
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 26 October 2022
Sec. Non-Neuronal Cells
Volume 16 - 2022 | https://doi.org/10.3389/fncel.2022.1028653
This article is part of the Research Topic The Role of Glial Cells in the Autoimmune Diseases of Nervous System View all 5 articles
 Zhao-Qing Li†
Zhao-Qing Li† Tong-Xin Li†
Tong-Xin Li† Miao Tian†
Miao Tian† Ze-Sheng Ren
Ze-Sheng Ren Chen-Yi Yuan
Chen-Yi Yuan Rui-Kun Yang
Rui-Kun Yang Su-Juan Shi
Su-Juan Shi Hui Li*
Hui Li* Zhen-Zhen Kou*
Zhen-Zhen Kou*Neurologic autoimmune disorders affect people’s physical and mental health seriously. Glial cells, as an important part of the nervous system, play a vital role in the occurrence of neurologic autoimmune disorders. Glial cells can be hyperactivated in the presence of autoantibodies or pathological changes, to influence neurologic autoimmune disorders. This review is mainly focused on the roles of glial cells in neurologic autoimmune disorders and the influence of autoantibodies produced by autoimmune disorders on glial cells. We speculate that the possibility of glial cells might be a novel way for the investigation and therapy of neurologic autoimmune disorders.
Neurologic autoimmune disorders are caused by an adaptive immune response directed against an antigen expressed within the nervous system, mainly involving young and middle-aged people with high mortality and disability (Rubin et al., 2018; Bhagavati, 2021). It is reported that environmental genetic susceptibility and various stress factors, including virus infection, childbirth, overwork, trauma, emotional agitation, and vaccination contribute to the development of neurologic autoimmune disorders (Fujinami, 2001). To date, there are more than 30 known autoimmune diseases of the nervous system. The most common of them are multiple sclerosis (MS) and Guillain–Barre syndrome (GBS).
It has been known that the etiology of these neurologic autoimmune disorders might have three potential mechanisms: (1) the common autoimmune response to myelin antigens or epitopes in the central nervous system (CNS) and peripheral nervous system (PNS); (2) the high general susceptibility to autoimmune diseases, for instance, autoimmune diseases may be caused or exacerbated by immunomodulatory therapy; and (3) the co-occurrence in both CNS and PNS could be a coincidence (Kamm and Zettl, 2012). Almost every structure of the CNS or PNS may become the target of autoimmune attacks. However, the exact causes of the neurologic autoimmune disorders remain unclear.
Glial cells play a key role in brain physiology, metabolism, development, and even nervous system diseases (Herculano-Houzel, 2014). Glial cells are involved in neuroinflammation and synaptic homeostasis, which are important for maintaining the physiological function of the nervous system. Recently, great attention has been paid to glial cells in nervous system diseases, especially neurologic autoimmune disorders (Brambilla, 2019; Voet et al., 2019). Myelination of axons is formed by oligodendrocytes and Schwann cells, which form an electrical insulator capable of rapid signal transmission. The importance of oligodendrocytes in the pathology of demyelinating diseases such as MS has been recognized, because myelin loss directly affects nerve transmission. Moreover, oligodendrocyte regulates neuronal metabolic support, which attributes to neuronal existence or death. In response to injury and inflammation, microglia and astrocytes could be activated immediately, or even overactive, which might be the potential reason for the occurrence of neurologic autoimmune disorders (Liddelow et al., 2020).
Nervous system autoantibodies mediate neurologic autoimmune disorders. According to the distribution of their target antigens, these nervous system autoantibodies could be roughly divided into PNS antibodies and CNS antibodies. In general, autoantibodies are stimulated by inflammation in specific organs and tissues. In some cases, cross-reactivity with microbial antigens could be detected. In specific conditions, such as virus infection, exposure to certain toxic chemicals, or neoplasms, antibodies targeting organs and tissue would trigger pathogenic status, which is always temporary when exposure is reduced or eliminated (Geng et al., 2020), resulting in the progression of autoimmune diseases. Autoantibodies, self-antigens, and other immune factors form an immune complex in the CNS and PNS, resulting in the dysfunction or destruction of neurons and glia (Kleopa, 2011).
In line with the clinical manifestations, the nervous system autoantibodies can be divided into: (1) autoimmune encephalitis-related antibodies: N-methyl-D-aspartate receptor (NMDAR), α-amino-3-hydroxy-5-metnyl-4-isoxazolepropionic acid receptor (AMPAR), gamma-aminobutyric acid B receptor (GABABR), voltage-gated potassium channel (VGKC), glycine receptor, metabotropic glutamate receptor 1 (mGluR1), metabotropic glutamate receptor 5 (mGluR5); (2) neuromyelitis pedigree-related antibodies: including myelin oligodendrocyte glycoprotein (MOG), myelin basic protein (MBP); (3) immune-mediated peripheral neuropathy-related antibodies: ganglioside M1 (GM1), ganglioside M2 (GM2), ganglioside M3 (GM3), ganglioside D1a (GD1a), ganglioside D1b (GD1b), ganglioside T1b (GT1b), ganglioside Q1B (GQ1b), myelin-associated glycoprotein (MAG), and thioester; and (4) immune-mediated neuromuscular junction disease-related antibodies: acetylcholine receptor (AChR) (Lalive, 2008; Zhang and Popovich, 2011; Gold et al., 2012; Pollak et al., 2016; Dalmau et al., 2017; Karim and Jacob, 2018). These autoantibodies not only attack neurons but also glial cells, leading to the imbalance of neural networks, disturbance of homeostasis, and inflammation hyperreaction, those further aggravate the destruction of neurons. In addition, the autoantibodies produced by autoimmunity or the autoantigens could recruit more glial cells in response to autoimmune reactions, resulting in neurologic autoimmune disorders in the nervous system. Autoantibodies targeting the aquaporin-4 (AQP4) channel, which is enriched on the surface of astrocytes and is involved in maintaining the stability of the blood–brain barrier (BBB), for example, cause neuromyelitis optica spectrum disorders (NMODSs).
Taken together, glial cells, as an important part of the nervous system, could be hyperactivated in the presence of autoantibodies or pathological changes, affecting the occurrence of neurologic autoimmune disorders. This review will focus on the roles of glial cells in neurologic autoimmune disorders and the influence of autoantibodies produced by autoimmune disorders on glial cells.
As an important factor in the balance of the neural network, glial cells play a role in maintaining the stability and functional integrity of the nervous system (Casella et al., 2020; Healy et al., 2020). The types of glial cells mainly include astrocytes, oligodendrocytes, and microglia in the CNS. The Schwann cells and satellite glia are presented in the PNS.
Astrocytes are the most widely distributed glial cells in the CNS and have star-shaped cell bodies with numerous long and branching processes, which are important for supporting neurons through tight metabolic coupling (Ioannou et al., 2019). The fine morphology of the processes of astrocytes makes close contact with neuronal synapses, therefore, directly affecting local neurotransmission. Most importantly, astrocytes are critical for propagating inflammation, which contributes to disease development. On the other side, in response to immune reactions, as a crucial part of BBB, astrocytes prevent the inflammatory cells from entering the CNS during disease and injury (Heneka et al., 2018).
Oligodendrocytes are found throughout the gray and white matter in the CNS. In the adult brain, the cell body of oligodendrocytes is round or oval, with fewer protrusions wrapped around the neuronal axons and are responsible for the formation of myelin sheath in the CNS. Its function is crucial for forming an insulating cover around the axon, thus improving the transmission speed of electrical signals. Myelinating oligodendrocytes originate from oligodendrocyte progenitor cells (OPCs), which are maintained in the adult CNS. The abilities of proliferation and differentiation from OPCs into myelinating oligodendrocytes are critical for the progress of neurologic autoimmune disorders, for instance, MS (Falcao et al., 2018).
Microglia are the smallest glial cell type in the CNS. The cell body of microglia is small and short rod-shaped. The protrusions from the cell body are long and thin, and there are many small spinous processes on the surface. According to the function and status, microglia can be defined as three types, process-bearing, highly ramified myeloid cells, and tissue-resident macrophages (Greter et al., 2015). In response to injury and disorders, microglia are highly dynamic and activating (Nimmerjahn et al., 2005). Particularly in synapses of neurons, microglia are involved in complement-mediated synapse elimination, which is critical for neuronal circuit development (Schafer et al., 2012).
Interestingly, in the PNS, macrophages have similar functions as microglia in the CNS, for instance, presentation of antigens and cytokines production (Cao and He, 2013). Microglia have been shown to be derived from the embryonic yolk sac and migrate into the CNS (Ginhoux et al., 2010). Macrophages have two different origins, one part is from embryonic progenitors in the yolk sac and fetal liver, and another part is derived from hematopoietic stem cells (HSCs) in the bone marrow and blood monocytes (Crotti and Ransohoff, 2016). There is an overlap in the origin of microglia with that of tissue macrophages.
During inflammatory demyelination, macrophages from HSCs are thought to differentiate from two types of monocytes, the inflammatory monocytes, and the resident monocytes, and two groups exhibit important migratory and functional differences between mice and humans. Reports indicated that the inflammatory monocytes are pro-inflammatory and involved in demyelination in MS, whereas the resident group act as patrolling cells (Fogg et al., 2006).
Schwann cells form the myelin sheath around the axons of neurons in the PNS. In the mature myelinated nerve axons, Schwann cells surround the segments between two Ranvier nodes of the nerve fibers, thus greatly accelerating the speed of neurotransmission in peripheral nerves. Therefore, when the myelin sheath of the PNS is damaged in neurologic autoimmune disorders, it ultimately affects the function of Schwann cells and axons. GBS is a rare neurologic autoimmune disease affecting the Schwann cells in the PNS (McGonigal et al., 2022).
Satellite cells surround the neuronal bodies of ganglion cells in the ganglia (such as sensory ganglia, sympathetic ganglia, and parasympathetic ganglia) of the PNS. Satellite cells have been found to play a variety of roles, including controlling the microenvironment of the sympathetic ganglia. They are considered to have similar functions to astrocytes in the central nervous system, providing support, nutrition, and protection for the surrounding neurons. In addition, satellite cells express a variety of receptors (such as purinergic receptors) that can interact with neuroactive substances (Kuang and Rudnicki, 2008).
Multiple sclerosis is an autoimmune disease characterized by chronic inflammation, demyelination, and gliosis in the CNS. The most affected regions by MS in the CNS are the periventricular area, subcortical area, optic nerve, spinal cord, brainstem, and cerebellum (Filippi et al., 2018). The pathogenesis of MS is involved in the aberrant immune response directly against myelin antigen, such as the immune attack against MBP, resulting in the loss of myelin sheath in the white matter of the CNS. In the progress of MS, glial cells play an important role in the pathogenesis of MS autoimmunity (Figure 1). Pathological studies also suggested that the process of multiple demyelination in white matter is often accompanied by reactive gliosis (van Wageningen et al., 2022).
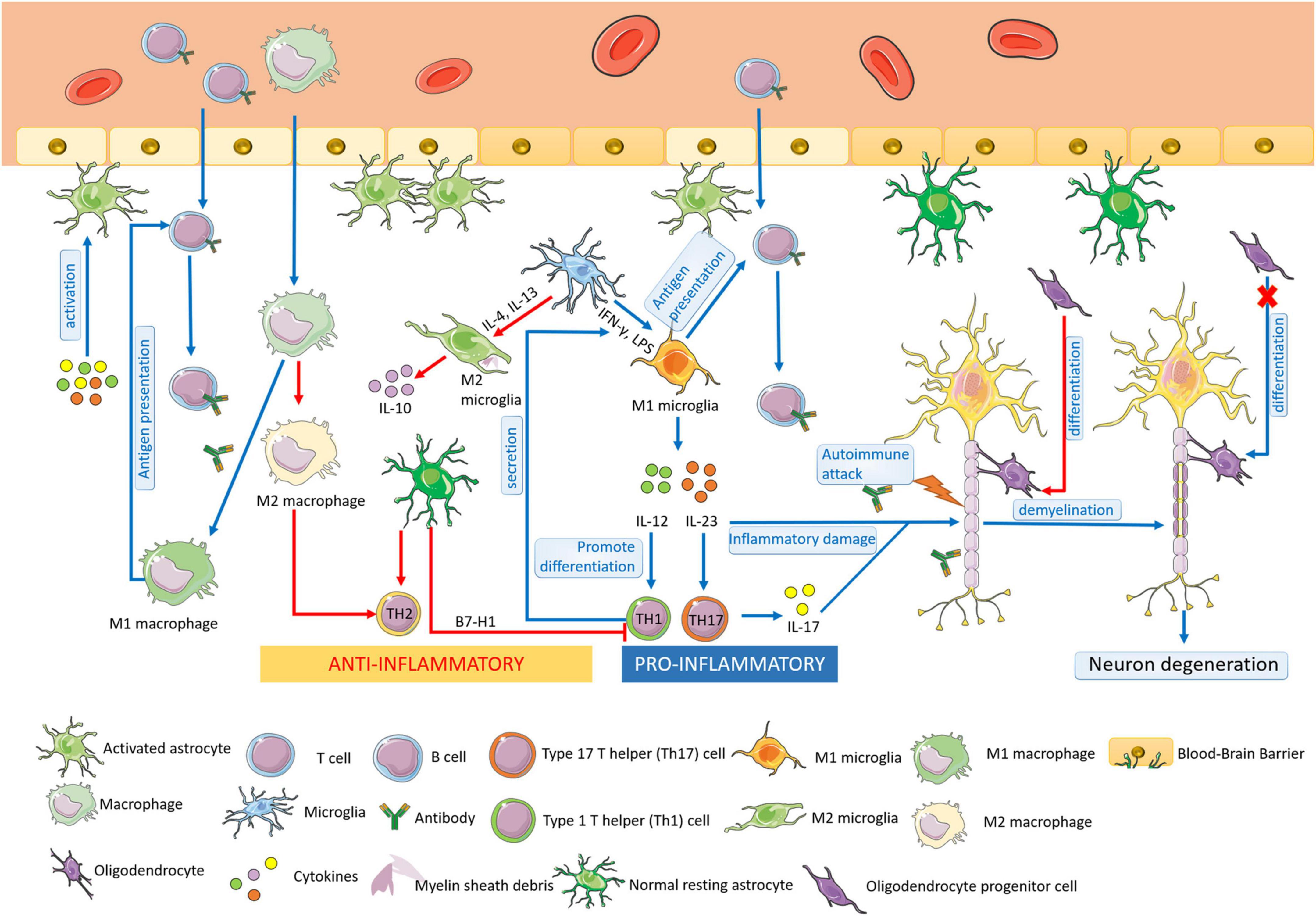
Figure 1. The pathogenesis of MS. Multiple sclerosis (MS) is an autoimmune disease characterized by chronic inflammation, demyelination, and gliosis in the CNS. At the early stage of MS, microglia could be activated and proliferated, which act as antigen-presenting cells. M1 microglia are differentiated by IFN-γ and LPS, secreting IL-12 and IL-23, promoting the differentiation of Th1 cells and Th 17 cells, leading to the aggravation of the inflammatory response of MS. M2 microglia are activated by IL-4/IL-13 and play a role in anti-inflammation. Macrophages have similar functions as microglia, including the presentation of antigens and cytokines production. M1 macrophages have the antigen-presenting ability the same as M1 microglia, resulting in the demyelination of nerves in MS. M2 macrophages increases Th2 cell differentiation to protect oligodendrocytes and neurons from damage. In MS, astrocytes can be activated, enhancing the permeability of BBB, and recruiting lymphocytes into the CNS. Moreover, astrocytes participate in the formation of BBB and secret immune inhibitory factors, acting in an anti-inflammatory role. The resting astrocytes inhibit the proliferation and promote apoptosis of Th1 cells and promote the secretion of anti-inflammatory cytokines by Th2 cells. Oligodendrocytes form the myelin sheath of CNS axons, providing nutrition and protecting nerve axons. At the early stage of MS, the regeneration and self-healing of nerve myelin sheath lesions are completed by the differentiation of OPCs into mature oligodendrocytes, but the differentiation ability is finally lost in progressive MS.
Microglia have the functions of phagocytosis, antigen presentation, and cytokine production. At the early stage of MS, microglia could be activated and proliferated, which act as antigen-presenting cells. The activated microglia can be observed in the white matter of brain tissue in patients with early MS (Singh et al., 2013).
Studies show that microglia play a dual role in MS, which can not only promote an inflammatory response that aggravates tissue damage but also have neuroprotective and repair effects. The activated microglia could be divided into M1 and M2, which are attributed to their roles in MS, respectively. Cytokine interferon-γ (IFN-γ), lipopolysaccharide (LPS) induces microglia to differentiate into M1, which secretes interleukin-12 (IL-12) and interleukin-23 (IL-23). IL-12 is the main factor promoting the differentiation of type 1 T helper (Th1) cells; IL-23 promotes the type 17 T helper (Th17) cell differentiation, leading to the production of the inflammatory factor interleukin-17 (IL-17) secretion. Thus, the M1 microglia aggravate the inflammatory response of MS (Kong et al., 2016; Thompson and Tsirka, 2017). Microglia can be stimulated by interleukin-4 (IL-4)/ interleukin-13 (IL-13) to an M2 phenotype for the resolution of inflammation and tissue repair. In MS, M2 microglia have neuroprotective effects. The activation and proliferation of M2 microglia can remove extracellular oxidized proteins, phagocytic debris, and degenerated myelin sheath in brain tissue, provide a relatively suitable microenvironment for neuronal repair, and help to mend the damaged nervous system (Jackle et al., 2020).
Macrophages are as same important as microglia in the pathogenesis of MS. M1 macrophages have the antigen-presenting ability the same as M1 microglia, resulting in the demyelination of nerves in MS. Moreover, M2 macrophages play an anti-inflammatory role with microglia by increasing type 2 T helper (Th2) cell differentiation, therefore, protecting oligodendrocytes and neurons from damage (Cao and He, 2013).
Astrocytes not only account for the majority of the CNS in quantity but also play a critical role in immune diseases of the CNS. Astrocytes can be activated by inflammatory factors, ischemia, hypoxia, and other stimuli. The permeability of BBB could be increased by activated astrocytes, facilitating the passage of lymphocytes to participate in the occurrence and development of lesions. Simultaneously, activated astrocytes directly or indirectly activate and recruit peripheral activated immune cells to the CNS by secreting cytokines, which is an important factor in the pathogenesis of MS (Farina et al., 2007; Aharoni et al., 2021). It was reported that the blockade of astrocyte CD38 activity suppressed pro-inflammatory transcriptional reprogramming, leading to the decreased expressions of pro-inflammatory transcriptional modules, which might contribute to CNS pathology in MS.
Moreover, astrocytes are not only involved in the occurrence of inflammation in the CNS but also act as important anti-inflammatory cells. At rest, as a maintainer of the stability of the CNS, astrocytes participate in the formation of BBB with vascular endothelial cells through its rich foot process structure, but also play a role in the biological barrier through the secretion of immune inhibitory factors. Studies have reported that normal resting astrocytes may inhibit the proliferation and promote apoptosis of T cells by expressing B7-H1 (Lipp et al., 2007). The proliferation of antigen-specific Th1 cells could be inhibited by astrocytes, which promote the secretion of anti-inflammatory cytokines by Th2 cells as well, thus inhibiting the development of inflammation (Liddelow et al., 2020). Therefore, the roles of astrocytes in the pathogenesis of MS still need further research.
Oligodendrocytes form the myelin sheath of CNS axons, providing nutrition and protecting nerve axons. As demonstrated, the pathological basis of MS is chronic inflammatory demyelination and neuronal degeneration of the CNS. At the early stage of MS, demyelinating lesions formed sclerotic plaques and had the function of nerve myelin regeneration and self-healing. However, with the development of MS, the functions of regeneration weaken and lose, eventually, the degeneration of nerve axons and neurons leads to brain parenchyma atrophy and severe disability (Brown et al., 2013). It is noted that the regeneration and self-healing of nerve myelin sheath at the early stage of MS lesions are completed by the differentiation of OPCs into mature glial cells (Meijer et al., 2022). Although OPCs always exist in the CNS, this differentiation ability in the lesions is finally lost in progressive MS (Ghorbani et al., 2022).
The pathology and the course of MS are sophisticated, suggesting that its clinical diagnosis and therapy might require targeting multiple biological processes. Although MS is generally considered to be triggered by autoimmune-mediated attacks of CNS myelin sheath, the anti-inflammatory treatments could not completely alleviate and prevent the development of MS, which is shown to be more and extensive axonal demyelination and degeneration (Trapp et al., 1998).
In response to neurologic autoimmune disorders, glial cells might be involved and subsequently connected to numerous biomarkers, which are critical and useful for diagnosis and therapy. In patients with MS, several myelin proteins including MBP and MOG have been detected and explored in cerebrospinal fluid (CSF) and serum, which are potential biomarkers for diagnosis.
Lenaldekar, an effective inhibitor of activated T cell proliferation, has been suggested to be a novel therapeutic approach for patients with MS. Moreover, in an experimental autoimmune encephalomyelitis (EAE) rodent model of MS, Lenaldekar treatment inhibited relapse severity, indicating a perspective application (Cusick et al., 2012). However, it is noted that no available animal model recapitulates the complicated and divergent pathogenesis of MS currently.
From the pathology of MS, preventing demyelination and promoting the regeneration of myelin sheath represent another potential therapeutic strategy for the treatment of MS. It is found that oligodendrocytes also produce nerve growth factor, neuregulin, glial-derived neurotrophic factor, and transforming growth factor (Acosta et al., 2013). These factors bind to corresponding receptors on neurons and glial cells and then affect the development and survival of neurons, astrocytes, and even oligodendrocytes themselves. In a word, oligodendrocytes play an important role in the progression and repair of MS disease (Zuchero and Barres, 2015). The enhancement of oligodendrocyte differentiation could be a way to help the regeneration of the myelin sheath (Rittchen et al., 2015); although many OPCs are in lesions sites at the early stage of MS, only rare maturing progenitors are found in chronic MS lesions, indicating that the differentiation capacity of OPCs is impaired in MS lesions (Kuhlmann et al., 2008).
Guillain–Barre syndrome is an acute immune-mediated polyneuropathy. Its pathophysiology can be divided into acute inflammatory demyelinating polyneuropathy (AIDP), acute sensory axonal neuropathy (ASAN), acute motor axonal neuropathy (AMAN), and acute motor and sensory axonal neuropathy (AMSAN). The main pathophysiological mechanism is complement-mediated nerve injury caused by antibody–antigen interaction in peripheral nerves. The anti-ganglioside antibody is the most common autoantibody in the development of GBS. GBS is induced by some immune trigger factors, most of which are precursory infections caused by pathogens, such as Campylobacter jejuni, Haemophilus influenzae, Mycoplasma pneumoniae, or Cytomegalovirus, and the current epidemic SARS-CoV-2 is also one of the infectious factors causing GBS (Kaida, 2019).
In the PNS, axons are myelinated by Schwann cells. Gangliosides, a family of glycosphingolipids containing sialic acid, ensure the rapid transmission of myelinated axons by nerve fibers in the nervous system by regulating the caliber of axons and organizing ion channels in the Ranvier node and prevent axon regeneration after injury. The anti-ganglioside antibody can simulate this inhibitory effect on nerve repair (Lopez and Baez, 2018). Gangliosides GM1, GD1a, or GD1b are highly enriched at or near the Ranvier nodes (Kaida and Kusunoki, 2009) in axons myelinated by Schwann cells.
In human AMAN and AMSAN, early pathological features include the expansion of the Ranvier nodes, the deposition of complement products, and the complement system playing a central role in the immune response to eliminate invasive pathogens (Ramaglia et al., 2008). Therefore, in immune-mediated neuropathy associated with GM1, GD1a, or GD1b autoantibodies, complement-mediated Ranvier nodes destruction and activation of peripheral Schwann cells may be the common mechanism of these anti-ganglioside antibody-mediated neuropathies, explaining the continuous spectrum of AMAN, AMSAN, and ASAN (Susuki et al., 2012).
Virus infection is also one of the main causes of GBS, and the effects of Zika virus and SARS-CoV-2 on glial cells in GBS are mainly discussed here. Some studies have shown that the Zika virus can replicate in microglia and cause cytopathy, resulting in a high degree of immune activation of microglia to resist pathogens in tissues. At the same time, it will also destroy normal tissues and cause neuropathy (Martinez Viedma and Pickett, 2018). Similarly, SARS-CoV-2 can also invade astrocytes, macrophages, and microglia in the CNS and induce the inflammatory response and increase the production of inflammatory mediators, including IL-1β and IL-2 (Raony et al., 2020). In addition, SARS-CoV-2 induces the expression of inflammatory cytokines in glial cells in vitro, such as IL-6, IL-12, IL-15, and TNF-α (Wu et al., 2020).
In conclusion, autoimmunity is the key to the pathophysiology of GBS. Glial cells present bacterial or viral particles to lymphocytes in the PNS, and the resulting immune response is accompanied by molecular simulation between pathogen epitopes and host gangliosides, such as N-acetylgalactosamine GD1a (GalNAc-GD1a), GD1a, GM1, ganglioside M1B (GM1b), and myelin proteins can lead to acute nerve injury and thus cause GBS (Mohammadi et al., 2020).
The progressive development of GBS is acute and serious, affecting the myelin sheath and the related axons. These damaged structures release biomarkers into the CSF. The candidate biomarkers for the diagnosis of GBS include MBP, axonal damage markers (neurofilaments, tau, anti-ganglioside antibodies), glial markers (S100B), and immunological markers (chemokines and complement factors) (Brettschneider et al., 2009). The applied potency of biomarkers in GBS is depending on several aspects: (1) the relevance to act as a surrogate for the disease development; (2) the practical results of prognostic accuracy; and (3) the potential to be predictors.
Intravenous immunoglobulin (IVIg) and plasma exchange have increasingly been used for the treatment of GBS (Yuki and Hartung, 2012). In addition, anti-GM1 IgG antibodies cause complement-mediated disruption of clusters of voltage-gated Na+ (Nav) channels at Ranvier nodes in peripheral motor nerve fibers of GBS animal models (Susuki et al., 2007). Application of complement inhibitors might reduce the formation of the membrane attack complex, prevent the disruption of sodium channel clusters, and lessen nerve injury in AMAN (Phongsisay et al., 2008).
Neuromyelitis optica spectrum disorders are destructive inflammatory diseases of the CNS induced by autoantibodies, often causing demyelination of the CNS, mainly affecting the spinal cord, optic nerve, and brainstem. The typical manifestations are recurrent optic neuritis, longitudinal generalized myelitis, brainstem, diencephalon, and brain syndrome (Valencia-Sanchez and Wingerchuk, 2021). For many years, optic neuromyelitis has been considered a type of MS. Until 2004, researchers found that the main pathogenic autoantibody of NMODS in patients with optic neuromyelitis was AQP4 immunoglobulin G (AQP4-IgG) targeting AQP4 on the foot process membrane of astrocytes. For this reason, the disease is also known as CNS autoimmune astrocyte disease.
AQP4 immunoglobulin G is a specific biomarker that can distinguish patients with NMODS from patients with MS. In addition, some patients with NMODS are AQP4-IgG seronegative. Therefore, the latest diagnostic criteria divide NMODS into AQP4-IgG seropositive and AQP4-IgG seronegative diseases (Paul et al., 2021). AQP4 is most strongly expressed in the CNS, mainly in astrocytes of brain, spinal cord, and optic nerve, especially on the pia mater and ependymal surface in contact with CSF (Rash et al., 1998). But it also exists in the epithelial cells of kidney (collecting duct), stomach (parietal cells), airway, gland, and skeletal muscle. It has been found that AQP4 is related to nerve excitation, astrocyte migration, and neuroinflammation, and the related contents have been summarized (Verkman, 2012).
In mice and in vitro experiments, the combination of AQP4-IgG and AQP4 activates complement-dependent cytotoxicity (CDC) and antibody-dependent cytotoxicity (ADCC) when killer cells are present. CDC may be the main mechanism of optic neuromyelitis. Some studies have also shown that the combination of AQP4-IgG and AQP4 can inhibit the aquaporin action or internalization of AQP4, because the loss of AQP4 expression in astrocytes can be observed in the early stage of NMODS (Carnero Contentti and Correale, 2021).
After AQP4-IgG enters the CNS, it first causes astrocyte damage through CDC. A series of inflammatory reactions will then occur, leading to the infiltration of inflammatory cells, such as granulocytes, eosinophils, and lymphocytes, followed by the infiltration of macrophages and microglia. It can be detected that there is a high concentration of granulocyte colony-stimulating factors (Papadopoulos and Verkman, 2012) in the CNS at the time of the lesion. A large number of activated macrophages causes axonal damage by phagocytizing myelin and secreting pro-inflammatory cytokines, free radicals, glutamate, and metalloproteinases, resulting in a large number of deaths of oligodendrocytes, and finally leading to neuronal demyelination and necrosis (Hendriks et al., 2005).
Aquaporin-4 is the first target antigen of autoimmune diseases of the CNS to be identified, but not all patients with NMODS have serum AQP4-IgG. The study found that 10–27% of patients with NMO had negative serum AQP4-IgG, but a large proportion (42%) of these patients with negative serum AQP4-IgG could detect MOG antibodies, especially in patients with recurrent optic neuritis (Hamid et al., 2017). MOG is a myelin protein located in the outermost layer of myelin sheath. Pathological studies of MOG-related diseases showed that there were well-defined areas of myelin loss in the lesion, and astrocytes and axons were relatively less damaged. The perivascular inflammatory infiltration was mainly CD4+ T cells and some B cells, and the deposition of complement could also be found. Due to different pathophysiology, serum biomarkers, clinical and radiological manifestations, and disease outcomes, MOG-related diseases are now regarded as unique disease entities (Paul et al., 2021).
In conclusion, the injury of astrocytes is the pathological basis of optic neuromyelitis, and then the infiltration of inflammatory cells and macrophages further leads to the necrosis of oligodendrocytes and finally the demyelination of neurons. This shows that demyelination is not the pathological feature of optic neuromyelitis, but a secondary phenomenon of astrocyte injury.
It has been demonstrated that up to several thousand times higher levels of glial fibrillary acidic protein (GFAP) could be detected in the CSF of patients suffering from NMODS compared to patients with MS (Takano et al., 2010). Therefore, except for AQP4-IgG, CSF GFAP could be a biomarker for examining astrocyte injury in patients.
Myelin oligodendrocyte glycoprotein is a highly conserved protein. It is only slightly expressed on the cell body of oligodendrocytes and the outer surface of the myelin sheath in the CNS (accounting for 0.05% of the total myelin protein). The exact role of MOG in the CNS remains unclear. By observing the extracellular domain of MOG, the researchers found that MOG exists in the form of dimer in natural or aqueous solution, so they speculated that MOG is an affinity adhesion receptor (Clements et al., 2003). In addition, MOG may also have the functions of stabilizing microtubules, activating complements, and participating in the transmission of information between cells (Johns and Bernard, 1999).
Myelin oligodendrocyte glycoprotein antibody-related disease (MOGAD) is a kind of autoimmune disease recently found (Dubey et al., 2019; Xie et al., 2022). The pathological manifestation of MOGAD is the demyelination of the CNS. Studies have shown that the most common clinical manifestation of MOGAD is optic neuromyelitis, in which AQP4-IgG serum negative optic neuromyelitis is the most common phenotype (Inan et al., 2020). Other clinical phenotypes include acute demyelinating encephalomyelitis and cortical encephalitis. Although there are overlapping clinical phenotypes between MOGAD, MS, and NMOSD, a large number of experiments and data show that MOGAD is a heterogeneous disease with characteristic clinical and imaging features (Shahriari et al., 2021).
The specific role of MOG antibodies (MOG-Abs) in the pathogenesis of MOGAD has not been clarified. At present, it is considered that most of the MOG-Abs are IgG I subtypes. By analyzing the complement activation in patients with MOGAD and healthy people, the researchers found that the proteins related to the classical complement activation pathway and alternative complement activation pathway increased significantly in patients with MOGAD (Keller et al., 2021), suggesting that complement activation is a prominent feature of MOGAD. The massive production of complement proteins will further activate CDC, resulting in extensive damage to oligodendrocytes in the CNS, which is manifested as neuronal demyelination. In addition to activating the complement pathway, denatured MOG protein can also activate T cell immunity (Ambrosius et al., 2020). The infiltration of inflammatory cells, the production of inflammatory factors, and the activation of macrophages will further aggravate the damaged myelinated axons by oligodendrocytes of the CNS.
In conclusion, oligodendrocyte injury leading to neuronal axon demyelination is the pathological basis and feature of MOGAD. The pathogenesis of MOGAD has not been fully clarified. It has been confirmed that the complement system will be activated at the onset, resulting in oligodendrocyte damage. At the same time, microglia and/or macrophages will also be activated to phagocytize myelin fragments and secrete inflammatory factors. The roles of other glial cells in the pathogenesis of MOGAD need further study.
Peripheral neuropathy is a kind of disease with the structure and dysfunction of motor sensation and autonomic nerve. Immune-mediated peripheral neuropathy is a neurological injury and dysfunction caused by the synergistic effect of humoral and cellular immunity. It has been found that the common mechanisms of several immune-mediated neurological diseases are as follows: (1) the body produces autoantibodies against myelin or protein in the Ranvier nodes; (2) the activated T cells can secrete chemokines and proteases, leading to the destruction of the blood–nerve barrier (Kieseier et al., 2018); and (3) macrophages break through the damaged blood–nerve barrier and destroy the nerve myelin sheath. The well-accepted hypothesis is the autoantibodies of nerve cell membrane and myelin. For example, injection of GM1 antibody into rabbits can cause a reversible intracellular block of the sciatic nerve (Pollard and Armati, 2011).
Schwann cells are the main glial cells in the PNS, which are divided into myelinated Schwann cells and unmyelinated Schwann cells (Armati and Mathey, 2014). Schwann cells express many receptors, which recognize risk factors and activate local immune response by providing antigens and secreting cytokines and chemokines, attracting immune cells to the injury site. In order to avoid inflammatory overreaction, Schwann cells can also produce anti-inflammatory cytokine interleukin-10 (IL-10), which can inhibit the inflammatory response (Ydens et al., 2013).
Satellite cells envelop sensory neurons in the ganglia, which suggest the communication between satellite cells and neurons through gap junctions without synaptic structures. Gap junctional coupling among satellite cells was found to be increased in pain models (Tracey, 2021). Interestingly, pain is a serious and common problem in patients suffering from MS, although the underlying mechanism is still unclear (Stenager et al., 1991). Even while MS has always been thought of as a CNS disease, research suggested that PNS may possibly be involved (Teixeira et al., 2020). Satellite cells activate and act through gap junctional coupling with neurons in the dorsal root ganglions (DRGs) in the peripheral component of pain syndrome in experimental MS animal models (Warwick et al., 2014). Further electrophysiological evidence also demonstrates that the membrane hyperexcitability of sensory neurons of DRG was found in animal models of MS, providing more clues for the sensitization of PNS in MS, but the mechanism and the roles of satellite cells in MS still need further investigations (Yousuf et al., 2019).
Although adaptive immunity to myelin antigens is essential in the pathogenesis of neurologic autoimmune disorders, innate immune mechanisms are likely involved in the initiation and perpetuation stages. Gut microbiota is closely related to the innate immune (Berer et al., 2011). Dysbiotic microbiota increases gut permeability and produces pro-inflammatory cytokines, stimulating inflammatory T cells, especially of the Th17 subtype, which is the same as that of myelin-destructive T cells. Therefore, T cells with cross-reactivity to myelin could become involved (Wekerle et al., 2013). In turn, circulating endotoxin may activate microglia, causing myelin sheath damage (Bergner et al., 2019). Therefore, the next question to ask is if there is a link between gut microbiota and neurologic autoimmune disorders (Zhang et al., 2020)? Studies in MS animal models suggest that the overactivated T cells are in line with the development of MS, which seems to require the presence of gut microbiota. It is reported that probiotic administration suppressed peripheral inflammatory responses in patients with MS (Tankou et al., 2018). Although a direct role for the gut immune system still needs to confirm, these clues for communication between gut microbiota and neurologic autoimmune disorders through glial cells suggest that the environmental factors might also be involved but needs further investigation (Hanninen, 2017).
To sum up, glial cells, as an important part of the nervous system, play an important role in the occurrence and development of autoimmune diseases in the nervous system. Glial cells are over-activated in the presence of autoantibody or pathology, altering the internal environment of neurons, releasing cytokines, and causing excessive synaptic pruning and extinction, resulting in neuroautoimmune diseases. In this process, autoantibodies are one of the important links. The autoantibodies produced by the body due to infection, environmental change, or virus invasion are often the cause of neurologic autoimmune disorders, and the injury of glial cells is also an important way to cause the disease. However, there are few studies on autoantibodies acting on glial cells, which need to be further explored and found. Second, the research on glial cells for neuroautoimmune diseases also suggests that we can treat such diseases through glial cells. For example, the use of anti AQP4 antibody can reduce the damage of astrocytes to neurons, to control the NMODS disease. We speculate the possibility of glial cells as a new direction for the study of neurologic autoimmune disorders, which provides a new idea for the potential clinical diagnosis and treatment.
HL and Z-ZK designed and constructed the manuscript. Z-QL, T-XL, MT, Z-SR, C-YY, R-KY, and S-JS reviewed the literature and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81971035 to Z-ZK) and by Shaanxi (2019SF-083 to Z-ZK).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
MS, multiple sclerosis; GBS, Guillain–Barre syndrome; NMODS, neuromyelitis optica spectrum disorders; CNS, central nervous system; PNS, peripheral nervous system; NMDAR, N-methyl -D-aspartate receptor; AMPAR, α -amino-3-hydroxy-5-metnyl-4-isoxazolepropionic acid receptor; GABABR, gamma-aminobutyric acid B receptor; VGKC, voltage-gated potassium channel; mGluR1, metabotropic glutamate receptor 1; mGluR5, metabotropic glutamate receptor 5; MOG, myelin oligodendrocyte glycoprotein; MBP, myelin basic protein; GM1, ganglioside M1; GM2, ganglioside M2; GM3, ganglioside M3; GD1a, ganglioside D1a; GD1b, ganglioside D1b; GT1b, ganglioside T1b; GQ1b, ganglioside Q1B; MAG, myelin-associated glycoprotein; AChR, acetylcholine receptor; AQP4, aquaporin-4; BBB, blood brain barrier; OPCs, oligodendrocyte progenitor cells; HSCs, hematopoietic stem cells; IFN-γ, cytokine interferon-γ; LPS, lipopolysaccharide; IL-12, interleukin-12; IL-23, interleukin-23; Th1, type 1 T helper; Th17, type 17 T helper; IL-17, interleukin-17; Th2, type 2 T helper; CSF, cerebrospinal fluid; AIDP, acute inflammatory demyelinating polyneuropathy; ASAN, acute sensory axonal neuropathy; AMAN, acute motor axonal neuropathy; AMSAN, acute motor and sensory axonal neuropathy; GalNAc-GD1a, N-acetylgalactosamine GD1a; GM1b, Ganglioside M1B; IVIg, intravenous immunoglobulin; Nav, voltage-gated Na+; CDC, complement-dependent cytotoxicity; ADCC, antibody-dependent cytotoxicity; GFAP, glial fibrillary acidic protein; MOGAD, myelin oligodendrocyte glycoprotein antibody-related disease; MOG-Abs, MOG antibodies; IL-10, interleukin-10; DRGs, dorsal root ganglions.
Acosta, C. M., Cortes, C., MacPhee, H., and Namaka, M. P. (2013). Exploring the role of nerve growth factor in multiple sclerosis: Implications in myelin repair. CNS Neurol. Disord. Drug Targets 12, 1242–1256. doi: 10.2174/18715273113129990087
Aharoni, R., Eilam, R., and Arnon, R. (2021). Astrocytes in multiple sclerosis-essential constituents with diverse multifaceted functions. Int. J. Mol. Sci. 22:5904. doi: 10.3390/ijms22115904
Ambrosius, W., Michalak, S., Kozubski, W., and Kalinowska, A. (2020). Myelin oligodendrocyte glycoprotein antibody-associated disease: Current insights into the disease pathophysiology, diagnosis and management. Int. J. Mol. Sci. 22:100. doi: 10.3390/ijms22010100
Armati, P. J., and Mathey, E. K. (2014). Clinical implications of Schwann cell biology. J. Peripher. Nerv. Syst. 19, 14–23. doi: 10.1111/jns5.12057
Berer, K., Mues, M., Koutrolos, M., Rasbi, Z. A., Boziki, M., Johner, C., et al. (2011). Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479, 538–541. doi: 10.1038/nature10554
Bergner, C. G., van der Meer, F., Winkler, A., Wrzos, C., Turkmen, M., Valizada, E., et al. (2019). Microglia damage precedes major myelin breakdown in X-linked adrenoleukodystrophy and metachromatic leukodystrophy. Glia 67, 1196–1209. doi: 10.1002/glia.23598
Bhagavati, S. (2021). Autoimmune disorders of the nervous system: Pathophysiology, clinical features, and therapy. Front. Neurol. 12:664664. doi: 10.3389/fneur.2021.664664
Brambilla, R. (2019). The contribution of astrocytes to the neuroinflammatory response in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. 137, 757–783. doi: 10.1007/s00401-019-01980-7
Brettschneider, J., Petzold, A., Sussmuth, S., and Tumani, H. (2009). Cerebrospinal fluid biomarkers in Guillain-Barre syndrome–where do we stand? J. Neurol. 256, 3–12. doi: 10.1007/s00415-009-0097-x
Brown, R. A., Narayanan, S., and Arnold, D. L. (2013). Segmentation of magnetization transfer ratio lesions for longitudinal analysis of demyelination and remyelination in multiple sclerosis. Neuroimage 66, 103–109. doi: 10.1016/j.neuroimage.2012.10.059
Cao, L., and He, C. (2013). Polarization of macrophages and microglia in inflammatory demyelination. Neurosci. Bull. 29, 189–198. doi: 10.1007/s12264-013-1324-0
Carnero Contentti, E., and Correale, J. (2021). Neuromyelitis optica spectrum disorders: From pathophysiology to therapeutic strategies. J. Neuroinflammation 18:208. doi: 10.1186/s12974-021-02249-1
Casella, G., Rasouli, J., Boehm, A., Zhang, W., Xiao, D., Ishikawa, L. L. W., et al. (2020). Oligodendrocyte-derived extracellular vesicles as antigen-specific therapy for autoimmune neuroinflammation in mice. Sci. Transl. Med. 12:eaba0599. doi: 10.1126/scitranslmed.aba0599
Clements, C. S., Reid, H. H., Beddoe, T., Tynan, F. E., Perugini, M. A., Johns, T. G., et al. (2003). The crystal structure of myelin oligodendrocyte glycoprotein, a key autoantigen in multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 100, 11059–11064. doi: 10.1073/pnas.1833158100
Crotti, A., and Ransohoff, R. M. (2016). Microglial physiology and pathophysiology: Insights from genome-wide transcriptional profiling. Immunity 44, 505–515. doi: 10.1016/j.immuni.2016.02.013
Cusick, M. F., Libbey, J. E., Trede, N. S., Eckels, D. D., and Fujinami, R. S. (2012). Human T cell expansion and experimental autoimmune encephalomyelitis inhibited by Lenaldekar, a small molecule discovered in a zebrafish screen. J. Neuroimmunol. 244, 35–44. doi: 10.1016/j.jneuroim.2011.12.024
Dalmau, J., Geis, C., and Graus, F. (2017). Autoantibodies to synaptic receptors and neuronal cell surface proteins in autoimmune diseases of the central nervous system. Physiol. Rev. 97, 839–887. doi: 10.1152/physrev.00010.2016
Dubey, D., Pittock, S. J., Krecke, K. N., Morris, P. P., Sechi, E., Zalewski, N. L., et al. (2019). Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol. 76, 301–309. doi: 10.1001/jamaneurol.2018.4053
Falcao, A. M., van Bruggen, D., Marques, S., Meijer, M., Jakel, S., Agirre, E., et al. (2018). Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 24, 1837–1844. doi: 10.1038/s41591-018-0236-y
Farina, C., Aloisi, F., and Meinl, E. (2007). Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28, 138–145. doi: 10.1016/j.it.2007.01.005
Filippi, M., Preziosa, P., Meani, A., Ciccarelli, O., Mesaros, S., Rovira, A., et al. (2018). Prediction of a multiple sclerosis diagnosis in patients with clinically isolated syndrome using the 2016 MAGNIMS and 2010 McDonald criteria: A retrospective study. Lancet Neurol. 17, 133–142. doi: 10.1016/S1474-4422(17)30469-6
Fogg, D. K., Sibon, C., Miled, C., Jung, S., Aucouturier, P., Littman, D. R., et al. (2006). A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 311, 83–87. doi: 10.1126/science.1117729
Fujinami, R. S. (2001). Can virus infections trigger autoimmune disease? J. Autoimmun. 16, 229–234. doi: 10.1006/jaut.2000.0484
Geng, G., Yu, X., Jiang, J., and Yu, X. (2020). Aetiology and pathogenesis of paraneoplastic autoimmune disorders. Autoimmun. Rev. 19:102422. doi: 10.1016/j.autrev.2019.102422
Ghorbani, S., Jelinek, E., Jain, R., Buehner, B., Li, C., Lozinski, B. M., et al. (2022). Versican promotes T helper 17 cytotoxic inflammation and impedes oligodendrocyte precursor cell remyelination. Nat. Commun. 13:2445. doi: 10.1038/s41467-022-30032-0
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Gold, M., Pul, R., Bach, J. P., Stangel, M., and Dodel, R. (2012). Pathogenic and physiological autoantibodies in the central nervous system. Immunol. Rev. 248, 68–86. doi: 10.1111/j.1600-065X.2012.01128.x
Greter, M., Lelios, I., and Croxford, A. L. (2015). Microglia versus myeloid cell nomenclature during brain inflammation. Front. Immunol. 6:249. doi: 10.3389/fimmu.2015.00249
Hamid, S. H., Elsone, L., Mutch, K., Solomon, T., and Jacob, A. (2017). The impact of 2015 neuromyelitis optica spectrum disorders criteria on diagnostic rates. Mult. Scler. 23, 228–233. doi: 10.1177/1352458516663853
Hanninen, A. (2017). Infections in MS: An innate immunity perspective. Acta Neurol. Scand. 136(Suppl. 201), 10–14. doi: 10.1111/ane.12838
Healy, L. M., Yaqubi, M., Ludwin, S., and Antel, J. P. (2020). Species differences in immune-mediated CNS tissue injury and repair: A (neuro)inflammatory topic. Glia 68, 811–829. doi: 10.1002/glia.23746
Hendriks, J. J., Teunissen, C. E., de Vries, H. E., and Dijkstra, C. D. (2005). Macrophages and neurodegeneration. Brain Res. Brain Res. Rev. 48, 185–195. doi: 10.1016/j.brainresrev.2004.12.008
Heneka, M. T., McManus, R. M., and Latz, E. (2018). Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 19, 610–621. doi: 10.1038/s41583-018-0055-7
Herculano-Houzel, S. (2014). The glia/neuron ratio: How it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia 62, 1377–1391. doi: 10.1002/glia.22683
Inan, B., Gocmen, R., Vural, A., Colpak, A. I., Meinl, E., Karabudak, R., et al. (2020). Myelin oligodendrocyte glycoprotein antibody associated central nervous system demyelinating disease: A tertiary center experience from Turkey. Mult. Scler. Relat. Disord 44:102376. doi: 10.1016/j.msard.2020.102376
Ioannou, M. S., Jackson, J., Sheu, S. H., Chang, C. L., Weigel, A. V., Liu, H., et al. (2019). Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell 177, 1522–1535.e14. doi: 10.1016/j.cell.2019.04.001
Jackle, K., Zeis, T., Schaeren-Wiemers, N., Junker, A., van der Meer, F., Kramann, N., et al. (2020). Molecular signature of slowly expanding lesions in progressive multiple sclerosis. Brain 143, 2073–2088. doi: 10.1093/brain/awaa158
Johns, T. G., and Bernard, C. C. (1999). The structure and function of myelin oligodendrocyte glycoprotein. J. Neurochem. 72, 1–9. doi: 10.1046/j.1471-4159.1999.0720001.x
Kaida, K. (2019). Guillain-barre syndrome. Adv. Exp. Med. Biol. 1190, 323–331. doi: 10.1007/978-981-32-9636-7_20
Kaida, K., and Kusunoki, S. (2009). Guillain-Barre syndrome: Update on immunobiology and treatment. Expert Rev. Neurother. 9, 1307–1319. doi: 10.1586/ern.09.77
Kamm, C., and Zettl, U. K. (2012). Autoimmune disorders affecting both the central and peripheral nervous system. Autoimmun. Rev. 11, 196–202. doi: 10.1016/j.autrev.2011.05.012
Karim, A. R., and Jacob, S. (2018). Experience with newer central nervous system autoantibodies. Ann. Clin. Biochem. 55, 7–17. doi: 10.1177/0004563217724818
Keller, C. W., Lopez, J. A., and Wendel, E. M. (2021). Complement activation is a prominent feature of MOGAD. Ann. Neurol. 90, 976–982. doi: 10.1002/ana.26226
Kieseier, B. C., Mathey, E. K., Sommer, C., and Hartung, H. P. (2018). Immune-mediated neuropathies. Nat. Rev. Dis. Primers 4:31. doi: 10.1038/s41572-018-0027-2
Kleopa, K. A. (2011). Autoimmune channelopathies of the nervous system. Curr. Neuropharmacol. 9, 458–467. doi: 10.2174/157015911796557966
Kong, W., Hooper, K. M., and Ganea, D. (2016). The natural dual cyclooxygenase and 5-lipoxygenase inhibitor flavocoxid is protective in EAE through effects on Th1/Th17 differentiation and macrophage/microglia activation. Brain Behav. Immun. 53, 59–71. doi: 10.1016/j.bbi.2015.11.002
Kuang, S., and Rudnicki, M. A. (2008). The emerging biology of satellite cells and their therapeutic potential. Trends Mol. Med. 14, 82–91. doi: 10.1016/j.molmed.2007.12.004
Kuhlmann, T., Miron, V., Cui, Q., Wegner, C., Antel, J., and Bruck, W. (2008). Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 131(Pt 7), 1749–1758. doi: 10.1093/brain/awn096
Lalive, P. H. (2008). Autoantibodies in inflammatory demyelinating diseases of the central nervous system. Swiss Med. Wkly. 138, 692–707.
Liddelow, S. A., Marsh, S. E., and Stevens, B. (2020). Microglia and astrocytes in disease: Dynamic duo or partners in crime? Trends Immunol. 41, 820–835. doi: 10.1016/j.it.2020.07.006
Lipp, M., Brandt, C., Dehghani, F., Kwidzinski, E., and Bechmann, I. (2007). PD-L1 (B7-H1) regulation in zones of axonal degeneration. Neurosci. Lett. 425, 156–161. doi: 10.1016/j.neulet.2007.07.053
Lopez, P. H. H., and Baez, B. B. (2018). Gangliosides in axon stability and regeneration. Prog. Mol. Biol. Transl. Sci. 156, 383–412. doi: 10.1016/bs.pmbts.2018.03.001
Martinez Viedma, M. D. P., and Pickett, B. E. (2018). Characterizing the different effects of Zika Virus infection in placenta and microglia cells. Viruses 10:649. doi: 10.3390/v10110649
McGonigal, R., Campbell, C. I., Barrie, J. A., Yao, D., Cunningham, M. E., Crawford, C. L., et al. (2022). Schwann cell nodal membrane disruption triggers bystander axonal degeneration in a Guillain-Barre syndrome mouse model. J. Clin. Invest. 132:e158524. doi: 10.1172/JCI158524
Meijer, M., Agirre, E., Kabbe, M., van Tuijn, C. A., Heskol, A., Zheng, C., et al. (2022). Epigenomic priming of immune genes implicates oligodendroglia in multiple sclerosis susceptibility. Neuron 110, 1193–1210.e13. doi: 10.1016/j.neuron.2021.12.034
Mohammadi, S., Moosaie, F., and Aarabi, M. H. (2020). Understanding the immunologic characteristics of neurologic manifestations of SARS-CoV-2 and potential immunological mechanisms. Mol. Neurobiol. 57, 5263–5275. doi: 10.1007/s12035-020-02094-y
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Papadopoulos, M. C., and Verkman, A. S. (2012). Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 11, 535–544. doi: 10.1016/s1474-4422(12)70133-3
Paul, S., Mondal, G. P., Bhattacharyya, R., Ghosh, K. C., and Bhat, I. A. (2021). Neuromyelitis optica spectrum disorders. J. Neurol. Sci. 420:117225. doi: 10.1016/j.jns.2020.117225
Phongsisay, V., Susuki, K., Matsuno, K., Yamahashi, T., Okamoto, S., Funakoshi, K., et al. (2008). Complement inhibitor prevents disruption of sodium channel clusters in a rabbit model of Guillain-Barre syndrome. J. Neuroimmunol. 205, 101–104. doi: 10.1016/j.jneuroim.2008.09.016
Pollak, T. A., Beck, K., Irani, S. R., Howes, O. D., David, A. S., and McGuire, P. K. (2016). Autoantibodies to central nervous system neuronal surface antigens: Psychiatric symptoms and psychopharmacological implications. Psychopharmacology (Berl) 233, 1605–1621. doi: 10.1007/s00213-015-4156-y
Pollard, J. D., and Armati, P. J. (2011). CIDP - the relevance of recent advances in Schwann cell/axonal neurobiology. J. Peripher. Nerv. Syst. 16, 15–23. doi: 10.1111/j.1529-8027.2011.00323.x
Ramaglia, V., Daha, M. R., and Baas, F. (2008). The complement system in the peripheral nerve: Friend or foe? Mol. Immunol. 45, 3865–3877. doi: 10.1016/j.molimm.2008.06.018
Raony, I., de Figueiredo, C. S., Pandolfo, P., Giestal-de-Araujo, E., Oliveira-Silva Bomfim, P., and Savino, W. (2020). Psycho-neuroendocrine-immune interactions in COVID-19: Potential Impacts on mental health. Front. Immunol. 11:1170. doi: 10.3389/fimmu.2020.01170
Rash, J. E., Yasumura, T., Hudson, C. S., Agre, P., and Nielsen, S. (1998). Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc. Natl. Acad. Sci. U.S.A. 95, 11981–11986. doi: 10.1073/pnas.95.20.11981
Rittchen, S., Boyd, A., Burns, A., Park, J., Fahmy, T. M., Metcalfe, S., et al. (2015). Myelin repair in vivo is increased by targeting oligodendrocyte precursor cells with nanoparticles encapsulating leukaemia inhibitory factor (LIF). Biomaterials 56, 78–85. doi: 10.1016/j.biomaterials.2015.03.044
Rubin, D. B., Batra, A., Vaitkevicius, H., and Vodopivec, I. (2018). Autoimmune neurologic disorders. Am. J. Med. 131, 226–236. doi: 10.1016/j.amjmed.2017.10.033
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Shahriari, M., Sotirchos, E. S., Newsome, S. D., and Yousem, D. M. (2021). MOGAD: How it differs from and resembles other neuroinflammatory disorders. AJR Am. J. Roentgenol. 216, 1031–1039. doi: 10.2214/ajr.20.24061
Singh, S., Metz, I., Amor, S., van der Valk, P., Stadelmann, C., and Bruck, W. (2013). Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol. 125, 595–608. doi: 10.1007/s00401-013-1082-0
Stenager, E., Knudsen, L., and Jensen, K. (1991). Acute and chronic pain syndromes in multiple sclerosis. Acta Neurol. Scand. 84, 197–200. doi: 10.1111/j.1600-0404.1991.tb04937.x
Susuki, K., Rasband, M. N., Tohyama, K., Koibuchi, K., Okamoto, S., Funakoshi, K., et al. (2007). Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J. Neurosci. 27, 3956–3967. doi: 10.1523/JNEUROSCI.4401-06.2007
Susuki, K., Yuki, N., Schafer, D. P., Hirata, K., Zhang, G., Funakoshi, K., et al. (2012). Dysfunction of nodes of Ranvier: A mechanism for anti-ganglioside antibody-mediated neuropathies. Exp. Neurol. 233, 534–542. doi: 10.1016/j.expneurol.2011.11.039
Takano, R., Misu, T., Takahashi, T., Sato, S., Fujihara, K., and Itoyama, Y. (2010). Astrocytic damage is far more severe than demyelination in NMO: A clinical CSF biomarker study. Neurology 75, 208–216. doi: 10.1212/WNL.0b013e3181e2414b
Tankou, S. K., Regev, K., Healy, B. C., Tjon, E., Laghi, L., Cox, L. M., et al. (2018). A probiotic modulates the microbiome and immunity in multiple sclerosis. Ann. Neurol. 83, 1147–1161. doi: 10.1002/ana.25244
Teixeira, N. B., Picolo, G., Giardini, A. C., Boumezbeur, F., Pottier, G., Kuhnast, B., et al. (2020). Alterations of peripheral nerve excitability in an experimental autoimmune encephalomyelitis mouse model for multiple sclerosis. J. Neuroinflammation 17:266. doi: 10.1186/s12974-020-01936-9
Thompson, K. K., and Tsirka, S. E. (2017). The diverse roles of microglia in the neurodegenerative aspects of Central Nervous System (CNS) autoimmunity. Int. J. Mol. Sci. 18:504. doi: 10.3390/ijms18030504
Tracey, K. J. (2021). From human to mouse and back offers hope for patients with fibromyalgia. J. Clin. Invest. 131:e150382. doi: 10.1172/JCI150382
Trapp, B. D., Peterson, J., Ransohoff, R. M., Rudick, R., Mork, S., and Bo, L. (1998). Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 338, 278–285. doi: 10.1056/NEJM199801293380502
Valencia-Sanchez, C., and Wingerchuk, D. M. (2021). Emerging targeted therapies for neuromyelitis optica spectrum disorders. BioDrugs 35, 7–17. doi: 10.1007/s40259-020-00460-9
van Wageningen, T. A., Gerrits, E., Brouwer, N., Breve, J. J. P., Geurts, J. J. G., Eggen, B. J. L., et al. (2022). Distinct gene expression in demyelinated white and grey matter areas of patients with multiple sclerosis. Brain Commun. 4:fcac005. doi: 10.1093/braincomms/fcac005
Verkman, A. S. (2012). Aquaporins in clinical medicine. Annu. Rev. Med. 63, 303–316. doi: 10.1146/annurev-med-043010-193843
Voet, S., Prinz, M., and van Loo, G. (2019). Microglia in Central Nervous System Inflammation and Multiple Sclerosis Pathology. Trends Mol. Med. 25, 112–123. doi: 10.1016/j.molmed.2018.11.005
Warwick, R. A., Ledgerwood, C. J., Brenner, T., and Hanani, M. (2014). Satellite glial cells in dorsal root ganglia are activated in experimental autoimmune encephalomyelitis. Neurosci. Lett. 569, 59–62. doi: 10.1016/j.neulet.2014.03.033
Wekerle, H., Berer, K., and Krishnamoorthy, G. (2013). Remote control-triggering of brain autoimmune disease in the gut. Curr. Opin. Immunol. 25, 683–689. doi: 10.1016/j.coi.2013.09.009
Wu, Y., Xu, X., Chen, Z., Duan, J., Hashimoto, K., Yang, L., et al. (2020). Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 87, 18–22. doi: 10.1016/j.bbi.2020.03.031
Xie, H., Shao, Y., Du, J., Song, Y., Li, Y., Duan, R., et al. (2022). Comparative analysis of clinical and imaging data between patients with myelin oligodendrocyte glycoprotein antibody disease and patients with aquaporin 4 antibody-positive neuromyelitis optica spectrum disorder. J. Neurol. 269, 1641–1650. doi: 10.1007/s00415-021-10749-6
Ydens, E., Lornet, G., Smits, V., Goethals, S., Timmerman, V., and Janssens, S. (2013). The neuroinflammatory role of Schwann cells in disease. Neurobiol. Dis. 55, 95–103. doi: 10.1016/j.nbd.2013.03.005
Yousuf, M. S., Noh, M. C., Friedman, T. N., Zubkow, K., Johnson, J. C., Tenorio, G., et al. (2019). Sensory neurons of the dorsal root ganglia become hyperexcitable in a T-cell-mediated MOG-EAE model of multiple sclerosis. eNeuro 6, 1–15. doi: 10.1523/ENEURO.0024-19.2019
Yuki, N., and Hartung, H. P. (2012). Guillain-Barre syndrome. N. Engl. J. Med. 366, 2294–2304. doi: 10.1056/NEJMra1114525
Zhang, X., Chen, B. D., Zhao, L. D., and Li, H. (2020). The gut microbiota: Emerging evidence in autoimmune diseases. Trends Mol Med 26, 862–873. doi: 10.1016/j.molmed.2020.04.001
Zhang, Y., and Popovich, P. (2011). Roles of autoantibodies in central nervous system injury. Discov. Med. 11, 395–402.
Keywords: neurologic autoimmune disorders, glial cells, autoantibody, multiple sclerosis, Guillain–Barre syndrome, neuromyelitis optica spectrum disorders, myelin oligodendrocyte glycoprotein antibody-related diseases
Citation: Li Z-Q, Li T-X, Tian M, Ren Z-S, Yuan C-Y, Yang R-K, Shi S-J, Li H and Kou Z-Z (2022) Glial cells and neurologic autoimmune disorders. Front. Cell. Neurosci. 16:1028653. doi: 10.3389/fncel.2022.1028653
Received: 26 August 2022; Accepted: 03 October 2022;
Published: 26 October 2022.
Edited by:
Mingqin Zhu, First Affiliated Hospital of Jilin University, ChinaReviewed by:
Peng Lei, Sichuan University, ChinaCopyright © 2022 Li, Li, Tian, Ren, Yuan, Yang, Shi, Li and Kou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhen-Zhen Kou, cGx1dG9taW5nQGZtbXUuZWR1LmNu; Hui Li, bGlfaHVpQGZtbXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.