 Kaige Ma1†
Kaige Ma1† Shan Xing2†
Shan Xing2† Yan Luan1
Yan Luan1 Chenglin Zhang3
Chenglin Zhang3 Yingfei Liu1
Yingfei Liu1 Yulang Fei4
Yulang Fei4 Zhichao Zhang 1
Zhichao Zhang 1 Yong Liu1
Yong Liu1 Xinlin Chen1*
Xinlin Chen1*- 1Institute of Neurobiology, Department of Neurobiology, Xi’an Jiaotong University Health Science Center, Xi’an, China
- 2Department of Neonatology, Children’s Hospital Affiliated to Zhengzhou University, Henan Children’s Hospital, Zhengzhou Children’s Hospital, Zhengzhou, China
- 32018 Grade, Zonglian College, Xi’an Jiaotong University Health Science Center, Xi’an, China
- 4Medical College, Xijing University, Xi’an, China
Neural stem cell (NSC) damage has been reported in patients with Alzheimer’s disease. Intracellular Aβ plays a vital role in NSC damage. Heparan sulfate proteoglycans are potent mediators of Aβ enrichment in the brain. We hypothesized the heparan sulfate proteoglycan glypican 4 (Gpc4) regulates Aβ internalization by NSCs. We evaluated Gpc4 expression in NSCs from P0–P2 generations using immunofluorescence. Adenovirus and lentivirus were used to regulate Gpc4 expression in NSCs and APP/PS1 mice, respectively. Co-immunoprecipitation was used to determine the relationship between Gpc4, Aβ, and low-density lipoprotein receptor-related protein 1 (LRP1). Intracellular Aβ concentrations were detected using enzyme-linked immunosorbent assay and immunofluorescence. The role of Gpc4/LRP1 on toxic/physical Aβ-induced effects was evaluated using the JC-1 kit, terminal deoxynucleotidyl transferase dUPT nick end labeling, and western blotting. Gpc4 was stably expressed in NSCs, neurons, and astrocytes. Gpc4 was upregulated by Aβ in NSCs and regulated Aβ internalization. Gpc4 attenuation reduced Aβ uptake; Gpc4 overexpression increased Aβ uptake. Gpc4 regulated Aβ internalization through LRP1 and contributed to Aβ internalization and toxic/physical concentrations of Aβ-induced mitochondrial membrane potential and cell apoptosis, partly via LRP1. Therefore, Gpc4 is a key regulator of Aβ enrichment in NSCs. Inhibiting Gpc4 rescued the Aβ-induced toxic effect and attenuated the nontoxic Aβ enrichment into intracellular toxic concentrations. Gpc4 contributed to Aβ internalization and toxic/physical concentrations of Aβ-induced mitochondrial membrane potential damage and cell apoptosis, partly via LRP1. These findings suggest a potential role of Gpc4 in treating Alzheimer’s disease at an early stage, by targeting NSCs.
Introduction
Neural stem cell (NSC) damage has been reported in the brains of Alzheimer’s disease (AD) patients (Wang et al., 2016; Huang et al., 2021). Cognitive decline in AD patients may rise because differentiation of NSCs in the adult brain cannot compensate for neuronal loss.
How does impairment of neurogenesis occur in patients with AD? Intracellular β-amyloid peptide (Aβ) aggregation occurs before the extracellular deposition and plaque formation (Bloom, 2014; Tiwari et al., 2019). Intracellular accumulation disrupts the cytoskeletal structure and induces cell apoptosis (Mohamed and Posse de Chaves, 2011). Aβ also induces neuronal toxicity in the early stages of AD (Oakley et al., 2006). Therefore, understanding the intracellular effects of Aβ will be beneficial for identifying potential targets for AD treatment. Manipulation of Aβ transport into NSCs is a promising line of research for the development of AD therapies.
Heparan sulfate proteoglycans accumulate abundantly in conjunction with Aβ (Timmer et al., 2010; Fu et al., 2016), and are particularly present in senile plaques (Ghazale et al., 2018). For two decades, the mRNA expression of glypican 4 (Gpc4) has been known to be upregulated in the brains of patients with AD, but its role in the disease remains to be elucidated.
Gpc4 is widely expressed in NSCs, astrocytes, and neurons. Its expression pattern suggests it has multiple roles during nervous system development. Interestingly, the heparan sulfate on the extracellular surface of Gpc4 has a high affinity for Aβ and its receptors, such as low-density lipoprotein receptor-related protein 1 (LRP1) and apolipoprotein E (ApoE; Liu et al., 2013, 2016). This suggests Gpc4 may play an important role in Aβ internalization in NSCs.
In this study, we hypothesized that Gpc4 may assist in Aβ internalization in NSCs, reducing their viability and facilitating the induction of apoptosis. Our results suggest this may be the case in the early stages of AD. Manipulating the transportation of Aβ into NSCs at an early stage by interfering with Gpc4 is a promising approach for AD therapeutic studies.
Materials and Methods
All animal experimental protocols were devised in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Xi’an Jiaotong University and. All efforts were made to minimize the number of animals used and their suffering.
NSC Culture
NSCs were isolated from mouse embryos (E14.5) as previously described (Zhang et al., 2015; Jiao et al., 2016; Ghazale et al., 2018). Epidermal growth factor, fibroblast growth factor, B27, and N2 were added to Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 as the NSC culture medium. NSCs from the P2 generation were plated at a density of 9 × 107 cells/well. NSCs were transfected with Gpc4 siRNA adenovirus (AdV), according to the manufacturer’s instructions (GeneChem, Shanghai, China). NSCs were treated with AdV for 24 h and then the medium with AdV was changed to complete medium without AdV. After 72 h of infection, Aβ was added. Internalization was detected after 6 h, and toxic effects were detected after 12 h.
Cell Viability Assay
Cell viability was evaluated using the Cell Counting Kit-8 (CCK-8, Sigma-Aldrich, St. Louis, MO, USA) assay. Cells were plated in 96-well plates at 20,000 cells/well density, 24 h before the experiments. The cells were then treated with different concentrations of Aβ (0, 200 nM, 1 μM, 2 μM, 5 μM, 10 μM, and 20 μM) for 12 h. Next, 20 μl of CCK-8 were added to each well and the cells were incubated for 2 h at 37°C. Absorbance was measured at 490 nm using a microplate spectrophotometer (BioTek, Winooski, VT, USA). Triplicate parallel wells were examined in each experiment, and data were collected from three independent experiments. The results are presented as a percentage of the absorbance compared to the control group.
Terminal Deoxynucleotidyl Transferase dUPT Nick End Labeling (TUNEL) Staining
To investigate the toxic effect of Aβ on NSC apoptosis mechanism, a terminal deoxynucleotidyl transferase dUPT nick end labeling (TUNEL) assay was performed according to the manufacturer’s instructions (Roche Diagnostics, Basel, Switzerland). Cells were fixed on cover slides in 4% paraformaldehyde for 15 min. The cells were then permeabilized in 0.3% Triton X-100 in phosphate-buffered saline for 5 min on ice. Cells were incubated in the TUNEL reaction mixture for 1 h at 37°C. Subsequently, the cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; 1:3,000, Sigma-Aldrich) before mounting. Images were taken within 2 h. Nine random fields were counted for each group with a 40× objective. The ratio was calculated as the percentage of TUNEL-positive cells compared to that of DAPI-positive cells.
Flow Cytometry Analysis
Flow cytometry analysis was used for the apoptosis tests in this study. The propidium iodide (PI)/Annexin V apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ, USA) was used according to the manufacturer’s instructions. Cells were dissociated, washed in cold phosphate-buffered saline, and resuspended in 1× binding buffer to obtain a concentration of 1 × 106 cells/ml. The cells were incubated with PI/Annexin V for 15 min in the dark. Subsequently, the cell solution was analyzed using FACSCalibur (BD Biosciences) for PI/Annexin V. The percentage of apoptotic cells in this experiment was calculated as the ratio of Annexin V-positive and PI-negative cells from the lower right quarter.
Immunohistochemistry
For immunohistochemistry performed in APP/PS1 mice, mouse brains were fixed with 4% paraformaldehyde and frozen coronal slices (30 μm) were obtained using a frozen section machine (Leica, Wetzlar, Germany). NSCs were fixed in 4% paraformaldehyde. The sections were treated with 0.3% Triton for 30 min. The cells were then blocked with 5% bovine serum albumin for 30 min. The sections were incubated with primary antibodies overnight at 4°C. The cells were then incubated with the secondary antibodies, goat-anti-rabbit-IgG-488 and goat-anti-mouse-IgG-594 (Invitrogen, Carlsbad, CA, USA), for 2 h at room temperature. Slides were cover-slipped using a mounting medium containing DAPI (blue). The primary antibodies used in this experiment were: mouse polyclonal anti-nestin antibody (1:200, Novus, St. Louis, MO, USA), rabbit polyclonal anti-Gpc4 antibody (1:100, Abcam, Cambridge, UK), rabbit polyclonal anti-sox2 antibody (1:200, Abcam), rabbit polyclonal anti-Ki67 antibody (1:200, Abcam, Cambridge, UK), mouse polyclonal anti-sox2 antibody (1:200, Abcam, Cambridge, UK), mouse polyclonal anti-LRP1 antibody (1:1,000, Abcam, Cambridge, UK), goat-anti-rabbit-IgG-488, goat-anti-mouse-IgG-594 (Invitrogen, Carlsbad, CA, USA), and DAPI (1:3,000, Sigma-Aldrich, USA). For 3,3’-diaminobenzidine staining, we followed the manufacturer’s instructions using a secondary antibody staining kit (ZSGB-BIO Inc., Beijing, China).
Western Blotting
NSCs were homogenized in radioimmunoprecipitation assay buffer supplemented with a protease inhibitor and centrifuged (15,000× g) for 30 min at 4°C. The protein concentration was quantified using the BCA assay (Beyotime, Nanjing, China). Protein (10 μg) was added to each sample, electrophoresed on 10% sodium dodecyl sulfate polyacrylamide gels, and transferred to a polyvinylidene fluoride membrane (Millipore, Burlington, MA, USA). The membranes were then blocked in 10% skimmed milk for 2 h and incubated with the following primary antibodies: rabbit anti-Gpc4 (1:2,000), rabbit anti-Cyt c (1:500), rabbit anti-Bax (1:500), rabbit anti-Bcl 2 (1:500), rabbit anti-GAPDH (1:10,000), or β-actin (1:10,000) (Abcam, Cambridge, UK). The primary antibody was dissolved in 10% skimmed milk, and the membrane was incubated at 4°C overnight. The membrane was then washed and incubated with horseradish peroxidase-conjugated secondary antibody. Membranes were developed using enhanced chemiluminescence and exposed to photographic films. All results were obtained from three independent experiments. The results were analyzed using ImageJ software (Yang et al., 2015).
Virus Infection
siGpc4, overexpressed Gpc4 (overGpc4), and siLRP1 were coated with AdV (4 × 1012 μg/ml) to infect NSC cell cultures. The overGpc4 sequence was obtained from Gene_ID:14735, GenBank: NM_008150. The forward siGpc4 sequence was 5-GGAUGGCAGU GGAUGACUUTT-3 and the reverse was 5-AAGU CAUCCACUGCCAUCCTT-3. The forward and reverse siLRP1 sequences were 5-GCCCAUUGGAUGAGUUUCATT-3 and 5-UGAAACUCAUCCAAUGGGCTT-3, respectively. The lentivirus vector for Gpc4 overexpression was GV367 Ubi-MCS-SV40-EGFP-IRES-pruromycin (GENE, Shanghai, China), and the lentivirus vector for siGpc4 was hU6-MCS-Ubiquitin-FGFP-IRES-puromycin (GENE, Shanghai, China). AdV was plated into 6-well plates at a density of 2 × 106 cells/ml; 10 μl of 2 × 1012 μg/ml AdV was added to each well in NSC complete medium (GENE, Shanghai, China). Twenty-four hours after infection, the medium was replaced with a complete medium without AdV. The cells were used for the experiment 72 h after infection.
Enzyme-Linked Immunosorbent Assay (ELISA)
10 μl of 2 × 1012 μg/ml AdV (overGpc4 and si Gpc4) was plated into 6-well plates at a density of 2 × 106 cells/ml (GENE, Shanghai, China). Twenty-four hours after infection, the medium was replaced with a complete medium without AdV. Aβ was added into each 6-well plates cells at a working concentration of 200 nM or 2 μM, 72 h after Adv infection. NSC samples were collected 12 h after Aβ treatment. Mouse Aβ enzyme-linked immunosorbent assay (ELISA) Kit (R&D Systems, Minneapolis, MN, USA; Ma et al., 2016) and quantification experiments were conducted according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN, USA). Protein samples in each group were detected three times in the 96T ELISA kit. Aβ concentrations were calculated by the standard curve. This experiment was repeated three times.
APP/PS1 Mice
The APP/PS1 mice used in this study were double transgenic mice bred by crossing two lines of commercial simple transgenic mice: APPSWE (Tg2576 on a B6J background) and PS1dE9 on a C57BL6SJL background. Acute crossing of these two lines produces an accelerated mouse model of AD on a B6J/B6SJL background combining cognitive and amyloid pathologies starting at 4 months old. Genotypes were confirmed by a polymerase chain reaction from a tail biopsy. All wild type mice in this study are of B6J/B6SJL littermates. All APP/PS1 mice were APP-and PS1 positive. The animals were obtained by Yang Wei-Na from Xi’an Jiaotong University, housed in an animal facility with two mice per cage in temperature- (22 ± 1°C) and humidity-controlled (50 ± 10%) conditions under a 12 h light/dark cycle. The mice had access to food and water ad libitum for 4 months before being used in the experiment.
Gpc4 siRNA lentivirus was injected into each mouse’s brain via stereotaxic surgery under deep isoflurane anesthesia (n = 6). Gpc4 siRNA lentivirus was injected using stereotaxic coordinates and a microsyringe. Virus was injected into the lateral ventricles and the CA1 hippocampal subfield (rostrocaudal: 1.0 mm, mediolateral: 1.30 mm, dorsoventral: 1.8 mm vs. Bregma) of both sides (rostrocaudal: 1.80 mm, mediolateral: 1.00 mm, dorsoventral: 3.10 mm vs. Bregma). The injection speed was 0.2 μl/min, and the total volume/side was 1 μl, 2 × 108/μl. The control virus was also injected into APP/PS1 mice (n = 5). After surgery, the animals were given 2 months for recovery and lentivirus expression.
Thioflavin-S Staining
After the brain was perfusion-fixed and sectioned at 30 μm on a freezing microtome, the selected series were mounted onto gelatine-coated glass slides and air-dried. The tissue was hydrated by passing it through a series of ethyl alcohol solutions (100%, 95%, 80%, and 70%, 1–2 min in each) before staining with 1% Thioflavin-S for 30 min (Oakley et al., 2006). The slides were subsequently rinsed in distilled water and transferred through 80% and 95%, and two sets of 100%, ethanol for 30 s before cover-slipping with mounting media and storage under refrigeration. Thioflavin-S staining was viewed under a green fluorescence microscope (480/525 nm).
Immunoprecipitation Experiment
Immunoprecipitation (IP; Proteintech, Chicago, IL, USA) was performed as previously described (Ma et al., 2018). Eluted proteins that bind to any protein complex containing Gpc4 were separated via sodium dodecyl-sulfate polyacrylamide gel electrophoresis. Gpc4, LRP1, and Aβ were detected using western blotting. Briefly, APP/PS1 mouse brain tissue supernatants containing 4 mg protein (in approximately 1 ml lysate) were pre-cleared for 1–4 h at 4°C with 100 μl of 80–100 protein G. Precleared supernatants were incubated overnight at 4°C with the desired IP antibody (MOAB2, 1:200; Gpc4, 1:100; goat anti-rabbit IgG, 1:100) and 200 μl of protein G beads. IP complexes were washed five times with wash buffer and eluted twice with 40 μl elution buffer. Protein complexes (80 μl) were obtained and used for western blotting.
JC-1 for Mitochondrial Membrane Potential (MMP)
NSCs were stained with JC-1 to determine the mitochondrial membrane potential (MMP). JC-1 is a membrane-permeable lipophilic dye that exists as JC-1 aggregates in the mitochondrial matrix (red) or as monomers in the cytoplasm (green). During mitochondrial depolarization, the red JC-1 aggregates form green monomers due to a change in proton motive force (ΔΨ). Thus, depolarization can be measured as an increase in the green fluorescent/red fluorescent intensity ratio. The JC-1 assay was performed as follows. Solutions containing 5 μg/ml JC-1 were added for AdV and Aβ treated NSCs for 30 min. The stained cells were rinsed twice with the culture medium. After the fresh media was added, the cells were analyzed under a green fluorescence microscope (480/525 nm).
Statistical Analyses
All data were analyzed with GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA, USA). For normal distribution data, one-way analysis of variance (ANOVA) followed with Tukey’s multiple comparisons test were performed. P-value <0.05 was regarded as significant. t-test was used to compare between two groups. All in vitro experiments have been independently repeated at least three times. For in vivo experiments, in each group at least five mice were used for statistical analysis.
Results
Gpc4 Is Expressed in NSCs
Neural stem cells derived from the cortex of embryonic 14.5 (E14.5) mice were observed. Approximately 5 days after primary culture (P0), the isolated NSCs were passed for the next generation (P1) and then for P2 (Figure 1A). We observed that neural spheres from P0-P2 stably expressed Gpc4 and nestin (Figure 1B). After NSC differentiation, we also detected high colocalization of Gpc4 and both neurons and astrocytes (Figures 1C,D).
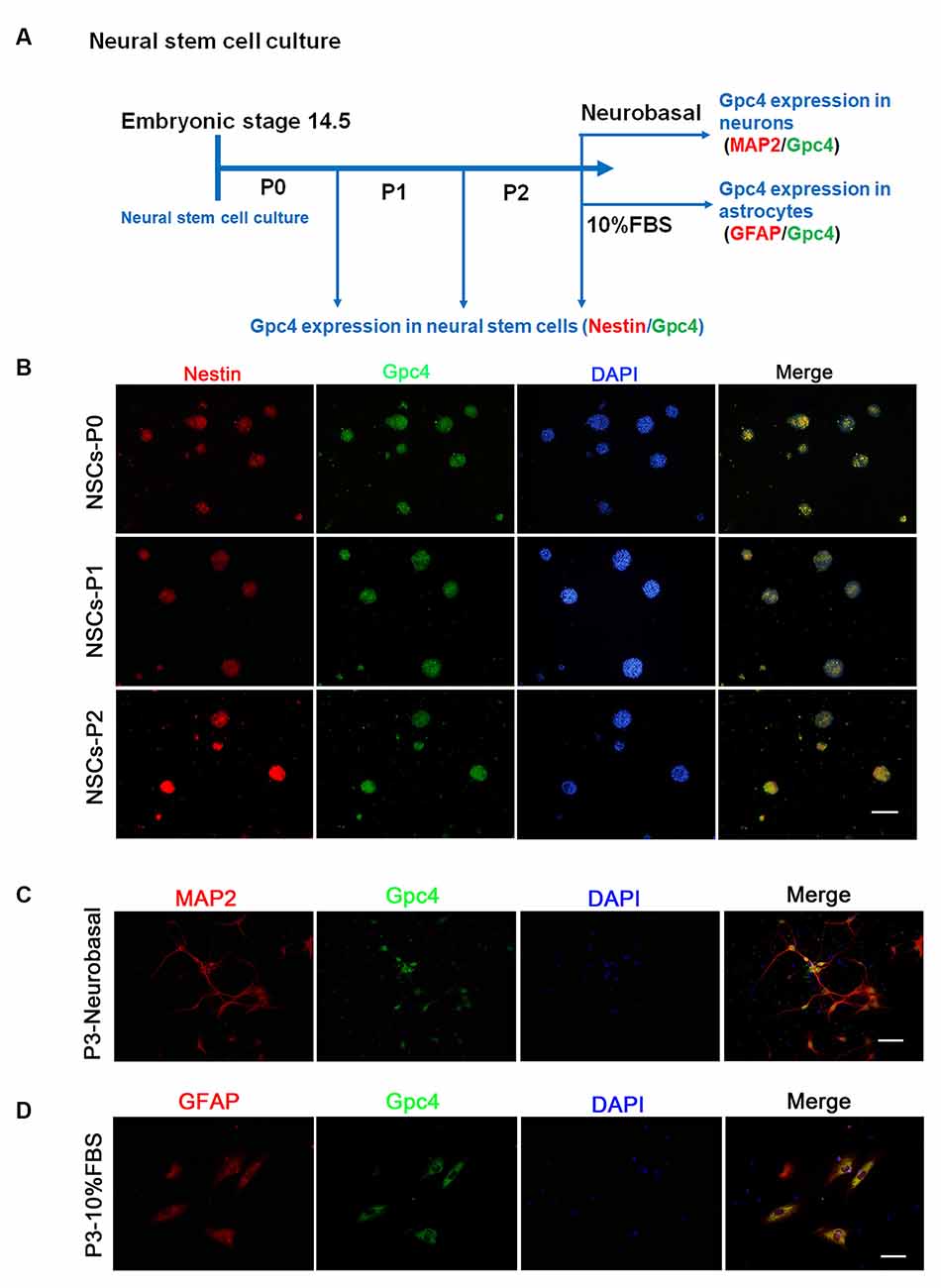
Figure 1. Expression of Gpc4 in neural stem cells (NSCs). (A) The schedule for detecting Gpc4 expression in NSCs and differentiated neurons and astrocytes. (B) At P0, P1, and P2, Gpc4 (green) colocalized with nestin (red) in the neural spheres. (C,D) NSCs were differentiated using neural basal medium containing B27 or DMEM/F12 containing 10% FBS. MAP2 (red) and GFAP (red) were used to illustrate Gpc4 (green) expression in neurons and astrocytes, respectively. Gpc4, glypican 4; DMEM/F12, Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12.
Different Concentrations of Aβ Affect NSC Viability and Apoptosis
This experiment aimed to determine the toxic dose of Aβ in NSCs. We found that 200 nM Aβ induced cell viability of NSCs, but that 1–20 μM Aβ was toxic to NSCs (Figure 2A). Therefore, 200 nM Aβ and 2 μM Aβ were used in this study. TUNEL staining showed that the number of apoptotic cells increased in the group treated with 2 μM Aβ compared to the 0 μM Aβ group (Figures 2B,C). We then used flow cytometry to detect PI/AnnexinV stained cells. The percentage of living cells in the group treated with 200 nM Aβ was significantly higher than that in the control group (0 μM Aβ). The percentage of living cells in the 2 μM Aβ group was significantly lower than that in the control group (0 μM Aβ). The apoptotic and living cell percentage in different concentrations of Aβ on NSCs is contrasting (Figures 2D,E). We used ELISA to detect the intracellular Aβ level. NSCs in the 200 nM group absorbed more Aβ than those in the control group. Cells treated with 2 μM Aβ showed a higher uptake of Aβ compared to the other groups (Figure 2F).
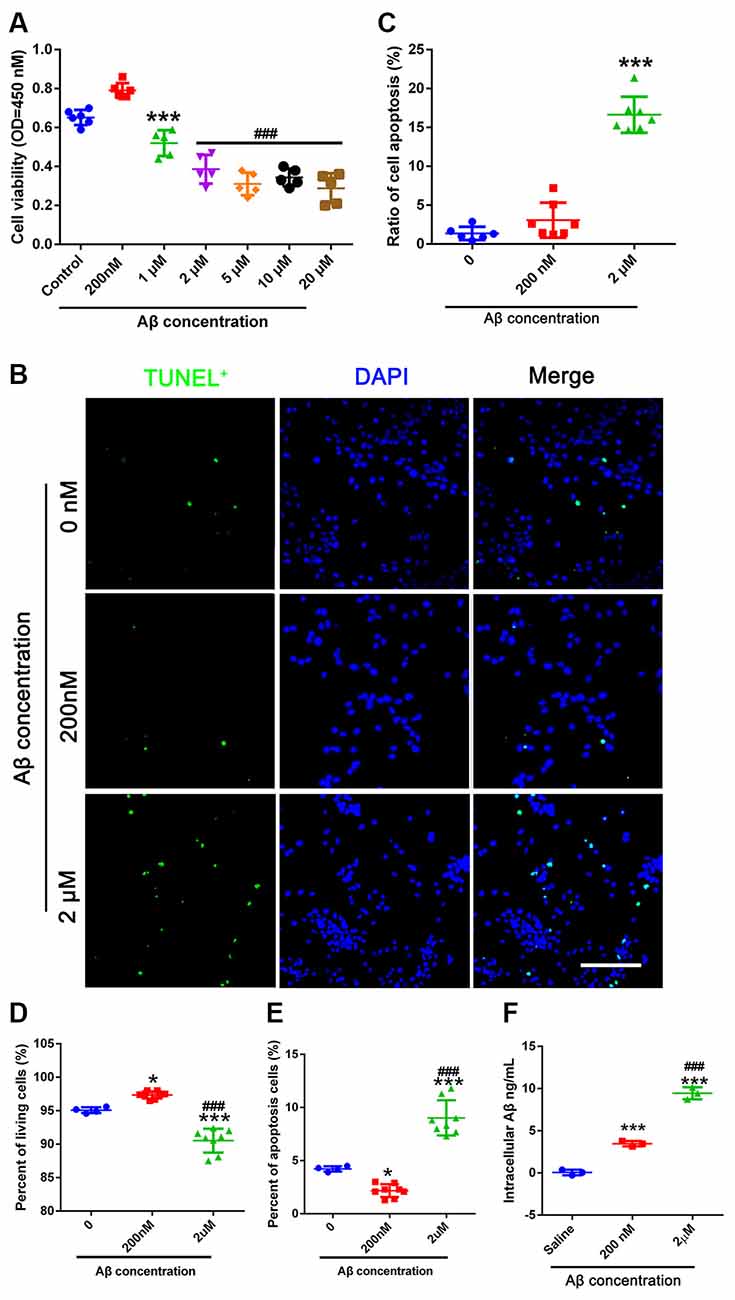
Figure 2. Different concentrations of Aβ affect NSC apoptosis. (A) CCK-8 was used to detect cell viability. NSCs were passed for 2–4 generations and plated into 96-well plates. Different concentrations of Aβ oligo were added for 24 h before the CCK-8 test. *** represents 1 μM vs. control group, P = 0.011. ### represents 2–20 μM vs. control group, P < 0.0001. (B) TUNEL was used to obtain apoptotic cell numbers in 0 μM Aβ-, 200 nM Aβ-, and 2 μM Aβ-treated NSCs. (C) The ratio of TUNEL (green) positive cells compared to DAPI (blue) was calculated. *** represents 2 μM Aβ vs. 0 μM Aβ, P < 0.0001. Scale bar = 50 μm. (D) NSCs treated with 0 μM Aβ, 200 nM Aβ, and 2 μM Aβ were harvested 24 h after Aβ treatment and stained with PI/AnnexinV for flow cytometry analysis. The percentage of living cells was calculated. * represents 200 nM Aβ vs. 0 μM Aβ, P = 0.0181, *** represents 2 μM Aβ vs. 0 μM Aβ, P < 0.0001, ### represents 2 μM Aβ vs. 200 nM Aβ, P < 0.0001. (E) Cells were treated as mentioned in (D). The percentage of apoptotic cells was calculated. * represents 200 nM Aβ vs. 0 μM Aβ, P = 0.0236, *** represents 2 μM Aβ vs. 0 μM Aβ, P < 0.0001, ### represents 2 μM Aβ vs. 200 nM Aβ, P < 0.0001. (F) 0 μM, 200 nM, and 2 μM Aβ were added to NSCs for 6 h. Intracellular Aβ was detected using the Aβ1–42-ELISA kit. *** represents 200 nM Aβ vs. 0 μM Aβ, P = 0.0003, *** represents 2 μM Aβ vs. 0 μM Aβ, P < 0.0001, ### represents 2 μM Aβ vs. 200 nM Aβ. NSC, neural stem cells; CCK-8, cell counting kit 8; TUNEL, terminal deoxynucleotidyl transferase dUPT nick end labeling; DAPI, 4′,6-diamidino-2-phenylindole; PI, propidium iodide; ELISA, enzyme-linked immunosorbent assay.
Aβ Upregulates Gpc4 Expression in NSCs
To identify the role of Gpc4 in NSCs in AD, we examined Gpc4 expression patterns in NSCs. Gpc4 (green) colocalized with cells positive for Ki67 or nestin (red), which proves the expression of Gpc4 in NSCs. Gpc4 was widely expressed in NSCs. Double-positive Gpc4/Ki67 represents NSCs undergoing cell division (Figure 3A, yellow arrow). We also observed some Gpc4-negative Ki67-positive NSCs (red arrow) and Gpc4-positive Ki67-negative NSCs (green arrow). Furthermore, we assessed whether the expression of Gpc4 was regulated by Aβ treatment. NSCs were incubated with 200 nM and 2 μM Aβ for 12 h. Our results from immunofluorescence staining and western blotting illustrated that Gpc4 expression increased with Aβ concentration (Figures 3B–D).
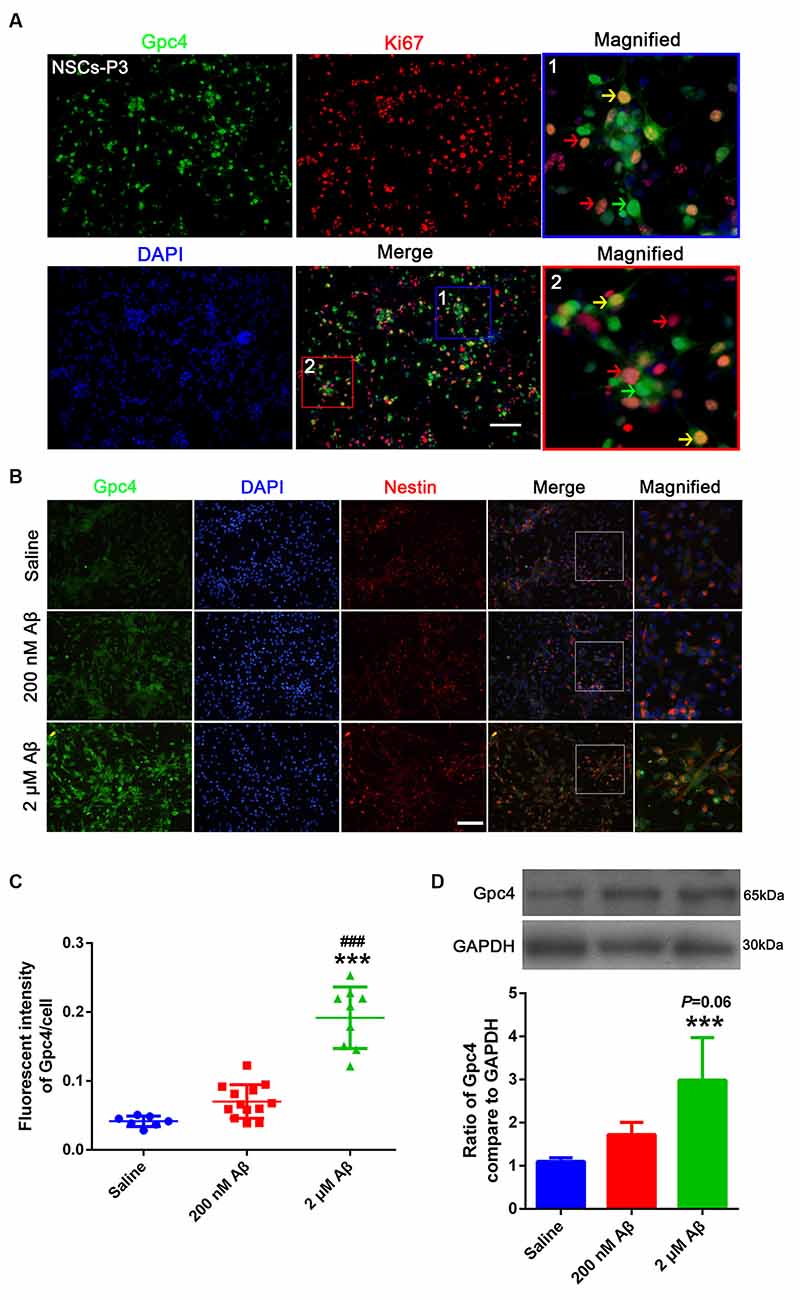
Figure 3. Aβ upregulated Gpc4 expression in NSCs. (A) Immunofluorescence analysis revealing the protein expression of Gpc4 (green) with Ki67 (red) in NSCs. Nuclei were counterstained with DAPI (blue). Gpc4 was widely expressed in NSCs (green). Double positive Gpc4/Ki67 NSCs (yellow arrow), Gpc4-negative Ki67-positive NSCs (red arrow), and Gpc4-positive Ki67-negative NSCs (green arrow) were also observed. Scale bar = 50 μm. (B,C) NSCs were treated with 0, 200 nM, and 2 μM Aβ for 24 h. Immunofluorescence analysis revealed the expression of Gpc4 (green), nestin (red), and DAPI (blue). Gpc4 colocalized with nestin-positive NSCs. *** represents 2 μM Aβ vs. saline, P < 0.0001, ### represents 2 μM Aβ vs. 200 nM Aβ, P < 0.0001. Scale bar = 50 μm. (D) NSCs were treated with 0, 200 nM, and 2 μM Aβ for 24 h before western blotting. Western blotting was used to detect Gpc4 (56 kDa) and GAPDH (35 kDa) expression levels. *** represents 2 μM Aβ vs. saline, P = 0.007. Gpc4, glypican 4; NSCs, neural stem cells; DAPI, 4′,6-diamidino-2-phenylindole; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Gpc4 Regulates NSC Uptake of Aβ
Co-IP was performed to detect the relationship between Gpc4 and Aβ. We found that Gpc4 forms a protein complex with Aβ in the APP/PS1 mouse brain (Figure 4A). To further determine the role of Gpc4 in Aβ internalization, AdV and lentivirus were used to either overexpress or attenuate Gpc4 expression in NSCs and APP/PS1 mice, respectively. The AdV transfection ratio was 72% for siGpc4-AdV and 70% for overGpc4-AdV (Figure 4B). The timing for the experimental procedure of Gpc4 and LRP1 AdV and Aβ treatment is laid out in Figure 4C. Furthermore, cy3-Aβ (200 nM and 2 μM) was added to siGpc4-AdV-, Gpc4-AdV-, or control-AdV-infected NSCs for 6 h. Subsequently, the Aβ particles per cell were counted. Attenuating Gpc4 decreased the number of Aβ particles in the 200 nM and 2 μM groups. Overexpression of Gpc4 was able to upregulate the Aβ particle number in the 200 nM group but had no significant effect in the 2 μM group (Figures 4D,E).
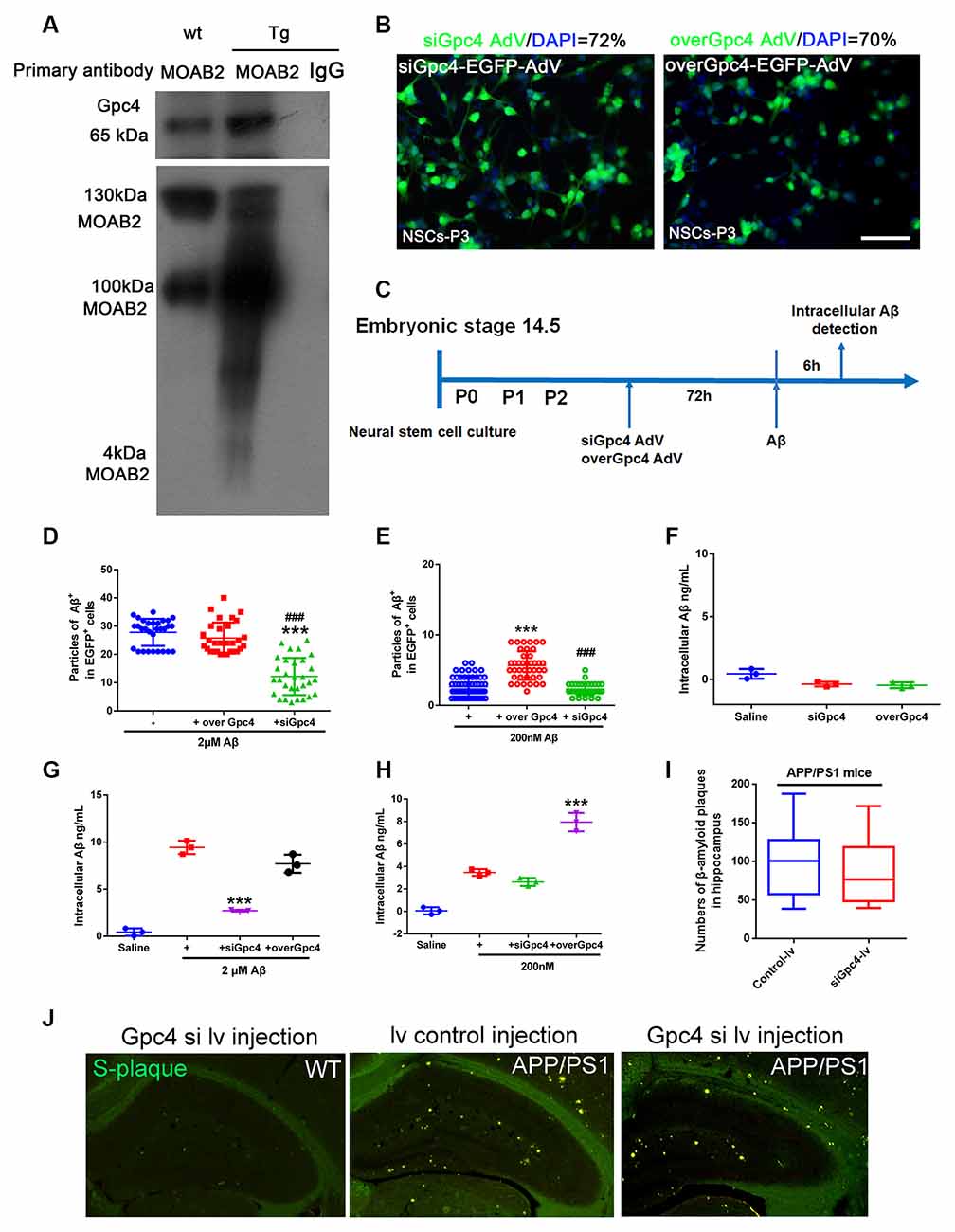
Figure 4. Gpc4 regulates Aβ internalization in NSCs. (A) Co-immunoprecipitation result from protein lysis extracted from 6-month-old APP/PS1 mice. Protein samples were incubated with the primary antibody MOAB2. Protein complexes able to bind to MOAB2 (Aβ42) were eluted. Western blotting was used to detect MOAB2 (Aβ42) and Gpc4 expression. (B) NSCs were plated in complete medium, and 2×1012 siGpc4 and overGpc4 AdV expression using enhanced green fluorescent protein (green) was added for 24 h. The medium was changed to complete medium, and cells were incubated for 48 h. Nuclei were marked with DAPI (blue). The ratio for AdV infection was calculated. (C) The schedule of virus infection and Aβ treatment on NSC cells. Seventy-two hours after virus infection, Aβ was added. Six hours after adding cy3-Aβ, all particles in AdV-positive cells were counted and compared among groups. (D) Six hours after adding 2 μM cy3-Aβ, all particles in AdV-positive cells were counted and compared among groups. *** represents siGpc4+ 2 μM Aβ vs. 2 μM Aβ, P < 0.0001, ### represents siGpc4+ 2 μM Aβ vs. overGpc4+ 2 μM Aβ, P < 0.0001. (E) Six hours after adding 200 nM cy3-Aβ, all particles in AdV-positive cells were counted and compared among groups. *** represents overGpc4+ 200 nM Aβ vs. 200 nM Aβ, P < 0.0001, ### represents siGpc4+ 200 nM Aβ vs. overGpc4+ 200 nM Aβ, P < 0.0001. (F) AdV was used to infect NSCs, and intracellular Aβ42 in NSCs was detected using ELISA. (G) NSCs were treated as previously described in (C). Instead of cy3-Aβ, the Aβ42 were used in this experiment. Six hours after adding 2 μM Aβ42, Aβ42 levels were detected using ELISA. *** represents siGpc4+ 2 μM Aβ vs. 2 μM Aβ, P < 0.0001. (H) NSCs were treated as previously described in (G). Six hours after adding 200 nM Aβ42, Aβ42 levels were detected using ELISA. *** represents overGpc4+ 200 nM Aβ vs. 200 nM Aβ, P < 0.0001. (I,J) siGpc4/overGpc4 lentivirus was injected into both lateral ventricles and the hippocampus of 4-month-old (APP/PS1) mice. Two months after lentivirus injection, mice were sacrificed for Thioflavin staining. β-amyloid plaques were stained in green. All particles were counted and compared against those in the hippocampus. NSCs, neural stem cells; Gpc4, glypican 4; AdV, adenovirus; DAPI, 4′,6-diamidino-2-phenylindole; ELISA, enzyme-linked immunosorbent assay.
Next, cells were monitored to detect whether Gpc4 induced Aβ production. No significant differences were observed between the siGpc4 and overGpc4 adenovirus groups (Figure 4F). overGpc4 had no significant effect on 2 μM Aβ internalization, but siGpc4 significantly attenuated the internalized Aβ level (Figure 4G). In the 200 nM Aβ-treated group, overGpc4-AdV increased NSC Aβ internalization, but siGpc4-AdV had no significant effect on blocking Aβ internalization (Figure 4H). siGpc4/overGpc4 lentivirus were injected into the hippocampus of 4-month old (APP/PS1) mice brains, and 2 months after injection, mice were sacrificed for S-plaque detection. Aβ plaques were stained using Thioflavin staining. Plaque numbers in the siGpc4 and overGpc4 groups did not change at this stage in APP/PS1 mice (Figures 4I,J). These results suggest an important dose-dependent effect of Gpc4 on Aβ internalization in NSCs at the early stage of AD.
LRP1 Is Expressed in NSCs and Regulates the Aβ Internalization
LRP1 is an Aβ receptor that plays a role in Gpc4-induced Aβ internalization. We first identified the expression of LRP1 in NSCs by double staining NSCs with Ki67 and LRP1. LRP1 colocalized with Ki67-positive cells (Figure 5A). Double-positive LRP1/Ki67 (yellow arrow), LRP1-negative Ki67-positive (red arrow), and LRP1-positive Ki67-negative NSCs (green arrow) were all observed (Figure 5A). Moreover, the transfection ratio of siLRP1 was 81% (Figure 5B. The effect of LRP1 on Aβ internalization was analyzed by double staining of LRP1 with the Aβ antibody MOAB2 in AdV control and AdV-siLRP1-treated groups. siLRP1 remarkably decreased Aβ levels in NSCs (Figures 5C,D), suggesting LRP1 is responsible for Aβ transportation in NSCs. However, the relationship between LRP1 and Gpc4 remains unclear.
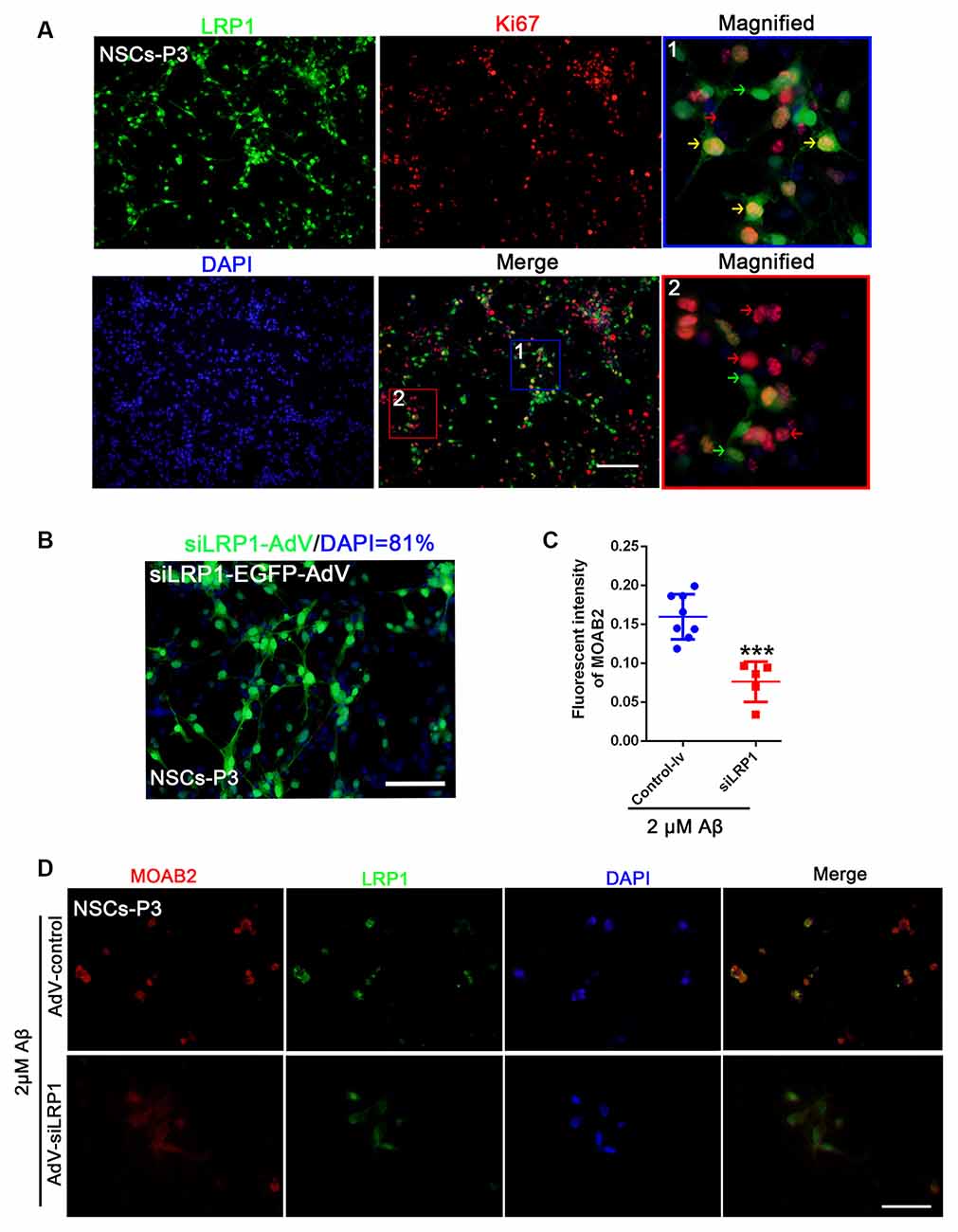
Figure 5. LRP1 is expressed in NSC and regulates the internalization of Aβ. (A) Immunofluorescence analysis revealing the expression of LRP1 (green) with Ki67 (red) in NSCs. Nuclei were counterstained with DAPI (blue). LRP1 was widely expressed in NSCs (green). Double positive LRP1/Ki67 (yellow arrow), LRP1-negative Ki67-positive NSCs (red arrow), and LRP1-positive Ki67-negative NSCs (green arrow) were also observed. Scale bar = 50 μm. (B) NSCs were plated in complete medium, and then siLRP1 adenovirus expression using enhanced green fluorescent protein (green) was added (2×1012) for 24 h for transfection. The medium was then changed for 48 h. Nuclei were marked with DAPI (blue). The ratio of adenovirus transfection was calculated. (C,D) NSCs were plated in complete medium, and then control-lentivirus and siLRP1 lentivirus were added 24 h for transfection. The medium was then changed for 48 h. Twelve hours after adding 2 μM Aβ, fluorescence intensities of MOAB2 and LRP1 were measured and compared. *** represents siLRP1+ 2 μM Aβ vs. 2 μM Aβ, P < 0.0001. LRP1, low-density lipoprotein receptor-related protein 1; NSCs, neural stem cells; DAPI, 4′,6-diamidino-2-phenylindole.
Gpc4 Regulates Aβ Internalization Partially via LRP1 in NSCs
To more precisely define the relationship between Gpc4 and LRP1, co-IP was performed on the cell lysis of APP/PS1 mice. Gpc4 was able to bind to LRP1, forming a protein complex in the brains of AD mice (Figures 6A,B). These observations suggest that LRP1 plays a role in Gpc4-induced Aβ internalization.
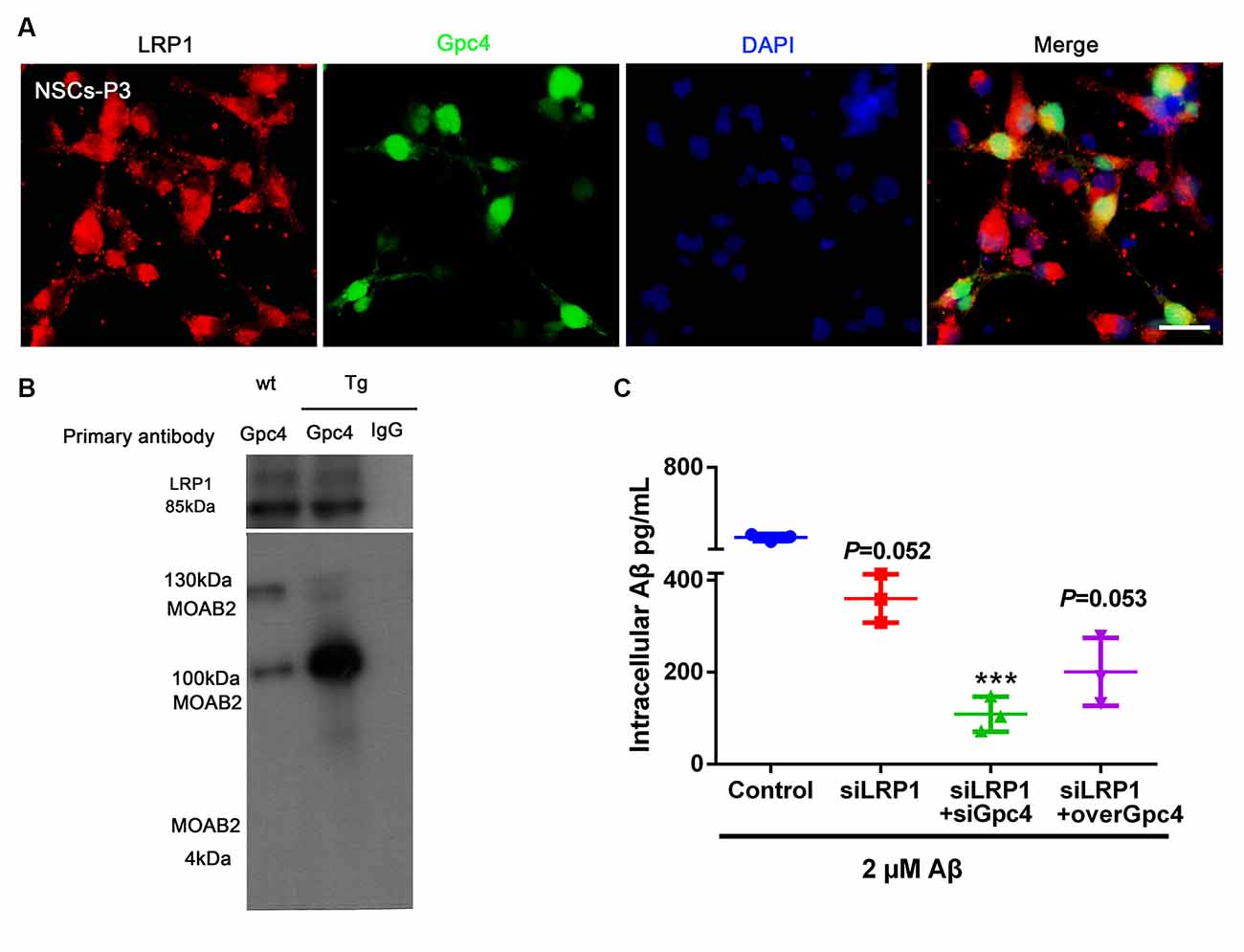
Figure 6. Gpc4 regulates Aβ internalization partially via LRP1 in NSCs. (A) Immunofluorescence revealed LRP1 (red) abundantly colocalized with Gpc4 (green) in NSCs. Nuclei were counterstained with DAPI (blue), scale bar = 20 μm. (B) Co-immunoprecipitation result from protein lysis extracted from 6-month-old APP/PS1 mice. Protein samples were incubated with primary antibody Gpc4. Protein complexes able to bind to Gpc4 were then eluted. Western blotting was used to detect the MOAB2- (Aβ42) and LRP1-positive binds. (C) Seventy-two hours after adenovirus transfection, 2 μM Aβ42 was added to the control, siLRP1, siLRP1+siGpc4, and siLRP1+overGpc4 groups. The levels of Aβ42 were detected using ELISA. *** represents siLRP1+ 2 μM Aβ vs. siGpc4+siLRP1+ 2 μM Aβ, P = 0.0011. GPc4, glypican 4; LRP1, low-density lipoprotein receptor related protein 1; NSCs, neural stem cells; DAPI, 4′,6-diamidino-2-phenylindole; ELISA, enzyme-linked immunosorbent assay.
We next aimed to reveal the action of Gpc4 and LRP1 in Aβ internalization. NSCs were transfected with control, siLRP1, siLRP1+siGpc4, and siLRP1+overGpc4 AdV for 3 days, and then Aβ was added for 6 h before cells were harvested for ELISA. siLRP1 remarkably decreased Aβ uptake by NSCs. In the siLRP1 and siGpc4 groups, the amount of internalized Aβ was much less compared to the siLRP1-treated group. Furthermore, in the siLRP1+overGpc4-treated group, the internalized Aβ level was increased compared to the siLRP1+siGpc4 group (Figure 6C).
Gpc4 Regulates Cell Apoptosis via LRP1 in NSCs
Control, siLRP1, overGpc4, and overGpc4+siLRP1 AdV were transfected into NSCs for 3 days, and then Aβ was added for 12 h. The JC-1 kit was used to detect the MMP in NSCs. JC-1 monomers (green) and aggregates (red) were compared and normalized to those of the control group. In 2 μM Aβ experiments, JC-1, TUNEL, and western blotting were used to determine whether Gpc4/LRP1 regulates the toxic effect of Aβ on NSCs. siLRP1 significantly decreased the ratio of JC-1 monomers/aggregates compared to the control group. Moreover, overGpc4 increased MMP damage compared to siLRP1. overGpc4+siLRP1 significantly increased the MMP compared to siLRP1; however, the MMP damage in the overGpc4+siLRP1 group was lower than that in the overGpc4 group (Figures 7A,B). Moreover, the level of Cytc was decreased in the siLRP1 and siLRP1+siGpc4 groups, and higher in the siLRP1+overGpc4 group (Figure 7C). Furthermore, TUNEL staining was used to detect whether Gpc4 and LRP1 regulate Aβ-induced cell death. Compared to the control group, siLRP1 attenuated NSC apoptosis while overGpc4 increased it. siLRP1 decreased the effect caused by overGpc4 (Figures 7D,E). The expression of Bax was also detected using western blotting, which is consistent with the results of TUNEL staining (Figures 7D–F).
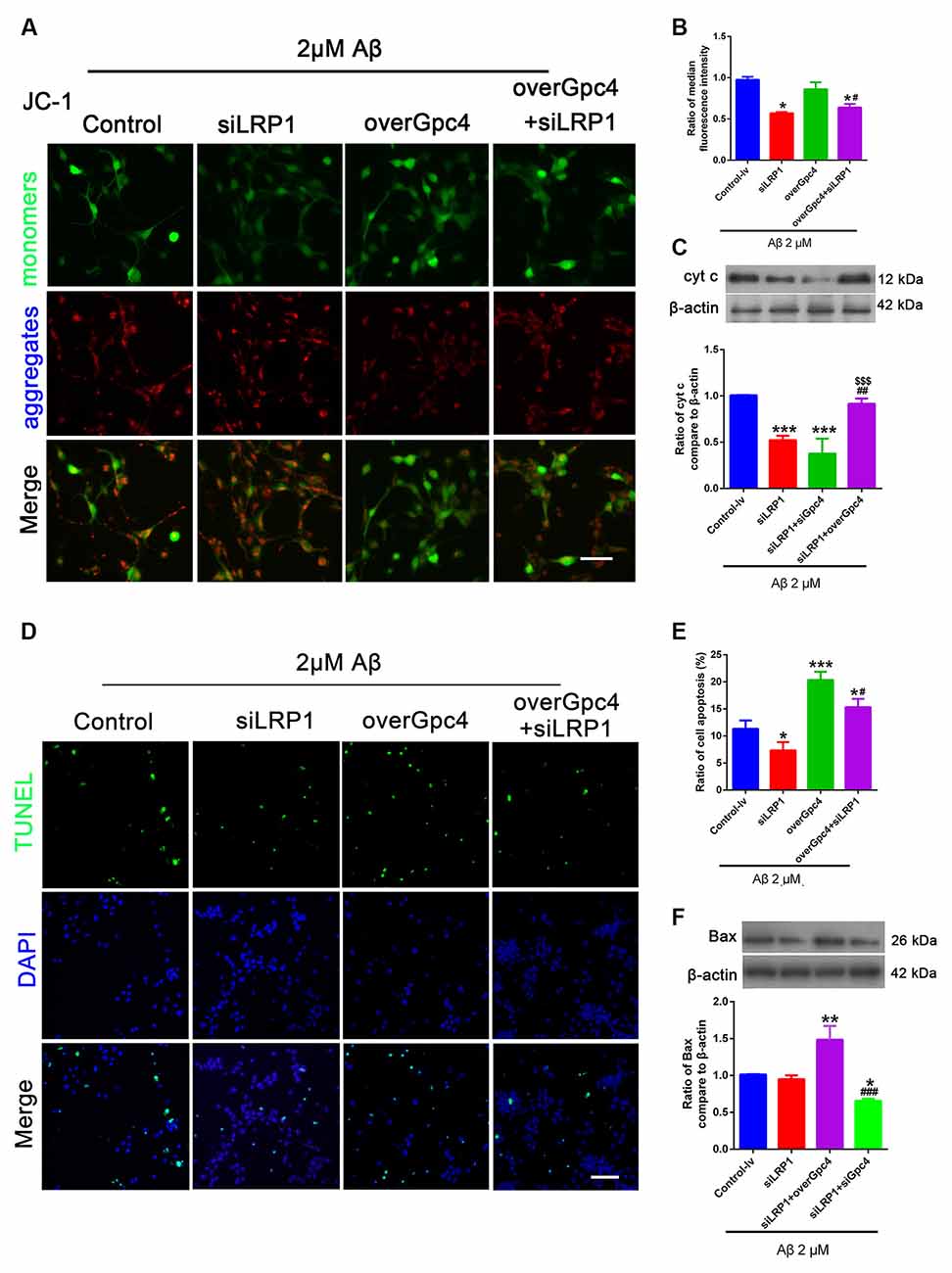
Figure 7. Gpc4 regulates cell apoptosis partly via LRP1 in NSCs. (A,B) NSCs were infected with control, siLRP1, overGpc4, and overGpc4+siLRP1 AdV for 3 days, and 2 μM Aβ was added for 12 h. The JC-1 kit was used to detect the mitochondrial membrane potential in NSCs. JC-1 monomers (green) and aggregates (red) were compared and normalized to the control group. The ratio of monomers to aggregates was calculated. (C) Seventy-two hours after AdV infection, 2 μM Aβ42 was added to the control, siLRP1, siLRP1+siGpc4, and siLRP1+overGpc4. The levels of Cytc were determined using western blotting. *** represents siLRP1+siGpc4 vs. control, P = 0.0001, siLRP1 vs. control, P = 0.0007; siLRP1+overGpc4 vs. siLRP1, P = 0.0028. $$$siLRP1+siGpc4 vs. siLRP1+overGpc4, P = 0.0252, *siLRP1+siGpc4 vs. siLRP1+overGpc4, P = 0.0003. (D,E) NSCs were infected with control, siLRP1, overGpc4, and overGpc4+siLRP1 AdV for 3 days, and Aβ was added for 12 h. TUNEL staining was used to detect whether Gpc4 regulated Aβ-induced cell death via LRP1. The ratio of apoptotic cells was calculated based on TUNEL positive cells (green) and DAPI (blue). (F) The expression of Bax was also detected using western blotting, ** represents siLRP1+overGpc4 vs. siLRP1, P = 0.0007, *siLRP1+siGpc4 vs. siLRP1, P = 0.0252, *siLRP1+siGpc4 vs. siLRP1+ overGpc4, P < 0.0001. GPc4, glypican 4; LRP1, low-density lipoprotein receptor related protein 1; NSCs, neural stem cells; DAPI, 4′,6-diamidino-2-phenylindole; AdV, adenovirus; TUNEL, terminal deoxynucleotidyl transferase dUPT nick end labeling.
In 200 nM Aβ experiments, JC-1 and TUNEL assays were used to determine whether Gpc4 induces toxic effects by enriching the nontoxic effect of Aβ on NSCs. Initially, 200 nM Aβ had no toxic effect on NSCs, but overGpc4 in 200 nM Aβ-treated NSCs increased MMP damage (Figures 8A,B). Furthermore, TUNEL staining was used to detect whether Gpc4/LRP1 regulated Aβ-induced cell death. No significant changes were observed among the four groups (Figures 8C,D).
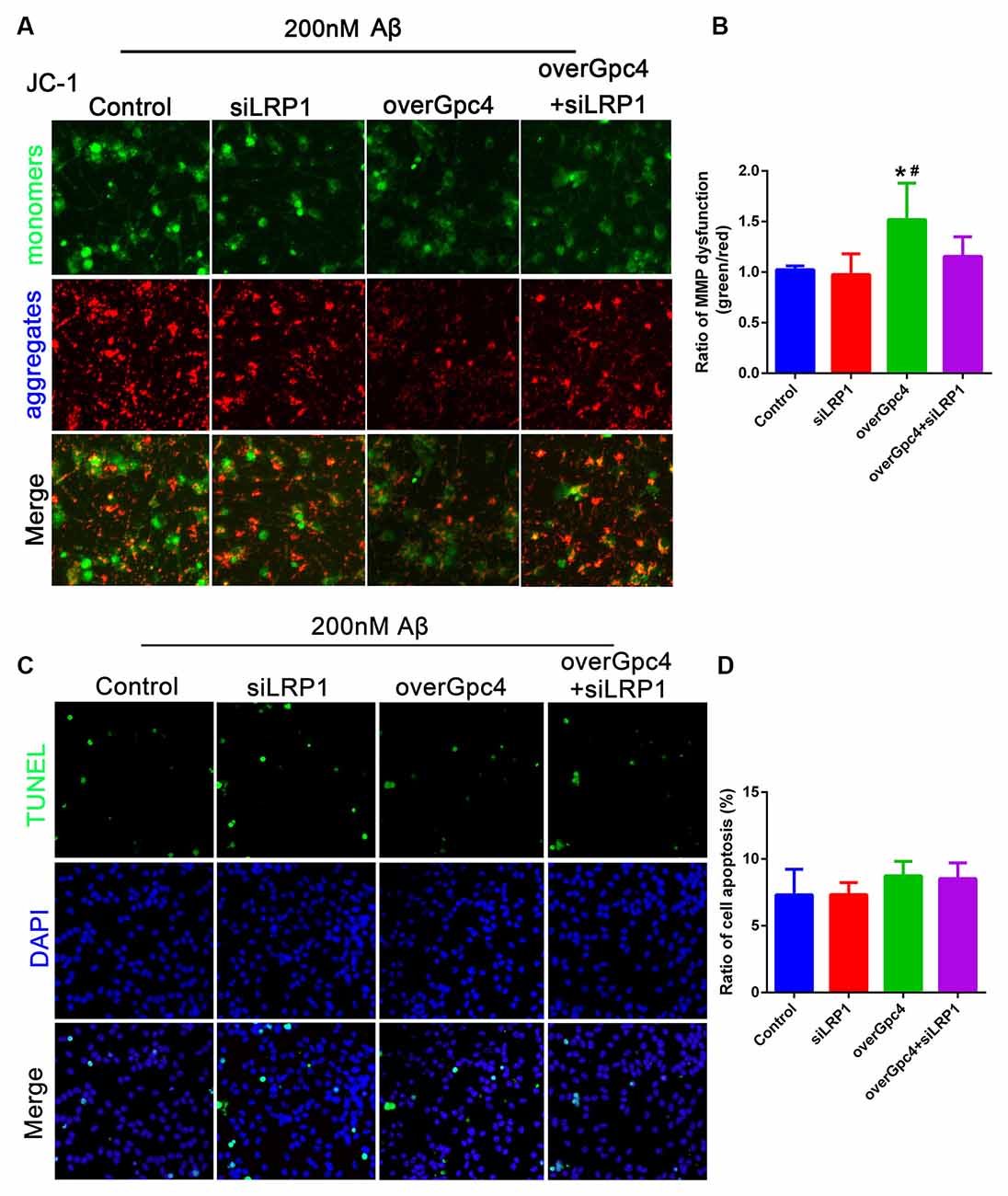
Figure 8. Role of Gpc4 and LRP1 on 200 nM Aβ on NSCs. (A,B) NSCs were infected with control, siLRP1, overGpc4, and overGpc4+siLRP1 AdV for 3 days, and then 200 nM Aβ was added for 12 h. The JC-1 kit was used to detect the mitochondrial membrane potential in NSCs. JC-1 monomers (green) and aggregates (red) were compared and normalized to those of the control group. The ratio of monomers/aggregates was calculated. * represents overGpc4 vs. control, P = 0.0036, # represents overGpc4 vs. siLRP1, P = 0.0080. (C,D) NSCs were infected with control, siLRP1, overGpc4, and overGpc4+siLRP1 AdV for 3 days, and 200 nM Aβ was added for 12 h. TUNEL staining was used to detect whether Gpc4 regulated the Aβ-induced cell death via LRP1. The ratio of apoptotic cells was calculated based on TUNEL positive cells (green) and DAPI (blue). NSCs, neural stem cells; GPc4, glypican 4; LRP1, low-density lipoprotein receptor related protein 1; AdV, adenovirus; TUNEL, terminal deoxynucleotidyl transferase dUPT nick end labeling; DAPI, 4′,6-diamidino-2-phenylindole.
Discussion
The dysregulation of adult neurogenesis in AD occurs at an early stage of the disease and the mechanism has not been completely elucidated (Boese et al., 2020). Heparan sulfate proteoglycan, a highly conserved protein expressed in many species and tissues, is involved in basic cellular processes, such as cell proliferation, cell growth, axon guidance, and synapse formation (Rawson et al., 2005; Stewart and Sanderson, 2014; Jiao et al., 2016; Condomitti and de Wit, 2018), and is highly related to AD pathology. Twenty years ago, Gpc4, a heparan sulfate proteoglycan, was reported to be increased in the brains of AD patients and was initially found to be responsible for neurogenesis (Gysi et al., 2013; Blanchette et al., 2015; Zhang et al., 2015). This suggests that abnormal expression of Gpc4 in AD patients may contribute to NSC damage. We confirmed that Gpc4 is highly expressed in NSCs (Luxardi et al., 2007; Yu et al., 2017). Gpc4 expression was significantly higher in Aβ-treated NSCs than in control group NSCs. Gpc4 also regulated the internalization of Aβ by NSCs and enriched low concentrations of Aβ into NSCs. The overexpression of Gpc4 suggests that it may play a role in Aβ-induced toxicity in NSCs.
Heparan sulfate proteoglycans have been proposed to be responsible for Aβ enrichment. We found that Gpc4 expression was increased in Aβ-treated NSCs in a concentration-dependent manner. Nevertheless, considering neurogenesis deficiency is an important aspect of memory loss, Gpc4 may regulate Aβ enrichment in NSCs. The average concentration of Aβ42 in the brain white matter of patients with AD is 60 pM/L (Collins-Praino et al., 2014; Fu et al., 2016). While the concentration of Aβ in the brain extracellular fluid, such as the interstitial fluid and cerebrospinal fluid, is low (10−10–10−9 M; Ma and Qian, 2019), in vitro studies have suggested that the critical concentration for spontaneous aggregation of Aβ is in the μM range. Therefore, Aβ concentrations in vivo would have to be increased by at least three orders of magnitude for spontaneous aggregation. Several mechanisms have been proposed to explain this large concentration gap. The most likely hypotheses are that the concentration could be increased through membrane association (Gorbenko and Kinnunen, 2006; Aisenbrey et al., 2008) or macromolecular crowding (Munishkina et al., 2004; Hu et al., 2009; Fernandez-Perez et al., 2020; Wang et al., 2020; Wu et al., 2020). However, the mechanism underlying Aβ enrichment in NSCs remains unknown.
The role of Gpc4 in regulating different concentrations of Aβ internalization in NSCs is Aβ dose-dependent. siGpc4 decreased NSC uptake of 2 μM Aβ, and overGpc4 increased NSC uptake of 200 nM Aβ. This different role of Gpc4 on 2 μM Aβ and 200 nM Aβ transportation indicates that Gpc4 is functional at nanomolar Aβ concentrations; however, the transportation efficiency of Gpc4 reaches its saturation point at micromolar Aβ concentrations. Gpc4 is a key regulator of Aβ enrichment at nanomolar concentrations of Aβ. This illustrates why siGpc4 lentivirus had no effect on β-amyloid plaque numbers. Therefore, Gpc4 is a key regulator of Aβ enrichment during the early stages of AD, but not in the later stages.
Heparan sulfate, the extracellular portion of Gpc4, has a high affinity for Aβ and its receptors (Yamada and Hamaguchi, 2018; Du et al., 2020). Thus, heparan sulfate on Gpc4 may play a role in regulating Aβ internalization. Heparan sulfate also has a high affinity for LRP1 and the cellular prion protein (Taylor et al., 2009; Gao et al., 2019; Hu et al., 2019), which are also key receptors for Aβ transportation. This suggests that Gpc4 may gather the receptor and Aβ to the cell surface during the early stage of AD. However, the relationship between Gpc4 and the classic Aβ receptor LRP1 is yet to be determined.
LRP1 is an important receptor for the induction of Aβ internalization in astrocytes. Previous studies have shown that LRP1 clears 73.8% of Aβ injected into the brain towards blood circulation through the blood-brain barrier (Ma et al., 2018; Van Gool et al., 2019). Through the LRP1-related pathway, Aβ is internalized by neurons and localized in lysosomes, endosomes, and mitochondria, with little recycling (Lillis et al., 2005). ApoE lipid rafts (Gilat-Frenkel et al., 2014; Rushworth and Megson, 2014), the ERK1/2 pathway, Wnt3a, JNK, and other heparan sulfate proteoglycans may also play a role in Aβ internalization (Lai et al., 2010; Shi et al., 2011; Li et al., 2019; Mii and Takada, 2020). First, decreased LRP1 allowed more ApoE4-coated Aβ to pass through the NSC membrane via Gpc4 overexpression, leading to an upregulation of intracellular Aβ levels. Second, ERK1/2, as the downstream target of both LRP1 and Gpc4, may also regulate the expression of other Aβ receptors. siLRP1 decreased the expression of ERK1/2; however, Gpc4 overexpression activated ERK1/2, subsequently activating other receptors related to Aβ internalization. Lastly, Gpc4 is responsible for many cellular growth processes, such as cell division and cell metabolism (Farhy-Tselnicker et al., 2017; McGough and Vecchia, 2020). It is possible that overGpc4+siLRP1 may activate these processes and therefore upregulate Aβ transportation.
We have previously shown that internalized Aβ is transported into the mitochondria in vitro and in vivo (Yang et al., 2015; Ma et al., 2016, 2018). In this study, we found that Gpc4 regulates mitochondrial dysfunction and cell apoptosis in NSCs partly via LRP1. We used the JC-1 kit to detect the role of internalized Aβ on MMP, which is also called mitochondrial depolarization. We found that in toxic Aβ-treated groups, attenuating Gpc4/LRP1 expression decreased intracellular Aβ, MMP damage, and NSC apoptosis. In the physical Aβ-treated groups, Gpc4 overexpression caused MMP damage but had no effect on cell apoptosis. These results indicate that internalized Aβ is harmful to both mitochondrial function and cell viability.
Conclusion
We demonstrated that Gpc4 regulates Aβ internalization at an early stage of AD. This process is dose-dependent, and occurs partly via the well-known Aβ receptor, LRP1. However, many questions remain unanswered. Heparan sulfate on the extracellular surface of Gpc4 is important for receiving and binding to Aβ and its receptors. Gpc4 was initially reported to have a major effect on synapse formation and neurogenesis; therefore, it is also important to determine whether Gpc4 mediates NSC-derived neurogenesis by regulating Aβ internalization in NSCs.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee at Xi’an Jiaotong University.
Author Contributions
KM performed most of the experiments, analyzed data, and wrote the rough draft. SX performed experiments, data analysis, and redrafting. YLu and YiLi performed NSCs cell culture. CZ performed the animal housing. ZZ and YF took part in the virus infection experiment. ZZ and YoLi participated in the experiment design. XC designed the research and finalized the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China, NSFC (No. 82001493 and No. 81901156); the China Postdoctoral Science Foundation (No. 2019M653662); Natural Science Basic Research Program of Shaanxi (No. 2021JQ-881); and Scientific Research Program Funded by Shaanxi Provincial Education Department (No. 20JK0958).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AD, Alzheimer’s disease; AdV, adenovirus; ApoE, apolipoprotein E; CCK-8, Cell Counting Kit-8; DAPI, 4′,6-diamidino-2-phenylindole; ELISA, enzyme-linked immunosorbent assay; Gpc4, glypican 4; IP, immunoprecipitation; LRP1, low-density lipoprotein receptor-related protein 1; MMP, mitochondrial membrane potential; NSC, neural stem cells; PI, propidium iodide; TUNEL, terminal deoxynucleotidyl transferase dUPT nick end labeling.
Acknowledgments
We sincerely thank Prof. Wei-Na Yang for her generous sharing of the AD transgenetic mice used in this experiment.
References
Aisenbrey, C., Borowik, T., Byström, R., Bokvist, M., Lindström, F., Misiak, H., et al. (2008). How is protein aggregation in amyloidogenic diseases modulated by biological membranes. Eur. Biophys. J. 37, 247–255. doi: 10.1007/s00249-007-0237-0
Blanchette, C. R., Perrat, P. N., Thackeray, A., and Bénard, C. Y. (2015). Glypican is a modulator of netrin-mediated axon guidance. PLoS Biol. 13:e1002183. doi: 10.1371/journal.pbio.1002183
Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Boese, A. C., Hamblin, M. H., and Lee, J. P. (2020). Neural stem cell therapy for neurovascular injury in Alzheimer’s disease. Exp. Neurol. 324:113112. doi: 10.1016/j.expneurol.2019.113112
Collins-Praino, L. E., Francis, Y. I., Griffith, E.Y., Wiegman, A. F., Urbach, J., Lawton, A., et al. (2014). Soluble amyloid beta levels are elevated in the white matter of Alzheimer’s patients, independent of cortical plaque severity. Acta Neuropathol. Commun. 2:83. doi: 10.1186/s40478-014-0083-0
Condomitti, G., and de Wit, J. (2018). Heparan sulfate proteoglycans as emerging players in synaptic specificity. Front. Mol. Neurosci. 11:14. doi: 10.3389/fnmol.2018.00014
Du, Y. Y., Liu, X. L., Zhu, X. L., Liu, Y., Wang, X. R., and Wu, X. P. (2020). Activating transcription factor 6 reduces A beta 1–42 and restores memory in Alzheimer’s disease model mice. Int. J. Neurosci. 130, 1015–1023. doi: 10.1080/00207454.2020.1715977
Farhy-Tselnicker, I., van Casteren, A. C. M., Lee, A., Chang, V. T., Aricescu, A. R., and Allen, N. J. (2017). Astrocyte-Secreted glypican 4 regulates release of neuronal pentraxin 1 from axons to induce functional synapse formation. Neuron 96, 428–445.e13. doi: 10.1016/j.neuron.2017.09.053
Fernandez-Perez, E. J., Gallegos, S., Armijo-Weingart, L., Araya, A., Riffo-Lepe, N. O., Cayuman, F., et al. (2020). Changes in neuronal excitability and synaptic transmission in nucleus accumbens in a transgenic Alzheimer’s disease mouse model. Sci. Rep. 10:19606. doi: 10.1038/s41598-020-76456-w
Fu, Y., Zhao, J., Atagi, Y., Nielsen, H. M., Liu, C. C., Zheng, H., et al. (2016). Apolipoprotein E lipoprotein particles inhibit amyloid-β uptake through cell surface heparan sulphate proteoglycan. Mol. Neurodegener. 11:37. doi: 10.1186/s13024-016-0099-y
Gao, Y. L., Liu, Q., Xu, L. L., Zheng, N., He, X. M., and Xu, F. Q. (2019). Imaging and spectral characteristics of amyloid plaque autofluorescence in brain slices from the APP/PS1 mouse model of Alzheimer’s disease. Neurosci. Bull. 35, 1126–1137. doi: 10.1007/s12264-019-00393-6
Ghazale, H., Ramadan, N., Mantash, S., Zibara, K., El-Sitt, S., Darwish, H., et al. (2018). Docosahexaenoic acid (DHA) enhances the therapeutic potential of neonatal neural stem cell transplantation post-traumatic brain injury. Behav. Brain Res. 340, 1–13. doi: 10.1016/j.bbr.2017.11.007
Gilat-Frenkel, M., Boehm-Cagan, A., Liraz, O., Xian, X., Herz, J., and Michaelson, D. M. (2014). Involvement of the Apoer2 and Lrp1 receptors in mediating the pathological effects of ApoE4 in vivo. Curr. Alzheimer Res. 11, 549–557. doi: 10.2174/1567205010666131119232444
Gorbenko, G. P., and Kinnunen, P. K. (2006). The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem. Phys. Lipids 141, 72–82. doi: 10.1016/j.chemphyslip.2006.02.006
Gysi, S., Rhiner, C., Flibotte, S., Moerman, D. G., and Hengartner, M. O. (2013). A network of HSPG core proteins and HS modifying enzymes regulates netrin-dependent guidance of D-type motor neurons in Caenorhabditis elegans. PLoS One 8:e74908. doi: 10.1371/journal.pone.0074908
Hu, X., Crick, S. L., Bu, G., Frieden, C., Pappu, R. V., and Lee, J. M. (2009). Amyloid seeds formed by cellular uptake, concentration and aggregation of the amyloid-beta peptide. Proc. Natl. Acad. Sci. U S A 106, 20324–20329. doi: 10.1073/pnas.0911281106
Hu, Y. R., Xing, S. L., Chen, C., Shen, D. Z., and Chen, J. L. (2019). Tiaoxin Recipe, a Chinese herbal formula, inhibits microRNA-34a expression in the APPswe/PS1 Delta E9 mouse model of Alzheimer’s disease. J. Integr. Medicine 17, 404–409. doi: 10.1016/j.joim.2019.09.002
Huang, D., Cao, Y., Yang, X., Liu, Y., Zhang, Y., Li, C., et al. (2021). A nanoformulation-mediated multifunctional stem cell therapy with improved beta-amyloid clearance and neural regeneration for alzheimer’s disease. Adv. Mater 33:e2006357. doi: 10.1002/adma.202006357
Jiao, Q., Wang, L., Zhang, Z. C., Wang, Y. Y., Yan, H. Q., Ma, W., et al. (2016). Dynamic expression of srGAP2 in cell nuclei and cytoplasm during the differentiation of rat neural stem cells in vitro. Mol. Med. Rep. 14, 4599–4605. doi: 10.3892/mmr.2016.5795
Lai, J. P., Sandhu, D. S., Yu, C., Moser, C. D., Hu, C., Shire, A. M., et al. (2010). Sulfatase 2 protects hepatocellular carcinoma cells against apoptosis induced by the PI3K inhibitor LY294002 and ERK and JNK kinase inhibitors. Liver Int. 30, 1522–1528. doi: 10.1111/j.1478-3231.2010.02336.x
Li, N., Wei, L., Liu, X., Bai, H., Ye, Y., Li, D., et al. (2019). A frizzled-like cysteine-rich domain in glypican-3 mediates wnt binding and regulates hepatocellular carcinoma tumor growth in mice. Hepatology 70, 1231–1245. doi: 10.1002/hep.30646
Lillis, A. P., Mikhailenko, I., and Strickland, D. K. (2005). Beyond endocytosis: LRP function in cell migration, proliferation and vascular permeability. J. Thromb. Haemost. 3, 1884–1893. doi: 10.1111/j.1538-7836.2005.01371.x
Liu, C. C., Liu, C. C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Liu, C. C., Zhao, N., Yamaguchi, Y., Cirrito, J. R., Kanekiyo, T., Holtzman, D. M., et al. (2016). Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-β clearance and aggregation in Alzheimer’s disease. Sci. Transl. Med. 8:332ra344. doi: 10.1126/scitranslmed.aad3650
Luxardi, G., Galli, A., Forlani, S., Lawson, K., Maina, F., and Dono, R. (2007). Glypicans are differentially expressed during patterning and neurogenesis of early mouse brain. Biochem. Biophys. Res. Commun. 352, 55–60. doi: 10.1016/j.bbrc.2006.10.185
Ma, K. G., Lv, J., Hu, X. D., Shi, L. L., Chang, K. W., Chen, X. L., et al. (2016). The p38 mitogen-activated protein kinase signaling pathway is involved in regulating low-density lipoprotein receptor-related protein 1-mediated β-amyloid protein internalization in mouse brain. Int. J. Biochem. Cell Biol. 76, 75–86. doi: 10.1016/j.biocel.2016.04.019
Ma, K. G., Lv, J., Yang, W. N., Chang, K. W., Hu, X. D., Shi, L. L., et al. (2018). The p38 mitogen activated protein kinase regulates β-amyloid protein internalization through the α7 nicotinic acetylcholine receptor in mouse brain. Brain Res. Bull. 137, 41–52. doi: 10.1016/j.brainresbull.2017.11.006
Ma, K. G., and Qian, Y. H. (2019). Alpha 7 nicotinic acetylcholine receptor and its effects on Alzheimer’s disease. Neuropeptides 73, 96–106.
McGough, I. J., and Vecchia, L. (2020). Glypicans shield the Wnt lipid moiety to enable signalling at a distance. Nature 585, 85–90. doi: 10.1038/s41586-020-2498-z
Mii, Y., and Takada, S. (2020). Heparan sulfate proteoglycan clustering in wnt signaling and dispersal. Front. Cell Dev. Biol. 8:631. doi: 10.3389/fcell.2020.00631
Mohamed, A., and Posse de Chaves, E. (2011). Abeta internalization by neurons and glia. Int. J. Alzheimers Dis. 2011:127984. doi: 10.4061/2011/127984
Munishkina, L. A., Cooper, E. M., Uversky, V. N., and Fink, A. L. (2004). The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J. Mol. Recognit. 17, 456–464. doi: 10.1002/jmr.699
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
Rawson, J. M., Dimitroff, B., Johnson, K. G., Rawson, J. M., Ge, X., Van Vactor, D., et al. (2005). The heparan sulfate proteoglycans dally-like and syndecan have distinct functions in axon guidance and visual-system assembly in Drosophila. Curr. Biol. 15, 833–838. doi: 10.1016/j.cub.2005.03.039
Rushworth, G. F., and Megson, I. L. (2014). Existing and potential therapeutic uses for N-acetylcysteine: the need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 141, 150–159. doi: 10.1016/j.pharmthera.2013.09.006
Shi, Z. D., Wang, H., and Tarbell, J. M. (2011). Heparan sulfate proteoglycans mediate interstitial flow mechanotransduction regulating MMP-13 expression and cell motility via FAK-ERK in 3D collagen. PLoS One 6:e15956. doi: 10.1371/journal.pone.0015956
Stewart, M. D., and Sanderson, R. D. (2014). Heparan sulfate in the nucleus and its control of cellular functions. Matrix Biol. 35, 56–59. doi: 10.1016/j.matbio.2013.10.009
Taylor, D. R., Whitehouse, I. J., and Hooper, N. M. (2009). Glypican-1 mediates both prion protein lipid raft association and disease isoform formation. PLoS Pathog. 5:e1000666. doi: 10.1371/journal.ppat.1000666
Timmer, N. M., Herbert, M. K., Kleinovink, J. W., Kiliaan, A. J., De Waal, R. M., and Verbeek, M. M. (2010). Limited expression of heparan sulphate proteoglycans associated with Aβ deposits in the APPswe/PS1dE9 mouse model for Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 36, 478–486. doi: 10.1111/j.1365-2990.2010.01091.x
Tiwari, S., Atluri, V., Kaushik, A., Yndart, A., and Nair, M. (2019). Alzheimer’s disease: pathogenesis, diagnostics and therapeutics. Int. J. Nanomed. 14, 5541–5554. doi: 10.2147/IJN.S200490
Van Gool, B., Storck, S. E., Reekmans, S. M., Lechat, B., Gordts, P., Pradier, L., et al. (2019). LRP1 has a predominant role in production over clearance of Aβ in a mouse model of Alzheimer’s disease. Mol. Neurobiol. 56, 7234–7245. doi: 10.1007/s12035-019-1594-2
Wang, C. Y., Cai, X. Y., Wang, R. C., Zhai, S. Y., Zhang, Y. F., Hu, W. J., et al. (2020). Neuroprotective effects of verbascoside against Alzheimer’s disease via the relief of endoplasmic reticulum stress in A beta-exposed U251 cells and APP/PS1 mice. J. Neuroinflammation 17:309. doi: 10.1186/s12974-020-01976-1
Wang, Y., Zhang, R., and Huang, Y. L. (2016). Propofol ameliorates ischemia/reperfusion induced cerebral injury by upregulation of microRNA-206 expression. Int. J. Clinn. Exp. Med. 9, 10256–10264. Available online at: https://www.webofscience.com/wos/alldb/full-record/WOS:000379156000161.
Wu, Q., Li, Q. F., Zhang, X., Ntim, M., Wu, X. F., Li, M., et al. (2020). Treatment with bifidobacteria can suppress A beta accumulation and neuroinflammation in APP/PS1 mice. PeerJ. 8:e10262. doi: 10.7717/peerj.10262
Yamada, M., and Hamaguchi, T. (2018). The sulfation code for propagation of neurodegeneration. J. Biol. Chem. 293, 10841–10842. doi: 10.1074/jbc.H118.003970
Yang, W. N., Ma, K. G., Qian, Y. H., Zhang, J. S., Feng, G. F., Shi, L. L., et al. (2015). Mitogen-activated protein kinase signaling pathways promote low-density lipoprotein receptor-related protein 1-mediated internalization of beta-amyloid protein in primary cortical neurons. Int. J. Biochem. Cell Biol. 64, 252–264. doi: 10.1016/j.biocel.2015.04.013
Yu, C., Griffiths, L. R., and Haupt, L. M. (2017). Exploiting heparan sulfate proteoglycans in human neurogenesis-controlling lineage specification and fate. Front. Integr. Neurosci. 11:28. doi: 10.3389/fnint.2017.00028
Zhang, Z. C., Ma, W., Wang, L., Gong, H. S., Tian, Y. M., Zhang, J. S., et al. (2015). Activation of type 4 metabotropic glutamate receptor attenuates oxidative stress-induced death of neural stem cells with inhibition of JNK and p38 MAPK signaling. Stem Cells Dev. 24, 2709–2722. doi: 10.1089/scd.2015.0067
Keywords: glypican 4, neural stem cells (NSCs), low density lipoprotein receptor related protein 1, β-amyloid internalization, Alzheimer’s disease
Citation: Ma K, Xing S, Luan Y, Zhang C, Liu Y, Fei Y, Zhang Z, Liu Y and Chen X (2021) Glypican 4 Regulates Aβ Internalization in Neural Stem Cells Partly via Low-Density Lipoprotein Receptor-Related Protein 1. Front. Cell. Neurosci. 15:732429. doi: 10.3389/fncel.2021.732429
Received: 29 June 2021; Accepted: 16 August 2021;
Published: 06 September 2021.
Edited by:
Ulises Gomez-Pinedo, Instituto de Investigación Sanitaria del Hospital Clínico San Carlos, SpainReviewed by:
Ahmed Elkamhawy, Mansoura University, EgyptLaura Garcia-Toscano, Complutense University of Madrid, Spain
Copyright © 2021 Ma, Xing, Luan, Zhang, Liu, Fei, Zhang, Liu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinlin Chen, Y2hlbnhsQHhqdHUuZWR1LmNu
† These authors have contributed equally to this work