 Griffith Kyle Otazu
Griffith Kyle Otazu Mojtaba Dayyani
Mojtaba Dayyani Behnam Badie*
Behnam Badie*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 16 December 2021
Sec. Cellular Neuropathology
Volume 15 - 2021 | https://doi.org/10.3389/fncel.2021.770472
This article is part of the Research Topic Brain Tumors and Neuroinflammation View all 6 articles
Gliomas, the most common form of brain cancer, can range from relatively slow-growing low-grade to highly aggressive glioblastoma that has a median overall survival of only 15 months despite multimodal standard therapy. Although immunotherapy with checkpoint inhibitors has significantly improved patient survival for some cancers, to date, these agents have not shown consistent efficacy against malignant gliomas. Therefore, there is a pressing need to better understand the impact of host inflammatory responses on the efficacy of emerging immunotherapy approaches for these resistant tumors. RAGE is a multi-ligand pattern recognition receptor that is activated in various inflammatory states such as diabetes, Alzheimer’s disease, cystic fibrosis, and cancer. Low levels of RAGE can be found under normal physiological conditions in neurons, immune cells, activated endothelial, and vascular smooth muscle cells, but it is over-expressed under chronic inflammation due to the accumulation of its ligands. RAGE binds to a range of damage-associated molecular pattern molecules (DAMPs) including AGEs, HMGB1, S100s, and DNA which mediate downstream cellular responses that promote tumor growth, angiogenesis, and invasion. Both in vitro and in vivo studies have shown that inhibition of RAGE signaling can disrupt inflammation and cancer progression and metastasis. Here, we will review our current understanding of the role of RAGE pathway on glioma progression and how it could be exploited to improve the efficacy of immunotherapy approaches.
Gliomas are classified as the most common type of brain cancer, accounting for about 80% of all diagnosed malignant brain tumors (Behnan et al., 2019). Based on the recent updates of cIMPACT−Utrecht committee on CNS tumor classification, gliomas are categorized into four grades (grade 1–4), among which grades 4s, and diffuse or anaplastic astrocytic gliomas with IDH-wildtype 4 are considered as “glioblastoma” (Gonzalez Castro and Wesseling, 2020; Louis et al., 2020). The overall age-adjusted incidence rates for all gliomas range from 4.67 to 5.73 per 100,000 persons, while for glioblastoma it ranges from 0.59 to 3.69 per 100,000 persons (Ostrom et al., 2014). For a low-grade glioma, the 10-year survival rate is 47% with a median survival time of 11.6 years (Xu et al., 2020) whereas patients with glioblastoma have a median overall survival of only 15 months (Stupp et al., 2005; Tamimi and Juweid, 2017).
Most patients with gliomas undergo surgical resection of the tumor, followed by radiation and chemotherapy (Davis, 2016; Banerjee et al., 2021). The goal of the surgery is to remove most of tumor without damaging the surrounding normal brain tissue. For low-grade gliomas, the extent of resection has been associated with better overall survival (Young et al., 2015; Morshed et al., 2019); but for higher-grade gliomas, survival benefit from aggressive surgery is modest (Young et al., 2015). Following surgery, patients are then treated with radiation and chemotherapy (typically temozolomide) as an adjuvant treatment (Banerjee et al., 2021; Singh et al., 2021). While advancements in these therapies have marginally extended the overall survival for patients with glioblastoma, tumor relapse is inevitable. A number of factors account for glioblastoma resistance to standard therapies. These include: tumor invasion into normal brain tissue, resistance to chemo/radiation therapy, molecular heterogeneity, and immunosuppressive stromal cells that promote evasion of host anti-tumor immune responses (Razavi et al., 2016; Medikonda et al., 2021).
Among the glioma-associated stromal cells, infiltrating microglia (MG) and macrophages (MP), (also known as tumor-associated macrophages or TAMs) are well known due to their involvement in glioma escape from anti-angiogenic agents (Piao et al., 2012; Chaudhary et al., 2021). TAMs, as a component of the innate immune system, are derived from both resident brain MG and myeloid-derived monocytes. A wide range of pattern recognition receptors that are expressed by TAMs, continuously monitor changes in tumor microenvironment. Among these, receptor for advanced glycation end-products (RAGE) is known as a membrane protein that binds glycosylated macromolecules and implicated in various human diseases (Ramasamy et al., 2009; Sorci et al., 2013; Shen et al., 2020). For instance, in conditions like diabetes, chronic inflammation or neurodegenerative disorders, RAGE expression is increased dramatically in vasculature, hematopoietic cells, and the central nervous system (CNS), while in normal physiological conditions it is expressed at high levels in the lungs, and at lower levels in different cell types including neurons, immune cells, activated endothelial and vascular smooth muscle cells (Sorci et al., 2013). Coupling of RAGE by its ligands activates several downstream signaling cascades that ultimately lead to the upregulation of cytokines, chemokines, adhesion molecules, and other molecular pathways involved in cell proliferation, differentiation, migration, survival, phagocytosis, and authophagy via the activation of NF-κB, ERK1/2, p38 and STAT3 (Sorci et al., 2013; Erusalimsky, 2021). Activation of these downstream regulatory pathways is one reason that RAGE signaling is considered to be involved in tumorigenesis. In this review, we provide an overview of the role of RAGE and its ligands on inflammatory responses to gliomas.
Glioblastoma is known to foster an inflammatory immune response that could shift the tumor microenvironment into a pro-tumorigenic milieu (Fan et al., 2020). Acute and transient inflammation may inhibit tumor growth (Fan et al., 2020) by upregulating pro-inflammatory cytokines such as TNFα, IL-1β, and IL-6 that are part of the initial inflammatory cascade and recruit other downstream targets to enhance anti-tumor responses. However, if inflammation becomes a chronic occurrence, the same inflammatory processes can exhaust the immune system’s ability to fend off glioma cells. These complex microenvironmental forces, combined with mutations in tumor oncogenes create a tumor milieu that can evade immune recognition that spreads throughout the brain parenchyma. While the presence of pro-inflammatory cytokines clearly affects the immune system in gliomas, their expression also contributes to the development of various neoplastic processes. For example, TNFα has been implicated as an inducer of angiogenesis, mainly through EGFR upregulation (Al-Kharboosh et al., 2020). Also, TNFα has been shown to facilitate glioma invasion, not by upregulating MMP2 or MMP9, but through modulation of MEK-ERK1/2 pathway (Ramaswamy et al., 2019). Similarly, increased expression of both IL-1β and IL-6 has been correlated with poorer survival rates in patients with glioblastoma (Al-Kharboosh et al., 2020), possibly through activation of STAT3 signaling and/or, NF-κB pathway. TNFα and IL-1β are known activators of NF-κB, and promote downstream inflammatory pathways by inducing NLRP3 inflammasome (Liu et al., 2017). IL-6, on the other hand, is not an activator of NF-κB, but is a cytokine released upon NF-κB activation (Mostofa et al., 2017). These complex inflammatory responses, although well-characterized in other inflammatory or pathological conditions, continue to be analyzed in glioblastoma models in order to improve the efficacy of immunotherapy approaches. The goal of this review is to examine how RAGE and its ligands alter glioma inflammatory responses and anti-tumor immune response.
RAGE is a multiligand receptor that is heavily involved in inflammatory responses. This receptor is classified as a transmembrane protein that belongs to the immunoglobulin (Ig) superfamily (Mulrennan et al., 2015). RAGE is also considered a pattern recognition receptor (PRR) due to its ability to recognize multiple classes of molecules (Chavakis et al., 2004). The most well-categorized ligands that interact with RAGE are AGEs, HMGB1, and members of the S100 family (Mulrennan et al., 2015), and modulate a range of immunological and inflammatory signaling pathways by activating RAGE.
RAGE is typically expressed on membranes of immune cells such as monocytes/macrophages, neutrophils, lymphocytes, and dendritic cells (Chuah et al., 2013). Additionally, RAGE is observed on fully differentiated neurons, and prolongs their survival during embryonic development (Lee and Park, 2013). During embryogenesis, the expression of both RAGE and its ligands is the highest, but tapers off to baseline levels in mature cells. The other well-characterized instance of RAGE activation is in chronic inflammation (Schmidt et al., 2001), which underscores its role as an inflammatory modulator.
RAGE receptors are composed of three subunits: an extracellular region that contains a V-type region that interacts with ligands, a transmembrane region, and a cytoplasmic region that allows signal transduction in the cell (Lee and Park, 2013). In addition to full-length RAGE, there are RAGE variants that are generated through different splicing patterns. These variants include N-truncated, dominant-negative, and soluble RAGE. The soluble RAGE variant (sRAGE) is of particular therapeutic interest. This isoform arises through either cleavage by proteases or by mRNA splicing (Chuah et al., 2013). As a result, sRAGE lacks a transmembrane domain and is capable of detaching from the cell surface but still contains its ligand-binding V-domain which allows it to potentially bind RAGE ligands and antagonize RAGE activation (Lee and Park, 2013).
RAGE is well-positioned to influence both inflammatory responses (due to its expression on a wide array of immune cells) and signaling processes in the CNS as a result of its localization in neurons and their proximity to other cell types within the brain. One common characteristic of RAGE and its ligands is that they participate in a positive feedback loop where upregulation of ligands will stimulate greater expression of RAGE receptors, thereby potentiating receptor-ligand interactions (Schmidt et al., 1999). This positive feedback loop has been observed in neurons expressing RAGE, and in turn leads to the activation of ERK1/2 (Kierdorf and Fritz, 2013). Furthermore, RAGE engagement can lead to the activation of the MAPK pathway and the production of reactive oxygen species (ROS), which are known to cause cell death. In examining Jurkat E6 leukemia cells and blood-derived mononuclear cells (monocytes, macrophages, and dendritic cells), one of the earlier studies found that high levels of S100B (RAGE ligand secreted by reactive astrocytes) potentiates RAGE expression and triggers the release of pro-inflammatory cytokines like TNFα and IL-1β (Hofmann et al., 1999; Figure 1).
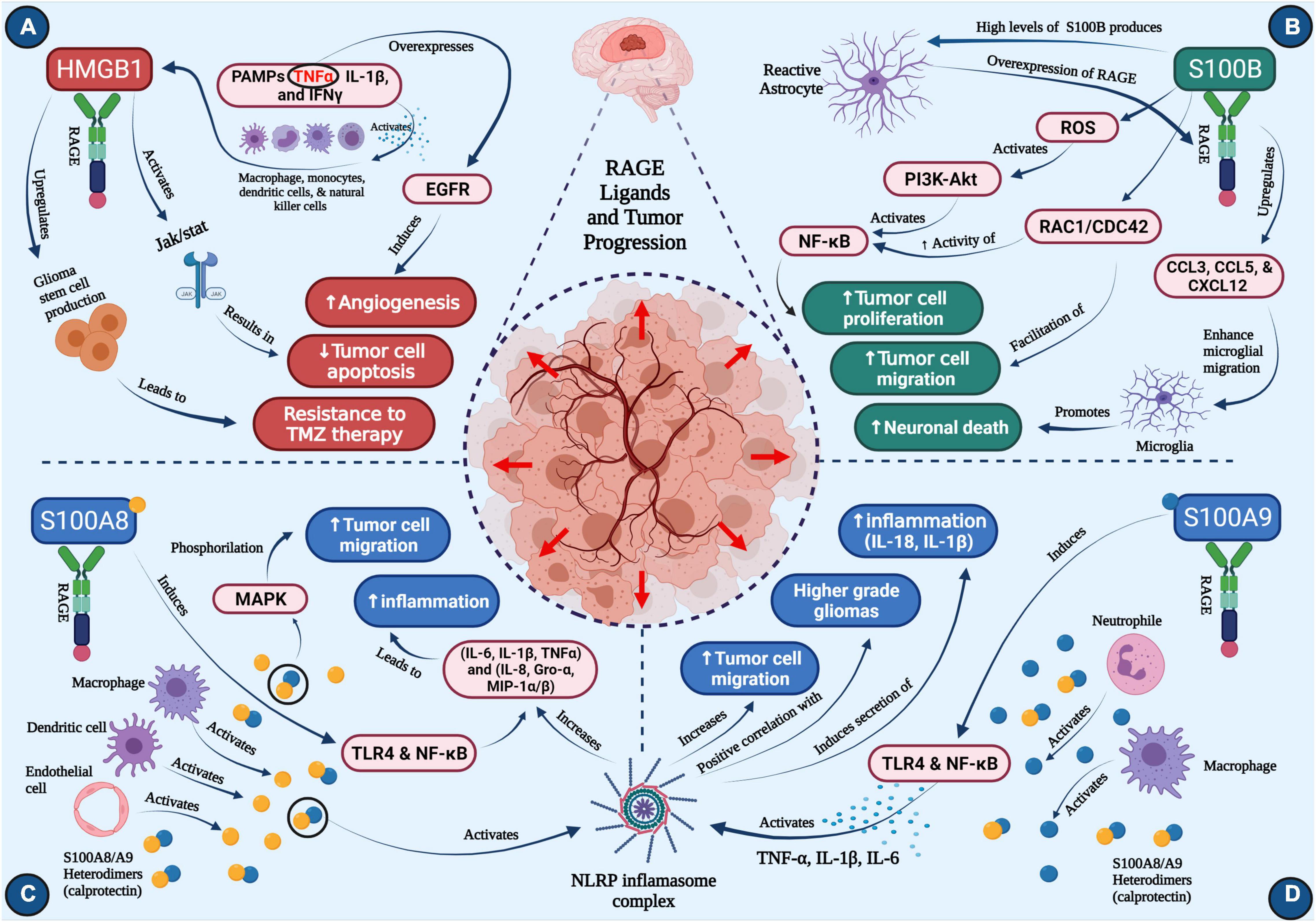
Figure 1. Roles of RAGE ligands in glioma progression (A) HMGB1, (B) S100B, (C) S100A8, (D) S100A9. Concept map depicts the major roles of four critical ligands of RAGE on glioma progression. Each section depicts pathways that activate downstream cascades important in neuro-inflammation, tumor cell migration, neuronal death, angiogenesis, temozolomide resistance, ultimately resulting in tumor progression (created with BioRender.com).
The most well-known ligands of RAGE are often characterized as a damage-associated molecular patterns or DAMP proteins, which alert the immune system of infection, cellular damage, or other injury (Roh and Sohn, 2018). In contrast to pathogen-associated molecular patterns (PAMPs) RAGE ligands are endogenous signals and do not stem from outside the body. As DAMPs, the role of RAGE ligands in inflammation is well-known, but in gliomas, their role continues to be elucidated. As mentioned, gliomas exploit the host inflammatory responses to promote their migration/progression, and thus, understanding the role of each RAGE ligand on tumor microenvironment will be critical in developing more effective therapies.
The high-mobility group box 1 (HMGB1) protein belongs to the HMGB superfamily and is expressed within the nucleus as a mediator of transcriptional processes. Additionally, HMGB1 is found throughout the cytoplasm where it modulates autophagy (Magna and Pisetsky, 2014). However, its interaction with RAGE becomes most pertinent upon its translocation outside the cell. Extracellular HMGB1 assumes the role of a DAMP molecule (Harris et al., 2012), and in doing so, becomes a key regulator of immune processes through upregulation of pro-inflammatory cytokines and chemokines.
HMGB1 translocation typically occurs in the context of inflammation or immunogenicity, and is linked to disease states in which these processes are common. More specifically, HMGB1 is known to be released into extracellular space upon the body’s immune response to foreign agents (Andersson and Tracey, 2011). One well-characterized route that enables this translocation is the activation of the innate immune cells, such as macrophages, monocytes, dendritic cells, and natural killer (NK) cells, by the sensing of PAMPs or pro-inflammatory cytokines like TNFα, IL-1β, and IFNγ (Figure 1A). LPS is another stimulant that is capable of inducing prolonged secretion of HMGB1 into extracellular space, following an increase in HMGB1 mRNA expression (Andersson and Tracey, 2011; Magna and Pisetsky, 2014).
The other mechanism of HMGB1 translocation into the extracellular space is through cell death (Yang et al., 2020), however, the specific mechanism of cell death is important in this process. In necrosis, HMGB1 can be passively released and displays a heightened mobility within the nucleus that, when paired with its relatively weak bonding with chromatin, enabled HMGB1 to relocate to the outside of the cell (Scaffidi et al., 2002; Xue et al., 2021). Conversely, the opposite may occur in apoptosis where HMGB1 intranuclear mobility is suppressed (Scaffidi et al., 2002). Based on these observations, it seems that the increased presence of HMGB1 through translocation may serve as a more reliable indicator of pathogenic processes since such events are usually characterized by necrosis instead of apoptosis (which is more commonly associated with physiological responses). For cells that do not express HMGB1, their death does not stimulate the production of inflammatory cytokines (Rovere-Querini et al., 2004). These observations suggest that HMGB1 plays a crucial role within immune responses due to its ability to mediate inflammation in the context of necrosis, a hallmark of glioblastoma.
Once HMGB1 has translocated to extracellular space, it exerts its tumorigenic properties. While well characterized in its ability to bind to RAGE, HMGB1 can also interact with TLR-2, TLR-4, and TLR-9 (Magna and Pisetsky, 2014; Xue et al., 2021). The RAGE-HMGB1 axis is a major aspect of immune signaling in pathogenic conditions like glioma and inflammatory diseases. However, by binding to multiple toll-like receptors, HMGB1 can mediate a greater number of pro-inflammatory pathways. For example, TLR-4 engagement activates TNF-α release from macrophages (Yang et al., 2010; Xue et al., 2021). Another HMGB1 signaling pathway is the activation of JAK/STAT (Figure 1A), which enables modulation of cell growth and differentiation, and migration of immune cells (Seidu et al., 2017). Increased expression of STAT has been known to inhibit apoptosis of tumor cells, and therefore, HMGB1 activation of STAT is considered pro-tumorigenic. Other potential downstream HMGB1 targets include MAPK, p38, and NF-κB (Xue et al., 2021). HMGB1 not only induces the release of pro-inflammatory cytokines, but synergizes with LPS by binding to RAGE (Qin et al., 2009; Rauvala and Rouhiainen, 2010).
Due to its implications in neuro-inflammation, HMGB1 has been considered as a therapeutic target in gliomas. Hong et al. (2019) examined the effects of HMGB1 on the efficacy of oncolytic herpes simplex virus (oHSV), and demonstrated that the use of HMGB1-blocking antibodies increased the survival of glioma-bearing animals. Additionally, they analyzed the activation of tumor endothelial cells by showing significant decrease in their permeability with anti-HMGB1 antibodies. Since endothelial cell activation is positively correlated with cerebral edema, they observed a decrease in cerebral edema in mice treated with both oHSV and anti-HMGB1 antibodies compared to mice treated with a control antibody.
HMGB1 has also been implicated as a key mediator of glioma resistance to TMZ (Gao et al., 2021). As previously mentioned, TMZ resistance is a common setback in treating patients with malignant gliomas and is an inherent characteristic that ultimately drives relapse of primary neoplasms. A well-characterized mechanism by which treatment resistance occurs is through the production and differentiation of glioma stem cells (GSCs). Classified as tumor-initiating cells, GSCs can not only stimulate tumor growth, but they may also contribute to treatment failure due to their resistance to both drugs and radiotherapy (Auffinger et al., 2014; Garnier et al., 2018). Further studies revealed that HMGB1 upregulates GSC production, and thereby resistance to TMZ, via activation of the Wnt/β-catenin pathway (Gao et al., 2021). This work suggests that successful targeting of HMGB1 may improve glioma treatment responses to TMZ (Figure 1A).
The S100 protein family consists of 25 unique members, each of which contributes to a multitude of processes ranging from maintenance of calcium balance and cell growth to migration (Chen et al., 2014; Allgöwer et al., 2020). However, in the context of this review, the behavior of S100 proteins as cytokine-like agents is of particular interest, especially within RAGE signaling pathways. RAGE interactions with S100B and S100A9 are well characterized within glioma, but in addition to these two proteins, 11 other S100 members have also been demonstrated to interact with RAGE (Leclerc et al., 2009).
According to The Human Protein Atlas, the brain is one of a few organs that exhibits high localization of S100B (Donato et al., 2013). At basal physiological states, S100B, has been shown to be neuroprotective. However, increased levels of S100B have been shown to upregulate pro-inflammatory pathways that can play a role in the pathogenesis of not only gliomas but also Alzheimer’s and epilepsy (Bianchi et al., 2011). Elevated S100B concentrations are typically caused by brain injury, which leads to subsequent release of S100B into extracellular space. As a result, S100B has been proposed to act as a DAMP (Villarreal et al., 2014), much like HMGB1.
RAGE-S100B interaction can activate the PI3K-AKT signaling pathway (Allgöwer et al., 2020), thus improving survival of neuroblastoma cell (Leclerc et al., 2007). In microglia, NF-κB (an essential regulator of inflammatory responses) is also a downstream target of S100B that is mediated by RAGE (Figure 1B), suggesting its role in promoting neuro-inflammation following brain injury (Leclerc et al., 2009). Finally, S100B upregulates CCL3, CCL5, and CXCL12 chemokines in a RAGE-dependent manner (Bianchi et al., 2011; Figure 1B), which then increases microglia trafficking into the brain.
As previously mentioned, astrocytes are a primary producer of S100B, and as such, may be involved in the deregulation of S100B expression and activity in the tumor-adjacent brain or gliomas. As tumors that arise from astrocytes, most astrocytomas express high levels of S100B. Cultured astrocytes have been shown to harness a variety of molecular pathways upon exposure to S100B. Both NF-κB and AKT were activated, but so were other RAGE-dependent pathways like Rac1 and CDC42 (Villarreal et al., 2014). These two proteins were required for S100B-mediated to cause astroglial stellation. The outgrowth of astrocytic cells then enabled cell motility, as evidenced by increased invasion in a wound-healing assay.
The link between S100B and glioma is being investigated by our group. One of our first studies that analyzed S100B in relation to gliomas, found that S100B modulated STAT3 activity in microglia (Zhang et al., 2011; Wang et al., 2013). Also, S100B fostered a cellular microenvironment that upregulates the CCL2 chemokine, allowing for migration of TAMs into gliomas (Wang et al., 2013). We also confirmed that increased S100B expression led to RAGE upregulation, thus confirming the presence of a positive feedback loop between RAGE and its ligands in gliomas. Further studies by our group showed that inhibiting S100B via administration of duloxetine (an SSNRI) abrogated glioma growth and reduced TAM infiltration (Gao et al., 2018). In addition, it has been demonstrated that poor prognosis in recurrent glioma patients correlates with high S100B levels (Holla et al., 2016). Together, these findings suggest S100B to be an important modulator of glioblastoma microenvironment and potentially be used as a therapeutic target.
S100A8 and S100A9 are another pair of members of the S100 calcium-binding family that, like S100B, are characterized as common RAGE ligands. These two S100 subtypes exist predominantly in a heterodimeric form (which is also known as calprotectin) upon release from neutrophils and monocytes (Wang et al., 2018). The S100A8/S100A9 heterodimer form is much more stable than S100A8 and S100A9 alone, however, it has also been observed that S100A9 homodimers are able to form in inflammatory environments and maintain their role in regulating inflammation (Riva et al., 2013; Mondet et al., 2021).
Both S100A8 and S100A9 are constitutively expressed by myeloid cell types, and may act as alarmins upon onset of pathological conditions, which include secretion by necrotic cells or activated immune cells (Crowe et al., 2019). S100A8 activation is also observed in cells involved in inflammatory responses, namely macrophages, dendritic cells, and endothelial cells (Wang et al., 2018; Figure 1C). Similarly, S100A9 is highly expressed in neutrophils and macrophages (Figure 1D). Interestingly, S100A8 appears to foster myeloid cell differentiation and inhibit telomerase activity, but S100A9 actually opposes the S100A8-induced reduction in telomerase activity, in addition to inhibiting myeloid differentiation (Donato et al., 2013; Mondet et al., 2021). This implies a potential regulatory interaction between these two proteins in pathological conditions.
One established downstream mediator of S100A8/S100A9 signaling is MAPK, which upon phosphorylation can induce tumor cell migration (Srikrishna, 2012); this is of particular interest because S100A8 is a chemotactic agent. Much like S100B, these two S100A subtypes are also integrated into the inflammatory response as DAMP molecules. The regulation of pro-inflammatory cytokine release is also well established, as S100A8 and S100A9 bind to TLR4 and activate NF-κB (Mondet et al., 2021). S100A8/S100A9 are also implicated in the activation of the NLRP3 inflammasome (Simard et al., 2013), which has been shown to play a role in glioma progression.
The S100A8/S100A9 subtypes may be promising biomarkers for glioblastoma. Not only were transcript levels of these genes upregulated in higher-grade gliomas, but overall survival rates were also lower in patients with greater expression of either form (Kan et al., 2020). Further studies have corroborated the presence of increased S100A8 and S100A9 levels in glioblastoma (Popescu et al., 2014; Arora et al., 2019). However, despite showing greater expression, serum levels of S100A9 were not a sufficient prognostic measure of glioblastoma at higher transcript levels, while only S100A8 maintained a significant correlation as a marker (Arora et al., 2019). One potential limitation that may affect the utility of S100A8 as a glioblastoma marker is its overexpression in other neurological and inflammatory diseases (Arora et al., 2019).
As alluded to previously, S100A8/S100A9 are implicated in the activation of NLRP3 inflammasome. The inflammasome unit is known to respond to various pathological conditions, such as bacterial and viral infections and DAMP-mediated inflammation (Swanson et al., 2019). Within inflammatory responses, NLRP3’s role centers around inducing the maturation and subsequent secretion of pro-inflammatory cytokines like IL-1β and IL-18 (Lamkanfi and Dixit, 2014). Analysis of tumor protein expression revealed a significantly positive correlation between NLRP3 levels and WHO glioma grade (Yin et al., 2018). Activation of NLRP3 expression by S100A8/S100A9 inhuman PBMCs increased the secretion of pro-inflammatory cytokines (IL-6, IL-1β, TNFα) and chemokines (IL-8, Gro-α, MIP-1α/β) that are prevalent in gliomas (Simard et al., 2013). Further research into the interactions between S100A8/S100A9, NLRP3, and common inflammatory cytokines may provide potential therapeutic targets for gliomas (Figures 1C,D).
Glioblastomas, in general, are considered to be immunologically “cold” tumors and not prone to immunotherapy. Although immunotherapies have improved the management of many solid tumors and hematologic malignancies, phase III studies of PD-1 checkpoint inhibition in both newly diagnosed and recurrent glioblastoma patients showed no efficacy (Reardon et al., 2020). Factors that have been proposed for glioblastoma resistance to checkpoint inhibitors include: high tumor heterogeneity, low mutational burden, systemic immunosuppression, and local immune dysfunction (Razavi et al., 2016; Medikonda et al., 2021). One of the hallmarks of the glioblastoma local immune dysfunction is T cell exhaustion, in which recurrent or prolonged antigen exposure impair T cell anti-tumor function (Medikonda et al., 2021). Furthermore, glioblastomas upregulate multiple immune checkpoints such as PD-1, TIM-3, LAG-3, and CTLA-4, that facilitate T cell exhaustion (Woroniecka et al., 2018). Also, recent evidence indicates a relation between RAGE ligands and T cell exhaustion. In a polytrauma rat model study investigating the association of HMGB1 levels in mediating dysregulated immune responses and its effects on the cellular levels of RAGE and toll-like receptor 4 (TLR4); it has been revealed that HMGB1 surge is responsible for the onset of T cell exhaustion and dysfunction, resulting in diminished RAGE and TLR4 surface expression and possibly preventing T cell function (Muire et al., 2021).
Considering the role of RAGE and its ligands on promoting a pro-inflammatory tumor microenvironment, it’s likely that alterations of RAGE signaling pathway may also impact the efficacy of immunotherapies for malignant gliomas. Our analysis of the human glioma TCGA database (Squatrito, 2015–2021) suggest a direct correlation between AGER (RAGE gene) and a number of lymphocyte markers in glioblastoma (Figure 2). Although the expression of CD3, CD8, appear to directly correlate with AGER, the expression of T cell inhibitory genes, such as PD-1 and LAG3 were also higher in RAGE-over-expressing glioblastomas. This finding confirms that RAGE activation does indeed promote a pro-inflammatory immune environment, but at the same time, may be associated with T cell exhaustion. Also, expression of RAGE ligands in tumor and non-tumor brain tissue in TCGA database revealed substantial expression of RAGE ligands in glioblastoma (Figure 3). Whether RAGE inhibition could impact the efficacy of immunotherapies by altering T cell trafficking and/or exhaustion needs further investigation.
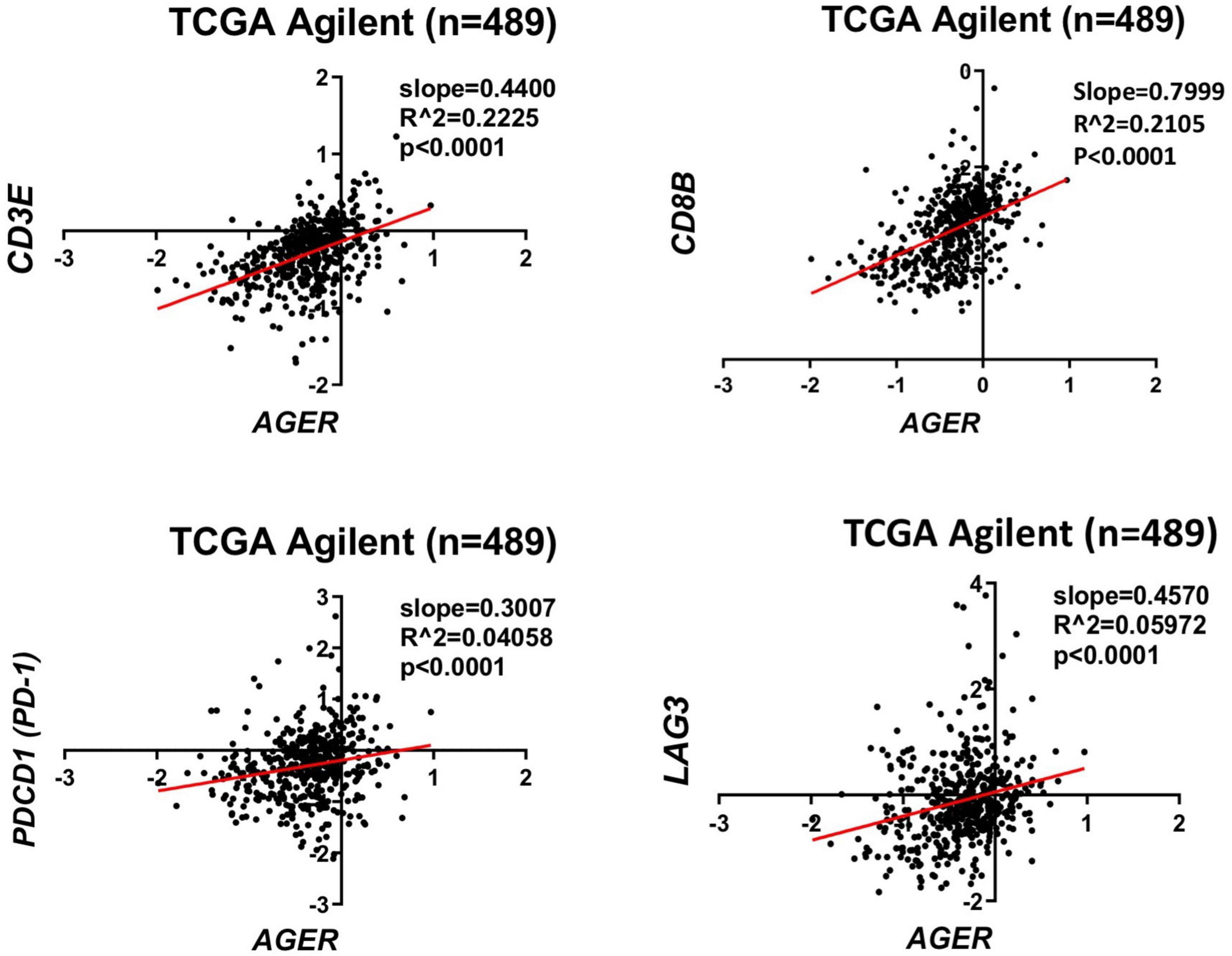
Figure 2. Correlation of RAGE expression to inflammatory markers in glioblastoma. TCGA analysis confirms a direct correlation between CD3 and CD8 lymphocyte markers with AGER. Expression of T cell exhaustion markers PD-1 and LAG3 also correlates with AGER expression.
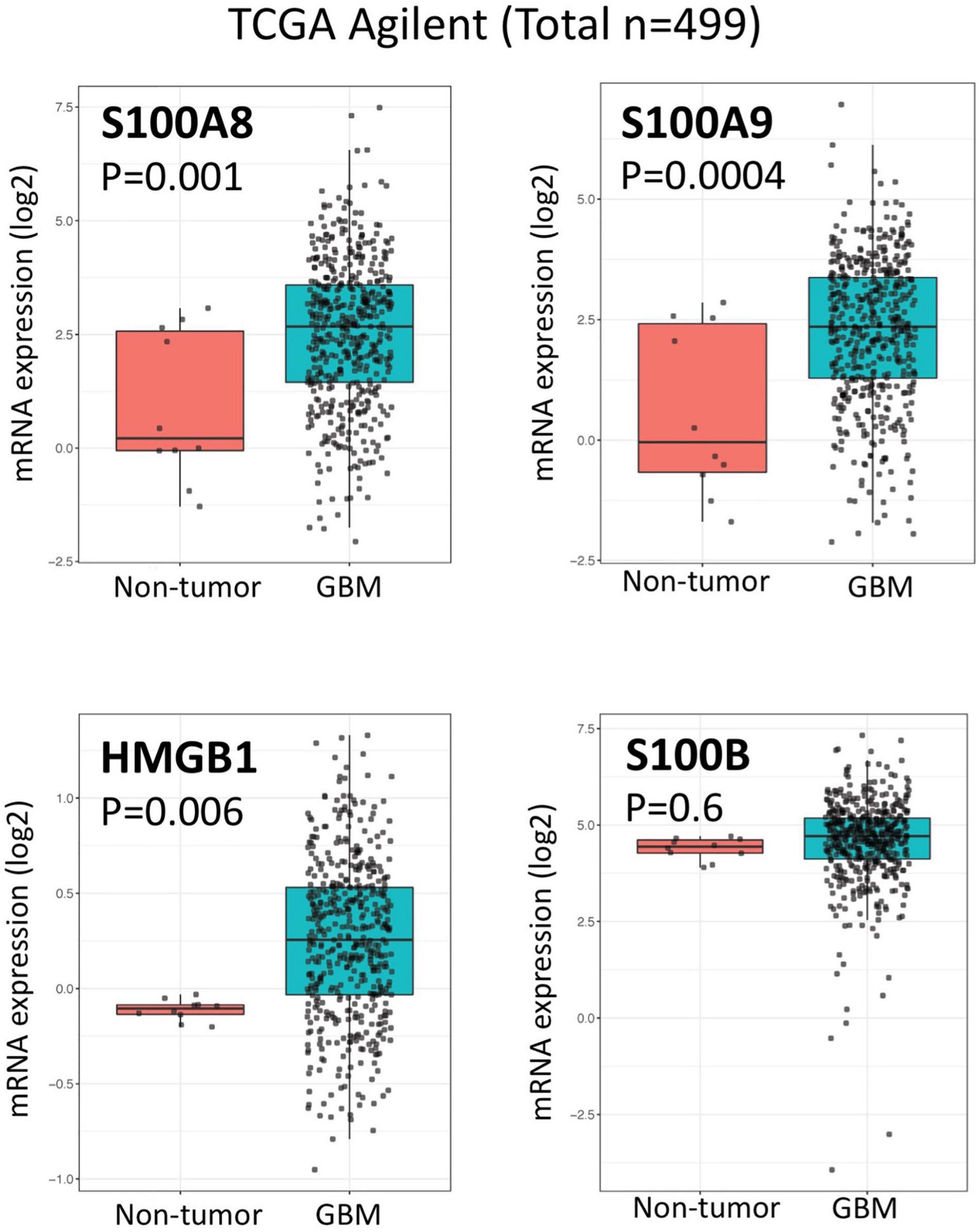
Figure 3. Expression of RAGE ligands in glioblastoma and non-tumor samples. Box plots of TCGA analysis demonstrate significant expression of RAGE ligands (HMGB1, S100A8, and S100A9) in glioblastoma.
Although the efficacy of small molecule RAGE inhibitors on immunotherapy responses have not been studied yet, others have suggested that such an approach may be reasonable. For example, tasquinimod, a second-generation oral quinoline-3-carboxamide that has been shown to bind S100A9 in the tumor microenvironment, blocks engagement of S100A9 with RAGE, and thus, prevents activation of downstream signaling pathways that lead to production of pro-inflammatory cytokines (Kierdorf and Fritz, 2013; Chen et al., 2014; Wang et al., 2018). Although the anti-tumor effects of tasquinimod were initially documented in a wide range of preclinical tumor models (Isaacs et al., 2006; Jennbacken et al., 2012), phase II and III trials in metastatic prostate cancer, hepatocellular, ovarian, gastric, and renal cell carcinomas showed no clinical benefit as a monotherapy (Bratt et al., 2009; Pili et al., 2011; Sternberg et al., 2016; Escudier et al., 2017). Others, however, have shown tasquinimod to block the trafficking of myeloid derived suppressive cells (which inhibit the anti-tumor activity of CD8 and CD4 cells) (Raymond et al., 2014), change the polarization of tumor-associated macrophages into M1 phenotype (Olsson et al., 2015), and improve efficacy of PD-L1 blockade (Shen et al., 2015; Nakhle et al., 2016). These findings support further evaluation of RAGE and RAGE inhibitors as immune modulators for glioblastoma immunotherapy.
RAGE signaling has been implicated in the pathogenesis of a variety of cancers including gliomas. Activation of RAGE by a plethora of ligands can modulate cellular properties that are involved in tumor proliferation, angiogenesis, invasion, metastasis and immune modulation. Thus, RAGE could be an attractive therapeutic target for many cancers, especially those that harness multiple oncogenic pathways for growth and invasion. Although small molecule RAGE inhibitors have been developed and tested for treatment of Alzheimer’s disease, these inhibitors have not been critically evaluated in glioblastoma models. Available data suggest that such inhibitors may have clinical utility when used alone or in combination with other glioma therapies.
GO: literature search, drafting of the manuscript and final approval of the version to be published. MD: critical revision of the content for intellectual material, designing the concept map, and final approval of the version to be published. BB: conception, critical revision of the content for intellectual material, supervision, and final approval of the version to be published. All authors contributed to the article and approved the submitted version.
This work was supported by R01CA155769, R01CA236500, and R01CA254271, Liam McGee Brain Tumor Fund, and the Joan Traver Walsh Family Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AGE, Advanced glycation end products; AKT, Protein kinase B; CNS, central nervous system; DAMPs, damage-associated molecular pattern molecules; ERK, Extracellular-signal-regulated kinase; HMGB1, high-mobility group box 1; JAK, Janus kinase; MG, microglia; MP, macrophages; MAPK, Mitogen-activated protein kinase; NK, natural killer; NF-kB, Nuclear factor kappa B; oHSV, oncolytic herpes simplex virus; PAMPs, pathogen-associated molecular patterns; PD-L1, Programmed death-ligand 1; PRR, pattern recognition receptor; PI3K, Phosphoinositide 3-kinases; RAGE, receptor for advanced glycation end-products; ROS, reactive oxygen species; sRAGE, soluble RAGE; SSNRI, selective serotonin and norepinephrine reuptake inhibitors; TAMs, tumor-associated macrophages; TLRs, Toll-like receptors; TMZ, temozolomide.
Al-Kharboosh, R., ReFaey, K., Lara-Velazquez, M., Grewal, S. S., Imitola, J., and Quiñones-Hinojosa, A. (2020). Inflammatory mediators in glioma microenvironment play a dual role in gliomagenesis and mesenchymal stem cell homing: implication for cellular therapy. Mayo Clin. Proc. Innov. Qual. Outcomes 4, 443–459. doi: 10.1016/j.mayocpiqo.2020.04.006
Allgöwer, C., Kretz, A.-L., von Karstedt, S., Wittau, M., Henne-Bruns, D., and Lemke, J. (2020). Friend or foe: S100 proteins in cancer. Cancers 12:2037. doi: 10.3390/cancers12082037
Andersson, U., and Tracey, K. J. (2011). HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29, 139–162. doi: 10.1146/annurev-immunol-030409-101323
Arora, A., Patil, V., Kundu, P., Kondaiah, P., Hegde, A. S., Arivazhagan, A., et al. (2019). Serum biomarkers identification by iTRAQ and verification by MRM: S100A8/S100A9 levels predict tumor-stroma involvement and prognosis in Glioblastoma. Sci. Rep. 9:2749. doi: 10.1038/s41598-019-39067-8
Auffinger, B., Tobias, A. L., Han, Y., Lee, G., Guo, D., Dey, M., et al. (2014). Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 21, 1119–1131. doi: 10.1038/cdd.2014.31
Banerjee, K., Núñez, F. J., Haase, S., McClellan, B. L., Faisal, S. M., Carney, S. V., et al. (2021). Current approaches for glioma gene therapy and virotherapy. Front. Mol. Neurosci. 14:621831. doi: 10.3389/fnmol.2021.621831
Behnan, J., Finocchiaro, G., and Hanna, G. (2019). The landscape of the mesenchymal signature in brain tumours. Brain 142, 847–866. doi: 10.1093/brain/awz044
Bianchi, R., Kastrisianaki, E., Giambanco, I., and Donato, R. (2011). S100B protein stimulates microglia migration via RAGE-dependent up-regulation of chemokine expression and release. J. Biol. Chem. 286, 7214–7226. doi: 10.1074/jbc.M110.169342
Bratt, O., Haggman, M., Ahlgren, G., Nordle, O., Bjork, A., and Damber, J. E. (2009). Open-label, clinical phase I studies of tasquinimod in patients with castration-resistant prostate cancer. Br. J. Cancer 101, 1233–1240. doi: 10.1038/sj.bjc.6605322
Chaudhary, R., Morris, R. J., and Steinson, E. (2021). The multifactorial roles of microglia and macrophages in the maintenance and progression of glioblastoma. J. Neuroimmunol. 357:577633. doi: 10.1016/j.jneuroim.2021.577633
Chavakis, T., Bierhaus, A., and Nawroth, P. P. (2004). RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microbes Infect. 6, 1219–1225. doi: 10.1016/j.micinf.2004.08.004
Chen, X., Zhang, L., Zhang, I. Y., Liang, J., Wang, H., Ouyang, M., et al. (2014). RAGE expression in tumor-associated macrophages promotes angiogenesis in glioma. Cancer Res. 74, 7285–7297. doi: 10.1158/0008-5472.CAN-14-1240
Chuah, Y. K., Basir, R., Talib, H., Tie, T. H., and Nordin, N. (2013). Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int. J. Inflam. 2013:403460. doi: 10.1155/2013/403460
Crowe, L. A. N., McLean, M., Kitson, S. M., Melchor, E. G., Patommel, K., Cao, H. M., et al. (2019). S100A8 & S100A9: alarmin mediated inflammation in tendinopathy. Sci. Rep. 9:1463. doi: 10.1038/s41598-018-37684-3
Davis, M. E. (2016). Glioblastoma: overview of disease and treatment. Clin. J. Oncol. Nurs. 20(5 Suppl.), S2–S8. doi: 10.1188/16.cjon.s1.2-8
Donato, R., Cannon, B. R., Sorci, G., Riuzzi, F., Hsu, K., Weber, D. J., et al. (2013). Functions of S100 proteins. Curr. Mol. Med. 13, 24–57.
Erusalimsky, J. D. (2021). The use of the soluble receptor for advanced glycation-end products (sRAGE) as a potential biomarker of disease risk and adverse outcomes. Redox Biol. 42:101958. doi: 10.1016/j.redox.2021.101958
Escudier, B., Faivre, S., Van Cutsem, E., Germann, N., Pouget, J. C., Plummer, R., et al. (2017). A phase II multicentre, open-label, proof-of-concept study of tasquinimod in hepatocellular, ovarian, renal cell, and gastric cancers. Target Oncol. 12, 655–661. doi: 10.1007/s11523-017-0525-2
Fan, W., Song, Y., Ren, Z., Cheng, X., Li, P., Song, H., et al. (2020). Glioma cells are resistant to inflammation-induced alterations of mitochondrial dynamics. Int. J. Oncol. 57, 1293–1306. doi: 10.3892/ijo.2020.5134
Gao, H., Zhang, I. Y., Zhang, L., Song, Y., Liu, S., Ren, H., et al. (2018). S100B suppression alters polarization of infiltrating myeloid-derived cells in gliomas and inhibits tumor growth. Cancer Lett. 439, 91–100. doi: 10.1016/j.canlet.2018.07.034
Gao, X. Y., Zang, J., Zheng, M. H., Zhang, Y. F., Yue, K. Y., Cao, X. L., et al. (2021). Temozolomide treatment induces HMGB1 to promote the formation of glioma stem cells via the TLR2/NEAT1/Wnt pathway in glioblastoma. Front. Cell Dev. Biol. 9:620883. doi: 10.3389/fcell.2021.620883
Garnier, D., Meehan, B., Kislinger, T., Daniel, P., Sinha, A., Abdulkarim, B., et al. (2018). Divergent evolution of temozolomide resistance in glioblastoma stem cells is reflected in extracellular vesicles and coupled with radiosensitization. Neuro Oncol. 20, 236–248. doi: 10.1093/neuonc/nox142
Gonzalez Castro, L. N., and Wesseling, P. (2020). The cIMPACT-now updates and their significance to current neuro-oncology practice. Neuro Oncol. Pract. 8, 4–10. doi: 10.1093/nop/npaa055
Harris, H. E., Andersson, U., and Pisetsky, D. S. (2012). HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 8, 195–202. doi: 10.1038/nrrheum.2011.222
Hofmann, M. A., Drury, S., Fu, C., Qu, W., Taguchi, A., Lu, Y., et al. (1999). RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97, 889–901. doi: 10.1016/s0092-8674(00)80801-6
Holla, F. K., Postma, T. J., Blankenstein, M. A., van Mierlo, T. J. M., Vos, M. J., Sizoo, E. M., et al. (2016). Prognostic value of the S100B protein in newly diagnosed and recurrent glioma patients: a serial analysis. J. Neurooncol. 129, 525–532. doi: 10.1007/s11060-016-2204-z
Hong, B., Muili, K., Bolyard, C., Russell, L., Lee, T. J., Banasavadi-Siddegowda, Y., et al. (2019). Suppression of HMGB1 released in the glioblastoma tumor microenvironment reduces tumoral edema. Mol. Ther. Oncolytics 12, 93–102. doi: 10.1016/j.omto.2018.11.005
Isaacs, J. T., Pili, R., Qian, D. Z., Dalrymple, S. L., Garrison, J. B., Kyprianou, N., et al. (2006). Identification of ABR-215050 as lead second generation quinoline-3-carboxamide anti-angiogenic agent for the treatment of prostate cancer. Prostate 66, 1768–1778. doi: 10.1002/pros.20509
Jennbacken, K., Welen, K., Olsson, A., Axelsson, B., Torngren, M., Damber, J. E., et al. (2012). Inhibition of metastasis in a castration resistant prostate cancer model by the quinoline-3-carboxamide tasquinimod (ABR-215050). Prostate 72, 913–924. doi: 10.1002/pros.21495
Kan, L. K., Drummond, K., Hunn, M., Williams, D., O’Brien, T. J., and Monif, M. (2020). Potential biomarkers and challenges in glioma diagnosis, therapy and prognosis. BMJ Neurol. Open 2:e000069. doi: 10.1136/bmjno-2020-000069
Kierdorf, K., and Fritz, G. (2013). RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 94, 55–68. doi: 10.1189/jlb.1012519
Lamkanfi, M., and Dixit, V. M. (2014). Mechanisms and functions of inflammasomes. Cell 157, 1013–1022. doi: 10.1016/j.cell.2014.04.007
Leclerc, E., Fritz, G., Vetter, S. W., and Heizmann, C. W. (2009). Binding of S100 proteins to RAGE: an update. Biochim. Biophys. Acta 1793, 993–1007. doi: 10.1016/j.bbamcr.2008.11.016
Leclerc, E., Fritz, G., Weibel, M., Heizmann, C. W., and Galichet, A. (2007). S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J. Biol. Chem. 282, 31317–31331. doi: 10.1074/jbc.M703951200
Lee, E. J., and Park, J. H. (2013). Receptor for advanced glycation endproducts (RAGE), its ligands, and soluble RAGE: potential biomarkers for diagnosis and therapeutic targets for human renal diseases. Genomics Inform. 11, 224–229. doi: 10.5808/gi.2013.11.4.224
Liu, T., Zhang, L., Joo, D., and Sun, S. C. (2017). NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2:17023. doi: 10.1038/sigtrans.2017.23
Louis, D. N., Wesseling, P., Aldape, K., Brat, D. J., Capper, D., Cree, I. A., et al. (2020). cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 30, 844–856. doi: 10.1111/bpa.12832
Magna, M., and Pisetsky, D. S. (2014). The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol. Med. 20, 138–146. doi: 10.2119/molmed.2013.00164
Medikonda, R., Dunn, G., Rahman, M., Fecci, P., and Lim, M. (2021). A review of glioblastoma immunotherapy. J. Neurooncol. 151, 41–53. doi: 10.1007/s11060-020-03448-1
Mondet, J., Chevalier, S., and Mossuz, P. (2021). Pathogenic roles of S100A8 and S100A9 proteins in acute myeloid and lymphoid leukemia: clinical and therapeutic impacts. Molecules 26:1323. doi: 10.3390/molecules26051323
Morshed, R. A., Young, J. S., Hervey-Jumper, S. L., and Berger, M. S. (2019). The management of low-grade gliomas in adults. J. Neurosurg. Sci. 63, 450–457. doi: 10.23736/s0390-5616.19.04701-5
Mostofa, A. G., Punganuru, S. R., Madala, H. R., Al-Obaide, M., and Srivenugopal, K. S. (2017). The process and regulatory components of inflammation in brain oncogenesis. Biomolecules 7:34. doi: 10.3390/biom7020034
Muire, P. J., Schwacha, M. G., and Wenke, J. C. (2021). Systemic T cell exhaustion dynamics is linked to early high mobility group box protein 1 (HMGB1) driven hyper-inflammation in a polytrauma rat model. Cells 10:1646. doi: 10.3390/cells10071646
Mulrennan, S., Baltic, S., Aggarwal, S., Wood, J., Miranda, A., Frost, F., et al. (2015). The role of receptor for advanced glycation end products in airway inflammation in CF and CF related diabetes. Sci. Rep. 5:8931. doi: 10.1038/srep08931
Nakhle, J., Pierron, V., Bauchet, A. L., Plas, P., Thiongane, A., Meyer-Losic, F., et al. (2016). Tasquinimod modulates tumor-infiltrating myeloid cells and improves the antitumor immune response to PD-L1 blockade in bladder cancer. Oncoimmunology 5:e1145333. doi: 10.1080/2162402X.2016.1145333
Olsson, A., Nakhle, J., Sundstedt, A., Plas, P., Bauchet, A. L., Pierron, V., et al. (2015). Tasquinimod triggers an early change in the polarization of tumor associated macrophages in the tumor microenvironment. J. Immunother. Cancer 3:53. doi: 10.1186/s40425-015-0098-5
Ostrom, Q. T., Bauchet, L., Davis, F. G., Deltour, I., Fisher, J. L., Langer, C. E., et al. (2014). The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol. 16, 896–913. doi: 10.1093/neuonc/nou087
Piao, Y., Liang, J., Holmes, L., Zurita, A. J., Henry, V., Heymach, J. V., et al. (2012). Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol. 14, 1379–1392. doi: 10.1093/neuonc/nos158
Pili, R., Haggman, M., Stadler, W. M., Gingrich, J. R., Assikis, V. J., Bjork, A., et al. (2011). Phase II randomized, double-blind, placebo-controlled study of tasquinimod in men with minimally symptomatic metastatic castrate-resistant prostate cancer. J. Clin. Oncol. 29, 4022–4028. doi: 10.1200/JCO.2011.35.6295
Popescu, I. D., Codrici, E., Albulescu, L., Mihai, S., Enciu, A. M., Albulescu, R., et al. (2014). Potential serum biomarkers for glioblastoma diagnostic assessed by proteomic approaches. Proteome Sci. 12:47. doi: 10.1186/s12953-014-0047-0
Qin, Y. H., Dai, S. M., Tang, G. S., Zhang, J., Ren, D., Wang, Z. W., et al. (2009). HMGB1 enhances the proinflammatory activity of lipopolysaccharide by promoting the phosphorylation of MAPK p38 through receptor for advanced glycation end products. J. Immunol. 183, 6244–6250. doi: 10.4049/jimmunol.0900390
Ramasamy, R., Yan, S. F., and Schmidt, A. M. (2009). RAGE: therapeutic target and biomarker of the inflammatory response–the evidence mounts. J. Leukoc. Biol. 86, 505–512. doi: 10.1189/jlb.0409230
Ramaswamy, P., Goswami, K., Dalavaikodihalli Nanjaiah, N., Srinivas, D., and Prasad, C. (2019). TNF-α mediated MEK-ERK signaling in invasion with putative network involving NF-κB and STAT-6: a new perspective in glioma. Cell Biol. Int. 43, 1257–1266. doi: 10.1002/cbin.11125
Rauvala, H., and Rouhiainen, A. (2010). Physiological and pathophysiological outcomes of the interactions of HMGB1 with cell surface receptors. Biochim. Biophys. Acta 1799, 164–170. doi: 10.1016/j.bbagrm.2009.11.012
Raymond, E., Dalgleish, A., Damber, J. E., Smith, M., and Pili, R. (2014). Mechanisms of action of tasquinimod on the tumour microenvironment. Cancer Chemother. Pharmacol. 73, 1–8. doi: 10.1007/s00280-013-2321-8
Razavi, S. M., Lee, K. E., Jin, B. E., Aujla, P. S., Gholamin, S., and Li, G. (2016). Immune evasion strategies of glioblastoma. Front. Surg. 3:11. doi: 10.3389/fsurg.2016.00011
Reardon, D. A., Brandes, A. A., Omuro, A., Mulholland, P., Lim, M., Wick, A., et al. (2020). Effect of Nivolumab vs Bevacizumab in patients with recurrent glioblastoma: the checkmate 143 phase 3 randomized clinical trial. JAMA Oncol. 6, 1003–1010. doi: 10.1001/jamaoncol.2020.1024
Riva, M., He, Z., Källberg, E., Ivars, F., and Leanderson, T. (2013). Human S100A9 protein is stabilized by inflammatory stimuli via the formation of proteolytically-resistant homodimers. PLoS One 8:e61832. doi: 10.1371/journal.pone.0061832
Roh, J. S., and Sohn, D. H. (2018). Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 18:e27. doi: 10.4110/in.2018.18.e27
Rovere-Querini, P., Capobianco, A., Scaffidi, P., Valentinis, B., Catalanotti, F., Giazzon, M., et al. (2004). HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 5, 825–830. doi: 10.1038/sj.embor.7400205
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195. doi: 10.1038/nature00858
Schmidt, A. M., Yan, S. D., Wautier, J. L., and Stern, D. (1999). Activation of receptor for advanced glycation end products: a mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ. Res. 84, 489–497. doi: 10.1161/01.res.84.5.489
Schmidt, A. M., Yan, S. D., Yan, S. F., and Stern, D. M. (2001). The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Invest. 108, 949–955. doi: 10.1172/jci14002
Seidu, R. A., Wu, M., Su, Z., and Xu, H. (2017). Paradoxical role of high mobility group box 1 in glioma: a suppressor or a promoter? Oncol. Rev. 11:325. doi: 10.4081/oncol.2017.325
Shen, C.-Y., Lu, C.-H., Wu, C.-H., Li, K.-J., Kuo, Y.-M., Hsieh, S.-C., et al. (2020). The development of maillard reaction, and advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling inhibitors as novel therapeutic strategies for patients with AGE-related diseases. Molecules 25:5591. doi: 10.3390/molecules25235591
Shen, L., Sundstedt, A., Ciesielski, M., Miles, K. M., Celander, M., Adelaiye, R., et al. (2015). Tasquinimod modulates suppressive myeloid cells and enhances cancer immunotherapies in murine models. Cancer Immunol. Res. 3, 136–148. doi: 10.1158/2326-6066.CIR-14-0036
Simard, J. C., Cesaro, A., Chapeton-Montes, J., Tardif, M., Antoine, F., Girard, D., et al. (2013). S100A8 and S100A9 induce cytokine expression and regulate the NLRP3 inflammasome via ROS-dependent activation of NF-κB(1). PLoS One 8:e72138. doi: 10.1371/journal.pone.0072138
Singh, N., Miner, A., Hennis, L., and Mittal, S. (2021). Mechanisms of temozolomide resistance in glioblastoma – a comprehensive review. Cancer Drug Resist. 4, 17–43. doi: 10.20517/cdr.2020.79
Sorci, G., Riuzzi, F., Giambanco, I., and Donato, R. (2013). RAGE in tissue homeostasis, repair and regeneration. Biochim. Biophys. Acta 1833, 101–109. doi: 10.1016/j.bbamcr.2012.10.021
Squatrito, M. (2015–2021). GlioVis Data Visualization Tools for Brain Tumor Datasets. Boston, MA: Free Software Foundation.
Srikrishna, G. (2012). S100A8 and S100A9: new insights into their roles in malignancy. J. Innate Immun. 4, 31–40. doi: 10.1159/000330095
Sternberg, C., Armstrong, A., Pili, R., Ng, S., Huddart, R., Agarwal, N., et al. (2016). Randomized, double-blind, placebo-controlled phase III study of tasquinimod in men with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 34, 2636–2643. doi: 10.1200/JCO.2016.66.9697
Stupp, R., Mason, W. P., van den Bent, M. J., Weller, M., Fisher, B., Taphoorn, M. J., et al. (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996. doi: 10.1056/NEJMoa043330
Swanson, K. V., Deng, M., and Ting, J. P. (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489. doi: 10.1038/s41577-019-0165-0
Tamimi, A. F., and Juweid, M. (2017). “Epidemiology and outcome of glioblastoma,” in Glioblastoma, ed. S. De Vleeschouwer (Brisbane, QLD: Codon Publications).
Villarreal, A., Seoane, R., González Torres, A., Rosciszewski, G., Angelo, M. F., Rossi, A., et al. (2014). S100B protein activates a RAGE-dependent autocrine loop in astrocytes: implications for its role in the propagation of reactive gliosis. J. Neurochem. 131, 190–205. doi: 10.1111/jnc.12790
Wang, H., Zhang, L., Zhang, I. Y., Chen, X., Da Fonseca, A., Wu, S., et al. (2013). S100B promotes glioma growth through chemoattraction of myeloid-derived macrophages. Clin. Cancer Res. 19, 3764–3775. doi: 10.1158/1078-0432.ccr-12-3725
Wang, S., Song, R., Wang, Z., Jing, Z., Wang, S., and Ma, J. (2018). S100A8/A9 in inflammation. Front. Immunol. 9:1298. doi: 10.3389/fimmu.2018.01298
Woroniecka, K., Chongsathidkiet, P., Rhodin, K., Kemeny, H., Dechant, C., Farber, S. H., et al. (2018). T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin. Cancer Res. 24, 4175–4186. doi: 10.1158/1078-0432.ccr-17-1846
Xu, S., Tang, L., Li, X., Fan, F., and Liu, Z. (2020). Immunotherapy for glioma: current management and future application. Cancer Lett. 476, 1–12. doi: 10.1016/j.canlet.2020.02.002
Xue, J., Suarez, J. S., Minaai, M., Li, S., Gaudino, G., Pass, H. I., et al. (2021). HMGB1 as a therapeutic target in disease. J. Cell. Physiol. 236, 3406–3419. doi: 10.1002/jcp.30125
Yang, H., Hreggvidsdottir, H. S., Palmblad, K., Wang, H., Ochani, M., Li, J., et al. (2010). A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. U.S.A. 107, 11942–11947. doi: 10.1073/pnas.1003893107
Yang, H., Wang, H., and Andersson, U. (2020). Targeting inflammation driven by HMGB1. Front. Immunol. 11:484. doi: 10.3389/fimmu.2020.00484
Yin, X.-F., Zhang, Q., Chen, Z.-Y., Wang, H.-F., Li, X., Wang, H.-X., et al. (2018). NLRP3 in human glioma is correlated with increased WHO grade, and regulates cellular proliferation, apoptosis and metastasis via epithelial-mesenchymal transition and the PTEN/AKT signaling pathway. Int. J. Oncol. 53, 973–986. doi: 10.3892/ijo.2018.4480
Young, R. M., Jamshidi, A., Davis, G., and Sherman, J. H. (2015). Current trends in the surgical management and treatment of adult glioblastoma. Ann. Transl. Med. 3:9.
Keywords: cancer, glioma, inflammation, HMGB1, S100A8, S100A9, S100B
Citation: Otazu GK, Dayyani M and Badie B (2021) Role of RAGE and Its Ligands on Inflammatory Responses to Brain Tumors. Front. Cell. Neurosci. 15:770472. doi: 10.3389/fncel.2021.770472
Received: 03 September 2021; Accepted: 22 November 2021;
Published: 16 December 2021.
Edited by:
Myriam Catalano, Sapienza University of Rome, ItalyReviewed by:
Nihal Karakas, Istanbul Medipol University, TurkeyCopyright © 2021 Otazu, Dayyani and Badie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Behnam Badie, YmJhZGllQGNvaC5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.