I., Stys, P. K., et al. (2020). Ferroptosis mediates cuprizone-induced loss of oligodendrocytes and demyelination. J. Neurosci. 40, 9327–9341. doi: 10.1523/JNEUROSCI.1749-20.2020
Jin, Z., Gao, W., Liao, S., Yu, T., Shi, Q., Yu, S., et al. (2021). Paeonol inhibits the progression of intracerebral haemorrhage by mediating the HOTAIR/UPF1/ACSL4 axis. ASN Neuro. 13:17590914211010647. doi: 10.1177/17590914211010647
Kagan, V. E., Mao, G., Qu, F., Angeli, J. P., Doll, S., Croix, C. S., et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90. doi: 10.1038/nchembio.2238
Kamezaki, T., Yanaka, K., Nagase, S., Fujita, K., Kato, N., and Nose, T. (2002). Increased levels of lipid peroxides as predictive of symptomatic vasospasm and poor outcome after aneurysmal subarachnoid hemorrhage. J Neurosurg 97, 1302–1305. doi: 10.3171/jns.2002.97.6.1302
Kapralov, A. A., Yang, Q., Dar, H. H., Tyurina, Y. Y., Anthonymuthu, T. S., Kim, R., et al. (2020). Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 16, 278–290. doi: 10.1038/s41589-019-0462-8
Karuppagounder, S. S., Alin, L., Chen, Y., Brand, D., Bourassa, M. W., Dietrich, K., et al. (2018). N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E(2) to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann. Neurol. 84, 854–872. doi: 10.1002/ana.25356
Katsarou, A., and Pantopoulos, K. (2020). Basics and principles of cellular and systemic iron homeostasis.. Mol. Aspects Med. 75:100866. doi: 10.1016/j.mam.2020.100866
Koppula, P., Zhuang, L., and Gan, B. (2021). Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 12, 599–620. doi: 10.1007/s13238-020-00789-5
Krafft, P. R., Rolland, W. B., Duris, K., Lekic, T., Campbell, A., Tang, J., et al. (2012). Modeling intracerebral hemorrhage in mice: Injection of autologous blood or bacterial collagenase. J. Vis. Exp. 67:e4289. doi: 10.3791/4289
Kusaka, G., Ishikawa, M., Nanda, A., Granger, D. N., and Zhang, J. H. (2004). Signaling pathways for early brain injury after subarachnoid hemorrhage. J. Cereb Blood Flow Metab. 24, 916–925. doi: 10.1097/01.WCB.0000125886.48838.7E
Laird, M. D., Wakade, C., Jr, C. H., and Dhandapani, K. M. (2008). Hemin-induced necroptosis involves glutathione depletion in mouse astrocytes. Free Radic. Biol. Med. 45, 1103–1114. doi: 10.1016/j.freeradbiomed.2008.07.003
Lee, J., Keep, R. F., He, Y., Sagher, O., Hua, Y., and Xi, G. (2010). Hemoglobin and iron handling in brain after subarachnoid hemorrhage and the effect of deferoxamine on early brain injury. J. Cereb Blood Flow Metab. 30, 1793–1803. doi: 10.1038/jcbfm.2010.137
Lei, B., Rolland, W. B., Duris, K., Lekic, T., Campbell, A., Tang, J., et al. (2014). Intrastriatal injection of autologous blood or clostridial collagenase as murine models of intracerebral hemorrhage. J. Vis. Exp. 89, 1–7. doi: 10.3791/51439
Lei, G., Zhuang, L., and Gan, B. (2022). Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 22, 381–396. doi: 10.1038/s41568-022-00459-0
Lekic, T., Hartman, R., Rojas, H., Manaenko, A., Chen, W., Ayer, R., et al. (2010). Protective effect of melatonin upon neuropathology, striatal function, and memory ability after intracerebral hemorrhage in rats. J. Neurotrauma 27, 627–637. doi: 10.1089/neu.2009.1163
Li, D., and Li, Y. (2020). The interaction between ferroptosis and lipid metabolism in cancer. Signal Transd. Target Ther. 5:108. doi: 10.1038/s41392-020-00216-5
Li, F., Long, H., Zhou, Z., Luo, H., Xu, S., and Gao, L. (2022). System X(c) (-)/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front. Pharmacol. 13:910292. doi: 10.3389/fphar.2022.910292
Li, J., Li, M., Ge, Y., Chen, J., Ma, J., Wang, C., et al. (2022). β-amyloid protein induces mitophagy-dependent ferroptosis through the CD36/PINK/PARKIN pathway leading to blood-brain barrier destruction in Alzheimer’s disease. Cell Biosci. 12:69. doi: 10.1186/s13578-022-00807-5
Li, N., Worthmann, H., Deb, M., Chen, S., and Weissenborn, K. (2011). Nitric oxide (NO) and asymmetric dimethylarginine (ADMA): Their pathophysiological role and involvement in intracerebral hemorrhage. Neurol. Res. 33, 541–548. doi: 10.1179/016164111X13007856084403
Li, Q., Han, X., Lan, X., Gao, Y., Wan, J., Durham, F., et al. (2017a). Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2:e90777. doi: 10.1172/jci.insight.90777
Li, Q., Li, Q., Jia, J., Sun, Q., Zhou, H., Jin, W., et al. (2019). Baicalein exerts neuroprotective effects in fecl(3)-induced posttraumatic epileptic seizures via suppressing ferroptosis. Front. Pharmacol. 10:638. doi: 10.3389/fphar.2019.00638
Li, Q., Wan, J., Lan, X., Han, X., Wang, Z., and Wang, J. (2017b). Neuroprotection of brain-permeable iron chelator VK-28 against intracerebral hemorrhage in mice. J. Cereb Blood Flow Metab. 37, 3110–3123. doi: 10.1177/0271678X17709186
Li, Q., Weiland, A., Chen, X., Lan, X., Han, X., Durham, F., et al. (2018). Ultrastructural characteristics of neuronal death and white matter injury in mouse brain tissues after intracerebral hemorrhage: Coexistence of ferroptosis. autophagy, and necrosis. Front. Neurol. 9:581. doi: 10.3389/fneur.2018.00581
Li, S., Zhou, C., Zhu, Y., Chao, Z., Sheng, Z., Zhang, Y., et al. (2021). Ferrostatin-1 alleviates angiotensin II (Ang II)- induced inflammation and ferroptosis in astrocytes. Int. Immunopharmacol. 90:107179. doi: 10.1016/j.intimp.2020.107179
Li, Y., Liu, Y., Wu, P., Tian, Y., Liu, B., Wang, J., et al. (2021). Inhibition of ferroptosis alleviates early brain injury after subarachnoid hemorrhage in vitro and in vivo via reduction of lipid peroxidation. Cell Mol. Neurobiol. 41, 263–278. doi: 10.1007/s10571-020-00850-1
Lin, C., Jeng, A. Y., Howng, S., and Kwan, A. (2004). Endothelin and subarachnoid hemorrhage-induced cerebral vasospasm: Pathogenesis and treatment. Curr. Med. Chem. 11, 1779–1791. doi: 10.2174/0929867043364919
Liu, L., Li, Y., Cao, D., Qiu, S., Li, Y., Jiang, C., et al. (2021). SIRT3 inhibits gallbladder cancer by induction of AKT-dependent ferroptosis and blockade of epithelial-mesenchymal transition. Cancer Lett. 510, 93–104. doi: 10.1016/j.canlet.2021.04.007
Liu, P., Feng, Y., Li, H., Chen, X., Wang, G., Xu, S., et al. (2020). Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol. Biol. Lett. 25:10. doi: 10.1186/s11658-020-00205-0
Liu, Z., Zhou, Z., Ai, P., Zhang, C., Chen, J., and Wang, Y. (2022). Astragaloside IV attenuates ferroptosis after subarachnoid hemorrhage via Nrf2/HO-1 signaling pathway. Front. Pharmacol. 13:924826. doi: 10.3389/fphar.2022.924826
Louandre, C., Ezzoukhry, Z., Godin, C., Barbare, J., Mazière, J., Chauffert, B., et al. (2013). Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 133, 1732–1742. doi: 10.1002/ijc.28159
Lu, H., Zhang, D., Chen, H., Lin, Y., Hang, C., Yin, H., et al. (2009). N-acetylcysteine suppresses oxidative stress in experimental rats with subarachnoid hemorrhage. J. Clin. Neurosci. 16, 684–688. doi: 10.1016/j.jocn.2008.04.021
Luchetti, F., Carloni, S., Nasoni, M. G., Reiter, R. J., and Balduini, W. (2023). Melatonin, tunneling nanotubes, mesenchymal cells, and tissue regeneration. Neural. Regen. Res. 18, 760–762. doi: 10.4103/1673-5374.353480
Luo, Z., Harada, T., London, S., Gajdusek, C., and Mayberg, M. R. (1995). Antioxidant and iron-chelating agents in cerebral vasospasm. Neurosurgery 37, 1154–8;discussion1158–9. doi: 10.1097/00006123-199512000-00015
Ma, H., Liu, Y., Miao, Z., Cheng, S., Zhu, Y., Wu, Y., et al. (2022). Neratinib inhibits proliferation and promotes apoptosis of acute myeloid leukemia cells by activating autophagy-dependent ferroptosis. Drug Dev. Res. 83, 1641 1653. doi: 10.1002/ddr.21983
Magid-Bernstein, J., Girard, R., Polster, S., Srinath, A., Romanos, S., Awad, I. A., et al. (2022). Cerebral hemorrhage: Pathophysiology. Treatment, and future directions. Circ Res 130, 1204–1229. doi: 10.1161/CIRCRESAHA.121.319949
Mendelow, A. D., Gregson, B. A., Rowan, E. N., Murray, G. D., Gholkar, A., Mitchell, P. M., et al. (2013). Early surgery versus initial conservative treatment in patients with spontaneous supratentorial lobar intracerebral haematomas (STICH II): A randomised trial. Lancet 382, 397–408. doi: 10.1016/S0140-6736(13)60986-1
Miriyala, S., Thippakorn, C., Chaiswing, L., Xu, Y., Noel, T., Tovmasyan, A., et al. (2016). Novel role of 4-hydroxy-2-nonenal in AIFm2-mediated mitochondrial stress signaling. Free Radic. Biol. Med. 91, 68–80. doi: 10.1016/j.freeradbiomed.2015.12.002
Mishima, E., and Conrad, M. (2022). Nutritional and metabolic control of ferroptosis. Annu. Rev. Nutr. 42, 275–309. doi: 10.1146/annurev-nutr-062320-114541
Mou, Y., Wang, J., Wu, J., He, D., Zhang, C., Duan, C., et al. (2019). Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 12:34. doi: 10.1186/s13045-019-0720-y
Nakamura, T., Keep, R. F., Hua, Y., Schallert, T., Hoff, J. T., and Xi, G. (2004). Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J. Neurosurg. 100, 672–678. doi: 10.3171/jns.2004.100.4.0672
Nakamura, T., Xi, G., Park, J., Hua, Y., Hoff, J. T., and Keep, R. F. (2005). Holo-transferrin and thrombin can interact to cause brain damage. Stroke 36, 348–352. doi: 10.1161/01.STR.0000153044.60858.1b
Nobuta, H., Yang, N., Han, Y., Marro, S. G., Sabeur, K., and Chavali, M. (2019). Oligodendrocyte death in pelizaeus-merzbacher disease is rescued by iron chelation. Cell Stem Cell 25, 531–541. doi: 10.1016/j.stem.2019.09.003
O’Connell, D. M., and Watson, I. D. (2003). Definitive angiographic detection of subarachnoid haemorrhage compared with laboratory assessment of intracranial bleed in CT-negative patients. Ann. Clin. Biochem. 40(Pt 3), 269–273. doi: 10.1258/000456303321610592
Park, M. W., Cha, H. W., Kim, J., Kim, J. H., Yang, H., Yoon, S., et al. (2021). NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. 41:101947. doi: 10.1016/j.redox.2021.101947
Peng, C., Fu, X., Wang, K., Chen, L., Luo, B., Huang, N., et al. (2022). Dauricine alleviated secondary brain injury after intracerebral hemorrhage by upregulating GPX4 expression and inhibiting ferroptosis of nerve cells. Eur. J. Pharmacol. 914:174461. doi: 10.1016/j.ejphar.2021.174461
Qin, X., Zhang, J., Wang, B., Xu, G., Yang, X., Zou, Z., et al. (2021). Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy 17, 4266–4285. doi: 10.1080/15548627.2021.1911016
Qu, W., Cheng, Y., Peng, W., Wu, Y., Rui, T., Luo, C., et al. (2022). Targeting iNOS alleviates early brain injury after experimental subarachnoid hemorrhage via promoting ferroptosis of M1 microglia and reducing neuroinflammation. Mol. Neurobiol. 59, 3124–3139. doi: 10.1007/s12035-022-02788-5
Qu, X., Liang, T., Wu, D., Lai, N., Deng, R., Ma, C., et al. (2021). Acyl-CoA synthetase long chain family member 4 plays detrimental role in early brain injury after subarachnoid hemorrhage in rats by inducing ferroptosis. CNS Neurosci. Ther. 27, 449–463. doi: 10.1111/cns.13548
Reed, A., Ichu, T., Milosevich, N., Melillo, B., Schafroth, M. A., Otsuka, Y., et al. (2022). LPCAT3 inhibitors remodel the polyunsaturated phospholipid content of human cells and protect from ferroptosis. ACS Chem. Biol. 17, 1607–1618. doi: 10.1021/acschembio.2c00317
Ren, S., Chen, Y., Wang, L., and Wu, G. (2022). Neuronal ferroptosis after intracerebral hemorrhage. Front. Mol. Biosci. 9:966478. doi: 10.3389/fmolb.2022.966478
Rodgers, S. J., Jones, E., Mitchell, C. A., and McGrath, M. J. (2022). Sequential conversion of PtdIns3P to PtdIns(3,5)P(2) via endosome maturation couples nutrient signaling to lysosome reformation and basal autophagy. Autophagy. doi: 10.1080/15548627.2022.2124499 [Epub ahead of print].
Sakaki, S., Kuwabara, H., and Ohta, S. (1986). Biological defence mechanism in the pathogenesis of prolonged cerebral vasospasm in the patients with ruptured intracranial aneurysms. Stroke 17, 196–202. doi: 10.1161/01.STR.17.2.196
Sekerdag, E., Solaroglu, I., and Gursoy-Ozdemir, Y. (2018). Cell death mechanisms in stroke and novel molecular and cellular treatment options. Curr. Neuropharmacol. 16, 1396–1415. doi: 10.2174/1570159X16666180302115544
Selim, M., Yeatts, S., Goldstein, J. N., Gomes, J., Greenberg, S., Morgenstern, L. B., et al. (2011). Safety and tolerability of deferoxamine mesylate in patients with acute intracerebral hemorrhage. Stroke 42, 3067–3074. doi: 10.1161/STROKEAHA.111.617589
Sha, R., Xu, Y., Yuan, C., Sheng, X., Wu, Z., Peng, J., et al. (2021). Predictive and prognostic impact of ferroptosis-related genes ACSL4 and GPX4 on breast cancer treated with neoadjuvant chemotherapy. EBioMedicine 71:103560. doi: 10.1016/j.ebiom.2021.103560
Shao, Z., Tu, S., and Shao, A. (2019). Pathophysiological mechanisms and potential therapeutic targets in intracerebral hemorrhage. Front. Pharmacol. 10:1079. doi: 10.3389/fphar.2019.01079
Shen, D., Wu, W., Liu, J., Lan, T., Xiao, Z., Gai, K., et al. (2022). Ferroptosis in oligodendrocyte progenitor cells mediates white matter injury after hemorrhagic stroke. Cell Death Dis. 13:259. doi: 10.1038/s41419-022-04712-0
Shen, L., Lin, D., Li, X., Wu, H., Lenahan, C., Pan, Y., et al. (2020). Ferroptosis in acute central nervous system injuries: The future direction? Front. Cell Dev. Biol. 8:594. doi: 10.3389/fcell.2020.00594
Smith, S. L., Scherch, H. M., and Hall, E. D. (1996). Protective effects of tirilazad mesylate and metabolite U-89678 against blood-brain barrier damage after subarachnoid hemorrhage and lipid peroxidative neuronal injury. J. Neurosurg. 84, 229–233. doi: 10.3171/jns.1996.84.2.0229
Stockwell, B. R., Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: A regulated cell death nexus linking metabolism. Redox Biol. Dis. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Sukumari-Ramesh, S., Laird, M. D., Singh, N., Vender, J. R., Jr, C. H., and Dhandapani, K. M. (2010). Astrocyte-derived glutathione attenuates hemin-induced apoptosis in cerebral microvascular cells. Glia 58, 1858–1870. doi: 10.1002/glia.21055
Sun, Y., Berleth, N., Wu, W., Schlütermann, D., Deitersen, J., Stuhldreier, F., et al. (2021). Fin56-induced ferroptosis is supported by autophagy-mediated GPX4 degradation and functions synergistically with mTOR inhibition to kill bladder cancer cells. Cell Death Dis. 12:1028. doi: 10.1038/s41419-021-04306-2
Suzuki, H., Muramatsu, M., Kojima, T., and Taki, W. (2003). Intracranial heme metabolism and cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke 34, 2796–2800. doi: 10.1161/01.STR.0000103743.62248.12
Suzuki, N., Nakamura, T., Imabayashi, S., Ishikawa, Y., Sasaki, T., and Asano, T. (1983). Identification of 5-hydroxy eicosatetraenoic acid in cerebrospinal fluid after subarachnoid hemorrhage. J. Neurochem. 41, 1186–1189. doi: 10.1111/j.1471-4159.1983.tb09071.x
Tao, Q., Qiu, X., Li, C., Zhou, J., Gu, L., Zhang, L., et al. (2022). S100A8 regulates autophagy-dependent ferroptosis in microglia after experimental subarachnoid hemorrhage. Exp. Neurol. 357:114171. doi: 10.1016/j.expneurol.2022.114171
Troadec, M., Ward, D. M., Lo, E., Kaplan, J., and Domenico, I. D. (2010). Induction of FPN1 transcription by MTF-1 reveals a role for ferroportin in transition metal efflux. Blood 116, 4657–4664. doi: 10.1182/blood-2010-04-278614
Tuo, Q., Lei, P., Jackman, K. A., Li, X., Xiong, H., Li, X., et al. (2017). Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 22, 1520–1530. doi: 10.1038/mp.2017.171
Uselis, J. (1970). [Iron-binding capacity of serum proteins and the degree of their saturation by iron in a selected group of shipyard welders]. Pol. Tyg Lek. 25, 755–757.
Utkan, T., Sarioglu, Y., Kaya, T., Akgün, M., Göksel, M., and Solak, O. (1996). Effect of deferoxamine and sympathectomy on vasospasm following subarachnoid hemorrhage. Pharmacology 52, 353–361. doi: 10.1159/000139402
Vogt, A. S., Arsiwala, T., Mohsen, M., Vogel, M., Manolova, V., and Bachmann, M. F. (2021). On iron metabolism and its regulation. Int. J. Mol. Sci. 22:4591. doi: 10.3390/ijms22094591
Wang, B., Zhang, X., Zhong, J., Wang, S., Zhang, C., Li, M., et al. (2022). Dexpramipexole attenuates white matter injury to facilitate locomotion and motor coordination recovery via reducing ferroptosis after intracerebral hemorrhage. Oxid. Med. Cell Longev. 2022:6160701. doi: 10.1155/2022/6160701
Wang, G., Hu, W., Tang, Q., Wang, L., Sun, X., Chen, Y., et al. (2016). Effect comparison of both iron chelators on outcomes. iron deposit, and iron transporters after intracerebral hemorrhage in rats. Mol. Neurobiol. 53, 3576–3585. doi: 10.1007/s12035-015-9302-3
Wang, G., Li, T., Duan, S., Dong, L., Sun, X., and Xue, F. (2018). PPAR-γ promotes hematoma clearance through haptoglobin-hemoglobin-CD163 in a rat model of intracerebral hemorrhage. Behav. Neurol. 2018:7646104. doi: 10.1155/2018/7646104
Wang, G., Yang, Q., Li, G., Wang, L., Hu, W., Tang, Q., et al. (2011). Time course of heme oxygenase-1 and oxidative stress after experimental intracerebral hemorrhage. Acta Neurochir (Wien) 153, 319–325. doi: 10.1007/s00701-010-0750-2
Wang, H., Lin, D., Yu, Q., Li, Z., Lenahan, C., Dong, Y., et al. (2021). A promising future of ferroptosis in tumor therapy. Front. Cell Dev. Biol. 9:629150. doi: 10.3389/fcell.2021.629150
Wang, P., Cui, Y., Liu, Y., Li, Z., Bai, H., Zhao, Y., et al. (2022). Mitochondrial ferritin alleviates apoptosis by enhancing mitochondrial bioenergetics and stimulating glucose metabolism in cerebral ischemia reperfusion. Redox Biol. 57:102475. doi: 10.1016/j.redox.2022.102475
Weiland, A., Wang, Y., Wu, W., Lan, X., Han, X., Li, Q., et al. (2019). Ferroptosis and its role in diverse brain diseases. Mol. Neurobiol. 56, 4880–4893. doi: 10.1007/s12035-018-1403-3
Wenzel, S. E., Tyurina, Y., Zhao, J., Cm, S. C., Dar, H. H., Mao, G., et al. (2017). PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171, 628–641e26. doi: 10.1016/j.cell.2017.09.044
Wu, T. Y., Campbell, B. C., Strbian, D., Yassi, N., Putaala, J., Tatlisumak, T., et al. (2016). Impact of pre-stroke sulphonylurea and metformin use on mortality of intracerebral haemorrhage. Eur. Stroke J. 1, 302–309. doi: 10.1177/2396987316666617
Wu, T., Liang, X., Liu, X., Li, Y., Wang, Y., Kong, L., et al. (2020). Induction of ferroptosis in response to graphene quantum dots through mitochondrial oxidative stress in microglia. Part Fibre Toxicol. 17:30. doi: 10.1186/s12989-020-00363-1
Xu, C., Li, J., Jiang, S., Wan, L., Zhang, X., Xia, L., et al. (2022). CD47 blockade accelerates blood clearance and alleviates early brain injury after experimental subarachnoid hemorrhage. Front. Immunol. 13:823999. doi: 10.3389/fimmu.2022.823999
Xu, Y., Chen, A., Wu, J., Wan, Y., You, M., Gu, X., et al. (2022). Nanomedicine: An emerging novel therapeutic strategy for hemorrhagic stroke. Int. J. Nanomed. 17, 1927–1950. doi: 10.2147/IJN.S357598
Yadav, P., Sharma, P., Sundaram, S., Venkatraman, G., Bera, A. K., and Karunagaran, D. (2021). SLC7A11/xCT is a target of miR-5096 and its restoration partially rescues miR-5096-mediated ferroptosis and anti-tumor effects in human breast cancer cells. Cancer Lett. 522, 211–224. doi: 10.1016/j.canlet.2021.09.033
Yang, G., Qian, C., Zhang, C., Bao, Y., Liu, M., Jiang, F., et al. (2021). Hepcidin attenuates the iron-mediated secondary neuronal injury after intracerebral hemorrhage in rats. Transl. Res. 229, 53–68. doi: 10.1016/j.trsl.2020.09.002
Yang, W. S., and Stockwell, B. R. (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245. doi: 10.1016/j.chembiol.2008.02.010
Yang, W. S., and Stockwell, B. R. (2016). Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 26, 165–176. doi: 10.1016/j.tcb.2015.10.014
Yao, Y., Chen, Z., Zhang, H., Chen, C., Zeng, M., Yunis, J., et al. (2021). Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat. Immunol. 22, 1127–1139. doi: 10.1038/s41590-021-00996-0
Yuan, B., Zhao, X., Shen, J., Chen, S., Huang, H., Zhou, X., et al. (2022). Activation of SIRT1 alleviates ferroptosis in the early brain injury after subarachnoid hemorrhage. Oxid. Med. Cell Longev. 2022:9069825. doi: 10.1155/2022/9069825
Yuan, Y., Yucai, L., Lu, L., Hui, L., Yong, P., and Haiyang, Y. (2022). Acrylamide induces ferroptosis in HSC-T6 cells by causing antioxidant imbalance of the XCT-GSH-GPX4 signaling and mitochondrial dysfunction. Toxicol Lett. 368, 24–32. doi: 10.1016/j.toxlet.2022.08.007
Zhang, H., Ostrowski, R., Jiang, D., Zhao, Q., Liang, Y., Che, X., et al. (2021). Hepcidin promoted ferroptosis through iron metabolism which is associated with DMT1 signaling activation in early brain injury following subarachnoid hemorrhage. Oxid Med. Cell Longev. 2021:9800794. doi: 10.1155/2021/9800794
Zhang, H., Tu, X., Song, S., Liang, R., and Shi, S. (2020). Baicalin reduces early brain injury after subarachnoid hemorrhage in rats. Chin. J. Integr. Med. 26, 510–518. doi: 10.1007/s11655-020-3183-7
Zhang, Y., Kong, Y., Ma, Y., Ni, S., Wikerholmen, T., Xi, K., et al. (2021). Loss of COPZ1 induces NCOA4 mediated autophagy and ferroptosis in glioblastoma cell lines. Oncogene 40, 1425–1439. doi: 10.1038/s41388-020-01622-3
Zhang, Z., Wu, Y., Yuan, S., Zhang, P., Zhang, J., Li, H., et al. (2018). Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 1701, 112–125. doi: 10.1016/j.brainres.2018.09.012
Zhao, F., Hua, Y., He, Y., Keep, R. F., and Xi, G. (2011). Minocycline-induced attenuation of iron overload and brain injury after experimental intracerebral hemorrhage. Stroke 42, 3587–3593. doi: 10.1161/STROKEAHA.111.623926
Zheng, Y., Tan, X., and Cao, S. (2022). The critical role of erythrolysis and microglia/macrophages in clot resolution after intracerebral hemorrhage: A review of the mechanisms and potential therapeutic targets. Cell Mol. Neurobiol. doi: 10.1007/s10571-021-01175-3 [Epub ahead of print].
Zhu, F., Zi, L., Yang, P., Wei, Y., Zhong, R., Wang, Y., et al. (2021). Efficient Iron and ROS Nanoscavengers for brain protection after intracerebral hemorrhage. ACS Appl. Mater Interfaces 13, 9729–9738. doi: 10.1021/acsami.1c00491
Keywords: stroke, intracerebral hemorrhage, subarachnoid hemorrhage, ferroptosis, target, lipid peroxidation
Citation: Pan F, Xu W, Ding J and Wang C (2023) Elucidating the progress and impact of ferroptosis in hemorrhagic stroke. Front. Cell. Neurosci. 16:1067570. doi: 10.3389/fncel.2022.1067570
Received: 12 October 2022; Accepted: 13 December 2022;
Published: 11 January 2023.
Edited by:
Zhen-Ni Guo, First Affiliated Hospital of Jilin University, ChinaReviewed by:
Frederick Colbourne, University of Alberta, CanadaCopyright © 2023 Pan, Xu, Ding and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chencen Wang,  dzExNTA5ODhAMTYzLmNvbQ==
dzExNTA5ODhAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.