 Noriko Shinjyo
Noriko Shinjyo Wataru Kagaya
Wataru Kagaya Marcela Pekna
Marcela Pekna- 1Laboratory of Immune Homeostasis, WPI Immunology Frontier Research Center, Osaka University, Osaka, Japan
- 2School of Tropical Medicine and Global Health, Nagasaki University, Nagasaki, Japan
- 3Department of Parasitology and Research Center for Infectious Disease Sciences, Graduate School of Medicine, Osaka City University, Osaka, Japan
- 4Laboratory of Regenerative Neuroimmunology, Center for Brain Repair, Department of Clinical Neuroscience, Institute of Neuroscience and Physiology, Sahlgrenska Academy at the University of Gothenburg, Gothenburg, Sweden
- 5Florey Institute of Neuroscience and Mental Health, Parkville, VIC, Australia
As part of the innate immune system, complement plays a critical role in the elimination of pathogens and mobilization of cellular immune responses. In the central nervous system (CNS), many complement proteins are locally produced and regulate nervous system development and physiological processes such as neural plasticity. However, aberrant complement activation has been implicated in neurodegeneration, including Alzheimer’s disease. There is a growing list of pathogens that have been shown to interact with the complement system in the brain but the short- and long-term consequences of infection-induced complement activation for neuronal functioning are largely elusive. Available evidence suggests that the infection-induced complement activation could be protective or harmful, depending on the context. Here we summarize how various infectious agents, including bacteria (e.g., Streptococcus spp.), viruses (e.g., HIV and measles virus), fungi (e.g., Candida spp.), parasites (e.g., Toxoplasma gondii and Plasmodium spp.), and prion proteins activate and manipulate the complement system in the CNS. We also discuss the potential mechanisms by which the interaction between the infectious agents and the complement system can play a role in neurodegeneration and dementia.
Introduction
The Complement System
The complement system is an evolutionarily conserved proteolytic cascade, consisting of over 40 components (Schartz and Tenner, 2020). Complement plays a key role as part of the innate immunity, eliminating pathogens and damaged cells, directly killing bacteria by forming the so-called membrane attack complex (MAC), and promoting inflammatory responses via the generation of anaphylatoxins (Yanamadala and Friedlander, 2010). The activation of the complement system can be triggered via three pathways: the classical pathway, the lectin pathway, and the alternative pathway (Veerhuis et al., 2011; Figure 1).
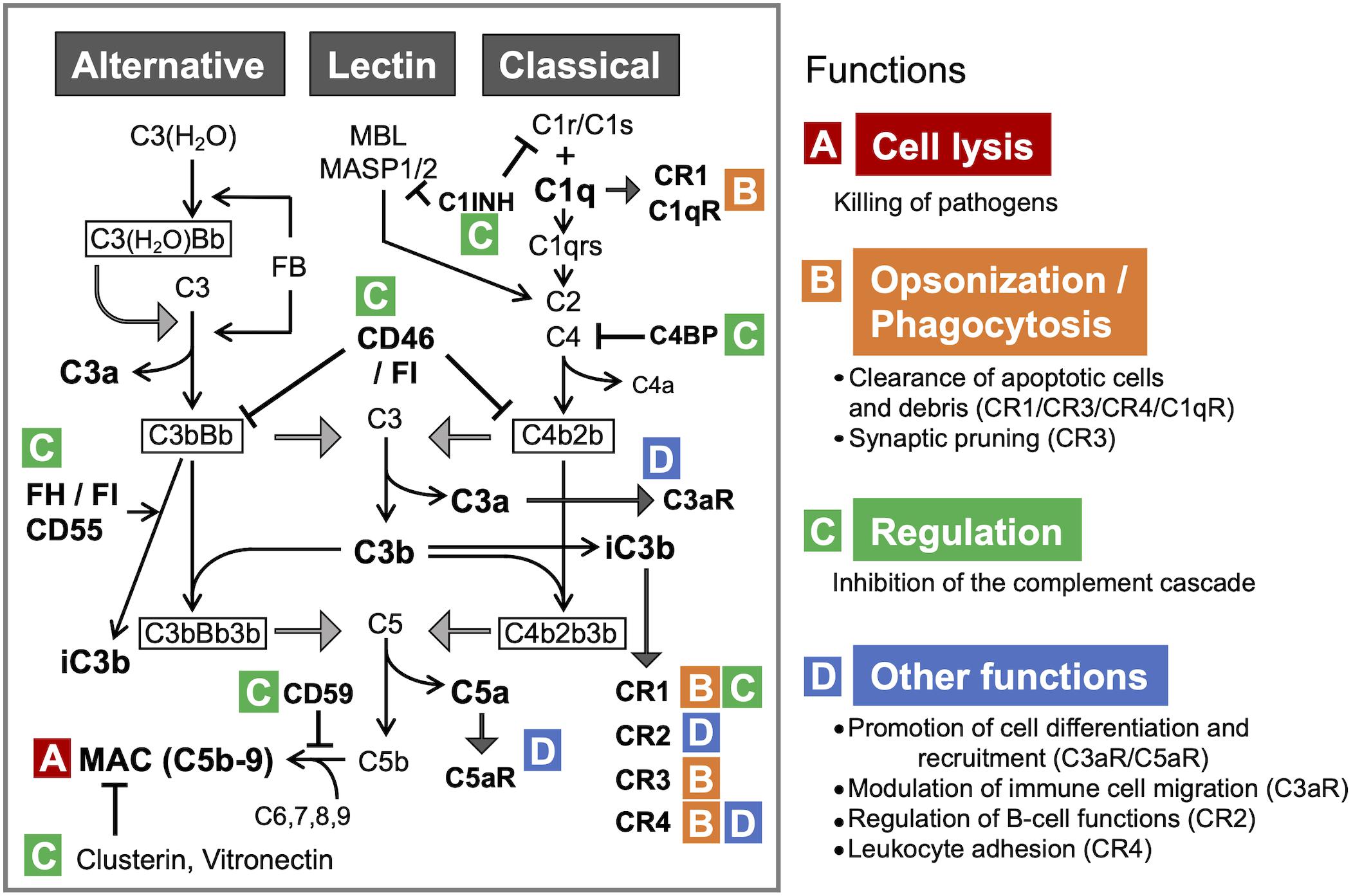
Figure 1. The complement system: activation pathways and functions in the CNS. All three pathways converge at C3. The formation of C3 convertase [C3(H2O)Bb and C3bBb in the alternative pathway or C4b2b in the classical and lectin pathways], followed by C3 cleavage, leads to the formation of MAC, the terminal complement complex (A). C1q and iC3b also facilitate immune responses via opsonization and promoting phagocytosis through binding to the receptors (CR1 and C1qR for C1q; CR1, CR2, and CR3 for iC3b) (B). Under physiological conditions, complement activation is tightly controlled by the regulators of complement activation (RCAs) (C). C3a and C5a, the smaller fragments generated through the proteolytic activation of C3 and C5, respectively, regulate differentiation and mobilization of immune and non-immune cells through the cognate receptors C3aR and C5aR, respectively (D). C3 and C5 can be also cleaved by other proteases via the so-called extrinsic pathway. C1qR, C1q receptor; C3bBb3b, C5 convertase formed by binding of additional C3b to C3bBb (C3 convertase); C3(H2O), hydrated C3; C3(H2O)Bb, the initiating C3 convertase of alternative pathway, consisting of C3(H2O) and Bb (Factor B [FB] cleavage product); CR1, complement receptor type 1; CR2, complement receptor type 2; CR3, complement receptor type 3; CR4, complement receptor type 4; FH, complement factor H; FI, complement factor I. C4BP, C4-binding protein; C1INH, C1-inhibitor.
Each of the activation pathways leads to the formation of C3 convertase (C4bC2b or C3bBb) that cleaves the third complement component (C3) into the C3a and C3b fragments, the latter of which functions as an opsonin, forms the C3 convertase of the alternative pathway (C3bBb) and takes part in the next step of the activation cascade, i.e., proteolytic cleavage of C5. C5 activation results in the release of C5a and the formation of MAC, a.k.a. the terminal complement complex, C5b-9. C3a and C5a are peptides with anaphylatoxin properties and function as fluid-phase inflammatory mediators. They exert their biological effects mainly through the cognate membrane-bound receptors C3a receptor (C3aR) and C5a receptor (C5aR1), respectively. C5a can also bind to the second C5a-like receptor (C5L2, C5aR2). C5aR1 can propagate proinflammatory signals to induce cytokine/chemokine release and mediate the chemoattraction and activation of neutrophils and macrophages (Mathern and Heeger, 2015). C3aR signaling is immunomodulatory, exerting pro- or anti-inflammatory effects in a context-dependent manner (Coulthard and Woodruff, 2015). For example, C3aR activation enhances phagocytosis by macrophages (Wu et al., 2019) and suppresses injury-induced mobilization of neutrophils (Wu et al., 2013; Brennan et al., 2019). Besides its C3aR-mediated actions, C3a has a direct anti-microbial activity (Pasupuleti et al., 2007), indicating its multiple modes of action as an anti-infective agent. Even without antibodies, complement can neutralize pathogens (Speth et al., 2002). In addition, there are links between complement and adaptive immunity, such as the robust augmentation of antibody responses mediated by the C3d fragment (proteolytically generated from C3b) and complement receptor 2 (CR2) (Dempsey et al., 1996) and the regulation of T cell activation by C3a/C5a (Lalli et al., 2008). Thus, the complement system has crucial functions in both innate and adaptive immune responses (Mathern and Heeger, 2015). In addition, recent reports show that a number of cell types store intracellular C3 (Elvington et al., 2016), which has been proposed to have an important role in the regulation of cellular homeostasis (Liszewski et al., 2017). For example, cathepsin L-mediated intracellular C3 cleavage and C3a generation are required for homeostatic T cell survival (Liszewski et al., 2013). Airway epithelial cells can de novo synthesize C3, and intracellular C3 protects these cells from stress-induced cell death (Kulkarni et al., 2019). Intracellular C3 is also required for autophagic turnover of pancreatic β cells during diabetic stress (King et al., 2019). Notably, C3 is an evolutionarily ancient protein; it is considered to be the first element of the original complement system, whose primary role was to guard intracellular environment and homeostasis from various stimuli (Elvington et al., 2016). While complement is widely acknowledged as part of vertebrate humoral immunity, the significance of intracellular complement is less recognized. The potential role for intracellular C3 in self-defense against infection warrants further investigation.
The classical pathway is initiated by binding of C1 (C1qrs) to activators such as immune complexes, microbes, apoptotic cells, and some specific proteins including fibrillar amyloid β (Aβ) (Veerhuis et al., 2011). The lectin pathway is activated by binding of mannose-binding lectin (MBL) and ficolin to carbohydrates on microorganisms or dying cells (Veerhuis et al., 2011). The classical and lectin pathways converge at the proteolytic cleavage of C4 and C2 and the formation of C4bC2b, the C3 convertase shared by these pathways. The alternative pathway continuously generates low levels of C3-derived activation products and C3 convertase C3bBb, providing a natural immunity to microbes (Farriesa and Atkinson, 1991). The activation of alternative pathway can also occur as a result of direct interaction between C3b and pathogenic molecules, including fibrillar Aβ, further amplifying the downstream of complement cascade (Tenner, 2020). C3 and C5 can be cleaved also by MASP 1 (Matsushita and Fujita, 1995), neutrophil elastase (Yuan et al., 2015), cathepsins (Liszewski et al., 2013; Yuan et al., 2015), granulocyte proteases (Johnson et al., 1976), lysosomal enzymes, kallikrein, coagulation factors XIa, Xa, IXa, thrombin, and plasmin (Markiewski and Lambris, 2007; Amara et al., 2010). This non-convertase mode of activation is also called the extrinsic pathway.
Under physiological conditions, complement activity is tightly regulated by regulators of complement activation (RCA) to safeguard against autologous tissue injury. The RCAs that limit complement activation both in time and space include factor H (FH), factor I, C1-inhibitor, C4-binding protein, clusterin, vitronectin, CD46 (membrane cofactor protein, MCP), CD55 (decay accelerating factor, DAF), and complement receptor 1 (CR1, CD35). Properdin (FP), on the other hand, acts as a positive regulator that stabilizes the alternative pathway C3-convertase (Figure 1).
The Complement System in the CNS
Although the liver is the major source of circulating soluble complement proteins (Perlmutter and Colten, 1986), it is now established that complement components are locally synthesized throughout the body, including the brain (Woodruff et al., 2010; Tenner et al., 2018). Complement proteins, including the regulatory factors and receptors, are produced by glia and neurons (Gasque et al., 1998; Ischenko et al., 1998; Nataf et al., 1999), and recent studies have revealed a range of non-immune functions of the complement system in the CNS. For example, complement deposition on synapses, followed by CR3-dependent microglial engulfment and elimination of C1q- and C3b/iC3b-tagged synapses, plays a crucial role in synaptic pruning during normal development (Schafer et al., 2012). C3aR and C5aR are expressed by neurons and glia in the brain under normal and pathological conditions (Barnum et al., 2002). C3aR signaling regulates neuronal migration during CNS development (Gorelik et al., 2017), controls neural progenitor cell proliferation (Coulthard et al., 2018), differentiation and migration in vitro (Shinjyo et al., 2009), and promotes basal as well as injury-induced neurogenesis in the adult mouse brain (Rahpeymai et al., 2006; Shinjyo et al., 2009; Stokowska and Pekna, 2018). Notably, mice lacking C3 exhibit excessive number of synapses as young adults (Stevens et al., 2007; Perez-Alcazar et al., 2014) and neuronal migration defects characteristic of autism spectrum disorder (Gorelik et al., 2017), and are protected against age-related loss of synapses in the hippocampus (Shi et al., 2015). Higher expression of C4 in the brain has been implicated in excessive loss of synapses and development of schizophrenia (Sekar et al., 2016). C3aR deficiency is associated with altered morphology of the hippocampus and amygdala, mild cognitive impairment and hyperactive behavior in unchallenged mice (Coulthard et al., 2018; Pozo-Rodrigálvarez et al., 2021) as well as reduced neural plasticity responses after ischemic brain injury (Stokowska et al., 2017). Thus, the complement system is required for the normal development and function of the CNS.
Considering the broad range of neurodevelopmental and neuroregulatory functions of complement in the brain, it is not surprising that dysfunction or aberrant activation of the complement system have been implicated in the pathogenesis of a number of brain disorders (Gasque et al., 2000; Morgan, 2015). More specifically, complement activation has been observed in human neurodegenerative diseases, such as Alzheimer’s disease (AD), prion disease, chromosome 13 dementias (Eikelenboom et al., 2002; Rostagno et al., 2002; Morgan, 2018), as well as other neurological disorders associated with the blood-brain barrier (BBB) dysfunctions such as cerebrovascular disease, multiple sclerosis (MS) (Storch et al., 1998), and meningitis (Koelman et al., 2019). In infection, while providing protection against pathogens, excessive or prolonged activation of the complement system can lead to exaggerated inflammation and unfavorable outcomes (Ross and Densen, 1984; Figure 1).
Complement and Dementia
Dementia is a broad term for conditions with progressive memory impairment, executive and behavioral anomaly, and emotional problems, affecting the ability to perform day-to-day activities. Complement activation is implicated in AD, the most prevalent form of dementia (Rubio-Perez and Morillas-Ruiz, 2012). Together with glial activation and pro-inflammatory cytokine release, complement activation occurs during the early stage of the disease progression (Aiyaz et al., 2012). As described above, amyloid proteins stimulate complement activity (Veerhuis et al., 2011; Tenner, 2020), leading to the activation of the surrounding glial cells and local chronic inflammation (Cadman and Puttfarcken, 1997; Veerhuis et al., 2011). Complement proteins are present in amyloid plaques in the brain (Aiyaz et al., 2012), and co-localize with Aβ in the brain capillaries of cerebral amyloid angiopathy (Matsuo et al., 2017), suggesting that complement activation is a key event associated with amyloid deposition. In addition, complement is likely involved in other forms of dementia, including chromosome 13 dementias (familial British dementia FBD and familial Danish dementia FDD). Similar to AD, chromosome 13 dementias are associated with neurodegeneration and amyloid deposition in the CNS. In spite of the structural differences in the disease-associated amyloid proteins (Aβ in AD, ABri in FBD, ADan in FDD), neurodegeneration and amyloid deposition, associated with glial activation and local inflammation, are the common hallmark in all of these dementias (Saul et al., 2013). Importantly, complement activation products co-localize with amyloid plaques and related deposits in FBD and FDD (Rostagno et al., 2002). ABri and ADan synthetic peptides activate classical and alternative complement pathways, leading to the formation of the terminal complex (C5b-9). These data suggest that chronic complement activation associated with amyloid deposits may be a contributing factor to dementia progression in chromosome 13 dementias as well as in AD. While experimental evidence points to the critical function of complement in Aβ clearance (Wyss-Coray et al., 2002), and C3- and CR3-dependent microglial phagocytosis appears to play a beneficial role in the elimination of foreign pathogens as well as Aβ (Fu et al., 2012), CR3 also limits Aβ clearance from the brain interstitial fluid (Czirr et al., 2017), and is involved in Aβ-induced microglia-mediated loss of synapses in AD (Hong et al., 2016). The complexity of the involvement of complement in AD type of neurodegeneration is further highlighted by findings in the C3 deficient mice carrying the AD-associated mutations in the amyloid polypeptide and presenilin 1 genes. In this model, the C3 deficiency was protective against plaque-related synapse and neuron loss as well as cognitive decline, despite higher plaque burden (Shi et al., 2017). In addition, C1q and CR3 are required for the clearance of neuronal debris by microglia, at least in the context of injury-induced neurodegeneration (Norris et al., 2018). By virtue of its proinflammatory functions, C5a plays a significant role in neurodegeneration, including AD (Landlinger et al., 2015). Jointly, these reports point to the role of the complement system in Aβ and debris clearance as well as synapse elimination, Aβ-induced neurodegeneration and dementia.
Complement and Brain Infection
In the presence of an intact BBB, circulating immune cells, such as B- and T-lymphocytes, have limited access to the CNS. As a result, the defense of brain tissue against invading pathogens is largely dependent on local innate immunity, in particular the complement system. A number of pathogens escape the peripheral immune responses and enter the CNS. The interaction between pathogens and complement can lead to the clearance of pathogens, however, it may also contribute to tissue damage. Here we summarize the interactions between complement and infectious organisms, including bacteria, viruses, fungi, and parasites (Table 1), and discuss the potential impact of these interactions on CNS function. The interactions between complement and prion proteins are also discussed.
Interactions Between Infectious Agents and Complement in the CNS
Bacteria
Genetic defects in the complement system increase the susceptibility to bacterial infection (Brouwer et al., 2009). C2, factor D (FD), and FP deficiencies are associated with predisposition to meningococcal disease (Fijen et al., 1999; Jönsson et al., 2005; Sprong et al., 2006), while single-nucleotide polymorphisms (SNPs) in MBL are associated with pneumococcal disease (Brouwer et al., 2009). FH genotype 496C/C is associated with meningococcal disease (Haralambous et al., 2006) as is a locus in the FH (CFH) region (Davila et al., 2010). C3a exhibits anti-microbial activity against Gram-negative and Gram-positive bacteria (Pasupuleti et al., 2007). These data show that complement-dependent immune response is crucial in the protection against bacterial infection.
The evidence for the involvement of the complement system in bacterial meningitis is summarized in Table 1.1. Bacterial meningitis is an acute inflammation caused by infection in the meninges that envelope and protect the brain and spinal cord. Commonly caused by Streptococcus pneumoniae, Neisseria meningitidis, and Listeria monocytogenes, bacterial meningitis is a major health threat, the morbidity and mortality of which are driven by dysregulated host immune reactions (Mook-Kanamori et al., 2011; van de Beek et al., 2016).
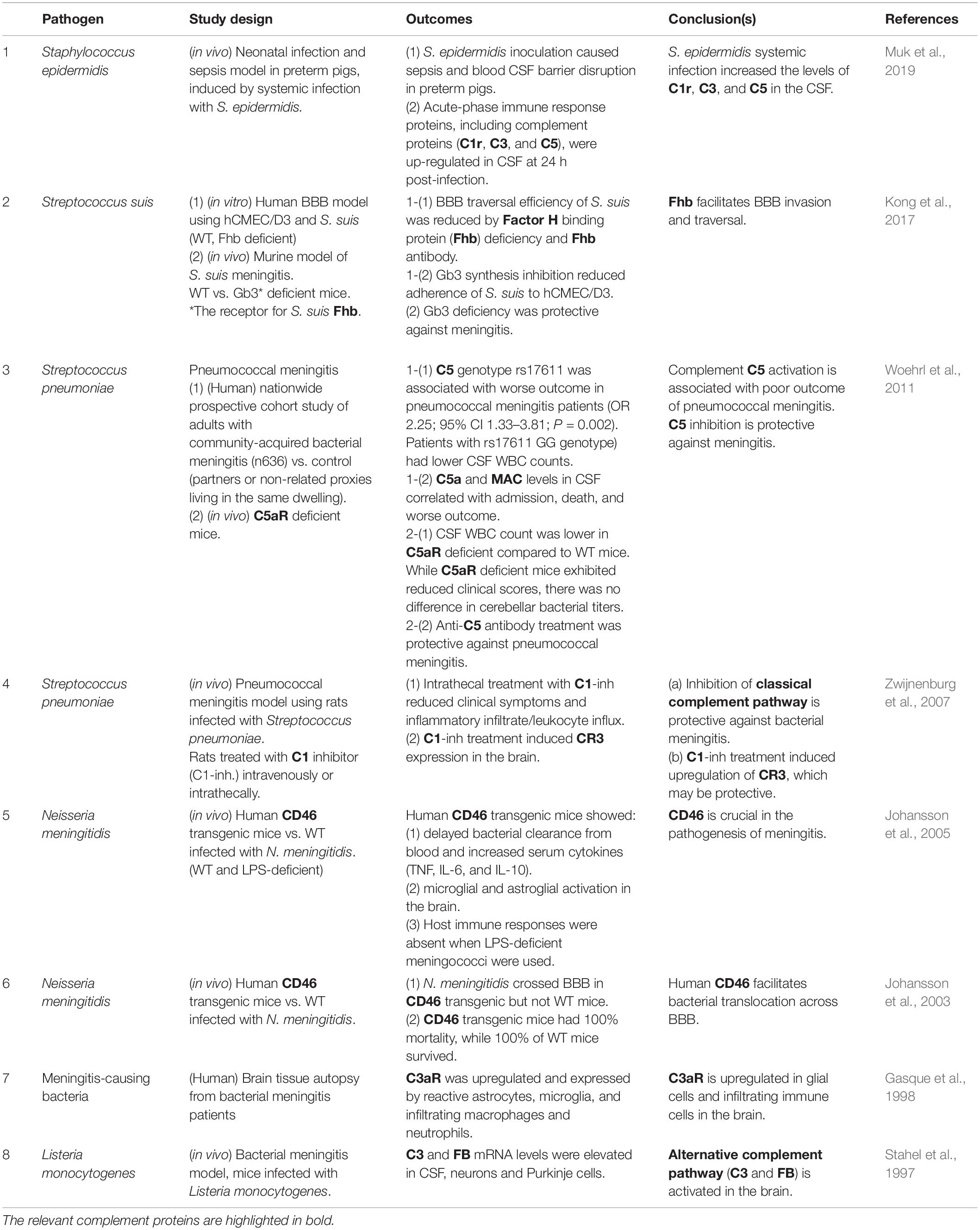
Table 1.1. Interactions between complement and bacteria in the CNS.
As the long-term sequelae of bacterial meningitis include cognitive impairment, infection-induced inflammation may have lasting or even permanent impact on brain function (van de Beek et al., 2004), and there is strong experimental and clinical evidence for the detrimental role of complement in meningitis. Inhibition of the classical pathway and of C5 were both protective against meningitis caused by S. pneumoniae in rodents (Zwijnenburg et al., 2007; Woehrl et al., 2011), and a single nucleotide polymorphism of C5 (rs17611, encoding V802I) was associated with poor outcome in pneumococcal meningitis patients (Woehrl et al., 2011), indicating that the activation of the classical complement pathway plays a key role in the pathogenesis. In murine bacterial meningitis model, L. monocytogenes infection caused the up-regulation of C3 and FB mRNA levels in the cerebrospinal fluid (CSF) as well as in parenchymal neurons (Stahel et al., 1997), implicating local activation of the alternative pathway in the brain during meningitis. C3aR is up-regulated in glial cells and infiltrating immune cell populations in the brain tissue autopsy from bacterial meningitis patients (Gasque et al., 1998), and C5aR deficient mice were less susceptible to meningitis development despite unaltered bacterial titers in the brain (Woehrl et al., 2011), suggesting the important roles for anaphylatoxin receptor signaling in infection-induced neuroinflammation.
FH and CD46, the negative regulators of the complement system, contribute to or even enable of bacterial dissemination into the CNS. FH binding protein (Fhb) is a meningococcal protein that binds to human FH. The interaction between Fhb and host FH enables the bacteria to evade the innate immune attack, thereby facilitating systemic dissemination. Streptococcus suis, a zoonotic bacterium causing bacterial meningitis (Dominguez-Punaro et al., 2007), expresses Fhb, which contributes to the S. suis virulence via inhibition of C3b/iC3b deposition on the bacterial surface (Pian et al., 2012). In addition, Fhb can contribute to the development of severe meningitis by enhancing the traversal of S. suis across the BBB, as suggested by in vivo and in vitro experiments (Kong et al., 2017). Similarly, CD46 acts as the entry receptor for a number of microbes, including N. meningitidis, an exclusively human pathogen. While CD46 is ubiquitously expressed in human tissues, it is only expressed in the eye and spermatozoa in mouse (Liszewski et al., 2000; Lyzogubov et al., 2014). N. meningitidis does not enter the brain in wild-type mice. However, in genetically engineered mice that express human CD46 (human CD46 transgenic), N. meningitidis traversed BBB, leading to significantly higher mortality (Johansson et al., 2003). In addition to delayed bacterial clearance from the circulation, microglial and astroglial activation was observed in the brain of CD46 transgenic mice (Johansson et al., 2005). These data suggest that N. meningitidis utilizes CD46 to facilitate its own entry into the CNS.
Systemic infection and sepsis can sensitize the brain to inflammation via complement activation, even without the bacteria entering the CNS. In preterm infants, S. epidermidis triggers cerebral inflammation, white matter injury and impaired neurodevelopment (Bi et al., 2015). While Toll-like receptor 2 (TLR2) is thought to be the major contributor to neonatal infection caused by Gram-positive bacteria, the observation that the CSF levels of C1r, C3, and C5 were rapidly increased in S. epidermidis infection-induced sepsis model in pigs (Muk et al., 2019) suggests that systemic infection may cause CNS injury via aberrant activation of the complement cascade. Bacterial lipopolysaccharide-induced long-term depression via microglial CR3 activation offers a specific mechanistic link between neuroinflammation, complement activation, synaptic dysfunction, and memory impairments (Zhang et al., 2014).
While complement protects the host against bacterial infection, excessive complement activation can contribute to CNS injury and long lasting or permanent dysfunction. In addition, several complement components and regulators are exploited by bacteria to facilitate their entry into the CNS.
Viruses
A large number of brain disease-causing viruses, including herpes viruses (human herpes virus 1 [HSV-1], human herpesvirus 6 [HHV-6], and γ-herpesviruses), HIV, measles virus, Borna disease virus (BDV), Theiler’s murine encephalitis virus (TMEV), Venezuelan equine encephalitis virus (VEEV), West Nile virus (WNV), and Zika virus (ZIKV), interact with the complement system in the CNS (Table 1.2) and this interaction may play a role in neurodegeneration. Latent HSV-1 infection is associated with increased risk for AD (Itzhaki, 2018). HHV-6A and HHV-6B are frequently found in patients with neuroinflammatory diseases including MS (Alvarez-Lafuente et al., 2006) and AD (Readhead et al., 2018). γ-herpesviruses, including human Epstein–Barr virus (EBV or HHV-4) and Kaposi’s sarcoma-associated herpesvirus (KSHV or HHV-8), also establish life-long latency in the host, which is associated with increased risk of diverse neurological conditions, including meningitis, encephalitis, HIV-related CNS lymphoma, and MS (Said et al., 1997; Bossolasco et al., 2006; Serafini et al., 2007). Seropositivity for EBV is associated with clinical AD (Carbone et al., 2014). In HIV patients, KSHV infection can manifest in the CNS (Andrews et al., 2011; Baldini et al., 2013; Tso et al., 2017). In addition, HIV can cause cognitive impairment, so called HIV-associated neurocognitive disorders (HAND, NeuroAIDS). Indeed, while antiretroviral therapy effectively inhibits viral replication and reduces mortality, more than 50% of HIV-positive individuals suffer from cognitive impairment (Clifford, 2017; Olivier et al., 2018). Even though antiretroviral therapy suppresses viral replication, neurotoxic HIV proteins, such as Tat, continue to be produced in the CNS, leading to persistent inflammation (Liu et al., 2014; Avalos et al., 2017). WNV is a neuroinvasive pathogen that causes significant neuronal loss, inflammation, and microglial activation (Clarke et al., 2014). WNV targets hippocampal neurons (Armah et al., 2007), and patients recovering from WNV disease often suffer from cognitive impairment (Sejvar et al., 2003; Sadek et al., 2010). ZIKV is the causative agent of Zika fever, congenital infection which leads to severe developmental neurodevelopmental defects, including neonatal microcephaly (Rasmussen et al., 2016; Russo et al., 2017). The fact that ZIKV infection in adults can lead to neurological complications such as acute myelitis, encephalomyelitis, encephalitis, meningoencephalitis, and sensory polyneuropathy (Mécharles et al., 2016; Brito Ferreira et al., 2017), further strengthens the notion that ZIKV is highly neurotropic (Souza et al., 2019). VEEV is endemic to Central and South America and periodically emerges from the enzootic cycle to cause infection in human and equid populations (Weaver and Barrett, 2004), occasionally causing severe encephalomyelitis (Weaver et al., 2004). Measles virus infects over 40 million individuals per year (McChesney and Oldstone, 1989). Acute infection can cause encephalomyelitis, the neurological sequelae of which include subacute sclerosing panencephalitis (SSPE) and measles inclusion body encephalitis (MIBE) (Griffin, 2020).

Table 1.2. Interactions between complement and viruses in the CNS.
The complement system provides the first line of defense against viruses, including HSV-1 via the lectin pathway (Bibert et al., 2019), and γ-herpesvirus, VEEV, and WNV via C3-dependent elimination mechanisms (Kapadia et al., 2002; Mehlhop et al., 2005; Brooke et al., 2012). On the other hand, the virus-induced up-regulation of complement components in the CNS may lead to BBB dysfunction and contribute to tissue damage in infection with HIV and TMEV (Speth et al., 2004; Woollard et al., 2014; Libbey et al., 2017). For example, HIV-Tat protein upregulated C3 expression in human brain microvascular endothelial cells (HBMEC) in vitro (Woollard et al., 2014), C1q is up-regulated in the brain of Simian immunodeficiency virus (SIV)-infected rhesus macaques (Speth et al., 2004; Depboylu et al., 2005) and mice after HIV-Tat injection into the cerebral cortex (Hammond et al., 2018), two in vivo models of NeuroAIDS. Increased C1q in the CSF is associated with cognitive impairment in HIV-infected individuals (McGuire et al., 2016). BDV induced local C1q mRNA expression in the hippocampus and cortex (Dietzschold et al., 1995) and ZIKV infection enhanced expression of C1q and C3, associated with microglial activation and hippocampal synaptic damage and memory impairment in a murine infection model using ZIKV intracerebroventricular infusion (Figueiredo et al., 2019). In murine model induced by intracranial infection of WNV, C1q protein colocalized with microglia adjacent to WNV-infected neurons, possibly causing synaptic terminal loss (Vasek et al., 2016). These data suggest that local complement activation may induce cognitive impairment via the disruption of neurotransmission.
Some viruses increase their virulence by manipulating complement regulatory proteins. HSV-1 infection caused the down-regulation of FH in human primary neuroglia in vitro (Hill et al., 2009), potentially leading to enhanced activity of the alternative pathway. As suggested by studies using CD46 transgenic mice, CD46 is involved in the dissemination of measles virus into the CNS, increasing the risk of measles encephalitis (Rall et al., 1997; Manchester et al., 1999; Oldstone et al., 1999; Evlashev et al., 2000). In postmortem SSPE brain tissues, strong CD46 signal was observed on cerebral endothelium throughout the brain, as well as ependymal cells lining the ventricles and choroid plexus (McQuaid and Cosby, 2002), while CD46 expression was suppressed by measles virus infection in SSPE lesions (Ogata et al., 1997; McQuaid and Cosby, 2002). In addition, CD46 expression was observed in primary human glial cells (oligodendrocytes, astrocytes, and microglia) and CD46 inhibition by anti-CD46 antibody suppressed the fusion of glia and measles virus-infected T cells in vitro, suggesting that measles virus may spread in the brain parenchyma via binding to CD46 on glia (Cassiani-Ingoni et al., 2005). In HHV-6 infection models in vitro, using neuronal and glial cell lines, CD46 also mediated HHV-6 infection-induced transactivation of Multiple Sclerosis-Associated Retrovirus (MSRV) (Charvet et al., 2018). In a murine γ-herpesviruses infection model, C3 deficiency caused higher susceptibility to viral latency in the CNS (Kapadia et al., 2002). Several γ-herpesviruses encode homologues of RCAs (Farriesa and Atkinson, 1991), to inhibit complement activation (Fodor et al., 1995; Kapadia et al., 1999, 2002) thereby preventing complement-mediated elimination (Liszewski et al., 1996; Russo et al., 1996; Virgin et al., 1997). Like bacteria, some viruses can utilize and mimic host RCA proteins for their dissemination and entry into the CNS.
Sustained complement activation is associated with neuroinflammation and neurodegeneration in a number of neurological disorders (Eikelenboom et al., 1989; Rogers et al., 1992). Of note, COVID-19 patients reportedly suffer from diverse neurological complications (Stracciari et al., 2021). Although there is no direct evidence thus far to indicate causality between SARS-CoV-2 infection and neuropathology (Maury et al., 2021), future research will show whether there is a causal link between overactivation of complement in response to SARS-CoV-2 and neurological symptoms or long-term complications of COVID-19 (Garred et al., 2021).
Fungi
Fungal infection and dissemination into the brain has been proposed to contribute to the etiology of AD (Alonso et al., 2014, 2018). Candida is one of the most common commensal fungi, and can cause systemic infections which frequently affect the CNS (Lionakis et al., 2011; Alonso et al., 2014, 2018). Mice with invasive candidiasis exhibit microglial activation and local inflammation (Lionakis et al., 2011). Aspergillus and Cryptococcus are also fungal species that can cause invasive CNS infection, especially in immunocompromised individuals (Panackal and Williamson, 2015). Similarly, cerebral aspergillosis is caused by Aspergillus spp. that mainly affect immunocompromised individuals, such as AIDS patients and those under immunosuppressive treatment regimens (Ruhnke et al., 2007). Cryptococcosis, caused by the encapsulated fungus Cryptococcus neoformans, frequently occurs in AIDS, organ transplant recipients and cancer patients, and is the leading cause of mortality in immunocompromised individuals (Rajasingham et al., 2017). These fungal species can also cause meningoencephalitis (Gottfredsson and Perfect, 2000; Góralska et al., 2018), suggesting that fungal infection-induced local inflammation and meningoencephalitis may lead to long-term sequelae including cognitive dysfunction.
The complement system plays pivotal roles in the susceptibility to fungal CNS infection and infection-induced neuropathology (Table 1.3). A study using murine Candida albican infection models showed that C5 could be a major determinant of CNS infection susceptibility (Tuite et al., 2005). Neutrophils play a major protective role against dissemination of C. neoformans (Lovchik and Lipscomb, 1993; Graybill et al., 1997), and C5aR signaling is required for infection-induced neutrophil recruitment to the brain as suggested by data obtained in a murine infection model using C5 deficiency and inhibitory anti-C5aR antibodies (Sun et al., 2016). C5 and its cleavage product C5a are required for pulmonary accumulation of neutrophils upon infection and intravascular clearance of C. neoformans (Lovchik and Lipscomb, 1993; Sun et al., 2016), further confirming the crucial roles for C5 and C5a in neutrophil-mediated killing of C. neoformans. Jointly, these data suggest that complement activation and C5aR signaling are protective against C. neoformans-induced meningoencephalitis. In postmortem brain tissues from subjects with cerebral aspergillosis, C1q, C4, C3, and C5 were up-regulated in the brain, and co-localized with neurons, astrocytes, oligodendrocytes, and infiltrating macrophages (Rambach et al., 2008).
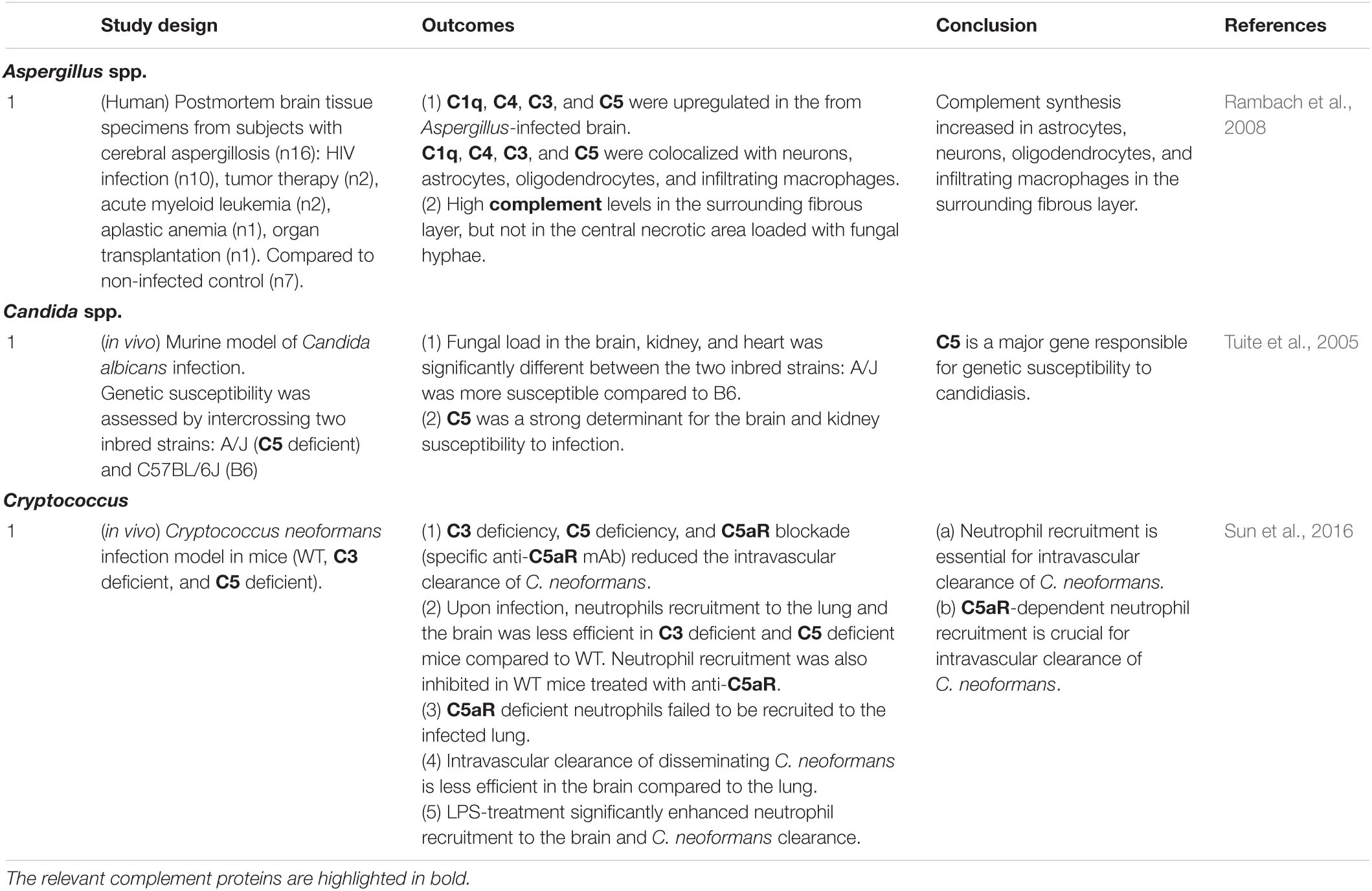
Table 1.3. Interactions between complement and fungi in the CNS.
While the specific mechanistic links between fungal infection and dementia remain to be elucidated (Alonso et al., 2014, 2018), complement may be protective against neurodegenerative disorders via limiting fungal invasion into the CNS.
Parasites
Protozoan parasites including Toxoplasma gondii and Plasmodium spp. are obligate intracellular organisms that can potentially cause dysfunction and parenchymal injury in the brain. While T. gondii infects all nucleated cells of warm-blooded vertebrates, erythrocytes are the main target in Plasmodium infection in vertebrates. Host immune responses to these eukaryotes are very complex (Ivanova et al., 2019). The interactions between the complement system and these protozoa, with relevance for brain pathologies and the potential links to dementia, are summarized in Table 1.4.

Table 1.4. Interactions between complement and parasites in the CNS.
Plasmodium
Plasmodium spp. are the agents causing malaria, one of the deadliest infectious diseases worldwide. Cerebral malaria (CM), caused by P. falciparum, is the most severe and life-threatening condition characterized by diffuse encephalopathy. CM is frequently accompanied by seizures and coma, and accounts for the majority of childhood deaths from malaria in endemic regions (Schmutzhard and Gerstenbrand, 1984; Idro et al., 2010; Riggle et al., 2020). In addition, more than 10% of children who survive CM have persistent neurological sequelae, including those affecting cognition and behavior (Frevert and Nacer, 2014). Among the features of CM is the adherence of Plasmodium-infected red blood cells (iRBCs) to brain vascular endothelium and BBB dysfunction (Yusuf et al., 2017; Pais and Penha-Gonçalves, 2019). Several mechanisms were suggested to explain the severe endothelial damage and vascular leakage in CM, and the proposed hypotheses include hemodynamic hypothesis (Van der Heyde et al., 2006; Wassmer and Grau, 2017), inflammation hypothesis (Silamut et al., 1999; Van der Heyde et al., 2006; Wassmer et al., 2006; Dunst et al., 2017), coagulation dysfunction hypothesis (Grau et al., 2003; Francischetti, 2008; Bridges et al., 2010; OSullivan et al., 2016), and innate immune hypothesis (Pais and Penha-Gonçalves, 2019). However, the exact mechanism remains elusive.
There is evidence suggesting that the complement system plays a significant role in CM pathogenesis. In an early study using murine CM model with P. berghei ANKA (Finley et al., 1982), T cell-deficient (nu/nu) mice were protected against CM compared to nu/+ mice, despite comparable parasitemia. The rapid decrease in serum C3 and concomitant increase in serum immune complexes observed in wildtype mice were absent in nu/nu mice, indicating the interaction between the complement system and cellular immunity in CM (Finley et al., 1982). C1q and C5 protein levels were also significantly higher in the brain of CM mice compared to non-CM mice, suggesting the activation of the classical pathway (Lackner et al., 2008). The roles for the complement system in CM were further examined using genetically modified mice, including mice deficient in C5 (Patel et al., 2008; Ramos et al., 2011; Buckingham et al., 2014), C5aR (Ramos et al., 2011; Kim et al., 2014; McDonald et al., 2015), C3 (Ramos et al., 2012), C4 (Ramos et al., 2012), FB (Ramos et al., 2012), and C3aR (Ramos et al., 2011). Notably, C5 deficient mice were fully protected from CM and CM-associated seizures (Ramos et al., 2011; Buckingham et al., 2014), while C5aR deficient mice were only moderately protected (Ramos et al., 2011; Kim et al., 2014). Mice lacking FB, C4, or C3aR showed outcomes comparable to wildtype mice (Ramos et al., 2011, 2012). In addition, C9 deposits were observed in the CNS during CM, and C9 inhibition (neutralizing anti-C9 antibody injection) significantly delayed CM development (Ramos et al., 2011). Results from clinical studies also show complement activation during CM. Serum C5a levels were significantly higher in children with CM compared to those without CM (Kim et al., 2014). Postmortem CM frontal lobes exhibited activation of complement, the coagulation cascade, and platelets (Kumar et al., 2018). These data suggest that MAC formation plays a critical role in CM pathogenesis. In addition, persistent cognitive deficits caused in the offspring by in utero exposure to malaria were dependent on maternal C5a-C5aR signaling (McDonald et al., 2015). These findings point to the role of C5a in the initiation of neuroinflammation together with the dysregulation of angiogenesis and synaptogenesis (McDonald et al., 2013). Surprisingly, the formation of conventional C5 convertase via C3 activation was not required for CM progression (Ramos et al., 2012), which suggests that C5 was activated by coagulation factors or other non-complement proteases (Ramos et al., 2012; Figure 2). These observations support the role for the complement system as a link between immune responses and dysregulated coagulation during CM.
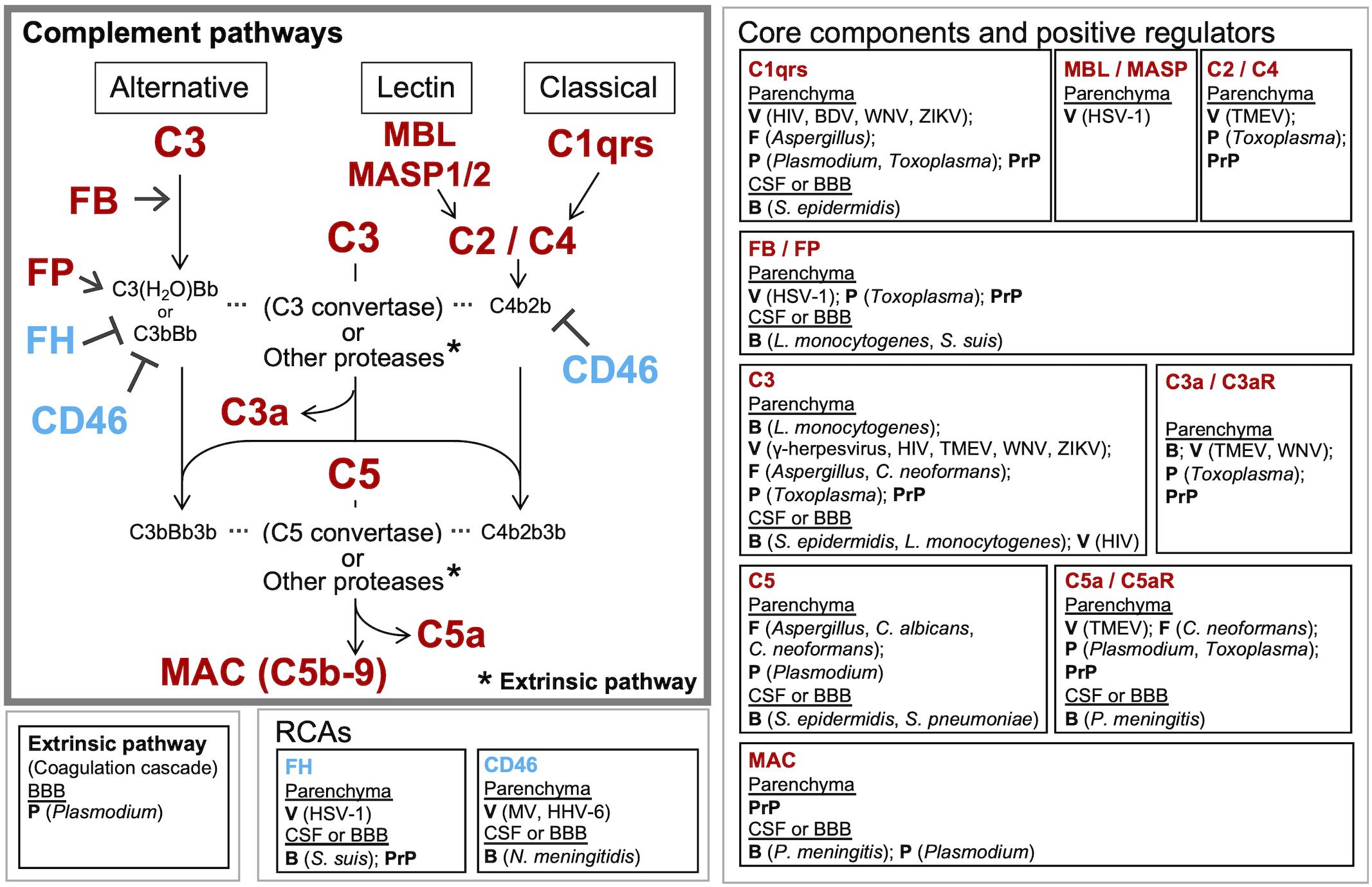
Figure 2. Known and potential interactions between complement proteins and infectious agents. Infectious agents can modulate the expression or activity of complement proteins and receptors in the brain parenchyma, CSF, and BBB. Highlighted are the core complement components/positive regulators (red) and negative regulators (RCAs) (blue), which are potentially affected by infectious agents. Infection-induced activation of the coagulation cascade can also lead to C3 and C5 cleavage via the extrinsic pathway. Pathogens known to interact with the respective complement proteins are presented in the boxes to the right or below the pathway diagram. B, bacteria; V, viruses; F, fungi; P, parasites; PrP, prion proteins.
The interactions between Plasmodium and the complement system are possibly even more intricate (Schmidt et al., 2015; Kennedy et al., 2016, 2017; Rosa et al., 2016). Blood stage P. falciparum (free merozoites as well as intraerythrocytic schizonts) evades complement-mediated destruction by recruiting FH, a major negative regulator of complement activation, on the cell surface (Kennedy et al., 2016; Rosa et al., 2016). As mentioned above, several other pathogens, including Neisseria meningitidis (Granoff et al., 2009; Schneider et al., 2009) and Borrelia burgdorferi (Kraiczy et al., 2001), use FH for the same purpose, suggesting that it is an evolutionarily conserved strategy to circumvent complement-mediated elimination. Plasmodium utilizes another complement regulatory protein CR1 as a receptor for red blood cell invasion (Tham et al., 2010; Spadafora et al., 2010). In addition, mobilization of CR1 on the surface of Plasmodium-infected red blood cells is required for rosetting (Rowe et al., 1997), a process implicated in vascular obstruction during severe malaria (Carlson, 1993). CR1 polymorphisms could explain the association between altered efficiency of rosetting and malaria severity (Cockburn et al., 2004; Schmidt et al., 2015).
In conclusion, overactivation of complement cascade triggered by the parasite conceivably contributes to CM pathogenesis, and the inhibition of the complement cascade at the level or downstream of C5 activation, and/or the inhibition of CR1 could be a beneficial treatment strategy for CM.
Toxoplasma gondii
Toxoplasma gondii infects approximately one-third of the world population. It is an opportunistic infection that in individuals with immunodeficiency can cause severe diseases, including Toxoplasma encephalitis (Marra, 2018). Infection during pregnancy can lead to congenital toxoplasmosis, the severity of which depends on the stage of pregnancy (Bigna et al., 2020). While Toxoplasma rarely causes symptoms in healthy individuals with effective immunity, it can establish latent infection in the brain and other tissues (Pittman and Knoll, 2015). It is also notable that the virulence can vary depending on Toxoplasma genotype (Howe and Sibley, 1995; Behnke et al., 2011; Pomares et al., 2018; Taniguchi et al., 2018). Once disseminated into the brain, the parasite transforms into bradyzoite form (cyst) and establishes a life-long infection in neurons and glia (Ólafsson and Barragan, 2020). The presence of Toxoplasma cysts in the CNS has been linked to various neuropsychiatric disorders, such as schizophrenia (Elsheikha et al., 2016; Del Grande et al., 2017; Fuglewicz et al., 2017; Fond et al., 2018; Stepanova et al., 2019; Tyebji et al., 2019a, b), possibly via the direct modulation of dopaminergic/serotonergic signaling as well as neuroinflammation (Henriquez et al., 2009; Mahmoud et al., 2017). T cells and type II interferon (IFN-γ)-dependent immune responses are an essential part of the host defense against Toxoplasma infection (Sasai et al., 2018). Type I interferons (IFNs) also play an important regulatory role in the innate immune response during protozoan infection, including toxoplasmosis (Silva-Barrios and Stäger, 2017).
Toxoplasma is classified into three groups: virulent type I, which causes acute infection, and avirulent type II and type III, which are responsible for chronic infection. Several studies have suggested the involvement of the complement system during acute Toxoplasma (type I) infection. Toxoplasma lytic activity of human sera was dependent on classical pathway components (C1q, C2, C4, C5, C6, C7, and C8) in a study using a virulent Toxoplasma strain in vitro (Schreiber and Feldman, 1980). Animal sera (pig, rabbit, and dog), with the exception of cat serum, effectively killed Toxoplasma tachyzoites of a virulent strain via C1q and natural IgM-dependent complement activation (Kaneko et al., 2004). Toxoplasma tachyzoites of the same strain were resistant to complement-mediated lysis via the formation of iC3b, the inactive form of C3b (Fuhrman and Joiner, 1989). Toxoplasma tachyzoites of both virulent and avirulent strains mobilized complement regulatory proteins (FH and C4BP) on their surface to evade complement-mediated parasite killing in vitro, whereas C3 deficient mice suffered higher parasite burden in the brain and other organs in vivo, suggesting complex host-parasite interactions (Sikorski et al., 2020). Of note, C5 deficiency rendered either protection or enhanced mortality depending on the genetic background of the murine host (Araujo et al., 1975). These data suggest that, while the complement system plays a pivotal role in the protection against acute Toxoplasma infection, the parasite has evolved strategies to manipulate the complement system to survive and propagate in the host.
The interaction between Toxoplasma and the complement system in the brain has recently been elucidated. In murine Toxoplasma infection models, cerebral C1q is upregulated during chronic infection with virulent and avirulent strains (Xiao et al., 2016), and persistent infection with a virulent Toxoplasma strain led to the upregulation of C1q, C1r, C3, and C4 mRNA levels and deposition of complement component proteins (C1q and C3) in the brain. These changes were associated with neurodegeneration (Li et al., 2019). An avirulent Toxoplasma strain also caused upregulation of complement proteins, including C3, C4b, and C1q, in the mouse brain (Huang et al., 2019). Furthermore, mRNAs for FB and FP, C3aR, and C5aR were up-regulated in the brain of mice chronically infected with an avirulent Toxoplasma strain (Shinjyo et al., 2021), suggesting that the alternative pathway is activated in the brain during chronic infection. The Toxoplasma infection-induced up-regulation of FB, FP, and C5aR mRNAs occurred in primary murine glial cells in vitro in a microglia-dependent manner (Shinjyo et al., 2021). These data suggest that, while complement-dependent clearance is essential in the initial, peripheral phase of infection, chronic Toxoplasma infection can cause persistent complement activation in the CNS.
Prion Proteins
Transmissible spongiform encephalopathies (TSEs, prion diseases) are fatal neurodegenerative diseases, which include bovine spongiform encephalopathy (BSE) in cattle, scrapie in sheep and goat, Creutzfeldt-Jakob disease (CJD), fatal familial insomnia, and Gerstmann-Sträussler-Scheinker syndrome in humans (Prusiner et al., 1998; Prusiner, 1998). Most neurodegenerative diseases, including prion diseases and AD, share two common features: the accumulation and self-propagation of misfolded proteins (Gomez-Gutierrez and Morales, 2020). TSE-causing infectious prion proteins (PrPSc) transmit the disease-associated conformation to normal prion proteins (PrPC), leading to further propagation of PrPSc. Following peripheral exposure and prior to neuroinvasion, PrPSc accumulates in lymphoid tissues, including lymph nodes and Peyer’s patches. Early PrPSc accumulation occurs within the germinal center on follicular dendritic cells (FDCs) (Brown et al., 1999) as well as within tingible body macrophages (McGovern and Jeffrey, 2007). From the lymphoid tissues, transmission to the CNS occurs via the peripheral nervous system (Glatzel et al., 2001). The central event in prion disease progression is the accumulation of PrPSc in the CNS accompanied by neuronal loss and spongiform neuropils, suggesting aggressive destruction of neural networks (Budka, 2000).
The involvement of the complement system in prion-induced neuropathology has been observed in different types of prion diseases (Bonifati and Kishore, 2007; Table 1.5). In scrapie-infected rodents, total complement activity in the brain was significantly increased at the terminal stage (Lv et al., 2014). In addition, C1q upregulation (Dandoy-Dron et al., 1998, 2000; Carroll et al., 2018), C3 upregulation and colocalization with neurons, and MAC deposition on neurons were observed in infected human brain (Kovacs et al., 2004) as well as in scrapie model in mice (Lv et al., 2014), suggesting the activation of the classical and terminal pathways. C1q also colocalized with infected neurons in vitro, and throughout the brain C1q distribution overlapped with PrP (Hasebe et al., 2012). In addition, FB and FP levels significantly increased in the brain of scrapie-infected mice, while FB and C3 colocalized with neurons and activated microglia (Chen et al., 2020), suggesting the activation of the alternative pathway and microglia. Notably, mice deficient in C1q or FB/C2 were protected against encephalopathy after intraperitoneal prion exposure, but C3 deficiency was not protective (Klein et al., 2001). Interestingly, reactive astrocytes in prion diseases are characterized by C3 up-regulation and mixed A1/A2 phenotype (Hartmann et al., 2019) distinct from neurotoxic astrocytes observed in other neurodegenerative diseases (Kwon and Koh, 2020). The abolishment of C3-positive astrocytes led to prion disease acceleration (Hartmann et al., 2019). These data suggest that C3 plays multiple roles in prion disease progression, depending on the context. In contrast, complement hemolytic activity was significantly lower in the CSF of CJD patients, particularly in genetic CJD cases (Chen et al., 2016). In addition, the protein levels of some complement components, including C3, C4, and C9, decreased in the CSF of sporadic CJD (sCJD) patients compared to non-CJD group (Chen et al., 2016). While the explanation conceivably lies in the increased consumption of complement in the brain parenchyma, the neuropathologic implications of these findings are currently elusive.
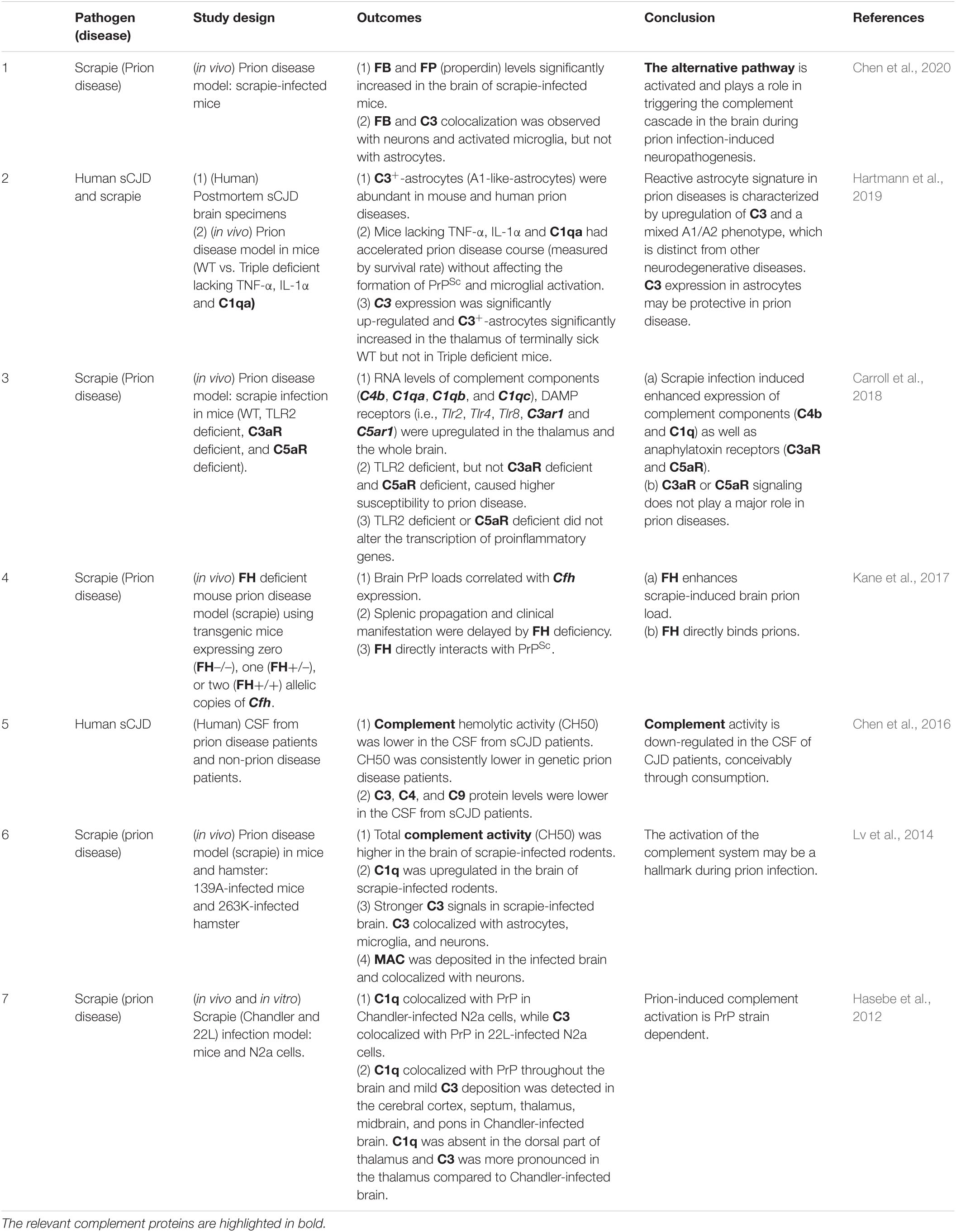
Table 1.5. Interactions between complement and prion proteins in the CNS.
Given that FH deficiency was protective against PrP propagation (Kane et al., 2017), prion proteins may escape complement-mediated clearance by manipulating FH-dependent complement inhibition, possibly via direct interaction between PrPSc and FH (Kane et al., 2017). Thus, RCAs seem important target molecules exploited by infectious agents, including prion proteins, to evade innate immunity and propagate throughout the body including the CNS.
Complement: A Double Edge Sword in the Infected Brain
The consequences of local complement activation by infectious agents for brain tissue integrity and function are highly context dependent. Complement has a role in the regulation of normal neuronal functioning and homeostasis. It may be neuroprotective and play a critical role in the elimination of various pathogens, but can also be a major mediator of neurodegeneration and tissue damage. The complement system can also be hijacked by some pathogens and play a complicit role in their dissemination. Figure 2 illustrates the potential interactions between the complement system and infectious agents, and Table 2 summarizes the responses of selected complement proteins to the individual pathogens, including up- or down-regulation, deposition, and the impact of genetic polymorphism or genetic manipulations.
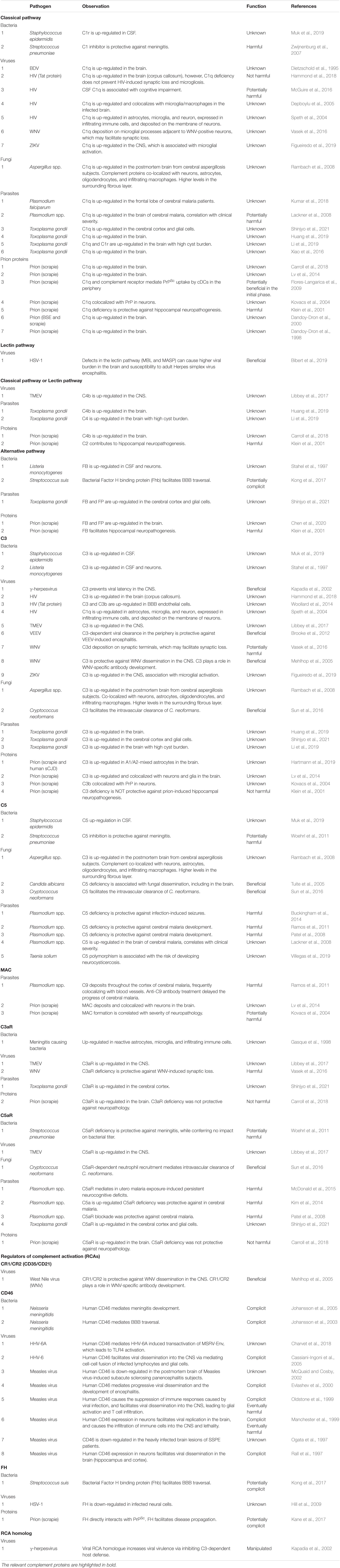
Table 2. The functions of specific complement proteins and in CNS infection.
In the CNS, C1q plays potentially harmful roles in bacterial (S. pneumoniae-induced bacterial meningitis) (Zwijnenburg et al., 2007), viral (HIV-associated cognitive impairment) (McGuire et al., 2016), parasite (Plasmodium, CM) (Lackner et al., 2008), and prion (Klein et al., 2001) infections, while it may be protective during the initial phase of prion disease (Flores-Langarica et al., 2009). Jointly these findings indicate the deleterious consequences of overactivation of the classical pathway. The lectin pathway is protective against herpes simplex virus encephalitis by inhibiting viral dissemination into the CNS (Bibert et al., 2019). While C3-mediated pathogen clearance is beneficial in the periphery (Mehlhop et al., 2005; Brooke et al., 2012; Sun et al., 2016), C3d deposition on synaptic terminals may facilitate synaptic loss in WNV infection (Vasek et al., 2016). Likewise, the net outcome of downstream complement activation seems to be context-dependent. C5 is important for fungal clearance in the vasculature (Sun et al., 2016) and inhibits fungal dissemination into the CNS (Tuite et al., 2005). In contrast, in the Plasmodium infection, C5 deficiency is protective against CM (Patel et al., 2008; Ramos et al., 2011; Buckingham et al., 2014). Inhibition of MAC formation was also protective against CM (Ramos et al., 2011), confirming the crucial role of terminal complement pathway in CM pathogenesis. C3aR was up-regulated in the brain upon viral infection (Libbey et al., 2017) and C3aR deficiency was protective against viral infection-induced synaptic loss (Vasek et al., 2016). C5aR deficiency was also protective against CM development (Patel et al., 2008; Kim et al., 2014; McDonald et al., 2015), as well as bacterial meningitis (Woehrl et al., 2011). However, neither C3aR deficiency nor C5aR deficiency were protective against prion-induced neuropathology (Carroll et al., 2018), indicating pathogen-specific roles of anaphylatoxin receptors in the CNS.
CD46, a negative regulator of the classical and lectin pathways, aids in the dissemination of bacteria (N. meningitidis) (Johansson et al., 2003, 2005) and viruses [Measles virus (Rall et al., 1997; Manchester et al., 1999; Oldstone et al., 1999; Evlashev et al., 2000) and HHV-6 (Cassiani-Ingoni et al., 2005)] into the CNS, leading to meningitis and encephalitis, respectively. While the use of CD46 by these pathogens could be an evasion strategy from the classical or lectin pathway-mediated clearance, CD46-dependent facilitation of immune cell infiltration into the CNS (Manchester et al., 1999; Oldstone et al., 1999) and cell-cell fusion of infected lymphocytes and glial cells (Cassiani-Ingoni et al., 2005) suggest higher mechanistic complexity of the interactions between complement and infectious agents. FH, a negative regulator of the alternative pathway, is also engaged in the dissemination of pathogens in bacterial (S. suis) (Kong et al., 2017) and prion infections (Kane et al., 2017), indicating that these infectious agents manipulate the alternative complement pathway for their propagation in the brain. The fact that a viral RCA homolog plays a facilitatory role in viral dissemination via the inhibition of C3-dependent host defense (Kapadia et al., 2002) suggests that infectious agents have evolved diverse molecular mechanisms to escape complement-mediated killing.
Discussion
Potential Link Between Infection-Induced Complement Activation and Dementia
Considering the link between cognitive impairment and aberrant complement activation in the CNS (Aiyaz et al., 2012; Rubio-Perez and Morillas-Ruiz, 2012), the most simplified view is that CNS infection could lead to dementia via the complement activation (Figure 3A: dotted rectangle). However, as discussed above, the mechanistic links between infectious agents, complement, and neurodegeneration are far more complex. Due to the multiple neurodevelopmental and homeostatic functions that the complement system and its individual components play in the brain (Figure 1), complement activation by infectious agents can cause an imbalance in the intricate web of interactions between complement proteins and the nervous system with deleterious consequences for CNS tissue integrity and function (Figure 3B: solid rectangle). This could explain the unpredictable and sometimes inconsistent observations in different studies in human patients as well as in animal models of CNS infection. The multiple functions of the complement system in the CNS together with the complexity of immune responses to infection call for well-informed and highly selective approaches when manipulating the complement system for therapeutic benefit. For example, inhibiting the down-stream of complement cascade (C5 and MAC) and/or C5aR could protect the brain from Plasmodium infection-induced tissue damage (Patel et al., 2008; Ramos et al., 2011; Buckingham et al., 2014; Kim et al., 2014), whereas inhibition restricted to the upstream components (C1q, C2, and FB) could be beneficial in prion diseases (Klein et al., 2001), as shown in animal models. However, these studies were conducted using constitutive inactivation of specific genes. As the complement system plays multiple roles in the CNS, roles which are distinct from those in the periphery, it is imperative to investigate CNS-specific roles of the complement system in the context of an infection by a specific pathogen. Furthermore, while complement proteins are locally produced in the brain, it is less clear which cell types are responsible, how complement protein production and responses are regulated, and how are these processes influenced by various stimuli under physiological and pathological conditions, including CNS infection. Of particular interest in this respect are the potential functions of intracellular C3 (Elvington et al., 2016), not only with regard to the interactions between infectious agents and the brain-resident cells but also the role of intracellular C3 in neurodegeneration. Considering the diverse roles of glial cells (astrocytes and microglia, the brain-resident macrophages) in the maintenance of CNS homeostasis, further studies are required to elucidate how infectious agents modulate complement generation in glial cells and how, in turn, it might affect the phenotypes of these cells.
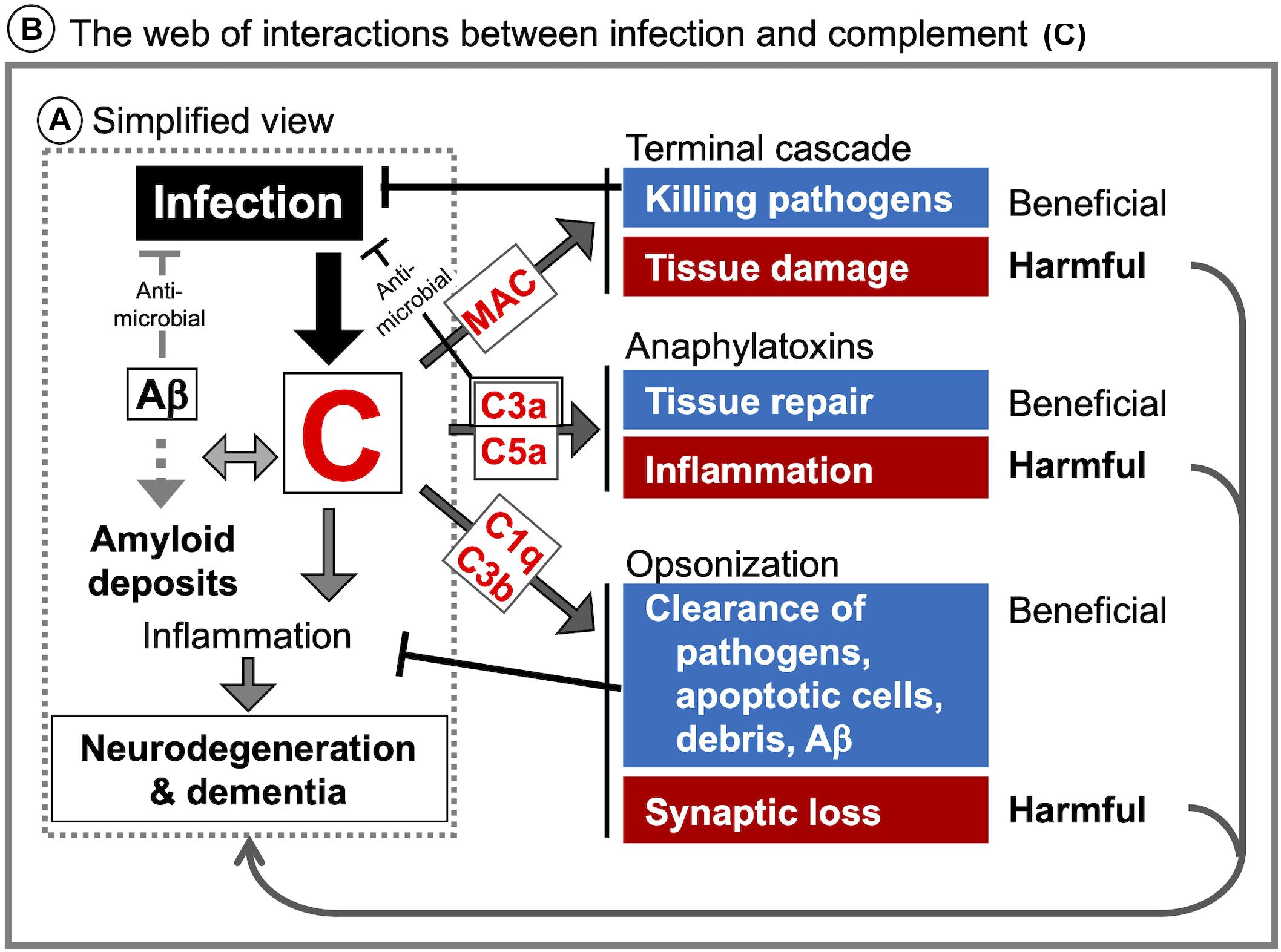
Figure 3. Infection-induced complement activation in the CNS and potential links to neurodegeneration and dementia. (A) The most simplified view of how CNS infection can lead to neurodegeneration and dementia is enclosed in the dotted rectangle. Infection-induced complement activation products may contribute to neurodegeneration and dementia via interaction with amyloid deposits and enhanced neuroinflammation. (B) A more detailed schematic representation of specific interactions between infectious agents and the complement system as a potential mechanistic link between CNS infection, neurodegeneration and dementia. The consequences of complement activation may be context-dependent. For example, MAC plays a role in pathogen elimination but can also contribute to tissue damage. C3a and C5a can support tissue repair and adaptive neural plasticity, but also promote potentially detrimental inflammation and leukocyte recruitment. Opsonization and phagocytosis are needed for clearance of pathogens, but the same mechanisms are involved in synapse elimination as the first step in neurodegeneration.
Conclusion
Of the approximately 50 million people with dementia worldwide, about 50% live in low- and middle-income countries (WHO, 2019). Considering higher risk of infection and infection-induced brain disorders in those countries (Fernandes and Caramelli, 2019; Singh et al., 2020), understanding of the potential mechanistic links between infection and neurodegenerative disorders including dementia, will inform the development of preventive and therapeutic strategies. While amyloidogenesis is widely accepted as the underlying cause of AD and other forms of neurodegenerative diseases, Aβ-targeting drug development has been so far unsuccessful (Robakis, 2020). Given that the presence of amyloid plaques does not necessarily indicate cognitive decline (Skaper, 2012; Robakis, 2020), alternative culprits and theories are needed. Indeed, there is a growing body of evidence showing that innate immunity and neuroinflammation play a pivotal role in dementia development (Leng and Edison, 2020).
Separated from the other parts of the body by the BBB and CSF barrier, the CNS is uniquely protected by special immune components, including the complement system and glial cells. While locally produced complement components play crucial homeostatic roles in the CNS, dysregulated complement activity could be detrimental to the function of the CNS. As the key element of brain-resident innate immunity, the complement system plays both protective and destructive roles in neurodegeneration (Kolev et al., 2009). Whereas aberrant complement activation is frequently associated with neuroinflammation and neurodegenerative disorders, the complement system exerts homeostatic functions via eliminating apoptotic cells, cell debris, and toxic substances, and supports plasticity and tissue repair. A broad range of infectious agents can potentially interact with the complement system in the brain and the net outcome of these interactions appears to depend on the specific pathogens, the target components, and the mode of interaction. It is therefore conceivable that pathogen-triggered and persistent activation of the complement system represents the missing mechanistic link between infection, in particular infection affecting the CNS, and neurodegeneration. We are fully aware that the simplified view presented in Figure 3 does not capture every aspect of the involvement of the complement system in infection-induced neuropathology and neurodegeneration, but only highlights the key components of the interplay between pathogens and complement activation induced–neurodegenerative processes that may lead to dementia. While the specific mechanistic links between CNS infection, neurodegeneration and dementia are still unclear, the involvement of the complement system and the pathogen- and disease stage-specific roles of the individual complement proteins warrant further intense investigation.
Author Contributions
NS wrote the first draft of the manuscript. MP and WK critically assessed the content. NS and MP jointly revised and finalized the manuscript. All the authors contributed to the article and approved the submitted version.
Funding
MP’s research was supported by the Swedish Research Council (2017-00991), Hjärnfonden/The Swedish Brain Foundation, the Swedish state under the agreement between the Swedish Government and the county councils, the ALF agreement (716591), E. Jacobson’s Foundation, and R. and U. Amlöv’s Foundation. NS’s research was supported by JSPS KAKENHI Grant No. 19K07839.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aiyaz, M., Lupton, M., Proitsi, P., Powell, J., and Lovestone, S. (2012). Complement activation as a biomarker for Alzheimer’s disease. Immunobiology 217, 204–215. doi: 10.1016/j.imbio.2011.07.023
Alonso, R., Marina, D., Morato, A., Rábano, E., Carrasco, A., and Pisa, L. (2014). Fungal infection in patients with Alzheimer’s disease. J. Alzheimers Dis. 41, 301–311. doi: 10.3233/JAD-132681
Alonso, R., Pisa, D., Fernández-Fernández, A., and Carrasco, L. (2018). Infection of fungi and bacteria in brain tissue from elderly persons and patients with Alzheimer’s disease. Front. Aging Neurosci. 10:159. doi: 10.3389/fnagi.2018.00159
Alvarez-Lafuente, R., García-Montojo, M., De las Heras, V., Bartolomé, M., and Arroyo, R. (2006). Clinical parameters and HHV-6 active replication in relapsing–remitting multiple sclerosis patients. J. Clin. Virol. 37, S24–S26. doi: 10.1016/S1386-6532(06)70007-5
Amara, U., Flierl, M., Rittirsch, D., Klos, A., Chen, H., Acker, B., et al. (2010). Molecular interacommunication between the complement and coagulation systems. J. Immunol. 185, 5628–5636. doi: 10.4049/jimmunol.0903678
Andrews, J., Cho-Park, Y., Ferry, J., Abramson, J., and Robbins, G. (2011). Kaposi’s sarcoma-associated herpesvirus-related solid lymphoma involving the heart and brain. AIDS Res. Treat. 2011:729854. doi: 10.1155/2011/729854
Araujo, F., Rosenberg, L., and Remington, J. (1975). Experimental Toxoplasma gondii infection in mice: the role of the fifth component of complement. Proc. Soc. Exp. Biol. Med. 149, 800–804. doi: 10.3181/00379727-149-38902
Armah, H., Wang, G., Omalu, B., Tesh, R., Gyure, K., Chute, D., et al. (2007). Systemic distribution of West Nile virus infection: postmortem immunohistochemical study of six cases. Brain Pathol. 17, 354–362. doi: 10.1111/j.1750-3639.2007.00080.x
Avalos, C., Abreu, C., Queen, S., Li, M., Price, S., Shirk, E., et al. (2017). Brain macrophages in simian immunodeficiency virus-infected, antiretroviral-suppressed macaques: a functional latent reservoir. mBio 8:e01186-17. doi: 10.1128/mBio.01186-17
Baldini, F., Baiocchini, A., Schininà, V., Agrati, C., Giancola, M., Alba, L., et al. (2013). Brain localization of Kaposi’s sarcoma in a patient treated by combination antiretroviral therapy. BMC Infect Dis. 13:600. doi: 10.1186/1471-2334-13-600
Barnum, S., Ames, R., Maycox, P., Hadingham, S., Meakin, J., Harrison, D., et al. (2002). Expression of the complement C3a and C5a receptors after permanent focal ischemia: an alternative interpretation. Glia 38, 169–173. doi: 10.1002/glia.10069
Behnke, M., Khan, A., Wootton, J., Dubey, J., Tang, K., and Sibley, L. (2011). Virulence differences in Toxoplasma mediated by amplification of a family of polymorphic pseudokinases. Proc. Natl. Acad. Sci. U.S.A. 108, 9631–9636. doi: 10.1073/pnas.1015338108
Bi, D., Qiao, L., Bergelson, I., Ek, C., Duan, L., Zhang, X., et al. (2015). Staphylococcus epidermidis bacteremia induces brain injury in neonatal mice via toll-like receptor 2-dependent and -independent pathways. J. Infect. Dis. 212, 1480–1490. doi: 10.1093/infdis/jiv231
Bibert, S., Piret, J., Quinodoz, M., Collinet, E., Zoete, V., Michielin, O., et al. (2019). Herpes simplex encephalitis in adult patients with MASP-2 deficiency. PLoS Pathog. 15:e1008168. doi: 10.1371/journal.ppat.1008168
Bigna, J., Tochie, J., Tounouga, D., Bekolo, A., Ymele, N., Youda, E., et al. (2020). Global, regional, and country seroprevalence of Toxoplasma gondii in pregnant women: a systematic review, modelling and meta-analysis. Sci. Rep. 10:12102. doi: 10.1038/s41598-020-69078-9
Bonifati, D., and Kishore, U. (2007). Role of complement in neurodegeneration and neuroinflammation. Mol. Immunol. 44, 999–1010. doi: 10.1016/j.molimm.2006.03.007
Bossolasco, S., Falk, K., Ponzoni, M., Ceserani, N., Crippa, F., Lazzarin, A., et al. (2006). Ganciclovir is associated with low or undetectable Epstein-Barr virus DNA load in cerebrospinal fluid of patients with HIV-related primary central nervous system lymphoma. Clin. Infect. Dis. 42, e21–e25.
Brennan, F., Jogia, T., Gillespie, E., Blomster, L., Li, X., Nowlan, B., et al. (2019). Complement receptor C3aR1 controls neutrophil mobilization following spinal cord injury through physiological antagonism of CXCR2. JCI Insight 4:e98254. doi: 10.1172/jci.insight.98254
Bridges, D., Bunn, J., van Mourik, J., Grau, G., Preston, R., and Molyneux, M. (2010). Rapid activation of endothelial cells enables Plasmodium falciparum adhesion to platelet-decorated von Willebrand factor strings. Blood 115, 1472–1474. doi: 10.1182/blood-2009-07-235150
Brito Ferreira, M., Antunes de Brito, C., Moreira, Á, de Morais Machado, M., Henriques-Souza, A., Cordeiro, M., et al. (2017). Guillain-Barré syndrome, acute disseminated encephalomyelitis and encephalitis associated with zika virus infection in Brazil: detection of viral RNA and isolation of virus during late infection. Am. J. Trop. Med. Hyg. 97, 1405–1409. doi: 10.4269/ajtmh.17-0106
Brooke, C., Schäfer, A., Matsushima, G., White, L., and Johnston, R. (2012). Early activation of the host complement system is required to restrict central nervous system invasion and limit neuropathology during Venezuelan equine encephalitis virus infection. J. Gen. Virol. 93, 797–806. doi: 10.1099/vir.0.038281-0
Brouwer, M., de Gans, J., Heckenberg, S., Zwinderman, A., van der Poll, T., and van de Beek, D. (2009). Host genetic susceptibility to pneumococcal and meningococcal disease: a systematic review and meta-analysis. Lancet Infect. Dis. 9, 31–44. doi: 10.1016/S1473-3099(08)70261-5
Brown, K., Stewart, K., Ritchie, D., Mabbott, N., Williams, A., Fraser, H., et al. (1999). Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat. Med. 5, 1308–1312. doi: 10.1038/15264
Buckingham, S., Ramos, T., and Barnum, S. (2014). Complement C5-deficient Mice are protected from seizures in experimental cerebral malaria. Epilepsia 55, e139–e142.
Budka, H. (2000). Histopathology and immunohistochemistry of human transmissible spongiform encephalopathies (TSEs). Arch. Virol. Suppl. 16, 135–142. doi: 10.1007/978-3-7091-6308-5_12
Cadman, E., and Puttfarcken, P. (1997). Beta-amyloid peptides initiate the complement cascade without producing a comparable effect on the terminal pathway in vitro. Exp. Neurol. 146, 388–394. doi: 10.1006/exnr.1997.6540
Carbone, I., Lazzarotto, T., Ianni, M., Porcellini, E., Forti, P., Masliah, E., et al. (2014). Herpes virus in Alzheimer’s disease: relation to progression of the disease. Neurobiol. Aging 35, 122–129. doi: 10.1016/j.neurobiolaging.2013.06.024
Carlson, J. (1993). Erythrocyte rosetting in Plasmodium falciparum malaria–with special reference to the pathogenesis of cerebral malaria. Scand. J. Infect. Dis. Suppl. 86, 1–79.
Carroll, J., Race, B., Williams, K., and Chesebro, B. (2018). Toll-like receptor 2 confers partial neuroprotection during prion disease. PLoS One 13:e0208559. doi: 10.1371/journal.pone.0208559
Cassiani-Ingoni, R., Greenstone, H., Donati, D., Fogdell-Hahn, A., Martinelli, E., Refai, D., et al. (2005). CD46 on glial cells can function as a receptor for viral glycoprotein-mediated cell-cell fusion. Glia 52, 252–258. doi: 10.1002/glia.20219
Charvet, B., Reynaud, J., Gourru-Lesimple, G., Perron, H., Marche, P., and Horvat, B. (2018). Induction of proinflammatory multiple sclerosis-associated retrovirus envelope protein by human herpesvirus-6A and CD46 receptor engagement. Front. Immunol. 9:2803. doi: 10.3389/fimmu.2018.02803
Chen, C., Lv, Y., Hu, C., Xu, X., Zhang, R., Xiao, K., et al. (2020). Alternative complement pathway is activated in the brains of scrapie-infected rodents. Med. Microbiol. Immunol. 209, 81–94. doi: 10.1007/s00430-019-00641-6
Chen, C., Lv, Y., Shi, Q., Zhou, W., Xiao, K., Sun, J., et al. (2016). Low activity of complement in the cerebrospinal fluid of the patients with various prion diseases. Infect. Dis. Poverty 5:35. doi: 10.1186/s40249-016-0128-7
Clarke, P., Leser, J., Quick, E., Dionne, K., Beckham, J., and Tyler, K. (2014). Death receptor-mediated apoptotic signaling is activated in the brain following infection with West Nile virus in the absence of a peripheral immune response. J. Virol. 88, 1080–1089. doi: 10.1128/JVI.02944-13
Clifford, D. (2017). HIV-associated neurocognitive disorder. Curr. Opin. Infect. Dis. 30, 117–122. doi: 10.1097/QCO.0000000000000328
Cockburn, I., Mackinnon, M., O’Donnell, A., Allen, S., Moulds, J., Baisor, M., et al. (2004). A human complement receptor 1 polymorphism that reduces Plasmodium falciparum rosetting confers protection against severe malaria. Proc. Natl. Acad. Sci. U.S.A. 101, 272–277. doi: 10.1073/pnas.0305306101
Coulthard, L., Hawksworth, O., Conroy, J., Lee, J., and Woodruff, T. (2018). Complement C3a receptor modulates embryonic neural progenitor cell proliferation and cognitive performance. Mol. Immunol. 101, 176–181. doi: 10.1016/j.molimm.2018.06.271
Coulthard, L., and Woodruff, T. (2015). Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J. Immunol. 194, 3542–3548. doi: 10.4049/jimmunol.1403068
Czirr, E., Castello, N., Mosher, K., Castellano, J., Hinkson, I., Lucin, K., et al. (2017). Microglial complement receptor 3 regulates brain Abeta levels through secreted proteolytic activity. J. Exp. Med. 214, 1081–1092. doi: 10.1084/jem.20162011
Dandoy-Dron, F., Benboudjema, L., Guillo, F., Jaegly, A., Jasmin, C., Dormont, D., et al. (2000). Enhanced levels of scrapie responsive gene mRNA in BSE-infected mouse brain. Brain Res. Mol. Brain Res. 76, 173–179. doi: 10.1016/s0169-328x(00)00028-0
Dandoy-Dron, F., Guillo, F., Benboudjema, L., Deslys, J., Lasmézas, C., Dormont, D., et al. (1998). Gene expression in scrapie. Cloning of a new scrapie-responsive gene and the identification of increased levels of seven other mRNA transcripts. J. Biol. Chem. 273, 7691–7697. doi: 10.1074/jbc.273.13.7691
Davila, S., Wright, V., Khor, C., Sim, K., Binder, A., Breunis, W., et al. (2010). Genome-wide association study identifies variants in the CFH region associated with host susceptibility to meningococcal disease. Nat. Genet. 42, 772–776. doi: 10.1038/ng.640
Del Grande, C., Galli, L., Schiavi, E., Dell’Osso, L., and Bruschi, F. (2017). Is Toxoplasma gondii a trigger of bipolar disorder? Pathogens 6:3. doi: 10.3390/pathogens6010003
Dempsey, P., Allison, M., Akkaraju, S., Goodnow, C., and Fearon, D. (1996). C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science 271, 348–350. doi: 10.1126/science.271.5247.348
Depboylu, C., Schäfer, M., Schwaeble, W., Reinhart, T., Maeda, H., Mitsuya, H., et al. (2005). Increase of C1q biosynthesis in brain microglia and macrophages during lentivirus infection in the rhesus macaque is sensitive to antiretroviral treatment with 6-chloro-2′,3′-dideoxyguanosine. Neurobiol. Dis. 20, 12–26. doi: 10.1016/j.nbd.2005.01.030
Dietzschold, B., Schwaeble, W., Schäfer, M., Hooper, D., Zehng, Y., Petry, F., et al. (1995). Expression of C1q, a subcomponent of the rat complement system, is dramatically enhanced in brains of rats with either Borna disease or experimental allergic encephalomyelitis. J. Neurol. Sci. 130, 11–16. doi: 10.1016/0022-510x(94)00269-t
Dominguez-Punaro, M., Segura, M., Plante, M., Lacouture, S., Rivest, S., and Gottschalk, M. (2007). Streptococcus suis serotype 2, an important swine and human pathogen, induces strong systemic and cerebral inflammatory responses in a mouse model of infection. J. Immunol. 179, 1842–1854. doi: 10.4049/jimmunol.179.3.1842
Dunst, J., Kamena, F., and Matuschewski, K. (2017). Cytokines and chemokines in cerebral malaria pathogenesis. Front. Cell Infect. Microbiol. 7:324. doi: 10.3389/fcimb.2017.00324
Eikelenboom, P., Bate, C., Van Gool, W., Hoozemans, J., Rozemuller, J., Veerhuis, R., et al. (2002). Neuroinflammation in Alzheimer’s disease and prion disease. Glia 40, 232–239. doi: 10.1002/glia.10146
Eikelenboom, P., Hack, C., Rozemuller, J., and Stam, F. (1989). Complement activation in amyloid plaques in Alzheimer’s dementia. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 56, 259–262. doi: 10.1007/BF02890024
Elsheikha, H., Büsselberg, D., and Zhu, X. (2016). The known and missing links between Toxoplasma gondii and schizophrenia. Metab. Brain Dis. 31, 749–759. doi: 10.1007/s11011-016-9822-1
Elvington, M., Liszewski, M., and Atkinson, J. (2016). Evolution of the complement system: from defense of the single cell to guardian of the intravascular space. Immunol. Rev. 274, 9–15. doi: 10.1111/imr.12474
Evlashev, A., Moyse, E., Valentin, H., Azocar, O., Trescol-Biémont, M., Marie, J., et al. (2000). Productive measles virus brain infection and apoptosis in CD46 transgenic mice. J. Virol. 74, 1373–1382. doi: 10.1128/jvi.74.3.1373-1382.2000
Farriesa, T., and Atkinson, J. (1991). Evolution of the complement system. Immunol. Today 12, 295–300. doi: 10.1016/0167-5699(91)90002-B
Fernandes, B., and Caramelli, P. (2019). Ischemic stroke and infectious diseases in low-income and middle-income countries. Curr. Opin. Neurol. 32, 43–48. doi: 10.1097/WCO.0000000000000641
Figueiredo, C., Barros-Aragão, F., Neris, R., Frost, P., Soares, C., Souza, I., et al. (2019). Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice. Nat. Commun. 10:3890. doi: 10.1038/s41467-019-11866-7
Fijen, C., van den Bogaard, R., Schipper, M., Mannens, M., Schlesinger, M., Nordin, F., et al. (1999). Properdin deficiency: molecular basis and disease association. Mol. Immunol. 36, 863–867. doi: 10.1016/s0161-5890(99)00107-8
Finley, R., Mackey, L., and Lambert, P. (1982). Virulent P. berghei malaria: prolonged survival and decreased cerebral pathology in cell-dependent nude mice. J. Immunol. 1219, 2213–2218.
Flores-Langarica, A., Sebti, Y., Mitchell, D., Sim, R., and MacPherson, G. (2009). Scrapie pathogenesis: the role of complement C1q in scrapie agent uptake by conventional dendritic cells. J. Immunol. 182, 1305–1313. doi: 10.4049/jimmunol.182.3.1305
Fodor, W., Rollins, S., Bianco-Caron, S., Rother, R., Guilmette, E., Burton, W., et al. (1995). The complement control protein homolog of herpesvirus saimiri regulates serum complement by inhibiting C3 convertase activity. J. Virol. 69, 3889–3892. doi: 10.1128/JVI.69.6.3889-3892.1995
Fond, G., Boyer, L., Schürhoff, F., Berna, F., Godin, O., Bulzacka, E., et al. (2018). Latent Toxoplasma infection in real-world schizophrenia: results from the national FACE-SZ cohort. Schizophr Res. S0920, 30262–30265. doi: 10.1016/j.schres.2018.05.007
Francischetti, I. (2008). Does activation of the blood coagulation cascade have a role in malaria pathogenesis? Trends Parasitol. 24, 258–263. doi: 10.1016/j.pt.2008.03.009
Frevert, U., and Nacer, A. (2014). Fatal cerebral malaria: a venous efflux problem. Front. Cell Infect. Microbiol. 4:155. doi: 10.3389/fcimb.2014.00155
Fu, H., Liu, B., Frost, J., Hong, S., Jin, M., Ostaszewski, B., et al. (2012). Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia 60, 993–1003. doi: 10.1002/glia.22331
Fuglewicz, A., Piotrowski, P., and Stodolak, A. (2017). Relationship between toxoplasmosis and schizophrenia: a review. Adv. Clin. Exp. Med. 26, 1031–1036.
Fuhrman, S., and Joiner, K. (1989). Toxoplasma gondii: mechanism of resistance to complement-mediated killing. J. Immunol. 142, 940–947.
Garred, P., Tenner, A., and Mollnes, T. (2021). Therapeutic targeting of the complement system: from rare diseases to pandemics. Pharmacol. Rev. 73, 792–827. doi: 10.1124/pharmrev.120.000072
Gasque, P., Dean, Y., McGreal, E., VanBeek, J., and Morgan, B. (2000). Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology 49, 171–186. doi: 10.1016/s0162-3109(00)80302-1
Gasque, P., Singhrao, S. K., Neal, J. W., Wang, P., Sayah, S., Fontaine, M., et al. (1998). The receptor for complement anaphylatoxin C3a is expressed by myeloid cells and nonmyeloid cells in inflamed human central nervous system: analysis in multiple sclerosis and bacterial meningitis. J. Immunol. 160, 3543–3554.
Glatzel, M., Heppner, F., Albers, K., and Aguzzi, A. (2001). Sympatheticinnervation of lymphoreticular organs is rate limiting for prion neuroinvasion. Neuron 31, 25–34. doi: 10.1016/s0896-6273(01)00331-2
Gomez-Gutierrez, R., and Morales, R. (2020). The prion-like phenomenon in Alzheimer’s disease: evidence of pathology transmission in humans. PLoS Pathog. 16:e1009004. doi: 10.1371/journal.ppat.1009004
Góralska, K., Blaszkowska, J., and Dzikowiec, M. (2018). Neuroinfections caused by fungi. Infection 46, 443–459. doi: 10.1007/s15010-018-1152-2
Gorelik, A., Sapir, T., Haffner-Krausz, R., Olender, T., Woodruff, T., and Reiner, O. (2017). Developmental activities of the complement pathway in migrating neurons. Nat. Commun. 8:15096. doi: 10.1038/ncomms15096
Gottfredsson, M., and Perfect, J. (2000). Fungal meningitis. Semin. Neurol. 20, 307–322. doi: 10.1055/s-2000-9394
Granoff, D., Welsch, J., and Ram, S. (2009). Binding of complement factor H (fH) to Neisseria meningitidis is specific for human fH and inhibits complement activation by rat and rabbit sera. Infect. Immun. 77, 764–769. doi: 10.1128/IAI.01191-08
Grau, G., Mackenzie, C., Carr, R., Redard, M., Pizzolato, G., Allasia, C., et al. (2003). Platelet accumulation in brain microvessels in fatal pediatric cerebral malaria. J. Infect. Dis. 187, 461–466. doi: 10.1086/367960
Graybill, J., Bocanegra, R., Lambros, C., and Luther, M. (1997). Granulocyte colony stimulating factor therapy of experimental cryptococcal meningitis. J. Med. Vet. Mycol. 35, 243–247. doi: 10.1080/02681219780001221
Griffin, D. (2020). Measles virus persistence and its consequences. Curr. Opin. Virol. 41, 46–51. doi: 10.1016/j.coviro.2020.03.003
Hammond, J., Qiu, W., Marker, D., Chamberlain, J., Greaves-Tunnell, W., Bellizzi, M., et al. (2018). HIV Tat causes synapse loss in a mouse model of HIV-associated neurocognitive disorder that is independent of the classical complement cascade component C1q. Glia 66, 2563–2574. doi: 10.1002/glia.23511
Haralambous, E., Dolly, S., Hibberd, M., Litt, D., Udalova, I., O’dwyer, C., et al. (2006). Factor H, a regulator of complement activity, is a major determinant of meningococcal disease susceptibility in UK Caucasian patients. Scand. J. Infect. Dis. 38, 764–771. doi: 10.1080/00365540600643203
Hartmann, K., Sepulveda-Falla, D., Rose, I., Madore, C., Muth, C., Matschke, J., et al. (2019). Complement 3(+)-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia. Acta Neuropathol. Commun. 7:83. doi: 10.1186/s40478-019-0735-1
Hasebe, R., Raymond, G., Horiuchi, M., and Caughey, B. (2012). Reaction of complement factors varies with prion strains in vitro and in vivo. Virology 423, 205–213. doi: 10.1016/j.virol.2011.11.017
Henriquez, S., Brett, R., Alexander, J., Pratt, J., and Roberts, C. (2009). Neuropsychiatric disease and Toxoplasma gondii infection. Neuroimmunomodulation 16, 122–133. doi: 10.1159/000180267
Hill, J., Zhao, Y., Clement, C., Neumann, D., and Lukiw, W. (2009). HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport 20, 1500–1505. doi: 10.1097/WNR.0b013e3283329c05
Hong, S., Beja-Glasser, V., Nfonoyim, B., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Howe, D., and Sibley, L. (1995). Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J. Infect. Dis. 172, 1561–1566. doi: 10.1093/infdis/172.6.1561
Huang, W., Wang, Y., Mahmmod, Y., Wang, J., Liu, T., Zheng, Y., et al. (2019). A double-edged sword: complement component 3 in Toxoplasma gondii infection. Proteomics 19, e1800271. doi: 10.1002/pmic.201800271
Idro, R., Marsh, K., John, C., and Newton, C. (2010). Europe PMC funders group cerebral malaria?; mechanisms of brain injury and strategies for improved neuro-cognitive outcome. Pediatr. Res. 68, 267–274. doi: 10.1203/00006450-201011001-00524
Ischenko, A., Sayah, S., Patte, C., Andreev, S., Gasque, P., Schouft, M. T., et al. (1998). Expression of a functional anaphylatoxin C3a receptor by astrocytes. J. Neurochem. 71, 2487–2496.
Itzhaki, R. (2018). Corroboration of a Major role for herpes simplex virus type 1 in Alzheimer’s disease. Front. Aging Neurosci. 10:324. doi: 10.3389/fnagi.2018.00324
Ivanova, D., Denton, S., Fettel, K., Sondgeroth, K., Munoz Gutierrez, J., Bangoura, B., et al. (2019). Innate lymphoid cells in protection, pathology, and adaptive immunity during apicomplexan infection. Front. Immunol. 10:196. doi: 10.3389/fimmu.2019.00196
Johansson, L., Rytkonen, A., Bergman, P., Albiger, B., Källström, H., Hökfelt, T., et al. (2003). CD46 in meningococcal disease. Science 301, 373–375. doi: 10.1126/science.1086476
Johansson, L., Rytkönen, A., Wan, H., Bergman, P., Plant, L., Agerberth, B., et al. (2005). Human-like immune responses in CD46 transgenic mice. J. Immunol. 175, 433–440. doi: 10.4049/jimmunol.175.1.433
Johnson, U., Ohlsson, K., and Olsson, I. (1976). Effects of granulocyte neutral proteases on complement components. Scand. J. Immunol. 5, 421–426. doi: 10.1111/j.1365-3083.1976.tb00296.x
Jönsson, G., Truedsson, L., Sturfelt, G., Oxelius, V., Braconier, J., and Sjöholm, A. (2005). Hereditary C2 deficiency in Sweden: frequent occurrence of invasive infection, atherosclerosis, and rheumatic disease. Medicine 84, 23–34.
Kane, S., Farley, T., Gordon, E., Estep, J., Bender, H., Moreno, J., et al. (2017). Complement regulatory protein factor H Is a soluble prion receptor that potentiates peripheral prion pathogenesis. J. Immunol. 199, 3821–3827. doi: 10.4049/jimmunol.1701100
Kaneko, Y., Takashima, Y., Xuaun, X., Igarashi, I., Nagasawa, H., Mikami, T., et al. (2004). Natural IgM antibodies in sera from various animals but not the cat kill Toxoplasma gondii by activating the classical complement pathway. Parasitology 128, 123–129. doi: 10.1017/s0031182003004414
Kapadia, S., Levine, B., Speck, S., and Virgin, H. IV (2002). Critical role of complement and viral evasion of complement in acute, persistent, and latent gamma-herpesvirus infection. Immunity 17, 143–155. doi: 10.1016/s1074-7613(02)00369-2
Kapadia, S., Molina, H., van Berkel, V., Speck, S., and Virgin, H. IV (1999). Murine gammaherpesvirus 68 encodes a functional regulator of complement activation. J. Virol. 73, 7658–7670. doi: 10.1128/JVI.73.9.7658-7670.1999
Kennedy, A., Schmidt, C., Thompson, J., Weiss, G., Taechalertpaisarn, T., Gilson, P., et al. (2016). Recruitment of factor H as a novel complement evasion strategy for blood-stage Plasmodium falciparum infection. J. Immunol. 196, 1239–1248. doi: 10.4049/jimmunol.1501581
Kennedy, A., Wijeyewickrema, L., Huglo, A., Lin, C., Pike, R., Cowman, A., et al. (2017). Recruitment of human C1 esterase inhibitor controls complement activation on blood stage Plasmodium falciparum merozoites. J. Immunol. 198, 4728–4737. doi: 10.4049/jimmunol.1700067
Kim, H., Erdman, L., Lu, Z., Serghides, L., Zhong, K., Dhabangi, A., et al. (2014). Functional roles for C5a and C5aR but Not C5L2 in the pathogenesis of human and experimental cerebral malaria. Infect. Immun. 82, 371–379. doi: 10.1128/IAI.01246-13
King, B., Renström, E., and Blom, A. (2019). Intracellular cytosolic complement component C3 regulates cytoprotective autophagy in pancreatic beta cells by interaction with ATG16L1. Autophagy 15, 919–921. doi: 10.1080/15548627.2019.1580515
Klein, M., Kaeser, P., Schwarz, P., Weyd, H., Xenarios, I., Zinkernagel, R., et al. (2001). Complement facilitates early prion pathogenesis. Nat. Med. 7, 488–492. doi: 10.1038/86567
Koelman, D., Brouwer, M., and van de Beek, D. (2019). Targeting the complement system in bacterial meningitis. Brain 142, 3325–3337. doi: 10.1093/brain/awz222
Kolev, M., Ruseva, M., Harris, C., Morgan, B., and Donev, R. (2009). Implication of complement system and its regulators in Alzheimer’s disease. Curr. Neuropharmacol. 7, 1–8. doi: 10.2174/157015909787602805
Kong, D., Chen, Z., Wang, J., Lv, Q., Jiang, H., Zheng, Y., et al. (2017). Interaction of factor H-binding protein of Streptococcus suis with globotriaosylceramide promotes the development of meningitis. Virulence 8, 1290–1302. doi: 10.1080/21505594.2017.1317426
Kovacs, G., Gasque, P., Ströbel, T., Lindeck-Pozza, E., Strohschneider, M., Ironside, J., et al. (2004). Complement activation in human prion disease. Neurobiol. Dis. 15, 21–28. doi: 10.1016/j.nbd.2003.09.010
Kraiczy, P., Skerka, C., Kirschfink, M., Brade, V., and Zipfel, P. (2001). Immune evasion of Borrelia burgdorferi by acquisition of human complement regulators FHL-1/reconectin and Factor H. Eur. J. Immunol. 31, 1674–1684. doi: 10.1002/1521-4141(200106)31:6<1674::aid-immu1674<3.0.co;2-2
Kulkarni, H., Elvington, M., Perng, Y., Liszewski, M., Byers, D., Farkouh, C., et al. (2019). Intracellular C3 protects human airway epithelial cells from stress-associated cell death. Am. J. Respir. Cell Mol. Biol. 60, 144–157. doi: 10.1165/rcmb.2017-0405OC
Kumar, M., Varun, C., Dey, G., Ravikumar, R., Mahadevan, A., Shankar, S., et al. (2018). Identification of host-response in cerebral malaria patients using quantitative proteomic analysis. Proteomics Clin. Appl. 12:e1600187. doi: 10.1002/prca.201600187
Kwon, H., and Koh, S. (2020). Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl. Neurodegener. 9:42. doi: 10.1186/s40035-020-00221-2
Lackner, P., Hametner, C., Beer, R., Burger, C., Broessner, G., Helbok, R., et al. (2008). Complement factors C1q, C3 and C5 in brain and serum of mice with cerebral malaria. Malar. J. 7:207. doi: 10.1186/1475-2875-7-207
Lalli, P., Strainic, M., Yang, M., Lin, F., Medof, M., and Heeger, P. (2008). Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood 112, 1759–1766. doi: 10.1182/blood-2008-04-151068
Landlinger, C., Oberleitner, L., Gruber, P., Noiges, B., Yatsyk, K., Santic, R., et al. (2015). Active immunization against complement factor C5a: a new therapeutic approach for Alzheimer’s disease. J. Neuroinflammation 12:50. doi: 10.1186/s12974-015-0369-6
Leng, F., and Edison, P. (2020). Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat. Rev. Neurol. 17, 157–172. doi: 10.1038/s41582-020-00435-y
Li, Y., Severance, E., Viscidi, R., Yolken, R., and Xiao, J. (2019). Persistent Toxoplasma infection of the brain induced neurodegeneration associated with activation of complement and microglia. Infect. Immun. 87:e00139-19. doi: 10.1128/IAI.00139-19
Libbey, J., Cusick, M., Doty, D., and Fujinami, R. (2017). Complement components are expressed by infiltrating macrophages/activated microglia early following viral infection. Viral Immunol. 30, 304–314. doi: 10.1089/vim.2016.0175
Lionakis, M., Lim, J., Lee, C., and Murphy, P. (2011). Organ-specific innate immune responses in a mouse model of invasive candidiasis. J. Innate Immun. 3, 180–199. doi: 10.1159/000321157
Liszewski, M., Elvington, M., Kulkarni, H., and Atkinson, J. (2017). Complement’s hidden arsenal: New insights and novel functions inside the cell. Mol. Immunol. 84, 2–9. doi: 10.1016/j.molimm.2017.01.004
Liszewski, M., Farries, T., Lublin, D., Rooney, I., and Atkinson, J. (1996). Control of the complement system. Adv. Immunol. 61, 201–283. doi: 10.1016/s0065-2776(08)60868-8
Liszewski, M., Kolev, M., Le Friec, G., Leung, M., Bertram, P., Fara, A., et al. (2013). Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 39, 1143–1157. doi: 10.1016/j.immuni.2013.10.018
Liszewski, M., Leung, M., Cui, W., Subramanian, V., Parkinson, J., Barlow, P., et al. (2000). Dissecting sites important for complement regulatory activity in membrane cofactor protein (MCP; CD46). J. Biol. Chem. 275, 37692–37701. doi: 10.1074/jbc.M004650200
Liu, F., Dai, S., Gordon, J., and Qin, X. (2014). Complement and HIV-I infection/HIV-associated neurocognitive disorders. J. Neurovirol. 20, 184–198. doi: 10.1007/s13365-014-0243-9
Lovchik, J., and Lipscomb, M. (1993). Role for C5 and neutrophils in the pulmonary intravascular clearance of circulating Cryptococcus neoformans. Am. J. Respir. Cell Mol. Biol. 9, 617–627. doi: 10.1165/ajrcmb/9.6.617
Lv, Y., Chen, C., Zhang, B., Xiao, K., Wang, J., Chen, L., et al. (2014). Remarkable activation of the complement system and aberrant neuronal localization of the membrane attack complex in the brain tissues of scrapie-infected rodents. Mol. Neurobiol. 52, 1165–1179. doi: 10.1007/s12035-014-8915-2
Lyzogubov, V., Wu, X., Jha, P., Tytarenko, R., Triebwasser, M., Kolar, G., et al. (2014). Complement regulatory protein CD46 protects against choroidal neovascularization in mice. Am. J. Pathol. 184, 2537–2548. doi: 10.1016/j.ajpath.2014.06.001
Mahmoud, M. E., Fereig, R., and Nishikawa, Y. (2017). Involvement of host defense mechanisms against Toxoplasma gondii infection in anhedonic and despair-like behaviors in mice. Infect. Immun. 85:e00007-17.
Manchester, M., Eto, D., and Oldstone, M. (1999). Characterization of the inflammatory response during acute measles encephalitis in NSE-CD46 transgenic mice. J. Neuroimmunol. 96, 207–217. doi: 10.1016/s0165-5728(99)00036-3
Markiewski, M. M., and Lambris, J. D. (2007). The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am. J. Pathol. 171, 715–727. doi: 10.2353/ajpath.2007.070166
Marra, C. (2018). Central nervous system infection with Toxoplasma gondii. Handb. Clin. Neurol. 152, 117–122. doi: 10.1016/B978-0-444-63849-6.00009-8
Mathern, D., and Heeger, P. (2015). Molecules great and small: the complement system. Clin. J. Am. Soc. Nephrol. 10, 1636–1650. doi: 10.2215/CJN.06230614
Matsuo, K., Shindo, A., Niwa, A., Tabei, K., Akatsu, H., Hashizume, Y., et al. (2017). Complement activation in capillary cerebral amyloid angiopathy. Dement. Geriatr. Cogn. Disord. 44, 343–353.
Matsushita, M., and Fujita, T. (1995). Cleavage of the third component of complement (C3) by mannose-binding protein-associated serine protease (MASP) with subsequent complement activation. Immunobiology 194, 443–448. doi: 10.1016/S0171-2985(11)80110-5
Maury, A., Lyoubi, A., Peiffer-Smadja, N., de Broucker, T., and Meppiel, E. (2021). Neurological manifestations associated with SARS-CoV-2 and other coronaviruses: a narrative review for clinicians. Rev. Neurol. 177, 51–64. doi: 10.1016/j.neurol.2020.10.001
McChesney, M., and Oldstone, M. (1989). Virus-induced immunosuppression: infections with measles virus and human immunodeficiency virus. Adv. Immunol. 45, 335–380. doi: 10.1016/s0065-2776(08)60696-3
McDonald, C., Cahill, L., Ho, K., Yang, J., Kim, H., Silver, K., et al. (2015). Experimental malaria in pregnancy induces neurocognitive injury in uninfected offspring via a C5a-C5a receptor dependent pathway. PLoS Pathog. 11:e1005140. doi: 10.1371/journal.ppat.1005140
McDonald, C., Elphinstone, R., and Kain, K. (2013). The impact of placental malaria on neurodevelopment of exposed infants: a role for the complement system? Trends Parasitol. 29, 213–219. doi: 10.1016/j.pt.2013.03.005
McGovern, G., and Jeffrey, M. (2007). Scrapie-specific pathology of sheep lymphoid tissues. PLoS One. 2:e1304. doi: 10.1371/journal.pone.0001304
McGuire, J., Gill, A., Douglas, S., Kolson, D., and CNS HIV Antiretroviral Therapy Effects Research (Charter) Group (2016). The complement system, neuronal injury, and cognitive function in horizontally-acquired HIV-infected youth. J. Neurovirol. 22, 823–830. doi: 10.1007/s13365-016-0460-5
McQuaid, S., and Cosby, S. (2002). An immunohistochemical study of the distribution of the measles virus receptors, CD46 and SLAM, in normal human tissues and subacute sclerosing panencephalitis. Lab. Invest. 82, 403–409. doi: 10.1038/labinvest.3780434
Mécharles, S., Herrmann, C., Poullain, P., Tran, T., Deschamps, N., Mathon, G., et al. (2016). Acute myelitis due to Zika virus infection. Lancet 387:1481. doi: 10.1016/S0140-6736(16)00644-9
Mehlhop, E., Whitby, K., Oliphant, T., Marri, A., Engle, M., and Diamond, M. (2005). Complement activation is required for induction of a protective antibody response against West Nile virus infection. J. Virol. 79, 7466–7477. doi: 10.1128/JVI.79.12.7466-7477.2005
Mook-Kanamori, B., Geldhoff, M., van der Poll, T., and van de Beek, D. (2011). Pathogenesis and pathophysiology of pneumococcal meningitis. Clin. Microbiol. Rev. 24, 557–591. doi: 10.1128/CMR.00008-11
Morgan, B. (2015). The role of complement in neurological and neuropsychiatric diseases. Expert Rev. Clin. Immunol. 11, 1109–1119. doi: 10.1586/1744666X.2015.1074039
Morgan, B. (2018). Complement in the pathogenesis of Alzheimer’s disease. Semin. Immunopathol. 40, 113–124. doi: 10.1007/s00281-017-0662-9
Muk, T., Stensballe, A., Pankratova, S., Nguyen, D., Brunse, A., Sangild, P., et al. (2019). Rapid Proteome changes in plasma and cerebrospinal fluid following bacterial infection in preterm newborn pigs. Front. Immunol. 10:2651. doi: 10.3389/fimmu.2019.02651
Nataf, S., Stahel, P., Davoust, N., and Barnum, S. (1999). Complement anaphylatoxin receptors on neurons: new tricks for old receptors? Trends Neurosci. 22, 397–402. doi: 10.1016/s0166-2236(98)01390-3
Norris, G., Smirnov, I., Filiano, A., Shadowen, H., Cody, K., Thompson, J., et al. (2018). Neuronal integrity and complement control synaptic material clearance by microglia after CNS injury. J. Exp. Med. 215, 1789–1801. doi: 10.1084/jem.20172244
Ogata, A., Czub, S., Ogata, S., Cosby, S., McQuaid, S., Budka, H., et al. (1997). Absence of measles virus receptor (CD46) in lesions of subacute sclerosing panencephalitis brains. Acta Neuropathol. 94, 444–449. doi: 10.1007/s004010050731
Ólafsson, E., and Barragan, A. (2020). The unicellular eukaryotic parasite Toxoplasma gondii hijacks the migration machinery of mononuclear phagocytes to promote its dissemination. Biol. Cell 112, 239–250. doi: 10.1111/boc.202000005
Oldstone, M., Lewicki, H., Thomas, D., Tishon, A., Dales, S., Patterson, J., et al. (1999). Measles virus infection in a transgenic model: virus-induced immunosuppression and central nervous system disease. Cell 98, 629–640. doi: 10.1016/s0092-8674(00)80050-1
Olivier, I., Cacabelos, R., and Naidoo, V. (2018). Risk factors and pathogenesis of HIV-associated neurocognitive disorder: the role of host genetics. Int. J. Mol. Sci. 19:3594. doi: 10.3390/ijms19113594
OSullivan, J., Preston, R., ORegan, N., and ODonnell, J. (2016). Emerging roles for hemostatic dysfunction in malaria pathogenesis. Blood 127, 2281–2288. doi: 10.1182/blood-2015-11-636464
Pais, T., and Penha-Gonçalves, C. (2019). Brain endothelium: the “innate immunity response hypothesis” in cerebral malaria pathogenesis. Front. Immunol. 9:3100. doi: 10.3389/fimmu.2018.03100
Panackal, A., and Williamson, P. (2015). Fungal infections of the central nervous system. Continuum (Minneap. Minn) 21, 1662–1678. doi: 10.1212/CON.0000000000000241
Pasupuleti, M., Walse, B., Nordahl, E., Mörgelin, M., Malmsten, M., and Schmidtchen, A. (2007). Preservation of antimicrobial properties of complement peptide C3a, from invertebrates to humans. J. Biol. Chem. 282, 2520–2528. doi: 10.1074/jbc.M607848200
Patel, S., Berghout, J., Lovegrove, F., Ayi, K., Conroy, A., Serghides, L., et al. (2008). C5 deficiency and C5a or C5aR blockade protects against cerebral malaria. J. Exp. Med. 205, 1133–1143. doi: 10.1084/jem.20072248
Perez-Alcazar, M., Daborg, J., Stokowska, A., Wasling, P., Björefeldt, A., Kalm, M., et al. (2014). Altered cognitive performance and synaptic function in the hippocampus of mice lacking C3. Exp Neurol. 253, 154–164. doi: 10.1016/j.expneurol.2013.12.013
Perlmutter, D., and Colten, H. (1986). Molecular immunobiology of complement biosynthesis: a model of single-cell control of effector-inhibitor balance. Annu. Rev. Immunol. 4, 231–251. doi: 10.1146/annurev.iy.04.040186.001311
Pian, Y., Gan, S., Wang, S., Guo, J., Wang, P., Zheng, Y., et al. (2012). Fhb, a novel factor H-binding surface protein, contributes to the antiphagocytic ability and virulence of Streptococcus suis. Infect. Immun. 80, 2402–2413. doi: 10.1128/IAI.06294-11
Pittman, K., and Knoll, L. (2015). Long-Term relationships: the complicated interplay between the host and the developmental stages of Toxoplasma gondii during acute and chronic infections. Microbiol. Mol. Biol. Rev. 79, 387–401. doi: 10.1128/MMBR.00027-15
Pomares, C., Devillard, S., Holmes, T., Olariu, T., Press, C., Ramirez, R., et al. (2018). Genetic characterization of Toxoplasma gondii DNA samples isolated from humans living in North America: an unexpected high prevalence of atypical genotypes. J. Infect. Dis. 218, 1783–1791. doi: 10.1093/infdis/jiy375
Pozo-Rodrigálvarez, A., Ollaranta, R., Skoog, J., Pekny, M., and Pekna, M. (2021). Hyperactive behavior and altered brain morphology in adult complement C3a receptor deficient mice. Front. Immunol. 12:604812. doi: 10.3389/fimmu.2021.604812
Prusiner, S. (1998). Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383. doi: 10.1073/pnas.95.23.13363
Prusiner, S., Scott, M., DeArmond, S., and Cohen, F. (1998). Prion protein biology. Cell 93, 337–348. doi: 10.1016/s0092-8674(00)81163-0
Rahpeymai, Y., Hietala, M. A., Wilhelmsson, U., Fotheringham, A., Davies, I., Nilsson, A. K., et al. (2006). Complement: a novel factor in basal and ischemia-induced neurogenesis. EMBO J. 25, 1364–1374.
Rajasingham, R., Smith, R., Park, B., Jarvis, J., Govender, N., Chiller, T., et al. (2017). Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect. Dis. 17, 873–881. doi: 10.1016/S1473-3099(17)30243-8
Rall, G., Manchester, M., Daniels, L., Callahan, E., Belman, A., and Oldstone, M. (1997). A transgenic mouse model for measles virus infection of the brain. Proc. Natl. Acad. Sci. U.S.A. 94, 4659–4663. doi: 10.1073/pnas.94.9.4659
Rambach, G., Maier, H., Vago, G., Mohsenipour, I., Lass-Flörl, C., Defant, A., et al. (2008). Complement induction and complement evasion in patients with cerebral aspergillosis. Microbes Infect. 10, 1567–1576. doi: 10.1016/j.micinf.2008.09.011
Ramos, T., Darley, M., Hu, X., Billker, O., Rayner, J., Ahras, M., et al. (2011). Cutting edge: the membrane attack complex of complement is required for the development of murine experimental cerebral malaria. J. Immunol. 186, 6657–6660. doi: 10.4049/jimmunol.1100603
Ramos, T., Darley, M., Weckbach, S., Stahel, P., Tomlinson, S., and Barnum, S. (2012). The C5 convertase is not required for activation of the terminal complement pathway in murine experimental cerebral malaria. J. Biol Chem. 287, 24734–24738. doi: 10.1074/jbc.C112.378364
Rasmussen, S., Jamieson, D., Honein, M., and Petersen, L. (2016). Zika virus and birth defects–reviewing the evidence for causality. N. Engl. J. Med. 374, 1981–1987. doi: 10.1056/NEJMsr1604338
Readhead, B., Haure-Mirande, J., Funk, C., Richards, M., Shannon, P., Haroutunian, V., et al. (2018). Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 99, 64–82.e7. doi: 10.1016/j.neuron.2018.05.023
Riggle, B., Miller, L., and Pierce, S. (2020). Desperately seeking therapies for cerebral malaria. J. Immunol. 204, 327–334. doi: 10.4049/jimmunol.1900829
Robakis, N. (2020). What do recent clinical trials teach us about the etiology of AD. Adv. Exp. Med. Biol. 1195:167. doi: 10.1007/978-3-030-32633-3_23
Rogers, J., Cooper, N., Webster, S., Schultz, J., McGeer, P., Styren, S., et al. (1992). Complement activation by beta-amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 89, 10016–10020. doi: 10.1073/pnas.89.21.10016
Rosa, T., Flammersfeld, A., Ngwa, C., Kiesow, R., Fischer, P., Zipfel, P., et al. (2016). The Plasmodium falciparum blood stages acquire factor H family proteins to evade destruction by human complemen. Cell. Microbiol. 18, 573–590. doi: 10.1111/cmi.12535
Ross, S., and Densen, P. (1984). Complement deficiency states and infection: epidemiology, pathogenesis and consequences of neisserial and other infections in an immune deficiency. Medicine 63, 243–273.
Rostagno, A., Revesz, T., Lashley, T., Tomidokoro, Y., Magnotti, L., Braendgaard, H., et al. (2002). Complement activation in chromosome 13 dementias. Similarities with Alzheimer’s disease. J. Biol. Chem. 277, 49782–49790. doi: 10.1074/jbc.M206448200
Rowe, J., Moulds, J., Newbold, C., and Miller, L. (1997). P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature 388, 292–295. doi: 10.1038/40888
Rubio-Perez, J., and Morillas-Ruiz, J. (2012). A review: inflammatory process in Alzheimer’s disease, role of cytokines. ScientificWorldJournal 2012:756357. doi: 10.1100/2012/756357
Ruhnke, M., Kofla, G., Otto, K., and Schwartz, S. (2007). CNS aspergillosis: recognition, diagnosis and management. CNS Drugs 21, 659–676. doi: 10.2165/00023210-200721080-00004
Russo, F., Jungmann, P., and Beltrão-Braga, P. (2017). Zika infection and the development of neurological defects. Cell Microbiol. 19:e12744. doi: 10.1111/cmi.12744
Russo, J., Bohenzky, R., Chien, M., Chen, J., Yan, M., Maddalena, D., et al. (1996). Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U.S.A. 93, 14862–14867. doi: 10.1073/pnas.93.25.14862
Sadek, J., Pergam, S., Harrington, J., Echevarria, L., Davis, L., Goade, D., et al. (2010). Persistent neuropsychological impairment associated with West Nile virus infection. J. Clin. Exp. Neuropsychol. 32, 81–87. doi: 10.1080/13803390902881918
Said, J., Tasaka, T., de Vos, S., and Koeffler, H. (1997). Kaposi’s sarcoma-associated herpesvirus/human herpesvirus type 8 encephalitis in HIV-positive and -negative individuals. AIDS 11, 1119–1122. doi: 10.1097/00002030-199709000-00006
Sasai, M., Pradipta, A., and Yamamoto, M. (2018). Host immune responses to Toxoplasma gondii. Int. Immunol. 30, 113–119.
Saul, A., Lashley, T., Revesz, T., Holton, J., Ghiso, J., Coomaraswamy, J., et al. (2013). Abundant pyroglutamate-modified ABri and ADan peptides in extracellular and vascular amyloid deposits in familial British and Danish dementias. Neurobiol. Aging 34, 1416–1425. doi: 10.1016/j.neurobiolaging.2012.11.014
Schafer, D., Lehrman, E., Kautzman, A., Koyama, R., Mardinly, A., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Schartz, N., and Tenner, A. (2020). The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J. Neuroinflammation 17:354. doi: 10.1186/s12974-020-02024-8
Schmidt, C., Kennedy, A., and Tham, W. (2015). More than just immune evasion: hijacking complement by Plasmodium falciparum. Mol. Immunol. 67, 71–84. doi: 10.1016/j.molimm.2015.03.006
Schmutzhard, E., and Gerstenbrand, F. (1984). Cerebral malaria in Tanzania. Its epidemiology, clinical symptoms and neurological long term sequelae in the light of 66 cases. Trans R. Soc. Trop. Med. Hyg. 78, 351–353. doi: 10.1016/0035-9203(84)90118-4
Schneider, M., Prosser, B., Caesar, J., Kugelberg, E., Li, S., Zhang, Q., et al. (2009). Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature 458, 890–893. doi: 10.1038/nature07769
Schreiber, R., and Feldman, H. (1980). Identification of the activator system for antibody to Toxoplasma as the classical complement pathway. J. Infect. Dis. 141, 366–369. doi: 10.1093/infdis/141.3.366
Sejvar, J., Haddad, M., Tierney, B., Campbell, G., Marfin, A., Van Gerpen, J., et al. (2003). Neurologic manifestations and outcome of West Nile virus infection. JAMA 290, 511–515. doi: 10.1001/jama.290.4.511
Sekar, A., Bialas, A., de Rivera, H., Davis, A., Hammond, T., Kamitaki, N., et al. (2016). Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183. doi: 10.1038/nature16549
Serafini, B., Rosicarelli, B., Franciotta, D., Magliozzi, R., Reynolds, R., Cinque, P., et al. (2007). Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J. Exp. Med. 204, 2899–2912.
Shi, Q., Chowdhury, S., Ma, R., Le, K., Hong, S., Caldarone, B., et al. (2017). Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 9:eaaf6295. doi: 10.1126/scitranslmed.aaf6295
Shi, Q., Colodner, K., Matousek, S., Merry, K., Hong, S., Kenison, J., et al. (2015). Complement C3-deficient mice fail to display age-related hippocampal decline. J. Neurosci. 35, 13029–13042. doi: 10.1523/JNEUROSCI.1698-15.2015
Shinjyo, N., Hikosaka, K., Kido, Y., Yoshida, H., and Norose, K. (2021). Toxoplasma infection induces sustained up-regulation of complement factor B and C5a receptor in the mouse brain via microglial activation: implication for the alternative complement pathway activation and anaphylatoxin signaling in cerebral toxoplasmosis. Front. Immunol. 11:603924. doi: 10.3389/fimmu.2020.603924
Shinjyo, N., Stahlberg, A., Dragunow, M., Pekny, M., and Pekna, M. (2009). Complement-derived anaphylatoxin C3a regulates in vitro differentiation and migration of neural progenitor cells in vitro. Stem Cells 27, 2824–2832. doi: 10.1002/stem.225
Sikorski, P., Commodaro, A., and Grigg, M. (2020). Toxoplasma gondii recruits factor H and C4b-Binding protein to mediate resistance to serum killing and promote parasite persistence in vivo. Front. Immunol. 10:3105. doi: 10.3389/fimmu.2019.03105
Silamut, K., Phu, N., Whitty, C., Turner, G., Louwrier, K., Mai, N., et al. (1999). A quantitative analysis of the microvascular sequestration of malaria parasites in the human brain. Am. J. Pathol. 155, 395–410. doi: 10.1016/S0002-9440(10)65136-X
Silva-Barrios, S., and Stäger, S. (2017). Protozoan parasites and type I IFNs. Front. Immunol. 8:14. doi: 10.3389/fimmu.2017.00014
Singh, G., Angwafor, S., Njamnshi, A., Fraimow, H., and Sander, J. (2020). Zoonotic and vector-borne parasites and epilepsy in low-income and middle-income countries. Nat. Rev. Neurol. 16, 333–345. doi: 10.1038/s41582-020-0361-3
Skaper, S. (2012). Alzheimer’s disease and amyloid: culprit or coincidence? Int. Rev. Neurobiol. 102, 277–316. doi: 10.1016/B978-0-12-386986-9.00011-9
Souza, I., Barros-Aragão, F., Frost, P., Figueiredo, C., and Clarke, J. (2019). Late neurological consequences of Zika Virus infection: risk factors and pharmaceutical approaches. Pharmaceuticals 12:60. doi: 10.3390/ph12020060
Spadafora, C., Awandare, G., Kopydlowski, K., Czege, J., Moch, J., Finberg, R., et al. (2010). Complement receptor 1 is a sialic acid-independent erythrocyte receptor of Plasmodium falciparum. PLoS Pathog. 6:e1000968. doi: 10.1371/journal.ppat.1000968
Speth, C., Dierich, M., and Gasque, P. (2002). Neuroinvasion by pathogens: a key role of the complement system. Mol. Immunol. 38, 669–679. doi: 10.1016/s0161-5890(01)00104-3
Speth, C., Williams, K., Hagleitner, M., Westmoreland, S., Rambach, G., Mohsenipour, I., et al. (2004). Complement synthesis and activation in the brain of SIV-infected monkeys. J. Neuroimmunol. 151, 45–54. doi: 10.1016/j.jneuroim.2004.02.013
Sprong, T., Roos, D., Weemaes, D., Neeleman, C., Geesing, C., Mollnes, T., et al. (2006). Deficient alternative complement pathway activation due to factor D deficiency by 2 novel mutations in the complement factor D gene in a family with meningococcal infections. Blood 107, 4865–4870. doi: 10.1182/blood-2005-07-2820
Stahel, P., Frei, K., Fontana, A., Eugster, H., Ault, B., and Barnum, S. (1997). Evidence for intrathecal synthesis of alternative pathway complement activation proteins in experimental meningitis. Am. J. Pathol. 151, 897–904.
Stepanova, E., Kondrashin, A., Sergiev, V., Morozova, L., Turbabina, N., Maksimova, M., et al. (2019). Toxoplasmosis and mental disorders in the Russian Federation (with special reference to schizophrenia). PLoS One 14:e0219454. doi: 10.1371/journal.pone.0219454
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Stokowska, A., Atkins, A., Morán, J., Pekny, T., Bulmer, L., Pascoe, M., et al. (2017). Complement peptide C3a stimulates neural plasticity after experimental brain ischemia. Brain 140, 353–369. doi: 10.1093/brain/aww314
Stokowska, A., and Pekna, M. (2018). “Complement C3a: shaping the plasticity of the post-stroke brain,” in Cellular and Molecular Approaches to Regeneration and Repair. Springer Series in Translational Stroke Research, eds P. Lapchak and J. Zhang (Cham: Springer), 521–541. doi: 10.1007/978-3-319-66679-2_26
Storch, M., Piddlesden, S., Haltia, M., Iivanainen, M., Morgan, P., and Lassmann, H. (1998). Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann. Neurol. 43, 465–471. doi: 10.1002/ana.410430409
Stracciari, A., Bottini, G., Guarino, M., Magni, E., Pantoni, L., and “Cognitive and Behavioral Neurology” Study Group of the Italian Neurological Society (2021). Cognitive and behavioral manifestations in SARS-CoV-2 infection: not specific or distinctive features? Neurol Sci. 42, 2273–2281. doi: 10.1007/s10072-021-05231-0
Sun, D., Zhang, M., Liu, G., Wu, H., Li, C., Zhou, H., et al. (2016). Intravascular clearance of disseminating Cryptococcus neoformans in the brain can be improved by enhancing neutrophil recruitment in mice. Eur. J. Immunol. 46, 1704–1714. doi: 10.1002/eji.201546239
Taniguchi, Y., Appiah-Kwarteng, C., Murakami, M., Fukumoto, J., Nagamune, K., Matsuo, T., et al. (2018). Atypical virulence in a type III Toxoplasma gondii strain isolated in Japan. Parasitol. Int. 67, 587–592.
Tenner, A. (2020). Complement-mediated events in Alzheimer’s disease: mechanisms and potential therapeutic targets. J. Immunol. 204, 306–315. doi: 10.4049/jimmunol.1901068
Tenner, A., Stevens, B., and Woodruff, T. (2018). New tricks for an ancient system: physiological and pathological roles of complement in the CNS. Mol. Immunol. 102, 3–13. doi: 10.1016/j.molimm.2018.06.264
Tham, W., Wilson, D., Lopaticki, S., Schmidt, C., Tetteh-Quarcoo, P., Barlow, P., et al. (2010). Complement receptor 1 is the host erythrocyte receptor for Plasmodium falciparum PfRh4 invasion ligand. Proc. Natl. Acad. Sci. U.S.A. 107, 17327–17332. doi: 10.1073/pnas.1008151107
Tso, F., Sawyer, A., Kwon, E., Mudenda, V., Langford, D., Zhou, Y., et al. (2017). Kaposi’s sarcoma-associated herpesvirus infection of neurons in HIV-positive patients. J. Infect. Dis. 215, 1898–1907. doi: 10.1093/infdis/jiw545
Tuite, A., Elias, M., Picard, S., Mullick, A., and Gros, P. (2005). Genetic control of susceptibility to Candida albicans in susceptible A/J and resistant C57BL/6J mice. Genes Immun. 6, 672–682. doi: 10.1038/sj.gene.6364254
Tyebji, S., Seizova, S., Garnham, A., Hannan, A., and Tonkin, C. (2019a). Impaired social behaviour and molecular mediators of associated neural circuits during chronic Toxoplasma gondii infection in female mice. Brain Behav. Immun. 80, 88–108. doi: 10.1016/j.bbi.2019.02.028
Tyebji, S., Seizova, S., Hannan, A., and Tonkin, C. (2019b). Toxoplasmosis: a pathway to neuropsychiatric disorder. Neurosci. Biobehav. Rev. 96, 72–92. doi: 10.1016/j.neubiorev.2018.11.012
van de Beek, D., Brouwer, M., Hasbun, R., Koedel, U., Whitney, C., and Wijdicks, E. (2016). Community-acquired bacterial meningitis. Nat. Rev. Dis. Primers 2:16074. doi: 10.1038/nrdp.2016.74
van de Beek, D., de Gans, J., Spanjaard, L., Weisfelt, M., Reitsma, J., and Vermeulen, M. (2004). Clinical features, complications, and outcome in adults with pneumococcal meningitis: a prospective case series. N. Engl. J. Med. 351, 1849–1859. doi: 10.1056/NEJMoa040845
Van der Heyde, H., Nolan, J., Combes, V., Gramaglia, I., and Grau, G. (2006). A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 22, 503–508. doi: 10.1016/j.pt.2006.09.002
Vasek, M., Garber, C., Dorsey, D., Durrant, D., Bollman, B., Soung, A., et al. (2016). A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534, 538–543. doi: 10.1038/nature18283
Veerhuis, R., Nielsen, H., and Tenner, A. (2011). Complement in the brain. Mol. Immunol. 48, 1592–1603. doi: 10.1016/j.molimm.2011.04.003
Villegas, M., Sciutto, E., Rosetti, M., Fleury, A., and Fragoso, G. (2019). Association of TRAF1/C5 locus polymorphisms with epilepsy and clinical traits in mexican patients with neurocysticercosis. Infect Immun. 87, e347–e319. doi: 10.1128/IAI.00347-19
Virgin, H. IV, Latreille, P., Wamsley, P., Hallsworth, K., Weck, K., Dal Canto, A., et al. (1997). Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71, 5894–5904. doi: 10.1128/JVI.71.8.5894-5904.1997
Wassmer, S., de Souza, J., Frère, C., Candal, F., Juhan-Vague, I., and Grau, G. (2006). TGF-beta1 released from activated platelets can induce TNF-stimulated human brain endothelium apoptosis: a new mechanism for microvascular lesion during cerebral malaria. J. Immunol. 176, 1180–1184. doi: 10.4049/jimmunol.176.2.1180
Wassmer, S., and Grau, G. (2017). Severe malaria: what’s new on the pathogenesis front? Int. J. Parasitol. 47, 145–152. doi: 10.1016/j.ijpara.2016.08.002
Weaver, S., and Barrett, A. (2004). Transmission cycles, host range, evolution and emergence of arboviral disease. Nat. Rev. Microbiol. 2, 789–801. doi: 10.1038/nrmicro1006
Weaver, S., Ferro, C., Barrera, R., Boshell, J., and Navarro, J. (2004). Venezuelan equine encephalitis. Annu. Rev. Entomol. 49, 141–174. doi: 10.1146/annurev.ento.49.061802.123422
Woehrl, B., Brouwer, M., Murr, C., Heckenberg, S., Baas, F., Pfister, H., et al. (2011). Complement component 5 contributes to poor disease outcome in humans and mice with pneumococcal meningitis. J. Clin. Invest. 121, 3943–3953. doi: 10.1172/JCI57522
Woodruff, T. M., Ager, R. R., Tenner, A. J., Noakes, P. G., and Taylor, S. M. (2010). The role of the complement system and the activation fragment C5a in the central nervous system. Neuromolecular Med. 12, 179–192.
Woollard, S., Bhargavan, B., Yu, F., and Kanmogne, G. (2014). Differential effects of Tat proteins derived from HIV-1 subtypes B and recombinant CRF02_AG on human brain microvascular endothelial cells: implications for blood-brain barrier dysfunction. J. Cereb. Blood Flow Metab. 34, 1047–1059. doi: 10.1038/jcbfm.2014.54
Wu, K., Zhang, T., Zhao, G., Ma, N., Zhao, S., Wang, N., et al. (2019). The C3a/C3aR axis mediates anti-inflammatory activity and protects against uropathogenic E coli-induced kidney injury in mice. Kidney Int. 96, 612–627. doi: 10.1016/j.kint.2019.03.005
Wu, M., Brennan, F., Lynch, J., Mantovani, S., Phipps, S., Wetsel, R., et al. (2013). The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc. Natl. Acad. Sci. U.S.A. 110, 9439–9444. doi: 10.1073/pnas.1218815110
Wyss-Coray, T., Yan, F., Lin, A., Lambris, J., Alexander, J., Quigg, R., et al. (2002). Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc. Natl. Acad. Sci. U.S.A. 99, 10837–10842. doi: 10.1073/pnas.162350199
Xiao, J., Li, Y., Gressitt, K., He, H., Kannan, G., Schultz, T., et al. (2016). Cerebral complement C1q activation in chronic Toxoplasma infection. Brain Behav. Immun. 58, 52–56. doi: 10.1016/j.bbi.2016.04.009
Yanamadala, V., and Friedlander, R. (2010). Complement in neuroprotection and neurodegeneration. Trends Mol. Med. 16, 69–76. doi: 10.1016/j.molmed.2009.12.001
Yuan, X., Shan, M., You, R., Frazier, M. V., Hong, M. J., Wetsel, R. A., et al. (2015). Activation of C3a receptor is required in cigarette smoke-mediated emphysema. Mucosal Immunol. 8, 874–885. doi: 10.1038/mi.2014.118
Yusuf, F., Hafiz, M., Shoaib, M., and Ahmed, S. (2017). Cerebral malaria: insight into pathogenesis, complications and molecular biomarkers. Infect. Drug Resist. 10, 57–59. doi: 10.2147/IDR.S125436
Zhang, J., Malik, A., Choi, H., Ko, R., Dissing-Olesen, L., and MacVicar, B. (2014). Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron 82, 195–207. doi: 10.1016/j.neuron.2014.01.043
Keywords: complement, infection, dementia, Alzheimer’s disease, inflammation
Citation: Shinjyo N, Kagaya W and Pekna M (2021) Interaction Between the Complement System and Infectious Agents – A Potential Mechanistic Link to Neurodegeneration and Dementia. Front. Cell. Neurosci. 15:710390. doi: 10.3389/fncel.2021.710390
Received: 16 May 2021; Accepted: 09 July 2021;
Published: 02 August 2021.
Edited by:
Marco Bacigaluppi, San Raffaele Scientific Institute (IRCCS), ItalyReviewed by:
Faith H. Brennan, The Ohio State University, United StatesJudith Fraussen, Hasselt University, Belgium
Copyright © 2021 Shinjyo, Kagaya and Pekna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Noriko Shinjyo, bnNoaW5qeW9AaWZyZWMub3Nha2EtdS5hYy5qcA==