
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 15 December 2021
Sec. Cellular Neuropathology
Volume 15 - 2021 | https://doi.org/10.3389/fncel.2021.784833
This article is part of the Research Topic Molecular Mechanisms Underlying C9orf72 Neurodegeneration View all 8 articles
 Iris-Stefania Pasniceanu
Iris-Stefania Pasniceanu Manpreet Singh Atwal
Manpreet Singh Atwal Cleide Dos Santos Souza
Cleide Dos Santos Souza Laura Ferraiuolo
Laura Ferraiuolo Matthew R. Livesey*
Matthew R. Livesey*
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are characterized by degeneration of upper and lower motor neurons and neurons of the prefrontal cortex. The emergence of the C9ORF72 hexanucleotide repeat expansion mutation as the leading genetic cause of ALS and FTD has led to a progressive understanding of the multiple cellular pathways leading to neuronal degeneration. Disturbances in neuronal function represent a major subset of these mechanisms and because such functional perturbations precede degeneration, it is likely that impaired neuronal function in ALS/FTD plays an active role in pathogenesis. This is supported by the fact that ALS/FTD patients consistently present with neurophysiological impairments prior to any apparent degeneration. In this review we summarize how the discovery of the C9ORF72 repeat expansion mutation has contributed to the current understanding of neuronal dysfunction in ALS/FTD. Here, we discuss the impact of the repeat expansion on neuronal function in relation to intrinsic excitability, synaptic, network and ion channel properties, highlighting evidence of conserved and divergent pathophysiological impacts between cortical and motor neurons and the influence of non-neuronal cells. We further highlight the emerging association between these dysfunctional properties with molecular mechanisms of the C9ORF72 mutation that appear to include roles for both, haploinsufficiency of the C9ORF72 protein and aberrantly generated dipeptide repeat protein species. Finally, we suggest that relating key pathological observations in C9ORF72 repeat expansion ALS/FTD patients to the mechanistic impact of the C9ORF72 repeat expansion on neuronal function will lead to an improved understanding of how neurophysiological dysfunction impacts upon pathogenesis.
The underlying genetic and pathological causes of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) overlap extensively placing them on an ALS-FTD spectrum (Kato et al., 1993; Talbot et al., 1995; Lomen-Hoerth et al., 2002). Clinical observations of ALS-FTD patients reinforce linked pathogenicity where almost half of ALS patients develop FTD-related cognitive disturbances and up to 30% of FTD patients exhibit motor impairment (Christidi et al., 2018). The GGGGCC (G4C2) hexanucleotide repeat expansion mutation is found within intron 1 of the C9ORF72 gene (C9ORF72 repeat expansion, C9ORF72RE), is causal to both ALS and FTD and is the most common pathogenic mutation within the ALS-FTD spectrum. Degeneration is classically prominent within the pre-frontal cortex in FTD and the motor cortex, upper motor neurons (layer V cortical projection neurons) and lower motor neurons in ALS. Understanding how this mutation mechanistically leads to neuronal injury and degeneration is of key importance.
Healthy individuals typically present with 2–30 G4C2 repeats whereas ALS-FTD patients living with the repeat expansion typically have hundreds to thousands of repeats, with 65 repeats argued as the pathological repeat-length threshold (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Furthermore, they share TDP-43 pathology that manifests in approximately 98% of ALS patients and 45% of FTD patients (Arai et al., 2006; Neumann et al., 2006). The repeat expansion drives pathogenicity through at least one of two potential broad mechanisms; haploinsufficiency of C9ORF72 protein expression and toxic gain-of-function of the repeat expansion (Gendron et al., 2014; Mizielinska and Isaacs, 2014). The latter can be further subdivided into transcribed repeat expansion sense and antisense RNA foci and aberrant non-ATG (RAN) translation leading to the generation of five potential dipeptide-repeat proteins (DPRs): poly-GA, -GP, -GR, -PA, and -PR (Donnelly et al., 2013; Gendron et al., 2013; Mori et al., 2013). Attention is now focusing on how these mechanisms drive the cellular disturbances observed in ALS-FTD, where emerging research places emphasis on both exclusive and synergistic mechanisms involving haploinsufficiency and aspects of toxic-gain-of-function. Further complexity to our understanding is contributed by the fact we have an incomplete appreciation of the physiological role of the C9ORF72 protein (Smeyers et al., 2021). Importantly, several rodent models initially generated to study C9ORF72RE mechanisms do not always recapitulate motor dysfunction (Balendra and Isaacs, 2018), though more recent studies now describe motor deficits in a C9ORF72 haploinsufficiency model (Shao et al., 2019) and that motor deficits are exacerbated in a background of both haploinsufficiency and the repeat expansion (Zhu et al., 2020). Nonetheless, it is clear that a combination of direct mechanisms associated with the C9ORF72RE mutation and downstream impacted cellular processes, including prominent neurophysiological perturbations, collectively contribute to C9ORF72RE mediated-disease progression.
Neurophysiological dysfunction is established and prominent within the advanced stages of neurodegenerative disease patients where a complex combination of neuronal and synaptic loss in addition to neuronal dysfunction leads to a consensus systemic loss of function (Frere and Slutsky, 2018). However, in current years, the monitoring of non-symptomatic neurodegenerative patients, including C9ORF72RE patients, carrying familial mutations is beginning to present a scenario whereby neurophysiological perturbations are evident before any notable clinical symptoms arise (Benussi et al., 2016; Geevasinga et al., 2016; Styr and Slutsky, 2018). Critically, these perturbations present as highly plausible, core contributors to disease pathogenesis, via neuronal injury through excitotoxicity and reduced function by way of impaired neurotransmission. Understanding the sources of the neurophysiological function and mechanisms directly linking these features to the molecular pathogenesis of the C9ORF72RE, thus have an important role to play in understanding ALS-FTD. Typically, we consider the general excitability of neurons to underpin its physiological function and is ultimately dependent upon a complex myriad of several factors including synaptic function, morphology and altered intrinsic excitability, which is dependent upon the functional expression of ion channels associated with action potential generation. This review summarizes the current literature describing C9ORF72RE-mediated neuronal dysfunction mechanisms in both cortical and motor neurons, contrasting these with each other as well as other ALS-FTD genetic backgrounds. We also review how these neurons may be impacted by other non-cell autonomous mechanisms involving glial cells. Finally, we will discuss our current understanding around the molecular determinants of this dysfunction and how these are linked to haploinsufficiency and related to repeat expansion toxic gain-of-function.
Beyond established degeneration of the motor cortex, neurophysiological disturbances in the cortex of ALS patients represents a longstanding pathological hallmark of disease. Such clinical observations are consistent between both sporadic and familial backgrounds (Geevasinga et al., 2016), including those harboring the C9ORF72RE mutation (Williams et al., 2013; Benussi et al., 2016; Schanz et al., 2016; Nasseroleslami et al., 2019). Supported by extensive transcranial magnetic stimulation (TMS) (Vucic et al., 2013; Eisen et al., 2017) and resting state magnetoencephalography (MEG) studies (Proudfoot et al., 2016), cortical network dysfunction in ALS patients is found to manifest early, possibly prodromally, typically preceding lower motor neuron dysfunction leading to a possible staged continuum of pathogenesis consistent with a feed-forward mechanism of neurodegeneration (Geevasinga et al., 2016; Menon et al., 2017). Figure 1 summarizes this concept. Importantly cortical dysfunction is not limited to ALS, it is present in FTD patients (Lindau et al., 2003; Nishida et al., 2011) and is a common observation in other neurodegenerative diseases including Alzheimer’s, Parkinson’s and Huntington’s Disease (Palop et al., 2006; Styr and Slutsky, 2018; McColgan et al., 2020). Like for many other neurodegenerative diseases (Selkoe, 2002), functional synaptic perturbations at early disease stages are thought to drive cortical synaptic loss, which correlates with severe cognitive impairments observed in C9ORF72RE patients (Henstridge et al., 2018). Further, magnetic resonance imaging (MRI) studies in ALS-FTD have demonstrated structural changes in the motor cortex that correlate with cognitive and behavioral impairments (Agosta et al., 2016; Consonni et al., 2018), in addition to functional defects that impact on cortical and subcortical activity (Mohammadi et al., 2015). Cortical dysfunction is therefore thought to play a key role in early pathogenic events in ALS-FTD. A summary of studies investigating C9ORF72RE cortical dysfunction is presented in Table 1.
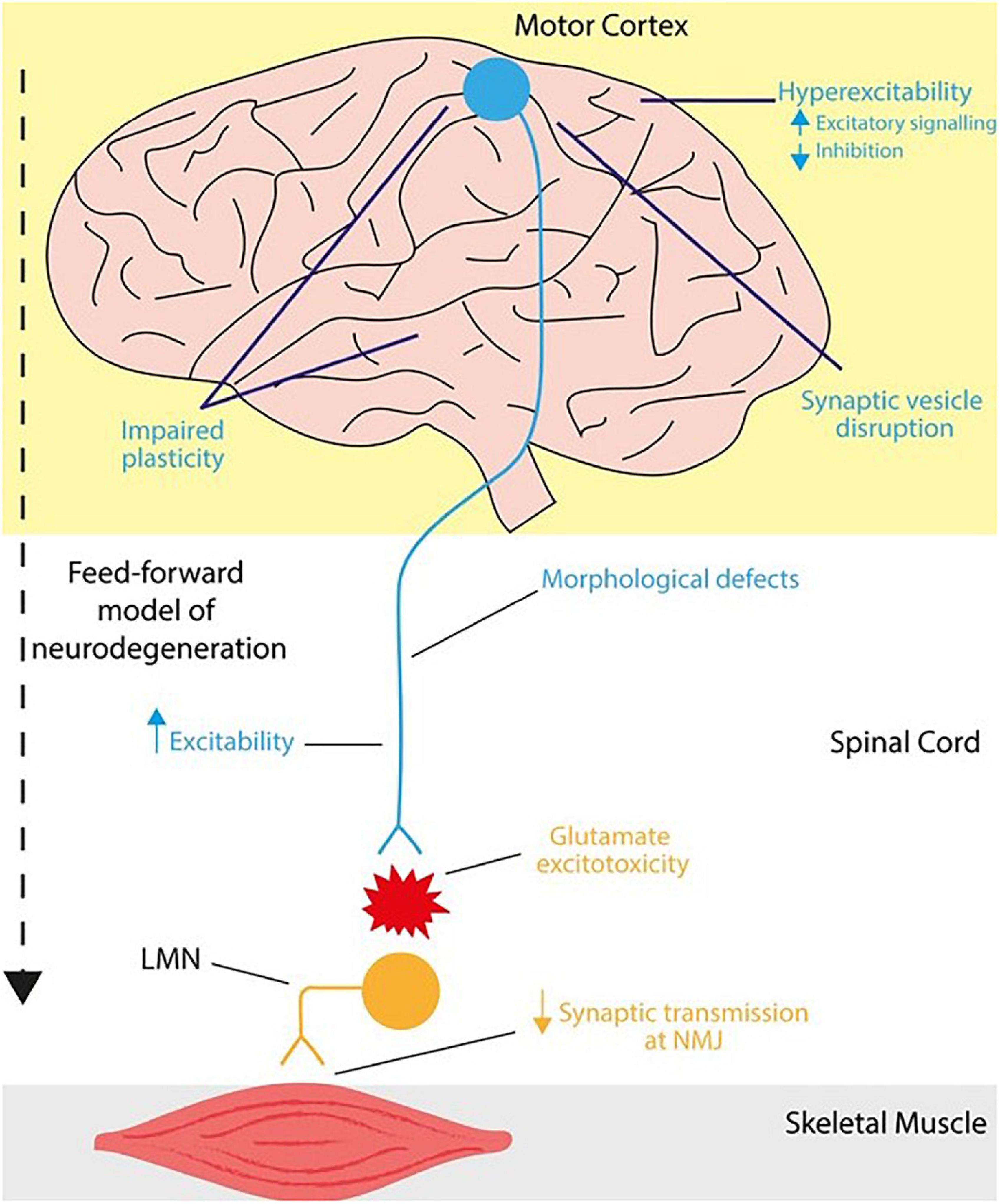
Figure 1. Mechanisms of neurophysiological impairments in the cortex and lower motor neurons in C9ORF72RE ALS. In humans, upper motor neurons (blue) descend from the motor cortex and project onto the brainstem and spinal cord via the corticospinal tract. These corticospinal neurons form a monosynaptic pathway (in primates and humans) that innervates lower motor neurons (orange), which in turn transmit motor signals to effector muscles. Together this forms the motor circuitry within humans. Neurophysiological impairments leading to ALS can arise in the corticospinal tract at various loci. In ALS, cortical dysfunction ranges from hyperexcitability (increased excitability) as a result of increased excitatory signaling or reduced inhibition, disruption of synaptic vesicle dynamics and impaired synaptic plasticity that also extends to cortico-hippocampal connections. Within the corticospinal tract, upper motor neurons are vulnerable to synaptic loss and dendrite pathology including loss of dendritic spines that may arise from increased hyperexcitability. In a feedforward mechanism of dysfunction, degeneration of lower motor neurons is mediated, at least in part, via glutamate-mediated excitotoxicity whereby, cortical dysfunction precedes that of lower motor neurons, potentially causing further neurophysiological impairments and injury in lower motor neurons.
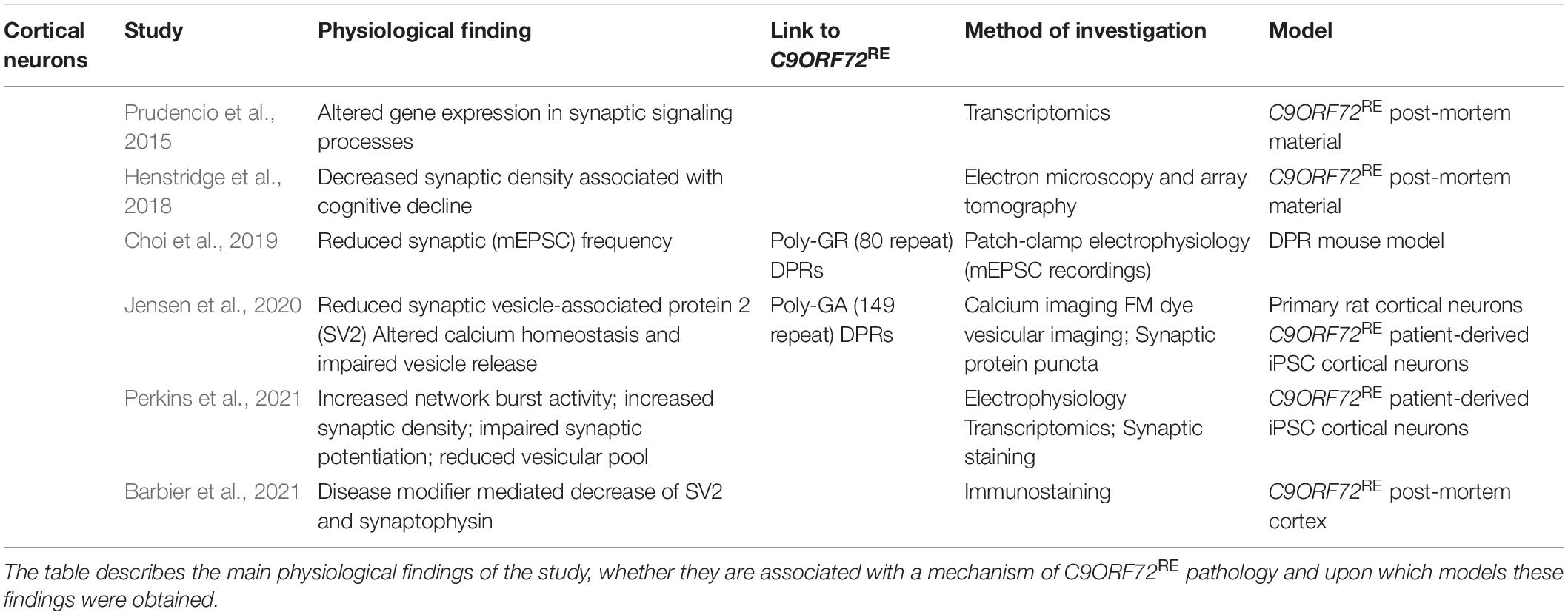
Table 1. Summary of physiological studies that have implication for the C9ORF72RE in cortical dysfunction.
Functional nervous system plasticity presents the critical ability to modify neuronal properties in response to environmental demands and may manifest in a number of structural and functional changes that impact upon neurons and glial cells (Turrigiano, 2012; Suminaite et al., 2019). Functional impairments in plasticity are considered major early features of neurodegenerative disease and are representative of altered homeostasis that precedes and, potentially, drives further neuronal dysfunction and/or loss (Milnerwood and Raymond, 2010; Starr and Sattler, 2018; Styr and Slutsky, 2018). Synaptic plasticity is the process by which synapses undergo activity-dependent changes in their efficacy, where long-term potentiation (LTP), long-term depression (LTD), and spike-time dependent plasticity are the cellular correlates of, inter alia, cognitive processes such as learning and memory (Malenka and Bear, 2004). Paired associative stimulation using TMS techniques has revealed striking LTP–like network plasticity impairments in asymptomatic C9ORF72RE patients indicative of early, widespread, cortical dysfunction of potential synaptic origins (Benussi et al., 2016). Benussi et al. (2016) predict that synaptic/network plasticity impairments present 15 years before symptomatic onset making these pathological observations some of the earliest evidenced in ALS-FTD patients. Direct evidence of impaired synaptic potentiation of mini excitatory post-synaptic currents was recently confirmed in induced pluripotent stem cell (iPSC)-derived cortical neurons generated from C9ORF72RE patients, a feature that was rescued in isogenic, gene-corrected lines (Perkins et al., 2021). Beyond this, functional investigations of impaired synaptic plasticity in ALS and FTD have been determined in hippocampal murine preparations: UBQLN2P497H (Gorrie et al., 2014); SOD1G93A (Spalloni et al., 2011) and TDP-43 transgenic mice (Koza et al., 2019), TDP-43 conditional knockout mice (Wu et al., 2019). Further, impaired hippocampal synaptic plasticity is observed in non-TDP-43 FTD models [progranulin knock out mice (Petkau et al., 2012) and MAPT knock out mice (Ahmed et al., 2014; Biundo et al., 2018)]. Also, impaired plasticity has been observed at the neuromuscular junction of Drosophila over-expressing C9ORF72RE (Perry et al., 2017). Broad cellular disruption affecting molecules and signaling processes relevant to synaptic plasticity are highlighted by transcriptional disturbances in both C9ORF72RE patient-derived cortical neurons (Perkins et al., 2021) and C9ORF72RE patient post-mortem cortex (Prudencio et al., 2015). Defined molecular pathological mechanisms of altered cortical synaptic plasticity in ALS-FTD remain to be elucidated. However, reduced LTP and LTD have been demonstrated in cortico-hippocampal connections of a murine C9ORF72 knockout model, which highlight a role for the C9ORF72 protein in synaptic plasticity mechanisms in the hippocampus, but also potentially suggest that haploinsufficiency of the C9ORF72 protein may underpin some synaptic plasticity deficits (Ho et al., 2020). Indeed, the knockout of putative interactors with C9ORF72 also yields notable impairments in LTP (Gerges et al., 2004; Niu et al., 2020).
Plasticity has close association with homeostatic function and the inability to modify neuronal function in response to external stimuli suggests that plasticity impairments may represent very early markers of disease onset where cells are unable to respond to, as yet unknown, chronic disease-mediated changes (Milnerwood and Raymond, 2010; Benussi et al., 2016; Starr and Sattler, 2018; Styr and Slutsky, 2018). Future work upon impaired plasticity in ALS-FTD cortical neurons and circuitry promises to yield leads into the early drivers of disease.
The neurophysiological profiling of ALS patients using transcranial magnetic stimulation has revealed considerable cortical and layer V projection neuron circuit perturbations that are consistent with a general increase in neuronal excitability within the motor cortex. Given these studies measure excitability from the motor cortex, early work did not show a correlation with (motor) cortical hyperexcitability being present in C9ORF72RE FTD patients (Schanz et al., 2016), however, recent work has shown that increased strength of cortical hyperexcitability in ALS patients is associated with increased cognitive impairments (Agarwal et al., 2021). Nonetheless, cortical hyperexcitability has been observed in FTD models (García-Cabrero et al., 2013), but the degree to which it plays a pathogenic role in FTD is less understood. The observation of reduced short interval intracortical inhibition (SICI), first reported by Kujirai et al. (1993) in ALS patients, is highly consistent amongst sporadic and familial cases (Geevasinga et al., 2016), including C9ORF72RE patients (Wainger and Cudkowicz, 2015; Schanz et al., 2016; Nasseroleslami et al., 2019), such that it is now considered a disease hallmark. Longitudinal assessments of ALS-FTD patients now indicate that reduced SICI manifests pre-symptomatically, preceding lower motor neuron dysfunction (Menon et al., 2015; Geevasinga et al., 2016) and becomes more pronounced with disease progression (Menon et al., 2020). Cortical circuits control upper motor neurons within the cortico-spinal tract, and hyperexcitability is associated with excitotoxicity, therefore cortical hyperexcitability is considered to be a pathogenic driver of motor neuron injury and dysfunction in ALS-FTD (Sahara Khademullah et al., 2020). Indeed, the degree of cortical hyperexcitability in ALS patients correlates with disease prognosis (Shibuya et al., 2016). Understanding the physiological and pathological determinants of cortical hyperexcitability in ALS-FTD is a key undertaking.
Physiological mechanisms explaining early cortical hyperexcitability are now emerging and center upon the deregulation of the complex synchronization of excitatory and inhibitory signaling within the cortex. Reduced cortical SICI argues toward a reduced inhibitory influence in the cortex of ALS-FTD patients. Accumulating evidence supports the involvement of inhibitory GABA-ergic interneurons, the predominant mediators of inhibitory activity in the networked circuitry of the cortex (Farrant and Nusser, 2005), as a pathological substrate in ALS-FTD patients. The loss of inhibitory signaling could be mediated via the loss of function or degeneration of interneurons thereby promoting cortical excitability. Recent work has demonstrated a reduction in parvalbumin interneurons, the major class of interneuron in the motor cortex (Estebanez et al., 2017), in a TDP-43Q331K model of ALS-FTD and C9ORF72RE ALS patient post-mortem suggesting the loss of inhibition may well also come from a selective vulnerability of this neuron class (Lin et al., 2021). In addition to this selective vulnerability, hippocampal interneurons appear to display considerable TDP-43 pathology in TDP-43 transgenic mice (Tsuiji et al., 2017). It may be therefore posited that cortical interneuron dysfunction is an early contributor to cortical hyperexcitability and that later interneuron degeneration contributes to a more pronounced hyperexcitability as the disease course progresses. However, to date there remains no data to assess the function of interneurons in the context of C9ORF72RE and our understanding must therefore be derived via other genetic models of ALS. Selective pharmacological rescue of cortical interneuron function in a mutant SOD1G93A mouse model preserves motor axon function and prolonged survival by rescuing reduced inhibitory input on to layer V projection neurons (Sahara Khademullah et al., 2020), suggesting that increasing interneuron function has the potential to reduce excitability in the motor cortex, thereby being neuroprotective to motor neurons. Although, we must also be careful in our assessment of long range impacts of cortical activity upon motor neuron function in ALS rodent models given that the monosynaptic cortico-spinal tract circuit is an anatomical feature that is exclusive to primates (Lemon, 2008). In contrast to the former study, Kim et al. revealed parvalbumin-expressing interneurons from neonatal and symptomatic SOD1G93A mice had increased intrinsic excitability compared to wild-type interneurons (Kim et al., 2017). However, data from a TDP-43A315T mouse model (Zhang et al., 2016) showed that young mice display sustained hyperexcitability in somatostatin-positive interneurons, but not in parvalbumin-positive neurons, which are hypoexcitable. Somatostatin interneurons regulate the excitability of parvalbumin interneurons, hence the hyperexcitability of somatostatin-expressing interneurons drives the hypoexcitable profile of parvalbumin interneurons, and in turn, causes hyperexcitability of the layer V projection neurons. In addition, recent studies have demonstrated that disturbances in the transcriptional landscape are consistent with an impact upon inhibitory synapses in FUSΔNLS/+ murine model (Sahadevan et al., 2021; Scekic-Zahirovic et al., 2021). Importantly, the interneuronal hypothesis also extends to non-TDP-43 FTD forms where interneuronal pathology is observed in murine FTD models (Lui et al., 2016) and noting that interneurons control cortical network synchronicity, may underlie altered EEG activity in FTD patients (Lindau et al., 2003; Nishida et al., 2011). The underlying molecular pathological mechanisms of interneuronal dysfunction and loss in ALS-FTD remains to be determined.
Excitatory neurons represent approximately 80% of the adult cortex and numerous pieces of evidence are converging toward the contribution of these neurons to abnormalities in cortical excitability in ALS-FTD patients. Perkins et al. (2021) demonstrated that cultures of excitatory cortical neurons derived from C9ORF72RE iPSCs displayed an enhanced network burst frequency compared to control derived neurons. These properties appear to be explained by the fact that C9ORF72RE excitatory neurons had an increased functional synaptic input due to increased synaptic density, but not altered intrinsic excitability. Interestingly, an increased synaptic input of excitatory cortical neurons was seen in the motor cortex of pre-symptomatic (at postnatal day 30) mutant TDP-43Q331K mice and SODG93A mice (Van Zundert et al., 2008; Fogarty et al., 2016a; Saba et al., 2016). Given that iPSC-derived cortical neurons are likely to reflect a physiologically early developmental status (Livesey et al., 2016) and that murine models show early disturbances, it is possible these studies are in line with an emerging consensus of increased excitatory synaptic activity as being a feature of cortical hyperexcitability. Importantly, this consensus may extend to other neurodegenerative diseases, such as Alzheimer’s Disease, where increased excitatory synaptic density and input early in disease is again observed in equivalent models (Šišková et al., 2014; Ghatak et al., 2019). Nonetheless, there are reports of excitatory input not changing in the TDP-43A315T model (Zhang et al., 2016) and a C9ORF72RE murine model, though this latter model does not display classical ALS-FTD pathology or neurodegeneration (Peters et al., 2015). Increased synaptic density in ALS-FTD is in clear contrast to the belief that neurodegeneration causes synaptic loss. Indeed, the degree of synaptic loss in the pre-frontal cortex of C9ORF72RE ALS-FTD patient post-mortem correlates with the degree of cognitive impairment displayed by the patients (Henstridge et al., 2019). Furthermore, later stage, symptomatic P60 TDP-43A315T mice exhibit layer V projection neurons with a decrease in synaptic input and spine density (Handley et al., 2017). These collective latter studies are therefore consistent with a trend that synaptic loss is restricted to latter stages of the disease course and accompanies the onset of symptomatic disease. The data would therefore suggest a shift from an early increased synaptic density property of C9ORF72RE excitatory cortical neurons, supporting cortical hyperexcitability observations, to a general decrease, which appear to be aligned to symptomatic onset.
Mechanisms promoting cortical synaptic density remain unreported but are associated with transcriptional dysregulation consistent with modified expression of synaptic architecture proteins (Prudencio et al., 2015; Perkins et al., 2021). Conversely, mechanisms supporting synaptic loss in C9ORF72RE cortical neurons are now emerging. Synaptic loss is observed in the prefrontal cortex of aged (4.5 months) transgenic mice expressing 80-repeat GR (GR80) DPRs (Choi et al., 2019). Furthermore, consistent with increasing reports of C9ORF72 localization at the synapse (Frick et al., 2018; Xiao et al., 2019), hippocampal regions of 3-month old C9ORF72 knockout mice show a reduction in synaptic density (Xiao et al., 2019), suggesting that haploinsufficiency may play a role in cortical synaptic loss. C9ORF72 is also highly expressed in microglia (Rizzu et al., 2016), and recent work has determined that loss of C9ORF72 exacerbates microglial synaptic pruning activity in the cortex, which correlates with cognitive impairments (Lall et al., 2021). Synaptic loss may therefore be driven by perturbed microglial function driven through C9ORF72 haploinsufficiency. Noting that microglia can equally sense and be regulated by neuronal excitability (Umpierre and Wu, 2021), how microglia contribute to cortical hyperexcitability or are potentially influenced by hyperexcitability will be a key question to resolve in how ALS-FTD progresses.
Layer V cortical projection neurons (aka upper motor neurons) are a vulnerable population in ALS that functionally connect the motor cortex to lower motor neuron populations in the spinal cord. Reduced inhibition onto layer V neurons appears to drive hyperexcitability in this neuronal population in TDP-43A135T mice (Zhang et al., 2016). Recent data from a rodent model in which hyperexcitability is chronically chemogenetically driven in upper motor neurons leads to the development of essential features of ALS, including upper and lower motor neuron degeneration, reactive gliosis and induced TDP-43 pathology (Haidar et al., 2021). Such data is consistent with the interrelation between hyperexcitability and the feed forward model of neurodegeneration. Furthermore, it appears that increased hyperexcitability can generate morphological changes. A study upon a nuclear localization sequence-deficient mouse model of TDP-43 identified that cytoplasmic mislocalization of TDP-43 drives intrinsic hyperexcitability and decreased excitatory synaptic inputs (Dyer et al., 2021). Indeed, hyperexcitability may drive continued functional synaptic loss, dendritic spine loss and dendrite pathology in upper motor neurons that are commonly observed features in upper motor neurons of ALS patient post-mortem tissue (Hammer et al., 1979; Genç et al., 2017) and other models, including TDP-43A315T (Handley et al., 2017), SOD1G93A (Fogarty et al., 2016b,2017), and FUSR521G (Sephtona et al., 2014). Clearly, data on C9ORF72RE remains scant for this cell type. Future work will be required to clarify whether impairments in layer V projection neurons are determined by intrinsic, cell autonomous mechanisms and/or are driven by altered input via cortical dysfunction which in turn drives hyperexcitability and synaptic loss.
Disease progression in neurodegenerative diseases is thought to reflect a stage of homeostatic adaptation, where disease-driven disturbances in network function are functionally tolerated for an undetermined period of time, but ultimately give way to network failure, where homeostasis mechanisms cannot viably maintain function (Frere and Slutsky, 2018). There is potential evidence for functional changes in ALS-FTD models that may reflect this early shifting landscape. Longitudinal assessment of synaptic and intrinsic excitability of SOD1G93A layer V cortical projections reveal a fluctuating reciprocal profile of altered intrinsic excitability and synaptic input that could reflect functional adaptation at the pre-symptomatic stage (Kim et al., 2017). Similarly, despite C9ORF72RE iPSC-derived cortical neurons exhibiting increased synaptic density, consistent with increased excitability (and burst frequency), neurons also display a reduced readily releasable pool of synaptic vesicles. As a result, these neurons display a reduced ability to maintain synaptic transmission and present a reduced burst duration (Perkins et al., 2021). These latter data are consistent with a putative role of C9ORF72 protein in vesicular trafficking within the trans-Golgi network (Snowden et al., 2012; Aoki et al., 2017; Frick et al., 2018) and that C9ORF72 haploinsufficiency may result in a reduction of the readily releasable pool of vesicles. Consistent with these data, mice engineered to express 149-repeat GA (GA149) DPRs also demonstrate a reduced expression of synaptic vesicle protein SV2 in addition to altered Ca2+ homeostasis and impaired vesicle release in cortical neurons (Jensen et al., 2020). In this regard, such reductions in general vesicular function may potentially reflect early homeostatic adaptations in response to increased synaptic density (or vice versa) driven by pathological C9ORF72RE-related mechanisms. Interestingly, potential modifiers of disease onset in C9ORF72RE FTD-mediated disease appear to be associated with altered expression of proteins with synaptic roles including synaptic vesicle dynamics (Barbier et al., 2021). Given that cortical function can be readily monitored in patients, establishing the earliest known physiological disturbances in cortical function in C9ORF72RE and the pathological drivers underpinning this may give us one of our earliest windows into understanding C9ORF72RE disease onset and progression.
In receiving monosynaptic innervation from upper motor neurons, lower spinal motor neurons represent the final effector component of the lower motor system, whose axons project to innervate skeletal muscle fibers (Burke, 1981). Lower motor neuron dysfunction has long been an established clinical observation, detected by nerve conduction and electromyography (EMG) and are key criteria in the diagnosis of ALS (Daube, 1985; Mogyoros et al., 1998; Geevasinga et al., 2015; de Carvalho and Swash, 2016) including C9ORF72RE patients (Geevasinga et al., 2015). Critically, altered lower motor neuron function in ALS patients is evidenced after that of cortical dysfunction and parallels the onset of patients developing muscle weakness, atrophy, fasciculation and cramps in ALS (Menon et al., 2015). Further, the development of fasciculation and cramps precede that of muscle weakness suggesting that hyperexcitability leading to progressive loss of function (hypoexcitability) is a feature of the lower motor neuron in ALS disease progression before eventual degeneration and loss (Bae et al., 2013). In this aspect of the review, we will discuss the mechanisms leading to the potential changes in excitability and neurophysiological mechanisms leading to excitotoxicity and cell death. These studies are summarized in Table 2.
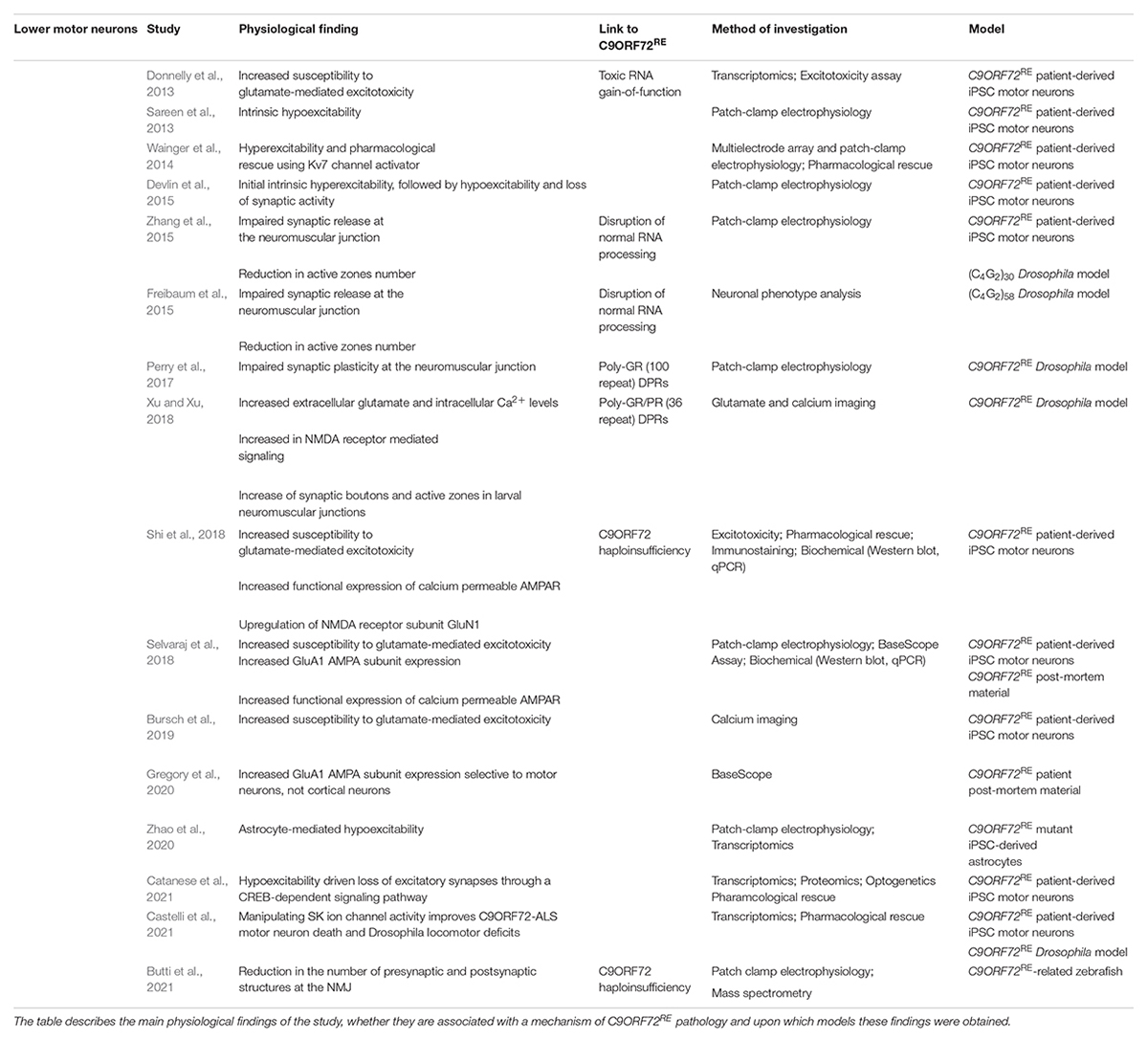
Table 2. Summary of physiological studies that have implication for the C9ORF72RE in lower motor neuron dysfunction.
Synaptic glutamatergic signaling links upper and lower motor neuron function, and, glutamate-mediated excitotoxicity is considered one of the main pathogenic mechanisms that contributes to the degeneration of motor neurons in ALS-FTD (Cleveland and Rothstein, 2001). The hypothesis is multifaceted and includes cell autonomous and non-cell autonomous mechanisms. Commensurate with cortical disease progression, synaptic loss in lower motor neurons is an established observation in the latter stages of ALS and is expected to be a major physiological determinant preventing lower motor neuron function in the later stages of disease (Sasaki and Maruyama, 1994). Synaptic loss accompanied by CREB-dependent transcriptomic and proteomic changes is observed in C9ORF72RE iPSC-derived motor neurons maintained for extended culture time (Catanese et al., 2021). A number of hypotheses center upon disturbances in glutamate-mediated signaling and altered excitability being major contributors to synaptic loss and other sites of lower motor neuron injury.
Lower motor neurons are responsive to synaptic glutamate via the synaptic expression of glutamatergic AMPA and NMDA receptors (Van Den Bosch et al., 2006). Early work determined an apparent intrinsic vulnerability of lower motor neurons to AMPA receptor-mediated excitotoxicity (Rothstein et al., 1990, 1992; Couratier et al., 1993; Rothstein, 1995; Cleveland and Rothstein, 2001). Elevated synaptic glutamate levels are predicted given the increased excitability of upper motor neurons. However, whether pre-synaptic terminal deficits in glutamate release from upper motor neurons exist remains to be determined. Nonetheless, the uptake of glutamate from the synaptic cleft is strongly hypothesized to be reduced given that the expression of astrocyte glutamate transporter (EAAT2) is widely reported to be attenuated in multiple ALS models (Rosenblum and Trotti, 2017). In the case of C9ORF72RE however, patient-derived astrocytes are not consistent with a reduction in EAAT2 expression or function (Allen et al., 2019b; Zhao et al., 2020). How such data are to be reconciled with other ALS models of glutamate transporter dysfunction and expression remains to be resolved.
Over stimulation of glutamate receptors gives rise to the possibility of an injurious, excitotoxic level of Ca2+ influx (Pina-Crespo et al., 2014) and iPSC-derived motor neurons obtained from C9ORF72RE patients exhibit enhanced vulnerability to glutamate receptor-mediated excitotoxicity (Donnelly et al., 2013; Selvaraj et al., 2018; Shi et al., 2018; Bursch et al., 2019). Interestingly, this vulnerability has been rescued pharmacologically in studies using an anticoagulation-deficient form of activated protein C (Shi et al., 2019) and antisense oligonucleotides against the repeat expansion (Donnelly et al., 2013). Mechanistically, this vulnerability has been shown to occur as a result of increased relative expression of Ca2+ permeable AMPA receptors in C9ORF72RE patient-derived motor neurons due to a greater expression of Ca2+ permeable AMPA receptor subunit GluA1 (Selvaraj et al., 2018; Shi et al., 2018; but see Moore et al., 2019). Further work on C9ORF72RE patient post-mortem demonstrated that the dysregulation of GluA1 is selective to C9ORF72RE lower motor neurons and is not present in the cortex, and thus providing an example of a regional specific degenerative mechanism (Selvaraj et al., 2018; Gregory et al., 2020). The dysregulation of GluA1 also appears conserved amongst other genetic ALS backgrounds including mutant TDP-43 motor neurons (Bursch et al., 2019), FUS (Udagawa et al., 2015) and in sporadic ALS patients, where the latter show further dysregulation of AMPA receptor subunits in the cortex (Gregory et al., 2020). In agreement, data from mSOD1 patients and models indicate a converging mechanism of vulnerability to glutamate-mediated excitotoxicity via Ca2+ permeable AMPA receptors (Shaw, 2005; Van Den Bosch et al., 2006), where such studies appear consistent with a reduction in the relative expression of GluA2 subunits, the master regulators of Ca2+ permeability. The GluA2 subunit achieves this because it predominantly presents in its post-transcriptionally edited form where a channel-lining, positively charged arginine side chain protrudes into the ion channel, presenting a charge block to Ca2+ flux (Traynelis et al., 2010) whereas, the pre-edited GluA2 form contains a non-charged glutamine side chain and permits Ca2+ flux. Notably, inefficient RNA editing of the GluA2 subunit, thus resulting in Ca2+-permeability, has been reported in sporadic ALS patient samples (Kawahara et al., 2004a,b). However, whilst appearing to impact upon the function of editing enzyme ADAR2, this mechanism does not appear to be the source of increased Ca2+-permeable AMPA receptors in C9ORF72RE patients (Selvaraj et al., 2018; Moore et al., 2019). Mechanisms of GluA1 upregulation in the context of C9ORF72RE has been associated with haploinsufficiency of C9ORF72 protein in lower motor neurons derived from patient iPSCs and C9ORF72 knockout mice (Shi et al., 2018) and also hippocampal neurons (Xiao et al., 2019). In support of this, the knockout of putative C9ORF72 interactor Rab39b in primary neuron culture results in increased GluA1 trafficking to dendrites (Mignogna et al., 2015, 2021). Interestingly, despite early work indicating the low impact of NMDA receptor-mediated excitotoxicity on motor neurons, recent studies have shown an upregulation of the NMDA receptor subunit GluN1 (Shi et al., 2018) that can be rescued along with GluA1 upregulation using small molecule inhibitors of phosphatidylinositol-5-kinase signaling (Staats et al., 2019). The role of NMDA receptors in glutamate-mediated excitotoxicity and the role of DPRs remains to be fully explored. However, recent Drosophila models presenting poly-GR and PR (GR36, PR36) constructs demonstrated an increase in NMDA receptor-mediated signaling in glutamatergic neurons, suggesting that DPRs may have a role in the dysregulation of glutamate receptors in C9ORF72RE motor neurons (Xu and Xu, 2018).
Clearly, a strong emphasis of research thus far has been given to the perturbations associated with glutamatergic signaling. The disruption of inhibitory GABA-ergic and glycinergic signaling in the spinal tract has been implicated in ALS, largely in mSOD1 models (Martin and Chang, 2012), though initial reports indicated this was a secondary event to motor neuron degeneration (Hossaini et al., 2011). Though, more recent work in SOD1G93A mice now implicates deficits in inhibitory signaling associated with V1 interneurons in the spinal tract that parallel motor disturbances, raising the possibility that increased excitatory signaling in ALS patients may also stem from a reduced impact of inhibitory influences (Allodi et al., 2021). The specific impact of the C9ORF72RE mutation on inhibitory signaling whether in the cortex or lower motor neuron remains to be determined.
Altered motor neuron excitability in C9ORF72RE patients is predicted to be underpinned by changes in intrinsic expression of ion channels that support action potential conduction (Geevasinga et al., 2015). Physiological mechanisms addressing lower motor neuron excitability in the context of C9ORF72RE have been widely investigated in vitro, employing iPSC-derived motor neurons from C9ORF72RE ALS patient fibroblasts. Patch-clamp studies have reported hyperexcitability at early stages of motor neuron differentiation (2–6 weeks in culture) where cells become intrinsically more excitable to depolarization (Devlin et al., 2015; Wainger and Cudkowicz, 2015). However, as cultures are maintained further (7–10 weeks), they become hypoexcitable, evidenced by a reduction in action potential generation in response to depolarization compared to motor neurons derived from healthy patients (Sareen et al., 2013; Zhang et al., 2013; Devlin et al., 2015; Naujock et al., 2016; Guo et al., 2017). No changes in cell survival were reported in these studies, which supports the idea that changes in excitability are early signs of functional loss of motor neurons prior to their degeneration, which is also supported by clinical studies of motor function in ALS patients (Iwai et al., 2016). Furthermore, these data are broadly consistent with the overall consensus nature of shifting excitability in mutant SOD1 mice motor neurons that display a period of early hyperexcitability before hypoexcitability (Leroy and Zytnicki, 2015), which in turn precedes motor neuron denervation (Martínez-Silva et al., 2018).
Key mechanisms that drive these excitability states are now emerging. Selective tuning of cortical inhibition in SOD1G93A mice to reduce potential cortical hyperexcitability has a protective impact upon lower motor neurons (Sahara Khademullah et al., 2020), suggesting that early hyperexcitability in lower motor neurons, at least in part, is driven by upstream cortical and upper motor neuron dysfunction and may drive injury or further pathological processes. On this note, increased depolarization of motor neurons, including via glutamate, has the ability to promote the formation of TDP-43 pathology (Weskamp et al., 2020) and drive DPR formation (Westergard et al., 2019). Interestingly, more recent data using improved iPSC-derived MN protocols yielding enriched, predominant neuronal cultures with very little glial differentiation does not exhibit any consistent differences in motor neuron excitability (Selvaraj et al., 2018; Zhao et al., 2020). This discrepancy from previous studies appears to be resolved by the fact that co-cultures of motor neurons with C9ORF72RE astrocytes are induced to be hypoexcitable and that previous studies used protocols with heterogeneous cellular specification including astrocytes (Zhao et al., 2020). Beyond other impacts upon motor neuron health (Serio et al., 2013; Meyer et al., 2014; Allen et al., 2019a), the roles of astrocytes are therefore likely to play critical non-cell autonomous roles in the modulation of motor neuron excitability. This mechanism may be related to a soluble transmissible factor given that cultures of murine motor neurons with conditioned medium derived from SOD1G93A expressing astrocytes, was found to alter ion channel function and motor neuron excitability (Fritz et al., 2013). Furthermore, recent data implicate a decreased expression of astrocyte KIR4.1-containing ion channels to adequately remove potassium extruded from active motor neuron axons in mSOD1 models indicating possible mechanisms impacting the ability of astrocytes to adequately maintain axonal homeostasis (Kelley et al., 2018). It therefore appears that numerous cell autonomous and non-autonomous mechanisms are at play here and not limited to intrinsic lower motor neuron processes.
The rescue of both hypoexcitability and hyperexcitability in motor neurons has been a pharmacological target in recent years. The promotion of increased excitability via pharmacological inhibition of small conductance calcium-activated potassium (SK) channels promotes survival and restores the activity-dependent transcriptional profiles and synaptic composition in C9ORF72RE iPSC-derived motor neurons, and furthermore, promotes locomotor function in a Drosophila model containing 36 hexanucleotide repeats (Castelli et al., 2021; Catanese et al., 2021). C9ORF72RE motor neurons also demonstrated an increase in the expression of SK channel subunits, which could be corrected using specific inhibition of the SRSF1-dependent nuclear export of pathological C9ORF72RE transcripts (Castelli et al., 2021). Contrastingly, hyperexcitability in lower motor neurons has been established in several other ALS models and studies have used pharmacological activators of Kv7 potassium ion channels to reduce hyperexcitability in C9ORF72RE-derived motor neurons with the possibility that they protect motor neurons from excitotoxicity (Wainger et al., 2014; Huang et al., 2021). These studies have now been translated into clinical trials (Wainger et al., 2021). Our current understanding of the shifting excitability in both cortical and motor neurons indicates that the pharmacological benefit of modulators of excitability to patients will need to be understood and carefully considered according to disease stage.
Motor neuron denervation from the neuromuscular junction precedes motor neuron loss. Given that motor activity is required for the maintenance of innervation, it is no surprise that loss of motor neuron pre-synaptic activity is associated with disease pathogenesis. Measurable loss of motor input is common to symptomatic ALS patients and consistent with pre-synaptic dysfunction of motor neurons (Maselli et al., 1993) that is observed primarily, and more aggressively, in association with neuromuscular junctions innervated by fast-twitch motor neurons in SOD1G93A mice (Cappello and Francolini, 2017). Beyond rodent models, reduced synaptic function has now been observed in several other models including Drosophila and zebrafish (Cappello and Francolini, 2017; Butti et al., 2021). Studies in fly models overexpressing hexanucleotide repeats (58 and 30 repeats) demonstrate impaired synaptic release at the neuromuscular junction and a reduced number of active zones in motor neurons (Freibaum et al., 2015; Zhang et al., 2015). Consistent with a reduction in synaptic activity over time, spontaneous post-synaptic current activity was shown to progressively decrease in C9ORF72RE iPSC-derived motor neurons and was directly associated with hypoexcitability, but not motor neuron loss (Devlin et al., 2015). The potential for impaired synaptic release may be therefore related to an inherent inability to generate sufficient action potential activity at the pre-synaptic terminal. Moreover, Jensen and colleagues recently reported aberrations in vesicle dynamics that coincide with the loss of vesicle protein SV2 and precede motor neuron loss in a poly-GA (GA149) animal model and C9ORF72RE motor neurons (Jensen et al., 2020). Interestingly, such observations align with investigations in cortical neurons and suggests that not only do poly-GA repeats interfere with the synaptic release mechanism but also the reduction in vesicular dynamics in the cortex may have mechanistic overlap with motor neurons (Jensen et al., 2020). This study also reports that altered vesicle dynamics are associated with elevated Ca2+ influx, which controls synaptic vesicular release (Jensen et al., 2020). It has therefore been hypothesized that the increased cytoplasmic Ca2+ may form a homeostatic mechanism to potentially rescue synaptic release. Indeed, the pharmacological rescue of C9ORF72RE model (GR100) via the induction of endogenous NMJ plasticity signaling can rescue synaptic function (Perry et al., 2017). Moreover, Coyne et al. report that synaptic vesicle cycling defects due to deficits in the post-transcriptional inhibition of Hsc70-4/HSPA8 expression are common to C9ORF72RE and mTDP-43 Drosophila models (Coyne et al., 2017), suggesting that vesicle depletion is at play at the NMJ. Importantly, this mechanism is linked to dynamin function, a key player in axonal transport, and therefore suggests that synaptic vesicle impairments and established impairments in axonal transport in ALS are potentially linked (Gunes et al., 2020).
Our review provides an overview of the key concepts of neurophysiological disturbances in C9ORF72RE-mediated ALS-FTD. We have provided details on the current mechanistic view of the sources of these perturbations, when these appear in disease and allude to their relevance to pathogenesis. Many aspects of neurophysiological dysfunction in the context of C9ORF72RE-mediated disease are currently inferred. In this respect, a general consensus of early cortical hyperexcitability progressing to general loss of function consistent with hypoexcitability in the symptomatic period appears to be consistent across patients and, importantly, several ALS-FTD models appear to replicate this progression, at least in aspects (summarized in Table 3). However, there are established examples of mechanistic disturbances that differ from other genetic backgrounds. Similarly, pathogenesis of cortical and motor dysfunction display overlapping dysfunctional features but also selective regional differences.
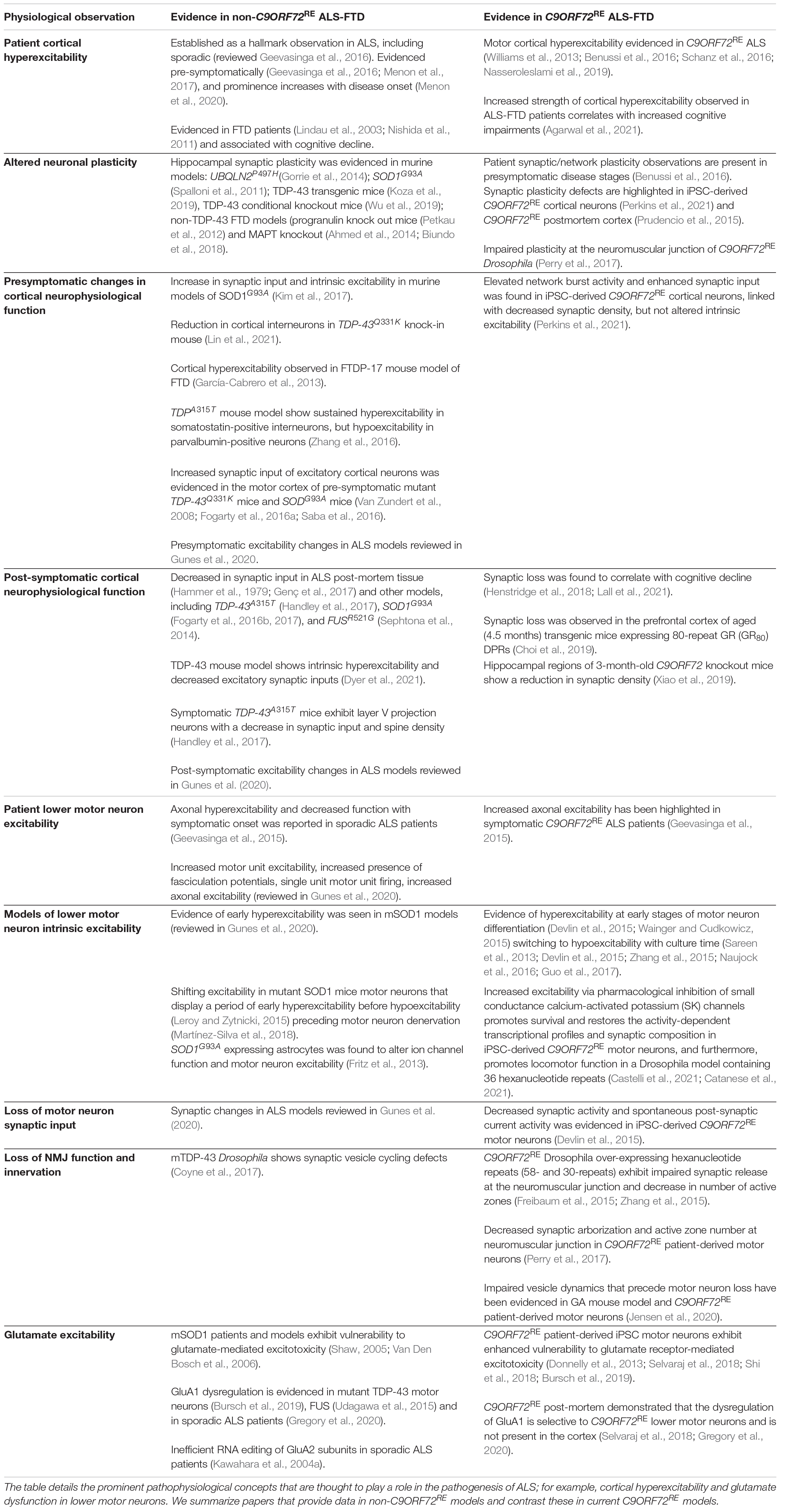
Table 3. Summary of the main physiological observations that are associated with ALS.
Current models of C9ORF72RE are broad, and are known to have both advantages and disadvantages especially in regards to their inability to fully capture the disease phenotype (Sances et al., 2016; Balendra and Isaacs, 2018). New improvements in disease modeling are needed to forward our understanding of disease pathogenesis and progress is now being made in this respect. For example, the ability to model the cortico-spinal tract in vitro in a human context is now documented (Andersen et al., 2020) and furthermore, it is now possible to examine the impact of native length DPR species in Drosophila (West et al., 2020). Importantly, the development of C9ORF72RE rodent models that successfully recapitulate major aspects of ALS-FTD remains ongoing (Balendra and Isaacs, 2018). Such tools will allow us to systematically define the mechanistic impact of the C9ORF72RE on cell types, as well as cell types upon each other.
In this regard, neurophysiological disturbances in ALS-FTD are now much more complex than previously believed. Beyond multiple molecular mechanisms associated with the C9ORF72RE, disturbances are likely to have an additional non-cell autonomous component relating to other dysfunctional cell-types that now include astrocytes and microglia. It is also becoming clear that for all neurodegenerative disease early functional changes may partially reflect homeostatic mechanisms that counteract disease-driven pathophysiology. On this note, emerging work is now beginning to consider the cortico-spinal circuit as a singular functional unit and this work will allow us to identify how each component can potentially impact each other. Such data will help stratify early mechanisms associated with disease progression for potential pharmacological benefit. Finally, we must consider that neurophysiological impairments may be causal to, or as a result of, a myriad of other equally known disease processes that include mitochondrial dysfunction, axonal transport dysregulation, impaired proteostasis and aberrant RNA metabolism. Although future studies have much to elucidate, it is now clear that altered neurophysiological function in C9ORF72RE ALS-FTD plays a key role in the pathogenesis of the disease.
ISP and ML wrote and edited the manuscript. MSA, CS, and LF edited the final version of the manuscript. All the authors contributed to the article and approved the submitted version.
This work was supported by a SITraN/University of Sheffield Ph.D. Studentship (ML and LF); a MNDA Ph.D. Studentship (Livesey/Oct20/900-792; ML and LF) a SITraN career track fellowship (ML), Rosetree’s Trust seed award (ML) and Royal Society project grant (ML); and MRC JPND (MR/V000470/1) grant to LF.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agarwal, S., Highton-Williamson, E., Caga, J., Howells, J., Dharmadasa, T., Matamala, J. M., et al. (2021). Motor cortical excitability predicts cognitive phenotypes in amyotrophic lateral sclerosis. Sci. Rep. 11, 1–9. doi: 10.1038/s41598-021-81612-x
Agosta, F., Ferraro, P. M., Riva, N., Spinelli, E. G., Chiò, A., Canu, E., et al. (2016). Structural brain correlates of cognitive and behavioral impairment in MND. Hum. Brain Mapp. 37:1614. doi: 10.1002/HBM.23124
Ahmed, T., Van der Jeugd, A., Blum, D., Galas, M. C., D’Hooge, R., Buee, L., et al. (2014). Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol. Aging 35, 2474–2478. doi: 10.1016/j.neurobiolaging.2014.05.005
Allen, S. P., Hall, B., Castelli, L. M., Francis, L., Woof, R., Siskos, A. P., et al. (2019a). Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 142, 586–605. doi: 10.1093/brain/awy353
Allen, S. P., Hall, B., Woof, R., Francis, L., Gatto, N., Shaw, A. C., et al. (2019b). C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain 142, 3771–3790. doi: 10.1093/brain/awz302
Allodi, I., Montañana-Rosell, R., Selvan, R., Löw, P., and Kiehn, O. (2021). Locomotor deficits in a mouse model of ALS are paralleled by loss of V1-interneuron connections onto fast motor neurons. Nat. Commun. 12, 1–18. doi: 10.1038/s41467-021-23224-7
Andersen, J., Revah, O., Miura, Y., Thom, N., Amin, N. D., Kelley, K. W., et al. (2020). Generation of Functional Human 3D Cortico-Motor Assembloids. Cell 183, 1913.e–1929.e. doi: 10.1016/j.cell.2020.11.017
Aoki, Y., Manzano, R., Lee, Y., Dafinca, R., Aoki, M., Douglas, A. G. L., et al. (2017). C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 140, 887–897. doi: 10.1093/brain/awx024
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Bae, J. S., Simon, N. G., Menon, P., Vucic, S., and Kiernan, M. C. (2013). The puzzling case of hyperexcitability in amyotrophic lateral sclerosis. J. Clin. Neurol. 9, 65–74. doi: 10.3988/jcn.2013.9.2.65
Balendra, R., and Isaacs, A. M. (2018). C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558. doi: 10.1038/s41582-018-0047-2
Barbier, M., Camuzat, A., Hachimi, K., El, Guegan, J., Rinaldi, D., et al. (2021). SLITRK2, an X-linked modifier of the age at onset in C9orf72 frontotemporal lobar degeneration. Brain 144, 2798–2811. doi: 10.1093/brain/awab171
Benussi, A., Cosseddu, M., Filareto, I., Dell’Era, V., Archetti, S., Sofia Cotelli, M., et al. (2016). Impaired long-term potentiation–like cortical plasticity in presymptomatic genetic frontotemporal dementia. Ann. Neurol. 80, 472–476. doi: 10.1002/ana.24731
Biundo, F., Del Prete, D., Zhang, H., Arancio, O., and D’Adamio, L. (2018). A role for tau in learning, memory and synaptic plasticity. Sci. Rep. 8, 1–13. doi: 10.1038/s41598-018-21596-3
Burke, R. E. (1981). Motor Units: Anatomy, Physiology, and Functional Organization. Compr. Physiol. 1981, 345–422. doi: 10.1002/cphy.cp010210
Bursch, F., Kalmbach, N., Naujock, M., Staege, S., Eggenschwiler, R., Abo-Rady, M., et al. (2019). Altered calcium dynamics and glutamate receptor properties in iPSC-derived motor neurons from ALS patients with C9orf72, FUS, SOD1 or TDP43 mutations. Hum. Mol. Genet. 28, 2835–2850. doi: 10.1093/hmg/ddz107
Butti, Z., Pan, Y. E., Giacomotto, J., and Patten, S. A. (2021). Reduced C9orf72 function leads to defective synaptic vesicle release and neuromuscular dysfunction in zebrafish. Commun. Biol. 4:792. doi: 10.1038/S42003-021-02302-Y
Cappello, V., and Francolini, M. (2017). Neuromuscular junction dismantling in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 18:ijms18102092. doi: 10.3390/ijms18102092
Castelli, L. M., Cutillo, L., Souza, C. D. S., Sanchez-Martinez, A., Granata, I., Lin, Y. H., et al. (2021). SRSF1-dependent inhibition of C9ORF72-repeat RNA nuclear export: genome-wide mechanisms for neuroprotection in amyotrophic lateral sclerosis. Mol. Neurodegener. 16:475–y. doi: 10.1186/s13024-021-00475-y
Catanese, A., Rajkumar, S., Sommer, D., Freisem, D., Wirth, A., Aly, A., et al. (2021). Synaptic disruption and CREB-regulated transcription are restored by K + channel blockers in ALS. EMBO Mol. Med. 13:e13131. doi: 10.15252/emmm.202013131
Choi, S. Y., Lopez-Gonzalez, R., Krishnan, G., Phillips, H. L., Li, A. N., Seeley, W. W., et al. (2019). C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci. 22, 851–862. doi: 10.1038/s41593-019-0397-0
Christidi, F., Karavasilis, E., Rentzos, M., Kelekis, N., Evdokimidis, I., and Bede, P. (2018). Clinical and radiological markers of extra-motor deficits in amyotrophic lateral sclerosis. Front. Neurol. 9:1005. doi: 10.3389/fneur.2018.01005
Cleveland, D. W., and Rothstein, J. D. (2001). From charcot to lou gehrig: deciphering selective motor neuron death in als. Nat. Rev. Neurosci. 2, 806–819. doi: 10.1038/35097565
Consonni, M., Contarino, V. E., Catricalà, E., Bella, E. D., Pensato, V., Gellera, C., et al. (2018). Cortical markers of cognitive syndromes in amyotrophic lateral sclerosis. Neuroimage 19:675. doi: 10.1016/J.NICL.2018.05.020
Couratier, P., Sindou, P., Hugon, J., Couratier, P., Hugon, J., Vallat, J. M., et al. (1993). Cell culture evidence for neuronal degeneration in amyotrophic lateral sclerosis being linked to glutamate AMPA/kainate receptors. Lancet 341, 265–268. doi: 10.1016/0140-6736(93)92615-Z
Coyne, A. N., Lorenzini, I., Chou, C. C., Torvund, M., Rogers, R. S., Starr, A., et al. (2017). Post-transcriptional Inhibition of Hsc70-4/HSPA8 Expression Leads to Synaptic Vesicle Cycling Defects in Multiple Models of ALS. Cell Rep. 21, 110–125. doi: 10.1016/J.CELREP.2017.09.028
Daube, J. R. (1985). Electrophysiologic studies in the diagnosis and prognosis of motor neuron diseases. Neurol. Clin. 3, 473–493. doi: 10.1016/s0733-8619(18)31017-x
de Carvalho, M., and Swash, M. (2016). Lower motor neuron dysfunction in ALS. Clin. Neurophysiol. 127, 2670–2681. doi: 10.1016/j.clinph.2016.03.024
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Devlin, A. C., Burr, K., Borooah, S., Foster, J. D., Cleary, E. M., Geti, I., et al. (2015). Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 6, 1–12. doi: 10.1038/ncomms6999
Donnelly, C. J., Zhang, P. W., Pham, J. T., Heusler, A. R., Mistry, N. A., Vidensky, S., et al. (2013). RNA Toxicity from the ALS/FTD C9ORF72 Expansion Is Mitigated by Antisense Intervention. Neuron 80, 415–428. doi: 10.1016/j.neuron.2013.10.015
Dyer, M. S., Woodhouse, A., and Blizzard, C. A. (2021). Cytoplasmic human tdp-43 mislocalization induces widespread dendritic spine loss in mouse upper motor neurons. Brain Sci. 11:brainsci11070883. doi: 10.3390/brainsci11070883
Eisen, A., Braak, H., Tredici, K., Del, Lemon, R., Ludolph, A. C., et al. (2017). Cortical influences drive amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 88, 917–924. doi: 10.1136/jnnp-2017-315573
Estebanez, L., Hoffmann, D., Voigt, B. C., and Poulet, J. F. A. (2017). Parvalbumin-Expressing GABAergic Neurons in Primary Motor Cortex Signal Reaching. Cell Rep. 20, 308–318. doi: 10.1016/j.celrep.2017.06.044
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: Phasic and tonic activation of GABA A receptors. Nat. Rev. Neurosci. 6, 215–229. doi: 10.1038/nrn1625
Fogarty, M. J., Klenowski, P. M., Lee, J. D., Drieberg-Thompson, J. R., Bartlett, S. E., Ngo, S. T., et al. (2016a). Cortical synaptic and dendritic spine abnormalities in a presymptomatic TDP-43 model of amyotrophic lateral sclerosis. Sci. Rep. 61, 1–13. doi: 10.1038/srep37968
Fogarty, M. J., Mu, E. W. H., Lavidis, N. A., Noakes, P. G., and Bellingham, M. C. (2017). Motor areas show altered dendritic structure in an amyotrophic lateral sclerosis mouse model. Front. Neurosci. 11:1–16. doi: 10.3389/fnins.2017.00609
Fogarty, M. J., Mu, E. W. H., Noakes, P. G., Lavidis, N. A., and Bellingham, M. C. (2016b). Marked changes in dendritic structure and spine density precede significant neuronal death in vulnerable cortical pyramidal neuron populations in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 4:347–y. doi: 10.1186/s40478-016-0347-y
Freibaum, B. D., Lu, Y., Lopez-Gonzalez, R., Kim, N. C., Almeida, S., Lee, K. H., et al. (2015). GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133. doi: 10.1038/nature14974
Frere, S., and Slutsky, I. (2018). Alzheimer’s Disease: From Firing Instability to Homeostasis Network Collapse. Neuron 97, 32–58. doi: 10.1016/j.neuron.2017.11.028
Frick, P., Sellier, C., Mackenzie, I. R. A., Cheng, C. Y., Tahraoui-Bories, J., Martinat, C., et al. (2018). Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol. Commun. 6:72. doi: 10.1186/s40478-018-0579-0
Fritz, E., Izaurieta, P., Weiss, A., Mir, F. R., Rojas, P., Gonzalez, D., et al. (2013). Mutant SOD1-expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability. J. Neurophysiol. 109, 2803–2814. doi: 10.1152/jn.00500.2012
García-Cabrero, A. M., Guerrero-López, R., Giráldez, B. G., Llorens-Martín, M., Ávila, J., Serratosa, J. M., et al. (2013). Hyperexcitability and epileptic seizures in a model of frontotemporal dementia. Neurobiol. Dis. 58, 200–208. doi: 10.1016/j.nbd.2013.06.005
Geevasinga, N., Menon, P., Howells, J., Nicholson, G. A., Kiernan, M. C., and Vucic, S. (2015). Axonal ion channel dysfunction in C9orf72 familial amyotrophic lateral sclerosis. JAMA Neurol. 72, 49–57. doi: 10.1001/jamaneurol.2014.2940
Geevasinga, N., Menon, P., Özdinler, P. H., Kiernan, M. C., and Vucic, S. (2016). Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat. Rev. Neurol. 12, 651–661. doi: 10.1038/nrneurol.2016.140
Genç, B. B., Jara, J. H., Lagrimas, A. K. B. B., Pytel, P., Roos, R. P., Mesulam, M. M., et al. (2017). Apical dendrite degeneration, a novel cellular pathology for Betz cells in ALS. Sci. Rep. 7, 1–10. doi: 10.1038/srep41765
Gendron, T. F., Belzil, V. V., Zhang, Y. J., and Petrucelli, L. (2014). Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol. 127, 359–376. doi: 10.1007/s00401-013-1237-z
Gendron, T. F., Bieniek, K. F., Zhang, Y.-J., Jansen-West, K., Ash, P. E. A., Caulfield, T., et al. (2013). Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844. doi: 10.1007/s00401-013-1192-8
Gerges, N. Z., Backos, D. S., and Esteban, J. A. (2004). Local control of AMPA receptor trafficking at the postsynaptic terminal by a small GTPase of the Rab family. J. Biol. Chem. 279, 43870–43878. doi: 10.1074/jbc.M404982200
Ghatak, S., Dolatabadi, N., Trudler, D., Zhang, X., Wu, Y., Mohata, M., et al. (2019). Mechanisms of hyperexcitability in alzheimer’s disease hiPSC-derived neurons and cerebral organoids vs. Isogenic control. Elife 8:50333. doi: 10.7554/ELIFE.50333
Gorrie, G. H., Fecto, F., Radzicki, D., Weiss, C., Shi, Y., Dong, H., et al. (2014). Dendritic spinopathy in transgenic mice expressing ALS/dementia-linked mutant UBQLN2. Proc. Natl. Acad. Sci. U S A. 111, 14524–14529. doi: 10.1073/pnas.1405741111
Gregory, J. M., Livesey, M. R., McDade, K., Selvaraj, B. T., Barton, S. K., Chandran, S., et al. (2020). Dysregulation of AMPA receptor subunit expression in sporadic ALS post-mortem brain. J. Pathol. 250, 67–78. doi: 10.1002/path.5351
Gunes, Z. I., Kan, V. W. Y., Ye, X. Q., and Liebscher, S. (2020). Exciting Complexity: The Role of Motor Circuit Elements in ALS Pathophysiology. Front. Neurosci. 14:573. doi: 10.3389/fnins.2020.00573
Guo, W., Naujock, M., Fumagalli, L., Vandoorne, T., Baatsen, P., Boon, R., et al. (2017). HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 8:911–y. doi: 10.1038/s41467-017-00911-y
Haidar, M., Viden, A., Cuic, B., Wang, T., Rosier, M., Tomas, D., et al. (2021). Cortical hyperexcitability drives dying forward ALS symptoms and pathology in mice. bioRxiv [Preprint]. doi: 10.1101/2021.08.13.456320
Hammer, R. P., Tomiyasu, U., and Scheibel, A. B. (1979). Degeneration of the human Betz cell due to amyotrophic lateral sclerosis. Exp. Neurol. 63, 336–346. doi: 10.1016/0014-4886(79)90129-8
Handley, E. E., Pitman, K. A., Dawkins, E., Young, K. M., Clark, R. M., Jiang, T. C., et al. (2017). Synapse Dysfunction of Layer v Pyramidal Neurons Precedes Neurodegeneration in a Mouse Model of TDP-43 Proteinopathies. Cereb. Cortex 27, 3630–3647. doi: 10.1093/cercor/bhw185
Henstridge, C. M., Sideris, D. I., Carroll, E., Rotariu, S., Salomonsson, S., Tzioras, M., et al. (2018). Synapse loss in the prefrontal cortex is associated with cognitive decline in amyotrophic lateral sclerosis. Acta Neuropathol. 135, 213–226. doi: 10.1007/s00401-017-1797-4
Henstridge, C. M., Tzioras, M., and Paolicelli, R. C. (2019). Glial contribution to excitatory and inhibitory synapse loss in neurodegeneration. Front. Cell. Neurosci. 13:63. doi: 10.3389/fncel.2019.00063
Ho, W. Y., Navakkode, S., Liu, F., Soong, T. W., and Ling, S. C. (2020). Deregulated expression of a longevity gene, Klotho, in the C9orf72 deletion mice with impaired synaptic plasticity and adult hippocampal neurogenesis. Acta Neuropathol. Commun. 8:155. doi: 10.1186/s40478-020-01030-4
Hossaini, M., Cano, S. C., Van Dis, V., Haasdijk, E. D., Hoogenraad, C. C., Holstege, J. C., et al. (2011). Spinal inhibitory interneuron pathology follows motor neuron degeneration independent of glial mutant superoxide dismutase 1 expression in SOD1-ALS mice. J. Neuropathol. Exp. Neurol. 70, 662–677. doi: 10.1097/NEN.0b013e31822581ac
Huang, X., Roet, K. C. D., Zhang, L., Brault, A., Berg, A. P., Jefferson, A. B., et al. (2021). Human amyotrophic lateral sclerosis excitability phenotype screen: Target discovery and validation. Cell Rep. 35:109224. doi: 10.1016/j.celrep.2021.109224
Iwai, Y., Shibuya, K., Misawa, S., Sekiguchi, Y., Watanabe, K., Amino, H., et al. (2016). Axonal dysfunction precedes motor neuronal death in amyotrophic lateral sclerosis. PLoS One 11:0158596. doi: 10.1371/journal.pone.0158596
Jensen, B. K., Schuldi, M. H., McAvoy, K., Russell, K. A., Boehringer, A., Curran, B. M., et al. (2020). Synaptic dysfunction induced by glycine-alanine dipeptides in C9orf72- ALS / FTD is rescued by SV 2 replenishment. EMBO Mol. Med. 12:201910722. doi: 10.15252/emmm.201910722
Kato, S., Hayashi, H., and Yagishita, A. (1993). Involvement of the frontotemporal lobe and limbic system in amyotrophic lateral sclerosis: As assessed by serial computed tomography and magnetic resonance imaging. J. Neurol. Sci. 116, 52–58. doi: 10.1016/0022-510X(93)90089-H
Kawahara, Y., Ito, K., Sun, H., Aizawa, H., Kanazawa, I., and Kwak, S. (2004a). RNA editing and death of motor neurons: There is a glutamate-receptor defect in patients with amyotrophic lateral sclerosis. Nature 427:801. doi: 10.1038/427801a
Kawahara, Y., Ito, K., Sun, H., Ito, M., Kanazawa, I., and Kwak, S. (2004b). Regulation of glutamate receptor RNA editing and ADAR mRNA expression in developing human normal and Down’s syndrome brains. Dev. Brain Res. 148, 151–155. doi: 10.1016/j.devbrainres.2003.11.008
Kelley, K. W., Ben Haim, L., Schirmer, L., Tyzack, G. E., Tolman, M., Miller, J. G., et al. (2018). Kir4.1-Dependent Astrocyte-Fast Motor Neuron Interactions Are Required for Peak Strength. Neuron 98, 306.e–319.e. doi: 10.1016/j.neuron.2018.03.010
Kim, J., Hughes, E. G., Shetty, A. S., Arlotta, P., Goff, L. A., Bergles, D. E., et al. (2017). Changes in the excitability of neocortical neurons in a mouse model of amyotrophic lateral sclerosis are not specific to corticospinal neurons and are modulated by advancing disease. J. Neurosci. 37, 9037–9053. doi: 10.1523/JNEUROSCI.0811-17.2017
Koza, P., Beroun, A., Konopka, A., Górkiewicz, T., Bijoch, L., Torres, J. C., et al. (2019). Neuronal TDP-43 depletion affects activity-dependent plasticity. Neurobiol. Dis. 130:104499. doi: 10.1016/j.nbd.2019.104499
Kujirai, T., Caramia, M. D., Rothwell, J. C., Day, B. L., Thompson, P. D., Ferbert, A., et al. (1993). Corticocortical inhibition in human motor cortex. J. Physiol. 471, 501–519. doi: 10.1113/jphysiol.1993.sp019912
Lall, D., Lorenzini, I., Mota, T. A., Bell, S., Mahan, T. E., Ulrich, J. D., et al. (2021). C9orf72 deficiency promotes microglial-mediated synaptic loss in aging and amyloid accumulation. Neuron 109, 2275.e–2291.e. doi: 10.1016/j.neuron.2021.05.020
Lemon, R. N. (2008). Descending pathways in motor control. Annu. Rev. Neurosci. 31, 195–218. doi: 10.1146/annurev.neuro.31.060407.125547
Leroy, F., and Zytnicki, D. (2015). Is hyperexcitability really guilty in amyotrophic lateral sclerosis? Neural Regen. Res. 10, 1413–1415. doi: 10.4103/1673-5374.165308
Lin, Z., Kim, E., Ahmed, M., Han, G., Simmons, C., Redhead, Y., et al. (2021). MRI-guided histology of TDP-43 knock-in mice implicates parvalbumin interneuron loss, impaired neurogenesis and aberrant neurodevelopment in amyotrophic lateral sclerosis-frontotemporal dementia. Brain Commun. 3:fcab114. doi: 10.1093/braincomms/fcab114
Lindau, M., Jelic, V., Johansson, S. E., Andersen, C., Wahlund, L. O., and Almkvist, O. (2003). Quantitative EEG abnormalities and cognitive dysfunctions in frontotemporal dementia and Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 15, 106–114. doi: 10.1159/000067973
Livesey, M. R., Magnani, D., Hardingham, G. E., Chandran, S., and Wyllie, D. J. A. (2016). Functional properties of in vitro excitatory cortical neurons derived from human pluripotent stem cells. J. Physiol. 594, 6573–6582. doi: 10.1113/JP270660
Lomen-Hoerth, C., Anderson, T., and Miller, B. (2002). The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59, 1077–1079. doi: 10.1212/WNL.59.7.1077
Lui, H., Zhang, J., Makinson, S. R., Cahill, M. K., Kelley, K. W., Huang, H. Y., et al. (2016). Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 165, 921–935. doi: 10.1016/j.cell.2016.04.001
Malenka, R. C., and Bear, M. F. (2004). LTP and LTD: An Embarrassment of Riches. Neuron 44, 5–21. doi: 10.1016/J.NEURON.2004.09.012
Martin, L. J., and Chang, Q. (2012). Inhibitory synaptic regulation of motoneurons: A new target of disease mechanisms in amyotrophic lateral sclerosis. Mol. Neurobiol. 45, 30–42. doi: 10.1007/s12035-011-8217-x
Martínez-Silva, M., de, L., Imhoff-Manuel, R. D., Sharma, A., Heckman, C. J., Shneider, N. A., et al. (2018). Hypoexcitability precedes denervation in the large fast-contracting motor units in two unrelated mouse models of ALS. Elife 7:30955. doi: 10.7554/eLife.30955
Maselli, R. A., Wollman, R. L., Leung, C., Distad, B., Palombi, S., Richman, D. P., et al. (1993). Neuromuscular transmission in amyotrophic lateral sclerosis. Muscle Nerve 16, 1193–1203. doi: 10.1002/mus.880161109
McColgan, P., Joubert, J., Tabrizi, S. J., and Rees, G. (2020). The human motor cortex microcircuit: insights for neurodegenerative disease. Nat. Rev. Neurosci. 21, 401–415. doi: 10.1038/s41583-020-0315-1
Menon, P., Geevasinga, N., van den Bos, M., Yiannikas, C., Kiernan, M. C., and Vucic, S. (2017). Cortical hyperexcitability and disease spread in amyotrophic lateral sclerosis. Eur. J. Neurol. 24, 816–824. doi: 10.1111/ene.13295
Menon, P., Higashihara, M., Bos, M., van den, Geevasinga, N., Kiernan, M. C., et al. (2020). Cortical hyperexcitability evolves with disease progression in ALS. Ann. Clin. Transl. Neurol. 7:733. doi: 10.1002/ACN3.51039
Menon, P., Kiernan, M. C., and Vucic, S. (2015). Cortical hyperexcitability precedes lower motor neuron dysfunction in ALS. Clin. Neurophysiol. 126, 803–809. doi: 10.1016/j.clinph.2014.04.023
Meyer, K., Ferraiuolo, L., Miranda, C. J., Likhite, S., McElroy, S., Renusch, S., et al. (2014). Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. U S A. 111, 829–832. doi: 10.1073/pnas.1314085111
Mignogna, M. L., Giannandrea, M., Gurgone, A., Fanelli, F., Raimondi, F., Mapelli, L., et al. (2015). The intellectual disability protein RAB39B selectively regulates GluA2 trafficking to determine synaptic AMPAR composition. Nat. Commun. 6, 1–15. doi: 10.1038/ncomms7504
Mignogna, M. L., Musardo, S., Ranieri, G., Gelmini, S., Espinosa, P., Marra, P., et al. (2021). RAB39B-mediated trafficking of the GluA2-AMPAR subunit controls dendritic spine maturation and intellectual disability-related behaviour. Mol. Psychiatry 2021, 1–19. doi: 10.1038/s41380-021-01155-5
Milnerwood, A. J., and Raymond, L. A. (2010). Early synaptic pathophysiology in neurodegeneration: Insights from Huntington’s disease. Trends Neurosci. 33, 513–523. doi: 10.1016/j.tins.2010.08.002
Mizielinska, S., and Isaacs, A. M. (2014). C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia: Gain or loss of function? Curr. Opin. Neurol. 27, 515–523. doi: 10.1097/WCO.0000000000000130
Mogyoros, I., Kiernan, M. C., Burke, D., and Bostock, H. (1998). Strength-duration properties of sensory and motor axons in amyotrophic lateral sclerosis. Brain 121, 851–859. doi: 10.1093/brain/121.5.851
Mohammadi, B., Kollewe, K., Cole, D. M., Fellbrich, A., Heldmann, M., Samii, A., et al. (2015). Amyotrophic lateral sclerosis affects cortical and subcortical activity underlying motor inhibition and action monitoring. Hum. Brain Mapp. 36, 2878–2889. doi: 10.1002/hbm.22814
Moore, S., Alsop, E., Lorenzini, I., Starr, A., Rabichow, B. E., Mendez, E., et al. (2019). ADAR2 mislocalization and widespread RNA editing aberrations in C9orf72-mediated ALS/FTD. Acta Neuropathol. 138:1999–w. doi: 10.1007/s00401-019-01999-w
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Nasseroleslami, B., Dukic, S., Broderick, M., Mohr, K., Schuster, C., Gavin, B., et al. (2019). Characteristic Increases in EEG Connectivity Correlate with Changes of Structural MRI in Amyotrophic Lateral Sclerosis. Cereb. Cortex 29, 27–41. doi: 10.1093/cercor/bhx301
Naujock, M., Stanslowsky, N., Bufler, S., Naumann, M., Reinhardt, P., Sterneckert, J., et al. (2016). 4-Aminopyridine Induced Activity Rescues Hypoexcitable Motor Neurons from Amyotrophic Lateral Sclerosis Patient-Derived Induced Pluripotent. Stem Cells 34, 1563–1575. doi: 10.1002/stem.2354
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nishida, K., Yoshimura, M., Isotani, T., Yoshida, T., Kitaura, Y., Saito, A., et al. (2011). Differences in quantitative EEG between frontotemporal dementia and Alzheimer’s disease as revealed by LORETA. Clin. Neurophysiol. 122, 1718–1725. doi: 10.1016/j.clinph.2011.02.011
Niu, M., Zheng, N., Wang, Z., Gao, Y., Luo, X., Chen, Z., et al. (2020). RAB39B Deficiency Impairs Learning and Memory Partially Through Compromising Autophagy. Front. Cell Dev. Biol. 8:1508. doi: 10.3389/fcell.2020.598622
Palop, J. J., Chin, J., and Mucke, L. (2006). A network dysfunction perspective on neurodegenerative diseases. Nature 443, 768–773. doi: 10.1038/nature05289
Perkins, E. M., Burr, K., Banerjee, P., Mehta, A. R., Dando, O., Selvaraj, B. T., et al. (2021). Altered network properties in C9ORF72 repeat expansion cortical neurons are due to synaptic dysfunction. Mol. Neurodegener. 16:13. doi: 10.1186/s13024-021-00433-8
Perry, S., Han, Y., Das, A., and Dickman, D. (2017). Homeostatic plasticity can be induced and expressed to restore synaptic strength at neuromuscular junctions undergoing ALS-related degeneration. Hum. Mol. Genet. 26, 4153–4167. doi: 10.1093/hmg/ddx304
Peters, O. M., Cabrera, G. T., Tran, H., Gendron, T. F., McKeon, J. E., Metterville, J., et al. (2015). Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 88, 902–909. doi: 10.1016/j.neuron.2015.11.018
Petkau, T. L., Neal, S. J., Milnerwood, A., Mew, A., Hill, A. M., Orban, P., et al. (2012). Synaptic dysfunction in progranulin-deficient mice. Neurobiol. Dis. 45, 711–722. doi: 10.1016/J.NBD.2011.10.016
Pina-Crespo, J. C., Sanz-Blasco, S., and Lipton, S. A. (2014). Concept of excitotoxicity via glutamate receptors. Handb. Neurotox. 2, 1015–1038. doi: 10.1007/978-1-4614-5836-4_125
Proudfoot, M., Rohenkohl, G., Quinn, A., Colclough, G. L., Wuu, J., Talbot, K., et al. (2016). Altered cortical beta-band oscillations reflect motor system degeneration in amyotrophic lateral sclerosis. Hum. Brain Mapp. 38, 237–254. doi: 10.1002/hbm.23357
Prudencio, M., Belzil, V. V., Batra, R., Ross, C. A., Gendron, T. F., Pregent, L. J., et al. (2015). Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 18:1175. doi: 10.1038/NN.4065
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Rizzu, P., Blauwendraat, C., Heetveld, S., Lynes, E. M., Castillo-Lizardo, M., Dhingra, A., et al. (2016). C9orf72 is differentially expressed in the central nervous system and myeloid cells and consistently reduced in C9orf72, MAPT and GRN mutation carriers. Acta Neuropathol. Commun. 4, 37. doi: 10.1186/s40478-016-0306-7
Rosenblum, L. T., and Trotti, D. (2017). EAAT2 and the molecular signature of amyotrophic lateral sclerosis. Adv. Neurobiol. 16, 117–136. doi: 10.1007/978-3-319-55769-4_6
Rothstein, J. D. (1995). Excitotoxic mechanisms in the pathogenesis of amyotrophic lateral sclerosis. Adv. Neurol. 68, 7–20.
Rothstein, J. D., Martin, L. J., and Kuncl, R. W. (1992). Decreased Glutamate Transport by the Brain and Spinal Cord in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 326, 1464–1468. doi: 10.1056/nejm199205283262204
Rothstein, J. D., Tsai, G., Kuncl, R. W., Clawson, L., Cornblath, D. R., Drachman, D. B., et al. (1990). Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann. Neurol. 28, 18–25. doi: 10.1002/ana.410280106
Saba, L., Viscomi, M. T., Caioli, S., Pignataro, A., Bisicchia, E., Pieri, M., et al. (2016). Altered Functionality, Morphology, and Vesicular Glutamate Transporter Expression of Cortical Motor Neurons from a Presymptomatic Mouse Model of Amyotrophic Lateral Sclerosis. Cereb. Cortex 26, 1512–1528. doi: 10.1093/cercor/bhu317
Sahadevan, S., Hembach, K. M., Tantardini, E., Pérez-Berlanga, M., Hruska-Plochan, M., Megat, S., et al. (2021). Synaptic FUS accumulation triggers early misregulation of synaptic RNAs in a mouse model of ALS. Nat. Commun. 12, 1–17. doi: 10.1038/s41467-021-23188-8
Sahara Khademullah, C., Aqrabawi, A. J., Place, K. M., Dargaei, Z., Liang, X., Pressey, J. C., et al. (2020). Cortical interneuron-mediated inhibition delays the onset of amyotrophic lateral sclerosis. Brain 143, 800–810. doi: 10.1093/brain/awaa034
Sances, S., Bruijn, L. I., Chandran, S., Eggan, K., Ho, R., Klim, J. R., et al. (2016). Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat. Neurosci. 19, 542–553. doi: 10.1038/nn.4273
Sareen, D., O’Rourke, J. G., Meera, P., Muhammad, A. K. M. G., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5:208ra149. doi: 10.1126/scitranslmed.3007529
Sasaki, S., and Maruyama, S. (1994). Immunocytochemical and ultrastructural studies of the motor cortex in amyotrophic lateral sclerosis. Acta Neuropathol. 87, 578–585. doi: 10.1007/BF00293318
Scekic-Zahirovic, J., Sanjuan-Ruiz, I., Kan, V., Megat, S., De Rossi, P., Dieterlé, S., et al. (2021). Cytoplasmic FUS triggers early behavioral alterations linked to cortical neuronal hyperactivity and inhibitory synaptic defects. Nat. Commun. 12, 1–19. doi: 10.1038/s41467-021-23187-9
Schanz, O., Bageac, D., Braun, L., Traynor, B. J., Lehky, T. J., and Floeter, M. K. (2016). Cortical hyperexcitability in patients with C9ORF72 mutations: Relationship to phenotype. Muscle Nerve 54, 264–269. doi: 10.1002/mus.25047
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Selvaraj, B. T., Livesey, M. R., Zhao, C., Gregory, J. M., James, O. T., Cleary, E. M., et al. (2018). C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 9, 1–14. doi: 10.1038/s41467-017-02729-0
Sephtona, C. F., Tangc, A. A., Kulkarnia, A., Westa, J., Brooksa, M., Stubblefielda, J. J., et al. (2014). Activity-dependent FUS dysregulation disrupts. Proc. Natl. Acad. Sci. U S A. 111, E4769–E4778. doi: 10.1073/pnas.1406162111
Serio, A., Bilican, B., Barmada, S. J., Ando, D. M., Zhao, C., Siller, R., et al. (2013). Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. U S A. 110, 4697–4702. doi: 10.1073/pnas.1300398110
Shao, Q., Liang, C., Chang, Q., Zhang, W., Yang, M., and Chen, J. F. (2019). C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose-dependent manner. Acta Neuropathol. Commun. 7:32. doi: 10.1186/s40478-019-0685-7
Shaw, P. J. (2005). Molecular and cellular pathways of neurodegeneration in motor neurone disease. J. Neurol. Neurosurg. Psychiatry 76, 1046–1057. doi: 10.1136/jnnp.2004.048652
Shi, Y., Hung, S. T., Rocha, G., Lin, S., Linares, G. R., Staats, K. A., et al. (2019). Identification and therapeutic rescue of autophagosome and glutamate receptor defects in C9ORF72 and sporadic ALS neurons. JCI Insight 4:127736. doi: 10.1172/jci.insight.127736
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W. H., Hung, S. T., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325. doi: 10.1038/nm.4490
Shibuya, K., Park, S. B., Geevasinga, N., Menon, P., Howells, J., Simon, N. G., et al. (2016). Motor cortical function determines prognosis in sporadic ALS. Neurology 87, 513–520. doi: 10.1212/WNL.0000000000002912
Šišková, Z., Justus, D., Kaneko, H., Friedrichs, D., Henneberg, N., Beutel, T., et al. (2014). Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of alzheimer’s disease. Neuron 84, 1023–1033. doi: 10.1016/j.neuron.2014.10.024
Smeyers, J., Banchi, E. G., and Latouche, M. (2021). C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell Neurosci. 15:109. doi: 10.3389/fncel.2021.661447
Snowden, J. S., Rollinson, S., Thompson, J. C., Harris, J. M., Stopford, C. L., Richardson, A. M. T., et al. (2012). Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 135, 693–708. doi: 10.1093/brain/awr355
Spalloni, A., Origlia, N., Sgobio, C., Trabalza, A., Nutini, M., Berretta, N., et al. (2011). Postsynaptic alteration of NR2A subunit and defective autophosphorylation of alpha CaMKII at Threonine-286 contribute to abnormal plasticity and morphology of upper motor neurons in presymptomatic SOD1 G93A mice, a murine model for amyotrophic lateral scl. Cereb. Cortex 21, 796–805. doi: 10.1093/cercor/bhq152
Staats, K. A., Seah, C., Sahimi, A., Wang, Y., Koutsodendris, N., Lin, S., et al. (2019). Small molecule inhibition of PIKFYVE kinase rescues gain- and loss-of-function C9ORF72 ALS/FTD disease processes in vivo. bioRxiv 2019:685800. doi: 10.1101/685800
Starr, A., and Sattler, R. (2018). Synaptic dysfunction and altered excitability in C9ORF72 ALS/FTD. Brain Res. 1693, 98–108. doi: 10.1016/j.brainres.2018.02.011
Styr, B., and Slutsky, I. (2018). Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nat. Neurosci. 21, 463–473. doi: 10.1038/s41593-018-0080-x
Suminaite, D., Lyons, D. A., and Livesey, M. R. (2019). Myelinated axon physiology and regulation of neural circuit function. Glia 67, 2050–2062. doi: 10.1002/glia.23665
Talbot, P. R., Goulding, P. J., Lloyd, J. J., Snowden, J. S., Neary, D., and Testa, H. J. (1995). Inter-relation between “classic” motor neuron disease and frontotemporal dementia: Neuropsychological and single photon emission computed tomography study. J. Neurol. Neurosurg. Psychiatry 58, 541–547. doi: 10.1136/jnnp.58.5.541
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., et al. (2010). Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 62, 405–496. doi: 10.1124/pr.109.002451
Tsuiji, H., Inoue, I., Takeuchi, M., Furuya, A., Yamakage, Y., Watanabe, S., et al. (2017). TDP-43 accelerates age-dependent degeneration of interneurons. Sci. Rep. 7:14966–w. doi: 10.1038/s41598-017-14966-w
Turrigiano, G. (2012). Homeostatic synaptic plasticity: Local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 4:a005736. doi: 10.1101/cshperspect.a005736
Udagawa, T., Fujioka, Y., Tanaka, M., Honda, D., Yokoi, S., Riku, Y., et al. (2015). FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat. Commun. 6, 1–13. doi: 10.1038/ncomms8098
Umpierre, A. D., and Wu, L. J. (2021). How microglia sense and regulate neuronal activity. Glia 69, 1637–1653. doi: 10.1002/glia.23961
Van Den Bosch, L., Van Damme, P., Bogaert, E., and Robberecht, W. (2006). The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 1762, 1068–1082. doi: 10.1016/j.bbadis.2006.05.002
Van Zundert, B., Peuscher, M. H., Hynynen, M., Chen, A., Neve, R. L., Brown, R. H., et al. (2008). Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J. Neurosci. 28, 10864–10874. doi: 10.1523/JNEUROSCI.1340-08.2008
Vucic, S., Ziemann, U., Eisen, A., Hallett, M., and Kiernan, M. C. (2013). Transcranial magnetic stimulation and amyotrophic lateral sclerosis: pathophysiological insights. J. Neurol. Neurosurg. Psychiatry 84, 1161–1170. doi: 10.1136/JNNP-2012-304019
Wainger, B. J., and Cudkowicz, M. E. (2015). Cortical hyperexcitability in amyotrophic lateral sclerosis C9ORF72 repeats. JAMA Neurol. 72, 1235–1236. doi: 10.1001/jamaneurol.2015.2197
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Steve, S. W., Berry, J. D., et al. (2014). Intrinsic membrane hyperexcitability of ALS patient-derived motor neurons. Cell Rep. 7, 1–11. doi: 10.1016/j.celrep.2014.03.019.Intrinsic
Wainger, B. J., Macklin, E. A., Vucic, S., McIlduff, C. E., Paganoni, S., Maragakis, N. J., et al. (2021). Effect of Ezogabine on Cortical and Spinal Motor Neuron Excitability in Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. JAMA Neurol. 78, 186–196. doi: 10.1001/jamaneurol.2020.4300
Weskamp, K., Tank, E. M., Miguez, R., McBride, J. P., Gómez, N. B., White, M., et al. (2020). Shortened TDP43 isoforms upregulated by neuronal hyperactivity drive TDP43 pathology in ALS. J. Clin. Invest. 130, 1139–1155. doi: 10.1172/JCI130988
West, R. J. H., Sharpe, J. L., Voelzmann, A., Munro, A. L., Hahn, I., Baines, R. A., et al. (2020). Co-expression of C9orf72 related dipeptide-repeats over 1000 repeat units reveals age-A nd combination-specific phenotypic profiles in Drosophila. Acta Neuropathol. Commun. 8, 1–19. doi: 10.1186/s40478-020-01028-y
Westergard, T., McAvoy, K., Russell, K., Wen, X., Pang, Y., Morris, B., et al. (2019). Repeat-associated non- AUG translation in C9orf72- ALS / FTD is driven by neuronal excitation and stress. EMBO Mol. Med. 11:201809423. doi: 10.15252/emmm.201809423
Williams, K. L., Fifita, J. A., Vucic, S., Durnall, J. C., Kiernan, M. C., Blair, I. P., et al. (2013). Pathophysiological insights into ALS with C9ORF72 expansions. J. Neurol. Neurosurg. Psychiatry 84, 931–935. doi: 10.1136/jnnp-2012-304529
Wu, L. S., Cheng, W. C., Chen, C. Y., Wu, M. C., Wang, Y. C., Tseng, Y. H., et al. (2019). Transcriptomopathies of pre- and post-symptomatic frontotemporal dementia-like mice with TDP-43 depletion in forebrain neurons. Acta Neuropathol. Commun. 7:50. doi: 10.1186/s40478-019-0674-x
Xiao, S., McKeever, P. M., Lau, A., and Robertson, J. (2019). Synaptic localization of C9orf72 regulates post-synaptic glutamate receptor 1 levels. Acta Neuropathol. Commun. 7:161. doi: 10.1186/s40478-019-0812-5
Xu, W., and Xu, J. (2018). C9orf72 dipeptide repeats cause selective neurodegeneration and cell-autonomous excitotoxicity in Drosophila glutamatergic neurons. J. Neurosci. 38, 7741–7752. doi: 10.1523/JNEUROSCI.0908-18.2018
Zhang, K., Donnelly, C. J., Haeusler, A. R., Grima, J. C., Machamer, J. B., Steinwald, P., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. doi: 10.1038/nature14973
Zhang, W., Zhang, L., Liang, B., Schroeder, D., Zhang, Z. W., Cox, G. A., et al. (2016). Hyperactive somatostatin interneurons contribute to excitotoxicity in neurodegenerative disorders. Nat. Neurosci. 19, 557–559. doi: 10.1038/nn.4257
Zhang, Z., Almeida, S., Lu, Y., Nishimura, A. L., Peng, L., Sun, D., et al. (2013). Downregulation of MicroRNA-9 in iPSC-Derived Neurons of FTD/ALS Patients with TDP-43 Mutations. PLoS One 8:0076055. doi: 10.1371/journal.pone.0076055
Zhao, C., Devlin, A. C., Chouhan, A. K., Selvaraj, B. T., Stavrou, M., Burr, K., et al. (2020). Mutant C9orf72 human iPSC-derived astrocytes cause non-cell autonomous motor neuron pathophysiology. Glia 68, 1046–1064. doi: 10.1002/glia.23761
Keywords: C9ORF72, neuron, synaptic, excitability, glutamate, physiology, ALS (amyotrophic lateral sclerosis), FTD (frontotemporal dementia)
Citation: Pasniceanu IS, Atwal MS, Souza CDS, Ferraiuolo L and Livesey MR (2021) Emerging Mechanisms Underpinning Neurophysiological Impairments in C9ORF72 Repeat Expansion-Mediated Amyotrophic Lateral Sclerosis/Frontotemporal Dementia. Front. Cell. Neurosci. 15:784833. doi: 10.3389/fncel.2021.784833
Received: 28 September 2021; Accepted: 10 November 2021;
Published: 15 December 2021.
Edited by:
Annakaisa Haapasalo, University of Eastern Finland, FinlandReviewed by:
Morwena Latouche, Paris Sciences et Lettres University, FranceCopyright © 2021 Pasniceanu, Atwal, Souza, Ferraiuolo and Livesey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew R. Livesey, TS5SLkxpdmVzZXlAU2hlZmZpZWxkLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.