 Mahmoud Suliman†
Mahmoud Suliman† Miriam L. Greenberg
Miriam L. Greenberg
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 31 August 2021
Sec. Cellular Neuropathology
Volume 15 - 2021 | https://doi.org/10.3389/fncel.2021.735622
This article is part of the Research TopicThe Role of Autophagy and the Unfolded Protein Response (Upr) in Proper and Improper Brain Function, in General, and in the Mechanism of Mood Stabilization, in ParticularView all 5 articles
Bipolar disorder (BD) is a mood disorder that affects millions worldwide and is associated with severe mood swings between mania and depression. The mood stabilizers valproate (VPA) and lithium (Li) are among the main drugs that are used to treat BD patients. However, these drugs are not effective for all patients and cause serious side effects. Therefore, better drugs are needed to treat BD patients. The main barrier to developing new drugs is the lack of knowledge about the therapeutic mechanism of currently available drugs. Several hypotheses have been proposed for the mechanism of action of mood stabilizers. However, it is still not known how they act to alleviate both mania and depression. The pathology of BD is characterized by mitochondrial dysfunction, oxidative stress, and abnormalities in calcium signaling. A deficiency in the unfolded protein response (UPR) pathway may be a shared mechanism that leads to these cellular dysfunctions. This is supported by reported abnormalities in the UPR pathway in lymphoblasts from BD patients. Additionally, studies have demonstrated that mood stabilizers alter the expression of several UPR target genes in mouse and human neuronal cells. In this review, we outline a new perspective wherein mood stabilizers exert their therapeutic mechanism by activating the UPR. Furthermore, we discuss UPR abnormalities in BD patients and suggest future research directions to resolve discrepancies in the literature.
Bipolar disorder (BD) is a mood disorder that is characterized by moods alternating between mania and depression (Baldessarini et al., 2020). BD affects 2% of the population and is associated with a high rate of suicide (Gonda et al., 2012; Baldessarini et al., 2020). While there is no single biological marker correlated with BD, there is strong evidence of heritability, and multiple genes have been found to be linked to increased risk for the disease (Grande et al., 2016; Stahl et al., 2019). Environmental factors also play a role in the onset of the disease (Vieta et al., 2018). Lithium (Li) and valproate (VPA) are among the primary drugs used to treat BD (Geddes and Miklowitz, 2013). Yet, these drugs are not effective for all patients and can cause serious side effects, including hepatoxicity, renal toxicity, teratogenicity, cognitive impairment, hair loss, and weight gain (Dreifuss et al., 1987; Pijl and Meinders, 1996; Mercke et al., 2000; Yonkers et al., 2004; Gitlin, 2016). Therefore, better drugs are needed to treat BD patients. However, the mechanism of action of BD drugs is not known, which hinders the development of effective drugs with minimal side effects.
Many studies have aimed to characterize the cellular effects of BD drugs in order to improve our understanding of their therapeutic mechanism, and numerous cellular targets have been proposed, including the neurotransmitter and neuromodulator systems, neuronal plasticity pathways, and myo-inositol metabolism (Berridge, 2014). Studies have also suggested that the unfolded protein response (UPR) pathway may be part of the pathophysiology of BD and that mood stabilizers could exert their therapeutic mechanism by activating the UPR (Wang et al., 1999; Bown et al., 2000; Chen et al., 2000; Kakiuchi et al., 2003; Shao et al., 2006; Kim et al., 2009; Pfaffenseller et al., 2014; Bengesser et al., 2016). The pathophysiology of BD is associated with mitochondrial dysfunction, oxidative stress, and abnormalities in calcium signaling, and the UPR plays a role in all of these pathways (Berk et al., 2011; Callaly et al., 2015; Vincenz-Donnelly and Hipp, 2017; Harrison et al., 2019). Therefore, deficient UPR activation could be a common mechanism underlying the array of cellular dysfunctions associated with BD. In support of this, several studies have reported a deficiency in UPR activation in lymphoblasts from BD patients (Table 1), as well as altered expression of UPR target genes following treatment with BD drugs (Table 2).
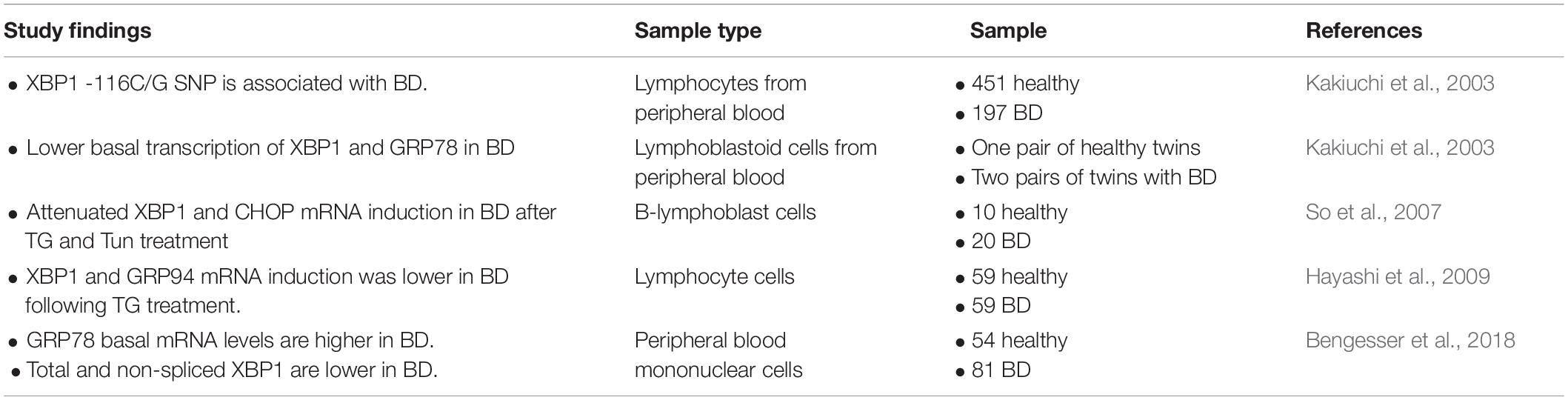
Table 1. Findings relevant to the role of UPR function in bipolar disorder.

Table 2. Mood stabilizers alter the expression of UPR target genes.
In this review, we highlight the existing data regarding UPR activation by mood stabilizers, discuss UPR abnormalities in BD patients, and suggest future research directions to clarify conflicting findings obtained from different studies. We analyze several mechanisms that could explain how mood stabilizers activate the UPR, focusing on a novel hypothesis wherein myo-inositol depletion serves as the mechanistic trigger for UPR activation.
The UPR is a stress response signaling pathway that is conserved from yeast to mammals (Foti et al., 1999). The UPR has a dual function, as it promotes homeostasis and cell survival under mild ER stress but can lead to apoptosis and cell death under intense, persistent stress (Chan et al., 2015; Hiramatsu et al., 2015). Various stressors activate the UPR, such as the accumulation of unfolded proteins in the ER lumen, lipid disequilibrium, calcium imbalance, nutrient limitation, and oxidative stress (Kaufman et al., 2002; Yan et al., 2008; Gardner and Walter, 2011; Hou et al., 2014; Krebs et al., 2015). The UPR activates downstream signaling cascades that induce genes functioning in protein folding, degradation, and translation arrest to reduce the protein load in the ER (Perri et al., 2015).
In yeast, UPR activation is mediated by IRE1, a type-1 transmembrane kinase, and endoribonuclease (Figure 1; Yamamoto et al., 2004; Korennykh et al., 2009). Activated IRE1 excises a 252 bp intronic region from HAC1 mRNA, which is translated to the active transcription factor Hac1 that translocates to the nucleus and activates UPR target genes (Kawahara et al., 1997; Foti et al., 1999). These genes, which contain UPR response elements (UPRE) in their promotors, include ER chaperones such as KAR2 (Figure 1; Kawahara et al., 1997; Foti et al., 1999). Chaperones assist in protein folding and maturation in the ER (Braakman and Hebert, 2013).
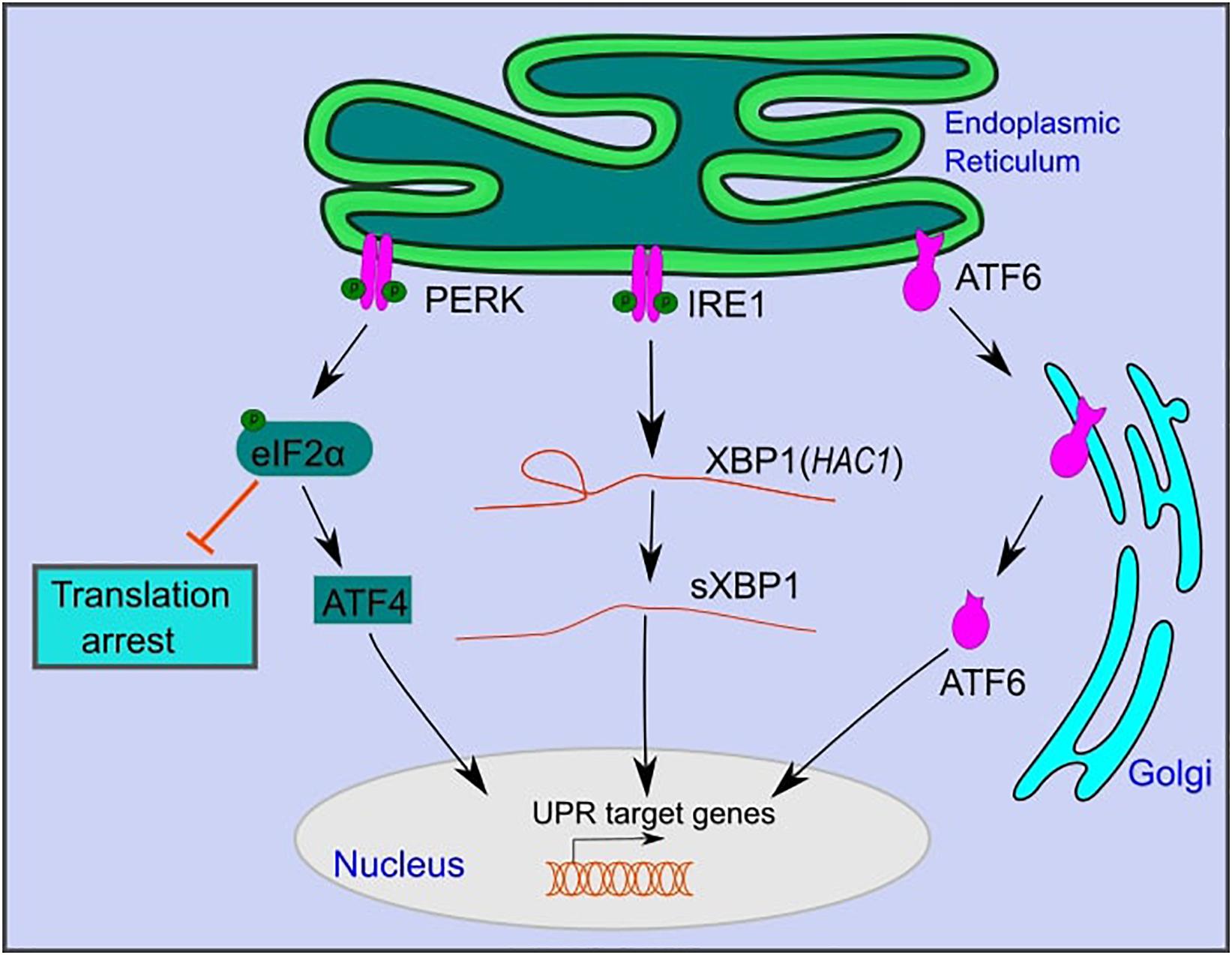
Figure 1. The mammalian unfolded protein response pathway. The UPR is activated upon ER stress caused by the accumulation of unfolded proteins in the lumen of the ER. UPR activation is mediated by three branches: PKR-like endoplasmic reticulum kinase (PERK), inositol-requiring kinase 1 (IRE1), and activating transcription factor 6 (ATF6). PERK and IRE1 are activated by autophosphorylation. Active PERK phosphorylates eIF2α, which inhibits overall protein translation, while selectively promoting the translation of activating transcription factor 4 (ATF4). Active IRE1 splices an intronic region from XBP1 mRNA (HAC1 in yeast) to form sXBP1, which is translated into an active transcription factor. ATF6 is translocated to the Golgi, where it is cleaved and further translocated to the nucleus. ATF6, sXBP1, and ATF4 activate downstream signaling cascades that increase the expression of genes that function in restoring ER homeostasis or inducing cell death under persistent ER stress.
IRE1 is the only UPR branch that is conserved from yeast to mammals (Yamamoto et al., 2004; Korennykh et al., 2009). Similar to yeast, upon ER stress, mammalian IRE1 is activated by autophosphorylation (Yamamoto et al., 2004; Korennykh et al., 2009). It catalyzes the splicing of a 26 bp intronic region of XBP1 mRNA to form spliced XBP1 (sXBP1), which is translated to an active transcription factor (Calfon et al., 2002; Hiramatsu et al., 2011). sXBP1 translocates to the nucleus and activates UPR target genes that contain conserved ER stress response elements (ERSE) in their promoters. These are similar to UPREs in yeast and include genes that function in protein folding, lipid metabolism, and ER-associated degradation (Yamamoto et al., 2004; Korennykh et al., 2009; Piperi et al., 2016).
In addition to IRE1, mammals have two additional UPR branches: activating transcription factor 6 (ATF6) and protein kinase RNA (PKR)-like ER kinase (PERK) (Figure 1; Perri et al., 2015). ATF6 is a member of the basic leucine zipper family of transcription factors (Wang et al., 2000). Under ER stress, ATF6 is translocated to the Golgi apparatus, where it is excised by site 1 and site 2 proteases to become an active transcription factor (Figure 1; Wang et al., 2000). Active ATF6 translocates to the nucleus and activates downstream target genes that function in protein folding and maturation, including glucose-regulated protein 78 (GRP78), glucose-regulated protein 94 (GRP94), and calreticulin (Schardt et al., 2010; Wang et al., 2016). ATF6 and XBP1 act to restore cell homeostasis and promote cell survival under ER stress (Yamamoto et al., 2004; Yoshida et al., 2006; Dadey et al., 2016).
PERK, a third branch of the UPR, functions to reduce the load of translated proteins that enter the ER and increases cell death under persistent ER stress (Lin et al., 2009; Kilberg et al., 2012). Following ER stress, PERK is oligomerized and activated by autophosphorylation (Harding et al., 2000). Active PERK phosphorylates eukaryotic translation initiation factor 2α (eIF2α) (Harding et al., 2000). Phosphorylated eIF2α inhibits eukaryotic translation initiation factor 2B (eIF2B) and decreases the assembly of the 43S initiation complex (Harding et al., 2000). This leads to translation arrest of most mRNAs while selectively allowing translation of specific proteins, such as activating transcription factor 4 (ATF4) (Harding et al., 2000). ATF4 regulates the expression of genes that function in amino acid metabolism and oxidative stress (Harding et al., 2003). Furthermore, ATF4 activates the transcription factor CCAAT/enhancer-binding protein homologous protein (CHOP), which plays a role in programmed cell death under persistent ER stress (Harding et al., 2000).
Several studies have reported a deficiency in UPR activation in lymphoblasts from BD patients (Kakiuchi et al., 2003; Kim et al., 2009; Pfaffenseller et al., 2014; Bengesser et al., 2016). Lymphoblast cells from BD patients have been used in many studies to characterize the pathology of BD, as access to live human brain tissue is not possible (Viswanath et al., 2015). An early study showed that the XBP1 single-nucleotide polymorphism (SNP) –116C→G is associated with an increased risk of developing BD (Kakiuchi et al., 2003). However, other studies failed to confirm the association of this SNP with BD (Hou et al., 2004; Kakiuchi et al., 2004). Additional work demonstrated that deficient UPR activation is caused by reduced transcription of the UPR target genes XBP1, GRP94, and CHOP in lymphoblast cells from BD patients after treatment with the ER stressors thapsigargin (TG) and tunicamycin (Tun) (So et al., 2007; Hayashi et al., 2009; Bengesser et al., 2018). This deficiency in UPR activation affects the ability to adapt to changes in the cellular environment, such as the accumulation of misfolded proteins in the ER, lipid overload, or changes in nutrient availability (Xu et al., 2005; Mandl et al., 2009). An inability of cells to adapt to ER stress results in increased cell death, and this has been demonstrated to occur in BD patient lymphocytes following treatment with Tun (Pfaffenseller et al., 2014). Increased neuronal cell death and neurodegeneration have been previously reported in BD patients (Karabulut et al., 2019; Gokcinar et al., 2020). Therefore, future studies should investigate whether ER stress and deficient UPR activation contribute to the neurodegeneration observed in BD patients.
Studies have suggested modulation of the UPR pathway as a therapeutic target of mood stabilizers. VPA and Li increase the expression of the UPR chaperones GRP78, GRP94, and calreticulin in rat brain samples and cultured rat cells (Table 2). In this way, VPA has been shown to protect cells from different stress situations by inducing the UPR (Zhang et al., 2011; Li et al., 2017). For example, VPA protects SH-SY5Y cells from ER stress-induced apoptosis following treatment with TG by increasing the pro-survival protein GRP78 and reducing the pro-apoptotic protein CHOP (Li et al., 2017). Similarly, VPA protects cells from ischemia-reperfusion injuries in rats by attenuating the increase in CHOP levels (Zhang et al., 2011). However, certain studies have also demonstrated no effect on the UPR pathway by mood stabilizers. Although VPA increases GRP78 protein levels in HEK293 cells, neither VPA nor Li has a significant impact on the expression of GRP78 in Neuro-2a cells (Kakiuchi et al., 2003, 2009; Shi et al., 2007). Additionally, VPA and Li do not increase XBP1 expression in SH-SY5Y and lymphoblastoid cells (Kakiuchi et al., 2003). Nonetheless, there is strong support for UPR activation by mood stabilizers in the majority of mammalian studies conducted to date.
Several mechanisms could explain how mood stabilizers activate the UPR. The first mechanism is through myo-inositol depletion and subsequent upregulation of ceramide levels. Abnormalities in myo-inositol levels have been observed in the brains of BD patients, and myo-inositol depletion has been hypothesized as part of the therapeutic mechanism of mood stabilizers (Shimon et al., 1997; Silverstone et al., 2005; Berridge, 2014). Studies have also reported alterations in the lipid profile, including changes in ceramide levels, in BD patients (Schwarz et al., 2008; Huang et al., 2018; Brunkhorst-Kanaan et al., 2019). An elegant study in yeast connected these observations and introduced a novel mechanism of UPR activation by the mood stabilizer VPA (Jadhav et al., 2016). Using yeast deficient in myo-inositol synthesis, Jadhav et al. (2016) demonstrated that depletion of intracellular myo-inositol by VPA upregulates ceramide levels and activates the UPR (Figure 2). In this study, it was shown that ceramide activates the UPR by inducing nutrient stress through the downregulation of plasma membrane amino acid transporters (Jadhav et al., 2016). It has also been shown that ceramide can activate the UPR pathway in human cells by inhibiting ER calcium uptake, suggesting that these ceramide-regulated mechanisms may work in tandem to induce the UPR following treatment with VPA (Liu et al., 2014). VPA-mediated activation of the UPR leads to increased expression of ER chaperones in yeast, including KAR2, the homolog of mammalian GRP78 (Jadhav et al., 2016). Upregulation of UPR chaperones indicates a protective effect of VPA. Therefore, myo-inositol depletion and UPR activation may be part of the same therapeutic mechanism employed by this drug. There is strong support for mood stabilizers leading to myo-inositol depletion in mammalian cells (Ye and Greenberg, 2015; Yu et al., 2017; Saiardi and Mudge, 2018). Additionally, it was shown that an increase in ceramide levels can activate the UPR pathway (Senkal et al., 2011; Liu et al., 2014). Future studies should characterize whether myo-inositol depletion activates the UPR in mammalian cells and whether this is dependent on an increase in ceramide levels.
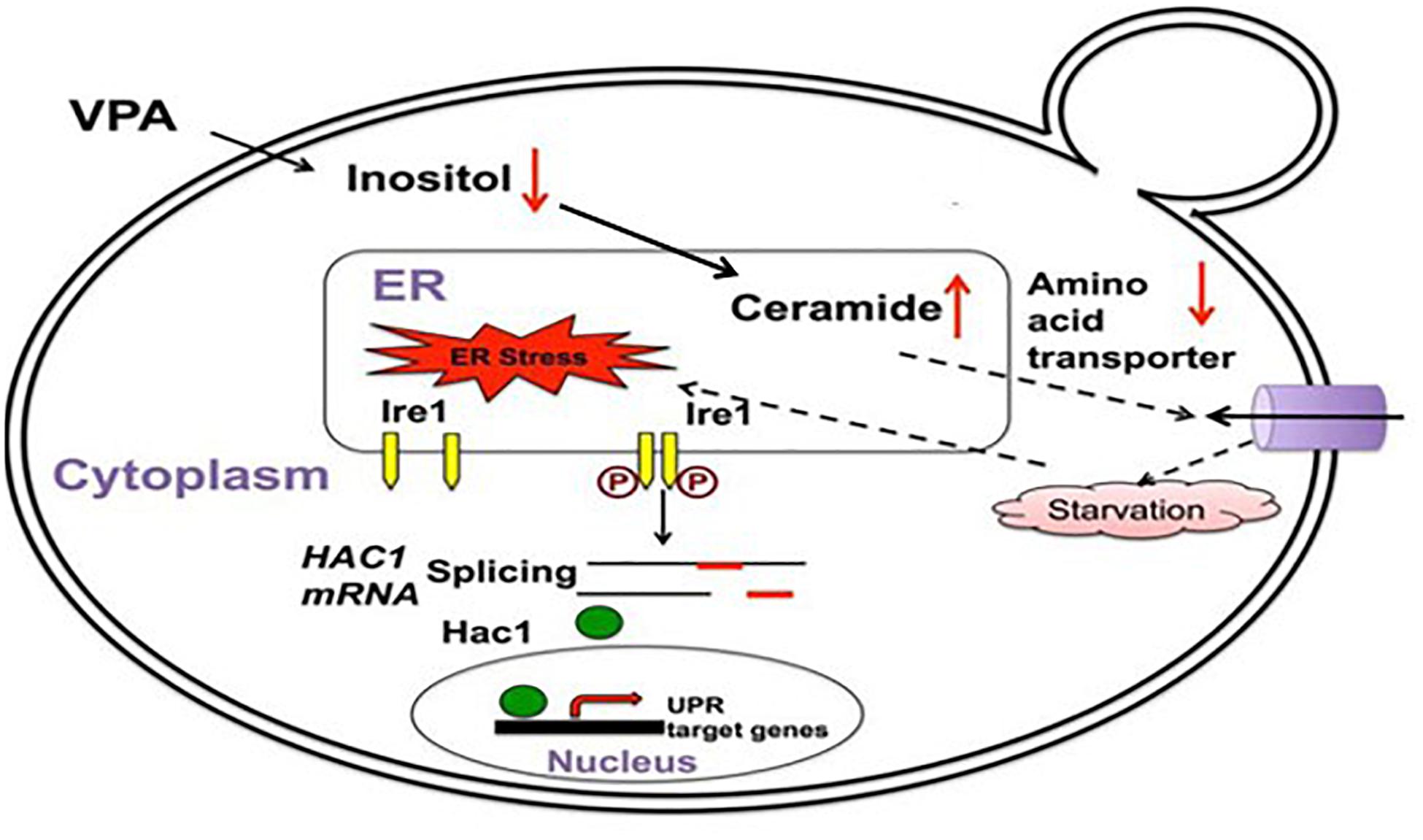
Figure 2. VPA induces the UPR pathway in yeast by increasing intracellular ceramide levels. VPA-mediated myo-inositol depletion results in elevated ceramide levels. Elevated ceramide results in the downregulation of amino acid transporters and subsequent UPR activation due to starvation stress. Figure adapted and modified from Jadhav et al. (2016).
A second mechanism is through inhibition of histone deacetylases (HDACs) (Shi et al., 2007). Support for this mechanism comes from a study showing that VPA, a known HDAC inhibitor, increases GRP78 expression, while VPA derivatives lacking the ability to inhibit HDACs do not increase GRP78 expression (Shi et al., 2007). In agreement with this, HDAC1 has been shown to bind to the promoter of GRP78 and repress its expression, suggesting that VPA may act at the transcriptional level to promote UPR activation by preventing repression by HDACs (Baumeister et al., 2009).
A third potential mechanism for UPR activation by mood stabilizers is through upregulation of the wolframin gene (WFS1) (Kakiuchi et al., 2009). WFS1 functions in mitigating ER stress, and WFS1 knockdown results in compensatory upregulation of GRP78, CHOP, and XBP1 in β-cells (Kakiuchi et al., 2009). In support of this mechanism, VPA has been shown to increase the expression of WFS1, leading to its dissociation from, and subsequent activation of, the ER chaperone GRP94 (Kakiuchi et al., 2009). Collectively, these studies support a protective role for the mood stabilizer VPA in the context of ER stress and highlight the possibility that VPA may act through more than one route to enhance the UPR response.
Regulation of the UPR pathway is linked to various aspects of brain function, and dysregulation is associated with the pathology of neurological disorders. GRP78 is an essential chaperone and a master regulator of the UPR, which functions in neuronal development (Weng et al., 2011). Abnormalities in GRP78 levels are associated with various neurological disorders such as Alzheimer’s and Parkinson’s diseases (Weng et al., 2011; Casas, 2017; Enogieru et al., 2019). Under normal conditions, GRP78 binds to the three UPR stress sensors, PERK, ATF6, and IRE1, and prevents their activation (Gong et al., 2017). However, under ER stress, GRP78 is released from these sensors by binding to unfolded proteins, resulting in sensor activation (Gong et al., 2017). Studies have suggested a role for the UPR in memory regulation, brain aging, neurotransmission, and in maintaining synaptic plasticity and structure in the central nervous system (Nosyreva and Kavalali, 2010; Freeman and Mallucci, 2016; Martínez et al., 2016; Miranda et al., 2020). Therefore, regulation of the UPR pathway may play a significant role in the pathophysiology and treatment of neurological disorders.
The UPR pathway may play a significant role in the pathology and treatment of BD, a severe mood disorder that disrupts the lives of patients and their families (Gonda et al., 2012; Baldessarini et al., 2020). Li and VPA are two of the primary drugs used to treat BD patients (Geddes and Miklowitz, 2013). However, their efficacy is not universal, and they can cause serious side effects (Dreifuss et al., 1987; Pijl and Meinders, 1996; Mercke et al., 2000; Yonkers et al., 2004; Gitlin, 2016). The therapeutic mechanism of these drugs is unknown, which poses a challenge for developing better and more effective medications. Several studies have suggested that a deficiency in UPR activation is connected to BD pathology and that mood stabilizers may activate the UPR pathway as part of their therapeutic mechanisms (Tables 1, 2).
There are multiple mechanisms that could potentially underlie UPR activation by mood stabilizers, including HDAC inhibition, upregulation of WFS1, and myo-inositol depletion (Shi et al., 2007; Kakiuchi et al., 2009; Jadhav et al., 2016). While these mechanisms are not mutually exclusive, the myo-inositol depletion mechanism is currently the best supported for several reasons. Studies have rigorously demonstrated that BD drugs induce myo-inositol depletion in both yeast and mammalian cells, and myo-inositol is known to be especially essential for brain function, where the concentration can reach levels 20 times higher than in the blood (Vaden et al., 2001; Ye and Greenberg, 2015; Bizzarri et al., 2016; Jadhav et al., 2016; Yu et al., 2017; Saiardi and Mudge, 2018). To date, the most complete mechanistic study of UPR activation by mood stabilizers provides strong evidence for a causative link between VPA treatment, myo-inositol depletion, increased ceramide levels, and UPR activation in yeast (Figure 2) (Jadhav et al., 2016), and this is further corroborated by studies showing that myo-inositol and ceramide levels are aberrant in BD patients (Shimon et al., 1997; Silverstone et al., 2005; Schwarz et al., 2008; Brunkhorst-Kanaan et al., 2019). However, this mechanism has yet to be tested in mammalian cells, and it is within reason to speculate that HDAC inhibition and WFS1 upregulation could also contribute to the therapeutic mechanism of VPA.
Several limitations and discrepancies in the studies in Tables 1, 2 warrant further investigation. First, the small sample size in many of these studies decreases the statistical power of the results due to variations among individuals and populations (So et al., 2007). Thus, there is a need to characterize the UPR pathway in larger, more diverse populations of BD patients. Differences in the disease stage between patients may also contribute to this variability, with advanced stages reported to have a higher deficiency in UPR activation (Pfaffenseller et al., 2014). However, a challenge in resolving this issue is that BD is a psychiatric disorder with no unique biological marker that allows accurate characterization of the disease and its severity (Sigitova et al., 2017). Conducting large-scale genome-wide association (GWA) studies may aid in identifying biological markers for BD (Chuang and Kuo, 2017). The third limitation is that UPR deficiency has mainly been characterized in lymphoblast cells from BD patients despite the fact that BD is a neuronal disorder. To address this issue, postmortem studies should be used to investigate whether UPR activation is impaired in brain tissue from BD patients. Another fundamental challenge is that BD patients are often treated simultaneously with multiple drugs, making it challenging to separate UPR phenotypes associated with the disease itself vs. those resulting from mood stabilizers or other medications (Pfaffenseller et al., 2014). This matter could be addressed by characterizing the UPR pathway in BD patients who are not treated by mood stabilizers. However, a caveat to this is that recruiting untreated BD patients may pose both logistical problems (e.g., small sample sizes) and ethical dilemmas, as the disease is associated with high rates of suicide, especially in untreated patients (Gonda et al., 2012; Baldessarini et al., 2020). Therefore, the best strategy to clarify existing data is to focus research efforts on collecting and analyzing larger, more diverse data sets, identifying biological markers for BD, and utilizing postmortem studies to investigate UPR activation in the brain.
Uncovering the role of the UPR in the pathophysiology and treatment of BD would facilitate the development of more effective drugs to treat this debilitating and widespread disease. Additionally, to characterize the extent of UPR deficiency in BD patients, studies should investigate whether downstream targets of UPR activation are also impaired in BD patients. For example, a deficiency in UPR activation may lead to abnormalities in lipid metabolism, mitochondrial function, protein secretion, or calcium signaling (Gong et al., 2017). Therefore, drugs could be designed to target specific signaling branches of the UPR pathway or act to resolve the downstream deficiencies of UPR activation. These drugs could be chemical chaperones such as 4-Phenylbutyric acid (PBA), which alleviates ER stress by assisting protein folding in the ER, and is already approved by the FDA to treat urea cycle disorder (Roy et al., 2015). Ultimately, specific and effective drugs with fewer side effects will reduce the severity of this disease and improve the lives of BD patients.
MSu, MSc, and MG wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was supported by the National Institutes of Health grant number R01GM125082 (to MG) and a Rumble Fellowship from the Department of Biological Sciences, Wayne State University (to MSu).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Baldessarini, R. J., Vazquez, G. H., and Tondo, L. (2020). Bipolar depression: a major unsolved challenge. Int. J. Bipolar Disord. 8:1. doi: 10.1186/s40345-019-0160-1
Baumeister, P., Dong, D., Fu, Y., and Lee, A. S. (2009). Transcriptional induction of GRP78/BiP by histone deacetylase inhibitors and resistance to histone deacetylase inhibitor-induced apoptosis. Mol. Cancer Ther. 8, 1086–1094. doi: 10.1158/1535-7163.MCT-08-1166
Bengesser, S. A., Fuchs, R., Lackner, N., Birner, A., Reininghaus, B., Meier-Allard, N., et al. (2016). Endoplasmic reticulum stress and bipolar disorder - almost forgotten therapeutic drug targets in the unfolded protein response pathway revisited. CNS Neurol. Disord. Drug Targets 15, 403–413. doi: 10.2174/1871527315666160321104613
Bengesser, S. A., Reininghaus, E. Z., Dalkner, N., Birner, A., Hohenberger, H., Queissner, R., et al. (2018). Endoplasmic reticulum stress in bipolar disorder? - BiP and CHOP gene expression- and XBP1 splicing analysis in peripheral blood. Psychoneuroendocrinology 95, 113–119. doi: 10.1016/j.psyneuen.2018.05.029
Berk, M., Kapczinski, F., Andreazza, A. C., Dean, O. M., Giorlando, F., Maes, M., et al. (2011). Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci. Biobehav. Rev. 35, 804–817. doi: 10.1016/j.neubiorev.2010.10.001
Berridge, M. J. (2014). Calcium signalling and psychiatric disease: bipolar disorder and schizophrenia. Cell Tissue Res. 357, 477–492. doi: 10.1007/s00441-014-1806-z
Bizzarri, M., Fuso, A., Dinicola, S., Cucina, A., and Bevilacqua, A. (2016). Pharmacodynamics and pharmacokinetics of inositol(s) in health and disease. Expert Opin. Drug Metab. Toxicol. 12, 1181–1196. doi: 10.1080/17425255.2016.1206887
Bown, C. D., Wang, J. F., and Young, L. T. (2000). Increased expression of endoplasmic reticulum stress proteins following chronic valproate treatment of rat C6 glioma cells. Neuropharmacology 39, 2162–2169. doi: 10.1016/s0028-3908(00)00029-0
Braakman, I., and Hebert, D. N. (2013). Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 5:a013201. doi: 10.1101/cshperspect.a013201
Brunkhorst-Kanaan, N., Klatt-Schreiner, K., Hackel, J., Schroter, K., Trautmann, S., Hahnefeld, L., et al. (2019). Targeted lipidomics reveal derangement of ceramides in major depression and bipolar disorder. Metabolism 95, 65–76. doi: 10.1016/j.metabol.2019.04.002
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., et al. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96. doi: 10.1038/415092a
Callaly, E., Walder, K., Morris, G., Maes, M., Debnath, M., and Berk, M. (2015). Mitochondrial dysfunction in the pathophysiology of bipolar disorder: effects of pharmacotherapy. Mini Rev. Med. Chem. 15, 355–365. doi: 10.2174/1389557515666150324122026
Casas, C. (2017). GRP78 at the centre of the stage in cancer and neuroprotection. Front. Neurosci. 11:177. doi: 10.3389/fnins.2017.00177
Chan, J. Y., Luzuriaga, J., Maxwell, E. L., West, P. K., Bensellam, M., and Laybutt, D. R. (2015). The balance between adaptive and apoptotic unfolded protein responses regulates beta-cell death under ER stress conditions through XBP1, CHOP and JNK. Mol. Cell Endocrinol. 413, 189–201. doi: 10.1016/j.mce.2015.06.025
Chen, B., Wang, J. F., and Young, L. T. (2000). Chronic valproate treatment increases expression of endoplasmic reticulum stress proteins in the rat cerebral cortex and hippocampus. Biol. Psychiatry 48, 658–664. doi: 10.1016/s0006-3223(00)00878-7
Chuang, L. C., and Kuo, P. H. (2017). Building a genetic risk model for bipolar disorder from genome-wide association data with random forest algorithm. Sci. Rep. 7:39943. doi: 10.1038/srep39943
Dadey, D. Y., Kapoor, V., Khudanyan, A., Urano, F., Kim, A. H., Thotala, D., et al. (2016). The ATF6 pathway of the ER stress response contributes to enhanced viability in glioblastoma. Oncotarget 7, 2080–2092. doi: 10.18632/oncotarget.6712
Dreifuss, F. E., Santilli, N., Langer, D. H., Sweeney, K. P., Moline, K. A., and Menander, K. B. (1987). Valproic acid hepatic fatalities: a retrospective review. Neurology 37, 379–385. doi: 10.1212/wnl.37.3.379
Enogieru, A. B., Omoruyi, S. I., Hiss, D. C., and Ekpo, O. E. (2019). GRP78/BIP/HSPA5 as a therapeutic target in models of Parkinson’s disease: a mini review. Adv. Pharmacol. Sci. 2019:2706783. doi: 10.1155/2019/2706783
Foti, D. M., Welihinda, A., Kaufman, R. J., and Lee, A. S. (1999). Conservation and divergence of the yeast and mammalian unfolded protein response. Activation of specific mammalian endoplasmic reticulum stress element of the grp78/BiP promoter by yeast Hac1. J. Biol. Chem. 274, 30402–30409. doi: 10.1074/jbc.274.43.30402
Freeman, O. J., and Mallucci, G. R. (2016). The UPR and synaptic dysfunction in neurodegeneration. Brain Res. 1648(Pt B), 530–537. doi: 10.1016/j.brainres.2016.03.029
Gardner, B. M., and Walter, P. (2011). Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 333, 1891–1894. doi: 10.1126/science.1209126
Geddes, J. R., and Miklowitz, D. J. (2013). Treatment of bipolar disorder. Lancet 381, 1672–1682. doi: 10.1016/S0140-6736(13)60857-0
Gitlin, M. (2016). Lithium side effects and toxicity: prevalence and management strategies. Int. J. Bipolar Disord. 4:27. doi: 10.1186/s40345-016-0068-y
Gokcinar, N. B., Buturak, S. V., Ozkal, F., Ozcicek, G., Yumusak, M. E., and Turgal, E. (2020). Optical coherence tomography neurodegenerative findings in patients with bipolar disorder. Asia Pac. Psychiatry 12:e12394. doi: 10.1111/appy.12394
Gonda, X., Pompili, M., Serafini, G., Montebovi, F., Campi, S., Dome, P., et al. (2012). Suicidal behavior in bipolar disorder: epidemiology, characteristics and major risk factors. J. Affect Disord. 143, 16–26. doi: 10.1016/j.jad.2012.04.041
Gong, J., Wang, X. Z., Wang, T., Chen, J. J., Xie, X. Y., Hu, H., et al. (2017). Molecular signal networks and regulating mechanisms of the unfolded protein response. J. Zhejiang Univ. Sci. B 18, 1–14. doi: 10.1631/jzus.B1600043
Grande, I., Berk, M., Birmaher, B., and Vieta, E. (2016). Bipolar disorder. Lancet 387, 1561–1572. doi: 10.1016/S0140-6736(15)00241-X
Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., et al. (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108. doi: 10.1016/s1097-2765(00)00108-8
Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. doi: 10.1016/s1097-2765(03)00105-9
Harrison, P. J., Hall, N., Mould, A., Al-Juffali, N., and Tunbridge, E. M. (2019). Cellular calcium in bipolar disorder: systematic review and meta-analysis. Mol. Psychiatry 1–11. doi: 10.1038/s41380-019-0622-y Available online at: https://www.nature.com/articles/s41380-019-0622-y
Hayashi, A., Kasahara, T., Kametani, M., Toyota, T., Yoshikawa, T., and Kato, T. (2009). Aberrant endoplasmic reticulum stress response in lymphoblastoid cells from patients with bipolar disorder. Int. J. Neuropsychopharmacol. 12, 33–43. doi: 10.1017/S1461145708009358
Hiramatsu, N., Chiang, W. C., Kurt, T. D., Sigurdson, C. J., and Lin, J. H. (2015). Multiple mechanisms of unfolded protein response-induced cell death. Am. J. Pathol. 185, 1800–1808. doi: 10.1016/j.ajpath.2015.03.009
Hiramatsu, N., Joseph, V. T., and Lin, J. H. (2011). Monitoring and manipulating mammalian unfolded protein response. Methods Enzymol. 491, 183–198. doi: 10.1016/B978-0-12-385928-0.00011-0
Hou, N. S., Gutschmidt, A., Choi, D. Y., Pather, K., Shi, X., Watts, J. L., et al. (2014). Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc. Natl. Acad. Sci. U.S.A. 111, E2271–E2280. doi: 10.1073/pnas.1318262111
Hou, S. J., Yen, F. C., Cheng, C. Y., Tsai, S. J., and Hong, C. J. (2004). X-box binding protein 1 (XBP1) C–116G polymorphisms in bipolar disorders and age of onset. Neurosci. Lett. 367, 232–234. doi: 10.1016/j.neulet.2004.06.012
Huang, Y. J., Tsai, S. Y., Chung, K. H., Chen, P. H., Huang, S. H., and Kuo, C. J. (2018). State-dependent alterations of lipid profiles in patients with bipolar disorder. Int. J. Psychiatry Med. 53, 273–281. doi: 10.1177/0091217417749786
Jadhav, S., Russo, S., Cottier, S., Schneiter, R., Cowart, A., and Greenberg, M. L. (2016). Valproate induces the unfolded protein response by increasing ceramide levels. J. Biol. Chem. 291, 22253–22261. doi: 10.1074/jbc.M116.752634
Kakiuchi, C., Ishigaki, S., Oslowski, C. M., Fonseca, S. G., Kato, T., and Urano, F. (2009). Valproate, a mood stabilizer, induces WFS1 expression and modulates its interaction with ER stress protein GRP94. PLoS One 4:e4134. doi: 10.1371/journal.pone.0004134
Kakiuchi, C., Iwamoto, K., Ishiwata, M., Bundo, M., Kasahara, T., Kusumi, I., et al. (2003). Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat. Genet. 35, 171–175. doi: 10.1038/ng1235
Kakiuchi, C., Nanko, S., Kunugi, H., and Kato, T. (2004). Reply to” Lack of support for a genetic association of the XBP1 promoter polymorphism with bipolar disorder in probands of European origin”. Nat. Genet. 36, 784–785. doi: 10.1038/ng0804-783
Karabulut, S., Tasdemir, I., Akcan, U., Kucukali, C. I., Tuzun, E., and Cakir, S. (2019). Inflammation and neurodegeneration in patients with early-stageand chronic bipolar disorder. Turk Psikiyatri Derg 30, 75–81.
Kaufman, R. J., Scheuner, D., Schroder, M., Shen, X., Lee, K., Liu, C. Y., et al. (2002). The unfolded protein response in nutrient sensing and differentiation. Nat. Rev. Mol. Cell. Biol. 3, 411–421. doi: 10.1038/nrm829
Kawahara, T., Yanagi, H., Yura, T., and Mori, K. (1997). Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol. Biol. Cell 8, 1845–1862. doi: 10.1091/mbc.8.10.1845
Kilberg, M. S., Balasubramanian, M., Fu, L., and Shan, J. (2012). The transcription factor network associated with the amino acid response in mammalian cells. Adv. Nutr. 3, 295–306. doi: 10.3945/an.112.001891
Kim, B., Kim, C. Y., Lee, M. J., and Joo, Y. H. (2009). Preliminary evidence on the association between XBP1-116C/G polymorphism and response to prophylactic treatment with valproate in bipolar disorders. Psychiatry Res. 168, 209–212. doi: 10.1016/j.psychres.2008.05.010
Korennykh, A. V., Egea, P. F., Korostelev, A. A., Finer-Moore, J., Zhang, C., Shokat, K. M., et al. (2009). The unfolded protein response signals through high-order assembly of Ire1. Nature 457, 687–693. doi: 10.1038/nature07661
Krebs, J., Agellon, L. B., and Michalak, M. (2015). Ca2+ homeostasis and endoplasmic reticulum (ER) stress: an integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 460, 114–121. doi: 10.1016/j.bbrc.2015.02.004
Li, Z., Wu, F., Zhang, X., Chai, Y., Chen, D., Yang, Y., et al. (2017). Valproate attenuates endoplasmic reticulum stress-induced apoptosis in SH-SY5Y cells via the AKT/GSK3β signaling pathway. Int. J. Mol. Sci. 18:315. doi: 10.3390/ijms18020315
Lin, J. H., Li, H., Zhang, Y., Ron, D., and Walter, P. (2009). Divergent effects of PERK and IRE1 signaling on cell viability. PLoS One 4:e4170. doi: 10.1371/journal.pone.0004170
Liu, Z., Xia, Y., Li, B., Xu, H., Wang, C., Liu, Y., et al. (2014). Induction of ER stress-mediated apoptosis by ceramide via disruption of ER Ca(2+) homeostasis in human adenoid cystic carcinoma cells. Cell Biosci. 4:71. doi: 10.1186/2045-3701-4-71
Mandl, J., Meszaros, T., Banhegyi, G., Hunyady, L., and Csala, M. (2009). Endoplasmic reticulum: nutrient sensor in physiology and pathology. Trends Endocrinol. Metab. 20, 194–201. doi: 10.1016/j.tem.2009.01.003
Martínez, G., Vidal, R. L., Mardones, P., Serrano, F. G., Ardiles, A. O., Wirth, C., et al. (2016). Regulation of memory formation by the transcription factor XBP1. Cell Rep. 14, 1382–1394. doi: 10.1016/j.celrep.2016.01.028
Mercke, Y., Sheng, H., Khan, T., and Lippmann, S. (2000). Hair loss in psychopharmacology. Ann. Clin. Psychiatry 12, 35–42. doi: 10.1023/a:1009074926921
Miranda, F. C., Tamburini, G., Martinez, G., Medinas, D., Gerakis, Y., Miedema, T., et al. (2020). Control of mammalian brain aging by the unfolded protein response (UPR). Biorxiv [Preprint] doi: 10.1101/2020.04.13.039172
Nosyreva, E., and Kavalali, E. T. (2010). Activity-dependent augmentation of spontaneous neurotransmission during endoplasmic reticulum stress. J. Neurosci. 30, 7358–7368. doi: 10.1523/JNEUROSCI.5358-09.2010
Perri, E. R., Thomas, C. J., Parakh, S., Spencer, D. M., and Atkin, J. D. (2015). The unfolded protein response and the role of protein disulfide isomerase in neurodegeneration. Front. Cell Dev. Biol. 3:80. doi: 10.3389/fcell.2015.00080
Pfaffenseller, B., Wollenhaupt-Aguiar, B., Fries, G. R., Colpo, G. D., Burque, R. K., Bristot, G., et al. (2014). Impaired endoplasmic reticulum stress response in bipolar disorder: cellular evidence of illness progression. Int. J. Neuropsychopharmacol. 17, 1453–1463. doi: 10.1017/S1461145714000443
Pijl, H., and Meinders, A. E. (1996). Bodyweight change as an adverse effect of drug treatment. Mechanisms and management. Drug Saf. 14, 329–342. doi: 10.2165/00002018-199614050-00005
Piperi, C., Adamopoulos, C., and Papavassiliou, A. G. (2016). XBP1: a pivotal transcriptional regulator of glucose and lipid metabolism. Trends Endocrinol. Metab. 27, 119–122. doi: 10.1016/j.tem.2016.01.001
Roy, D., Kumar, V., James, J., Shihabudeen, M. S., Kulshrestha, S., Goel, V., et al. (2015). Evidence that chemical chaperone 4-phenylbutyric acid binds to human serum albumin at fatty acid binding sites. PLoS One 10:e0133012. doi: 10.1371/journal.pone.0133012
Saiardi, A., and Mudge, A. W. (2018). Lithium and fluoxetine regulate the rate of phosphoinositide synthesis in neurons: a new view of their mechanisms of action in bipolar disorder. Transl. Psychiatry 8, 1–12. doi: 10.1038/s41398-018-0235-2
Schardt, J. A., Eyholzer, M., Timchenko, N. A., Mueller, B. U., and Pabst, T. (2010). Unfolded protein response suppresses CEBPA by induction of calreticulin in acute myeloid leukaemia. J. Cell Mol. Med. 14, 1509–1519. doi: 10.1111/j.1582-4934.2009.00870.x
Schwarz, E., Prabakaran, S., Whitfield, P., Major, H., Leweke, F. M., Koethe, D., et al. (2008). High throughput lipidomic profiling of schizophrenia and bipolar disorder brain tissue reveals alterations of free fatty acids, phosphatidylcholines, and ceramides. J. Proteome Res. 7, 4266–4277. doi: 10.1021/pr800188y
Senkal, C. E., Ponnusamy, S., Manevich, Y., Meyers-Needham, M., Saddoughi, S. A., Mukhopadyay, A., et al. (2011). Alteration of ceramide synthase 6/C16-ceramide induces activating transcription factor 6-mediated endoplasmic reticulum (ER) stress and apoptosis via perturbation of cellular Ca2+ and ER/Golgi membrane network. J. Biol. Chem. 286, 42446–42458. doi: 10.1074/jbc.M111.287383
Shao, L., Sun, X., Xu, L., Young, L. T., and Wang, J. F. (2006). Mood stabilizing drug lithium increases expression of endoplasmic reticulum stress proteins in primary cultured rat cerebral cortical cells. Life Sci. 78, 1317–1323. doi: 10.1016/j.lfs.2005.07.007
Shi, Y., Gerritsma, D., Bowes, A. J., Capretta, A., and Werstuck, G. H. (2007). Induction of GRP78 by valproic acid is dependent upon histone deacetylase inhibition. Bioorganic Med. Chem. Lett. 17, 4491–4494. doi: 10.1016/j.bmcl.2007.06.006
Shimon, H., Agam, G., Belmaker, R. H., Hyde, T. M., and Kleinman, J. E. (1997). Reduced frontal cortex inositol levels in postmortem brain of suicide victims and patients with bipolar disorder. Am. J. Psychiatry 154, 1148–1150. doi: 10.1176/ajp.154.8.1148
Sigitova, E., Fisar, Z., Hroudova, J., Cikankova, T., and Raboch, J. (2017). Biological hypotheses and biomarkers of bipolar disorder. Psychiatry Clin. Neurosci. 71, 77–103. doi: 10.1111/pcn.12476
Silverstone, P. H., McGrath, B. M., and Kim, H. (2005). Bipolar disorder and myo-inositol: a review of the magnetic resonance spectroscopy findings. Bipolar Disord. 7, 1–10. doi: 10.1111/j.1399-5618.2004.00174.x
So, J., Warsh, J. J., and Li, P. P. (2007). Impaired endoplasmic reticulum stress response in B-lymphoblasts from patients with bipolar-I disorder. Biol. Psychiatry 62, 141–147. doi: 10.1016/j.biopsych.2006.10.014
Stahl, E. A., Breen, G., Forstner, A. J., McQuillin, A., Ripke, S., Trubetskoy, V., et al. (2019). Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 51, 793–803. doi: 10.1038/s41588-019-0397-8
Vaden, D. L., Ding, D., Peterson, B., and Greenberg, M. L. (2001). Lithium and valproate decrease inositol mass and increase expression of the yeast INO1 and INO2 genes for inositol biosynthesis. J. Biol. Chem. 276, 15466–15471. doi: 10.1074/jbc.M004179200
Vieta, E., Berk, M., Schulze, T. G., Carvalho, A. F., Suppes, T., Calabrese, J. R., et al. (2018). Bipolar disorders. Nat. Rev. Dis. Primers 4:18008. doi: 10.1038/nrdp.2018.8
Vincenz-Donnelly, L., and Hipp, M. S. (2017). The endoplasmic reticulum: a hub of protein quality control in health and disease. Free Radic. Biol. Med. 108, 383–393. doi: 10.1016/j.freeradbiomed.2017.03.031
Viswanath, B., Jose, S. P., Squassina, A., Thirthalli, J., Purushottam, M., Mukherjee, O., et al. (2015). Cellular models to study bipolar disorder: a systematic review. J. Affect Disord. 184, 36–50. doi: 10.1016/j.jad.2015.05.037
Wang, J. F., Bown, C., and Young, L. T. (1999). Differential display PCR reveals novel targets for the mood-stabilizing drug valproate including the molecular chaperone GRP78. Mol. Pharmacol. 55, 521–527.
Wang, X., Zhang, T., Mao, H., Mi, Y., Zhong, B., Wei, L., et al. (2016). Grass carp (Ctenopharyngodon idella) ATF6 (activating transcription factor 6) modulates the transcriptional level of GRP78 and GRP94 in CIK cells. Fish Shellfish Immunol. 52, 65–73. doi: 10.1016/j.fsi.2016.03.028
Wang, Y., Shen, J., Arenzana, N., Tirasophon, W., Kaufman, R. J., and Prywes, R. (2000). Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J. Biol. Chem. 275, 27013–27020. doi: 10.1074/jbc.M003322200
Weng, W. C., Lee, W. T., Hsu, W. M., Chang, B. E., and Lee, H. (2011). Role of glucose-regulated Protein 78 in embryonic development and neurological disorders. J. Formos Med. Assoc. 110, 428–437. doi: 10.1016/S0929-6646(11)60064-8
Xu, C., Bailly-Maitre, B., and Reed, J. C. (2005). Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Invest. 115, 2656–2664. doi: 10.1172/JCI26373
Yamamoto, K., Yoshida, H., Kokame, K., Kaufman, R. J., and Mori, K. (2004). Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 136, 343–350. doi: 10.1093/jb/mvh122
Yan, M., Shen, J., Person, M. D., Kuang, X., Lynn, W. S., Atlas, D., et al. (2008). Endoplasmic reticulum stress and unfolded protein response in Atm-deficient thymocytes and thymic lymphoma cells are attributable to oxidative stress. Neoplasia 10, 160–167. doi: 10.1593/neo.07935
Ye, C., and Greenberg, M. L. (2015). Inositol synthesis regulates the activation of GSK-3alpha in neuronal cells. J. Neurochem. 133, 273–283. doi: 10.1111/jnc.12978
Yonkers, K. A., Wisner, K. L., Stowe, Z., Leibenluft, E., Cohen, L., Miller, L., et al. (2004). Management of bipolar disorder during pregnancy and the postpartum period. Am. J. Psychiatry 161, 608–620. doi: 10.1176/appi.ajp.161.4.608
Yoshida, H., Nadanaka, S., Sato, R., and Mori, K. (2006). XBP1 is critical to protect cells from endoplasmic reticulum stress: evidence from Site-2 protease-deficient Chinese hamster ovary cells. Cell Struct. Funct. 31, 117–125. doi: 10.1247/csf.06016
Yu, W., Daniel, J., Mehta, D., Maddipati, K. R., and Greenberg, M. L. (2017). MCK1 is a novel regulator of myo-inositol phosphate synthase (MIPS) that is required for inhibition of inositol synthesis by the mood stabilizer valproate. PLoS One 12:e0182534. doi: 10.1371/journal.pone.0182534
Keywords: bipolar disorder, endoplasmic reticulum stress, mood disorder, unfolded protein response, valproate, lithium
Citation: Suliman M, Schmidtke MW and Greenberg ML (2021) The Role of the UPR Pathway in the Pathophysiology and Treatment of Bipolar Disorder. Front. Cell. Neurosci. 15:735622. doi: 10.3389/fncel.2021.735622
Received: 02 July 2021; Accepted: 09 August 2021;
Published: 31 August 2021.
Edited by:
Xin Qi, Case Western Reserve University, United StatesReviewed by:
Senthilkumar Rajagopal, Rayalaseema University, IndiaCopyright © 2021 Suliman, Schmidtke and Greenberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miriam L. Greenberg, bWdyZWVuYmVyZ0B3YXluZS5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.