 Katelyn J. Hoff
Katelyn J. Hoff Jeffrey K. Moore
Jeffrey K. Moore
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 02 November 2022
Sec. Cellular Neuropathology
Volume 16 - 2022 | https://doi.org/10.3389/fncel.2022.1023267
This article is part of the Research Topic Tubulinopathies: Fundamental and Clinical Challenges View all 7 articles
Heterozygous, missense mutations in both α- and β-tubulin genes have been linked to an array of neurodevelopment disorders, commonly referred to as “tubulinopathies.” To date, tubulinopathy mutations have been identified in three β-tubulin isotypes and one α-tubulin isotype. These mutations occur throughout the different genetic domains and protein structures of these tubulin isotypes, and the field is working to address how this molecular-level diversity results in different cellular and tissue-level pathologies. Studies from many groups have focused on elucidating the consequences of individual mutations; however, the field lacks comprehensive models for the molecular etiology of different types of tubulinopathies, presenting a major gap in diagnosis and treatment. This review highlights recent advances in understanding tubulin structural dynamics, the roles microtubule-associated proteins (MAPs) play in microtubule regulation, and how these are inextricably linked. We emphasize the value of investigating interactions between tubulin structures, microtubules, and MAPs to understand and predict the impact of tubulinopathy mutations at the cell and tissue levels. Microtubule regulation is multifaceted and provides a complex set of controls for generating a functional cytoskeleton at the right place and right time during neurodevelopment. Understanding how tubulinopathy mutations disrupt distinct subsets of those controls, and how that ultimately disrupts neurodevelopment, will be important for establishing mechanistic themes among tubulinopathies that may lead to insights in other neurodevelopment disorders and normal neurodevelopment.
Microtubules are a critical component of every eukaryotic cell, and they have highly regulated roles in multiple neuronal functions during neurodevelopment. During neurodevelopment, microtubules play a key role in the neurons that structure the sulci and gyri of the cerebral cortex. Mutations in the genes encoding the α- and β-tubulin subunits of microtubules are associated with a range of malformations of the cerebral cortex and constitute a class of disorders known as “tubulinopathies.” Cortical malformations encompass multiple types of abnormalities, including too many or too few brain folds, with or without a reduction in brain size. While the number of tubulinopathy mutants identified in patients continues to increase, the molecular mechanisms that connect tubulin mutations to neurodevelopment disorders remains an important area of investigation.
The goal of this review is to discuss the importance of understanding basic tubulin biology and regulatory mechanisms to better predict the neurodevelopmental impacts of individual tubulin mutants. We will discuss foundational literature as well as recent advances in understanding the interconnected role of tubulin structure, microtubules, and microtubule-associated proteins (MAPs) in neurodevelopment. Additionally, we will review the roles of MAPs and motors in both healthy and disease models to understand how microtubules are regulated during neurodevelopment, and how tubulinopathy mutations disrupt this important regulation. Finally, we attempt to identify mechanistic themes between types and locations of tubulinopathy mutations within the protein and the associated cortical malformation.
The cytoskeleton is comprised of various filamentous structures, including microtubules, F-actin, and intermediate filaments. Each of these structures plays critical roles in development, disease, and basic cell biology. Microtubules are polar, cylindrical structures that are highly dynamic and are composed of tubulin heterodimers, consisting of one α and one β monomer. Microtubules are the only cytoskeletal filaments that exhibit what is known as “dynamic instability,” the stochastic cycling between states of polymerization and depolymerization. This dynamic instability is an intrinsic feature of microtubules and can be reconstituted in vitro using purified tubulin proteins, independent of any accessory proteins (Mitchison and Kirschner, 1984). Under these conditions, the rate of microtubule polymerization depends on the concentration of free tubulin available in the system, while the rate of depolymerization is concentration-independent. These intrinsic properties of microtubule dynamics are controlled by tubulin’s ability to act as a switch that alternates between conformational states that either favor or disfavor microtubule assembly. In cells, this switch-like behavior of tubulin is further modulated by a host of MAPs and motors.
One way in which the tubulin heterodimer acts as a switch is via its nucleotide state. Tubulin binds to two GTP molecules at two distinct locations (Figure 1A). One of these sites is the non-exchangeable site, also known as the “N-site,” which is located at the intradimer interface between the α- and β-tubulin monomers (Nogales et al., 1998). As the name suggests, this GTP molecule is not hydrolyzed (Spiegelman et al., 1977). The other GTP-binding site is the exchangeable site, or “E-site.” The E-site is located on β-tubulin where it interacts with the α-tubulin of the incoming tubulin heterodimer during microtubule assembly and can be hydrolyzed from GTP to GDP (Carlier and Pantaloni, 1981; Nogales et al., 1998). Therefore, when referring to the nucleotide state of a heterodimer, it is in reference to the nucleotide state at the E-site, as that is the one that cycles between GTP and GDP and has consequences for microtubule dynamics. GTP-tubulin in solution is added on to the microtubule plus end and exhibits faster assembly activity than GDP-tubulin. As the GTP-tubulin heterodimer is further incorporated into the microtubule, GTP is hydrolyzed to GDP (Nogales et al., 1998; Carlier and Pantaloni, 1981). This process of hydrolysis is coupled to changes in tubulin structure and interaction with its neighbors, and it is primarily thought that GTP-tubulin stabilizes microtubules while GDP-tubulin is less stable (Alushin et al., 2014; Zhang et al., 2015). As GTP-tubulin assembles onto the microtubule plus end, it forms what is known as the “GTP-cap”, which is a stable structure that prevents microtubule depolymerization until the cap is hydrolyzed to GDP (Mitchison and Kirschner, 1984; O’Brien et al., 1987; Drechsel and Kirschner, 1994; Caplow and Shanks, 1996; Roostalu et al., 2020). The formation of the GTP-cap requires that the rate of heterodimer(s) addition at the plus-end is faster than the rate of GTP hydrolysis. Once the rate of GTP hydrolysis outpaces the addition of new heterodimer, the stable GTP-cap is extinguished and the exposed, unstable GDP heterodimers trigger microtubule catastrophe. Additionally, there are conformational changes undergone by the tubulin heterodimer as it undergoes GTP hydrolysis and is incorporated further into the microtubule lattice. Many studies have sought to understand whether tubulin compaction occurs within the microtubule lattice and whether compaction is influenced by nucleotide state and vice versa. For a more comprehensive analysis on tubulin compaction in the microtubule lattice, we direct readers toward previous works (Zhang et al., 2015; Estévez-Gallego et al., 2020; Cleary and Hancock, 2021).
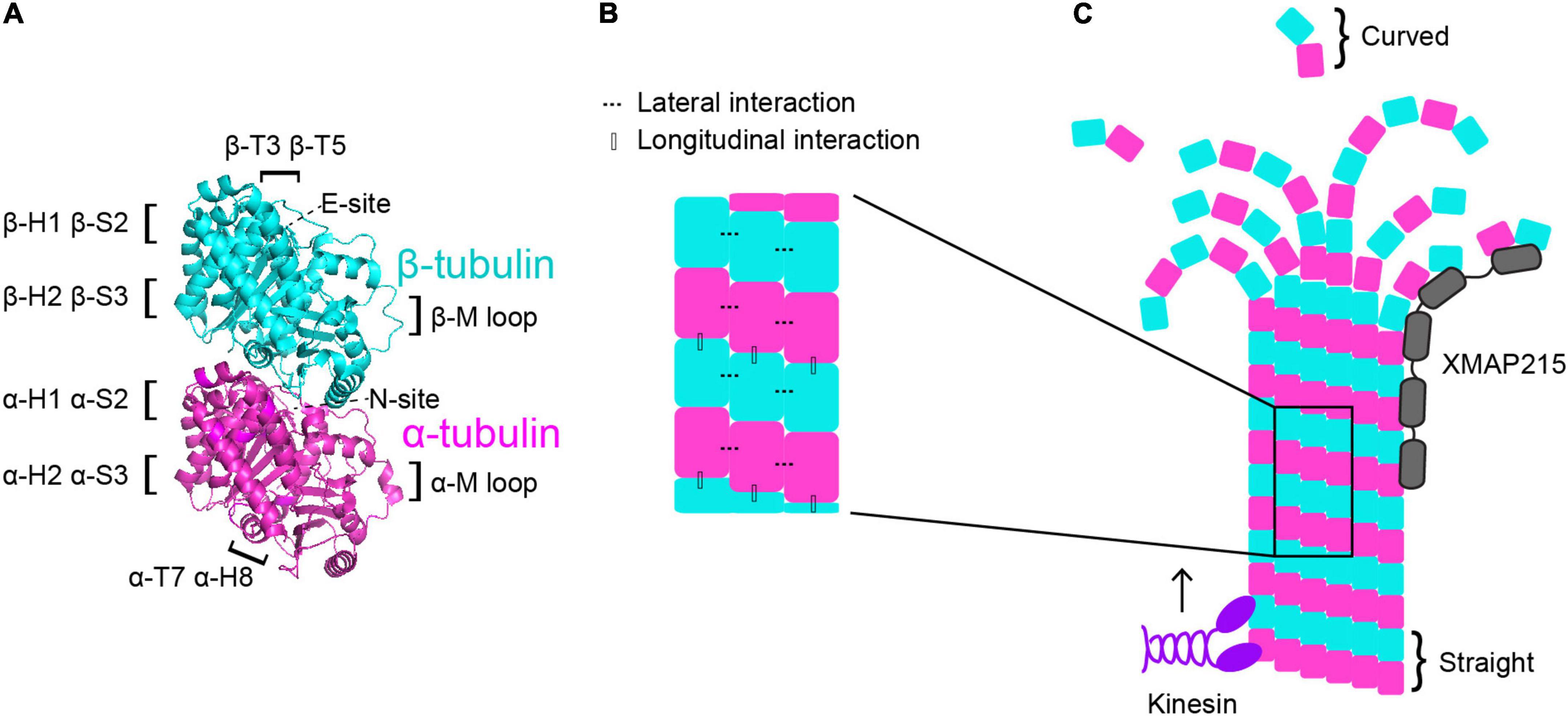
Figure 1. Microtubule dynamics are regulated at multiple control points. (A) α-tubulin is human TUBA1A in magenta and β-tubulin is human TUBB3 is in cyan (PDB structure: 5JCO). Regions that participate in longitudinal and lateral interactions are described along the sides of the structure. α- and β-tubulin each bind one GTP molecule. The non-exchangeable “N-site” on α-tubulin sits at the intradimer surface and the GTP is not hydrolyzed. The exchangeable “E-site” on β-tubulin is at the interdimer interface and the GTP molecule is hydrolyzed upon incorporation of the heterodimer into the microtubule lattice. (B) Lateral interactions occur between neighboring protofilaments and are illustrated by the dotted lines. Longitudinal interactions occur between at the interdimer interface between two stacked heterodimers. (C) Microtubule-associated proteins (MAPs) and motors can have specific preferences for associating with different segments of microtubules. For example, XMAP215 (gray) associates at microtubule plus ends and kinesins (purple) walk along the lattice toward the plus end. Tubulin undergoes a series of conformational changes. Free, curved heterodimer subsequently straightens as it is assembled into microtubule lattice.
A second way that the tubulin heterodimer acts like a switch to modulate microtubule dynamics involves nucleotide-independent conformational transitions (Figure 1C). Free tubulin heterodimers are curved in solution, while dimers within the microtubule lattice are straight. Early work postulated that the curved vs straight conformations of tubulin were dependent on the nucleotide state; that curved tubulin was GTP-bound and straight tubulin was GDP-bound (Melki et al., 1989). This model was supported by early electron cryomicroscopy work that indicated that depolymerizing microtubules, in which the heterodimers at the plus-end are GDP-bound, have splayed protofilaments, while GTP-capped polymerizing microtubules have straight ends (Mandelkow et al., 1991). However, more recent evidence indicates that the curvature of the free heterodimer is not appreciably influenced by the nucleotide state (Chrétien et al., 1995; Buey et al., 2006; Rice Luke et al., 2008; Nawrotek et al., 2011; Ayaz et al., 2012; Pecqueur et al., 2012). Additionally, other works have identified long, curving protofilament bundles at the plus-ends of polymerizing microtubules (Chrétien et al., 1995; Müller-Reichert et al., 1998; Vitre et al., 2008; Guesdon et al., 2016). More recent electron cryotomography work using microtubules assembled from purified tubulin in vitro, as well as microtubules in cells, reveals flared individual protofilaments at polymerizing microtubules plus-ends (McIntosh et al., 2018; Figure 1C). However, these pieces of work do not explicitly test whether straightening the heterodimer directly impacts nucleotide state. While the plus-end of the growing microtubule continues to be an active area of investigation, together these works indicate that the switch between curved and straight conformations is nucleotide-independent and may represent a separate point of microtubule regulation.
The curvature of the tubulin heterodimer plays a critical role in microtubule polymerization and depolymerization at plus ends because it facilitates the formation and breaking of interactions between tubulin heterodimers. The connections between heterodimers within the microtubule are critical in facilitating microtubule dynamics. When a free tubulin heterodimer lands at the plus end of a protofilament, it establishes longitudinal interactions with the previously added heterodimer. These longitudinal interactions occur at the interdimer interface and are established between the T7 loop and helix 8 (H8) of the incoming α-tubulin and the T3 and T5 loops of the β subunit at the microtubule plus end (Löwe et al., 2001; Figures 1A,B). Along with longitudinal interactions, the tubulin heterodimer also forms lateral interactions with heterodimers in neighboring protofilaments. On both α- and β-tubulin, the lateral contacts are mediated by H1, sheet 2 (S2), H2, and S3 on one subunit, and the M loop on the α or β subunit on the adjacent protofilament (Löwe et al., 2001; Figures 1A,B). Importantly, curved protofilaments that splay out from the microtubule plus end can only support longitudinal interactions, while straight protofilaments in the lattice support both longitudinal and lateral interactions. Establishment of these interactions facilitates the straightening of the tubulin heterodimer, making them a crucial intrinsic control in microtubule dynamics.
These features of the tubulin heterodimer reveal the dynamic instability properties that are unique to microtubules, and importantly, distinct from actin and intermediate filaments mentioned above. Additionally, each of these properties serves as a potential point of microtubule regulation. Indeed, there are a multitude of proteins in cells that bind and regulate microtubules to perform specific cellular functions. This next section will broadly discuss the role of a few key microtubule motors and MAPs. We will then focus on the importance of these intrinsic and extrinsic regulators in neurodevelopment.
Broadly, MAPs encompass the group of proteins that physically interact with microtubules. MAPs are further classified into subgroups depending on function and activity. This review will not fully capture the multitude and complexity of all MAPs, instead we will briefly describe a few key players that are highly involved in microtubule dynamics so that we may further investigate how these MAPs influence microtubule networks during neurodevelopment. For a more comprehensive review on MAPs, we point the reader to different reviews covering this topic (Goodson and Jonasson, 2018; Bodakuntla et al., 2019).
One of the key MAP subclasses is microtubule plus-end tracking proteins, also known as “+ TIPS.” As the name suggests, this group is comprised of MAPs that selectively associate with microtubule plus-ends, rather than along the microtubule lattice. +TIPs include proteins that play important roles in regulating microtubule dynamics, such as proteins in the XMAP215 and EB families. While each of these proteins reside at plus ends, they each bind to different tubulin structures and ultimately have vastly different functions. XMAP215 is a processive, concentration-dependent microtubule polymerase (Brouhard et al., 2008). Polymerase activity involves specific TOG (tumor-overexpressed gene) domains that are found in all XMAP215 proteins and recognize the curved conformation of tubulin (Ayaz et al., 2012; Al-Bassam et al., 2012; Byrnes and Slep, 2017). This unique conformation-preference enables TOG domains to preferentially bind to free tubulin heterodimers and to heterodimers at the distal tips of protofilaments (Ayaz et al., 2012; Al-Bassam et al., 2012; Byrnes and Slep, 2017; Figure 1C). EB1 is the most well-studied member of the EB protein family and regulates microtubule dynamics by increasing polymerization rates, catastrophe frequencies, and rescue frequencies of microtubules in vitro (Vitre et al., 2008). While EB1 does increase polymerization rates, its impact is rather mild compared to the effect of XMAP215 on microtubule polymerization. While XMAP215 activity is modulated by the curved tubulin conformation, EB1 is sensitive to the nucleotide state. Specifically, EB1 binds the microtubule lattice at the junction of four tubulin heterodimers that are in a transition state following GTP hydrolysis (Maurer et al., 2012). Importantly, MAPs do not act alone in cells, rather they may work with or against one another to regulate microtubules. To this end, when XMAP215 and EB1 are combined in vitro they synergistically increase microtubule polymerization rates beyond what either protein does on its own (Zanic et al., 2013). These +TIPS and others form a complex network of proteins that regulate microtubule dynamics to promote the formation, maintenance, or disassembly of microtubules at the right place and time.
Another subgroup of MAPs is microtubule motors, which are classified as either kinesins or dyneins (Figure 1C). Kinesins are a vast group of ATPases that are comprised of 44 individual kinesin genes divided into 16 subfamilies in humans (Miki et al., 2001, reviewed in Kalantari and Filges, 2020). Kinesins hydrolyze ATP to propel predominantly plus-end directed movement along the external surface of the microtubule. Kinesins are canonically considered to transport cargo along microtubule tracks, however certain kinesins also influence microtubule dynamics. For example, kinesin-5 serves as a microtubule polymerase (Chen and Hancock, 2015) and kinesin-8 and kinesin-13 motors serve as microtubule depolymerases (Desai et al., 1999; Hunter et al., 2003; Varga et al., 2006; Varga et al., 2009; Friel and Howard, 2011). Dynein is also an ATPase-dependent motor, however unlike kinesins, it primarily moves toward the microtubule minus ends. Additionally, while there are 44 different kinesin genes in humans, there is only one cytoplasmic dynein. Dynein activity is highly regulated by a suite of accessory chains, adaptors and MAPs that influence complex stability, speed, processivity, as well as selective binding to the various cargoes it transports (Reck-Peterson et al., 2018). Beyond cargo transport, dynein also plays crucial roles in cell division and migration (Dantas et al., 2016). Particularly important in neuronal migration, dynein generates pulling forces on the microtubule cage surrounding the nucleus and centrosome to ensure they are moved toward the leading process. Additionally, dynein can regulate microtubule dynamics by destabilizing microtubules (Laan et al., 2012; Estrem et al., 2017). Together, kinesin and dynein motors rely on microtubules to deliver cargo throughout the cell. However, they also serve important roles in regulating microtubule dynamics either directly, such as kinesin-5 or -8, or indirectly by influencing the binding of other MAPs to the microtubule. We will further explore the roles MAPs play in regulating microtubule dynamics, and how they interface with intrinsic tubulin properties, in the final section of this review.
In the next section we dive into how MAPs influence microtubule dynamics during neurodevelopment. Additionally, we describe the consequences of altering these extrinsic regulators during the critical time period of neurodevelopment and consider what disease-mutants can teach us about the requirements for proper brain development.
For proper neurodevelopment, the microtubule cytoskeleton plays a critical role in major cellular events, including progenitor proliferation, neuronal migration, and neuronal morphogenesis (Figure 2; reviewed in: Kriegstein and Noctor, 2004; Götz and Huttner, 2005). Microtubules are not simply structural scaffolds in these processes; they are actively involved in cargo transport, cell signaling, and dictating highly dynamic processes like symmetry breaking. While there are many factors that influence neurodevelopment, we will focus here on the importance of microtubules and their associated proteins in this process, as well as examples of diseases that occur as a result of mutations in MAPs and tubulin. Understanding microtubule activity and regulation at each step in the process of neurodevelopment is crucial for our understanding of proper neurodevelopment, and how it can go awry when these regulatory steps are perturbed.
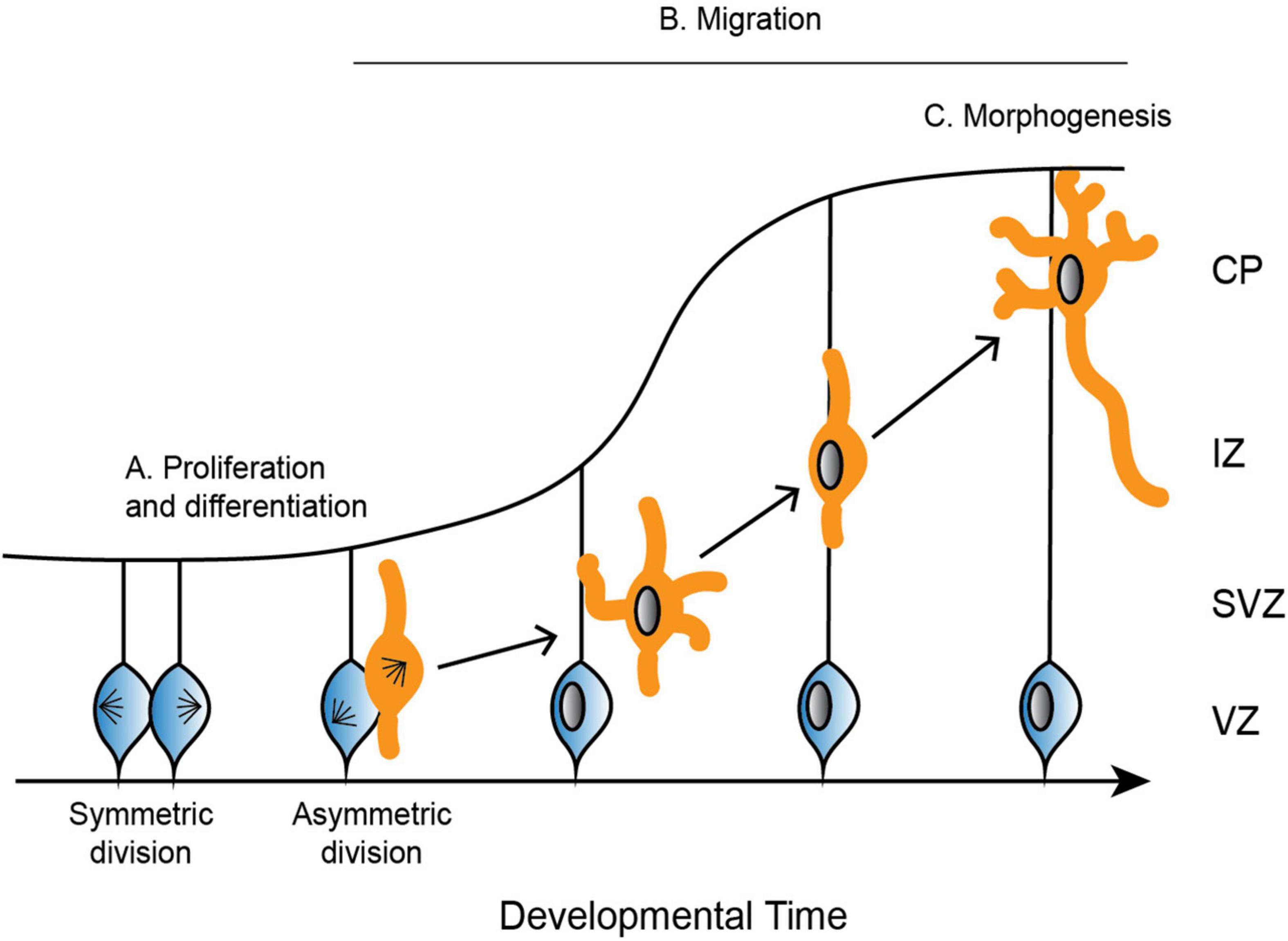
Figure 2. Depiction of the development of the cerebral cortex. (A) Neural progenitor cells (blue) undergo symmetric and asymmetric divisions before differentiating into specific cell populations, such as neurons (orange). (B) Neurons radially migrate along glial cells from the ventricular zone (VZ) to the cortical plate (CP). Neurons are bipolar in the VZ before switching to a multipolar state in the subventricular zone (SVZ). To continue radial migration, neurons then revert back to a bipolar state through the intermediate zone (IZ) until they reach the developing CP. (C) Upon reaching the CP, neurons undergo further morphogenesis, including maturation of the axon and extension of multiple dendrites.
In mammals, neurons are produced through a series of asymmetric and symmetric neural progenitor cell divisions (Figure 2A; Arai and Taverna, 2017). The differentiation of these neural progenitor cells is crucial for the specification of highly specialized cells that go on to form distinct structures of the brain. Defects in neural progenitor division can be catastrophic in either direction, as too many cell divisions may result in improper cellular differentiation, while delayed proliferation results in an under-production of specialized cell types. Aberrant divisions may give rise to incorrect cell types, or they may result in too many or too few progenitor cells at critical neurodevelopment time points.
Proliferation and differentiation require that progenitor cells form a bipolar mitotic spindle comprised of microtubules and an array of MAPs. Kinetochore microtubules capture and align chromosomes along the metaphase plate, and the chromosomes are separated by the force generated by the microtubules. This microtubule-generated force is highly regulated by a suite of microtubule motors and MAPs and relies on the coordination between many proteins to ensure proper chromosome segregation.
Following the differentiation of neural progenitor cells into neurons, different neuronal cell types migrate to specific regions of the developing brain (Reviewed in Marín and Rubenstein, 2003). Excitatory neurons radially migrate to the cortex and hippocampus, while inhibitory neurons tangentially migrate to the dorsal forebrain (Rakic, 1972; Altman and Bayer, 1990, reviewed in Kriegstein and Noctor, 2004). Excitatory neurons rely on multiple modes of transportation during radial migration, each of which requires dynamic and adaptable microtubule networks to remodel neuronal morphology. Newborn neurons undergo a brief period of bipolar locomotion in the ventricular zone before they move into the intermediate/subventricular zone and adopt a multipolar morphology with multiple dynamic processes (Figure 2B; Tabata and Nakajima, 2003). Neurons then transition back from a multipolar to a bipolar state. Axons are initiated during the multipolar stage and ultimately become the trailing process behind the stabilized pial surface-directed leading edge. Neurons then once again follow radial glial-guided locomotion until contacting the cortical surface and undergoing the terminal somal translocation, during which the apical dendrite is anchored to the marginal zone and the neuron detaches from the radial glia (Nadarajah et al., 2001; Santana and Marzolo, 2017).
The microtubule cytoskeleton is crucial throughout these stages of radial migration. During neuronal morphogenesis from bipolar to multipolar and back, microtubule dynamics are regulated via a complex signaling network (reviewed in Heng et al., 2010). Signaling pathways such as CDK5-driven phosphorylation of neuronal MAPs are crucial for adjusting the affinity, and thus the activity, between these MAPs and microtubules (reviewed in Heng et al., 2010). This tight regulation involving many MAPs are crucial in supporting the various process extension and retraction events that must occur during specific developmental time points. In the process of locomotion, microtubules are necessary to form a cage-like structure around the nucleus (Rivas and Hatten, 1995). The force generated to move the nucleus up the radial path to the cortical plate relies on dynein, as well as kinesins such as KIF1A and KIF2A (Homma et al., 2003; Tsai et al., 2007, 2010). Dynein, along with its regulator Lis1, generate the forces required to pull the nucleus and centrosome up the migrating cell toward the leading process (Tsai et al., 2007). Ultimately, each of these steps in neuronal migration requires properly regulated microtubules that respond to environmental signals to support the extension and retraction of processes, cargo transport, and signal transduction.
Once neurons have migrated to the upper cortical plate, they undergo further morphogenesis and differentiation as they mature the axon and extend many dendrites (Figure 2C). These elaborate morphologies are crucial for both sending signals through the axon, as well as receiving signals that are passed between thousands of other cells through the dendrites. Establishing neuronal polarity is therefore critical for proper brain function, and it is a complex process that relies on multiple intrinsic cellular and extrinsic environmental cues, many of which converge on the regulation of the microtubule cytoskeleton.
Neuronal polarity is established in part through microtubules. One of the defining distinctions in neuronal polarity is the plus-end out orientation of microtubules in the axon (Baas et al., 1988). In contrast, dendrites have mixed end microtubule polarity, meaning that a proportion of microtubules are plus-end out, while others are minus-end out (Baas et al., 1988; Burton, 1988). These differences in microtubule polarity have significant ramifications for cargo transport. Kinesins and dynein drive anterograde and retrograde transport, respectively, in axons (reviewed in Kevenaar and Hoogenraad, 2015). Dendrites on the other hand have dynein-driven anterograde transport (Kapitein et al., 2010). Microtubule polarity is a crucial part of proper cargo transport, which plays important roles in establishing proper neuronal polarity and morphology.
Much of our understanding of the importance of microtubule regulation in neurodevelopment stems from disease-associated mutations. For instance, mutations or deletions of MAPs, motors, or even tubulin itself results in neurodevelopment disorders. In this section we will discuss mutations in microtubule-related proteins that result in numerous neurodevelopment disorders, such as lissencephaly and pachygyria, polymicrogyria, and microcephaly. Each of these disorders present in patients with alterations to the folds (gyri) and grooves (sulci) that comprise the cortical structure. Lissencephaly is often referred to as “smooth brain” because patients have a total loss of folding patterns. Pachygyria is a milder version of lissencephaly as it still has folds and grooves, but the area of the folds is much broader, and the grooves are shallower. On the opposite end of the scale, polymicrogyria is characterized by having many gyri and sulci, and the folds are comparatively very small. Microcephaly is represented in patients with a smaller than typical brain. While each of these disorders are distinct, a single patient can present with a combination of these phenotypes, along with other neurological disorders not discussed here. This suggests that each of these disease-associated mutations may have multiple and nuanced effects on microtubule regulation, resulting in complex neurological phenotypes. Adding to this complexity, we know of some examples where mutations in different microtubule-related genes can give rise to similar phenotypes, but in other cases different mutations in the same gene are associated with vastly different disease states.
On the surface, this presents an interesting conundrum because cortical malformations cannot so easily be predicted from knowing the affected gene or location of the mutation within that gene. However, by examining what we know about microtubule regulation during neurodevelopment with what we know from in-depth studies of individual mutations, we may gain some understanding of mechanistic themes that link mutations to malformations. In this section, we highlight key mutations in microtubule-related proteins that result in one or multiple of these neurodevelopment disorders and discuss how studying these mutants can contribute to our understanding of proper neurodevelopment.
Studying the effect of mutations in MAPs that are linked with patient cortical malformations is important for several reasons when discussing the molecular mechanisms of tubulinopathies. The first reason is that studying MAP mutations in a disease context can provide great insight into how microtubules and microtubule regulators normally function in healthy contexts. Secondly, tubulinopathy mutations may lead to a loss of MAP binding and function, and therefore the tubulinopathy mutation may phenocopy mutations in MAPs that hinder their ability to properly regulate microtubule dynamics. And finally, some tubulinopathy mutations may alter the intrinsic properties of tubulin in a way that mimics constitutive MAP activity, ultimately subverting important local and temporal regulation that is normally provided by MAPs.
The first MAP to be specifically linked to lissencephalies is the gene lissencephaly 1 (LIS1) (Reiner et al., 1993). A homozygous Lis1 knockout in mice is lethal, and heterozygous Lis1-null mice have disorganized cortical layers, among other neurodevelopment defects (Hirotsune et al., 1998). LIS1 is typically studied in the brain and testes, but Lis1 mRNA is expressed in many murine cell types, and different cell types express various splice variants (Péterfy et al., 1998). In migrating neurons, LIS1 localizes primarily to the centrosome, and depletion of LIS1 in mice results in delayed and/or reduced neuronal migration (Hirotsune et al., 1998; Gambello et al., 2003; Tanaka et al., 2004). Importantly, LIS1 interacts and co-localizes with dynein and dynactin, and together these proteins pull the nucleus and centrosome toward the leading process during neuronal radial migration (Smith et al., 2000; Faulkner et al., 2000; Tsai et al., 2007). Additionally, LIS1, dynein, and dynactin localize to the growth cone and promote axon extension (Grabham et al., 2007). Depletion of LIS1 or dynein disrupts nuclear movement, further emphasizing the importance of these two proteins working together during neuronal migration (Shu et al., 2004; Tsai et al., 2007). Furthermore, LIS1 interacts directly with microtubules as well as tubulin heterodimers and reduces the frequency of microtubule catastrophe events in vitro (Sapir et al., 1997). Patient mutations have been identified across the LIS1 gene with no evident correlation between location within the gene and degree of lissencephaly severity observed in patients (Saillour et al., 2009). Furthering our understanding of how specific residue changes in LIS1 impact microtubules on a molecular level will be insightful in understanding changes observed in neuronal migration and ultimately tissue development.
A second critical MAP linked to lissencephaly is the protein doublecortin (DCX) (des Portes et al., 1998; Gleeson et al., 1998). DCX is located on the X chromosome and is specifically expressed during the proliferation of neuroprogenitors, and later during differentiation and neuronal migration (Francis et al., 1999; Gleeson et al., 1999; Kappeler et al., 2006; Koizumi et al., 2006). Its chromosome localization makes it a predominantly male-associated disease, though heterozygous female patients that have been identified have a milder, more mosaic phenotype. The DCX protein is localized at the leading edge of migrating neurons, consistent with interneurons derived from DCX knockout mice forming excessive branches with very short lifetimes (Friocourt et al., 2003; Kappeler et al., 2006). Additionally, these interneurons have disorganized migration patterns (Kappeler et al., 2006). DCX plays a crucial role in microtubule stabilization by promoting nucleation of 13 protofilament microtubules, as well as cooperatively binding and tracking of these 13 protofilament plus-ends (Moores et al., 2004; Bechstedt and Brouhard, 2012). Disease-associated mutations in DCX are found across the gene, with no obvious clustering in a particular domain that gives immediate insight into the effect of these mutations on microtubules and neurodevelopment (Bahi-Buisson et al., 2013). Presumably these mutations alter microtubule stability and/or binding of other MAPs to the microtubule. There has been substantial work uncovering the role of DCX in migrating neurons and ultimately neurodevelopment, however questions remain regarding how individual DCX residue positions and amino acid substitutions determine malformation severity (Reviewed in Bahi-Buisson et al., 2013; Ayanlaja et al., 2017).
Along with mutations identified in classical MAPs that are linked to neurodevelopment disorders, there have also been numerous cases linking mutations in microtubule motors to these disease states. Dynein cytoplasmic 1 heavy chain 1 (DYNC1H1), in this review referred to as dynein, is a minus-end directed motor that is expressed in a majority of human tissues. Patients harboring heterozygous mutations in dynein often present with polymicrogyria, which is identified as having many cortical folds that are abnormally small (Willemsen et al., 2012; Poirier et al., 2013; Fiorillo et al., 2014; Laquerriere et al., 2017; Amabile et al., 2020). Despite the functional intersection between dynein and LIS1, patient mutations in LIS1 are associated with lissencephaly, while dynein mutants are more often associated with polymicrogyria. Therefore, additional work in mouse models has been done to elucidate the role of dynein in neuron migration and why these patient mutations may result in unique patient phenotypes. Mice that are homozygous null for dynein are inviable, while heterozygous mice have no distinguishable defects (Harada et al., 1998). This suggests that one functional copy of dynein is sufficient for proper neurodevelopment in mice. Considering dynein is reliant on multiple regulators for proper function, it would be reasonable to consider that these regulators may help compensate for the depleted dynein levels in the heterozygous mice. Beyond its role in generating pulling forces on the nucleus and centrosome during neuronal migration, dynein serves as the primary retrograde motor for axonal transport, due to axonal microtubules being plus-end out (Heidemann et al., 1981; Baas et al., 1988). Dynein motors are necessary for plus-end out microtubule orientation, as well as trafficking specific cargos from the distal tip of the axon to the cell body (Zheng et al., 2008; Satoh et al., 2008; reviewed in Hirokawa et al., 2010). Multiple groups have thus used ENU-induced heterozygous missense mutations or RNAi depletion to study the effects of dynein in neurodevelopment and neurodegeneration. In these models, neuronal migration was perturbed, and dynein-directed retrograde axonal transport of key cargoes were delayed and, in some instances, significantly impaired (Hafezparast et al., 2003; Ori-McKenney and Vallee, 2011; Zhao et al., 2016). Interestingly, patient mutations have been identified across DYNC1H1, with varying degrees of patient phenotypes and associated severities (Harms et al., 2012; Hoang et al., 2017; Beecroft et al., 2017). However, patients that present with cortical malformations typically have mutations clustered in the motor domains (Harms et al., 2012; Hoang et al., 2017). It has been speculated that some of the mutants may interfere with destabilizing the conformational state of dynein during ATP hydrolysis (Hoang et al., 2017). Further work on multiple patient-associated dynein mutants located across the protein indicate that these mutations have many different effects, including in some cases defects in spindle positioning and altered motility in vitro (Marzo et al., 2019). Studying disease mutants, as well as studying the role of different dynein domains, played a crucial role in advancing our understanding of how patient mutations that occur throughout the protein contribute to impaired neurodevelopment (Niekamp et al., 2019). Applying ideas and studies such as these to tubulinopathy mutations may be crucially important to developing mechanistic themes about how tubulinopathies arise.
While dynein serves as the primary minus-end directed motor, the kinesin families principally function as plus-end directed motors. Similar to dynein, there are several mutations that have been identified in different kinesins that are associated with neurodevelopment disorders. In humans, there are 44 identified kinesin family member genes, which are further split into 16 subfamilies (Miki et al., 2001; Kalantari and Filges, 2020). While mutations in many kinesins are associated with neurological disorders, we focus here on three specific kinesins that have been associated with cortical malformations: KIF5C, KIF2A, and KIF21B.
KIF5C is a member of the kinesin-1 family and is particularly enriched in neuronal cells (Kanai et al., 2000). Patient mutations that have been identified in KIF5C have presented as pachygyria, characterized by broad folds and shallow grooves (Poirier et al., 2013; Willemsen et al., 2014; Jamuar et al., 2014; Cavallin et al., 2016; Michels et al., 2017). To date, five of the six KIF5C mutations identified occur at the glutamate residue 237, while the sixth mutation is located at alanine residue 268. Both residues reside in the microtubule-binding domain of KIF5C, which functional and structural studies have indicated is necessary for selective accumulation of KIF5C in the axon (Nakata et al., 2011; Morikawa et al., 2015). In accordance with these data, mutating glutamate 237 to a valine inhibits ATP hydrolysis and disrupts KIF5C localization at the cell cortex (Poirier et al., 2013). To date, the molecular impact of the other KIF5C mutations is unknown, however it is reasonable to consider that other mutants identified in the motor domain may have similar effects. Further work addressing the neuronal cellular impact of improper KIF5C localization and activity will further elucidate how these mutations result in cortical malformations such as pachygyria.
Another kinesin that has been identified to have mutations linked to neurodevelopment disorders is KIF2A, a member of the kinesin-13 family, which is expressed throughout human tissues. Patient phenotypes from different KIF2A mutants present as lissencephaly, pachygyria, and microcephaly, or a combination of these malformations (Poirier et al., 2013; Tian et al., 2016; Cavallin et al., 2016). In-depth studies of two of these mutants reveal that ectopic expression of disease-causing mutants disrupt interneuron migration, as well as the proliferation of progenitor cells (Broix et al., 2018). In developing neurons, KIF2A is localized to growth cones of developing axons and plays critical roles in axonal branching and pruning (Noda et al., 1995; Homma et al., 2003; Maor-Nof et al., 2013). Similar to the mutations identified in KIF5C, all mutations in KIF2A reported to date are located in the same region. However, in the case of KIF2A mutants, all are located in the motor domain (Poirier et al., 2013; Tian et al., 2016; Cavallin et al., 2016; Broix et al., 2018). KIF2A functions as a microtubule depolymerase, specifically by inducing a destabilizing conformational change at microtubule ends (Desai et al., 1999). Microtubule regulation via KIF2A’s microtubule depolymerase activity is clearly linked to proper cortical development, however future work will need to determine precisely when and where KIF2A activity is required.
KIF21B is a member of the kinesin-4 family and is predominantly expressed in the human testes, spleen, and is highly enriched in neurons (Marszalek et al., 1999). Mice that are homozygous null for KIF21B display microcephaly and isolated neuronal cultures have decreased dendritic complexity (Muhia et al., 2016; Kannan et al., 2017). To date, three patients have been identified with missense mutations in KIF21B. Interestingly, each patient presents with a different brain structure; one patient presents with microcephaly, one with complete agenesis of the corpus callosum, and the third with no notable brain structure abnormalities (Asselin et al., 2020). Further analysis into these three patient-associated mutations highlight that, to varying degrees, the KIF21B mutants impair neuronal migration by increasing KIF21B motor activity (Asselin et al., 2020). The results of recent molecular studies of KIF21B are complex. For example, the depletion of KIF21B by RNA interference in neuronal cultures results in increased microtubule polymerization rates (Ghiretti et al., 2016), but a full KIF21B knockout results in decreased rates (Muhia et al., 2016). However, in vitro work indicates that full-length KIF21B increases microtubule growth rates (Ghiretti et al., 2016), while other work indicates that the KIF21B motor domain alone decreases growth rates in vitro (van Riel et al., 2017). These seemingly contradictory pieces of work highlight the importance of understanding the contribution of individual protein domains and residues in regulating microtubule dynamics and cargo transport, and the ultimate effect of this on neurodevelopment.
Patient-associated mutations identified in both MAPs and motors have been crucial to our understanding of the important mechanisms that underlie proper neurodevelopment. The focus of the next section will be centered around patient-associated mutations in tubulin that are associated with neurodevelopment disorders, and whether the field can predict the molecular and tissue-level outcomes of this multitude of mutants.
Advancements in patient exome and whole genome sequencing have enabled the identification of single nucleotide polymorphisms (SNPs) linked to cortical malformation disorders. To date, mutations have been identified in four human β-tubulin isotypes and one of the α-tubulin isotypes (Bahi-Buisson et al., 2014; Maillard et al., 2022). While a subset of mutations leads to particular patient cortical malformations, other mutations, often in the same gene or even domain, result in other types of malformations. In this section, we discuss whether certain phenotypes can be attributed to the expression levels of certain isotypes during development. We also describe specific tubulin mutations and how those relate to the patient-observed cortical malformations. Whether these changes can be attributed to the type of change in amino acid, or if it is residue/domain specific, remains a large and unanswered question in the field.
Humans and many other species express multiple α- and β-tubulin genes, known as isotypes. Isotypes are differentially expressed according to cell type and developmental timing, and encode proteins that exhibit high levels of conservation, but an array of subtle sequence differences (Ludueña and Banerjee, 2008). A majority of these sequence differences exist in the C-terminal tails, the 10–20 amino acids at the carboxy-terminus of α- or β-tubulin. In some cases, these sequence differences have been shown to impart significant functional differences (Sirajuddin et al., 2014; Vemu et al., 2017). These different isotypes can co-polymerize together, forming diverse microtubules composed of multiple tubulin isotypes (Lewis et al., 1987). While the isotypes have the capacity to co-polymerize with one another, the varying expression levels of the isotypes in different cell types and throughout development indicate that not all isotypes may be involved in all cellular functions, at all times. Therefore, it remains an open question whether isotypes show selective enrichment into particular subsets of microtubules. Studying the expression and role of various isotypes in neurodevelopment may provide great insight into the importance of varied tubulin expression in neurodevelopment and disease.
A classic example of unique cell type and developmental time point expression of tubulin isotypes is the β-tubulin-III (TUBB3). In the brain, TUBB3 is classified as a neuron-specific β-isotype (Jiang and Oblinger, 1992; Burgoyne et al., 1988). Additionally, TUBB3 expression is high in neurons both in the fetus and at birth, then decreases throughout postnatal development (Denoulet et al., 1986; Jiang and Oblinger, 1992; Miller et al., 2014). Interestingly, despite being a neuron-specific isotype, Tubb3-/- mice have no discernible neurodevelopment defects or cortical malformations (Latremoliere et al., 2018). In neurons from the Tubb3 knockout mice there is a decrease in microtubule polymerization rates in growth cones, as well as a decrease in neurite outgrowth (Latremoliere et al., 2018). Importantly, there is an increase in other β-tubulin isotypes so that total β levels are unchanged, indicating that other isotypes are upregulated to compensate for the loss of Tubb3 (Latremoliere et al., 2018). Together, these data indicate that while TUBB3 is a neuron-specific isotype, it is not required to maintain the appropriate amount of total β-tubulin, nor is it required for proper neurodevelopment. However, the observed changes in microtubule polymerization rates are interesting and are consistent with different isotypes exhibiting various microtubule dynamics, and that isotypes co-polymerizing in different proportions are likely to alter dynamics (Vemu et al., 2017).
It is interesting to note that despite Tubb3-/- mice displaying relatively normal neurodevelopment, specific Tubb3 point mutations disrupt proper brain development in mice (Tischfield et al., 2010). There are a number of patient cases that have heterozygous, missense mutations in TUBB3 and present with severe neurodevelopment phenotypes (Poirier et al., 2010; Oegema et al., 2015; Park et al., 2021). This suggests that either point mutations act in a dominant manner that is distinct from the total loss of TUBB3, or that the requirement for TUBB3 function is higher for human brain development compared to mouse brain development. These disease-associated mutations have a range of clinical phenotypes; some have cortical malformations such as polymicrogyria, while others have a relatively well-developed cortex but display agenesis or hypoplasia of the corpus callosum or basal ganglia dysplasia (Poirier et al., 2010; Oegema et al., 2015; Park et al., 2021). Additionally, these mutants form heterodimer and incorporate into microtubules at various efficiencies (Poirier et al., 2010; Oegema et al., 2015). Comparing phenotypes between Tubb3-/- mice and TUBB3-tubulinopathy mutations indicates that proper neurodevelopment is achieved in animals completely lacking Tubb3, but not in animals or patients that harbor a missense mutation in the gene. Clearly the cells can compensate in the absence of Tubb3 with other β-isotypes, indicating that these TUBB3-associated tubulinopathies are acting in a different mechanism that cannot be solely explained by changes in isotype expression due to haploinsufficiency. Based on RNA-seq data gathered from the whole fetal brain, TUBB3 makes up approximately 17% of β-tubulin at this crucial developmental time point (Miller et al., 2014; Park et al., 2021). Therefore, how missense mutations in a β-tubulin isotype that represents only a portion of the tubulin pool can exert dominant effects on neurodevelopment will be critically important in future studies.
On the other side of the heterodimer is α-tubulin, which similarly to β-tubulin, has multiple isotypes in humans and other vertebrates, and these isotypes are expressed at various levels in different cell types and points during development (Ludueña and Banerjee, 2008). The most highly expressed α-tubulin isotype in post-mitotic developing neurons is TUBA1A (Miller et al., 1987; Gloster et al., 1994; Gloster et al., 1999). Unlike Tubb3, Tuba1a is seemingly indispensable in mouse neurodevelopment and Tuba1a-/- is lethal (Bittermann et al., 2019). Interestingly, there is only a slight reduction in survivability in mice that have only one copy of Tuba1a deleted (Bittermann et al., 2019). Analysis of the fetal forebrain indicates that Tuba1a-/- mice have an increase in ventricular zone width, and decreased intermediate zone and cortical plate widths, as compared to WT and Tuba1a heterozygous mice (Bittermann et al., 2019). Mice do not develop gyri and sulci as humans do, and therefore their brains are always smooth, making it difficult to do a side-to-side comparison with human cortical malformations, such as lissencephaly. However, the changes observed in the developing cortex regions in the Tuba1a-/- mice is consistent with neuronal migration defects that result in cortical malformations in patients (Keays et al., 2007). Compared to total protein levels at this developmental time point, total α-tubulin is decreased in brain lysates of Tuba1a-/- mice compared to WT (Bittermann et al., 2019). This indicates that for Tuba1a, other α-isotypes are not sufficiently upregulated in response to the knockout. This suggests that the requirement for TUBA1A in the human developing brain may be quite distinct from TUBB3.
Results from unbiased, ENU mutagenesis screens conducted in two labs further demonstrate the unique requirements of Tuba1a in mouse neurodevelopment. One study identified a Tuba1a-S140G mutant that disrupts neuronal radial migration, a key feature of many cortical malformations (Keays et al., 2007; Belvindrah et al., 2017). More recently, an analogous mutation in human TUBA1A has been identified in a patient that is documented to display microcephaly and aplasia/hypoplasia of the corpus callosum (from DECIPHER database, ref ID: 273321; Hebebrand et al., 2019). Further analysis reveals that Tuba1a-S140G has reduced GTP-binding and heterodimer formation, although the mutant heterodimer is still able to incorporate into microtubules (Keays et al., 2007). Summarizing these data, Tuba1a-/- and Tuba1a-S140G heterozygous mice both have impaired neuronal migration (Keays et al., 2007; Belvindrah et al., 2017; Bittermann et al., 2019), while Tuba1a+/- mice have no migration defects (Bittermann et al., 2019). These data cannot be explained simply by a reduction in the amount of Tuba1a expression but may rather be explained by the S140G mutant having a dominant effect on the microtubules in which it incorporates, and ultimately brain development.
A second ENU-generated mutation to note is Tuba1a-N102D, which was identified in a forward genetic screen searching for locomotor defects in mice (Gartz Hanson et al., 2016). Tuba1aND/ND mice are neonatal lethal, have small brains, and display defects in cortical layering in the cerebral cortex (Gartz Hanson et al., 2016). The heterozygous Tuba1aND/+ mice survive to adulthood, but do not form proper midline commissures (Buscaglia et al., 2022). Additionally, Tuba1aND/+ brains have significantly reduced levels of total α-tubulin at P0 (Buscaglia et al., 2020), and the mutant is unable to incorporate into microtubules in neuronal cultures (Buscaglia et al., 2022). At the molecular level, the N102D mutant disrupts heterodimer stability and incorporation into the microtubule lattice (Gartz Hanson et al., 2016). The N102D mutant is an interesting way to probe the importance of TUBA1A in the developing brain, particularly because it provides insight into the necessity of having not only sufficient TUBA1A protein levels, but also a level of TUBA1A function that is sufficient for microtubule dynamics, neuronal cell biology, and ultimately tissue formation.
It is clear from these two ENU-generated mouse models that Tuba1a plays a critical role in neurodevelopment, and that compensation for Tuba1a-/- by other α-isotypes does not occur in a similar manner as seen for Tubb3–/–. The molecular and phenotypic differences between the Tuba1a-S140G and Tuba1a-N102D heterozygous mice also point toward the possibility that neurodevelopment defects may have different molecular drivers, depending on specifics of the missense mutation such as what the mutation is, and where it is located. Therefore, it is interesting to probe further into TUBA1A specific tubulinopathy mutations and determine if there are particular patterns or themes that we can draw from these studies about how particular regions influence TUBA1A biology and ultimately neurodevelopment.
Much of the field’s understanding of how specific tubulinopathy-associated amino acid changes impact molecular and/or cellular function stem from a handful of studies of the mechanistic impact of a particular mutation. There are 119 heterozygous, missense mutations that have been identified in TUBA1A to date (Hebebrand et al., 2019). Therefore, while these individual studies can provide important insights into how a certain mutant alters microtubule dynamics and neuronal cell biology, it would be useful to identify mechanistic themes that apply across multiple of these mutations. Here, we summarize the work that has been done in these studies modeling patient mutations. Additionally, we attempt to identify themes that span across these studies in order to gain a greater understanding of how TUBA1A mutants impact molecular and cell biology that ultimately lead to cortical malformations.
The first study of a patient mutation, TUBA1A-R264C, suggested that the mutant may act through a haploinsufficiency mechanism. This mutant was first identified in a patient that presented with pachygyria (Keays et al., 2007). Further analysis revealed that the R264C mutation decreases the formation of α-/β-tubulin heterodimer in vitro, as a result of decreased affinity between the mutant α-tubulin and a tubulin chaperone, TBCB, that is important for tubulin heterodimer biogenesis (Tian et al., 2008). Despite the reduction in the biogenesis of heterodimers containing α-tubulin R264C, when a heterodimer is formed from this mutant tubulin it incorporates into microtubules, albeit at a reduced rate. Based on these results, the authors conclude that the decreased formation of heterodimer from α-tubulin R264C is consistent with a haploinsufficiency phenotype; essentially, there is insufficient functional heterodimer to support necessary microtubule structures and dynamics (Tian et al., 2008). While haploinsufficiency could be a cause of disease, it is important to note that this study does not address whether total α-tubulin levels are altered in neurons that express R264C. This is an important consideration, given the robust tubulin biogenesis pathways that produce heterodimers in cells. Furthermore, it would be important to investigate whether microtubules that do have R264C heterodimer incorporated have altered dynamics compared to microtubules containing wild-type tubulin. Answering questions such as these would provide the field valuable insight into whether these TUBA1A mutants have the capacity to dominantly disrupt microtubule dynamics and function, and to what extent cells may have the capacity to mitigate these effects.
Whether TUBA1A tubulinopathy mutants all act through a common mechanism of haploinsufficiency was called into question in 2010, when Tian et al. (2010) investigated nine additional tubulinopathy disease-associated mutants in TUBA1A [I188L, P263T, L286F, R402C, R402H, S419L, I238V, and L397P]. These mutants are spread across the TUBA1A gene with no apparent clustering that aligns with molecular or patient-level phenotype (Tian et al., 2010). The results of this study reveal that some mutants disrupt the tubulin heterodimer assembly pathway, while others are unstable in vitro, and others impact microtubule assembly (Tian et al., 2010). These results suggest that some mutants result in haploinsufficiency while others mutants may act dominantly to perturb microtubule dynamics. Understanding how these two different scenarios ultimately result in similar patient phenotypes is an outstanding question.
As stated above, it had been previously found that TUBA1A-R402C significantly, and -R402H slightly, reduces the amount heterodimer in vitro (Tian et al., 2010). However, they also find that these mutants do incorporate into microtubules (Tian et al., 2010). Although these mutants do not form heterodimer in vitro, it was later found that these mutants do form heterodimer in cells (Aiken et al., 2019). This study showed that TUBA1A-R402C and -R402H assemble into microtubule polymer and act dominantly to disrupt neuronal migration in mice (Aiken et al., 2019). Furthermore, when modeled in budding yeast, these mutations produce normal amounts of heterodimer that incorporates into microtubules. Strikingly, microtubules containing R402 mutant tubulins disrupt, but do not fully ablate, dynein activity (Aiken et al., 2019). This indicates an alternative model for tubulinopathies where if these mutant tubulins are able to assemble into microtubules, they may then alter the binding of MAPs.
Our recent work further highlights the importance of the interactions between tubulin subunits, MAPs, and the interplay between the two. Our recent investigation of TUBA1A-V409I and V409A highlights the ability of these mutants to polymerize into microtubules and dominantly disrupt neuronal migration and morphologies (Hoff et al., 2022). Similar to the above mentioned studies of R402C and R402H tubulinopathy mutants, the V409I and V409A mutants were modeled in cultured neurons and in budding yeast to investigate molecular and cellular-level effects. In this case, the two mutants disrupt the microtubule dynamics regulation conferred by XMAP215/Stu2, and the more severe V410A mutant intrinsically drives increased microtubule polymerization rates in in vitro reconstitution experiments with purified tubulin. These findings highlight the importance of studying points of both the intrinsic activity of tubulin proteins and extrinsic microtubule regulation by MAPs, and how these areas interact to control microtubule dynamics throughout development and disease.
While these individual examples do not encompass all of the work that has been done to decipher how tubulinopathy mutations result in such catastrophic cortical malformations, they provide a sample of how the field has expanded its thinking of the mechanistic themes underlying these diseases. While some work has indicated that TUBA1A tubulinopathy mutations result in haploinsufficiency, more recent works have highlighted how patient mutations dispersed across the α-tubulin protein can act dominantly from within microtubules to alter dynamics and interactions with MAPs (Figure 3A). The ability of many of these mutant tubulins to incorporate into microtubules and dominantly disrupt everything from MAP binding, microtubule dynamics, neuronal cell biology, and tissue structures indicates that the field should be considering these mutations at multiple levels.
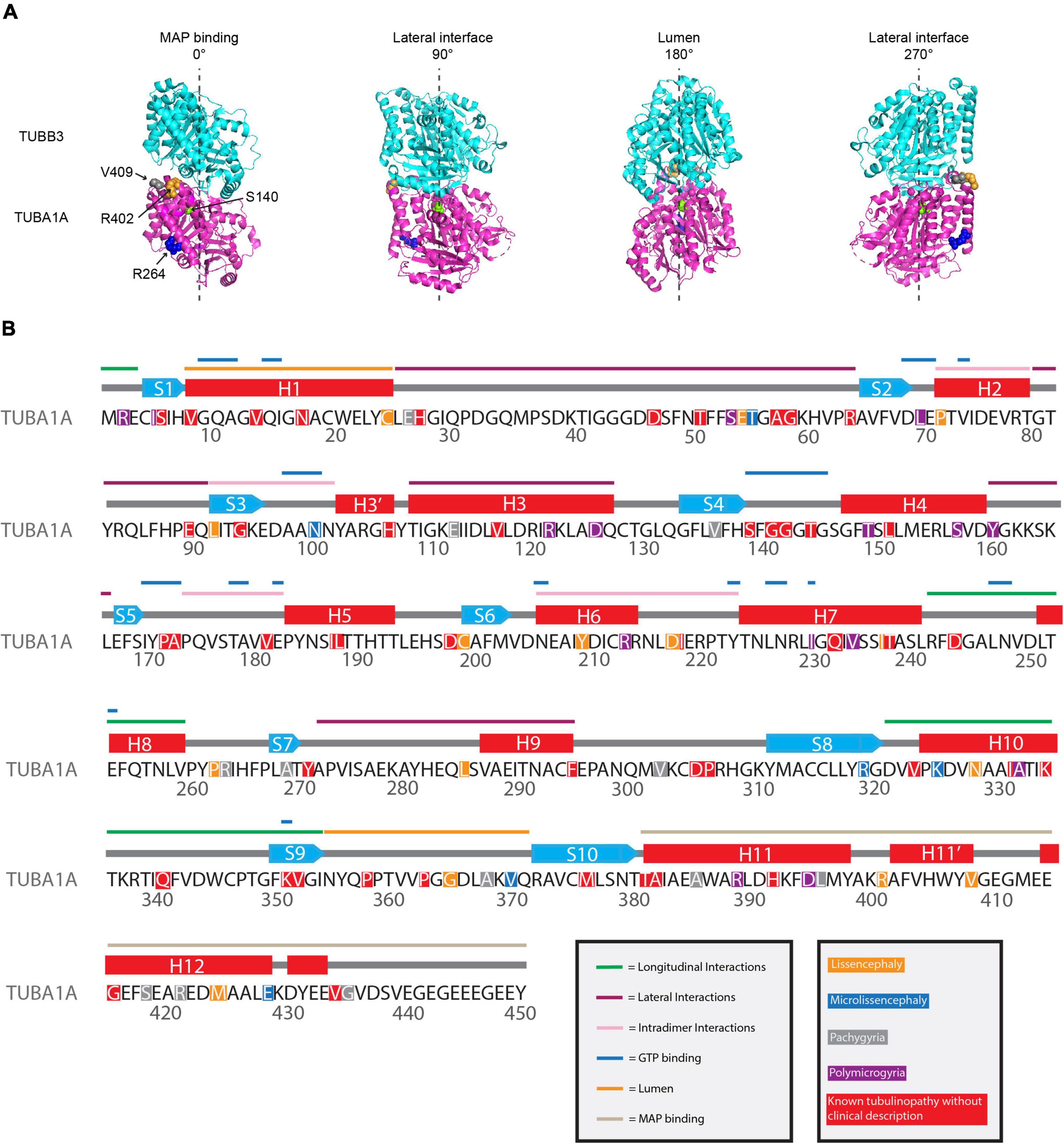
Figure 3. Mapping TUBA1A tubulinopathy mutations. (A) Mapping individual mutants described in section “Investigating specific residues affected by mutations in patients; can they tell us more about the etiology of tubulinopathies?” on TUBA1A (PDB structure: 5JCO). α-tubulin is human TUBA1A in magenta and β-tubulin is human TUBB3 in cyan. Spheres represent TUBA1A residues S140 (green), R264 (blue), R402 (orange), and V409 (gray). Four views of the structure are shown, each rotated 90° along the longitudinal axis. The 0° angle represents the outer surface of the microtubule that is considered the MAP binding region. 90° and 270° angles represent lateral interfaces with adjacent protofilaments. 180° represents the luminal side of the microtubule. (B) Known TUBA1A mutations to date mapped on primary sequence according to predominant cortical malformation. If no clinical imaging data is available to us, the mutation is highlighted in red. Functional regions described in section “Mapping the tubulinopathy mutations” are labeled in various colors above the secondary structures.
The increasing number of identified tubulinopathy-associated mutations emphasizes the need to identify broader mechanistic themes that link molecular level tubulin defects to tissue level disease. In the long term, identifying these links will be useful for predicting the phenotypic outcome of specific mutations and identifying new treatment options. As mentioned previously, the tubulin heterodimer has multiple interfaces that are the intrinsic regulatory elements of microtubule dynamics. These interfaces include the nucleotide N- and E-sites, as well as longitudinal and lateral interfaces. Despite these regions being recognized as key intrinsic regulators of microtubule dynamics, many tubulinopathy mutations reside outside these regions and yet still impact microtubule activity.
Other regions are predicted and/or have been identified to interact with various MAPs and motors. These MAPs and motors have primarily been identified to interact with the external sides of the microtubule. It is important to note that another class of microtubule binders have been identified, known as microtubule inner proteins (MIPs), although to date this class of proteins have only been identified to interact with microtubule doublets that are present in the axonemes of cilia and flagella (Nicastro et al., 2006, 2011). This class of proteins is much less studied compared to MAPs and motors; however, it is important to consider that there are residues within the microtubule lumen that may interact with these MIPs. Identifying whether TUBA1A mutations may impact one or multiple of these interacting sites will be important in understanding the functional consequences of these mutants and may provide further insight into the specific roles of microtubules in neurodevelopment.
To understand if we can decipher patterns between TUBA1A mutations and disease phenotype and/or severity, we mapped the tubulinopathy mutants in several ways. From a list of the 119 TUBA1A mutations identified to date, we classified each mutation with the predominant cortical malformation that has previously been described in the literature (Hebebrand et al., 2019). The major cortical malformations we identified in the literature and that we used for the remainder of the analysis in this section are lissencephaly, microlissencephaly, pachygyria, and polymicrogyria. If no clinical assessment was available to our knowledge, we classified the mutation as “not available”. Since one might expect mutations with similar malformations to affect similar regions of the tubulin protein, we mapped the mutants along the primary sequence. However, the distribution of mutations does not reveal any striking enrichment of mutations that result in similar malformations anywhere along the primary amino acid sequence (Figure 3B). Rather, the mutations that cause lissencephaly, microlissencephaly, pachygyria, and polymicrogyria appear to be randomly distributed across the gene.
The charge and hydrophobicity of amino acids are important for a multitude of protein functions Therefore, we considered whether different types of amino acid side chain changes were linked to specific cortical malformations. Of the 119 TUBA1A mutations identified, 59 of them do not have clinical neuroimaging data publicly available. Therefore, we excluded these mutations from the following analysis. We classified these types of amino acid changes as: (1) loss of charge or charge swap, (2) gain of charge, (3) loss or gain of hydrophobicity, and (4) no change in charge or hydrophobicity. Tubulin electrostatic charges are critical for interactions with MAPs and motors, therefore we reasoned that a loss of charge or a charge swap could have significant implications for microtubule interactors. On the other hand, a mutation that results in a gain of charge on a previously uncharged amino acid could potentially impact microtubule interactors, but it could likely have different effects depending on where in the protein it is located, and therefore we made a separate category. The loss or gain of hydrophobicity could have significant impacts on protein folding. The final classification of no change represents mutations that do not impact the charge or hydrophobicity from the original amino acid, calling into question how these seemingly benign mutations cause such devastating malformations. It is important to note that these categories are not all mutually exclusive. For example, one mutation could result in a loss of charge as well as a gain of hydrophobicity. We find that each malformation is associated with mutants with at least one of each type of amino acid change (Figure 4). Because these data are not mutually exclusive, we are unable to run statistics on the data set to determine whether the types of amino acid changes are significantly different than one another. However, because each malformation is associated with at least one mutant from each type of amino acid change, we asked whether we could identify common mechanisms of these mutants by categorizing by where the mutations lie within the protein structure.
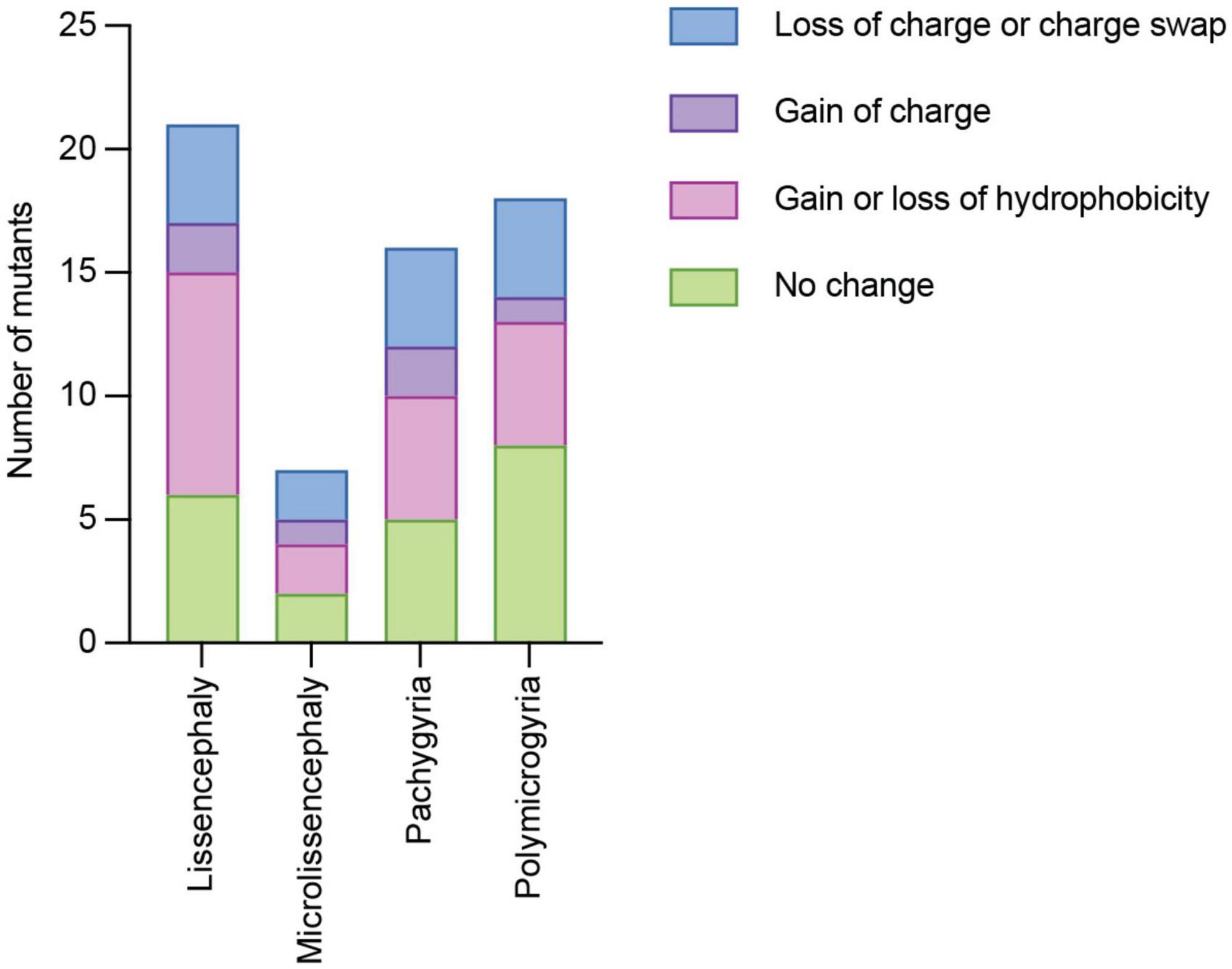
Figure 4. Quantifying TUBA1A mutations by type of amino acid side chain change. TUBA1A mutations were sorted by the type of amino acid change that occurs in patients (loss of charge or charge swap, gain of charge, gain or loss of hydrophobicity, or no change in charge or hydrophobicity). TUBA1A mutations were also sorted by the primary cortical malformation resulting from each mutation (lissencephaly, microlissencephaly, pachygyria, or polymicrogyria). For each cortical malformation, the number of mutations that qualify as one (or multiple) of these types of amino acid changes were reported.
Secondary structures play crucial roles in protein folding, structure, and function. TUBA1A secondary structure consists of approximately 39% helices, 12% sheets, and 49% loops (Löwe et al., 2001). Using all 119 TUBA1A mutants, regardless of whether there is publicly available clinical data, we asked whether tubulinopathy mutations occur more frequently in a particular secondary structure, potentially revealing a bias for disease-associated mutations. Based on a Chi-squared test, we find there is no significant enrichment of TUBA1A mutations in any of the secondary structures beyond what would be expected if the mutations were randomly distributed [Figure 5A; 5.99 critical value when α = 0.05; X2 (df = 2, N = 119) = 1.202; p = 0.55]. This indicates that there is not a general enrichment of mutations in the secondary structures. We next asked whether mutations associated with a particular cortical malformation were biased toward particular secondary structures. Therefore, for this analysis we used only the 53 TUBA1A mutations that have available clinical neuroimaging data. For each individual malformation category, we used a Fisher’s exact test to determine whether the number of mutants found in one secondary structure were significantly different than would be expected if the mutations were random. We find that the majority of TUBA1A mutations are not significantly enriched in a particular secondary structure. However, the number of polymicrogyria mutations found in helices is significantly higher than what would randomly be expected (Figure 5B; Table 1; p = 0.03). This indicates that polymicrogyria mutations in TUBA1A are more likely to be found in helices. Further work will be needed to investigate the link between disrupting helical structures and the high number of small gyri observed in polymicrogyria patients.
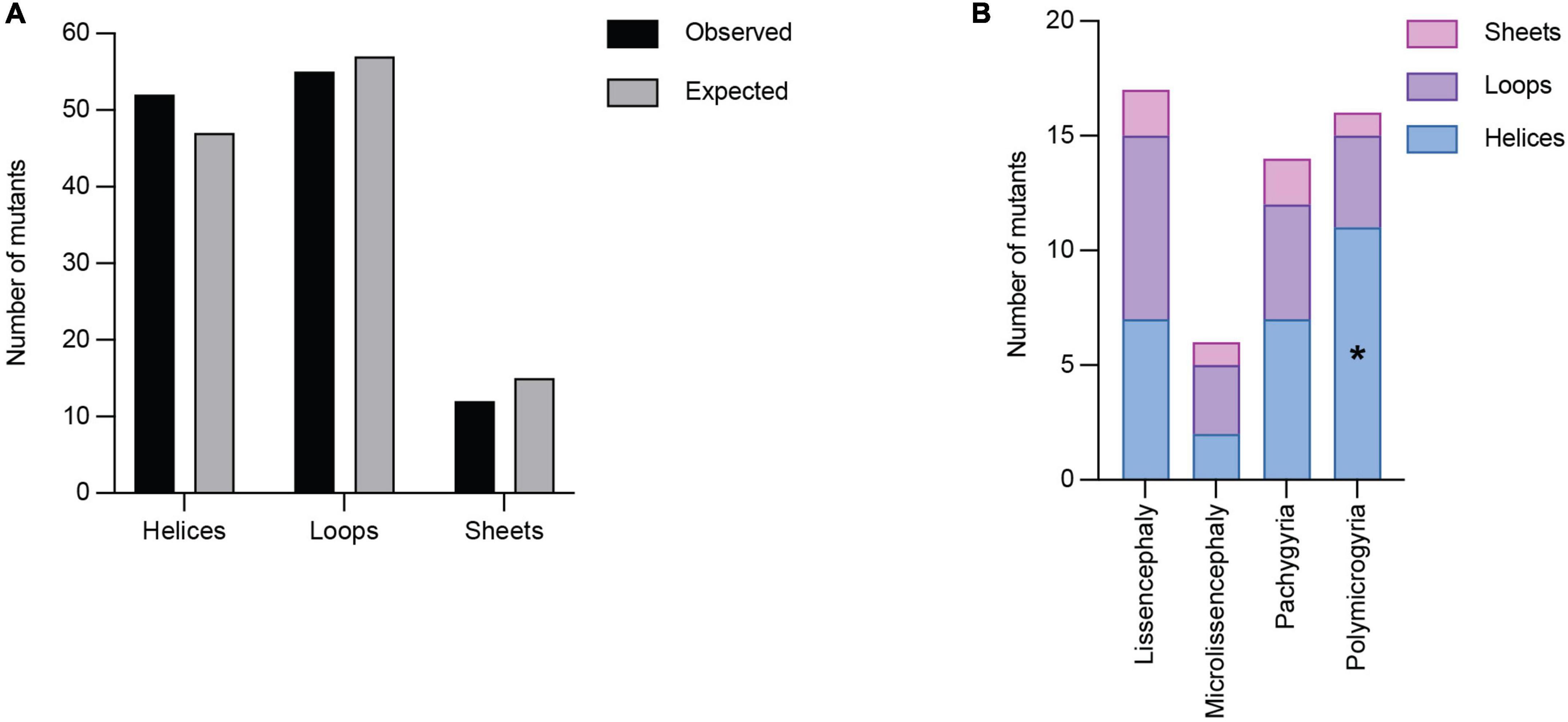
Figure 5. Quantifying TUBA1A mutations by secondary structure. (A) TUBA1A mutations were sorted by the secondary structural element (helix, loop, or sheet) in which they reside. Observed and expected number of mutations were compared using a Chi-squared test [5.99 critical value when α = 0.05; X2 (df = 2, N = 119) = 1.202; p = 0.55]. The expected number of mutations in each secondary structure was calculated by determining the percentage of amino acids that reside in each structure, then multiplying that by 119 (the number of TUBA1A missense mutations known to date). This value represents the number of mutations that would be expected to appear in each structure if all 119 mutations were randomly distributed. (B) TUBA1A mutations were sorted by the primary cortical malformation resulting from each mutation (lissencephaly, microlissencephaly, pachygyria, or polymicrogyria). For each cortical malformation, the number of mutations in each secondary structural element was reported. A Fisher’s exact test was run for each cortical malformation category to determine if mutations were enriched in one secondary structure over the others. Asterisk (*) on bar indicates p-value < 0.05 (polymicrogyria helices p = 0.03).
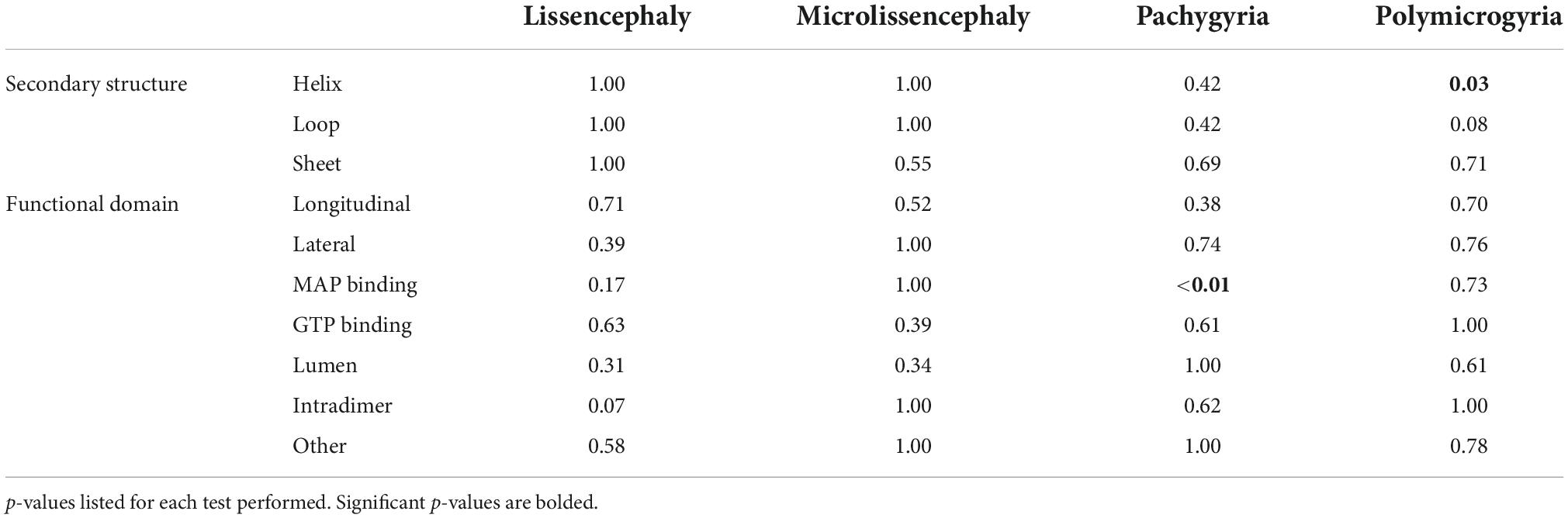
Table 1. Fisher’s exact tests were performed to determine whether the associations between cortical malformation mutants and either secondary structures or functional domains are non-random.
We next asked whether TUBA1A mutations are grouped in specific regions of the three-dimensional tubulin structure that are known to play important roles in tubulin function. Based on structural and functional studies, we classified each mutant into the following ‘functional domains’: longitudinal, lateral, MAP binding, GTP binding, lumen, intradimer, and other. It is important to note that a mutation that is in the GTP binding region will also overlap with one of the other six functional domains. For simplicity and to ensure each category was mutually exclusive, if a mutation was located at a GTP binding residue, we classified it only as GTP. To test whether there is an overall enrichment of the 119 TUBA1A mutants in one of these functional domains we again used a Chi-squared test. We find there is no significant difference in the number of mutations found in each functional domain compared to what we would expect if the mutations were random [Figure 6A; 12.59 critical value when α = 0.05; X2 (df = 6, N = 119) = 5.177; p = 0.52]. Next, we asked whether within each malformation category there were significant differences in the number of mutations found in each functional domain. We used the 53 TUBA1A mutations with publicly available clinical analysis to perform a Fisher’s exact test. We find that the number of pachygyria mutations found in the MAP binding domain was significantly higher than would be expected if the mutations were randomly distributed (Figure 6B; Table 1; p < 0.01). This indicates that pachygyria mutations are more likely to be found in MAP binding domains compared to the other functional domains that we classified. It will be interesting to investigate further whether these mutations all impact the same MAP(s), and how disrupting these MAPs may result specifically in pachygyria. Mapping the mutations in this manner calls into question how we define functional domains. While we used structural and functional studies to define these regions, it is interesting to consider that other portions of the protein may have long-range effects that can impact these domains. Therefore, it is plausible that a mutation in one domain may also impact a domain on the far other side of the protein.
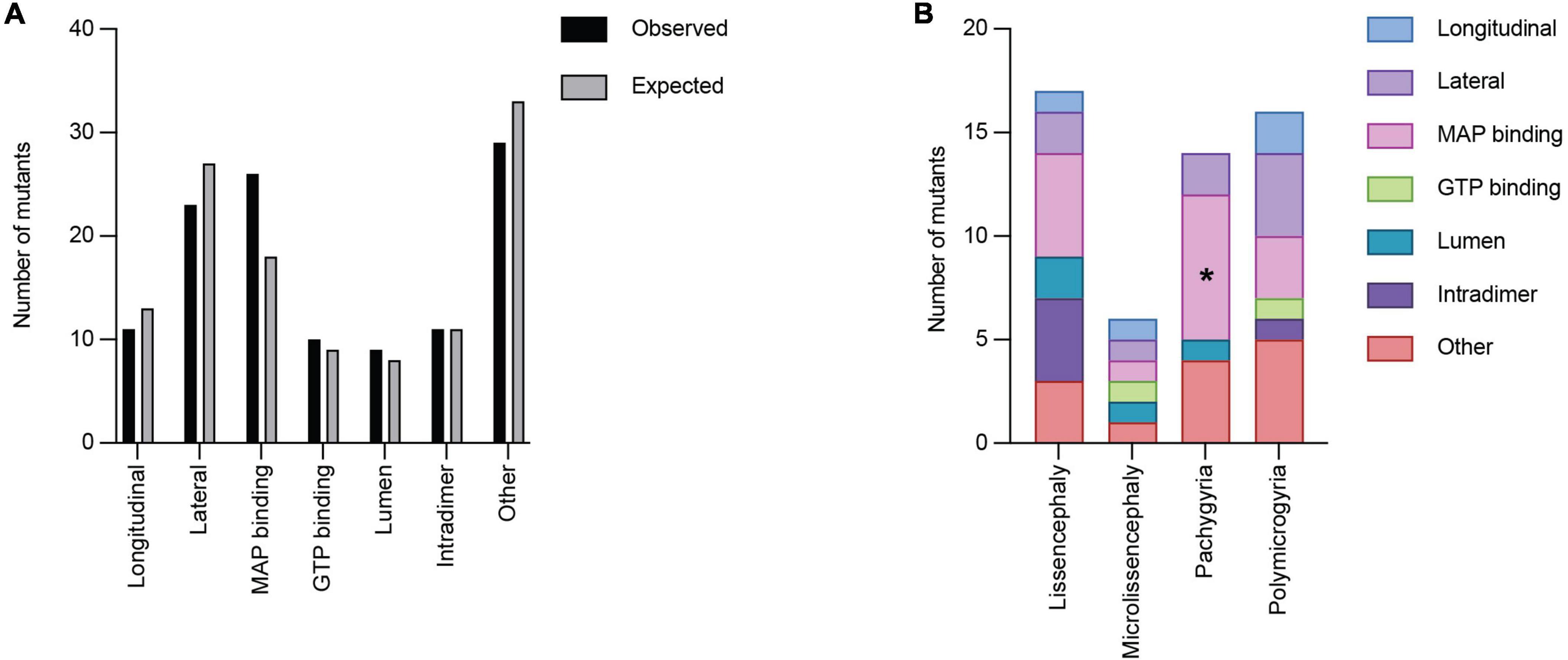
Figure 6. Quantifying TUBA1A mutations by functional region. (A) TUBA1A mutations were sorted by the functional domains (longitudinal, lateral, MAP binding, GTP binding, lumen, intradimer, or other) in which they reside. Observed and expected number of mutations were compared using a Chi-squared test [12.59 critical value when α = 0.05; X2 (df = 6, N = 119) = 5.177; p = 0.5]2). The expected number of mutations in each functional domain was calculated by determining the percentage of amino acids that reside in each domain, then multiplying that by 119 (the number of TUBA1A missense mutations known to date). This value represents the number of mutations that would be expected to appear in each domain if all 119 mutations were randomly distributed. (B) TUBA1A mutations were sorted by the primary cortical malformation resulting from each mutation (lissencephaly, microlissencephaly, pachygyria, or polymicrogyria). For each cortical malformation, the number of mutations in each functional domain was reported. A Fisher’s exact test was run for each cortical malformation category to determine if mutations were enriched in one functional domain over the others. Asterisk (*) on bar indicates p-value < 0.05 (pachygyria MAP binding p < 0.01).
Importantly, these analyses suggest that we may reconsider how we view tubulinopathies. Microtubule regulation is a complex process that involves many moving parts that are connected across the tubulin heterodimer, and the role of a particular amino acid may not be limited to its local secondary structures or interacting regions of tubulin. In the next section, we synthesize our understanding of microtubule dynamics and the roles of specific regions of the tubulin heterodimer, and we argue that increasing our understanding basic tubulin biology will be important for understanding the field of tubulinopathies.
The association of MAP and tubulin mutations with cortical malformations in patients underscores the critical role of proper microtubule function during neurodevelopment. Therefore, studying the role of extrinsic MAPs and intrinsic tubulin dynamics, at the molecular level as well as in the process of neurodevelopment, is important for predicting how these individual mutations will disrupt neuronal cell biology and cortical formation at the tissue level. In this section, we will focus on recent advancements in the field’s understanding of tubulin intrinsic and extrinsic regulatory mechanisms. Additionally, we will argue how understanding these fundamental microtubule regulation studies will help us uncover molecular mechanisms and themes that persist across tubulinopathies.
As discussed in the first section of this review, the series of conformational states adopted by the tubulin heterodimer are important aspects of both microtubule polymerization and depolymerization. The free, curved heterodimer in solution must add on to the microtubule plus-end (Buey et al., 2006; Rice Luke et al., 2008; Nawrotek et al., 2011), then subsequently straighten as it is assembled into the microtubule lattice (Nogales et al., 1999; Alushin et al., 2014) forming lateral bonds with neighboring protofilaments (Löwe et al., 2001). As these lateral bonds break apart during the process of depolymerization, tubulin heterodimers again splay out in a curved conformation and form the classically described “ram’s horns” (Mandelkow et al., 1991; Müller-Reichert et al., 1998). Understanding how the heterodimer shifts between these conformational states will thus be crucial in our understanding of intrinsic microtubule dynamics regulation.
The transition between these tubulin conformations states involves a network of structural rearrangements across the protein. Free α/β-tubulin heterodimer adopts an ∼12° bend at the intradimer interface, with a range between 9° and 18°, depending on the source of the tubulin structure (Campanacci et al., 2019). Recent work has identified various regions along the heterodimer that shift and/or contribute to these conformational states, with much of the work centered around specific β-tubulin rearrangements. Notably, there is a shift in helix 6 (H6) and H7, as well as the H6–H7 and T5 loops (Ravelli et al., 2004; Ayukawa et al., 2021). Additionally, a larger segment of β-tubulin containing the N-terminal domain and H11-H11’-H12 helices of the C-terminal domain also appear to shift with respect to one another (Ravelli et al., 2004). Further work has been done using mutations to disrupt β-tubulin H7 and has found that altering the structural regulation of this helix results in altered microtubule dynamics (Geyer et al., 2015; Ayukawa et al., 2021).
Additionally, a disease-causing mutant identified in the human β-tubulin isotype, TUBB3-D417H, results in a straightened heterodimer conformation and intrinsically faster microtubule polymerization rates in vitro (Ti et al., 2016). Similarly, disease-causing mutations identified in TUBB4A that result in dystonia and hypomyelination with atrophy of the basal ganglia, also impact tubulin conformation and microtubule dynamics (Krajka et al., 2022). However, these mutants had the opposite effect and instead promoted a curved tubulin conformation over a straightened state, with decreased microtubule polymerization rates in vitro and in cells (Krajka et al., 2022). These data highlight the significant contributions of individual regions of the heterodimer to conformation states, and how this can intrinsically alter microtubule dynamics.
In contrast to the progress made on the contributions of specific regions of β-tubulin to heterodimer conformation, decidedly less is known about α-tubulin’s influence. Our previous work indicates that a lissencephaly-causing mutation, TUBA1A-V409A, causes intrinsically faster microtubule polymerization rates despite decreased interaction with XMAP215/Stu2 TOG domains (Hoff et al., 2022). We propose a model in which the V409A mutation biases the heterodimer toward a straighter state that is more intrinsically favorable to microtubule polymerization and bypasses extrinsic regulation by TOG domains (Hoff et al., 2022) that recognize the curved tubulin state (Ayaz et al., 2012). This study likely indicates that there are regions of α-tubulin that are also crucial in establishing heterodimer conformations, however future in-depth studies will be required to identify and characterize these regions.
Particularly with the studies discussed regarding α-tubulin’s rule in dictating heterodimer conformation, it is clear that conformation states not only impact intrinsic microtubule dynamics, but also regulation conferred by extrinsic MAPs. In this next section we will discuss studies of MAPs that recognize and/or facilitate changes in tubulin conformation states, with a particular focus on how these two elements are significantly intertwined.
In recent years, an increasing number of studies have identified MAPs that either preferentially bind to particular conformation states and/or control the transitions that occur between the curved and straight states of the heterodimer. One particular MAP of interest is XMAP215. As mentioned previously, XMAP215 is a microtubule polymerase (Brouhard et al., 2008; Al-Bassam et al., 2012) comprised of multiple TOG domains that preferentially bind curved tubulin (Ayaz et al., 2012; Ayaz et al., 2014). Additionally, XMAP215 proteins have a C-terminal basic region that is required for localization and tracking to the microtubule plus-end (Widlund et al., 2011; Byrnes and Slep, 2017). The prevailing model is that the TOG domains preferentially bind curved tubulin and stabilize an intermediate state for multiple rounds of addition at the microtubule plus-end. Therefore, while tubulin is able to polymerize on its own in an in vitro setting, microtubule polymerization rates are substantially increased when tubulin is in the presence of XMAP215 (Brouhard et al., 2008). Thus, recognition and stabilization of the free tubulin heterodimer by XMAP215 regulates its ability to assemble into microtubule polymer.
Interestingly, in the absence of free tubulin, the addition of XMAP215 also increases the rate of depolymerization (Brouhard et al., 2008). Similarly, the overexpression of Alp14, the XMAP215 homolog in fission yeast, spurs microtubule depolymerization (Al-Bassam et al., 2012). While the mechanism of how XMAP215 can also promote microtubule depolymerization is less understood, it is reasonable to assume that similar to other depolymerases discussed below, it involves recognition of a tubulin conformation state.
Op18/stathmin is another MAP family that has been shown to inducing and/or stabilize tubulin curvature (Gigant et al., 2000; Steinmetz et al., 2000). Stathmin binds to two individual tubulin heterodimers (Gigant et al., 2000; Steinmetz et al., 2000) and significantly increases microtubule catastrophe frequency (Belmont and Mitchison, 1996). As stathmin preferentially interacts with free tubulin heterodimers (Belmont and Mitchison, 1996), its pro-catastrophe mechanism is often described as sequestering free tubulin and preventing continued microtubule polymerization. While this is part of the mechanism, multiple works argue that the level of increase in microtubule catastrophe frequency observed in the presence of stathmin is also due to stathmin specifically targeting microtubule plus-ends to trigger catastrophes (Belmont and Mitchison, 1996; Steinmetz et al., 2000). These works highlight another MAP family that utilizes the curved conformation to recognize and induce changes in microtubule dynamics.
Beyond these MAPs that have been characterized to interact with particular heterodimer conformations, there are MAPs such as TOG-containing CLASPs and other kinesin polymerases that need to be further studied and may also recognize and/or influence tubulin conformations. However, it is clear from these studies that there is substantial interplay between tubulin conformations and MAP regulations, particularly that either factor can influence the other, or both can influence one another. Furthering our understanding of how these intrinsic and extrinsic modes of microtubule regulations work in unison will help us better identify how disruptions in these regulatory elements may ultimately lead to improper development and disease.
Almost all tubulinopathy mutations identified in patients to date appear as heterozygous, missense mutations (Hebebrand et al., 2019). A persistent question is how a subset of mutant tubulin can dominantly perturb microtubule networks in a way that ultimately disrupts a number of critical neurodevelopment events. Particularly for the mutations that appear subtle and do not significantly disrupt the hydrophobicity or charge of the amino acid at that residue, it is curious to consider how these mutants can act in such a dominant fashion.
In the few studies of individual tubulinopathy mutations, there has been an emphasis on mutants that would be predicted to disrupt MAP binding. For example, the TUBA1A-R402C/H tubulinopathy mutations are in a region predicted to be important for dynein binding (Mizuno et al., 2004). Accordingly, modeling these mutants in budding yeast α-tubulin specifically disrupts dynein, but not kinesin, function and in mice they are sufficient to dominantly disrupt neuronal migration (Aiken et al., 2019). However, MAP-binding may in some cases be accompanied by other consequences for tubulin function. The two β-tubulin mutations, TUBB3-D417H and -R262H, are located at residues near the kinesin binding site (Uchimura et al., 2006; Ti et al., 2016). While these mutant microtubules do decrease kinesin binding, they also bias the tubulin heterodimer toward a straightened conformation and accelerate microtubule polymerization (Ti et al., 2016). TUBA1A-V409I/A mutants disrupt binding of TOG domains, key components of the microtubule polymerase XMAP215 (Byrnes and Slep, 2017), but also accelerate microtubule polymerization (Hoff et al., 2022). These two latter cases further emphasize the point that the effects of mutations may be complex and impact both intrinsic tubulin activity and regulation by extrinsic factors. Therefore, understanding how intrinsic and extrinsic effects are linked will provide insight into how tubulinopathy mutations, even seemingly subtle substitutions, may have outsized effect on microtubule networks.
The studies of TUBA1A-V409I/A and TUBB3-D417H and -R262H also bring up a second important point—how do mutations in one region of the heterodimer impact structures or conformational changes that are not immediately adjacent to the affected residue? One consideration is that the normal tubulin activity relies on waves of conformational changes that are propagated across the heterodimer. Therefore, point mutations can have long-ranging, allosteric effects throughout the tubulin heterodimer itself, as well as along the entirety of the microtubule polymer. One example of this is threonine 238 of β-tubulin. A T238A mutation has been shown to form hyperstable microtubules, yet it is buried within the core of the tubulin protein (Thomas et al., 1985; Machin et al., 1995; Geyer et al., 2015). Despite residue T238 not being in direct contact with the E-site nucleotide, the T238A mutation prevents a conformational change that typically occurs in the lattice following GTP hydrolysis (Geyer et al., 2015). Thus an amino acid change far from the E-site creates microtubules that constitutively mimic a GTP-like state. Similarly, it is reasonable to consider that tubulinopathy mutations, such as TUBA1A-V409I/A and TUBB3-D417H/-R262H may have allosteric effects across the heterodimer that impact tubulin conformations and interactions with MAPs. Additionally, as these mutations make up less than half of the total α- or β-tubulin pool in the cell, it is also important to consider that these mutants may have long-ranging effects that impact neighboring heterodimers in the microtubule. More work will be needed to test this hypothesis. Testing the potential allosteric effects of individual disease mutants will likely require a combination of computational and structural studies, along with studies of tubulin activity in vitro and in cells that blend mutant and wild-type tubulin.
Interestingly, disease-associated mutations in other proteins have also been implicated in altering protein conformations. For example, the human protein ferritin, which stores iron inside cells, has at least eight unique disease-associated mutations that have been identified in patients (Cooper et al., 2005). These mutations have been linked to iron misregulation, cataract syndrome, basal ganglia disease, and a number of neurodegenerative diseases (reviewed in Levi and Rovida, 2015). These eight mutations are spread across the protein; however, it was found that each resided in regions of high rigidity and altered protein conformation dynamics (Kumar et al., 2015). On the other hand, mutations in more flexible regions that were not associated with disease were less likely to impact conformations (Kumar et al., 2015). Another example is a mutation in the prion protein that is associated with Creutzfeldt-Jakob disease, a neurodegenerative disease (Cooper et al., 2005). This mutation causes a conformational change in the C-terminal helix that inhibits the ability of the prion to form a functional dimer (Prabantu et al., 2021). Beyond these examples, an increasing number of studies are focusing on the allosteric impacts of single point mutations and how these ultimately lead to disease states (reviewed in Naganathan, 2019). We argue that there are lessons and tools to be learned from these areas that can be applied to tubulinopathy mutations to better understand the molecular mechanisms of these mutants.
Here, we review the link between tubulin structure, microtubules, and MAPs, and the roles they play in regulating microtubule dynamics. Importantly, we discuss how tubulinopathy mutations may disrupt multiple points of dynamics regulation. A major goal in the field of tubulinopathies is to identify mechanistic themes that can be used to accurately predict the impact of individual mutations at the molecular, cellular, and tissue levels. It is clear that the location and identity of tubulinopathy mutations is insufficient to explain disease phenotype. Therefore, we must recognize and better our understanding of the complexity and interconnectedness of microtubule regulators.
We propose a holistic approach to studying tubulinopathy mutations using improved computational models to predict allosteric impacts across the heterodimer and with MAP interactions. These approaches will be useful both in understanding mutations’ potential long-range effects, as well as for generating ideas about how specific regions of tubulin can impact activity from a distance. While computational simulations are an important first step, these ideas must then be validated using both in vitro reconstitution and experiments in cells that capture the complexity of isotypes and tubulin proteostasis. Together, using these tools in concert will be invaluable to advancing the field of tubulinopathies to ultimately identify common mechanistic origins of these neurodevelopment disorders so that we can better inform diagnoses and treatments.
KH wrote the manuscript. AN helped write the manuscript. JM edited the manuscript and supervised the project. All authors contributed to the article and approved the submitted version.
This work was supported by Bolie Scholar Award (KH), T32 GM136444 (KH and AN), and NIH R35GM136253 (JM).
We are grateful to members of the Moore Lab for helpful discussions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aiken, J., Moore, J. K., and Bates, E. A. (2019). TUBA1A mutations identified in lissencephaly patients dominantly disrupt neuronal migration and impair dynein activity. Hum. Mol. Genet. 28, 1227–1243. doi: 10.1093/hmg/ddy416
Al-Bassam, J., Kim, H., Flor-Parra, I., Lal, N., Velji, H., and Chang, F. (2012). Fission yeast Alp14 is a dose-dependent plus end–tracking microtubule polymerase. Mol. Biol. Cell 23, 2878–2890. doi: 10.1091/mbc.e12-03-0205
Altman, J., and Bayer, S. A. (1990). Prolonged sojourn of developing pyramidal cells in the intermediate zone of the Hippocampus and their settling in the stratum pyramidale. J. Comp. Neurol. 301, 343–364. doi: 10.1002/cne.903010303
Alushin, G. M., Lander, G. C., Kellogg, E. H., Zhang, R., Baker, D., and Nogales, E. (2014). High-resolution microtubule structures reveal the structural transitions in αβ-tubulin upon GTP hydrolysis. Cell 157, 1117–1129. doi: 10.1016/j.cell.2014.03.053
Amabile, S., Jeffries, L., McGrath, J. M., Ji, W., Spencer-Manzon, M., Zhang, H., et al. (2020). DYNC1H1-related disorders: A description of four new unrelated patients and a comprehensive review of previously reported variants. Am. J. Med. Genet. Part A 182, 2049–2057. doi: 10.1002/ajmg.a.61729
Arai, Y., and Taverna, E. (2017). Neural progenitor cell polarity and cortical development. Front. Cell. Neurosci. 11:384. doi: 10.3389/fncel.2017.00384
Asselin, L., Rivera Alvarez, J., Heide, S., Bonnet, C. S., Tilly, P., Vitet, H., et al. (2020). Mutations in the KIF21B kinesin gene cause neurodevelopmental disorders through imbalanced canonical motor activity. Nat. Commun. 11, 2441–2441. doi: 10.1038/s41467-020-16294-6
Ayanlaja, A. A., Xiong, Y., Gao, Y., Ji, G., Tang, C., Abdikani Abdullah, Z., et al. (2017). Distinct features of doublecortin as a marker of neuronal migration and its implications in cancer cell mobility. Front. Mol. Neurosci. 10:199. doi: 10.3389/fnmol.2017.00199
Ayaz, P., Munyoki, S., Geyer, E. A., Piedra, F.-A., Vu, E. S., Bromberg, R., et al. (2014). A tethered delivery mechanism explains the catalytic action of a microtubule polymerase. Elife 3:e03069. doi: 10.7554/eLife.03069
Ayaz, P., Ye, X., Huddleston, P., Brautigam, C. A., and Rice, L. M. (2012). A TOG:αβ-tubulin complex structure reveals conformation-based mechanisms for a microtubule polymerase. Science 337, 857–860. doi: 10.1126/science.1221698
Ayukawa, R., Iwata, S., Imai, H., Kamimura, S., Hayashi, M., Ngo, K. X., et al. (2021). GTP-dependent formation of straight tubulin oligomers leads to microtubule nucleation. J. Cell Biol. 220:e202007033. doi: 10.1083/jcb.202007033
Baas, P. W., Deitch, J. S., Black, M. M., and Banker, G. A. (1988). Polarity orientation of microtubules in hippocampal neurons: Uniformity in the axon and nonuniformity in the dendrite. Proc. Natl. Acad. Sci. U.S.A. 85, 8335–8339. doi: 10.1073/pnas.85.21.8335
Bahi-Buisson, N., Poirier, K., Fourniol, F., Saillour, Y., Valence, S., Lebrun, N., et al. (2014). The wide spectrum of tubulinopathies: What are the key features for the diagnosis? Brain 137, 1676–1700. doi: 10.1093/brain/awu082
Bahi-Buisson, N., Souville, I., Fourniol, F. J., Toussaint, A., Moores, C. A., Houdusse, A., et al. (2013). New insights into genotype–phenotype correlations for the doublecortin-related lissencephaly spectrum. Brain 136, 223–244. doi: 10.1093/brain/aws323
Bechstedt, S., and Brouhard, G. J. (2012). Doublecortin recognizes the 13-protofilament microtubule cooperatively and tracks microtubule ends. Dev. Cell 23, 181–192. doi: 10.1016/j.devcel.2012.05.006
Beecroft, S. J., McLean, C. A., Delatycki, M. B., Koshy, K., Yiu, E., Haliloglu, G., et al. (2017). Expanding the phenotypic spectrum associated with mutations of DYNC1H1. Neuromuscul. Disord. 27, 607–615. doi: 10.1016/j.nmd.2017.04.011
Belmont, L. D., and Mitchison, T. J. (1996). Identification of a protein that interacts with tubulin dimers and increases the catastrophe rate of microtubules. Cell 84, 623–631. doi: 10.1016/S0092-8674(00)81037-5
Belvindrah, R., Natarajan, K., Shabajee, P., Bruel-Jungerman, E., Bernard, J., Goutierre, M., et al. (2017). Mutation of the α-tubulin Tuba1a leads to straighter microtubules and perturbs neuronal migration. J. Cell Biol. 216, 2443–2461. doi: 10.1083/jcb.201607074
Bittermann, E., Abdelhamed, Z., Liegel, R. P., Menke, C., Timms, A., Beier, D. R., et al. (2019). Differential requirements of tubulin genes in mammalian forebrain development. PLoS Genet. 15:e1008243. doi: 10.1371/journal.pgen.1008243
Bodakuntla, S., Jijumon, A. S., Villablanca, C., Gonzalez-Billault, C., and Janke, C. (2019). Microtubule-associated proteins: Structuring the cytoskeleton. Trends Cell Biol. 29, 804–819. doi: 10.1016/j.tcb.2019.07.004
Broix, L., Asselin, L., Silva, C. G., Ivanova, E. L., Tilly, P., Gilet, J. G., et al. (2018). Ciliogenesis and cell cycle alterations contribute to KIF2A-related malformations of cortical development. Hum. Mol. Genet. 27, 224–238. doi: 10.1093/hmg/ddx384
Brouhard, G. J., Stear, J. H., Noetzel, T. L., Al-Bassam, J., Kinoshita, K., Harrison, S. C., et al. (2008). XMAP215 is a processive microtubule polymerase. Cell 132, 79–88. doi: 10.1016/j.cell.2007.11.043
Buey, R. M., Díaz, J. F., and Andreu, J. M. (2006). The Nucleotide switch of tubulin and microtubule assembly: A polymerization-driven structural change. Biochemistry 45, 5933–5938. doi: 10.1021/bi060334m
Burgoyne, R. D., Cambray-Deakin, M. A., Lewis, S. A., Sarkar, S., and Cowan, N. J. (1988). Differential distribution of beta-tubulin isotypes in cerebellum. EMBO J. 7, 2311–2319. doi: 10.1002/j.1460-2075.1988.tb03074.x
Burton, P. R. (1988). Dendrites of mitral cell neurons contain microtubules of opposite polarity. Brain Res. 473, 107–115. doi: 10.1016/0006-8993(88)90321-6
Buscaglia, G., Northington, K. R., Aiken, J., Hoff, K. J., and Bates, E. A. (2022). Bridging the gap: The importance of TUBA1A α-tubulin in forming midline commissures. Front. Cell Dev. Biol. 9:789438. doi: 10.3389/fcell.2021.789438
Buscaglia, G., Northington, K. R., Moore, J. K., and Bates, E. A. (2020). Reduced TUBA1A tubulin causes defects in trafficking and impaired adult motor behavior. eNeuro 7. doi: 10.1523/ENEURO.0045-20.2020
Byrnes, A. E., and Slep, K. C. (2017). TOG–tubulin binding specificity promotes microtubule dynamics and mitotic spindle formation. J. Cell Biol. 216, 1641–1657. doi: 10.1083/jcb.201610090
Campanacci, V., Urvoas, A., Consolati, T., Cantos-Fernandes, S., Aumont-Nicaise, M., Valerio-Lepiniec, M., et al. (2019). Selection and characterization of artificial proteins targeting the tubulin α subunit. Structure 27, 497–506.e4. doi: 10.1016/j.str.2018.12.001
Caplow, M., and Shanks, J. (1996). Evidence that a single monolayer tubulin-GTP cap is both necessary and sufficient to stabilize microtubules. Mol. Biol. Cell 7, 663–675. doi: 10.1091/mbc.7.4.663
Carlier, M. F., and Pantaloni, D. (1981). Kinetic analysis of guanosine 5’-triphosphate hydrolysis associated with tubulin polymerization. Biochemistry 20, 1918–1924. doi: 10.1021/bi00510a030
Cavallin, M., Hubert, L., Cantagrel, V., Munnich, A., Boddaert, N., Vincent-Delorme, C., et al. (2016). Recurrent KIF5C mutation leading to frontal pachygyria without microcephaly. Neurogenetics 17, 79–82. doi: 10.1007/s10048-015-0459-8
Chen, Y., and Hancock, W. O. (2015). Kinesin-5 is a microtubule polymerase. Nat. Commun. 6:8160. doi: 10.1038/ncomms9160
Chrétien, D., Fuller, S. D., and Karsenti, E. (1995). Structure of growing microtubule ends: Two-dimensional sheets close into tubes at variable rates. J. Cell Biol. 129, 1311–1328. doi: 10.1083/jcb.129.5.1311
Cleary, J. M., and Hancock, W. O. (2021). Molecular mechanisms underlying microtubule growth dynamics. Curr. Biol. 31, R560–R573. doi: 10.1016/j.cub.2021.02.035
Cooper, D. N., Stenson, P. D., and Chuzhanova, N. A. (2005). The human gene mutation database (HGMD) and its exploitation in the study of mutational mechanisms. Curr. Protoc. Bioinform. 12, 1–13. doi: 10.1002/0471250953.bi0113s12
Dantas, T. J., Carabalona, A., Hu, D. J. K., and Vallee, R. B. (2016). Emerging roles for motor proteins in progenitor cell behavior and neuronal migration during brain development. Cytoskeleton 73, 566–576. doi: 10.1002/cm.21293
Denoulet, P., Eddé, B., and Gros, F. (1986). Differential expression of several neurospecific β-tubulin mRNAs in the mouse brain during development. Gene 50, 289–297. doi: 10.1016/0378-1119(86)90333-1
des Portes, V., Pinard, J. M., Billuart, P., Vinet, M. C., Koulakoff, A., Carrié, A., et al. (1998). A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell 92, 51–61. doi: 10.1016/S0092-8674(00)80898-3
Desai, A., Verma, S., Mitchison, T. J., and Walczak, C. E. (1999). Kin I kinesins are microtubule-destabilizing enzymes. Cell 96, 69–78. doi: 10.1016/S0092-8674(00)80960-5
Drechsel, D. N., and Kirschner, M. W. (1994). The minimum GTP cap required to stabilize microtubules. Curr. Biol. 4, 1053–1061. doi: 10.1016/S0960-9822(00)00243-8
Estévez-Gallego, J., Josa-Prado, F., Ku, S., Buey, R. M., Balaguer, F. A., Prota, A. E., et al. (2020). Structural model for differential cap maturation at growing microtubule ends. Elife 9:e50155. doi: 10.7554/eLife.50155
Estrem, C., Fees, C. P., and Moore, J. K. (2017). Dynein is regulated by the stability of its microtubule track. J. Cell Biol. 216, 2047–2058. doi: 10.1083/jcb.201611105
Faulkner, N. E., Dujardin, D. L., Tai, C.-Y., Vaughan, K. T., O’Connell, C. B., Wang, Y., et al. (2000). A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat. Cell Biol. 2, 784–791. doi: 10.1038/35041020
Fiorillo, C., Moro, F., Yi, J., Weil, S., Brisca, G., Astrea, G., et al. (2014). Novel dynein DYNC1H1 neck and motor domain mutations link distal spinal muscular atrophy and abnormal cortical development. Hum. Mutat. 35, 298–302. doi: 10.1002/humu.22491
Francis, F., Koulakoff, A., Boucher, D., Chafey, P., Schaar, B., Vinet, M.-C., et al. (1999). Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron 23, 247–256. doi: 10.1016/S0896-6273(00)80777-1
Friel, C. T., and Howard, J. (2011). The kinesin-13 MCAK has an unconventional ATPase cycle adapted for microtubule depolymerization. EMBO J. 30, 3928–3939. doi: 10.1038/emboj.2011.290
Friocourt, G., Koulakoff, A., Chafey, P., Boucher, D., Fauchereau, F., Chelly, J., et al. (2003). Doublecortin functions at the extremities of growing neuronal processes. Cereb. Cortex 13, 620–626. doi: 10.1093/cercor/13.6.620
Gambello, M. J., Darling, D. L., Yingling, J., Tanaka, T., Gleeson, J. G., and Wynshaw-Boris, A. (2003). Multiple dose-dependent effects of Lis1 on cerebral cortical development. J. Neurosci. 23:1719. doi: 10.1523/JNEUROSCI.23-05-01719.2003
Gartz Hanson, M., Aiken, J., Sietsema, D. V., Sept, D., Bates, E. A., Niswander, L., et al. (2016). Novel α-tubulin mutation disrupts neural development and tubulin proteostasis. Dev. Biol. 409, 406–419. doi: 10.1016/j.ydbio.2015.11.022
Geyer, E. A., Burns, A., Lalonde, B. A., Ye, X., Piedra, F.-A., Huffaker, T. C., et al. (2015). A mutation uncouples the tubulin conformational and GTPase cycles, revealing allosteric control of microtubule dynamics. Elife 4:e10113. doi: 10.7554/eLife.10113
Ghiretti, A. E., Thies, E., Tokito, M. K., Lin, T., Ostap, E. M., Kneussel, M., et al. (2016). Activity-dependent regulation of distinct transport and cytoskeletal remodeling functions of the dendritic kinesin KIF21B. Neuron 92, 857–872. doi: 10.1016/j.neuron.2016.10.003
Gigant, B., Curmi, P. A., Martin-Barbey, C., Charbaut, E., Lachkar, S., Lebeau, L., et al. (2000). The 4 Å X-Ray Structure of a tubulin:Stathmin-like domain complex. Cell 102, 809–816. doi: 10.1016/S0092-8674(00)00069-6
Gleeson, J. G., Allen, K. M., Fox, J. W., Lamperti, E. D., Berkovic, S., Scheffer, I., et al. (1998). Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92, 63–72. doi: 10.1016/S0092-8674(00)80899-5
Gleeson, J. G., Lin, P. T., Flanagan, L. A., and Walsh, C. A. (1999). Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 23, 257–271. doi: 10.1016/S0896-6273(00)80778-3
Gloster, A., El-Bizri, H., Bamji, S. X., Rogers, D., and Miller, F. D. (1999). Early induction of Tα1 α-tubulin transcription in neurons of the developing nervous system. J. Comp. Neurol. 405, 45–60. doi: 10.1002/(SICI)1096-9861(19990301)405:1<45::AID-CNE4<3.0.CO;2-M
Gloster, A., Wu, W., Speelman, A., Weiss, S., Causing, C., Pozniak, C., et al. (1994). The T alpha 1 alpha-tubulin promoter specifies gene expression as a function of neuronal growth and regeneration in transgenic mice. J. Neurosci. 14:7319. doi: 10.1523/JNEUROSCI.14-12-07319.1994
Goodson, H. V., and Jonasson, E. M. (2018). Microtubules and microtubule-associated proteins. Cold Spring Harb. Perspect. Biol. 10:a022608.
Götz, M., and Huttner, W. B. (2005). The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 6, 777–788. doi: 10.1038/nrm1739
Grabham, P. W., Seale, G. E., Bennecib, M., Goldberg, D. J., and Vallee, R. B. (2007). Cytoplasmic dynein and LIS1 are required for microtubule advance during growth cone remodeling and fast axonal outgrowth. J. Neurosci. 27:5823. doi: 10.1523/JNEUROSCI.1135-07.2007
Guesdon, A., Bazile, F., Buey, R. M., Mohan, R., Monier, S., García, R. R., et al. (2016). EB1 interacts with outwardly curved and straight regions of the microtubule lattice. Nat. Cell Biol. 18, 1102–1108. doi: 10.1038/ncb3412
Hafezparast, M., Klocke, R., Ruhrberg, C., Marquardt, A., Ahmad-Annuar, A., Bowen, S., et al. (2003). Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 300, 808–812. doi: 10.1126/science.1083129
Harada, A., Takei, Y., Kanai, Y., Tanaka, Y., Nonaka, S., and Hirokawa, N. (1998). Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J. Cell Biol. 141, 51–59. doi: 10.1083/jcb.141.1.51
Harms, M. B., Ori-McKenney, K. M., Scoto, M., Tuck, E. P., Bell, S., Ma, D., et al. (2012). Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 78:1714. doi: 10.1212/WNL.0b013e3182556c05
Hebebrand, M., Hüffmeier, U., Trollmann, R., Hehr, U., Uebe, S., Ekici, A. B., et al. (2019). The mutational and phenotypic spectrum of TUBA1A-associated tubulinopathy. Orphanet J. Rare Dis. 14:38. doi: 10.1186/s13023-019-1020-x
Heidemann, S. R., Landers, J. M., and Hamborg, M. A. (1981). Polarity orientation of axonal microtubules. J. Cell Biol. 91, 661–665. doi: 10.1083/jcb.91.3.661
Heng, J. I.-T., Chariot, A., and Nguyen, L. (2010). Molecular layers underlying cytoskeletal remodelling during cortical development. Trends Neurosci. 33, 38–47. doi: 10.1016/j.tins.2009.09.003
Hirokawa, N., Niwa, S., and Tanaka, Y. (2010). Molecular motors in neurons: Transport mechanisms and roles in brain function, development, and disease. Neuron 68, 610–638. doi: 10.1016/j.neuron.2010.09.039
Hirotsune, S., Fleck, M. W., Gambello, M. J., Bix, G. J., Chen, A., Clark, G. D., et al. (1998). Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat. Genet. 19, 333–339. doi: 10.1038/1221
Hoang, H. T., Schlager, M. A., Carter, A. P., and Bullock, S. L. (2017). DYNC1H1 mutations associated with neurological diseases compromise processivity of dynein–dynactin–cargo adaptor complexes. Proc. Natl. Acad. Sci. U.S.A. 114, E1597–E1606. doi: 10.1073/pnas.1620141114
Hoff, K. J., Aiken, J. E., Gutierrez, M. A., Franco, S. J., and Moore, J. K. (2022). TUBA1A tubulinopathy mutants disrupt neuron morphogenesis and override XMAP215/Stu2 regulation of microtubule dynamics. Elife 11:e76189. doi: 10.7554/eLife.76189
Homma, N., Takei, Y., Tanaka, Y., Nakata, T., Terada, S., Kikkawa, M., et al. (2003). Kinesin superfamily protein 2A (KIF2A) functions in suppression of collateral branch extension. Cell 114, 229–239. doi: 10.1016/S0092-8674(03)00522-1
Hunter, A. W., Caplow, M., Coy, D. L., Hancock, W. O., Diez, S., Wordeman, L., et al. (2003). The kinesin-related protein MCAK is a microtubule depolymerase that forms an ATP-hydrolyzing complex at microtubule ends. Mol. Cell 11, 445–457. doi: 10.1016/S1097-2765(03)00049-2
Jamuar, S. S., Lam, A.-T. N., Kircher, M., D’Gama, A. M., Wang, J., Barry, B. J., et al. (2014). Somatic mutations in cerebral cortical malformations. N. Engl. J. Med. 371, 733–743. doi: 10.1056/NEJMoa1314432
Jiang, Y. Q., and Oblinger, M. M. (1992). Differential regulation of beta III and other tubulin genes during peripheral and central neuron development. J. Cell Sci. 103, 643–651. doi: 10.1242/jcs.103.3.643
Kalantari, S., and Filges, I. (2020). ‘Kinesinopathies’: Emerging role of the kinesin family member genes in birth defects. J. Med. Genet. 57:797. doi: 10.1136/jmedgenet-2019-106769
Kanai, Y., Okada, Y., Tanaka, Y., Harada, A., Terada, S., and Hirokawa, N. (2000). KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci. 20, 6374–6384. doi: 10.1523/JNEUROSCI.20-17-06374.2000
Kannan, M., Bayam, E., Wagner, C., Rinaldi, B., Kretz, P. F., Tilly, P., et al. (2017). WD40-repeat 47, a microtubule-associated protein, is essential for brain development and autophagy. Proc. Natl. Acad. Sci. U.S.A. 114, E9308–E9317. doi: 10.1073/pnas.1713625114
Kapitein, L. C., Schlager, M. A., Kuijpers, M., Wulf, P. S., van Spronsen, M., MacKintosh, F. C., et al. (2010). Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr. Biol. 20, 290–299. doi: 10.1016/j.cub.2009.12.052
Kappeler, C., Saillour, Y., Baudoin, J.-P., Tuy, F. P. D., Alvarez, C., Houbron, C., et al. (2006). Branching and nucleokinesis defects in migrating interneurons derived from doublecortin knockout mice. Hum. Mol. Genet. 15, 1387–1400. doi: 10.1093/hmg/ddl062
Keays, D. A., Tian, G., Poirier, K., Huang, G.-J., Siebold, C., Cleak, J., et al. (2007). Mutations in α-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 128, 45–57. doi: 10.1016/j.cell.2006.12.017
Kevenaar, J. T., and Hoogenraad, C. C. (2015). The axonal cytoskeleton: From organization to function. Front. Mol. Neurosci. 8:44. doi: 10.3389/fnmol.2015.00044
Koizumi, H., Higginbotham, H., Poon, T., Tanaka, T., Brinkman, B. C., and Gleeson, J. G. (2006). Doublecortin maintains bipolar shape and nuclear translocation during migration in the adult forebrain. Nat. Neurosci. 9, 779–786. doi: 10.1038/nn1704
Krajka, V., Vulinovic, F., Genova, M., Tanzer, K., Jijumon, A. S., Bodakuntla, S., et al. (2022). H-ABC– and dystonia-causing TUBB4A mutations show distinct pathogenic effects. Sci. Adv. 8:eabj9229. doi: 10.1126/sciadv.abj9229
Kriegstein, A. R., and Noctor, S. C. (2004). Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 27, 392–399. doi: 10.1016/j.tins.2004.05.001
Kumar, A., Glembo, T. J., and Ozkan, S. B. (2015). The role of conformational dynamics and allostery in the disease development of human ferritin. Biophys. J. 109, 1273–1281. doi: 10.1016/j.bpj.2015.06.060
Laan, L., Pavin, N., Husson, J., Romet-Lemonne, G., van Duijn, M., López, M. P., et al. (2012). Cortical dynein controls microtubule dynamics to generate pulling forces that position microtubule asters. Cell 148, 502–514. doi: 10.1016/j.cell.2012.01.007
Laquerriere, A., Maillard, C., Cavallin, M., Chapon, F., Marguet, F., Molin, A., et al. (2017). Neuropathological hallmarks of brain malformations in extreme phenotypes related to DYNC1H1 mutations. J. Neuropathol. Exp. Neurol. 76, 195–205. doi: 10.1093/jnen/nlw124
Latremoliere, A., Cheng, L., DeLisle, M., Wu, C., Chew, S., Hutchinson, E. B., et al. (2018). Neuronal-specific TUBB3 Is not required for normal neuronal function but is essential for timely axon regeneration. Cell Rep. 24, 1865–1879.e9. doi: 10.1016/j.celrep.2018.07.029
Levi, S., and Rovida, E. (2015). Neuroferritinopathy: From ferritin structure modification to pathogenetic mechanism. Neurobiol. Dis. 81, 134–143. doi: 10.1016/j.nbd.2015.02.007
Lewis, S. A., Gu, W., and Cowan, N. J. (1987). Free intermingling of mammalian β-tubulin isotypes among functionally distinct microtubules. Cell 49, 539–548. doi: 10.1016/0092-8674(87)90456-9
Löwe, J., Li, H., Downing, K. H., and Nogales, E. (2001). Refined structure of αβ-tubulin at 3.5 Å resolution. J. Mol. Biol. 313, 1045–1057. doi: 10.1006/jmbi.2001.5077
Ludueña, R. F., and Banerjee, A. (2008). “The tubulin superfamily,” in The role of microtubules in cell biology, neurobiology, and oncology, ed. T. Fojo (Totowa, NJ: Humana Press), 177–191. doi: 10.1007/978-1-59745-336-3_7
Machin, N. A., Lee, J. M., and Barnes, G. (1995). Microtubule stability in budding yeast: Characterization and dosage suppression of a benomyl-dependent tubulin mutant. Mol. Biol. Cell 6, 1241–1259. doi: 10.1091/mbc.6.9.1241
Maillard, C., Roux, C. J., Charbit-Henrion, F., Steffann, J., Laquerriere, A., Quazza, F., et al. (2022). Tubulin mutations in human neurodevelopmental disorders. Semin. Cell Dev. Biol. doi: 10.1016/j.semcdb.2022.07.009
Mandelkow, E. M., Mandelkow, E., and Milligan, R. A. (1991). Microtubule dynamics and microtubule caps: A time-resolved cryo-electron microscopy study. J. Cell Biol. 114, 977–991. doi: 10.1083/jcb.114.5.977
Maor-Nof, M., Homma, N., Raanan, C., Nof, A., Hirokawa, N., and Yaron, A. (2013). Axonal pruning is actively regulated by the microtubule-destabilizing protein kinesin superfamily protein 2A. Cell Rep. 3, 971–977. doi: 10.1016/j.celrep.2013.03.005
Marín, O., and Rubenstein, J. L. R. (2003). Cell migration in the forebrain. Annu. Rev. Neurosci. 26, 441–483. doi: 10.1146/annurev.neuro.26.041002.131058
Marszalek, J. R., Weiner, J. A., Farlow, S. J., Chun, J., and Goldstein, L. S. B. (1999). Novel dendritic kinesin sorting identified by different process targeting of two related kinesins: KIF21A and KIF21B. J. Cell Biol. 145, 469–479. doi: 10.1083/jcb.145.3.469
Marzo, M. G., Griswold, J. M., Ruff, K. M., Buchmeier, R. E., Fees, C. P., and Markus, S. M. (2019). Molecular basis for dyneinopathies reveals insight into dynein regulation and dysfunction. Elife 8:e47246. doi: 10.7554/eLife.47246
Maurer, S. P., Fourniol, F. J., Bohner, G., Moores, C. A., and Surrey, T. (2012). EBs recognize a nucleotide-dependent structural cap at growing microtubule ends. Cell 149, 371–382. doi: 10.1016/j.cell.2012.02.049
McIntosh, J. R., O’Toole, E., Morgan, G., Austin, J., Ulyanov, E., Ataullakhanov, F., et al. (2018). Microtubules grow by the addition of bent guanosine triphosphate tubulin to the tips of curved protofilaments. J. Cell Biol. 217, 2691–2708. doi: 10.1083/jcb.201802138
Melki, R., Carlier, M. F., Pantaloni, D., and Timasheff, S. N. (1989). Cold depolymerization of microtubules to double rings: Geometric stabilization of assemblies. Biochemistry 28, 9143–9152. doi: 10.1021/bi00449a028
Michels, S., Foss, K., Park, K., Golden-Grant, K., Saneto, R., Lopez, J., et al. (2017). Mutations of KIF5C cause a neurodevelopmental disorder of infantile-onset epilepsy, absent language, and distinctive malformations of cortical development. Am. J. Med. Genet. Part A 173, 3127–3131. doi: 10.1002/ajmg.a.38496
Miki, H., Setou, M., Kaneshiro, K., and Hirokawa, N. (2001). All kinesin superfamily protein, KIF, genes in mouse and human. Proc. Natl. Acad. Sci. U.S.A. 98, 7004–7011. doi: 10.1073/pnas.111145398
Miller, F. D., Naus, C. C., Durand, M., Bloom, F. E., and Milner, R. J. (1987). Isotypes of alpha-tubulin are differentially regulated during neuronal maturation. J. Cell Biol. 105, 3065–3073. doi: 10.1083/jcb.105.6.3065
Miller, J. A., Ding, S.-L., Sunkin, S. M., Smith, K. A., Ng, L., Szafer, A., et al. (2014). Transcriptional landscape of the prenatal human brain. Nature 508, 199–206. doi: 10.1038/nature13185
Mitchison, T., and Kirschner, M. (1984). Dynamic instability of microtubule growth. Nature 312, 237–242. doi: 10.1038/312237a0
Mizuno, N., Toba, S., Edamatsu, M., Watai-Nishii, J., Hirokawa, N., Toyoshima, Y. Y., et al. (2004). Dynein and kinesin share an overlapping microtubule-binding site. EMBO J. 23, 2459–2467. doi: 10.1038/sj.emboj.7600240
Moores, C. A., Perderiset, M., Francis, F., Chelly, J., Houdusse, A., and Milligan, R. A. (2004). Mechanism of microtubule stabilization by doublecortin. Mol. Cell 14, 833–839. doi: 10.1016/j.molcel.2004.06.009
Morikawa, M., Yajima, H., Nitta, R., Inoue, S., Ogura, T., Sato, C., et al. (2015). X-ray and Cryo-EM structures reveal mutual conformational changes of Kinesin and GTP-state microtubules upon binding. EMBO J. 34, 1270–1286. doi: 10.15252/embj.201490588
Muhia, M., Thies, E., Labonté, D., Ghiretti, A. E., Gromova, K. V., Xompero, F., et al. (2016). The kinesin KIF21B regulates microtubule dynamics and is essential for neuronal morphology, synapse function, and learning and memory. Cell Rep. 15, 968–977. doi: 10.1016/j.celrep.2016.03.086
Müller-Reichert, T., Chrétien, D., Severin, F., and Hyman, A. A. (1998). Structural changes at microtubule ends accompanying GTP hydrolysis: Information from a slowly hydrolyzable analogue of GTP, guanylyl (α,β)methylenediphosphonate. Proc. Natl. Acad. Sci. U.S.A. 95, 3661–3666. doi: 10.1073/pnas.95.7.3661
Nadarajah, B., Brunstrom, J. E., Grutzendler, J., Wong, R. O. L., and Pearlman, A. L. (2001). Two modes of radial migration in early development of the cerebral cortex. Nat. Neurosci. 4, 143–150. doi: 10.1038/83967
Naganathan, A. N. (2019). Modulation of allosteric coupling by mutations: From protein dynamics and packing to altered native ensembles and function. Curr. Opin. Struct. Biol. 54, 1–9. doi: 10.1016/j.sbi.2018.09.004
Nakata, T., Niwa, S., Okada, Y., Perez, F., and Hirokawa, N. (2011). Preferential binding of a kinesin-1 motor to GTP-tubulin–rich microtubules underlies polarized vesicle transport. J. Cell Biol. 194, 245–255. doi: 10.1083/jcb.201104034
Nawrotek, A., Knossow, M., and Gigant, B. (2011). The Determinants that govern microtubule assembly from the atomic structure of GTP-tubulin. J. Mol. Biol. 412, 35–42. doi: 10.1016/j.jmb.2011.07.029
Nicastro, D., Fu, X., Heuser, T., Tso, A., Porter, M. E., and Linck, R. W. (2011). Cryo-electron tomography reveals conserved features of doublet microtubules in flagella. Proc. Natl. Acad. Sci. U.S.A. 108, E845–E853. doi: 10.1073/pnas.1106178108
Nicastro, D., Schwartz, C., Pierson, J., Gaudette, R., Porter, M. E., and McIntosh, J. R. (2006). The molecular architecture of axonemes revealed by cryoelectron tomography. Science 313, 944–948. doi: 10.1126/science.1128618
Niekamp, S., Coudray, N., Zhang, N., Vale, R. D., and Bhabha, G. (2019). Coupling of ATPase activity, microtubule binding, and mechanics in the dynein motor domain. EMBO J. 38:e101414. doi: 10.15252/embj.2018101414
Noda, Y., Sato-Yoshitake, R., Kondo, S., Nangaku, M., and Hirokawa, N. (1995). KIF2 is a new microtubule-based anterograde motor that transports membranous organelles distinct from those carried by kinesin heavy chain or KIF3A/B. J. Cell Biol. 129, 157–167. doi: 10.1083/jcb.129.1.157
Nogales, E., Whittaker, M., Milligan, R. A., and Downing, K. H. (1999). High-resolution model of the microtubule. Cell 96, 79–88. doi: 10.1016/S0092-8674(00)80961-7
Nogales, E., Wolf, S. G., and Downing, K. H. (1998). Structure of the αβ tubulin dimer by electron crystallography. Nature 391, 199–203. doi: 10.1038/34465
O’Brien, E. T., Voter, W. A., and Erickson, H. P. (1987). GTP hydrolysis during microtubule assembly. Biochemistry 26, 4148–4156. doi: 10.1021/bi00387a061
Oegema, R., Cushion, T. D., Phelps, I. G., Chung, S.-K., Dempsey, J. C., Collins, S., et al. (2015). Recognizable cerebellar dysplasia associated with mutations in multiple tubulin genes. Hum. Mol. Genet. 24, 5313–5325. doi: 10.1093/hmg/ddv250
Ori-McKenney, K. M., and Vallee, R. B. (2011). Neuronal migration defects in the Loa dynein mutant mouse. Neural Dev. 6:26. doi: 10.1186/1749-8104-6-26
Park, K., Hoff, K. J., Wethekam, L., Stence, N., Saenz, M., and Moore, J. K. (2021). Kinetically stabilizing mutations in beta tubulins create isotype-specific brain malformations. Front. Cell Dev. Biol. 9:765992. doi: 10.3389/fcell.2021.765992
Pecqueur, L., Duellberg, C., Dreier, B., Jiang, Q., Wang, C., Plückthun, A., et al. (2012). A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proc. Natl. Acad. Sci. U.S.A. 109, 12011–12016. doi: 10.1073/pnas.1204129109
Péterfy, M., Gyuris, T., Grosshans, D., Cuaresma, C. C., and Takács, L. (1998). Cloning and characterization of cDNAs and the gene encoding the mouse platelet-activating factor acetylhydrolase Ib α Subunit/Lissencephaly-1 Protein. Genomics 47, 200–206. doi: 10.1006/geno.1997.5121
Poirier, K., Lebrun, N., Broix, L., Tian, G., Saillour, Y., Boscheron, C., et al. (2013). Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 45, 639–647. doi: 10.1038/ng.2613
Poirier, K., Saillour, Y., Bahi-Buisson, N., Jaglin, X. H., Fallet-Bianco, C., Nabbout, R., et al. (2010). Mutations in the neuronal β-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum. Mol. Genet. 19, 4462–4473. doi: 10.1093/hmg/ddq377
Prabantu, V. M., Naveenkumar, N., and Srinivasan, N. (2021). Influence of disease-causing mutations on protein structural networks. Front. Mol. Biosci. 7:620554. doi: 10.3389/fmolb.2020.620554
Rakic, P. (1972). Mode of cell migration to the superficial layers of fetal monkey neocortex. J. Comp. Neurol. 145, 61–83. doi: 10.1002/cne.901450105
Ravelli, R. B. G., Gigant, B., Curmi, P. A., Jourdain, I., Lachkar, S., Sobel, A., et al. (2004). Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 428, 198–202. doi: 10.1038/nature02393
Reck-Peterson, S. L., Redwine, W. B., Vale, R. D., and Carter, A. P. (2018). The cytoplasmic dynein transport machinery and its many cargoes. Nat. Rev. Mol. Cell Biol. 19, 382–398. doi: 10.1038/s41580-018-0004-3
Reiner, O., Carrozzo, R., Shen, Y., Wehnert, M., Faustinella, F., Dobyns, W. B., et al. (1993). Isolation of a miller–dicker lissencephaly gene containing G protein β-subunit-like repeats. Nature 364, 717–721. doi: 10.1038/364717a0
Rice Luke, M., Montabana Elizabeth, A., and Agard David, A. (2008). The lattice as allosteric effector: Structural studies of αβ- and γ-tubulin clarify the role of GTP in microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 105, 5378–5383. doi: 10.1073/pnas.0801155105
Rivas, R., and Hatten, M. (1995). Motility and cytoskeletal organization of migrating cerebellar granule neurons. J. Neurosci. 15:981. doi: 10.1523/JNEUROSCI.15-02-00981.1995
Roostalu, J., Thomas, C., Cade, N. I., Kunzelmann, S., Taylor, I. A., and Surrey, T. (2020). The speed of GTP hydrolysis determines GTP cap size and controls microtubule stability. Elife 9:e51992. doi: 10.7554/eLife.51992
Saillour, Y., Carion, N., Quelin, C., Leger, P.-L., Boddaert, N., Elie, C., et al. (2009). LIS1-Related isolated lissencephaly: Spectrum of mutations and relationships with malformation severity. Arch. Neurol. 66, 1007–1015. doi: 10.1001/archneurol.2009.149
Santana, J., and Marzolo, M.-P. (2017). The functions of Reelin in membrane trafficking and cytoskeletal dynamics: Implications for neuronal migration, polarization and differentiation. Biochem. J. 474, 3137–3165. doi: 10.1042/BCJ20160628
Sapir, T., Elbaum, M., and Reiner, O. (1997). Reduction of microtubule catastrophe events by LIS1, platelet-activating factor acetylhydrolase subunit. EMBO J. 16, 6977–6984. doi: 10.1093/emboj/16.23.6977
Satoh, D., Sato, D., Tsuyama, T., Saito, M., Ohkura, H., Rolls, M. M., et al. (2008). Spatial control of branching within dendritic arbors by dynein-dependent transport of Rab5-endosomes. Nat. Cell Biol. 10, 1164–1171. doi: 10.1038/ncb1776
Shu, T., Ayala, R., Nguyen, M.-D., Xie, Z., Gleeson, J. G., and Tsai, L.-H. (2004). Ndel1 operates in a common pathway with LIS1 and Cytoplasmic dynein to regulate cortical neuronal positioning. Neuron 44, 263–277. doi: 10.1016/j.neuron.2004.09.030
Sirajuddin, M., Rice, L. M., and Vale, R. D. (2014). Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat. Cell Biol. 16, 335–344. doi: 10.1038/ncb2920
Smith, D. S., Niethammer, M., Ayala, R., Zhou, Y., Gambello, M. J., Wynshaw-Boris, A., et al. (2000). Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat. Cell Biol. 2, 767–775. doi: 10.1038/35041000
Spiegelman, B. M., Penningroth, S. M., and Kirschner, M. W. (1977). Turnover of tubulin and the N site GTP in chinese hamster ovary cells. Cell 12, 587–600. doi: 10.1016/0092-8674(77)90259-8
Steinmetz, M. O., Kammerer, R. A., Jahnke, W., Goldie, K. N., Lustig, A., and van Oostrum, J. (2000). Op18/stathmin caps a kinked protofilament-like tubulin tetramer. EMBO J. 19, 572–580. doi: 10.1093/emboj/19.4.572
Tabata, H., and Nakajima, K. (2003). Multipolar migration: The third mode of radial neuronal migration in the developing cerebral cortex. J. Neurosci. 23:9996. doi: 10.1523/JNEUROSCI.23-31-09996.2003
Tanaka, T., Serneo, F. F., Higgins, C., Gambello, M. J., Wynshaw-Boris, A., and Gleeson, J. G. (2004). Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J. Cell Biol. 165, 709–721. doi: 10.1083/jcb.200309025
Thomas, J. H., Neff, N. F., and Botstein, D. (1985). Isolation and characterization of mutations in the β-tubulin gene of saccharomyces cerevisiae. Genetics 111, 715–734. doi: 10.1093/genetics/111.4.715
Ti, S.-C., Pamula, M. C., Howes, S. C., Duellberg, C., Cade, N. I., Kleiner, R. E., et al. (2016). Mutations in human tubulin proximal to the Kinesin-Binding site alter dynamic instability at microtubule plus- and minus-ends. Dev. Cell 37, 72–84. doi: 10.1016/j.devcel.2016.03.003
Tian, G., Cristancho, A. G., Dubbs, H. A., Liu, G. T., Cowan, N. J., and Goldberg, E. M. (2016). A patient with lissencephaly, developmental delay, and infantile spasms, due to de novo heterozygous mutation of KIF2A. Mol. Genet. Genom. Med. 4, 599–603. doi: 10.1002/mgg3.236
Tian, G., Jaglin, X. H., Keays, D. A., Francis, F., Chelly, J., and Cowan, N. J. (2010). Disease-associated mutations in TUBA1A result in a spectrum of defects in the tubulin folding and heterodimer assembly pathway. Hum. Mol. Genet. 19, 3599–3613. doi: 10.1093/hmg/ddq276
Tian, G., Kong, X.-P., Jaglin, X. H., Chelly, J., Keays, D., and Cowan, N. J. (2008). A Pachygyria-causing α-Tubulin mutation results in inefficient cycling with CCT and a deficient interaction with TBCB. Mol. Biol. Cell 19, 1152–1161. doi: 10.1091/mbc.e07-09-0861
Tischfield, M. A., Baris, H. N., Wu, C., Rudolph, G., Van Maldergem, L., He, W., et al. (2010). Human TUBB3 Mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 140, 74–87. doi: 10.1016/j.cell.2009.12.011
Tsai, J.-W., Bremner, K. H., and Vallee, R. B. (2007). Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat. Neurosci. 10, 970–979. doi: 10.1038/nn1934
Tsai, J.-W., Lian, W.-N., Kemal, S., Kriegstein, A. R., and Vallee, R. B. (2010). Kinesin 3 and cytoplasmic dynein mediate interkinetic nuclear migration in neural stem cells. Nat. Neurosci. 13, 1463–1471. doi: 10.1038/nn.2665
Uchimura, S., Oguchi, Y., Katsuki, M., Usui, T., Osada, H., Nikawa, J., et al. (2006). Identification of a strong binding site for kinesin on the microtubule using mutant analysis of tubulin. EMBO J. 25, 5932–5941. doi: 10.1038/sj.emboj.7601442
van Riel, W. E., Rai, A., Bianchi, S., Katrukha, E. A., Liu, Q., Heck, A. J., et al. (2017). Kinesin-4 KIF21B is a potent microtubule pausing factor. Elife 6:e24746. doi: 10.7554/eLife.24746
Varga, V., Helenius, J., Tanaka, K., Hyman, A. A., Tanaka, T. U., and Howard, J. (2006). Yeast kinesin-8 depolymerizes microtubules in a length-dependent manner. Nat. Cell Biol. 8, 957–962. doi: 10.1038/ncb1462
Varga, V., Leduc, C., Bormuth, V., Diez, S., and Howard, J. (2009). Kinesin-8 Motors act cooperatively to mediate length-dependent microtubule depolymerization. Cell 138, 1174–1183. doi: 10.1016/j.cell.2009.07.032
Vemu, A., Atherton, J., Spector, J. O., Moores, C. A., and Roll-Mecak, A. (2017). Tubulin isoform composition tunes microtubule dynamics. Mol. Biol. Cell 28, 3564–3572. doi: 10.1091/mbc.e17-02-0124
Vitre, B., Coquelle, F. M., Heichette, C., Garnier, C., Chrétien, D., and Arnal, I. (2008). EB1 regulates microtubule dynamics and tubulin sheet closure in vitro. Nat. Cell Biol. 10, 415–421. doi: 10.1038/ncb1703
Widlund, P. O., Stear, J. H., Pozniakovsky, A., Zanic, M., Reber, S., Brouhard, G. J., et al. (2011). XMAP215 polymerase activity is built by combining multiple tubulin-binding TOG domains and a basic lattice-binding region. Proc. Natl. Acad. Sci. U.S.A. 108, 2741–2746. doi: 10.1073/pnas.1016498108
Willemsen, M. H., Ba, W., Wissink-Lindhout, W. M., de Brouwer, A. P. M., Haas, S. A., Bienek, M., et al. (2014). Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic function. J. Med. Genet. 51, 487–494. doi: 10.1136/jmedgenet-2013-102182
Willemsen, M. H., Vissers, L. E. L., Willemsen, M. A. A. P., van Bon, B. W. M., Kroes, T., de Ligt, J., et al. (2012). Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J. Med. Genet. 49, 179–183. doi: 10.1136/jmedgenet-2011-100542
Zanic, M., Widlund, P. O., Hyman, A. A., and Howard, J. (2013). Synergy between XMAP215 and EB1 increases microtubule growth rates to physiological levels. Nat. Cell Biol. 15, 688–693. doi: 10.1038/ncb2744
Zhang, R., Alushin, G. M., Brown, A., and Nogales, E. (2015). Mechanistic origin of microtubule dynamic instability and its modulation by EB proteins. Cell 162, 849–859. doi: 10.1016/j.cell.2015.07.012
Zhao, J., Wang, Y., Xu, H., Fu, Y., Qian, T., Bo, D., et al. (2016). Dync1h1 mutation causes proprioceptive sensory neuron loss and impaired retrograde axonal transport of dorsal root ganglion neurons. CNS Neurosci. Ther. 22, 593–601. doi: 10.1111/cns.12552
Keywords: tubulinopathy, microtubule, neurodevelopment, cytoskeleton, protein structure
Citation: Hoff KJ, Neumann AJ and Moore JK (2022) The molecular biology of tubulinopathies: Understanding the impact of variants on tubulin structure and microtubule regulation. Front. Cell. Neurosci. 16:1023267. doi: 10.3389/fncel.2022.1023267
Received: 19 August 2022; Accepted: 30 September 2022;
Published: 02 November 2022.
Edited by:
Georg Haase, INSERM U1106 Institut de Neurosciences des Systèmes, FranceReviewed by:
Jose R. Eguibar, Meritorious Autonomous University of Puebla, MexicoCopyright © 2022 Hoff, Neumann and Moore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeffrey K. Moore, SmVmZnJleS5Nb29yZUBDVUFuc2NodXR6LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.