
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 16 January 2023
Sec. Cellular Neuropathology
Volume 16 - 2022 | https://doi.org/10.3389/fncel.2022.1036313
This article is part of the Research Topic Crosstalk between Peripheral and Local Immune Response in the Pathophysiology of Stroke and Neurodegeneration Diseases View all 30 articles
 Kaijing Fu1†
Kaijing Fu1† Weilin Xu2†
Weilin Xu2† Cameron Lenahan3
Cameron Lenahan3 Yong Mo1
Yong Mo1 Jing Wen4
Jing Wen4 Teng Deng1
Teng Deng1 Qianrong Huang1
Qianrong Huang1 Fangzhou Guo1
Fangzhou Guo1 Ligen Mo1*
Ligen Mo1* Jun Yan1*‡
Jun Yan1*‡Intracerebral hemorrhage (ICH) is the second-largest stroke subtype and has a high mortality and disability rate. Secondary brain injury (SBI) is delayed after ICH. The main contributors to SBI are inflammation, oxidative stress, and excitotoxicity. Harmful substances from blood and hemolysis, such as hemoglobin, thrombin, and iron, induce SBI. When cells suffer stress, a critical protective mechanism called “autophagy” help to maintain the homeostasis of damaged cells, remove harmful substances or damaged organelles, and recycle them. Autophagy plays a critical role in the pathology of ICH, and its function remains controversial. Several lines of evidence demonstrate a pro-survival role for autophagy in ICH by facilitating the removal of damaged proteins and organelles. However, many studies have found that heme and iron can aggravate SBI by enhancing autophagy. Autophagy and inflammation are essential culprits in the progression of brain injury. It is a fascinating hypothesis that autophagy regulates inflammation in ICH-induced SBI. Autophagy could degrade and clear pro-IL-1β and apoptosis-associated speck-like protein containing a CARD (ASC) to antagonize NLRP3-mediated inflammation. In addition, mitophagy can remove endogenous activators of inflammasomes, such as reactive oxygen species (ROS), inflammatory components, and cytokines, in damaged mitochondria. However, many studies support the idea that autophagy activates microglia and aggravates microglial inflammation via the toll-like receptor 4 (TLR4) pathway. In addition, autophagy can promote ICH-induced SBI through inflammasome-dependent NLRP6-mediated inflammation. Moreover, some resident cells in the brain are involved in autophagy in regulating inflammation after ICH. Some compounds or therapeutic targets that regulate inflammation by autophagy may represent promising candidates for the treatment of ICH-induced SBI. In conclusion, the mutual regulation of autophagy and inflammation in ICH is worth exploring. The control of inflammation by autophagy will hopefully prove to be an essential treatment target for ICH.
Intracerebral hemorrhage (ICH), the second most common type of stroke, is characterized by non-traumatic brain parenchymal hemorrhage (Nobleza, 2021). It causes severe neurological dysfunction with high morbidity and mortality rates, accounting for 15% of strokes (Shao et al., 2019; Zhang and Liu, 2020). ICH-induced secondary brain injury (SBI) occurs because blood components and hemolysates from ruptured blood vessels contribute to brain tissue damage and cell death by activating inflammatory reactions, cytotoxicity, and excitatory toxicity (Zhu et al., 2019). At present, although removing hematomas via a minimally invasive operation can relieve the mechanical compression of hematomas on peripheral brain tissue, there are still no effective treatments for post-hemorrhage-mediated SBI (Shao et al., 2019).
Autophagy, sometimes called “self-eating,” is a cellular process of stress that is dynamic and complex (Yu et al., 2018). It can maintain the stability of the material and energy and ensure cells’ survival in starvation or other types of stimulations, positively contributing to stroke (Ghavami et al., 2014; Zhang and Liu, 2020). Under normal circumstances, autophagy occurs at a basic level in the brain. It is involved in many physiological activities in most cells, such as cell development and death, immune function decline, and anti-aging mechanism (Wang and Zhang, 2019). However, autophagic activity is significantly enhanced in ICH. In addition, iron, heme, thrombin, and other harmful substances play a role in ICH-induced SBI by regulating autophagy. Neuroinflammation, which plays a critical role in ICH-induced SBI, exacerbates the mass effect by increasing the permeability of the blood-brain barrier (BBB) around the hematoma, leading to cell death. Meanwhile, the cells that die release inflammatory mediators that further aggravate neuroinflammation.
Therefore, reduced neuroinflammation is particularly important for treating ICH (Xiao L. et al., 2020). Is there a relationship between autophagy and inflammation in ICH? It remains a fascinating topic for research that the mechanisms are interrelated but have yet to be fully explained. Obviously, multitarget neuroprotective compounds will be a promising strategy to alleviate ICH-induced SBI. Therefore, in this article, we summarized the fundamental mechanisms of autophagy following ICH and the interrelationship between autophagy and inflammation. We also summarize in detail the potential compounds and therapeutic targets of autophagy in regulating inflammation.
Intracerebral hemorrhage usually occurs deep in the brain (basal ganglia and thalamus) (Zhang et al., 2020; O’Carroll et al., 2021), and 30% of patients with ICH may develop hematoma dilation within the first 6 h after ICH (Tschoe et al., 2020). The leading cause is chronically elevated blood pressure (hypertension) and cerebral amyloid angiopathy (Nobleza, 2021). The pathophysiology of ICH is extremely complex, mainly including primary brain injury (PBI) caused by hematoma-related pathological reactions. However, the hematoma is the leading cause of PBI after ICH. It can aggravate SBI, leading to severe neurological dysfunction and even death (Zhang and Liu, 2020). Although these injury pathways are distinct in pathophysiology, the mechanisms by which they injure the brain parenchyma overlap (Magid-Bernstein et al., 2022). For example, inflammation-mediated inflammatory factors and leukocyte infiltration can also lead to BBB breakdown (mainly endothelial cell damage and deformation) after ICH. BBB disruption and increased permeability can also lead to further perihematomal edema, thus forming a vicious circle (Figure 1).
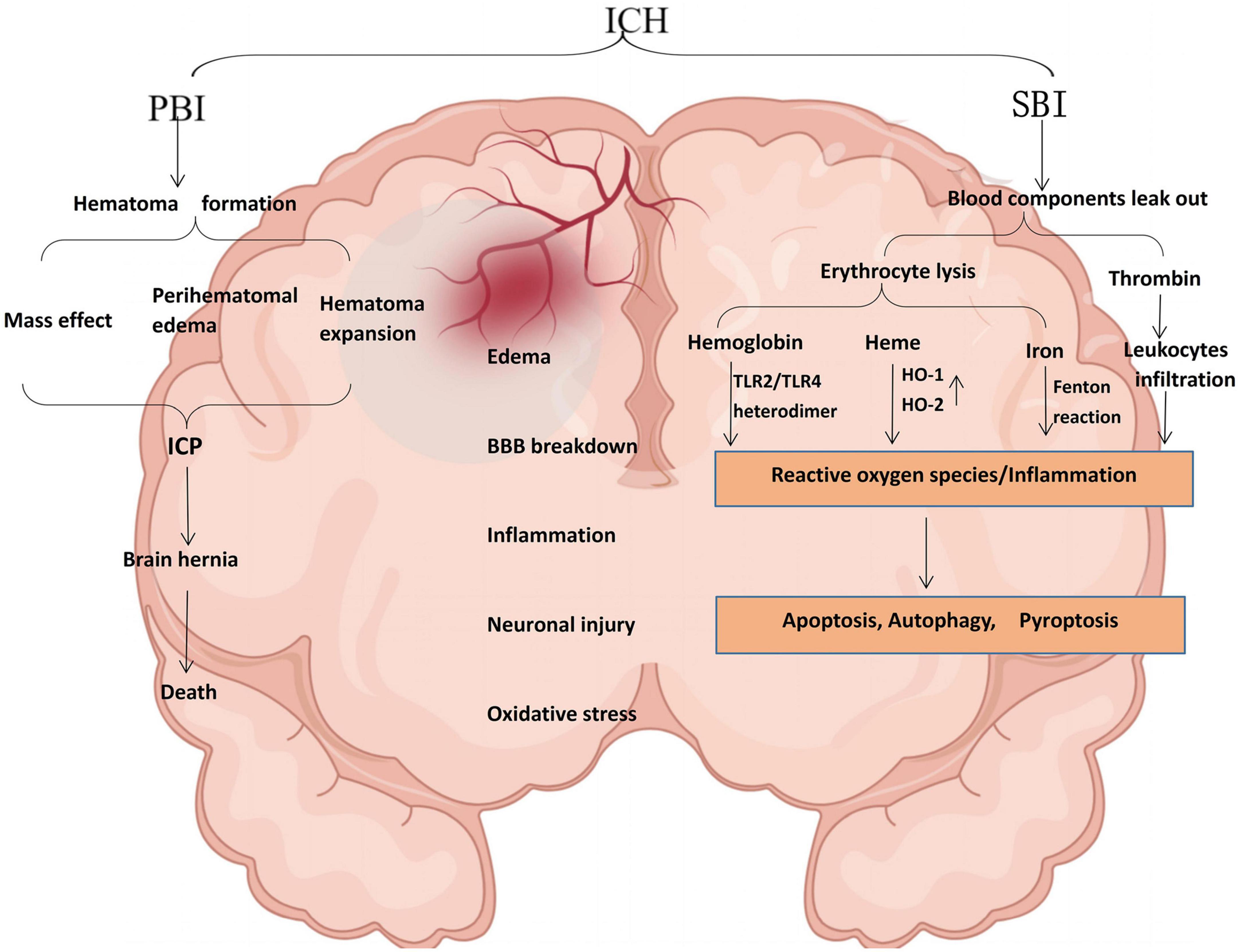
Figure 1. Schematic illustration of pathophysiology after ICH. PBI and SBI are not performed singly, leading to edema, BBB disruption, inflammation, neuronal damage, and oxidative stress. Thrombin can induce significant neuroinflammation in microglia. Most extravasated erythrocytes are trapped in the hematoma and phagocytosed or lysed by microglia/macrophages. In ICH, hemoglobin is a key factor in the inflammation of damaged erythrocyte leakage and is a potent activator of inflammation. Hemoglobin induces proinflammatory cytokine production in microglia through TLR2/TLR4 heterodimer. Heme is partially degraded by HO-1 in macrophages/microglia or HO-2 in neurons to form ferrous iron, carbon monoxide, and biliverdin. The released iron can produce a large amount of ROS through the Fenton reaction and cause oxidative stress damage to nearby tissues. TLR2/TLR4, toll-like receptor 2/4; HO-1, heme-oxygenase-1; ROS, reactive oxygen species (by Figdraw).
Occurring immediately within the first few days after bleeding, PBI is a direct injury caused by the acute mass effect of an intracerebral hematoma, which causes brain tissue deformation and intracranial pressure. PBI in ICH is characterized by hematoma formation and expansion, the mass effect that leads to greater intracranial pressure (Bautista et al., 2021). The severity of ICH is mainly affected by the site of the hemorrhage and the amount of bleeding. When the blood loss is extensive, the compression of the tissue intensifies. It causes intracranial hypertension (ICP), leading to mechanical damage to adjacent tissue and decreasing cerebral perfusion, eventually bringing severe irreversible neurological dysfunction to the tissue. In addition, mass effects may lead to midline displacement, brain herniation formation, and even death (Magid-Bernstein et al., 2022).
Previous studies have shown that SBI after ICH is a vital factor leading to neurological dysfunction and a significant determinant of the poor prognosis of patients (Shao et al., 2019). When blood components and damage-associated molecular patterns (DAMPs) are released from necrotic and damaged tissues, injury pathways are activated, including inflammation, blood cytotoxicity, and oxidative stress (Aronowski and Zhao, 2011). SBI after ICH is exceptionally complex, and the most severe consequences of SBI are neuronal cell death or endothelial damage (and subsequent vasogenic edema) (Duris et al., 2018). In addition, hemoglobin, thrombin, heme, and iron cause BBB hyperpermeability after ICH (Bian et al., 2022; Xia et al., 2022).
There are three types of autophagy, namely, macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy, also known as autophagy (‘self-eating’) is a major pathway involved in cytoplasmic content recycling and degradation in autophagosomes. Microautophagy can phagocytose cellular solutes into lysosomes and degrade them through the formation of characteristic invaginations of the lysosomal membrane. Unlike macroautophagy, microautophagy does not require the formation of autophagosomes, and its substrates are directly phagocytic and degraded by lysosomes. Chaperone-mediated autophagy is a highly selective pathway recognized only by soluble proteins containing chaperone recognition sites, such as the heat shock protein-70 (HSP70) complex (Kanno et al., 2022). Among the three kinds of autophagy, macroautophagy is the most widely studied and the most characteristic of autophagy. In general terms, “autophagy” refers to macroautophagy. The autophagy mentioned in this review is all macroautophagy (hereinafter, “autophagy”).
In mammalian cells, autophagy can also be classified as selective and non-selective (Li et al., 2021d). Selective autophagy is a major process in maintaining cellular homeostasis, in which lysosomes recognize and degrade cargo through the activity of selective autophagy receptors (Lamark and Johansen, 2021; Xu W. et al., 2021). Selective autophagy can be divided into the following categories according to the targeted organelles or particles such as some damaged organelles: mitophagy (impaired mitochondria), reticulophagy (endoplasmic reticulum), xenophagy (pathogens), or aggrephagy (misfolded proteins) (Faruk et al., 2021). Non-selective autophagy is a process in which cytoplasmic components are non-selectively wrapped by bilayer phagosomes to form autophagosomes and transported to lysosomes for degradation and recycling as essential metabolic substrates. It is a physiological reaction to maintain nutrient supplies by stimulating hunger and other behaviors (Li et al., 2021d).
As shown in Figure 2, the canonical autophagy process involves the sequential and selective recruitment of ATG proteins that can be divided into five sequential steps, including (1) the initiation and nucleation of phagophores; (2) double membrane formation and elongation; (3) the maturation and completion of autophagosomes; (4) lysosomes’ integration with autophagosomes to form autolysosomes; and (5) the degradation of intra-autophagosomal contents by lysosomal enzymes (Yu et al., 2018). In the autophagolysosome, many autophagy-related (ATG) proteins can regulate autophagy, playing an extremely crucial role in the formation of autophagy (Lõrincz and Juhász, 2020). When cells are subjected to various stimuli, such as starvation and energy deprivation, or the activity of the mammalian target of rapamycin (mTOR) complex is inhibited, the initiation of autophagy is mediated by the Unc-51-like autophagy-activating kinase (ULK1) complex, consisting of ULK1/2, Atg13, FIP200, and Atg101 (Jung et al., 2009; Mizushima, 2010; Zachari and Ganley, 2017). Nutritional conditions regulate this complex: When nutrients are plentiful, mTOR phosphorylates ULK1 and Atg13, inactivating the ULK1 complex (Wang and Zhang, 2019). When cells are starved or hypoxic, mTOR activity is inhibited, leading to dephosphorylation and the activation of the ULK1 complex (Fang et al., 2018). Therefore, the ULK1 complex completes the crucial step of inducing autophagy.
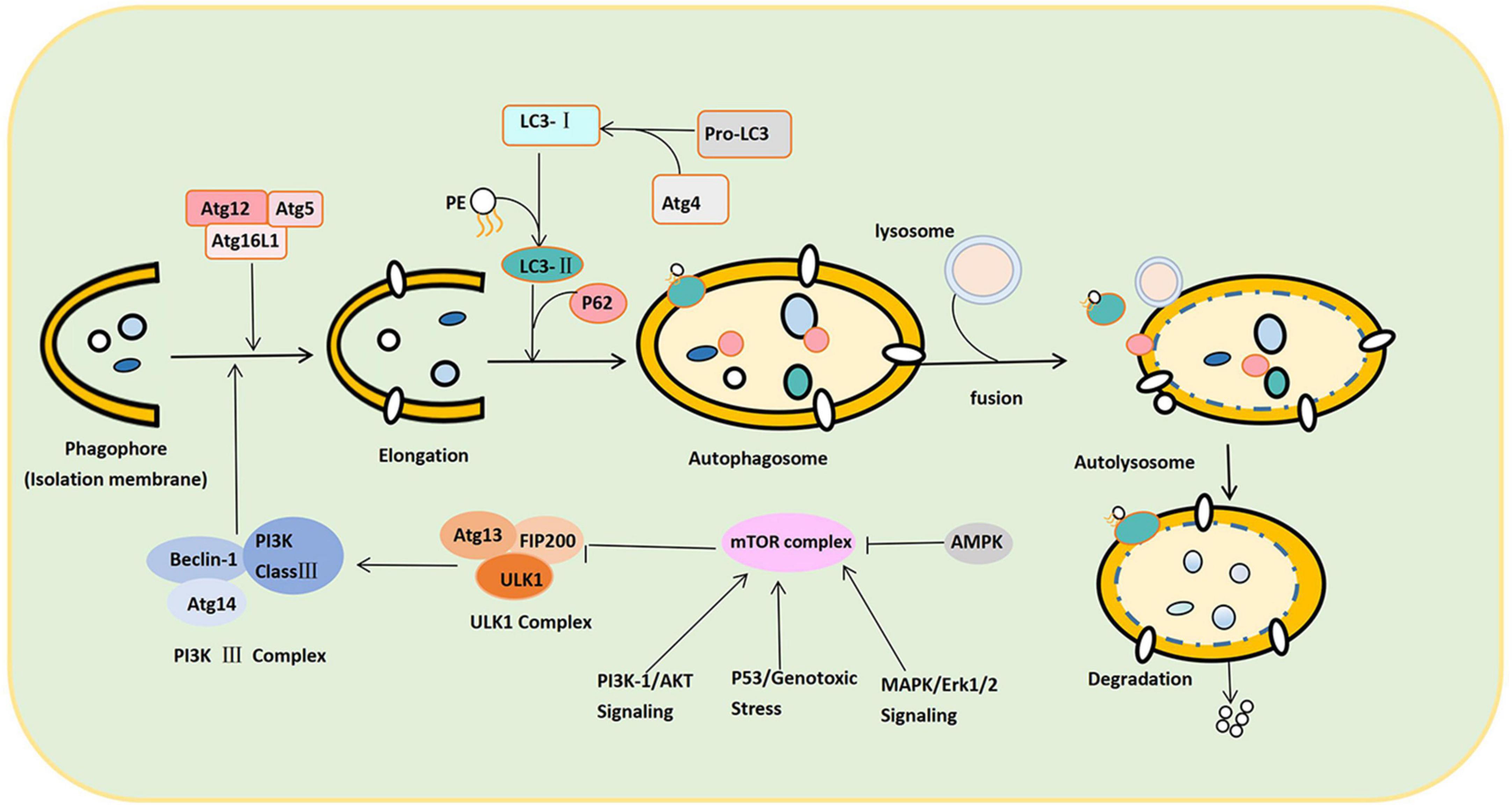
Figure 2. Schematic illustration of the autophagy process. The autophagy process can be divided into the following stages: Induction, nucleation of phagosomes, elongation of phagosomes, completion of autophagosomes, fusion with lysosomes to form autophagosomes, and content degradation. After LC3 binds to phospholipid ethanolamine (PE), LC3-II is anchored to the cell membrane, promoting the expansion and closure of phagosomes and finally forming autophagosomes. P62 links LC3-II to the substrate on the isolation membrane and separates the isolation substrate. In response to SNARE proteins, both ends of the sequestration membrane close and form a complete autophagosome with a bilayer structure. The autophagosome and lysosome fuse to form the autophagolysosome. Under the control of lysosomal-associated membrane proteins, the autophagy outer membrane fuses with the outer lysosomal membrane to form an autophagolysosome. Autophagolysosome degrades the substrate into small molecules such as lipids, nucleosides, and other substances used by the cell.
Second, the class III phosphatidylinositol 3-kinase (PI3K class III) complex consisting of Beclin-1-Atg14-PI3K class III is an essential component of nucleation and assembly (Wang X. et al., 2020). The PI3K class III complex generates phosphatidylinositol 3-phosphate and recruits additional ATG proteins to form the isolation membrane (also termed the “phagosome”) (Muller et al., 2017). Autophagosome formation and elongation depend on Atg5-Atg12-Atg16L and microtubule-associated protein 1A/1B light chain 3 (LC3) (Yang and Klionsky, 2010). ATG4 can transform Pro-LC3 into LC3. When the complex (ATG12-ATG5-ATG16L) binds LC3 to phosphatidylethanolamine (PE), the cytoplasmic form of LC3 (LC3-I) is anchored to the cell membrane as LC3-II (Stavoe and Holzbaur, 2019). LC3-II promotes the expansion and closure of the phagophore and, eventually, the formation of autophagosomes. Thus, the conversion of LC3-I to LC3-II marks autophagy initiation. After expansion and closure, all the ATGs escape from the autophagosome and return to the cytoplasm. Only the LC3-II is anchored to the autophagosome membrane. Meanwhile, substrate recognition and transport are completed by P62 (Galluzzi and Green, 2019). Hence, P62 can target and bind the substrate and LC3-II, connecting LC3-II with the substrate in the isolation membrane. Once a phagophore is formed, it isolates and coats the substrate (damaged intracellular proteins and organelles). Soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins transport vesicles and fuse autophagy membranes during autophagy formation. The two ends of the isolation membrane close and form a complete autophagosome with a bilayer membrane structure under the action of SNARE proteins (Tian et al., 2021).
Subsequently, the autophagosome moves along the microtubule system of the cytoskeleton to lysosomes containing cathepsin B and cathepsin D (Hossain et al., 2021). Under the control of lysosomal-associated membrane proteins, the autophagy outer membrane fuses with the outer lysosomal membrane. The inner membrane is degraded by lysosomal enzymes and eventually turns into an autophagolysosome. Hydrolase in the autophagolysosome degrades the substrates into small molecular substances and transports them to the cytoplasm for reuse (Fang et al., 2018).
The regulation process of autophagy can be divided into mTOR-dependent and mTOR-independent pathways. Under normal nutritional conditions, mTOR, which is highly active, recognizes and phosphorylates ULK1 and Atg13 to inactivate the ULK1 complex, which exerts a negative regulatory effect to prevent autophagy (Zachari and Ganley, 2017). In contrast, under starvation or hypoxia, mTOR activity is inhibited. The inhibition of the ULK complex by mTOR is then reduced, which effects the activation of ULK1 kinase and autophagy. The PI3K complex is a downstream regulator of the ULK complex, which can induce an autophagic membrane to turn into nucleation (Shen et al., 2021). There are four main regulatory pathways upstream of mTOR, namely, adenosine monophosphate-activated protein kinase (AMPK), PI3K/protein kinase B (Akt) signaling, MAPK/Erk1/2 signaling, and P53. The inhibition of autophagy occurs through PBK/Akt signaling and MAPK/Erk1/2 signaling, while the activation of autophagy occurs through AMPK and P53 (Xu D. et al., 2021). AMPK can also activate it with the phosphorylation of ULK1, thereby promoting autophagy (Li and Chen, 2019). In addition, p53 is usually present in the cytoplasm. However, when DNA is damaged, it is phosphorylated and translocated to the nucleus. Nonetheless, p53 can affect autophagy in both the cytoplasm and the nucleus (White, 2016). In the cytoplasm, p53 inhibits autophagy by inhibiting AMPK and activating mTOR. It can also directly inhibit FIP200 through a non-mTOR-dependent pathway, inhibiting the autophagy pathway. In the nucleus, though, p53 promotes the transcription of autophagy-related genes to promote autophagy (Tanida et al., 2008; Figure 2).
There is increasing evidence to support the claim that autophagy is activated and involved in the pathophysiology of ICH-induced SBI (Duris et al., 2018). It was found that autophagy was activated 6 h after ICH and peaked at 12 and 24 h (Zhang and Liu, 2020). It has also been revealed that autophagy occurred in the perihematomal area after ICH in rat models, specifically manifested in increased cathepsin D expression and vacuole formation and the conversion of LC3-I to LC3-II. In addition, 24 h after ICH, the protein levels of LC3 and Beclin-1 were increased, and the protein levels of SQSTM1/P62 were decreased, suggesting that autophagy is enhanced in the process of ICH-induced SBI. We have summarized the mechanism of autophagy in ICH (Figure 3A). Nevertheless, this mechanism still merits further exploration.
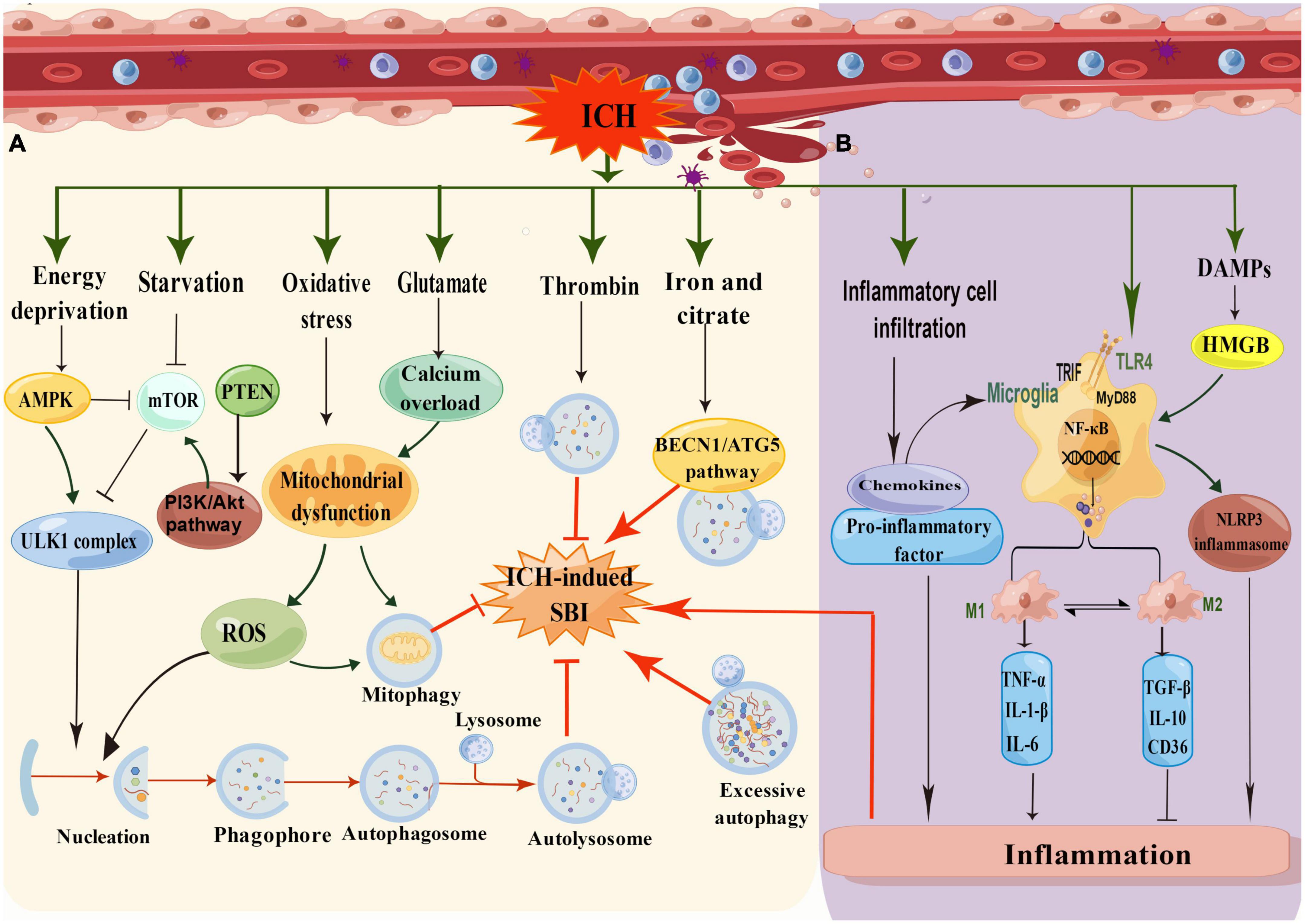
Figure 3. (A) Schematic illustration of the mechanism of autophagy in ICH. In low energy states, the increasing ratio of AMP/ATP leads to the activation of AMPK and subsequently inhibits mTOR for induction of autophagy. In addition, the AMPK pathway suppresses mTOR activity, while the cell undergoes induction of starvation or calcium signals. After ICH, oxidative stress produces many ROS, and glutamate causes calcium overload, resulting in mitochondrial damage and inducing mitophagy. Thrombin can reduce SBI by inducing autophagy, whereas iron and citrate can induce autophagy to aggravate SBI through the BECN1/ATG5 pathway. (B) Schematic illustration of the mechanism of inflammation in ICH. The inflammatory mechanisms after ICH mainly include microglia activation, inflammatory cell infiltration, toll-like receptor activation, and DMPs mode regulation (by Figdraw).
After ICH, the hematoma expands and presses on the surrounding tissue, leading to edema of the surrounding tissue and resulting in cell hypoxia and starvation. Autophagy is strongly induced by starvation and is an adaptive response to nutrient deficiency and hypoxia, which promotes nutrient recycling (Vargas et al., 2022). In response to a decrease in ATP levels and a rise in AMPK levels, the mTOR pathway was inhibited. Furthermore, energy deprivation, starvation, and other stimuli could initiate autophagy through the ULK1 complex pathway after ICH. In addition, oxidative stress and calcium overload caused by glutamate could lead to mitochondrial dysfunction, producing reactive oxygen species (ROS) to trigger autophagy or mitophagy pathways. It has been mentioned that erythrocyte lysis, iron toxicity, and thrombin play significant roles in ICH-mediated SBI (Zhang and Liu, 2020). Coagulation components or their products, such as thrombin and hemoglobin, participate in endogenous autophagy regulation during ICH. Hematoma-induced thrombin and iron overload accumulation are the leading causes of autophagy.
Many studies suggest that there is no unified theory on the role of autophagy in ICH (Li et al., 2018). The double-sided role of autophagy in ICH may be because different periods and phases of ICH are characterized by different roles in autophagy, mainly depending on the level of autophagy and whether the autophagy flux is complete (Duan et al., 2017).
Studies have confirmed that enhancing autophagy is beneficial for the repair of nervous system diseases (Xu W. et al., 2021). Thrombin is an essential part of the coagulation cascade. A large amount of thrombin is produced to induce hemostasis in the early stage after ICH, which induces leukocyte infiltration and brain edema formation, leading to SBI (Ye et al., 2021). Many studies have found that thrombin is a vital inducer of autophagy after ICH and that autophagy plays a beneficial role in thrombin-induced SBI (Wu et al., 2019). In astrocytes, autophagy was obviously activated 24 h after intracerebral thrombin injection in rats, and autophagy activity reached its peak 3 days after injection. In addition, in vitro and in vivo data have confirmed the beneficial aspects of autophagy in ICH after SBI induced by thrombin (Shao et al., 2019). The inhibition of thrombin-induced autophagy exacerbated cell death via the inhibition of autophagy with 3-MA (3-methyl-adenine), the autophagy inhibitor. Interestingly, ATG7 is an essential autophagy regulator but has no autophagy-unrelated functions. When ATG7 is knocked down using siRNA, the thrombin-induced expression of inflammatory molecules, such as IL-6, is significantly reduced (Shadab et al., 2020). Duan et al. (2016) found that autophagy and ER had distinct effects on ICH at disparate periods. At 6 h after ICH, the autophagy level is too high, and ER stress induction can enhance autophagy, leading to brain injury. However, after 7 days, autophagy may improve the protective effect of ER stress inhibitors by removing the cellular waste produced by early impaired autophagy flux. The primary possible reason is that autophagy flux was damaged 72 h after ICH and restored to integrity after 7 days (Duan et al., 2017). Furthermore, Wang et al. found that the knockdown of the p75 neurotrophin receptor (p75NTR) could promote neuronal autophagy by inactivating the mTOR pathway, presenting protective roles in a rat model of ICH (Wang L. et al., 2020).
The function of autophagy is usually to prevent cellular death. However, excessive autophagy can also cause cell death (Fricker et al., 2018). The excessive elevation of autophagy activity may lead to the accumulation of cellular solutes and the enhanced degradation of essential cellular components in autophagosomes. Excessive autophagy, which blindly hydrolyzes damaged or undamaged cytoplasm and organelles, may lead to increased lysosomal membrane permeability and the release of large amounts of cathepsin into the cytoplasm, thereby aggravating cell damage (Fang et al., 2018). Cathepsin B in lysosomes promotes the release of cytochrome C from mitochondria by regulating the proapoptotic protein Bid, activating caspase family members, and finally inducing apoptosis. Numerous studies have found in rats injected with ferrous citrate and iron that autophagy is highly active and that iron or citrate-induced autophagy has harmful effects on ICH-mediated SBI (Zhang and Liu, 2020). After hemorrhage, iron accumulation subsequently generates abundant ROS, which may induce autophagy and neurotoxicity. However, some studies have found that the inhibition of autophagy can reduce the ICH-mediated SBI induced by ferrous citrate. In addition, heme enhances neuronal autophagy through ER stress and induces cell death through the BECN1 (Beclin1)/ATG5 pathway, and the silencing of Beclin1 and Atg5 or the use of autophagy inhibitors can reduce the release of inflammatory factors in microglia induced by lysed erythrocytes in vitro, which is beneficial for alleviating brain injury after ICH (Yang et al., 2020).
There is a great deal of interesting speculation that the beneficial or detrimental effects of autophagy depend in large part on whether the autophagic flux is unimpeded (Li et al., 2018). A complete autophagic flux is essential for homeostasis in almost all cells, especially neurons. According to previous studies, autophagy intensity was primarily determined by the number of autophagosomes or LC3-II. In recent years, it has been found that an increase in autophagosomes is not essentially equivalent to an increase in autophagy levels. An increase in autophagosomes only reflects the induction of autophagy or the inhibition of autophagosome clearance (Li et al., 2018; Zhang and Liu, 2020). From the perspective of autophagic flux, the number of autophagosomes is affected by both formation and clearance. Meanwhile, the heightened activity of cathepsin D and acid phosphatase may be responsible for the greater autophagic flux. Autophagic flux can be demonstrated by the disappearance of the autophagic substrate p62/SQTM1 (Fricker et al., 2018). It is necessary to observe not only autophagosomes but also the entire autophagic flux dynamically to assess autophagy accurately and comprehensively to discern whether the whole autophagic flux process is stable.
Earlier studies classified autophagy as a non-selective process, but recent studies have suggested that the selectivity of targeted cargo is achieved through a series of autophagy receptors (Xu et al., 2015). In 2014, at the Vancouver Symposium on Autophagy, scholars focused on the importance of autophagy as a selective mechanism in pathological conditions, emphasizing the role of autophagy in the strict regulation and precise targeting of substrates (Li et al., 2018). It suggests that a complete understanding of the regulation of selective autophagy in disease may accelerate the development of autophagy-based therapies. Two kinds of selective autophagy, such as mitophagy and ferritinophagy, have been found to play a role in ICH. The roles of selective autophagy in stroke have been reported (Table 1). Currently, most literature on selective autophagy in ICH focuses on mitophagy.
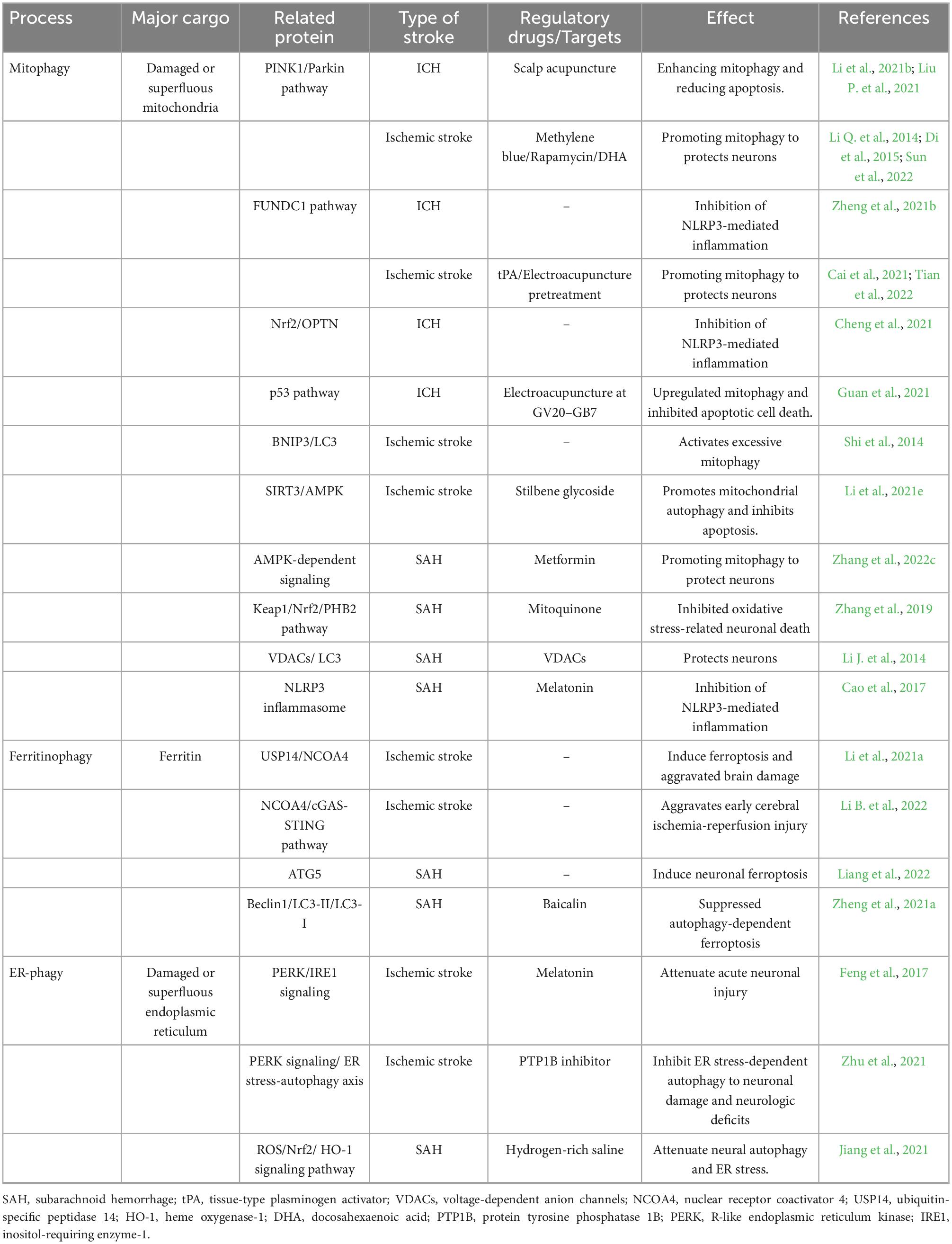
Table 1. Selective autophagy characteristics and effect in stroke.
Mitophagy is one of the most recognized types of selective autophagy and is the mechanism of the specific autophagic elimination of mitochondria (Lou et al., 2020). Mitophagy, as a mechanism of mitochondrial quality and quantity control, selectively identifies and eliminates dysfunctional or superfluous mitochondria by autophagy, thus playing a role in protecting the brain (Guan et al., 2018). Many mitophagy receptors link target mitochondria to autophagosomes for degradation in response to various environmental or intracellular stresses. It remains to be determined how mitophagy is regulated, whether it is inducted or inhibited, and its specific role in ICH. Mitophagy is restrictively controlled by several proteins, including phosphatase and tensin (PTEN)-induced putative kinase (PINK1) and FUN14 domain containing 1 (FUNDC1) (Li et al., 2021d). The decreased oxygen metabolism in hematomas after ICH leads to mitochondrial dysfunction. In damaged mitochondria, the significant decrease in intracellular ATP and Ca2+ overload and the increase in ROS production are essential factors aggravating the SBI caused by ICH (Guan et al., 2018; Bian et al., 2022). Excessive ROS in tissues or cells produced during oxidative stress can induce autophagy through various mechanisms to prevent further oxidative damage overload in the brain after a hemorrhage (Yao et al., 2021). In addition, PTEN-induced kinase 1 (PINK1) is a mitochondrial-targeted protein kinase. By encouraging mitophagy in microglia, PINK1 defends against ICH-induced SBI, and PINK1 overexpression can treat ICH-induced behavioral problems (Li et al., 2021b). As a vital component of the mitochondrial contact site and cristae junction organizing system, MIC60 plays a significant role in maintaining the mitochondrial structure and functioning. After ICH, mitochondrial damage, mitochondrial crest remodeling and the reduction of MIC60 protein after the PINK1 level decrease, and Parkin localization errors are observed. The overexpression of MIC60 could maintain mitochondrial structural integrity and reverse ICH-induced neuronal cell death and apoptosis (Deng et al., 2021).
As a selective autophagy type, ferritinophagy mediates the degradation of ferritin and the release of free iron and engages in the regulation of intracellular iron content (Vargas et al., 2022). Ferritin chelates free iron and ensures that iron homeostasis is maintained at tolerable levels within cells. However, an overload of ferritin can cause brain damage and cell death (Li et al., 2021a). The release of free iron after erythrocyte lysis and from ferritin stores may influence oxidative stress, glutamine release, and inflammation after hemorrhagic brain injury. Nuclear receptor coactivator 4 (NCOA4) mediates ferritin degradation via the autophagosome-lysosome pathway through ferritinophagy (Li B. et al., 2023). With the help of the autophagy proteins ULK112 and FIP200, Tax1-binding protein 1 binds directly to NCOA4 and promotes ferritin lysosomal localization and degradation under iron-depleted conditions (Zheng et al., 2021b).
As a type of selective autophagy, reticulophagy involves the selective degradation of ER fragments. Reticulophagy is induced by ER stress to maintain the stable state of ER, and intracellular reticulophagy is maintained at a low level under normal physiological conditions. The ER activates reticulophagy when it receives harmful signals, including nutritional deficiencies or unfolding protein aggregation (Wilkinson, 2019). Interestingly, selective degradation of the ER by autophagy has become one of the typical responses to ER stress (Thangaraj et al., 2020).
Neuroinflammation is a crucial factor in ICH-induced SBI and may develop shortly after ICH and peak a few days later (Li et al., 2021c). Inflammation caused by various proinflammatory factors, such as IL-1β, IL-18, and TNF-α, leads to the destruction of the BBB, exacerbated brain edema, and neuronal death (Tschoe et al., 2020; Li H. et al., 2022). There are many reactive inflammatory processes after ICH, including microglia activation, inflammatory cytokine release, and leukocyte infiltration (Figure 3B; Ye et al., 2021). After ICH, the hemolytic components of red blood cells (hemoglobin and iron) and plasma proteins (thrombin and fibrinogen) are released into the brain tissue, leading to inflammatory reactions via the activation of the immune, oxidative stress, hemostasis, and other systems (Fu et al., 2021a). After a hemorrhage, neuronal injury and DAMPs trigger proinflammatory responses that increase BBB permeability around the hematoma and worsen the mass effect (Wu et al., 2020a).
Neuroinflammation is characterized by the polarization of microglia toward the proinflammatory phenotype and the release of proinflammatory factors (Taylor et al., 2017). Neuroinflammation occurred on day 1 post-ICH, and changes in microglia size and morphology were observed. Activation of microglia leads to the infiltration of macrophages and T cells, which release inflammatory cytokines, chemokines, free radicals, and other potentially toxic chemicals, triggering a cascade of inflammatory responses. Shtaya’s study showed that the accumulation of neutrophils on the endothelial surface is considered a marker of intracerebral microvascular inflammation within 1–4 days after ICH (Shtaya et al., 2019). The proinflammatory factors (TNF-α and IL-1β) and ROS produced by aseptic inflammation after ICH promote the activation of endotheliocytes, which change their phenotype and connexin composition, and express more selectin and adhesion molecules, which is a crucial step in recruiting white blood cell from peripheral blood (Eser et al., 2022). The accumulation of astrocytes in the area surrounding the hematoma was observed 1–3 days after the onset of ICH (Scimemi, 2018). In addition, astrocytes secrete various proinflammatory factors (such as IL-1β and IL-6), which aggravate the neuronal injury and brain injury (Tschoe et al., 2020).
The inflammasome is a crucial component of innate immunity and is usually composed of the pattern recognition receptor (PRR) containing the pyrin domain, the apoptosis-associated spot-like protein (ASC), and the effector protease caspase-1 (Broz and Dixit, 2016). When stress factors, such as DAMPs, are detected, PRRs divide and activate caspase-1, which leads to the activation of interleukin-1β (IL-1β) and interleukin-18 (IL-18), ultimately resulting in inflammation (Chung et al., 2020). Nod-like receptors (NLRs) are PRRs involved in detecting invading pathogens and initiating innate immune responses. This section mainly describes the role of NLRP inflammasomes in ICH. As the most characteristic inflammasome, the NLRP3 inflammasome plays a crucial role in developing an immune response and disease. NLRP3, pro-caspase-1, and ASC aggregate to form NLRP3 inflammasomes (Huang et al., 2021; Gu et al., 2022). Studies have shown that the NLRP3 inflammasome expression is significantly elevated after ICH (Xiao L. et al., 2020). Ma et al. demonstrated that the NLRP3 inflammasome activation amplifies the inflammatory reaction, promotes neutrophil infiltration, aggravates brain edema, and worsens neurological functioning after ICH in a mouse model of autologous blood injections. There is increasing evidence that the NLRP3 inflammasome is a critical component of the inflammatory reaction in strokes (Gao et al., 2017; Luo et al., 2019). In addition, mature NLRP3 inflammasomes can promote IL-1β and IL-18 maturation and recruit peripheral immune cells, which can amplify an inflammatory reaction (Xiao L. et al., 2020).
The NLRP6 inflammasome is a critical intracellular innate immune sensor shown to regulate immune responses. NLRP6 recruits the adaptor apoptosis-associated ASC and inflammatory caspase-1 or caspase-11 to form an inflammasome (Zheng et al., 2021a). NLRP6 could regulate inflammation in inflammasome-dependent and non-inflammasome-independent ways. The expression of the NLRP6 inflammasome increased in perihematomal brain tissue from 6 h to 3 days and peaked 1 day after ICH. However, the role of NLRP6 in ICH remains controversial (Wang P. F. et al., 2017).
The autophagy mechanism is linked to most cellular stress response pathways, especially inflammatory pathways (Zheng et al., 2021e). Autophagy and inflammation are significant pathologies of ICH-induced SBI; however, their relationship remains unclear (Durocher et al., 2021). Many studies have illustrated that autophagy activation could block inflammation, inhibiting the body’s inflammatory response and reducing the disease in inflammatory tissue injury (Matsuzawa-Ishimoto et al., 2018). Nonetheless, microglial autophagy can also promote inflammation and aggravate SBI. What is the relationship between ICH-induced autophagy and ICH-induced inflammation, and is it an enemy or a friend?
Increasing evidence suggests that autophagy inhibits the neuroinflammation mediated by NLRP3 inflammasome activation and, correspondingly, attenuates inflammatory damage (Biasizzo and Kopitar-Jerala, 2020; Figure 4A). Recent studies have depicted that autophagy reduces caspase-1 production by inducing the selective clearance of damaged mitochondria in cells, thereby affecting NLRP3 inflammasome activation and NLRP3-ASC assembly (Plaza-Zabala et al., 2017). Moreover, p62 can recognize ubiquitinated ASC and induce autophagy to degrade inflammasome complexes selectively (Zhao et al., 2021). In addition, when autophagy is impaired, Beclin-1 reduction leads to the enhanced release of IL-1β and IL-18 from the microglia. Autophagy inhibits NLRP3 inflammasome overactivation by removing pro-IL-1β through the autophagic lysosomal pathway (Plaza-Zabala et al., 2017; Houtman et al., 2019). In conclusion, autophagy removes NLRP3 inflammasome activators, NLRP3 inflammasome components, and inflammatory cytokines, reducing inflammasome activation and inflammation (Biasizzo and Kopitar-Jerala, 2020). Additionally, the inflammation caused by ischemic stimulation can be inhibited by autophagy via the mTOR and AMPK pathways and by inhibiting inflammasome activation (Mo et al., 2020). However, the mechanism of autophagy regulating inflammation in ICH still necessitates detailed research.
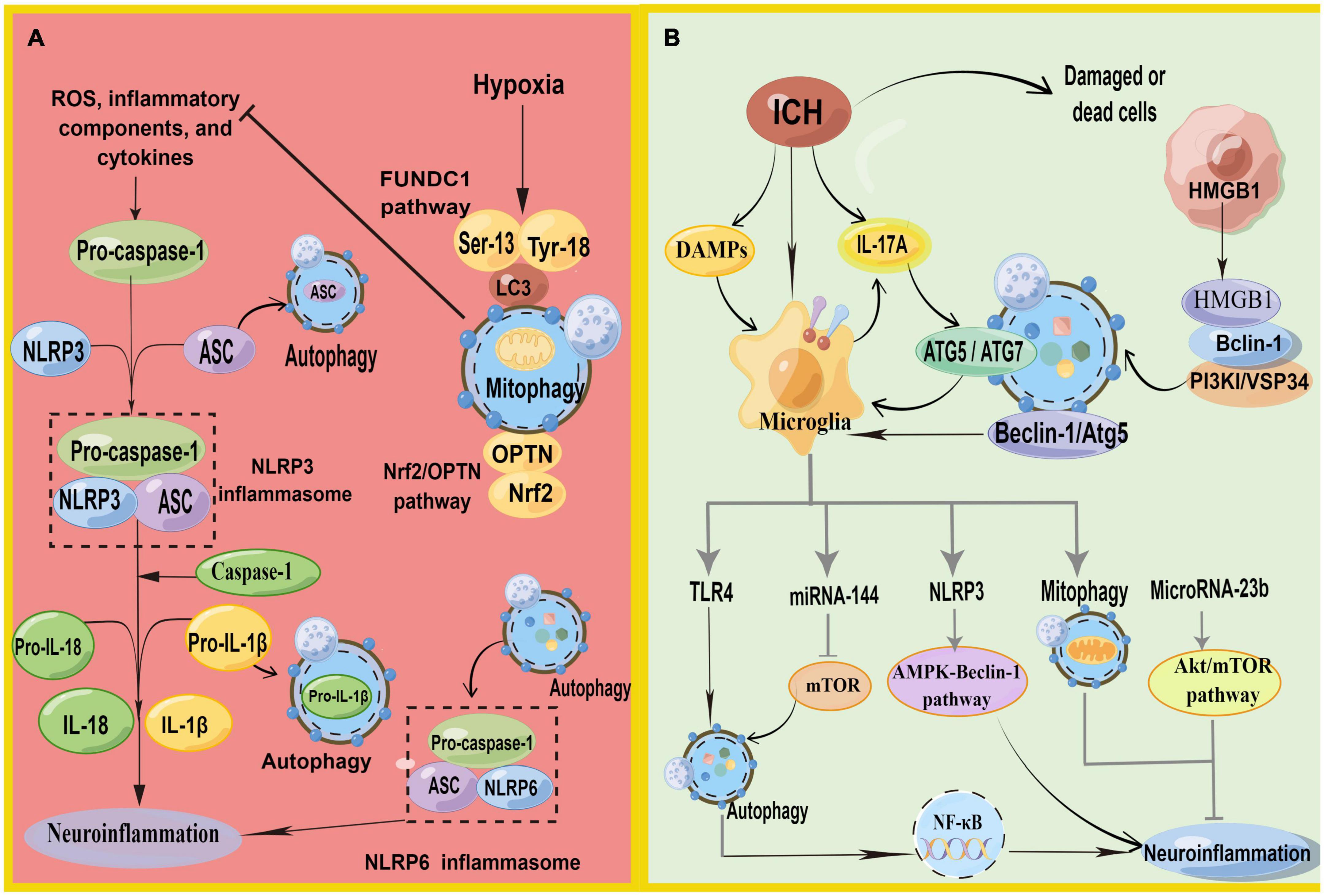
Figure 4. (A) Schematic illustration of autophagy regulates NLRP3 inflammasome and NLRP6 inflammasome. NLRP3, pro-caspase-1, and ASC aggregate to form NLRP3 inflammasome. Autophagy antagonizes inflammation mainly in three parts: (1) P62 can recognize ubiquitinated ASC and induce autophagy to degrade inflammasome adaptor protein ASC selectively. (2) Decomposition and removal of pro-IL-1β, the substrate of inflammatory mediators. (3) Can inhibit NLRP3 inflammasome activation by removing endogenous inflammasome activators, such as ROS. Meanwhile, mitophagy antagonizes neuroinflammation-mediated NLRP3 inflammasome via FUNDC1 and Nrf2/OPTN pathways. In addition, autophagy promotes inflammasome-dependent NLRP6-mediated inflammation after ICH. (B) Schematic illustration of autophagy in microglia regulates autophagy in microglia. HMGB1 combines with Beclin1 and PI3K I/Vsp34 to activate autophagy. Autophagy promotes microglial activation and microglia-mediated inflammation via the HMGB1/TLR4/MyD88, Beclin-1-Atg5 pathway. IL-17A-mediated autophagy activation induced microglial activation and microglial inflammation via ATG5/ATG7. In addition, microglial autophagy can regulate inflammation through TRL4, miRNA-144, NLRP3, mitophagy, and Microrna-23b (by Figdraw).
Xiao H. et al. (2020) found that NLRP6 can reduce brain damage by lessening inflammation and inhibiting autophagy after ICH with the NLRP6 gene knockout. They also demonstrated that autophagy promoted inflammasome-dependent NLRP6-mediated inflammation after ICH. However, the role of NLRP6 in ICH remains controversial (Wang P. F. et al., 2017).
Reactive oxygen species generation is a vital activation pathway of NLRP3. The primary source of ROS has damaged mitochondria. Mitochondria play a vital role in initiating and regulating NLRP3 inflammasomes and are the basis for NLRP3 inflammasome activation. Mitophagy can inhibit NLRP3 inflammasome activation by removing endogenous activators of the inflammasome, including ROS, inflammatory components, and cytokines (Biasizzo and Kopitar-Jerala, 2020; Zhao et al., 2021). The loss of autophagy leads to the accumulation of damaged mitochondria and increased ROS. Currently, mitophagy has been shown to inhibit neuroinflammatory-mediated NLRP3 inflammasome attenuation ICH-induced SBI through the FUNDC1 and nuclear factor E2-related factor 2 (Nrf2)/Optineurin- (OPTN-) mediated autophagy pathways (Cheng et al., 2021; Zheng et al., 2021c). FUNDC1 is a mammalian mitophagy receptor containing LC3-interacting regions capable of interacting with LC3. Hypoxia is one of the most common conditions for inducing the FUNDC1 pathway, and it is a common pathological state in tissues surrounding the ICH. The function of FUNDC1 is regulated by phosphorylation at Ser13, Tyr18, and Ser17. During hypoxia, phosphoglycerate mutase family member 5 phosphatase dephosphorylates Ser13 of FUNDC1, subsequently activating mitophagy by binding to LC3 (Chen et al., 2016; Wu et al., 2017). Zheng et al. found that FUNDC1 protein levels were upregulated and peaked 12 h after ICH (Zheng et al., 2021c). They first demonstrated that FUNDC1 alleviated ICH-induced inflammation by inhibiting NLRP3-mediated inflammation by promoting mitophagy. In addition, the inhibition of FUNDC1-mediated mitophagy markedly increased IL-1β production in vitro and in vivo (Huang et al., 2020). Optineurin is an autophagy receptor and an autophagy inducer that participates in the initiation of autophagosome membrane formation (Ying and Yue, 2016; Qiu et al., 2022). Cheng et al. found that Nrf2 could interact with the mitophagy receptor OPTN and regulate OPTN-mediated mitotic phagocytosis to effectively remove damaged mitochondria and inhibit NLRP3 inflammasome activation after ICH, thereby alleviating SBI after ICH by collecting human ICH brain flecked for iTRAQ-based quantitative proteomics (Cheng et al., 2021).
Surprisingly, more evidence suggests that autophagy does not always fight inflammation in ICH but promotes neuroinflammation and exacerbates ICH-induced SBI. The process of autophagy-promoting inflammation is inseparable from microglia (Figure 4B), especially TLR4, on the microglial membrane.
TLRs belong to a large family of pattern-recognition receptors. TLR4-mediated microglial activation is a pivotal factor in inflammatory injury in ICH (Lee et al., 2019). TLR4 is activated during ICH by several endogenous ligands with injury-related molecular patterns, including hemes and fibrinogen. The activation of TLR4 leads to downstream inflammatory signaling cascades, including the myeloid differentiation primary response 88(MyD88)/TRIF pathway (Gu et al., 2018). TLR4 was upregulated in the early ICH (Deng S. et al., 2022). Interestingly, ROS accumulation in the microglia can trigger the release of inflammatory mediators by activating signaling molecules, including mitogen-activated protein kinase and NF-κB. The activation of microglia and inflammation are associated with TLR4-mediated autophagy. TLR4 synuclein was accompanied by a notable increase in P62, while the levels of other autophagic receptors remained constant. This process is mediated by the activation of NF-κB and promotes the transcription of P62. Increasing evidence demonstrates that the inhibition of TLR4 and its downstream molecules can alleviate the brain injury caused by ICH.
Yang et al. (2015) treated microglia cultured in vitro with exogenous heme chloride and observed greater TLR4 expression, as well as proinflammatory cytokines, and proved that autophagy was involved in TLR4-mediated inflammatory damage and the migration of microglia. They also found that 3-MA reduced microglial activation and inflammatory damage caused by lysed red blood cells, thereby augmenting neurological functioning in ICH mice models. This mechanism may occur because autophagy promotes an inflammatory reaction through the high-mobility group box-1 (HMGB1)/TLR4/MyD88 pathway (Lei et al., 2022). HMGB1 has been widely reported to play a central role as a regulator of autophagy and in initiating proinflammatory cytokine production (Xu et al., 2019). After ICH, peripheral nerve cells are damaged, and HMGB1 can be passively released from necrotic or apoptotic cells and transported from the nucleus to the cytoplasm. HMGB1 in cytoplasm mainly promotes the binding of Beclin1 and PI3K I/Vsp34 to activate autophagy by binding to the autophagy factor Beclin1 (Zhang et al., 2022a). Some research has revealed that HMGB1 upregulates the expression of TLR4 and MyD88, causing neurological deficits, and autophagy promotes microglial activation and microglia-mediated inflammation through the HMGB1/TLR4/MyD88 axis in the acute phase of ICH (Lei et al., 2022).
IL-17A is one of the cytokine members of the IL-17 family, which is produced by a subset of T cells. IL-17A promotes the activation of microglia by stimulating the expression of ICH-induced cytokines, including IL-1β and IL-6, TNF-α, as well as downstream signaling molecules, including P65, TRIF, NF-κB, and MyD88. The inhibition of autophagy with RNA interference in essential autophagy genes (ATG5 and ATG7) reduced microglial autophagy and inflammation and ameliorated neurological functioning in a mice model of ICH (Shi et al., 2018). Consequently, IL-17A-mediated autophagy activation induced microglial activation and microglial inflammation.
miRNAs play a crucial role in inflammation by negatively regulating the expression and function of genes. In addition, miRNA-144 targets mTOR through direct interaction with the 3’-untranslated regions and mutations at the binding site cancel the responsiveness of miRNA-144. Yu et al. detected the autophagy activity, inflammation, and expression of miRNA-144 of microglia in ICH and knocked out endogenous miRNA-144 (Yu et al., 2017). Meanwhile, they demonstrated that miRNA-144 enhanced autophagy activity and the inflammation of microglia after ICH through the mTOR pathway. Furthermore, hemoglobin-mediated autophagy and the inflammation of microglia are mediated by miRNA-144. In addition, microRNA-23b plays an anti-inflammatory role in many diseases and is downregulated in patients with ICH (Wang Z. et al., 2017). MicroRNA-23b negatively regulates inositol polyphosphate multikinase-mediated autophagy, after which it participates in post-ICH anti-inflammatory effects via the Akt/mTOR pathway (Hu et al., 2019).
Interleukin-33 (IL-33) is a new member of the IL-1 family cytokines, and is mainly used as a novel serum prognostic marker in ICH. Its signals through a heterodimer comprising the IL-1 receptor-associated protein ST2L and IL-1RACP to play a biological function is mainly used as a novel serum prognostic marker in ICH (Miao et al., 2021). IL-33 plays a neuroprotective role in inhibiting autophagy and inflammation by increasing P62 and decreasing the expression of LC3-2 and Beclin-1 (Gao et al., 2017). IL-33 also reduces neuronal and white matter damage by promoting the M2-like microglia polarization of microglia after ICH, thereby optimizing neurological outcomes (Chen Z. et al., 2019).
Yuan et al. (2017) found that autophagy promoted microglial activation via the Beclin-1-Atg5 pathway in ICH. They also demonstrated that the inhibition of autophagy with pharmacological inhibitors or RNA interference with essential autophagy genes reduced microglial activation and inflammation in ICH and reduced brain damage.
The modes of cell death following ICH can be categorized into autophagy, apoptosis, ferroptosis, necrosis, and pyroptosis (Table 2). Autophagy interacts with various forms of cell death, making it difficult to separate the roles of autophagy (Denton and Kumar, 2019). Therefore, the concept of cell death that is solely dependent on autophagy remains somewhat controversial.
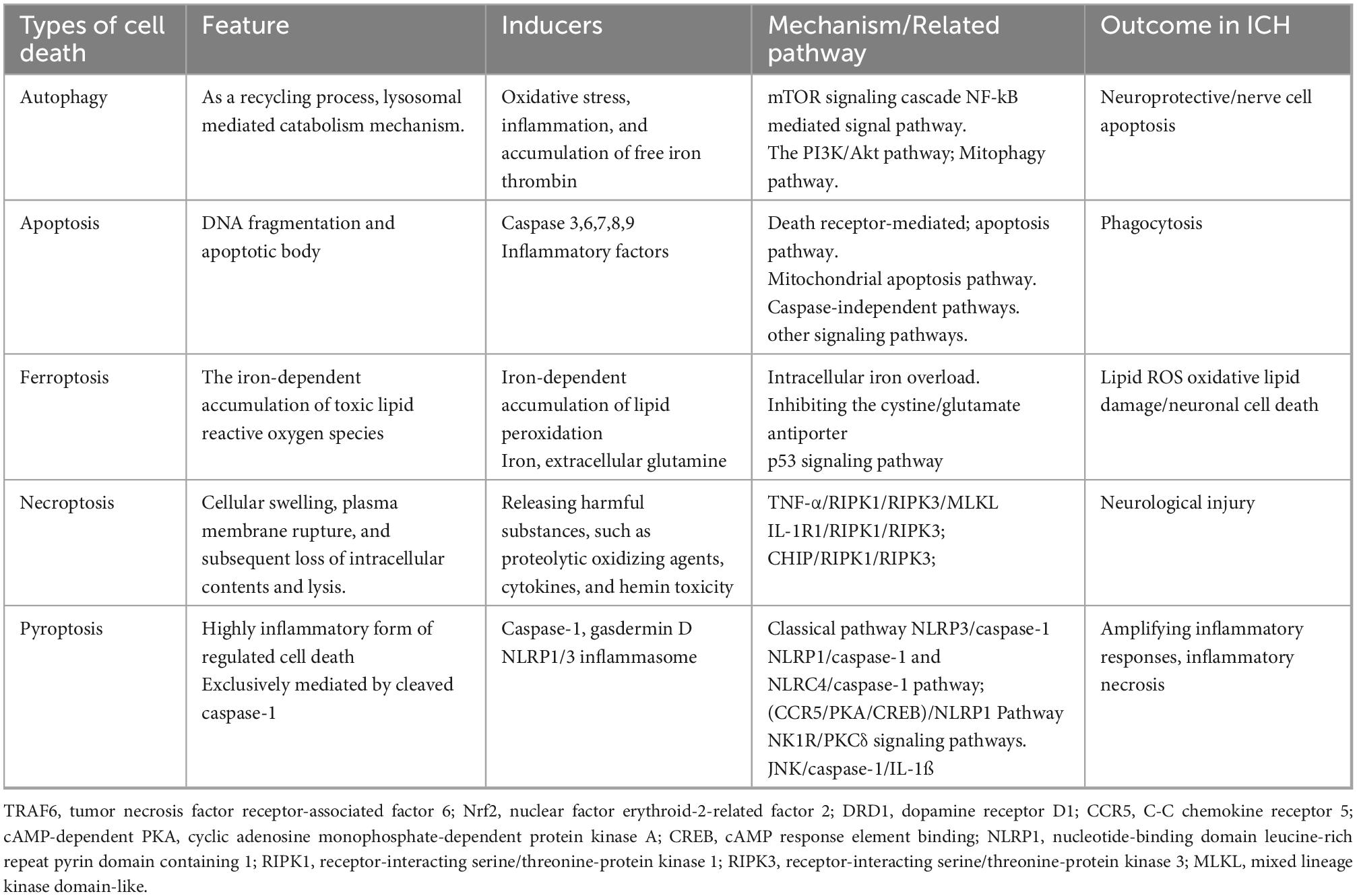
Table 2. Comparison of different ICH-induced cell death.
Apoptosis is a type of programmed cell death, which is a series of coordinated and progressive biochemical reactions that lead to the orderly decomposition of cells. The apoptosis of nerve cells exists in the perihematomal region of the brain, a crucial pathological process after ICH (Fricker et al., 2018). The toxic effects of hematoma components and their degradation products, oxidative stress surrounding hematomas, and thrombin release are the principal causes of ICH-induced apoptosis. In general, apoptosis can be divided into intrinsic (mediated by mitochondrial dysfunction or ER stress) and extrinsic (mediated by cell surface receptors, namely, tumor necrosis factor and apoptosis-related factor receptor system) pathways (Bobinger et al., 2018; Wu et al., 2018). The activation of a series of cysteine aspartate proteases is key to apoptosis, especially Caspase 3, a common downstream effect of multiple apoptotic pathways termed the “executive death protease”(Nagata, 2018).
Autophagy-dependent cell death is a unique mechanism that occurs independently of apoptosis or necrosis; however, autophagy-induced cell death is often accompanied or triggered by apoptosis (Denton and Kumar, 2019). There is complex crosstalk between ICH-induced apoptosis and autophagic cell death. Bcl-2 is a crucial protein in the regulation of apoptosis. Besides inhibiting apoptosis, Bcl-2 could also inhibit Beclin-1, thus blocking autophagy (Gao et al., 2017). When external pressure or injury destroys normal intracellular homeostasis, the decrease of Beclin-1 binding with Bcl-2 could lead to the induction of autophagy. Autophagy and apoptosis usually occur in the same cell, and studies have shown that autophagy mainly precedes apoptosis. In general, autophagy and apoptosis are mutually inhibitory processes (Maiuri et al., 2007). Autophagy usually inhibits the occurrence of apoptosis, and the caspase protein activated after the occurrence of apoptosis blocks autophagy (Maiuri et al., 2007). For example, an experiment suggested that heme-induced apoptosis was promoted by autophagy via the BECN1/ATG5 pathway (Yang et al., 2020).
Ferroptosis is an iron-dependent non-apoptotic cell death mode (Ren et al., 2022). Ferroptosis in ICH is closely related to increased lipid peroxidation, disorders of iron metabolism, and glutathione (GSH)-glutathione peroxidase 4 (GPX4) antioxidant systems (Zhang et al., 2018). Necrosis is a caspase-independent and regulated form of cell death. The mass effect of hematoma formation, including thrombin, hemoglobin, and their metabolites, can lead to the necrosis of peripheral nerve cells through the pathophysiological process. Pyroptosis, a programmed cell death mediated by NLRP1/NLRP3 inflammasomes and the caspase family, is an important mechanism of inflammation-induced neuronal cell death in ICH. Pyroptosis is accompanied by cell lysis, which promotes the release of proinflammatory factors, in turn triggering and amplifying inflammatory responses (Gou et al., 2021).
Various cells in the central nervous system (CNS), such as microglia, cerebral endothelial cells, and astrocytes, are involved in SBI after ICH. In particular, these cells play an essential role in both inflammation and autophagy, but the functional significance of their reactions is still unclear.
Neurons are the most basic structural and functional units of the nervous system that maintain cellular homeostasis via strict protein quality control and elimination of abnormal organelles (Zhao et al., 2021). Generally, neurons are divided into three compartments, namely, the soma, the axon, and the dendrites. Neuronal lysosomes and autophagosomes fuse in the soma of the neuron, and most autophagosomes form in the axon (Sedlackova et al., 2020). Interestingly, some studies have found that the production and transport of autophagosomes mainly occur in axons, so axonal transport is crucial for the clearance of damaged mitochondria. Autophagy is especially important in neurons and can directly affect neuronal death. Therefore, neuronal cell lines have been used as models to analyze autophagy (Benito-Cuesta et al., 2017). However, the role of autophagy in neurons is complex, and current research has achieved satisfactory results. Under physiological conditions, autophagy in the neuron is maintained at a low level, but when stimulated, autophagy is enhanced (Stavoe and Holzbaur, 2019).
Microglia, as resident immune cells of the CNS, are mainly responsible for clearing damaged tissues and pathogens and reshaping the extracellular matrix (Liu J. et al., 2021). In a normal brain, they are stationary (Eldahshan et al., 2019). Microglia are sensitive to a variety of transcription factors and growth factors, and they activate rapidly by changing morphology and polarization in response to various brain injuries. Activated microglia respond to stimuli to maintain CNS homeostasis by phagocytosing to remove harmful substances. Previously, we have discussed the interaction between microglial autophagy and inflammation after ICH. The regulation of microglia-mediated neuroinflammation by autophagy is not an isolated process but has complex relationships with other pathophysiological processes, such as apoptosis.
Endotheliocytes are actively involved in the complex process of inflammation in the CNS. Endotheliocytes are significant components of the BBB, a critical barrier separating the CNS from peripheral cells. The BBB maintains CNS homeostasis by restricting the entry of inflammatory molecules and peripheral immune cells into the CNS (Abdullahi et al., 2018). A disrupted BBB is a critical feature of neuroinflammation leading to harmful inflammatory cascades (Zheng et al., 2021d). Autophagy plays an indispensable role in vascular diseases as a regulator of endotheliocytes senescence and inflammation. Autophagy has beneficial effects on BBB integrity. For instance, autophagy in endotheliocytes has been shown to target lipid deposition in vascular walls through selective autophagy (lipid phagocytosis). When stimulated by hypoxia and low energy, vascular endothelial growth factor (VEGF) activates autophagy through AMPK-dependent mechanisms, further enhanced by rapamycin treatment, which increases zonula occludens-1 (ZO-1) levels and protects cells from ROS production. However, the role of autophagy in regulating BBB homeostasis remains controversial, as some studies have shown that the activation of autophagy promotes the degradation of BBB components, such as claudin-5 or occludin, in endotheliocytes and capillaries (Yang Z. et al., 2021).
Astrocytes, known as “support cells,” are the most numerous glial cells in the CNS. The accumulation of astrocytes in the area surrounding the hematoma is observed 1–3 days after the onset of ICH (Scimemi, 2018). Astrocytes exhibit a stronger autophagy response to hypoxia and starvation by activating AMPK-dependent autophagy protection (Murray et al., 2022). In cultured astrocytes, thrombin activates autophagy (enhancing the conversion of LC3-I to LC3-II) in the brain, showing that thrombin plays a role in ICH-induced autophagy. Astrocyte-derived proinflammatory cytokine IL-17A can inhibit autophagy-mediated microglial activation and neuroinflammation, and it decreases the expressions of TNF-α, IL-1β, and P65 (Shi et al., 2018).
Oligodendrocytes are the main targets of white matter injury during the stroke (Fu et al., 2021b) and may interfere with autophagy flux through oxidative stress and mitochondrial damage, thereby preventing autophagy from degrading α-synuclein (α-Syn). However, the effect of oligodendrocytes on the pathophysiology of SBI has not been fully revealed (Kang and Yao, 2019). Autophagy not only contributes to T cell homeostasis and affects T cell repertoires and polarization but also contributes to antigen presentation. Platelet hyperactivity is the hallmark of thrombosis, and a study has demonstrated that substantial autophagy is induced (above basal level) by hemostatic agonists, decreasing platelet aggregation (Banerjee et al., 2019; Paul et al., 2019).
Interestingly, since neuroinflammation has been clearly associated with the extent of ICH-induced SBI, the regulation of neuroinflammation by autophagy is a promising therapeutic strategy. Accordingly, it is exciting to search for compounds or multiple targets that modulate inflammation through autophagy in ICH. Further exploration of the molecular mechanisms underlying the neuroprotective effects of these compounds will help us understand the pathology of ICH and illuminate potential treatments for ICH. In this section, we focus on targets and compounds related to the autophagy regulation of the inflammation treatment of ICH.
Several targets modulate inflammation through autophagy in the ICH (Table 3). MCC950, an NLRP3 inflammasome-specific inhibitor, inhibits NLRP3 derived from microglia via the AMPK/Beclin-1 pathway to prevent excessive autophagy and reduce the release of IL-1β into the cell (Zhang et al., 2022b). Besides, alpha 7 nicotinic receptor (α-7nAChR) is a subtype of acetylcholine receptor primarily found in the CNS, and α-7nAChR is activated against inflammatory diseases by triggering cholinergic anti-inflammatory pathways. PNU-282987, an α7-nAChR agonist, activates α7-NachR to reduce inflammatory factors, promote the polarization of macrophages into anti-inflammatory subtypes, repair BBB injury, alleviate acute brain edema, and then recover neurological dysfunction via the AMPK-mTOR-P70S6K-associated autophagy pathway (Su et al., 2022). Protocatechuic acid (PCA) is an important phenolic acid in plants with antibacterial, anti-tumor, and anti-oxidation effects. PCA is inhibited mTOR signaling and therefore improves M1/M2 polarization switching and attenuated neuroinflammation by suppressing the activation of the microglia (Xi et al., 2021).
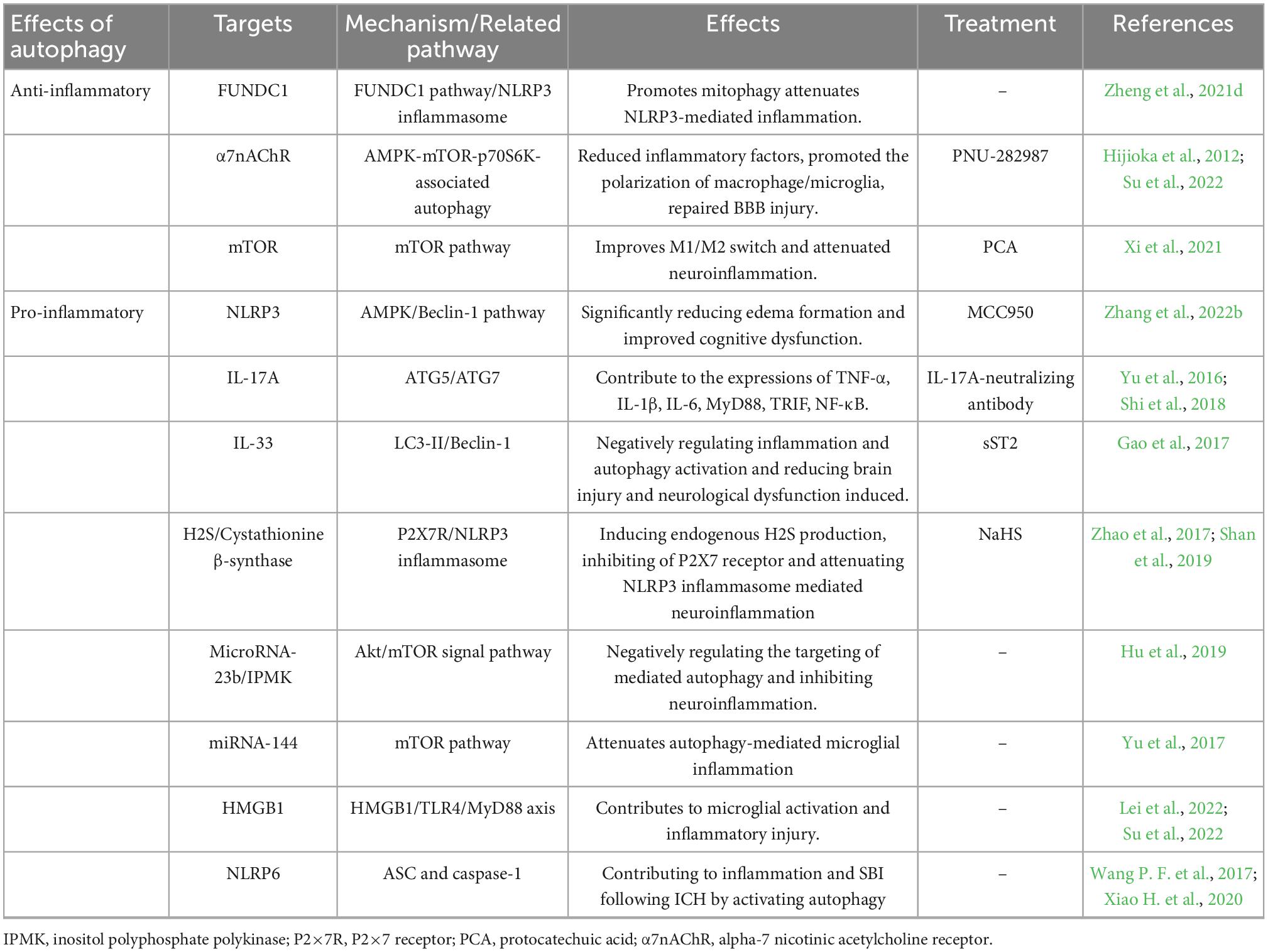
Table 3. Summary of potential targets related to autophagy and inflammation for ICH.
There are several poststroke autophagy drugs in clinical trials (Table 4). Although many autophagy-related drugs have been preclinically tested in the treatment of ICH, few have been tested for neuroprotective clinical efficacy.1 Only patients taking minocycline achieved beneficial results. Minocycline is a tetracycline class of oral antibiotics. After the injection of minocycline in rat models, Wu et al. found that Beclin-1 and LC3 II/I were suppressed and that minocycline could reduce brain injury via the anti-autophagy and anti-apoptosis pathways (Wu et al., 2016). Minocycline can reduce the iron overload caused by ICH, upregulate the expression of Bcl-2 protein, and restrict the flow of calcium ions into the mitochondrial membrane, thus inhibiting autophagy and heightening neural functioning. Statins such as lovastatin have been shown to reduce the risk of ICH, maintain the integrity of the BBB, and enhance neurological function (Chen C. J. et al., 2019). Hepcidin (a hepatic peptide hormone to regulate iron absorption) and hesperidin (a flavanone glycoside with a wide range of biological effects) could inhibit autophagy and reduce brain injury. However, hirudin (a thrombin inhibitor), luteolin (a group of flavonoids), and acupuncture could activate autophagy for neuroprotective effects.
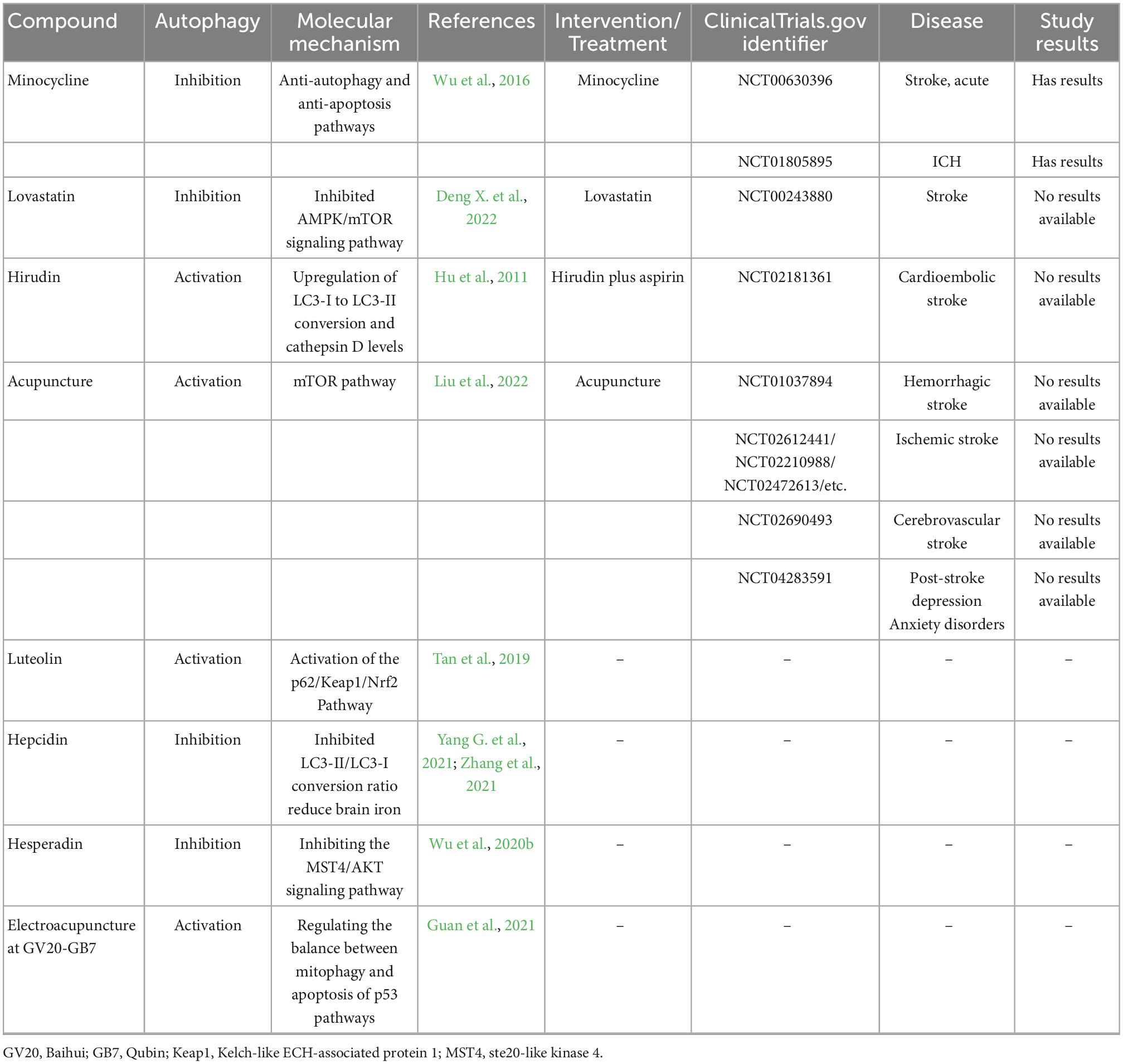
Table 4. Autophagy-related compounds in ICH and these compounds or treatment in clinical trials for stroke reported (https://www.clinicaltrials.gov/).
Intracerebral hemorrhage remains a worldwide concern and a serious risk to human longevity. However, the pathophysiology of ICH-induced SBI is extremely complex. Many studies have demonstrated that neuroinflammation and autophagy are promising targets for the treatment of strokes. Damaged organelles and misfolded proteins are degraded during autophagy, a cellular self-degradation process. However, due to the diversity of potential factors affecting autophagy, autophagy does not always act as an enemy against ICH-induced SBI. In contrast, autophagy sometimes contributes to inflammation as a friend of SBI. Is that because the activation of autophagy in ICH is excessive or the flow of autophagy is blocked? To better understand inflammation and autophagy in ICH, we consider it advisable to start with intracranial cells, such as microglia and neurons, although the current research on these aspects is not sufficient. The regulation of neuroinflammation by autophagy to treat ICH is an exciting area of research with many unanswered questions.
KF, WX, and JY designed the structure of the manuscript. CL, YM, TD, JW, QH, and FG managed the literature searches and analyses. KF wrote the manuscript. JY and LM assisted with the improvement of the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by grants from National Natural Science Foundation of China (nos. 82060225 and 82260239), Guangxi Natural Science Foundation (no. 2020GXNSFAA297154), and Guangxi Medical University Education and Teaching Reform Project (2020XJGA24) to JY.
We thank editage for the English language edition.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdullahi, W., Tripathi, D., and Ronaldson, P. T. (2018). Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol. Cell Physiol. 315, C343–C356. doi: 10.1152/ajpcell.00095.2018
Aronowski, J., and Zhao, X. (2011). Molecular pathophysiology of cerebral hemorrhage: Secondary brain injury. Stroke 42, 1781–1786. doi: 10.1161/STROKEAHA.110.596718
Banerjee, M., Huang, Y., Ouseph, M. M., Joshi, S., Pokrovskaya, I., Storrie, B., et al. (2019). Autophagy in platelets. Methods Mol. Biol. 1880, 511–528. doi: 10.1007/978-1-4939-8873-0_32
Bautista, W., Adelson, P. D., Bicher, N., Themistocleous, M., Tsivgoulis, G., and Chang, J. J. (2021). Secondary mechanisms of injury and viable pathophysiological targets in intracerebral hemorrhage. Ther. Adv. Neurol. Disord. 14:17562864211049208. doi: 10.1177/17562864211049208
Benito-Cuesta, I., Diez, H., Ordoñez, L., and Wandosell, F. (2017). Assessment of autophagy in neurons and brain tissue. Cells 6:25. doi: 10.3390/cells6030025
Bian, C., Wan, Y., Koduri, S., Hua, Y., Keep, R. F., and Xi, G. (2022). Iron-induced hydrocephalus: The role of choroid plexus stromal macrophages. Transl. Stroke Res. [Epub ahead of print]. doi: 10.1007/s12975-022-01031-6
Biasizzo, M., and Kopitar-Jerala, N. (2020). Interplay between NLRP3 inflammasome and autophagy. Front. Immunol. 11:591803. doi: 10.3389/fimmu.2020.591803
Bobinger, T., Burkardt, P., Huttner, H. B., and Manaenko, A. (2018). Programmed cell death after intracerebral hemorrhage. Curr. Neuropharmacol. 16, 1267–1281. doi: 10.2174/1570159X15666170602112851
Broz, P., and Dixit, V. M. (2016). Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420. doi: 10.1038/nri.2016.58
Cai, Y., Yang, E., Yao, X., Zhang, X., Wang, Q., Wang, Y., et al. (2021). FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol. 38:101792. doi: 10.1016/j.redox.2020.101792
Cao, S., Shrestha, S., Li, J., Yu, X., Chen, J., Yan, F., et al. (2017). Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci. Rep. 7:2417. doi: 10.1038/s41598-017-02679-z
Chen, M., Chen, Z., Wang, Y., Tan, Z., Zhu, C., Li, Y., et al. (2016). Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12, 689–702. doi: 10.1080/15548627.2016.1151580
Chen, Z., Xu, N., Dai, X., Zhao, C., Wu, X., Shankar, S., et al. (2019). Interleukin-33 reduces neuronal damage and white matter injury via selective microglia M2 polarization after intracerebral hemorrhage in rats. Brain Res. Bull. 150, 127–135. doi: 10.1016/j.brainresbull.2019.05.016
Chen, C. J., Ding, D., Ironside, N., Buell, T. J., Elder, L. J., Warren, A., et al. (2019). Statins for neuroprotection in spontaneous intracerebral hemorrhage. Neurology 93, 1056–1066. doi: 10.1212/WNL.0000000000008627
Cheng, Y., Liu, M., Tang, H., Chen, B., Yang, G., Zhao, W., et al. (2021). ITRAQ-based quantitative proteomics indicated Nrf2/OPTN-mediated mitophagy inhibits NLRP3 inflammasome activation after intracerebral hemorrhage. Oxidat. Med. Cell. Longev. 2021:6630281. doi: 10.1155/2021/6630281
Chung, C., Seo, W., Silwal, P., and Jo, E. K. (2020). Crosstalks between inflammasome and autophagy in cancer. J. Hematol. Oncol. 13:100. doi: 10.1186/s13045-020-00936-9
Deng, R., Wang, W., Xu, X., Ding, J., Wang, J., Yang, S., et al. (2021). Loss of MIC60 aggravates neuronal death by inducing mitochondrial dysfunction in a rat model of intracerebral hemorrhage. Mol. Neurobiol. 58, 4999–5013. doi: 10.1007/s12035-021-02468-w
Deng, S., Chen, X., Lei, Q., and Lu, W. (2022). AQP2 promotes astrocyte activation by modulating the TLR4/NFκB-p65 pathway following intracerebral hemorrhage. Front. Immunol. 13:847360. doi: 10.3389/fimmu.2022.847360
Deng, X., Yang, J., Qing, R., Yuan, H., Yue, P., and Tian, S. (2022). Suppressive role of lovastatin in intracerebral hemorrhage through repression of autophagy. Metab. Brain Dis. [Epub ahead of print]. doi: 10.1007/s11011-022-01101-6
Denton, D., and Kumar, S. (2019). Autophagy-dependent cell death. Cell Death Differ. 26, 605–616. doi: 10.1038/s41418-018-0252-y
Di, Y., He, Y. L., Zhao, T., Huang, X., Wu, K. W., Liu, S. H., et al. (2015). Methylene blue reduces acute cerebral ischemic injury via the induction of mitophagy. Mol. Med. 21, 420–429. doi: 10.2119/molmed.2015.00038
Duan, X., Wen, Z., Shen, H., Shen, M., and Chen, G. (2016). Intracerebral hemorrhage, oxidative stress, and antioxidant therapy. Oxid. Med. Cell. Longev. 2016:1203285. doi: 10.1155/2016/1203285
Duan, X. C., Wang, W., Feng, D. X., Yin, J., Zuo, G., Chen, D. D., et al. (2017). Roles of autophagy and endoplasmic reticulum stress in intracerebral hemorrhage-induced secondary brain injury in rats. CNS Neurosci. Ther. 23, 554–566. doi: 10.1111/cns.12703
Duris, K., Splichal, Z., and Jurajda, M. (2018). The role of inflammatory response in stroke associated programmed cell death. Curr. Neuropharmacol. 16, 1365–1374. doi: 10.2174/1570159X16666180222155833
Durocher, M., Knepp, B., Yee, A., Jickling, G., Rodriguez, F., Ng, K., et al. (2021). Molecular correlates of hemorrhage and edema volumes following human intracerebral hemorrhage implicate Inflammation, autophagy, mRNA splicing, and T cell receptor signaling. Transl. Stroke Res. 12, 754–777. doi: 10.1007/s12975-020-00869-y
Eldahshan, W., Fagan, S. C., and Ergul, A. (2019). Inflammation within the neurovascular unit: Focus on microglia for stroke injury and recovery. Pharmacol. Res. 147:104349. doi: 10.1016/j.phrs.2019.104349
Eser, P., Taskapilioglu, M. O., and Kocaeli, H. (2022). Targeting Mfsd2a in hemorrhagic cerebrovascular diseases. Transl. Stroke Res. 13, 861–862. doi: 10.1007/s12975-022-01015-6
Fang, Y., Chen, S., Reis, C., and Zhang, J. (2018). The role of autophagy in subarachnoid hemorrhage: An update. Curr. Neuropharmacol. 16, 1255–1266. doi: 10.2174/1570159X15666170406142631
Faruk, M. O., Ichimura, Y., and Komatsu, M. (2021). Selective autophagy. Cancer Sci. 112, 3972–3978. doi: 10.1111/cas.15112
Feng, D., Wang, B., Wang, L., Abraham, N., Tao, K., Huang, L., et al. (2017). Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE1 signalings. J. Pineal Res. 62, e12395. doi: 10.1111/jpi.12395
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Fu, X., Zeng, H., Zhao, J., Zhou, G., Zhou, H., Zhuang, J., et al. (2021a). Inhibition of dectin-1 ameliorates neuroinflammation by regulating microglia/macrophage phenotype after intracerebral hemorrhage in mice. Transl. Stroke Res. 12, 1018–1034. doi: 10.1007/s12975-021-00889-2
Fu, X., Zhou, G., Zhuang, J., Xu, C., Zhou, H., Peng, Y., et al. (2021b). White matter injury after intracerebral hemorrhage. Front. Neurol. 12:562090. doi: 10.3389/fneur.2021.562090
Galluzzi, L., and Green, D. R. (2019). Autophagy-independent functions of the autophagy machinery. Cell 177, 1682–1699. doi: 10.1016/j.cell.2019.05.026
Gao, Y., Ma, L., Luo, C. L., Wang, T., Zhang, M. Y., Shen, X., et al. (2017). IL-33 exerts neuroprotective effect in mice intracerebral hemorrhage model through suppressing inflammation/apoptotic/autophagic pathway. Mol. Neurobiol. 54, 3879–3892. doi: 10.1007/s12035-016-9947-6
Ghavami, S., Shojaei, S., Yeganeh, B., Ande, S. R., Jangamreddy, J. R., Mehrpour, M., et al. (2014). Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 112, 24–49. doi: 10.1016/j.pneurobio.2013.10.004
Gou, X., Xu, D., Li, F., Hou, K., Fang, W., and Li, Y. (2021). Pyroptosis in stroke-new insights into disease mechanisms and therapeutic strategies. J. Physiol. Biochem. 77, 511–529. doi: 10.1007/s13105-021-00817-w
Gu, C., Wu, Y., Fan, Z., and Han, W. (2018). Simvastatin improves intracerebral hemorrhage through NF-κB-mediated apoptosis via the MyD88/TRIF signaling pathway. Exp. Ther. Med. 15, 377–382. doi: 10.3892/etm.2017.5349
Gu, L., Sun, M., Li, R., Tao, Y., Luo, X., Xu, J., et al. (2022). Activation of RKIP binding ASC attenuates neuronal pyroptosis and brain injury via caspase-1/GSDMD signaling pathway after intracerebral hemorrhage in mice. Transl. Stroke Res. 13, 1037–1054. doi: 10.1007/s12975-022-01009-4
Guan, R., Li, Z., Dai, X., Zou, W., Yu, X., Liu, H., et al. (2021). Electroacupuncture at GV20-GB7 regulates mitophagy to protect against neurological deficits following intracerebral hemorrhage via inhibition of apoptosis. Mol. Med. Rep. 24:492. doi: 10.3892/mmr.2021.12131
Guan, R., Zou, W., Dai, X., Yu, X., Liu, H., Chen, Q., et al. (2018). Mitophagy, a potential therapeutic target for stroke. J. Biomed. Sci. 25:87. doi: 10.1186/s12929-018-0487-4
Hijioka, M., Matsushita, H., Ishibashi, H., Hisatsune, A., Isohama, Y., and Katsuki, H. (2012). α7 Nicotinic acetylcholine receptor agonist attenuates neuropathological changes associated with intracerebral hemorrhage in mice. Neuroscience 222, 10–19. doi: 10.1016/j.neuroscience.2012.07.024
Houtman, J., Freitag, K., Gimber, N., Schmoranzer, J., Heppner, F. L., and Jendrach, M. (2019). Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP3. EMBO J. 38:e99430. doi: 10.15252/embj.201899430
Hossain, M. I., Marcus, J. M., Lee, J. H., Garcia, P. L., Singh, V., Shacka, J. J., et al. (2021). Restoration of CTSD (cathepsin D) and lysosomal function in stroke is neuroprotective. Autophagy. 17:1330–1348. doi: 10.1080/15548627.2020.1761219
Hu, L., Zhang, H., Wang, B., Ao, Q., Shi, J., and He, Z. (2019). MicroRNA-23b alleviates neuroinflammation and brain injury in intracerebral hemorrhage by targeting inositol polyphosphate multikinase. Int. Immunopharmacol. 76:105887. doi: 10.1016/j.intimp.2019.105887
Hu, S., Xi, G., Jin, H., He, Y., Keep, R. F., and Hua, Y. (2011). Thrombin-induced autophagy: A potential role in intracerebral hemorrhage. Brain Res. 1424, 60–66. doi: 10.1016/j.brainres.2011.09.062
Huang, J., Zhu, T., Rong, R., You, M., Ji, D., and Li, H. (2020). FUN14 domain-containing 1-mediated mitophagy suppresses interleukin-1β production in macrophages. Int. Immunopharmacol. 88:106964. doi: 10.1016/j.intimp.2020.106964
Huang, Y., Xu, W., and Zhou, R. (2021). NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 18, 2114–2127. doi: 10.1038/s41423-021-00740-6
Jiang, B., Li, Y., Dai, W., Wu, A., Wu, H., and Mao, D. (2021). Hydrogen-rich saline alleviates early brain injury through regulating of ER stress and autophagy after experimental subarachnoid hemorrhage. Acta Cirurg. Brasil. 36:e360804. doi: 10.1590/ACB360804
Jung, C. H., Jun, C. B., Ro, S. H., Kim, Y. M., Otto, N. M., Cao, J., et al. (2009). ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003. doi: 10.1091/mbc.E08-12-1249
Kang, M., and Yao, Y. (2019). Oligodendrocytes in intracerebral hemorrhage. CNS Neurosci. Ther. 25, 1075–1084. doi: 10.1111/cns.13193
Kanno, H., Handa, K., Murakami, T., Aizawa, T., and Ozawa, H. (2022). Chaperone-mediated autophagy in neurodegenerative diseases and acute neurological insults in the central nervous system. Cells 11:1205. doi: 10.3390/cells11071205
Lamark, T., and Johansen, T. (2021). Mechanisms of selective autophagy. Annu. Rev. Cell Dev. Biol. 37, 143–169. doi: 10.1146/annurev-cellbio-120219-035530
Lee, J. W., Nam, H., Kim, L. E., Jeon, Y., Min, H., Ha, S., et al. (2019). TLR4 (toll-like receptor 4) activation suppresses autophagy through inhibition of FOXO3 and impairs phagocytic capacity of microglia. Autophagy 15, 753–770. doi: 10.1080/15548627.2018.1556946
Lei, C., Li, Y., Zhu, X., Li, H., and Chang, X. (2022). HMGB1/TLR4 induces autophagy and promotes neuroinflammation after intracerebral hemorrhage. Brain Res. 1792:148003. doi: 10.1016/j.brainres.2022.148003
Li, B., Wang, W., Li, Y., Wang, S., Liu, H., Xia, Z., et al. (2023). Cgas-STING pathway aggravates early cerebral ischemia-reperfusion injury in mice by activating NCOA4-mediated ferritinophagy. Exp. Neurol. 359:114269. doi: 10.1016/j.expneurol.2022.114269
Li, H., Tian, J., Yin, Y., Diao, S., Zhang, X., Zuo, T., et al. (2022). Interleukin-18 mediated inflammatory brain injury after intracerebral hemorrhage in male mice. J. Neurosci. Res. 100, 1359–1369. doi: 10.1002/jnr.25044
Li, W., He, P., Huang, Y., Li, Y. F., Lu, J., Li, M., et al. (2021d). Selective autophagy of intracellular organelles: Recent research advances. Theranostics 11, 222–256. doi: 10.7150/thno.49860
Li, J., Wu, X., He, Y., Wu, S., Guo, E., Feng, Y., et al. (2021b). PINK1 antagonize intracerebral hemorrhage by promoting mitochondrial autophagy. Ann. Clin. Transl. Neurol. 8, 1951–1960. doi: 10.1002/acn3.51425
Li, C., Sun, G., Chen, B., Xu, L., Ye, Y., He, J., et al. (2021a). Nuclear receptor coactivator 4-mediated ferritinophagy contributes to cerebral ischemia-induced ferroptosis in ischemic stroke. Pharmacol. Res. 174:105933. doi: 10.1016/j.phrs.2021.105933
Li, W., Chopp, M., Zacharek, A., Yang, W., Chen, Z., Landschoot-Ward, J., et al. (2021c). SUMO1 deficiency exacerbates neurological and cardiac dysfunction after intracerebral hemorrhage in aged mice. Transl. Stroke Res. 12, 631–642. doi: 10.1007/s12975-020-00837-6
Li, Y., Hu, K., Liang, M., Yan, Q., Huang, M., Jin, L., et al. (2021e). Stilbene glycoside upregulates SIRT3/AMPK to promotes neuronal mitochondrial autophagy and inhibit apoptosis in ischemic stroke. Adv. Clin. Exp. Med. 30, 139–146. doi: 10.17219/acem/130608
Li, H., Wu, J., Shen, H., Yao, X., Liu, C., Pianta, S., et al. (2018). Autophagy in hemorrhagic stroke: Mechanisms and clinical implications. Prog. Neurobiol. 163–164, 79–97. doi: 10.1016/j.pneurobio.2017.04.002
Li, J., Lu, J., Mi, Y., Shi, Z., Chen, C., Riley, J., et al. (2014). Voltage-dependent anion channels (VDACs) promote mitophagy to protect neuron from death in an early brain injury following a subarachnoid hemorrhage in rats. Brain Res. 1573, 74–83. doi: 10.1016/j.brainres.2014.05.021
Li, Q., Zhang, T., Wang, J., Zhang, Z., Zhai, Y., Yang, G. Y., et al. (2014). Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem. Biophys. Res. Commun. 444, 182–188. doi: 10.1016/j.bbrc.2014.01.032
Li, Y., and Chen, Y. (2019). AMPK and Autophagy. Adv. Exp. Med. Biol. 1206, 85–108. doi: 10.1007/978-981-15-0602-4_4
Liang, Y., Deng, Y., Zhao, J., Liu, L., Wang, J., Chen, P., et al. (2022). Ferritinophagy is involved in experimental subarachnoid hemorrhage-induced neuronal ferroptosis. Neurochem. Res. 47, 692–700. doi: 10.1007/s11064-021-03477-w
Liu, H., Zhang, B., Li, X. W., Du, J., Feng, P. P., Cheng, C., et al. (2022). Acupuncture inhibits mammalian target of rapamycin, promotes autophagy and attenuates neurological deficits in a rat model of hemorrhagic stroke. Acupunct. Med. 40, 59–67. doi: 10.1177/09645284211028873
Liu, J., Liu, L., Wang, X., Jiang, R., Bai, Q., and Wang, G. (2021). Microglia: A double-edged sword in intracerebral hemorrhage from basic mechanisms to clinical research. Front. Immunol. 12:675660. doi: 10.3389/fimmu.2021.675660
Liu, P., Yu, X., Dai, X., Zou, W., Yu, X., Niu, M., et al. (2021). Scalp acupuncture attenuates brain damage after intracerebral hemorrhage through enhanced mitophagy and reduced apoptosis in rats. Front. Aging Neurosci. 13:718631. doi: 10.3389/fnagi.2021.718631
Lőrincz, P., and Juhász, G. (2020). Autophagosome-lysosome fusion. J. Mol. Biol. 432, 2462–2482. doi: 10.1016/j.jmb.2019.10.028
Lou, G., Palikaras, K., Lautrup, S., Scheibye-Knudsen, M., Tavernarakis, N., and Fang, E. F. (2020). Mitophagy and neuroprotection. Trends Mol. Med. 26, 8–20. doi: 10.1016/j.molmed.2019.07.002
Luo, Y., Reis, C., and Chen, S. (2019). NLRP3 inflammasome in the pathophysiology of hemorrhagic stroke: A review. Curr. Neuropharmacol. 17, 582–589. doi: 10.2174/1570159X17666181227170053
Magid-Bernstein, J., Girard, R., Polster, S., Srinath, A., Romanos, S., Awad, A., et al. (2022). Cerebral hemorrhage: Pathophysiology, treatment, and future directions. Circul. Res. 130, 1204–1229. doi: 10.1161/CIRCRESAHA.121.319949
Maiuri, M. C., Zalckvar, E., Kimchi, A., and Kroemer, G. (2007). Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 8, 741–752.
Matsuzawa-Ishimoto, Y., Hwang, S., and Cadwell, K. (2018). Autophagy and inflammation. Annu. Rev. Immunol. 36, 73–101. doi: 10.1146/annurev-immunol-042617-053253
Miao, Y., Zhang, Z. X., Feng, X., and Sun, W. M. (2021). IL-33 as a novel serum prognostic marker of intracerebral hemorrhage. Oxidat. Med. Cell. Longev. 2021:5597790. doi: 10.1155/2021/5597790
Mizushima, N. (2010). The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 22, 132–139. doi: 10.1016/j.ceb.2009.12.004
Mo, Y., Sun, Y. Y., and Liu, K. Y. (2020). Autophagy and inflammation in ischemic stroke. Neural Regener. Res. 15, 1388–1396. doi: 10.4103/1673-5374.274331
Muller, S., Brun, S., René, F., de Sèze, J., Loeffler, J., and Jeltsch-David, H. (2017). Autophagy in neuroinflammatory diseases. Autoimmun. Rev. 16, 856–874. doi: 10.1016/j.autrev.2017.05.015
Murray, T. E., Richards, C. M., Robert-Gostlin, V. N., Bernath, A. K., Lindhout, A., and Klegeris, A. (2022). Potential neurotoxic activity of diverse molecules released by astrocytes. Brain Res. Bull. 189, 80–101. doi: 10.1016/j.brainresbull.2022.08.015
Nagata, S. (2018). Apoptosis and clearance of apoptotic cells. Annu. Rev. Immunol. 36, 489–517. doi: 10.1146/annurev-immunol-042617-053010
Nobleza, C. O. H. S. (2021). Intracerebral hemorrhage. Continuum 27, 1246–1277. doi: 10.1212/CON.0000000000001018
O’Carroll, C. B., Brown, B. L., and Freeman, W. D. (2021). Intracerebral hemorrhage: A common yet disproportionately deadly stroke subtype. Mayo Clin. Proc. 96, 1639–1654. doi: 10.1016/j.mayocp.2020.10.034
Paul, M., Hemshekhar, M., Kemparaju, K., and Girish, K. S. (2019). Aggregation is impaired in starved platelets due to enhanced autophagy and cellular energy depletion. Platelets 30, 487–497. doi: 10.1080/09537104.2018.1475630
Plaza-Zabala, A., Sierra-Torre, V., and Sierra, A. (2017). Autophagy and microglia: Novel partners in neurodegeneration and aging. Int. J. Mol. Sci. 18:598. doi: 10.3390/ijms18030598
Qiu, Y., Wang, J., Li, H., Yang, B., Wang, J., He, Q., et al. (2022). Emerging views of OPTN (optineurin) function in the autophagic process associated with disease. Autophagy 18, 73–85. doi: 10.1080/15548627.2021.1908722
Ren, S., Chen, Y., Wang, L., and Wu, G. (2022). Neuronal ferroptosis after intracerebral hemorrhage. Front. Mol. Biosci. 9:966478. doi: 10.3389/fmolb.2022.966478
Scimemi, A. (2018). Astrocytes and the warning signs of intracerebral hemorrhagic stroke. Neural Plast. 2018:7301623. doi: 10.1155/2018/7301623
Sedlackova, L., Kelly, G., and Korolchuk, V. I. (2020). The pROS of autophagy in neuronal health. J. Mol. Biol. 432, 2546–2559. doi: 10.1016/j.jmb.2020.01.020
Shadab, M., Millar, M. W., Slavin, S. A., Leonard, A., Fazal, F., and Rahman, A. (2020). Autophagy protein ATG7 is a critical regulator of endothelial cell inflammation and permeability. Sci. Rep. 10:13708. doi: 10.1038/s41598-020-70126-7
Shan, H., Qiu, J., Chang, P., Chu, Y., Gao, C., Wang, H., et al. (2019). Exogenous hydrogen sulfide offers neuroprotection on intracerebral hemorrhage injury through modulating endogenous hs metabolism in mice. Front. Cell. Neurosci. 13:349. doi: 10.3389/fncel.2019.00349
Shao, Z., Tu, S., and Shao, A. (2019). Pathophysiological mechanisms and potential therapeutic targets in intracerebral hemorrhage. Front. Pharmacol. 10:1079. doi: 10.3389/fphar.2019.01079
Shen, J. Z., Wu, G., and Guo, S. (2021). Amino acids in autophagy: Regulation and function. Adv. Exp. Med. Biol. 1332, 51–66. doi: 10.1007/978-3-030-74180-8_4
Shi, H., Wang, J., Wang, J., Huang, Z., and Yang, Z. (2018). IL-17A induces autophagy and promotes microglial neuroinflammation through ATG5 and ATG7 in intracerebral hemorrhage. J. Neuroimmunol. 323, 143–151. doi: 10.1016/j.jneuroim.2017.07.015
Shi, R. Y., Zhu, S. H., Li, V., Gibson, S. B., Xu, X. S., and Kong, J. M. (2014). BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 20, 1045–1055. doi: 10.1111/cns.12325
Shtaya, A., Bridges, L. R., Esiri, M. M., Lam-Wong, J., Nicoll, J. A. R., Boche, D., et al. (2019). Rapid neuroinflammatory changes in human acute intracerebral hemorrhage. Ann. Clin. Transl. Neurol. 6, 1465–1479. doi: 10.1002/acn3.50842
Stavoe, A. K. H., and Holzbaur, E. L. F. (2019). Autophagy in neurons. Annu. Rev. Cell Dev. Biol. 35, 477–500. doi: 10.1146/annurev-cellbio-100818-125242
Su, Y., Zhang, W., Zhang, R., Yuan, Q., Wu, R., Liu, X., et al. (2022). Activation of cholinergic anti-inflammatory pathway ameliorates cerebral and cardiac dysfunction after intracerebral hemorrhage through autophagy. Front. Immunol. 13:870174. doi: 10.3389/fimmu.2022.870174
Sun, E., Zhang, J., Deng, Y., Wang, J., Wu, Q., Chen, W., et al. (2022). Docosahexaenoic acid alleviates brain damage by promoting mitophagy in mice with ischaemic stroke. Oxidat. Med. Cell. Longev. 2022:3119649. doi: 10.1155/2022/3119649
Tan, X., Yang, Y., Xu, J., Zhang, P., Deng, R., Mao, Y., et al. (2019). Luteolin exerts neuroprotection modulation of the p62/Keap1/Nrf2 pathway in intracerebral hemorrhage. Front. Pharmacol. 10:1551. doi: 10.3389/fphar.2019.01551
Tanida, I., Ueno, T., and Kominami, E. (2008). LC3 and autophagy. Methods Mol. Biol. 445, 77–88. doi: 10.1007/978-1-59745-157-4_4
Taylor, R. A., Chang, C. F., Goods, B. A., Hammond, M. D., Mac Grory, B., Ai, Y., et al. (2017). TGF-β1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J. Clin. Investig. 127, 280–292. doi: 10.1172/JCI88647
Thangaraj, A., Sil, S., Tripathi, A., Chivero, E. T., Periyasamy, P., and Buch, S. (2020). Targeting endoplasmic reticulum stress and autophagy as therapeutic approaches for neurological diseases. Int. Rev. Cell Mol. Biol. 350, 285–325. doi: 10.1016/bs.ircmb.2019.11.001
Tian, W., Zhu, M., Zhou, Y., Mao, C., Zou, R., Cui, Y., et al. (2022). Electroacupuncture pretreatment alleviates cerebral ischemia-reperfusion injury by regulating mitophagy via mTOR-ULK1/FUNDC1 axis in rats. J. Stroke Cerebrovasc. Dis. 31:106202. doi: 10.1016/j.jstrokecerebrovasdis.2021.106202
Tian, X., Teng, J., and Chen, J. (2021). New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy 17, 2680–2688. doi: 10.1080/15548627.2020.1823124
Tschoe, C., Bushnell, C. D., Duncan, P. W., Alexander-Miller, M. A., and Wolfe, S. Q. (2020). Neuroinflammation after intracerebral hemorrhage and potential therapeutic targets. J. Stroke 22, 29–46. doi: 10.5853/jos.2019.02236
Vargas, J. N. S., Hamasaki, M., Kawabata, T., Youle, R. J., and Yoshimori, T. (2022). The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. [Epub ahead of print]. doi: 10.1038/s41580-022-00542-2
Wang, L., Tian, M., and Hao, Y. (2020). Role of p75 neurotrophin receptor in neuronal autophagy in intracerebral hemorrhage in rats through the mTOR signaling pathway. Cell Cycle 19, 376–389. doi: 10.1080/15384101.2019.1711318
Wang, P. F., Li, Z. G., Zhang, Y., Ju, X. H., Liu, X. W., Zhou, A. M., et al. (2017). NLRP6 inflammasome ameliorates brain injury after intracerebral hemorrhage. Front. Cell. Neurosci. 11:206. doi: 10.3389/fncel.2017.00206
Wang, X., Jiang, Y., Zhu, L., Cao, L., Xu, W., Rahman, S. U., et al. (2020). Autophagy protects PC12 cells against deoxynivalenol toxicity via the Class III PI3K/beclin 1/Bcl-2 pathway. J. Cell. Physiol. 235, 7803–7815. doi: 10.1002/jcp.29433
Wang, Y., and Zhang, H. (2019). Regulation of autophagy by mTOR signaling pathway. Adv. Exp. Med. Biol. 1206, 67–83. doi: 10.1007/978-981-15-0602-4_3
Wang, Z., Yuan, B., Fu, F., Huang, S., and Yang, Z. (2017). Hemoglobin enhances miRNA-144 expression and autophagic activation mediated inflammation of microglia via mTOR pathway. Sci. Rep. 7:11861. doi: 10.1038/s41598-017-12067-2
White, E. (2016). Autophagy and p53. Cold Spring Harbor Perspect. Med. 6:a026120. doi: 10.1101/cshperspect.a026120
Wilkinson, S. (2019). ER-phagy: Shaping up and destressing the endoplasmic reticulum. FEBS J. 286, 2645–2663. doi: 10.1111/febs.14932
Wu, C., Yan, X., Liao, Y., Liao, L., Huang, S., Zuo, Q., et al. (2019). Increased perihematomal neuron autophagy and plasma thrombin-antithrombin levels in patients with intracerebral hemorrhage: An observational study. Medicine 98:e17130. doi: 10.1097/MD.0000000000017130
Wu, X., Wu, F. H., Wu, Q., Zhang, S., Chen, S., and Sima, M. (2017). Phylogenetic and molecular evolutionary analysis of mitophagy receptors under hypoxic conditions. Front. Physiol. 8:539. doi: 10.3389/fphys.2017.00539
Wu, X., Luo, J., Liu, H., Cui, W., Guo, K., Zhao, L., et al. (2020a). Recombinant adiponectin peptide ameliorates brain injury following intracerebral hemorrhage by suppressing astrocyte-derived inflammation via the inhibition of drp1-mediated mitochondrial fission. Transl. Stroke Res. 11, 924–939. doi: 10.1007/s12975-019-00768-x
Wu, X., Wu, J., Hu, W., Wang, Q., Liu, H., Chu, Z., et al. (2020b). MST4 kinase inhibitor hesperadin attenuates autophagy and behavioral disorder via the MST4/AKT pathway in intracerebral hemorrhage mice. Behav. Neurol. 2020:2476861. doi: 10.1155/2020/2476861
Wu, Y., Wang, L., Hu, K., Yu, C., Zhu, Y., Zhang, S., et al. (2018). Mechanisms and therapeutic targets of depression after intracerebral hemorrhage. Front. Psychiatry 9:682. doi: 10.3389/fpsyt.2018.00682
Wu, Z., Zou, X., Zhu, W., Mao, Y., Chen, L., and Zhao, F. (2016). Minocycline is effective in intracerebral hemorrhage by inhibition of apoptosis and autophagy. J. Neurol. Sci. 371, 88–95. doi: 10.1016/j.jns.2016.10.025
Xi, Z., Xu, C., Chen, X., Wang, B., Zhong, Z., Sun, Q., et al. (2021). Protocatechuic acid suppresses microglia activation and facilitates M1 to M2 phenotype switching in intracerebral hemorrhage mice. J. Stroke Cerebrovasc. Dis. 30:105765. doi: 10.1016/j.jstrokecerebrovasdis.2021.105765
Xia, F., Keep, R. F., Ye, F., Holste, K. G., Wan, S., Xi, G., et al. (2022). The fate of erythrocytes after cerebral hemorrhage. Transl. Stroke Res. 13, 655–664. doi: 10.1007/s12975-021-00980-8
Xiao, H., Chen, H., Jiang, R., Zhang, L., Wang, L., Gan, H., et al. (2020). NLRP6 contributes to inflammation and brain injury following intracerebral haemorrhage by activating autophagy. J. Mol. Med. 98, 1319–1331. doi: 10.1007/s00109-020-01962-3
Xiao, L., Zheng, H., Li, J., Wang, Q., and Sun, H. (2020). Neuroinflammation mediated by NLRP3 inflammasome after intracerebral hemorrhage and potential therapeutic targets. Mol. Neurobiol. 57, 5130–5149. doi: 10.1007/s12035-020-02082-2
Xu, D., Kong, T., Zhang, S., Cheng, B., Chen, J., and Wang, C. (2021). Orexin-A protects against cerebral ischemia-reperfusion injury by inhibiting excessive autophagy through OX1R-mediated MAPK/ERK/mTOR pathway. Cell. Signal. 79:109839. doi: 10.1016/j.cellsig.2020.109839
Xu, T., Jiang, L., and Wang, Z. (2019). The progression of HMGB1-induced autophagy in cancer biology. Oncotargets Ther. 12, 365–377. doi: 10.2147/OTT.S185876
Xu, W., Ocak, U., Gao, L., Tu, S., Lenahan, C. J., Zhang, J., et al. (2021). Selective autophagy as a therapeutic target for neurological diseases. Cell. Mol. Life Sci. 78, 1369–1392. doi: 10.1007/s00018-020-03667-9
Xu, Z., Yang, L., Xu, S., Zhang, Z., and Cao, Y. (2015). The receptor proteins: Pivotal roles in selective autophagy. Acta Biochim. Biophys. Sin. 47, 571–580. doi: 10.1093/abbs/gmv055
Yang, G., Qian, C., Zhang, C., Bao, Y., Liu, M. Y., Jiang, F., et al. (2021). Hepcidin attenuates the iron-mediated secondary neuronal injury after intracerebral hemorrhage in rats. Transl. Res. 229, 53–68. doi: 10.1016/j.trsl.2020.09.002
Yang, Z., and Klionsky, D. J. (2010). Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22, 124–131. doi: 10.1016/j.ceb.2009.11.014
Yang, Z., Lin, P., Chen, B., Zhang, X., Xiao, W., Wu, S., et al. (2021). Autophagy alleviates hypoxia-induced blood-brain barrier injury via regulation of CLDN5 (claudin 5). Autophagy 17, 3048–3067. doi: 10.1080/15548627.2020.1851897
Yang, Z., Liu, B., Zhong, L., Shen, H., Lin, C., Lin, L., et al. (2015). Toll-like receptor-4-mediated autophagy contributes to microglial activation and inflammatory injury in mouse models of intracerebral haemorrhage. Neuropathol. Appl. Neurobiol. 41, e95–e106. doi: 10.1111/nan.12177
Yang, Z., Zhou, C., Shi, H., Zhang, N., Tang, B., and Ji, N. (2020). Heme induces BECN1/ATG5-mediated autophagic cell death via ER stress in neurons. Neurotox. Res. 38, 1037–1048. doi: 10.1007/s12640-020-00275-0
Yao, Z., Bai, Q., and Wang, G. (2021). Mechanisms of oxidative stress and therapeutic targets following intracerebral hemorrhage. Oxidat. Med. Cell. Longev. 2021:8815441. doi: 10.1155/2021/8815441
Ye, F., Garton, H. J. L., Hua, Y., Keep, R. F., and Xi, G. (2021). The role of thrombin in brain injury after hemorrhagic and ischemic stroke. Transl. Stroke Res. 12, 496–511. doi: 10.1007/s12975-020-00855-4
Ying, H., and Yue, B. Y. J. T. (2016). Optineurin: The autophagy connection. Exp. Eye Res. 144, 73–80. doi: 10.1016/j.exer.2015.06.029
Yu, A., Duan, H., Zhang, T., Pan, Y., Kou, Z., Zhang, X., et al. (2016). IL-17A promotes microglial activation and neuroinflammation in mouse models of intracerebral haemorrhage. Mol. Immunol. 73, 151–157. doi: 10.1016/j.m
Yu, A., Zhang, T., Zhong, W., Duan, H., Wang, S., Ye, P., et al. (2017). miRNA-144 induces microglial autophagy and inflammation following intracerebral hemorrhage. Immunol. Lett. 182, 18–23. doi: 10.1016/j.imlet.2017.01.002
Yu, L., Chen, Y., and Tooze, S. A. (2018). Autophagy pathway: Cellular and molecular mechanisms. Autophagy 14, 207–215. doi: 10.1080/15548627.2017.1378838
Yuan, B., Shen, H., Lin, L., Su, T., Zhong, L., and Yang, Z. (2017). Autophagy promotes microglia activation through beclin-1-Atg5 pathway in intracerebral hemorrhage. Mol. Neurobiol. 54, 115–124. doi: 10.1007/s12035-015-9642-z
Zachari, M., and Ganley, I. G. (2017). The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 61, 585–596. doi: 10.1042/EBC20170021
Zhang, C., Qian, C., Yang, G., Bao, Y. X., and Qian, Z. M. (2021). Hepcidin inhibits autophagy in intracerebral hemorrhage models in vitro and in vivo. Mol. Cell. Neurosci. 111:103589. doi: 10.1016/j.mcn.2021.103589
Zhang, L., Miao, S., Yang, Z., Li, Z., Fan, Y., Yu, K., et al. (2022a). Suppression of HMGB1 inhibits neuronal autophagy and apoptosis to improve neurological deficits in rats following intracerebral hemorrhage. Nan Fang Yi Ke Da Xue Xue Bao 42, 1050–1056. doi: 10.12122/j.issn.1673-4254.2022.07.13
Zhang, Z., Guo, P., Huang, S., Jia, Z., Chen, T., Liu, X., et al. (2022b). Inhibiting microglia-derived NLRP3 alleviates subependymal edema and cognitive dysfunction in posthemorrhagic hydrocephalus after intracerebral hemorrhage via AMPK/beclin-1 pathway. Oxidat. Med. Cell. Longev. 2022:4177317. doi: 10.1155/2022/4177317
Zhang, Y., Zhang, T., Li, Y., Guo, Y., Liu, B., Tian, Y., et al. (2022c). Metformin attenuates early brain injury after subarachnoid hemorrhage in rats via AMPK-dependent mitophagy. Exp. Neurol. 353:114055. doi: 10.1016/j.expneurol.2022.114055
Zhang, S., Hu, Z. W., Luo, H. Y., Mao, C. Y., Tang, M. B., Li, Y. S., et al. (2020). AAV/BBB-mediated gene transfer of chip attenuates brain injury following experimental intracerebral hemorrhage. Transl. Stroke Res. 11, 296–309. doi: 10.1007/s12975-019-00715-w
Zhang, T., Wu, P., Budbazar, E., Zhu, Q., Sun, C., Mo, J., et al. (2019). Mitophagy reduces oxidative stress via keap1 (kelch-like epichlorohydrin-associated protein 1)/Nrf2 (nuclear factor-E2-related factor 2)/PHB2 (prohibitin 2) pathway after subarachnoid hemorrhage in rats. Stroke 50, 978–988. doi: 10.1161/STROKEAHA.118.021590
Zhang, Y., and Liu, C. (2020). Autophagy and hemorrhagic stroke. Adv. Exp. Med. Biol. 1207, 135–147. doi: 10.1007/978-981-15-4272-5_8
Zhang, Z., Wu, Y., Yuan, S., Zhang, P., Zhang, J., Li, H., et al. (2018). Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 1701, 112–125. doi: 10.1016/j.brainres.2018.09.012
Zhao, H., Pan, P., Yang, Y., Ge, H., Chen, W., Qu, J., et al. (2017). Endogenous hydrogen sulphide attenuates NLRP3 inflammasome-mediated neuroinflammation by suppressing the P2X7 receptor after intracerebral haemorrhage in rats. J. Neuroinflamm. 14:163. doi: 10.1186/s12974-017-0940-4
Zhao, S., Li, X., Wang, J., and Wang, H. (2021). The role of the effects of autophagy on NLRP3 inflammasome in inflammatory nervous system diseases. Front. Cell Dev. Biol. 9:657478. doi: 10.3389/fcell.2021.657478
Zheng, D., Kern, L., and Elinav, E. (2021b). The NLRP6 inflammasome. Immunology 162, 281–289. doi: 10.1111/imm.13293
Zheng, B., Zhou, X., Pang, L., Che, Y., and Qi, X. (2021a). Baicalin suppresses autophagy-dependent ferroptosis in early brain injury after subarachnoid hemorrhage. Bioengineered 12, 7794–7804. doi: 10.1080/21655979.2021.1975999
Zheng, K., Dong, Y., Yang, R., Liang, Y., Wu, H., and He, Z. (2021c). Regulation of ferroptosis by bioactive phytochemicals: Implications for medical nutritional therapy. Pharmacol. Res. 168:105580. doi: 10.1016/j.phrs.2021.105580
Zheng, S., Jian, D., Gan, H., Wang, L., Zhao, J., and Zhai, X. (2021d). FUNDC1 inhibits NLRP3-mediated inflammation after intracerebral hemorrhage by promoting mitophagy in mice. Neurosci. Lett. 756:135967. doi: 10.1016/j.neulet.2021.135967
Zheng, Y., Zhou, Z., Han, F., and Chen, Z. (2021e). Special issue: Neuroinflammatory pathways as treatment targets in brain disorders autophagic regulation of neuroinflammation in ischemic stroke. Neurochem. Int. 148:105114. doi: 10.1016/j.neuint.2021.105114
Zhu, H., Wang, Z., Yu, J., Yang, X., He, F., Liu, Z., et al. (2019). Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. Prog. Neurobiol. 178:101610. doi: 10.1016/j.pneurobio.2019.03.003
Keywords: intracerebral hemorrhage, autophagy, secondary brain injury, neuroinflammatory, NLRP3 inflammasome, microglia, mitophagy
Citation: Fu K, Xu W, Lenahan C, Mo Y, Wen J, Deng T, Huang Q, Guo F, Mo L and Yan J (2023) Autophagy regulates inflammation in intracerebral hemorrhage: Enemy or friend? Front. Cell. Neurosci. 16:1036313. doi: 10.3389/fncel.2022.1036313
Received: 04 September 2022; Accepted: 19 December 2022;
Published: 16 January 2023.
Edited by:
Dirk M. Hermann, University of Duisburg-Essen, GermanyReviewed by:
Sahabuddin Ahmed, Yale University, United StatesCopyright © 2023 Fu, Xu, Lenahan, Mo, Wen, Deng, Huang, Guo, Mo and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Yan,  eWFuanVuQGd4bXUuZWR1LmNu; Ligen Mo, bGlnZW5tb0AxNjMuY29t
eWFuanVuQGd4bXUuZWR1LmNu; Ligen Mo, bGlnZW5tb0AxNjMuY29t
†These authors have contributed equally to this work
‡ORCID: Jun Yan, orcid.org/0000-0001-6847-2985
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.