 Yu Han
Yu Han Mei Yuan1,2
Mei Yuan1,2 Xia Bi
Xia Bi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 02 August 2021
Sec. Cellular Neuropathology
Volume 15 - 2021 | https://doi.org/10.3389/fncel.2021.704334
Endoplasmic reticulum (ER) is the main organelle for protein synthesis, trafficking and maintaining intracellular Ca2+ homeostasis. The stress response of ER results from the disruption of ER homeostasis in neurological disorders. Among these disorders, cerebral ischemia is a prevalent reason of death and disability in the world. ER stress stemed from ischemic injury initiates unfolded protein response (UPR) regarded as a protection mechanism. Important, disruption of Ca2+ homeostasis resulted from cytosolic Ca2+ overload and depletion of Ca2+ in the lumen of the ER could be a trigger of ER stress and the misfolded protein synthesis. Brain cells including neurons, glial cells and endothelial cells are involved in the complex pathophysiology of ischemic stroke. This is generally important for protein underfolding, but even more for cytosolic Ca2+ overload. Mild ER stress promotes cells to break away from danger signals and enter the adaptive procedure with the activation of pro-survival mechanism to rescue ischemic injury, while chronic ER stress generally serves as a detrimental role on nerve cells via triggering diverse pro-apoptotic mechanism. What’s more, the determination of some proteins in UPR during cerebral ischemia to cell fate may have two diametrically opposed results which involves in a specialized set of inflammatory and apoptotic signaling pathways. A reasonable understanding and exploration of the underlying molecular mechanism related to ER stress and cerebral ischemia is a prerequisite for a major breakthrough in stroke treatment in the future. This review focuses on recent findings of the ER stress as well as the progress research of mechanism in ischemic stroke prognosis provide a new treatment idea for recovery of cerebral ischemia.
Stroke is the second highest cause of death. Cerebral ischemia stemming from arterial occlusion is responsible for most of the stroke, a catastrophic illness leading to permanent disability in 80% of survivors (Moskowitz et al., 2010; Campbell et al., 2019; Jayaraj et al., 2019). In the face of such an intractable problem, relatively effective interventions to prevent death and improve brain cells recovery are in short supply today (Ekker et al., 2018; Zerna et al., 2018).
Endoplasmic reticulum served as a highly multifaceted organelle essentially differs from the surrounding cytoplasm both structurally and functionally (Sanderson et al., 2015). Scores of chaperones, enzymes, and cofactors associated with ER regulate precise folding of newly synthesized proteins and assist polypeptides in achieving final functional conformation (Chapman et al., 1998). Under the physiological environment, misfolded proteins are decreased and degraded by the accurate quality control mechanisms of proteasome, lysosome, and autophagy pathways to maintain the protein homeostasis called proteostasis in eukaryotic cells (Kaushik and Cuervo, 2015; Hetz and Saxena, 2017).
Apart from the dynamic integration of proteins, ER also plays an indispensable role in the intracellular Ca2+ homeostasis related to cell survival and neuronal plasticity (Mattson et al., 2000). The regulation of Ca2+ homeostasis by ER mainly depends on the dynamic balance of Ca2+ release and re-uptake in the lumen of the ER. Inositol 1,4,5-trisphosphate (IP3), a second messenger, mobilizes Ca2+ stored in ER directly and stimulates Ca2+ entry indirectly to regulate intracellular Ca2+ levels (Berridge and Irvine, 1989). IP3 receptor (IP3R) and ryanodine receptor (RyR) are part of Ca2+ release channels (Foskett et al., 2007; Van Petegem, 2012). Ca2+ pumps of sarcoplasmic/endoplasmic reticulum calcium ATPases (SERCA) are responsible for Ca2+ re-uptake (Takenaka et al., 1982). Most ER associated proteins are involved in maintaining ER Ca2+ homeostasis where Bip, GRP94, and cofactors are conducive to Ca2+ buffering in the ER lumen. Failure of the Ca2+ homeostasis by toxic stimuli such as ischemia, hypoxia, and hypertension (Marciniak and Ron, 2006; Walter and Ron, 2011) leads to ER stress which causes protein misfolding and generates a pathological state to cells in danger. When the accumulation of unfolded polypeptides or folding-incompetent proteins surpass the capacity of ER chaperones, a signal transduction pathway called UPR in ER lumen is preferentially activated ensure cells to restore homeostasis (Hetz, 2012; Hetz and Papa, 2018). However, a high level of insult above the regulation of UPR, sets a motion pro-apoptotic pathways making cells end up with death.
A variety of experimental studies have demonstrated that ER stress induced by cerebral ischemia could be a key element in complex pathology mechanisms involving neuronal cell, glial cells, and endothelial cells loss during or after cerebral ischemia (Rissanen et al., 2006; Zhao et al., 2018; Wang et al., 2019; Haupt et al., 2020). Most animal models used in experiments performed on transient cerebral ischemia models, which mimics the widespread clinical ischemia/reperfusion (I/R) where the occurrence of cortical spreading depression, periinfarct depolarization, and cerebral edema result in secondary neuronal damage and infarct expansion (Dohmen et al., 2008; Liebeskind et al., 2019). Sufficient evidence has suggested that targeted inhibition of ER stress and UPR can effectively improve experimental I/R injury. For instance, astragalin, a kind of flavonoid, significantly attenuated the expression levels of apoptotic proteins (Bax and cleaved-caspase-3) and the release of inflammatory cytokines, as well as the ER-related proteins, glucose-regulated protein to alleviate I/R injury via suppression of ER stress on rats after transient middle cerebral artery occlusion (MCAO). Similar results were obtained in vitro neuronal cell culture model (Liu et al., 2020). However, potential contamination, histological techniques, and methodological defects could be encountered in neuron cell culture technology (Molcanyi et al., 2013, 2014). Caution is required by interpreting any neuronal cell culture result. A recent study showed that acupuncture operated on Baihui (GV 20), and Qubin (GB7) acupoints in rats immediately after reperfusion can play a neuroprotection role to improve neurological scores and restrain autophagy and apoptosis induced by ER stress (Sun et al., 2020). Therefore, the mechanism of ER stress in cerebral ischemia is worth studying. This review aims to contribute to this growing area of research by summarizing the molecular pathways of ER stress controlling cell fate after cerebral ischemia.
The UPR served as stress sensors and regulators of downstream transcription factors monitors unfolded and misfolded proteins aggregation and recode genes to determine cell destiny (Chen and Brandizzi, 2013; Chow et al., 2015). This phenomenon is the main subsequent performance after ER stress. Initiation of UPR depends on the activation of three type-I transmembrane proteins namely protein kinase RNA-like ER kinase (PERK), inositol-requiring protein 1 (IRE1), and activating transcription factor-6 (ATF6). Under the condition of neuronal homeostasis, these three proteins are in devitalized state binding to glucose regulated protein 78 (GRP78), a major molecular ER chaperone. GRP78 separated from PERK, IRE1, and ATF6 bind to misfolded proteins upon ER dysfunction in pathological state caused by ischemic stroke, indicating UPR initiation. Phosphorylation of eukaryotic initiation factor 2α (eIF2α) induced by PERK inhibit protein synthesis to reduce the load of unfolded proteins on ER and upgrade the expression of ER chaperone to rectify misfolded proteins and restore folding order (DeGracia and Montie, 2004). Newly synthesized proteins ultimately identified as nonfunctional will be reversely transported from ER to cytosol. The process by which the substrates are subsequently polyubiquitinated and degraded by proteases is known as ER-associated degradation (ERAD) (Lopata et al., 2020). Typically, activated IRE1 and cleaved ATF6 involve in ERAD induced by X-box binding protein 1 (XBP1) (Chen and Brandizzi, 2013; Sun et al., 2015; Jung et al., 2019; Waldherr et al., 2019). Within a few hours after I/R, PERK and IRE1 will be activated to varying degrees. Various damage mechanisms related to cerebral ischemia control the fate of nerve cells by altering the expression of UPR induced mRNA (Figure 1; Deng et al., 2004). UPR down-regulates protein translation and up-regulates ER-related enzymes, cofactors, and chaperones alleviating ER stress to play a neuroprotective role in ischemic injury. During intense ER stress and prolonged UPR in cerebral ischemia, the early adaptive protection of UPR to cells could be replaced to cell programmed death.
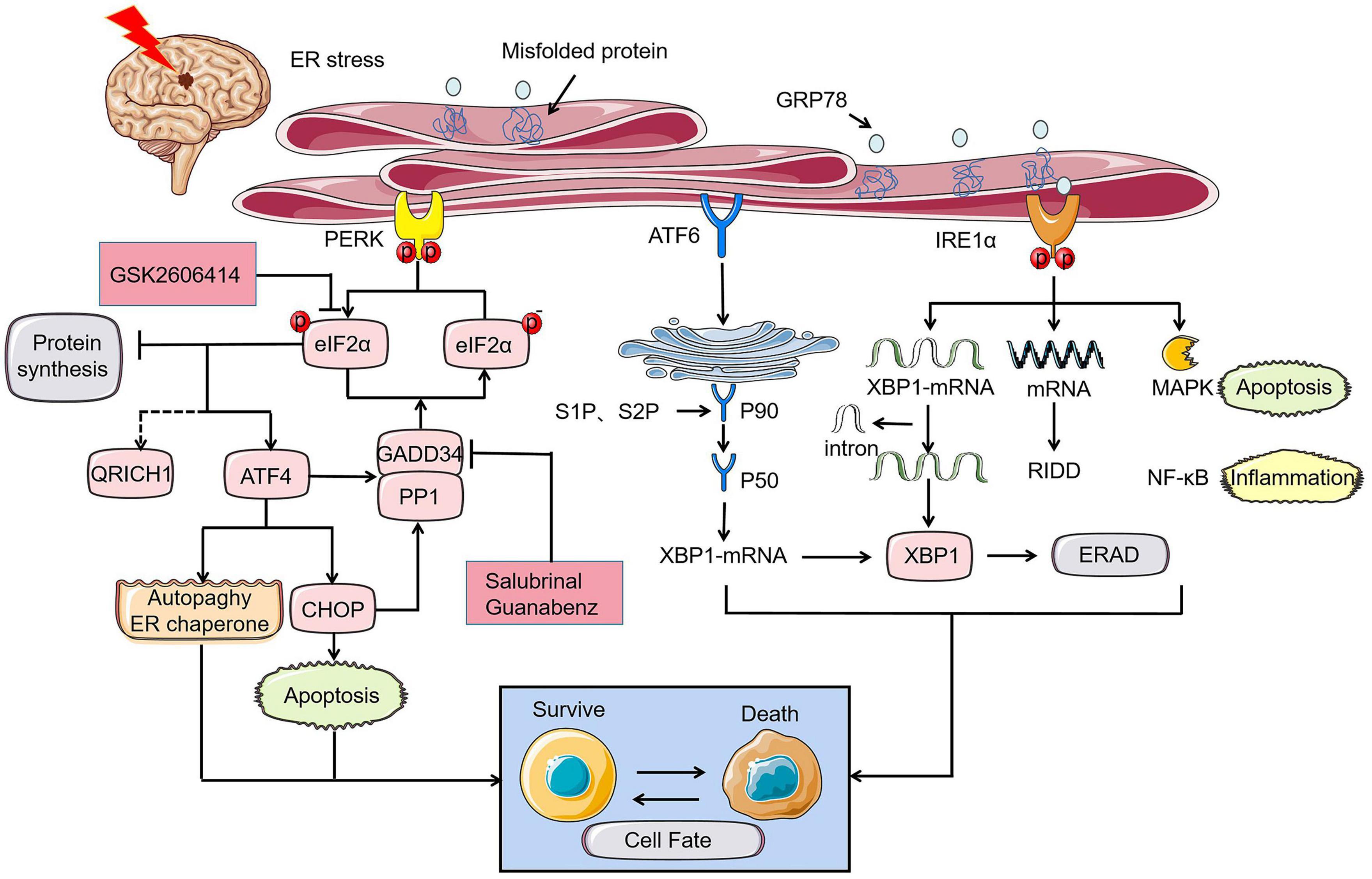
Figure 1. UPR of ER stress in cerebral ischemia determines cell fate through PERK, IRE1, and ATF6 pathways. Under the ischemic state, PERK-eIF2α-ATF4-CHOP axis controls cell fate by apoptosis where p- eyIF2α play a key role in inhibiting protein synthesis and its role will be reversed by PERK inhibitor GSK2606414. GADD34, which is simultaneously regulated by ATF4 and CHOP promotes promoting the dephosphorylation of eIF2α under prolonged ER stress. Similarly, the role of GADD34 can disappear with the intervention of salubrinal and guanabenz. The rescue of cell fate by ATF4 is mainly achieved by inhibiting autophagy and controlling the ER chaperones. The different expression of XBP1 induced by ATF6 and IRE1α can also bring disparate endings to the cell. MAPK apoptosis pathway and NF-κB inflammation related to IRE1α can directly determine cell survival.
Protein kinase RNA-like ER kinase, a type I transmembrane ER-resident protein, is a central part of UPR, as its signaling pathway regulates the control of protein synthesis and translation to a large extent. The occurrence of PERK activation suppresses the level of global protein synthesis momently through targeting phosphorylation eIF2α at serine residue 51 when misfolded and unfolded protein accumulates under the ER stress after transient global brain ischemia (Harding et al., 1999; Owen et al., 2005; Marciniak et al., 2006). eIF2 is essential for the initiation of mRNA translation (Kimball, 1999). Therefore, phosphorylation of eIF2α has been regarded as a specific mechanism to inhibit translation and decrease the protein load of ER. It skillfully links the upstream kinase of eIF2α with the downstream translation events.
The preservation of protein synthesis inhibition (PSI) is closely related to neuronal survival following brain ischemia and reperfusion. The phosphorylation of eIF2α, which appears in the short-lived acute stage of brain ischemia, is responsible for the first few hours of PSI and does not explain the continued prolongation of PSI. Recently, researchers found that Hes1 knockdown aggravated the neural apoptosis through the PERK pathway where the level of p-eIF2α enhanced dramatically in transient cerebral ischemia model because the increase of infarct size and a deterioration of the nervous system are counteracted with PERK-specific inhibitors GSK2606414 (Li Y. et al., 2020). In another experiment, PERK-specific knockout mice induced by hybridization and tamoxifen showed larger infarct volume and worse neurological function scores with low expression of p-eIF2α in 3-day and 3-week recovery after MCAO compared to control mice (Wang et al., 2020). This indicates that the lack of PERK-induced phosphorylation of eIF2α and subsequent inhibition of translation is an important factor leading to neurological damage in cerebral ischemia. Therefore, inhibition of protein synthesis is the main way for PERK to restore neurological function after cerebral ischemia. Interestingly, the knock-down and activation of PERK by different means both have aggravated the functional impairment after ischemia, which may associates with the duration of UPR. In the future, the targeted intervention of PERK could be a potential therapeutic direction for stroke.
Although PERK-mediated phosphorylation of eIF2α inhibits global protein translation, it specifically upregulates the translation of some mRNAs including the activating transcription factor 4 (ATF4) due to ATF4 mRNA open reading frames (ORF2) existing specific sequence (Harding et al., 2000). The expression level of ATF4 mRNA has been verified to be enhanced by anoxia in primary rat fibroblasts in Estes et al. (1995). ATF4 diverges into two branches. ATF4 is usually described as a transcriptional activator controlling the fate of cells under stress, which has received widespread attention in cerebral ischemic injury. Recently, the research has indicated that overexpression of ATF4 induced by adeno-associated virus protects against I/R injury, reduces the infarct volume and neurological scores with suppression of inflammatory response (He et al., 2019).
Activation of ATF4 downstream target transcription factors and association with other signaling pathways predict cellular adaptive regulation or death program involved in pro-apoptosis, autophagy, and protein synthesis. In terms of pro-survival mechanisms, the expression of various adaptive genes including ER chaperones, metabolic enzymes, AA transporters is promoted by ATF4 to relieve stress and fit cells into an adaptive environment. The brain tissue of animals in hibernation can mimic a variety of stress response states including ischemia, hypoxia, oxidative stress. in brain tissue from hibernating squirrels, ATF4 was found to transfer to the nucleus and form a compound with the nucleus co-factor CREB1 to which combined with the promoter region of GRP78 to support cell survive (Mamady and Storey, 2008). On the contrary, C/EBP homologous protein (CHOP), a pro-apoptotic factor is directly induced by ATF4 during ER stress guiding cell death (Harding et al., 2002; Marciniak et al., 2004). In the investigation of the neuroprotective mechanism of granulocyte colony stimulating factor (G-CSF)on ischemic stroke, it was found that the expression of ATF4, CHOP, caspase-12 related to the apoptosis pathway induced by ER stress decreased, and the mice showed signs of better ER homeostasis reconstruction and less acute neuronal degeneration of post-ischemic stroke (Modi et al., 2020).
Activating transcription factor 4 also indirectly interacts with autophagy and protein synthesis related pathways. In terms of autophagy regulation, ATF4 silencing attenuates the expression of PARK2, weakens the phagocytosis induced by PARK2, and reverses the neuroprotective effects of ER stress activators tunicamycin and thapsigargin in the vivo and vitro model of ischemia (Zhang X. et al., 2014). Protein synthesis regulation induced by ATF4 is accompanied through enhancing the translation of the growth-arrest-and DNA-damage-induced transcript 34 (GADD34) which dephosphorylates eIF2α to recover protein synthesis (Marciniak et al., 2004).
Whether autophagy or protein synthesis, once the regulatory intensity exceeds the threshold of suitable living context, it will lead to tragedy. Only maintenance of intracellular protein homeostasis and effective establishment of recycling of cellular material can save cells from the stress response.
C/EBP homologous protein, a key transcription factor downstream of perk- eIF2α-ATF4, plays an important role in apoptosis induced by ER stress. The expression of CHOP is at a low level under physiological conditions and elevates dramatically when cells are stimulated by stress. CHOP mRNA induction was observed in the rats’ hippocampus suffering from global cerebral ischemia (Paschen et al., 1998). CHOP is a pro-apoptotic factor. It cannot directly cause apoptosis but mediates apoptosis by regulating the expression of downstream apoptosis-related factors BCL-2 and caspase-12 (Oida et al., 2008). According to studies, CHOP promotes cell death still by increasing protein synthesis, which leads to oxidative stress (Marciniak et al., 2004; Han et al., 2013). The up-regulation of CHOP due to I/R with the TUNEL-positive neuron cells, which were co-located with CHOP and caspase-12, decayed (Zhao et al., 2018) is counteracted by the intervention of chrysophanol. Decrease of CHOP expression and increase of ER oxidoreductin-α (Ero1-α) induced by hypothermic reversed apoptosis in the models of ischemic stroke compared to normal temperature (Poone et al., 2015). CHOP is detected in the nuclei of damaged neurons after 24 h of bilateral common carotid artery occlusion. In vitro experiment, the primary hippocampal neurons of wild-type mice are more sensitive to apoptosis induced by oxygen and glucose deprivation (OGD)/reoxygenation compared with CHOP–/–mice (Tajiri et al., 2004). These all suggest that knock-down and inactivation of CHOP facilitate the nerve cells’ recovery in ER stress due to cerebral ischemia. Moreover, CHOP controls cell death, depends on the intensity of the stress. Only under intense stress, CHOP strongly dominates apoptosis factors and thus induces cell death, but under mild stress, it cannot add the intensity of stress and remarkably promote protein secretion with the result of low correlation with apoptosis (Southwood et al., 2002). It is worth noting that recently, p-eIF2α raises the translation of QRICH1 which accelerates the rate of cell death during ER stress in intestinal epithelial cells and also involves the regulation of transcription procedures that promote protein secretion. It can be inferred that QRICH1 may be a new transcription regulator on the PERK-eIF2α axis. However, the effects and location of QRICH1 require to be confirmed in cerebral ischemia (You et al., 2021).
Under ER stress, ATF4, CHOP, and a great deal of mRNA of encoding transcription factors together induce the expression of GADD34, which bound to the serine/threonine protein phosphatase 1 (PP1) is an integral component for eIF2α phosphatase complex. There has been controversy about the decision of GADD34 on cell fate in cerebral ischemia. In early studies, it was found that GADD34 was up-regulated in neurons and microglia in the peri-infarct region of cerebral ischemia rodents and the human brain after cardiac arrest (Imai et al., 2002; White et al., 2004). The increase in GADD34 expression can restore protein synthesis inhibition and participate in DNA repair to promote cell survival. Unexpectedly, The cerebral ischemia model mice injected with the viral vector containing the full-length GADD34 showed more severe infarction volume than no insert full-length GADD34 (McCabe et al., 2008). Restoring protein synthesis by dephosphorylation of eIF2α is an important step in reversing damage, but at the same time, it will enhance the accumulation of misfolded proteins and cause further damage. The molecular mechanism of mammalian cells restoring normal protein synthesis after ER stress associated with ischemia is explained by the role of GADD34:PP1 complex dephosphorylation. The experiment has demonstrated that inactivation of GADD34 decreased the damage during ER stress without excessive protein accumulation (Marciniak et al., 2004).
Salubrinal, an eIF2α dephosphorylation inhibitor, is equivalent to reach the p-eIF2α state in disguise by dissociating GADD34:PP1 complex to preserve nutrition and lessen ER chaperones to neutralize damage of ER stress (Boyce et al., 2005). The reduction of CA1 cell death and blood-brain barrier damage is found in the salubrinal-treated mice model of global cerebral ischemia (Nakka et al., 2010; Anuncibay-Soto et al., 2016). However, some studies have found that the intervention of salubrinal can aggravate neuronal apoptosis and degeneration in vivo and in vitro, and exacerbate neurobehavioral damage in intracerebral hemorrhage models (Meng et al., 2018). Furthermore, there is a standpoint that salubrinal is regarded as an inhibitor of the eIF2α phosphatases which lacks an experimental basis (Boyce et al., 2005). The production of the paradox and contradictory experimental results may attribute to the dissimilar time node and duration of eIF2α phosphorylation under different stress levels. The specific evidence-based basis needs further scientific exploration. However, guanabenz complex selectively disrupting dephosphorylation of eIF2α by binding to GADD34:PP1 complex regulates the rate of protein synthesis within the controllable range of the ER chaperones to protect cells from the pressure of protein misfolding (Tsaytler et al., 2011). Phosphorylation of eIF2α as a hub to alleviate ER stress will become a heated issue in the treatment of neurological diseases in the future.
IRE1α, a most conserved transducer of UPR, distributes abundantly in various tissues in response to ER stress and contains a serine/threonine kinase domain and an endoribonuclease domain (Tirasophon et al., 1998; Kim et al., 2008). Similar to PERK, GRP78 dissociates from IRE1α and approaches to dimerization and autophosphorylation of IRE1 under ER stress. Subsequently, the endonuclease activity of IRE1 specifically splices the mRNA encoding the transcription factor X-box binding protein 1 (XBP1) to remove a 26-nucleotide intron from the coding region of XBP1 mRNA (Calfon et al., 2002). Translocation of XBP1 mRNA open reading frame prompt it to be translated into XBP1s protein, an active transcription factor upregulating pro-survival signaling (Paschen et al., 2003). XBP1s specifically induces the expression of target genes involved in ERAD and protein folding and has completed related events that control protein homeostasis to unload ER lumen (Hetz et al., 2015).
The result that the increased translation of GRP78, GRP94, and other glucose regulatory proteins due to XBP1s bind to unfolded protein contributes to the recovery of ER stress (Lee A.-H. et al., 2003). In the in vitro model of I/R, overexpression of XBP1 induced by adenovirus suppressed cell death induced by OGD/R stress, relieving the damnification of ER stress (Ibuki et al., 2012). Icariin enhanced the cell viability of primary cortical neurons and weakened inflammation levels after OGD/R injury via restraining the IRE1α-XBP1 signaling pathway where the expression of XBP1s, IL-β, IL-6, TNF-α, the ratio of p-IRE1α/IRE1αare decreased (Mo Z.-T. et al., 2020). Melatonin protected more neurons survival in mice with cerebral ischemia (Lin et al., 2018) via inhibiting the PERK pathway. Interestingly, Melatonin treatment before ischemia can inhibit endoplasmic reticulum stress-dependent autophagy through the PERK and IRE1 pathways to relieve cerebral edema and neuronal apoptosis (Feng et al., 2017).
After the mitigation of ER stress, similar to the principle of eIF2α dephosphorylation, spliced XBP1 mRNA is gradually replaced by an unspliced form. A great deal of mRNA and micro RNAs regulated by the activated IRE1α plays a role in multiple signaling pathways, mediating inflammation, apoptosis, lipid synthesis, and protein secretion, which is known as IRE1α-dependent mRNA decay (RIDD) (Hollien and Weissman, 2006). However, the research of RIDD in ischemic diseases is relatively few. In addition, activated IRE1 recruits binding protein associated with stress pathways, including mitogen-activated protein kinase (MAPK) pathway inducing apoptosis. Inflammation pathway mainly governed by nuclear factor-κB (NF-κB), and these cascade events have been validated in cerebral ischemia (Borsello et al., 2003; Hu et al., 2006; Luo et al., 2008; Zhang et al., 2016). In short, as a branch of UPR, IRE1 participates in the mechanism program of various cell survival and determines cell fate under cerebral ischemia.
Activating transcription factor-6 is another type-I transmembrane protein in ER lumen besides PERK and IRE1. ATF6 disassociated from GRP78 is translocated to Golgi apparatus where inactive 90 kDa ATF6 cleaved by endopeptidase S1P and S2P is converted into activated 50 kDa ATF6 and then transported to nucleus regulating gene expression (Ye et al., 2000). ATF6 also has two cell control mechanisms, pro-apoptosis, and pro-survival. ATF6 cooperates with IRE1 to control the translation of XBP1 and improves the ERAD by inducing α-mannosidase-like protein 1 (EDEM1) and GRP78 (Yamamoto et al., 2007). Not only in cerebral ischemia models, but also myocardial ischemia, renal ischemia, and other ischemic models, the protective effect appears in the cell homeostasis of the pharmacological ATF6 reprogramming protein induced by a compound called 147 and disappears after the absence of specific ATF6. In this experiment, the expression of GRP78 and catalase which recently proved to be a principal members of the novel antioxidant gene program induced by ATF6 were both elevated (Blackwood et al., 2019). Treatment strategy based on protein homeostasis induced by ATF6 may restore the injury of insecure proteostasis cerebral ischemia (Thiebaut et al., 2019). Moreover, a study indicates that ATF6 is the chief protein that enhances neuroprotection within 2 h before I/R (Urban et al., 2009). On the contrary, the role of taurine in alleviating ER stress is to inhibit ATF6, which is considered to be a detrimental factor in this experiment in MCAO and neuron culture models (Gharibani et al., 2013). The result may be attributed to the fact that ATF6 is also present in the promoter of gene encoding CHOP in addition to ATF4 and XBP1. The clear nature of ATF6 is still controversial. Cleaved ATF6 is a powerful marker of UPR activation. Unfortunately, there are fewer studies on the ATF6 branch in the ER stress after cerebral ischemia than the PERK and IRE1 arm given its difficult to detect. All the above-mentioned studies related to UPR pathway have been summarized (Table 1).
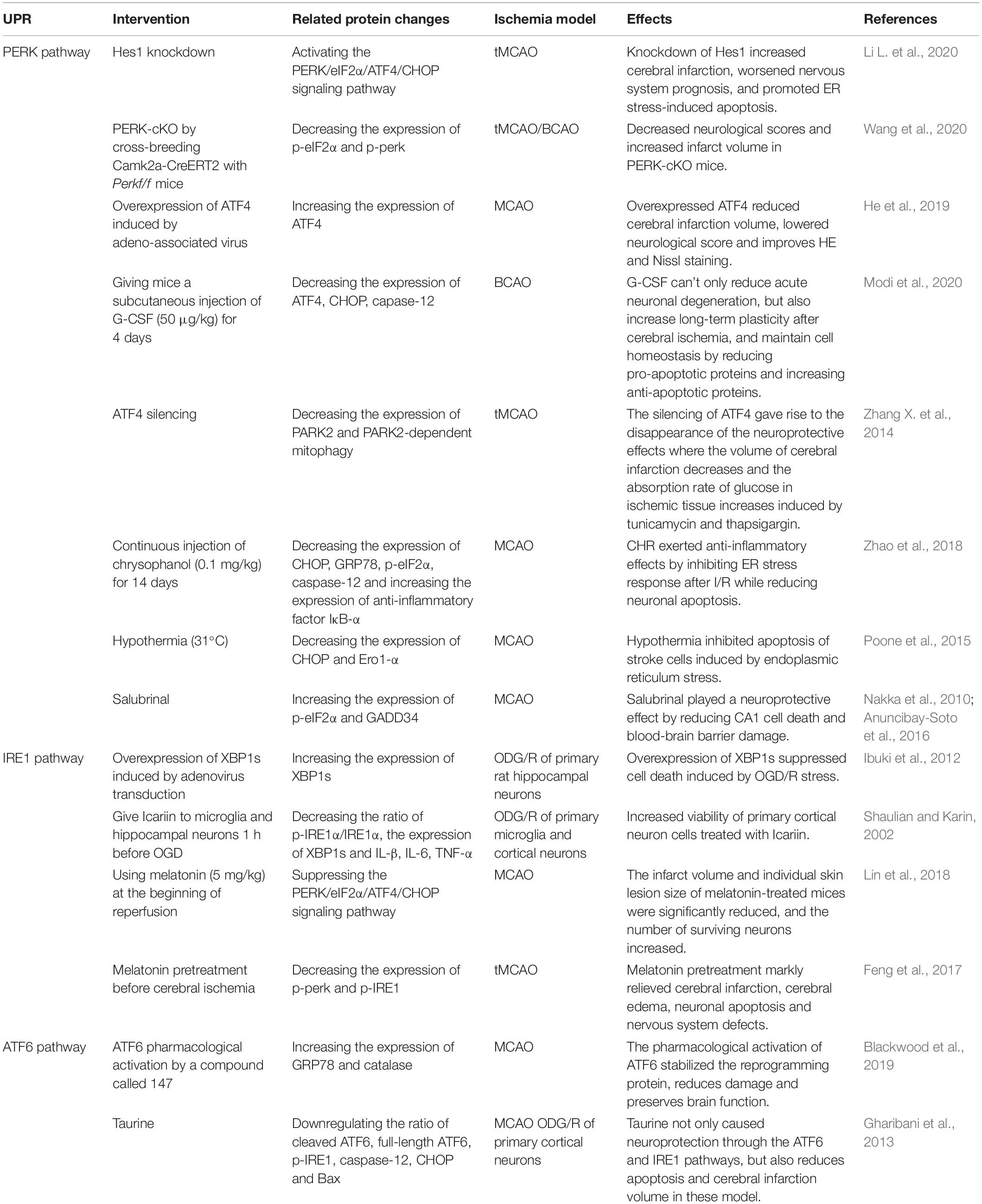
Table 1. Studies on intervention of UPR in cerebral ischemia.
Ca2+ homeostasis makes a difference in integrating multiple brain functions in the nerve cells across the cytomembrane and between the cytoplasm and intracellular organelles (Zündorf and Reiser, 2011). Under normal physiological conditions, ER lumen provides a high Ca2+ place where the concentration is higher than cytoplasm for ER-client enzymes and chaperones to maintain ER functions with an efficient operation (Mattson et al., 2000; Michalak et al., 2002; Coe and Michalak, 2009) and involve in rapid Ca2+ signaling events associated with chemical and electrical cell stimulation.
As mentioned earlier, Ca2+ homeostasis in the ER benefits from the dynamic balance of Ca2+ release and re-uptake. The generation of IP3 is a secondary event induced by the agonist-dependent hydrolysis of the lipid (Berridge, 1989). The stimulation of phospholipase C hydrolyzing the membrane lipid phosphatidylinositol 4,5-bisphosphate to gain IP3, which is the follow-up reaction of cell surface receptors activation due to extracellular agonists (Furuichi et al., 1989). IP3Rs are expressed in all cell types and active during above-mentioned state. RyRs, another calcium release channel, are mainly distributed in neurons and skeletal muscle, which discharge Ca2+ from ER lumen back into cytoplasm (Foskett et al., 2007; Gong et al., 2019) to cut down the concentration within ER together with IP3Rs (Santulli et al., 2017). In parallel, SERCA pumps Ca2+ from the cytoplasm into ER lumen to recovery of Ca2+ concentration in the ER. A recent study showed that SERCA2, a P-type ion-motive ATPase that resides on the ER membrane, is regarded as a gatekeeper of neuronal calcium homeostasis in the central nervous system (CNS) (Britzolaki et al., 2018).
In addition to the source of cytoplasm, Ca2+ supplement also involves extracellular pathways. ER Ca2+ from the extracellular environment through the plasma membrane (PM)cannot do without the store operated Ca2+ entry (SOCE) along with the activation of neuronal SOC channels (Chang, 2006; Smyth et al., 2010; Mohammed Thangameeran et al., 2020). The conception of SOCE dates back to Putney (1986) but the exact regulatory mechanism is unclear. SOCE is executed by the stromal interaction molecule 1 and 2 (STIM1/2), which act as Ca2+ sensors located in ER membrane and plasma membrane protein ORAI1, which function as pore-forming subunits of SOCE channels (Smyth et al., 2010). After depletion of ER Ca2+ induced by IP3R due to physiological extracellular stimuli, Ca2+ separates oneself from the STIM1/2 N-terminal domain, which leads to STIM1/2 undergoing complex conformational changes and oligomerization, allowing oligomer to activate and bine ORAI1 in ER-PM junctions. The best understood of SOCE is the Ca2+ release-activated Ca2+ (CRAC) channel trapped in the ER-PM junctions in response to depletion of ER Ca2+ to trigger Ca2+ influx (Lewis, 2020).
Both dysregulation of Ca2+ homeostasis in the ER and Ca2+ overload in cytoplasm plays a key role in cell apoptosis under an ischemic state. Ca2+ imbalance and ER stress are inseparable where various chaperones, enzymes, and cofactors cannot assist in synthesizing protein normally due to depletion of Ca2+ in the ER. The interruption of energy supply due to oxygen and glucose deprivation following cerebral ischemia can give rise to the deficiency of ATP ion channels and sustained neuronal depolarization. Under the state of abnormal electrochemical gradient and cells depolarization, over-opening voltage-dependent Ca2+ channels (VDCCs) and unintentional release of the neurotoxic excitatory neurotransmitter glutamate which activates N-methyl-d-aspartate receptors (NMDARs) leading to incremental intracellular Ca2+ concentration and excitotoxicity (Szydlowska and Tymianski, 2010; Singh et al., 2019). Neurons and oligodendrocytes are intensively sensitive to NMDA mediated excitotoxicity, which is responsible for their fatal death at the early stage.
Although astrocytes and endothelial cells have a longer survival period in ODG, they eventually die after reoxygenation due to energy compensation mechanism where the compensatory hypoxic protective response of astrocyte system can provide sufficient oxygen supply and energy substrate for neurons (Lee D.R. et al., 2003).
Ca2+ overload involves several initiations of biochemical and physiological reactions and actives Ca2+-associated proteins, which include phospholipases, endonucleases, caspase, calpains, and other kinases causing a series of neuronal cell death (Singh et al., 2019). One of the typical events of the following excitotoxicity after cerebral ischemia is pathologic activation of calpains. The cleavage of neuronal substrates via calpain adversely affects the structure and function of neurons. Various proteins located in membrane (L-type calcium channels, PMCA, and NCX) and ER (IP3R, RyR, and SERCA) involving in Ca2+ homeostasis have been discovered to be cleaved by calpain with its greater activation in vivo or in vitro under the excitotoxicity (Vosler et al., 2008). For example, calpain inhibits IP3 metabolism to make it act on IP3R for a longer time, which undergirds Ca2+ efflux from ER (Bevers and Neumar, 2008). An intervention to calpain inhibition has been proved to be a useful treatment for I/R. Calpeptin and MDL28170 termed calpain inhibitors reduced neuroinflammation and neuronal apoptosis after focal cerebral I/R injury in rats (Peng et al., 2011; Wang et al., 2021).
To make matters worse, the initiation of SOCE engenders Ca2+ influx in response to the overstimulation of glutamate receptors (Berna-Erro et al., 2009). As thus double whammy of Ca2+ inflow from the activation of NMDARs and SOCE may raise the concentration of Ca2+ to a critical level in which brain cells are extremely prone to death (Serwach and Gruszczynska-Biegala, 2019). Various components of SOCE playing a key role in cerebral ischemia have been shown under extensive researches. For capacitive Ca2+ entry (CCE) and Ca2+ overload induced by ischemia, the regulating effect of STIM2 is more significant than STIM1 in neuronal hypoxic cell death (Berna-Erro et al., 2009). Compared with wild-type mice, hippocampal neurons from STIM2 KO mice have a higher survival rate and lower level of ER Ca2+ concentration under hypoxic space both in culture and in acute hippocampal slices (Berna-Erro et al., 2009). Exhaustion of STIM2 is better protected against ischemic damage obviously (Berna-Erro et al., 2009). Even so, the importance of STIM1 cannot be overlooked in terms of improving neurological function. Intracellular high Ca2+ concentration due to the increased STIM1 and ORAI1 levels that occur in global ischemia rats dropped significantly with the injection of STIM1 siRNA, and then the rats had fewer neurological deficits compared with control (Zhang M. et al., 2014). ORAI1 is essential for pathological thrombus formation. Analogously, mice with deletion of ORAI1 were resistant to ischemic brain infarction (Braun et al., 2009). Those findings show that pharmacologic intervention of SORE to reduce or treat the formation of thrombus may be a potential therapeutic strategy to manage cerebrovascular events (Mammadova-Bach et al., 2019).
Apart from ER, Ca2+ regulation of mitochondria in the occurrence and development of cerebral ischemia is indispensable when it comes to ion homeostasis. The close connection between ER and mitochondria is indicated by the uptake of Ca2+ from the ER to mitochondria with the aid of mitochondria-associated ER membranes (MAMs). Mitochondria, an organelle that sustains energy in living organisms, can be transformed into cell killers under massive harmful stimuli. Generally, mitochondria and ER interact to form a closely related whole that controls cell fate through the regulation of Ca2+ signaling pathways.
In cerebral ischemia, mitochondrial dysfunction caused by mitochondrial oxidative stress leads to neuronal death, and Ca2+ imbalance is the primary factor in this result. Extreme intracellular Ca2+ release from ER lumen and activation of NMDA following ischemia cause Ca2+ overload in mitochondrial matrix. Subsequently, excessive Ca2+ entering the mitochondria opens the mitochondrial permeability transition pore (MPTP), which induces the release of cytochrome c and initiates cell apoptotic program by activating caspases-3 and caspases-9 (Celsi et al., 2009). Cyclophilin D (CypD) recognized as a regulator of MPTP has been targeted by most MPTP inhibitors (Briston et al., 2019). Significant reduction in brain infarct size and neuroprotection shown in CypD-deficient mice after acute middle cerebral artery occlusion and reperfusion indicated that CypD plays a key role in ischemic models referring to Ca2+ overload and oxidative stress (Schinzel et al., 2005). Moreover, ER Ca2+ release and mitochondrial Ca2+ uptake are prevented with the participation of c- Jun N- terminal kinase1/2 (JNK1/2) small siRNA, which suggests JNK also contributes to the opening of MPTP and cell death (Verma et al., 2013). Therefore, the effective control of Ca2+ homeostasis to restrict the opening of MPTP may become the direction of efforts to reduce neuronal death.
Besides Ca2+, reactive oxygen species (ROS) generation is also a critical mechanism in ER–mitochondria crosstalk to regulate cell death (Marchi et al., 2018). On the one hand, high intracellular Ca2+ concentration augments the synthesis of nitric-oxide (NO), which enhances the production of ROS via disturbing the electron transport of mitochondria (Moncada and Erusalimsky, 2002). SERCA pump is presented at weak activity by the formation of NO and ischemic absence of ATP hydrolysis (Parsons et al., 1999). RyR2 is overactivated due to the excessive ROS in ischemia (Bull et al., 2008), which contributes to the higher intracellular Ca2+ concentration. On the other hand, mitochondrial Ca2+ is combined with cardiolipin to disintegrate respiratory chain complex II resulting in ROS generation and oxidative stress (Hwang et al., 2014). Superfluous ROS will bring about accumulation of oxidatively modified and misfolded proteins in the ER, which destroys ER homeostasis and causes ER stress (Brunet et al., 2000). In brain cells, astrocytes have better resistance to ROS, while endothelial cells are vulnerabal to ROS (Ouyang et al., 2007; Hertz, 2008).
Destruction of Ca2+ homeostasis due to the absence of energy in ischemia state will lead to ER stress which operates UPR to eliminate the accumulation of misfolded protein in the ER. Regulation of UPR is related to Ca2+ to a certain degree. For example, Calreticulin, an ER resident Ca2+ buffer can respond to Ca2+ depletion to stop assisting the combination of GRP78 and ATF6 to activate ATF6 (Hong et al., 2004). Not only abnormal activation of SOCE but also PDIA6, an ER resident oxidoreductase interacting on the domain of IRE1α will enhance IRE1α activity under ER stress. It is vital to show that the activity of PERK pathway will not be impacted when IRE1α activity is promoted by PDAI6 (Groenendyk et al., 2014). Those outcomes suggest that the activation of UPR branches is related to distinct Ca2+ regulation components and keeps relatively independent. Nowadays, thapsigargin and tunicamycin have been widely used as ER stress agonists in experiments by inhibiting SERCA and protein glycosylation, respectively (Zhang X. et al., 2014). In summary, starting from the mechanism of Ca2+ homeostasis destroyed by cerebral ischemia and restoring it could inhibit ER stress and save cell life.
The pathways of apoptosis induced by ER stress are different from those of mitochondria and death receptors, but they are connected in tandem to form a whole apoptotic network. CHOP, ASK1-JNK, and Caspase-12 pathway activation are recognized as three main apoptosis pathways under ER stress (Figure 2; Kaufman, 2002).
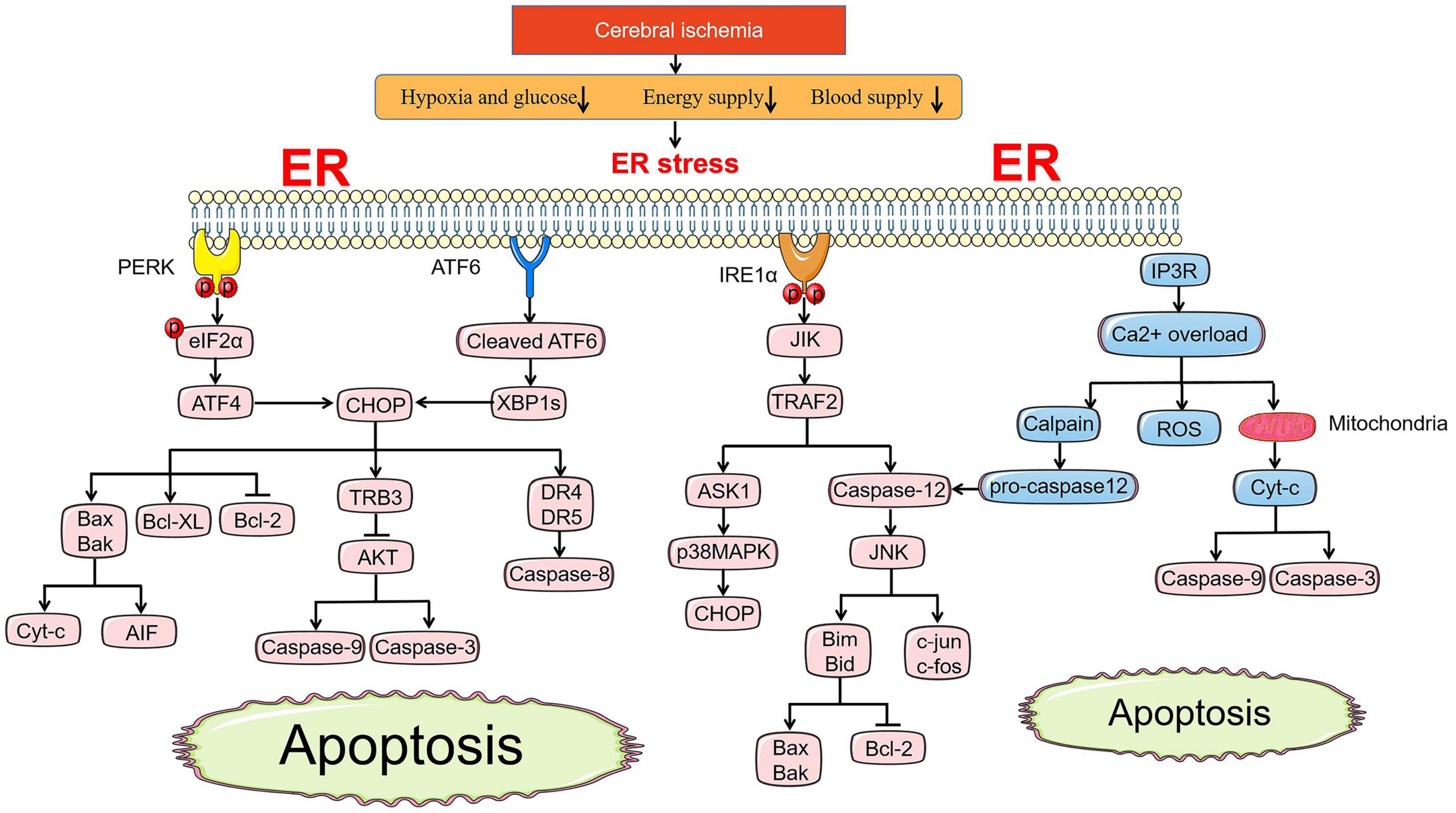
Figure 2. ER stress-mediated apoptosis pathway under cerebral ischemia. Under ischemic state, PERK and ATF6 both involve in apoptosis pathways induced by CHOP. BCL family, TRB3-AKT axis and death receptor DR4/5 are all regulated by CHOP to promote the release of Cyc-c and AIF, active the caspase-9, caspase-3, and caspase-8, respectively. JIK-TRAF2 apoptosis pathway induced by IRE1α and Ca2+ overload promote the activation of caspase-12, which accelerates the pro-apopotic protein of BCL family by controlling JNK. Ca2+ overload leads to the release of ROS directly and Cyt-c indirectly activating caspase-9 and caspase-3 via cross-talk with mitochondria.
Protein kinase RNA-like ER kinase, IRE1, and ATF6 both induce the transcription of CHOP leading to cell apoptosis with prolonged ER stress. To a certain extent, the current researches are only relevant to the upstream regulation mechanism of CHOP associated with apoptosis and the downstream regulation mechanism is still vague. The expression of the TRB3 gene is upregulated by CHOP inhibiting the phosphorylation of Akt, a crucial anti-apoptotic signaling molecule, which successively activates the caspase-9 and caspase-3 (Datta et al., 1997; Ohoka et al., 2005). The level of Bcl2-family proteins is regulated by CHOP to induce apoptosis in ER stress. CHOP can upregulate the pro-apoptotic gene Bax and Bak, which enhances the release of cytochrome c (Cyt-C) and apoptosis inducing factor (AIF) involving in the mitochondrial apoptosis pathway and can suppress the expression of anti-apoptotic protein Bcl-2 and Bcl-XL promoting apoptosis (Fu et al., 2010; Tuzlak et al., 2016). Furthermore, CHOP mediates apoptosis in tandem with the death receptor pathway where the caspase-8-mediated cascade event happens after the elevated death receptor 4 (DR4) and DR5 due to the regulation of CHOP (Lu et al., 2014). The downstream apoptotic pathway of CHOP is not limited to ER stress alone, and more attention could be paid to the overall apoptosis formed by its cross-talk with mitochondrial pathways and death receptors in exploring the treatment of cerebral ischemia.
Apoptosis signal-regulating kinase 1-JNK pathway is another apoptotic pathway in ER stress where tumor necrosis factor receptor associated factor 2 (TRAF2) is deemed as a mainstay. First of all, under physiological conditions, TRAF2 combines with procaspase-12 to form a stable complex, while under ER stress, IRE1 activates C-jun-N-terminal-inhibiting kinase (JIK) to phosphorylate TRAF2, which separates procaspase-12 from TRAF2 and activates caspase-12 via homodimerization, causing cell apoptosis (Noguchi et al., 1999; Yoneda et al., 2001). On the other hand, the JIK-IRE1 complex recruits TRAF2 and leads to apoptosis signal-regulating kinase 1 (ASK1) activation acting on the downstream JNK and p38 mitogen activated protein kinases (p38MAPK) which phosphorylation modifies CHOP to regulate the apoptosis (Hatai et al., 2000). Pharmacological regulation of p38MAPK can reduce cell injury and apoptosis under ER stress after I/R (Li L. et al., 2020). Activated JNK is translocated to the nucleus to phosphorylate c-jun and c-fos, and then induces the expression of downstream apoptotic genes and ligand proteins such as FASL and TNF to initiate apoptosis pathway of death receptor via a transcription dependent manner (Shin et al., 1998). The role of c-Jun in the onset of apoptosis death in neurons has been demonstrated in cerebral ischemia (Xiao et al., 2018). Moreover, similar to the CHOP pathway in I/R models, JNK also upregulates the expression of pro-apoptotic BH3-only proteins Bim and Bid which inhibits the activity of Bcl-2 anti-apoptotic members or activates Bax/Bak-like pro-apoptotic members to mediate the mitochondrial pathway of apoptosis (Putcha et al., 2003; Guan et al., 2006).
Caspases-12 is located in the outer membrane of the ER and is only activated under ER stress. It is a key molecule that mediates ER stress and apoptosis. Calpain associated with mitochondrial dysfunction is activated owing to excessive Ca2+ in the cytoplasm, which cleaves the procaspase-12 located in ER and assists in the release activated caspase-12 into the cytoplasm (Nakagawa and Yuan, 2000; Chan et al., 2002). The above mentioned TRAF2 is directly related to the activation of caspase-12. A study has shown that the intervention of JNK inhibitors in rats after MCAO reduces not only the expression of p-JNK but also the caspase-12, which means JNK may be involved in the activation of caspase-12 (Zhu et al., 2012). According to the report, caspase-7 moved to the surface of the ER can cleave procaspase-12 of pro-domain to engender activated caspase-12 (Rao et al., 2001). Caspase-7 mutants and the addition of caspase-7 inhibitor in hippocampal cultured neurons with glucose deprivation significantly decline the expression of caspase-12 (de la Cadena et al., 2014). Finally, caspase-12 is specifically activated and activates caspase-9 in collaboration with other ER stress molecules, which then cause fine apoptosis through caspase-3. In addition to apoptosis induced by caspase-12, a research found that caspase-4 can be seen in humans as ER stress-specific caspase and may be involved in the pathogenesis of Alzheimer’s disease (Hitomi et al., 2004). MCAO rats treated by Tanshinone IIA (TSA)showed a reduction in infarction area, accompanied by a decrease in caspase-3 and caspase-8 compared with the control (Zhou et al., 2015). The relationship between the caspase family and apoptosis is quite consanguineous and complex. It is necessary to clarify the roles of initiator and effector in the upstream and downstream of death signal transduction.
In addition to regulating the apoptosis pathway by coordinating ER stress-related genes to control cell survival, UPR seeks to inform cells in a dangerous context through an inflammatory pathway that is significant for the innate immune response to alleviate ER stress (Figure 3). Similarly, the qualitative release of inflammatory signaling depends on the intensity of stress where inflammatory signaling assists in relieve cell injury under transient ER stress, while prolonged stress will directly link inflammation with cell death. Inflammatory signaling pathways in cerebral ischemia include toll-like receptors (TLR) signaling pathway, MAPK signaling pathway, and NF-κB signaling pathway, the latter two are mainly driven by UPR under ER stress (Marsh et al., 2009; Lanzillotta et al., 2013; Mo Y. et al., 2020).
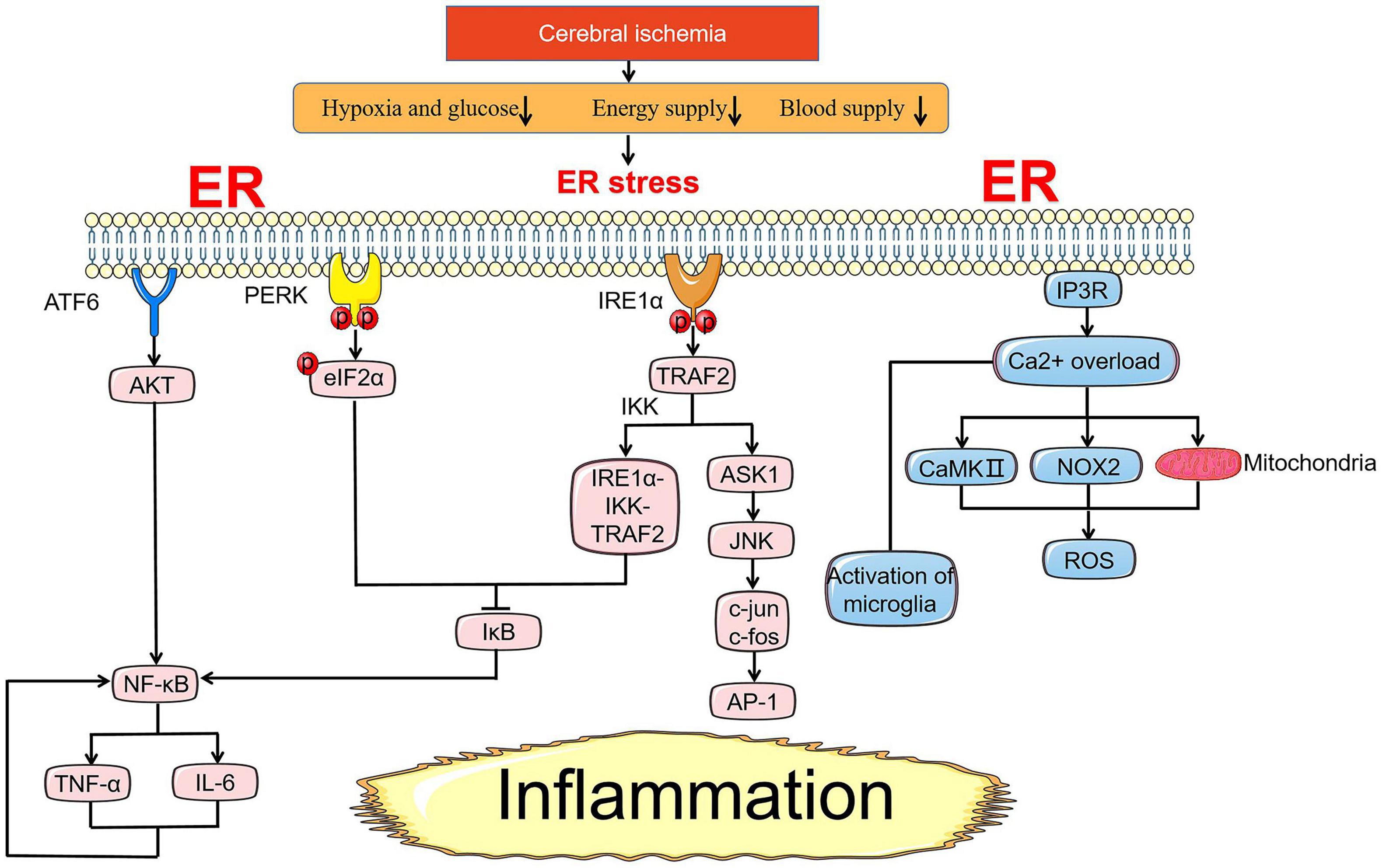
Figure 3. ER stress-mediated inflammation pathway under cerebral ischemia. Under ischemic state, NF-κB is an essential pivot in the inflammation pathway induced by ER stress. NF-κB is controlled by the activation of AKT and suppression of inhibitory IκB proteins. NF-κB and TNF-α promote each other casing nausea cycle of inflammation response. In addition to NF-κB, the release of inflammatory factors induced by IRE1α-TRAF2-JNK axis is also a crux part of the injury mechanism in cerebral ischemia. Ca2+ overload from the ER through NOX2-dependent, subsequent activation of CaMKII and mitochondria pathway lead to the generation of ROS involving in inflammation response. Reactive microglia are mainly expressed as M1 pro-inflammatory type with Ca2+ overload after I/R injury.
After cerebral ischemia, the NF-κB inflammatory pathway is most relevant to ER stress, which precisely stems from the cell-autonomous pro-inflammatory transcription programs mediated by the three arms of the UPR all gathering to the generation of pro-inflammatory factors with the location of NF-κB in nucleus. Under cells homeostasis state, the incorporation of NF-κB and several inhibitory IκB proteins isolates inactivated NF-κB in the cytoplasm (Gilmore, 2006). With initiation of UPR under ER stress, the decreased expression of IκB and activated NF-κB is detected in cells as PERK phosphorylates eIF2α reducing the overall protein synthesis (Deng et al., 2004). This suggests that eIF2α phosphorylation excites NF-κB by suppressing the level of IκB where the mechanism of activation is ambiguous. Some scholars inferred that the occurrence of this phenomenon may be the increased proportion of NF-κB to IκB promoting more NF-κB transfer to the nucleus due to a shorter half-life of IκB compared with NF-κB (Sprenkle et al., 2017). The active IRE1α recruits NF-κB kinase inhibitors IκB kinase (IKK) near the ER to form an IRE1α-IKK-TRAF2 complex which causes phosphorylation and ubiquitination of IκB. Consequently, IκB is degraded by protease leading to NF-κB dissociation and translocation to the nucleus, which induces the production of pro-inflammatory cytokine TNF - α and IL-6 accelerating stress-induced cell death (Hu et al., 2006). In addition, the existence of cleaved ATF6 also makes sense in the activation of NF-κB. Transient phosphorylation of AKT was discovered before subtilase cytotoxin (SubAB) activated NF-κB and among the three signaling pathways of UPR, only gene and pharmacological inhibition of ATF6 can reduce the expression of p-AKT and the activation of NF-κB (Yamazaki et al., 2009). ROS as a core mediator of inflammatory response is responsible for the stimulation of NF-κB by ER stress (Pahl and Baeuerle, 1996). The source of ROS in the body is excessive Ca2+ in the cytoplasm. High Ca2+ concentration in mitochondria results in mitochondrial dysfunction releasing ROS abnormally (Malhotra et al., 2008). On the other hand, Ca2+ efflux from the ER through NOX2-dependent and subsequent activation of calcium dependent protein kinases (CaMKII) lead to the generation of ROS (Li et al., 2010). In tMCAO models, treatment of cerebral ischemia with Xyloketal B (Xyl-B) can dramatically reduce the excessive production of ROS in brain tissue, suppress the expression of NF-κB and inducible nitric oxide synthase (iNOS), and down-regulate the mRNA levels of pro-inflammatory cytokines, including IL-1β, TNF-α, IL-6, and IFN-γ (Pan et al., 2017). Substantial evidence indicates that anti-inflammatory therapy focusing on NF-κB after cerebral ischemia requires focusing on the regulation of UPR and Ca2+ homeostasis.
In MAPK signaling pathway, JNK pathway induced by IRE1α provides a manner by which not only apoptosis signaling, inflammatory signaling is also free. AP-1, a heterodimer composed of c-Fos and c-Jun, deemed as an activating transcription in cells is stimulated to accelerate the transcription of pro-inflammatory cytokines after activation of JNK regulated by ASK1 (Tang et al., 2001; Shaulian and Karin, 2002). Furthermore, the release of p38 MAPK due to ASK1 also acts directly on inflammation mediated by cerebral damage (Sun and Nan, 2016). Robenacoxib, an anti-inflammation drug, has been demonstrated to have a deleterious effect that accelerates neuronal death. Interestingly, the side effects of using robenacoxib are offset with the combination of ER stress inhibitor salubrinal and robenacoxib to treat cerebral ischemia. The neuroprotective effect is reflected in the decreased activation of glial cells, especially microglia, which reduces the degeneration and loss of neurons caused by inflammation (Anuncibay-Soto et al., 2018). This indicates that ER stress and microglia may have an unknown connection to control inflammation.
In some non-ischemic models, other inflammatory pathways related to ER stress have been found. It has been shown that the reciprocity of nucleotide-binding oligomerization domain 1 and 2 (NOD1/2) and serine/threonine protein kinase 2 (RIPK2) could be enhanced by IRE-1α-TRAF2 axis leading to the activation of NF-κB under ER stress induced by brucella abortus infection (Keestra-Gounder et al., 2016). A research actualized via animal and clinical trials suggested that conjugated bile acids (CBAs) mainly combine with ATF6α alleviating airway inflammation with the attenuation of the IL-4, IL-5, IL-13 production (Nakada et al., 2019).
Actually, the conduction of inflammatory signaling from ER stress is bidirectional rather than a one-way street. The release of inflammatory mediators such as IL-6 and TNF-α will also accelerate the activation of UPR and release more inflammatory factors to produce a nausea cycle of inflammatory response where the mechanism involved may be related to the cascade events caused by overload Ca2 including mitochondrial dysfunction and accumulation of misfolded proteins in ER lumen (Xue et al., 2005).
As mentioned earlier, the involvement of neuronal cells in ER stress after cerebral ischemia is concernful certainly. However, the risk of stroke depends not only on the neuron in the brain but also on the function and health of endothelial and glial cells in general.
The consequences of early ischemic injury to endothelial cells may be wider scope than indicated by the small volume they occupy. Subtle changes are discovered in microvessels after ischemic injury. Adhesion molecules and regulatory cytokines located at the membranes of endothelial cells have interaction with activating leukocytes, which activates platelets and promotes micro thrombosis, thereby aggrandizing the infarct range in the ischemic area (Garcia et al., 1994). Clinically, brain edema after cerebral ischemia is a common cause of death. Ischemic stroke with poor prognosis largely depends on the formation of severe edema. The damage caused by increased intracranial pressure is irreversible (Liebeskind et al., 2019). Endothelial cells are the main component of whole blood-brain barrier (BBB). During cerebral ischemia, endothelial cells contracted, endothelial space expanded, BBB permeability increased, resulting in vasogenic brain edema (Petito et al., 1982). In addition to endothelial cell swelling, the increase of the terminal volume of astrocytes surrounding endothelial cells may potentially compromise the BBB function. Subsequently, the leakage of BBB will contribute to the fortified blood protein extravasation and further aggravate the angiogenic edema. With the aid of a reasonable amount of lithium, the damaged endothelial cells are repaired, which enhances the vasodilation ability and contribution to the stabilization of BBB and alleviation of edema (Bosche et al., 2016a,b; Haupt et al., 2020). Mechanisms underlying brain swelling are not clear but Ca2+ overload in endothelial cells has been identified as the main factor (Olson et al., 1990). The augment of Ca2+ concentration in endothelial cells is mainly due to the release of ER Ca2+ channel, which implies that endothelial cells could have a certain inevitable connection with ER stress. Fortunately, despite the small number of relevant studies, some have shown that endothelial cells are directly involved in the complex pathological mechanism of ER stress induced by cerebral ischemia. Pretreatment with siRNA-vascular endothelial growth factor (VEGF) before OGD/R changed the biological characteristics of brain-derived Endothelial cells 3 (BEND3), decreasing the expression level of XBP1, CHOP, and GRP78 (Feng et al., 2019). Astragaloside IV can effectively reduce the permeability of BBB by inhibiting the apoptosis of endothelial cells mediated by ER stress after I/R (Hou et al., 2020). Those researches indicate that endothelial cells are certainly correlated with ER stress. However, more sufficient experiments and arguments are needed to clarify the regulatory mechanism.
Glial cells are divided into astrocytes, oligodendrocytes, and microglia cells, which are activated in different degrees in response to cerebral ischemia. After 3 days of cerebral ischemia, GRP78 expression was highly correlated with the activation of microglia around infarction and the activation of astrocytes in the peri-infarct area (Jin et al., 2018). As just noted, the interaction between ER stress and inflammation is regulated by phenotype of reactive microglia. Astrocytes and pyramidal neurons in CA1 region emerged ER stress dependent apoptosis after ischemia. Notably, ameliorated CHOP in neurons remained unchanged in astrocytes (Osada et al., 2010). Similarly, neurovascular unit presents a structure-dependent response to ischemia with the salubrinal treatment. Descendent GFAP, a marker of activated astrocytes were detected in CA1 but no change in CA3. Outcomes of this research also revealed that endothelial cells are highly responsive to salubrinal, while astrocytes have limitations to responsiveness (Anuncibay-Soto et al., 2016). Therefore, decreased intensity of ER stress after cerebral ischemia hinges on differential responses state from different neurovascular units. Oligodendrocytes are sensitive to excitotoxicity and ER stress induced by ischemia and eventually produce oxidative stress and apoptosis. Regretfully, whether oligodendrocytes are involved in the regulation of ER stress and its specific mechanisms need to be further explored (Ruiz et al., 2020).
The concept of the neurovascular unit takes the integrity of brain function and the interaction between different structures into account, which provides a new mentality for clinical treatment of ischemic stroke.
The evidence for activation of pathways associated with ER stress after cerebral ischemia is strong and the life of brain cells cannot be separated from the UPR propagated or induced by ER stress. Various enzymes and transcription factors downstream of UPR will control cell fate according to the intensity and duration of ER stress. However, there is still controversy regarding the increased or decreased expression of transcription factors such as ATF4, GADD34, XBP1s, ATF6. downstream of UPR in the neuroprotective effect of cerebral ischemia. Therefore, the critical point and clear molecular mechanism of UPR’s reversal need to be further discussed. Therefore, breakthrough research on related small molecule inhibitors under ER stress will be what stroke patients and neurologists are eager to see.
Although the positive effect of small molecule inhibitors of targeted ER stress in experimental cerebral ischemia has been proven, such as dantrolene (Li et al., 2005), salubrinal, 4-PBA, and guanabenz, the successful verification of these in clinical trials is rarely reported. The ER is closely related to calcium ions, and the most urgent affair to alleviate ER stress after cerebral ischemia is to restore Ca2+ homeostasis. It is a good idea to save cell fate from the apoptosis pathway and inflammation pathway related to ER stress. The treatment and mechanism of cerebral ischemia of different cell groups should be taken into consideration rather than limited to neurons. What remains to come up with is a complete and relatively mature response plan to deal with nerve damage after cerebral ischemia.
A better understanding of the diversiform mechanisms involved in ER stress will contribute to identify useful cerebral ischemia treatments. Further studies ought to address the unknown downstream molecular mechanisms, the potential for digging deep into known pathways of ER stress response and their implications in neurological diseases beside cerebral ischemia.
MY and Y-SG designed the structure of the manuscript. X-YS and Z-KG managed the literature searches and analyses. YH wrote the manuscript. XB assisted with the improvement of the manuscript. All authors contributed to and have approved the final manuscript.
This study was supported by the Scientific Research Projects of Shanghai Municipal Health Bureau in China, No. 201940031 (to XB) and the Leading Personnel Training Project of Shanghai Pudong New District Municipal Health Bureau in China, No. PWR12018-04 (to XB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Anuncibay-Soto, B., Pérez-Rodríguez, D., Santos-Galdiano, M., Font, E., Regueiro-Purriños, M., and Fernández-López, A. (2016). Post-ischemic salubrinal treatment results in a neuroprotective role in global cerebral ischemia. J. Neurochem. 138, 295–306. doi: 10.1111/jnc.13651
Anuncibay-Soto, B., Pérez-Rodriguez, D., Santos-Galdiano, M., Font-Belmonte, E., Ugidos, I. F., Gonzalez-Rodriguez, P., et al. (2018). Salubrinal and robenacoxib treatment after global cerebral ischemia. Exploring the interactions between ER stress and inflammation. Biochem. Pharmacol. 151, 26–37. doi: 10.1016/j.bcp.2018.02.029
Berna-Erro, A., Braun, A., Kraft, R., Kleinschnitz, C., Schuhmann, M. K., Stegner, D., et al. (2009). STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2:ra67. doi: 10.1126/scisignal.2000522
Berridge, M. J. (1989). The Albert Lasker medical awards. Inositol trisphosphate, calcium, lithium, and cell signaling. JAMA 262, 1834–1841. doi: 10.1001/jama.262.13.1834
Berridge, M. J., and Irvine, R. F. (1989). Inositol phosphates and cell signalling. Nature 341, 197–205. doi: 10.1038/341197a0
Bevers, M. B., and Neumar, R. W. (2008). Mechanistic role of calpains in postischemic neurodegeneration. J. Cereb. Blood Flow Metab. 28, 655–673. doi: 10.1038/sj.jcbfm.9600595
Blackwood, E. A., Azizi, K., Thuerauf, D. J., Paxman, R. J., Plate, L., Kelly, J. W., et al. (2019). Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat. Commun. 10:187. doi: 10.1038/s41467-018-08129-2
Borsello, T., Clarke, P. G. H., Hirt, L., Vercelli, A., Repici, M., Schorderet, D. F., et al. (2003). A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 9, 1180–1186. doi: 10.1038/nm911
Bosche, B., Molcanyi, M., Noll, T., Rej, S., Zatschler, B., Doeppner, T. R., et al. (2016a). A differential impact of lithium on endothelium-dependent but not on endothelium-independent vessel relaxation. Prog. Neuro Psychopharmacol. Biol. Psychiatry 67, 98–106. doi: 10.1016/j.pnpbp.2016.02.004
Bosche, B., Molcanyi, M., Rej, S., Doeppner, T. R., Obermann, M., Müller, D. J., et al. (2016b). Low-dose lithium stabilizes human endothelial barrier by decreasing MLC phosphorylation and universally augments cholinergic vasorelaxation capacity in a direct manner. Front. Physiol. 7:593. doi: 10.3389/fphys.2016.00593
Boyce, M., Bryant, K. F., Jousse, C., Long, K., Harding, H. P., Scheuner, D., et al. (2005). A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307, 935–939. doi: 10.1126/science.1101902
Braun, A., Varga-Szabo, D., Kleinschnitz, C., Pleines, I., Bender, M., Austinat, M., et al. (2009). Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood 113, 2056–2063. doi: 10.1182/blood-2008-07-171611
Briston, T., Selwood, D. L., Szabadkai, G., and Duchen, M. R. (2019). Mitochondrial permeability transition: a molecular lesion with multiple drug targets. Trends Pharmacol. Sci. 40, 50–70. doi: 10.1016/j.tips.2018.11.004
Britzolaki, A., Saurine, J., Flaherty, E., Thelen, C., and Pitychoutis, P. M. (2018). The SERCA2: a gatekeeper of neuronal calcium homeostasis in the brain. Cell. Mol. Neurobiol. 38, 981–994. doi: 10.1007/s10571-018-0583-8
Brunet, S., Thibault, L., Lepage, G., Seidman, E. G., Dubé, N., and Levy, E. (2000). Modulation of endoplasmic reticulum-bound cholesterol regulatory enzymes by iron/ascorbate-mediated lipid peroxidation. Free Radic. Biol. Med. 28, 46–54. doi: 10.1016/s0891-5849(99)00197-5
Bull, R., Finkelstein, J. P., Gálvez, J., Sánchez, G., Donoso, P., Behrens, M. I., et al. (2008). Ischemia enhances activation by Ca2+ and redox modification of ryanodine receptor channels from rat brain cortex. J. Neurosci. 28, 9463–9472. doi: 10.1523/JNEUROSCI.2286-08.2008
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., et al. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96. doi: 10.1038/415092a
Campbell, B. C. V., De Silva, D. A., Macleod, M. R., Coutts, S. B., Schwamm, L. H., Davis, S. M., et al. (2019). Ischaemic stroke. Nat. Rev. Dis. Primers 5:70. doi: 10.1038/s41572-019-0118-8
Celsi, F., Pizzo, P., Brini, M., Leo, S., Fotino, C., Pinton, P., et al. (2009). Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim. Biophys. acta 1787, 335–344. doi: 10.1016/j.bbabio.2009.02.021
Chan, S. L., Culmsee, C., Haughey, N., Klapper, W., and Mattson, M. P. (2002). Presenilin-1 mutations sensitize neurons to DNA damage-induced death by a mechanism involving perturbed calcium homeostasis and activation of calpains and caspase-12. Neurobiol. Dis. 11, 2–19. doi: 10.1006/nbdi.2002.0542
Chang, W.-c (2006). Store-operated calcium channels and pro-inflammatory signals. Acta Pharmacol. Sin. 27, 813–820. doi: 10.1111/j.1745-7254.2006.00395.x
Chapman, R., Sidrauski, C., and Walter, P. (1998). Intracellular signaling from the endoplasmic reticulum to the nucleus. Annu. Rev. Cell Dev. Biol. 14, 459–485. doi: 10.1146/annurev.cellbio.14.1.459
Chen, Y., and Brandizzi, F. (2013). IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 23, 547–555. doi: 10.1016/j.tcb.2013.06.005
Chow, C. Y., Wang, X., Riccardi, D., Wolfner, M. F., and Clark, A. G. (2015). The genetic architecture of the genome-wide transcriptional response to ER stress in the mouse. PLoS Genet. 11:e1004924. doi: 10.1371/journal.pgen.1004924
Coe, H., and Michalak, M. (2009). Calcium binding chaperones of the endoplasmic reticulum. Gen. Physiol. Biophys. 28 Spec No Focus, F96–F103.
Datta, S. R., Dudek, H., Tao, X., Masters, S., Fu, H., Gotoh, Y., et al. (1997). Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91, 231–241. doi: 10.1016/s0092-8674(00)80405-5
de la Cadena, S. G., Hernández-Fonseca, K., Camacho-Arroyo, I., and Massieu, L. (2014). Glucose deprivation induces reticulum stress by the PERK pathway and caspase-7- and calpain-mediated caspase-12 activation. Apoptosis 19, 414–427. doi: 10.1007/s10495-013-0930-7
DeGracia, D. J., and Montie, H. L. (2004). Cerebral ischemia and the unfolded protein response. J. Neurochem. 91, 1–8. doi: 10.1111/j.1471-4159.2004.02703.x
Deng, J., Lu, P. D., Zhang, Y., Scheuner, D., Kaufman, R. J., Sonenberg, N., et al. (2004). Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 24, 10161–10168. doi: 10.1128/mcb.24.23.10161-10168.2004
Dohmen, C., Sakowitz, O. W., Fabricius, M., Bosche, B., Reithmeier, T., Ernestus, R.-I., et al. (2008). Spreading depolarizations occur in human ischemic stroke with high incidence. Ann. Neurol. 63, 720–728. doi: 10.1002/ana.21390
Ekker, M. S., Boot, E. M., Singhal, A. B., Tan, K. S., Debette, S., Tuladhar, A. M., et al. (2018). Epidemiology, aetiology, and management of ischaemic stroke in young adults. Lancet Neurol. 17, 790–801. doi: 10.1016/S1474-4422(18)30233-3
Estes, S. D., Stoler, D. L., and Anderson, G. R. (1995). Normal fibroblasts induce the C/EBP beta and ATF-4 bZIP transcription factors in response to anoxia. Exp. Cell Res. 220, 47–54. doi: 10.1006/excr.1995.1290
Feng, D., Wang, B., Wang, L., Abraham, N., Tao, K., Huang, L., et al. (2017). Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE1 signalings. J. Pineal Res. 62:e12395. doi: 10.1111/jpi.12395
Feng, S.-Q., Zong, S.-Y., Liu, J.-X., Chen, Y., Xu, R., Yin, X., et al. (2019). VEGF antagonism attenuates cerebral ischemia/reperfusion-induced injury via inhibiting endoplasmic reticulum stress-mediated apoptosis. Biol. Pharm. Bull. 42, 692–702. doi: 10.1248/bpb.b18-00628
Foskett, J. K., White, C., Cheung, K.-H., and Mak, D.-O. D. (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658. doi: 10.1152/physrev.00035.2006
Fu, H. Y., Okada, K.-i., Liao, Y., Tsukamoto, O., Isomura, T., Asai, M., et al. (2010). Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 122, 361–369. doi: 10.1161/CIRCULATIONAHA.109.917914
Furuichi, T., Yoshikawa, S., Miyawaki, A., Wada, K., Maeda, N., and Mikoshiba, K. (1989). Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature 342, 32–38. doi: 10.1038/342032a0
Garcia, J. H., Liu, K. F., Yoshida, Y., Lian, J., Chen, S., and del Zoppo, G. J. (1994). Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat). Am. J. Pathol. 144, 188–199.
Gharibani, P. M., Modi, J., Pan, C., Menzie, J., Ma, Z., Chen, P.-C., et al. (2013). The mechanism of taurine protection against endoplasmic reticulum stress in an animal stroke model of cerebral artery occlusion and stroke-related conditions in primary neuronal cell culture. Adv. Exp. Med. Biol. 776, 241–258. doi: 10.1007/978-1-4614-6093-0_23
Gilmore, T. D. (2006). Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 25, 6680–6684. doi: 10.1038/sj.onc.1209954
Gong, D., Chi, X., Wei, J., Zhou, G., Huang, G., Zhang, L., et al. (2019). Modulation of cardiac ryanodine receptor 2 by calmodulin. Nature 572, 347–351. doi: 10.1038/s41586-019-1377-y
Groenendyk, J., Peng, Z., Dudek, E., Fan, X., Mizianty, M. J., Dufey, E., et al. (2014). Interplay between the oxidoreductase PDIA6 and microRNA-322 controls the response to disrupted endoplasmic reticulum calcium homeostasis. Sci. Signal. 7:ra54. doi: 10.1126/scisignal.2004983
Guan, Q.-H., Pei, D.-S., Xu, T.-L., and Zhang, G.-Y. (2006). Brain ischemia/reperfusion-induced expression of DP5 and its interaction with Bcl-2, thus freeing Bax from Bcl-2/Bax dimmers are mediated by c-Jun N-terminal kinase (JNK) pathway. Neurosci. Lett. 393, 226–230. doi: 10.1016/j.neulet.2005.09.075
Han, J., Back, S. H., Hur, J., Lin, Y.-H., Gildersleeve, R., Shan, J., et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490. doi: 10.1038/ncb2738
Harding, H. P., Calfon, M., Urano, F., Novoa, I., and Ron, D. (2002). Transcriptional and translational control in the mammalian unfolded protein response. Annu. Rev. Cell Dev. Biol. 18, 575–599. doi: 10.1146/annurev.cellbio.18.011402.160624
Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., et al. (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108. doi: 10.1016/s1097-2765(00)00108-8
Harding, H. P., Zhang, Y., and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274. doi: 10.1038/16729
Hatai, T., Matsuzawa, A., Inoshita, S., Mochida, Y., Kuroda, T., Sakamaki, K., et al. (2000). Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by the mitochondria-dependent caspase activation. J. Biol. Chem. 275, 26576–26581. doi: 10.1074/jbc.m003412200
Haupt, M., Zechmeister, B., Bosche, B., Lieschke, S., Zheng, X., Zhang, L., et al. (2020). Lithium enhances post-stroke blood-brain barrier integrity, activates the MAPK/ERK1/2 pathway and alters immune cell migration in mice. Neuropharmacology 181:108357. doi: 10.1016/j.neuropharm.2020.108357
He, Q., Li, Z., Meng, C., Wu, J., Zhao, Y., and Zhao, J. (2019). Parkin-dependent mitophagy is required for the inhibition of ATF4 on NLRP3 inflammasome activation in cerebral ischemia-reperfusion injury in rats. Cells 8:897. doi: 10.3390/cells8080897
Hertz, L. (2008). Bioenergetics of cerebral ischemia: a cellular perspective. Neuropharmacology 55, 289–309. doi: 10.1016/j.neuropharm.2008.05.023
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Hetz, C., Chevet, E., and Oakes, S. A. (2015). Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829–838. doi: 10.1038/ncb3184
Hetz, C., and Papa, F. R. (2018). The unfolded protein response and cell fate control. Mol. Cell 69, 169–181. doi: 10.1016/j.molcel.2017.06.017
Hetz, C., and Saxena, S. (2017). ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 13, 477–491. doi: 10.1038/nrneurol.2017.99
Hitomi, J., Katayama, T., Eguchi, Y., Kudo, T., Taniguchi, M., Koyama, Y., et al. (2004). Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and abeta-induced cell death. J. Cell Biol. 165, 347–356. doi: 10.1083/jcb.200310015
Hollien, J., and Weissman, J. S. (2006). Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107. doi: 10.1126/science.1129631
Hong, M., Luo, S., Baumeister, P., Huang, J.-M., Gogia, R. K., Li, M., et al. (2004). Underglycosylation of ATF6 as a novel sensing mechanism for activation of the unfolded protein response. J. Biol. Chem. 279, 11354–11363. doi: 10.1074/jbc.m309804200
Hou, B., Liu, R., Wu, Y., and Huang, S. (2020). Astragaloside IV reduces cerebral ischemia/reperfusion-induced blood-brain barrier permeability in rats by inhibiting ER stress-mediated apoptosis. Evid. Based Complement. Alternat. Med. 2020:9087873. doi: 10.1155/2020/9087873
Hu, P., Han, Z., Couvillon, A. D., Kaufman, R. J., and Exton, J. H. (2006). Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol. Cell. Biol. 26, 3071–3084. doi: 10.1128/mcb.26.8.3071-3084.2006
Hwang, M. S., Schwall, C. T., Pazarentzos, E., Datler, C., Alder, N. N., and Grimm, S. (2014). Mitochondrial Ca(2+) influx targets cardiolipin to disintegrate respiratory chain complex II for cell death induction. Cell Death Differ. 21, 1733–1745. doi: 10.1038/cdd.2014.84
Ibuki, T., Yamasaki, Y., Mizuguchi, H., and Sokabe, M. (2012). Protective effects of XBP1 against oxygen and glucose deprivation/reoxygenation injury in rat primary hippocampal neurons. Neurosci. Lett. 518, 45–48. doi: 10.1016/j.neulet.2012.04.053
Imai, H., Harland, J., McCulloch, J., Graham, D. I., Brown, S. M., and Macrae, I. M. (2002). Specific expression of the cell cycle regulation proteins, GADD34 and PCNA, in the peri-infarct zone after focal cerebral ischaemia in the rat. Eur. J. neurosci. 15, 1929–1936. doi: 10.1046/j.1460-9568.2002.02025.x
Jayaraj, R. L., Azimullah, S., Beiram, R., Jalal, F. Y., and Rosenberg, G. A. (2019). Neuroinflammation: friend and foe for ischemic stroke. J. Neuroinflammation 16:142. doi: 10.1186/s12974-019-1516-2
Jin, X., Kim, D. K., Riew, T.-R., Kim, H. L., and Lee, M.-Y. (2018). Cellular and subcellular localization of endoplasmic reticulum chaperone GRP78 Following transient focal cerebral ischemia in rats. Neurochem. Res. 43, 1348–1362. doi: 10.1007/s11064-018-2550-7
Jung, K. I., Ko, D.-H., Shin, N., Pyo, C. W., and Choi, S.-Y. (2019). Endoplasmic reticulum-associated degradation potentiates the infectivity of influenza A virus by regulating the host redox state. Free Radic. Biol. Med. 135, 293–305. doi: 10.1016/j.freeradbiomed.2019.03.021
Kaufman, R. J. (2002). Orchestrating the unfolded protein response in health and disease. J. clin. Invest. 110, 1389–1398. doi: 10.1172/jci0216886
Kaushik, S., and Cuervo, A. M. (2015). Proteostasis and aging. Nat. Med. 21, 1406–1415. doi: 10.1038/nm.4001
Keestra-Gounder, A. M., Byndloss, M. X., Seyffert, N., Young, B. M., Chávez-Arroyo, A., Tsai, A. Y., et al. (2016). NOD1 and NOD2 signalling links ER stress with inflammation. Nature 532, 394–397. doi: 10.1038/nature17631
Kim, I., Xu, W., and Reed, J. C. (2008). Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 7, 1013–1030. doi: 10.1038/nrd2755
Kimball, S. R. (1999). Eukaryotic initiation factor eIF2. Int. J. Biochem. Cell Biol. 31, 25–29. doi: 10.1016/s1357-2725(98)00128-9
Lanzillotta, A., Pignataro, G., Branca, C., Cuomo, O., Sarnico, I., Benarese, M., et al. (2013). Targeted acetylation of NF-kappaB/RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol. Dis. 49, 177–189. doi: 10.1016/j.nbd.2012.08.018
Lee, A.-H., Iwakoshi, N. N., and Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459. doi: 10.1128/mcb.23.21.7448-7459.2003
Lee, D. R., Helps, S. C., Gibbins, I. L., Nilsson, M., and Sims, N. R. (2003). Losses of NG2 and NeuN immunoreactivity but not astrocytic markers during early reperfusion following severe focal cerebral ischemia. Brain Res. 989, 221–230. doi: 10.1016/s0006-8993(03)03373-0
Lewis, R. S. (2020). Store-operated calcium channels: from function to structure and back again. Cold Spring Harb. Perspect. Biol. 12:a035055. doi: 10.1101/cshperspect.a035055
Li, F., Hayashi, T., Jin, G., Deguchi, K., Nagotani, S., Nagano, I., et al. (2005). The protective effect of dantrolene on ischemic neuronal cell death is associated with reduced expression of endoplasmic reticulum stress markers. Brain Res. 1048, 59–68. doi: 10.1016/j.brainres.2005.04.058
Li, G., Scull, C., Ozcan, L., and Tabas, I. (2010). NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J. Cell Biol. 191, 1113–1125. doi: 10.1083/jcb.201006121
Li, L., Li, Y., Miao, C., Liu, Y., and Liu, R. (2020). Coriolus versicolor polysaccharides (CVP) regulates neuronal apoptosis in cerebral ischemia-reperfusion injury via the p38MAPK signaling pathway. Ann. Transl. Med. 8:1168. doi: 10.21037/atm-20-5759
Li, Y., Zhang, Y., Fu, H., Huang, H., Lu, Q., Qin, H., et al. (2020). Hes1 knockdown exacerbates ischemic stroke following tMCAO by increasing ER stress-dependent apoptosis via the PERK/eIF2α/ATF4/CHOP signaling pathway. Neurosci. Bull. 36, 134–142. doi: 10.1007/s12264-019-00411-7
Liebeskind, D. S., Jüttler, E., Shapovalov, Y., Yegin, A., Landen, J., and Jauch, E. C. (2019). Cerebral edema associated with large hemispheric infarction. Stroke 50, 2619–2625. doi: 10.1161/STROKEAHA.118.024766
Lin, Y. W., Chen, T. Y., Hung, C. Y., Tai, S. H., Huang, S. Y., Chang, C. C., et al. (2018). Melatonin protects brain against ischemia/reperfusion injury by attenuating endoplasmic reticulum stress. Int. J. Mol. Med. 42, 182–192. doi: 10.3892/ijmm.2018.3607
Liu, D., Gu, Y., Wang, W., and Chen, W. (2020). Astragalin alleviates ischemia/reperfusion-induced brain injury via suppression of endoplasmic reticulum stress. Mol. Med. Rep. 22, 4070–4078. doi: 10.3892/mmr.2020.11448
Lopata, A., Kniss, A., Löhr, F., Rogov, V. V., and Dötsch, V. (2020). Ubiquitination in the ERAD process. Int. J. Mol. Sci. 21:5369. doi: 10.3390/ijms21155369
Lu, M., Lawrence, D. A., Marsters, S., Acosta-Alvear, D., Kimmig, P., Mendez, A. S., et al. (2014). Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 345, 98–101. doi: 10.1126/science.1254312
Luo, D., He, Y., Zhang, H., Yu, L., Chen, H., Xu, Z., et al. (2008). AIP1 is critical in transducing IRE1-mediated endoplasmic reticulum stress response. J. biol. Chem. 283, 11905–11912. doi: 10.1074/jbc.M710557200
Malhotra, J. D., Miao, H., Zhang, K., Wolfson, A., Pennathur, S., Pipe, S. W., et al. (2008). Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. U.S.A. 105, 18525–18530. doi: 10.1073/pnas.0809677105
Mamady, H., and Storey, K. B. (2008). Coping with the stress: expression of ATF4, ATF6, and downstream targets in organs of hibernating ground squirrels. Arch. Biochem. Biophys. 477, 77–85. doi: 10.1016/j.abb.2008.05.006
Mammadova-Bach, E., Nagy, M., Heemskerk, J. W. M., Nieswandt, B., and Braun, A. (2019). Store-operated calcium entry in thrombosis and thrombo-inflammation. Cell Calcium 77, 39–48. doi: 10.1016/j.ceca.2018.11.005
Marchi, S., Patergnani, S., Missiroli, S., Morciano, G., Rimessi, A., Wieckowski, M. R., et al. (2018). Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69, 62–72. doi: 10.1016/j.ceca.2017.05.003
Marciniak, S. J., Garcia-Bonilla, L., Hu, J., Harding, H. P., and Ron, D. (2006). Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J. Cell Biol. 172, 201–209. doi: 10.1083/jcb.200508099
Marciniak, S. J., and Ron, D. (2006). Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 86, 1133–1149. doi: 10.1152/physrev.00015.2006
Marciniak, S. J., Yun, C. Y., Oyadomari, S., Novoa, I., Zhang, Y., Jungreis, R., et al. (2004). CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077. doi: 10.1101/gad.1250704
Marsh, B., Stevens, S. L., Packard, A. E. B., Gopalan, B., Hunter, B., Leung, P. Y., et al. (2009). Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J. Neurosci. 29, 9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009
Mattson, M. P., LaFerla, F. M., Chan, S. L., Leissring, M. A., Shepel, P. N., and Geiger, J. D. (2000). Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 23, 222–229. doi: 10.1016/s0166-2236(00)01548-4
McCabe, C., White, F., Brown, S. M., and Macrae, I. M. (2008). GADD34 gene restores virulence in viral vector used in experimental stroke study. J. Cereb. Blood Flow Metab. 28, 747–751. doi: 10.1038/sj.jcbfm.9600565
Meng, C., Zhang, J., Dang, B., Li, H., Shen, H., Li, X., et al. (2018). PERK pathway activation promotes intracerebral hemorrhage induced secondary brain injury by inducing neuronal apoptosis both in vivo and in vitro. Front. Neurosci. 12:111. doi: 10.3389/fnins.2018.00111
Michalak, M., Robert Parker, J. M., and Opas, M. (2002). Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 32, 269–278. doi: 10.1016/s0143416002001884
Mo, Y., Sun, Y.-Y., and Liu, K.-Y. (2020). Autophagy and inflammation in ischemic stroke. Neural Regen. Res. 15, 1388–1396. doi: 10.4103/1673-5374.274331
Mo, Z.-T., Liao, Y.-L., Zheng, J., and Li, W.-N. (2020). Icariin protects neurons from endoplasmic reticulum stress-induced apoptosis after OGD/R injury via suppressing IRE1α-XBP1 signaling pathway. Life Sci. 255:117847. doi: 10.1016/j.lfs.2020.117847
Modi, J., Menzie-Suderam, J., Xu, H., Trujillo, P., Medley, K., Marshall, M. L., et al. (2020). Mode of action of granulocyte-colony stimulating factor (G-CSF) as a novel therapy for stroke in a mouse model. J. Biomed. Sci. 27:19. doi: 10.1186/s12929-019-0597-7
Mohammed Thangameeran, S. I., Tsai, S.-T., Hung, H.-Y., Hu, W.-F., Pang, C.-Y., Chen, S.-Y., et al. (2020). A role for endoplasmic reticulum stress in intracerebral hemorrhage. Cells 9:750. doi: 10.3390/cells9030750
Molcanyi, M., Bosche, B., Kraitsy, K., Patz, S., Zivcak, J., Riess, P., et al. (2013). Pitfalls and fallacies interfering with correct identification of embryonic stem cells implanted into the brain after experimental traumatic injury. J. Neurosci. Methods 215, 60–70. doi: 10.1016/j.jneumeth.2013.02.012
Molcanyi, M., Mehrjardi, N. Z., Schäfer, U., Haj-Yasein, N. N., Brockmann, M., Penner, M., et al. (2014). Impurity of stem cell graft by murine embryonic fibroblasts–implications for cell-based therapy of the central nervous system. Front. Cell. Neurosci. 8:257. doi: 10.3389/fncel.2014.00257
Moncada, S., and Erusalimsky, J. D. (2002). Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat. Rev. Mol. Cell Biol. 3, 214–220. doi: 10.1038/nrm762
Moskowitz, M. A., Lo, E. H., and Iadecola, C. (2010). The science of stroke: mechanisms in search of treatments. Neuron 67, 181–198. doi: 10.1016/j.neuron.2010.07.002
Nakada, E. M., Bhakta, N. R., Korwin-Mihavics, B. R., Kumar, A., Chamberlain, N., Bruno, S. R., et al. (2019). Conjugated bile acids attenuate allergen-induced airway inflammation and hyperresponsiveness by inhibiting UPR transducers. JCI Insight 4:e98101. doi: 10.1172/jci.insight.98101
Nakagawa, T., and Yuan, J. (2000). Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 150, 887–894. doi: 10.1083/jcb.150.4.887
Nakka, V. P., Gusain, A., and Raghubir, R. (2010). Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox. Res. 17, 189–202. doi: 10.1007/s12640-009-9110-5
Noguchi, K., Kitanaka, C., Yamana, H., Kokubu, A., Mochizuki, T., and Kuchino, Y. (1999). Regulation of c-Myc through phosphorylation at Ser-62 and Ser-71 by c-Jun N-terminal kinase. J. Biol. Chem. 274, 32580–32587.
Ohoka, N., Yoshii, S., Hattori, T., Onozaki, K., and Hayashi, H. (2005). TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 24, 1243–1255. doi: 10.1038/sj.emboj.7600596
Oida, Y., Shimazawa, M., Imaizumi, K., and Hara, H. (2008). Involvement of endoplasmic reticulum stress in the neuronal death induced by transient forebrain ischemia in gerbil. Neuroscience 151, 111–119. doi: 10.1016/j.neuroscience.2007.10.047
Olson, J. E., Fleischhacker, D., Murray, W. B., and Holtzman, D. (1990). Control of astrocyte volume by intracellular and extracellular Ca2+. Glia 3, 405–412. doi: 10.1002/glia.440030512
Osada, N., Kosuge, Y., Ishige, K., and Ito, Y. (2010). Characterization of neuronal and astroglial responses to ER stress in the hippocampal CA1 area in mice following transient forebrain ischemia. Neurochem. Int. 57, 1–7. doi: 10.1016/j.neuint.2010.03.017
Ouyang, Y.-B., Voloboueva, L. A., Xu, L.-J., and Giffard, R. G. (2007). Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J. Neurosci. 27, 4253–4260. doi: 10.1523/jneurosci.0211-07.2007
Owen, C. R., Kumar, R., Zhang, P., McGrath, B. C., Cavener, D. R., and Krause, G. S. (2005). PERK is responsible for the increased phosphorylation of eIF2alpha and the severe inhibition of protein synthesis after transient global brain ischemia. J. Neurochem. 94, 1235–1242. doi: 10.1111/j.1471-4159.2005.03276.x
Pahl, H. L., and Baeuerle, P. A. (1996). Activation of NF-kappa B by ER stress requires both Ca2+ and reactive oxygen intermediates as messengers. FEBS Lett. 392, 129–136. doi: 10.1016/0014-5793(96)00800-9
Pan, N., Lu, L.-Y., Li, M., Wang, G.-H., Sun, F.-Y., Sun, H.-S., et al. (2017). Xyloketal B alleviates cerebral infarction and neurologic deficits in a mouse stroke model by suppressing the ROS/TLR4/NF-κB inflammatory signaling pathway. Acta Pharmacol. Sin. 38, 1236–1247. doi: 10.1038/aps.2017.22
Parsons, J. T., Churn, S. B., and DeLorenzo, R. J. (1999). Global ischemia-induced inhibition of the coupling ratio of calcium uptake and ATP hydrolysis by rat whole brain microsomal Mg(2+)/Ca(2+) ATPase. Brain Res. 834, 32–41. doi: 10.1016/s0006-8993(99)01504-8
Paschen, W., Aufenberg, C., Hotop, S., and Mengesdorf, T. (2003). Transient cerebral ischemia activates processing of xbp1 messenger RNA indicative of endoplasmic reticulum stress. J. Cereb. Blood Flow Metab. 23, 449–461. doi: 10.1097/00004647-200304000-00009
Paschen, W., Gissel, C., Linden, T., Althausen, S., and Doutheil, J. (1998). Activation of gadd153 expression through transient cerebral ischemia: evidence that ischemia causes endoplasmic reticulum dysfunction. Brain Res. Mol. Brain Res. 60, 115–122. doi: 10.1016/s0169-328x(98)00180-6
Peng, S., Kuang, Z., Zhang, Y., Xu, H., and Cheng, Q. (2011). The protective effects and potential mechanism of Calpain inhibitor Calpeptin against focal cerebral ischemia-reperfusion injury in rats. Mol. Biol. Rep. 38, 905–912. doi: 10.1007/s11033-010-0183-2
Petito, C. K., Pulsinelli, W. A., Jacobson, G., and Plum, F. (1982). Edema and vascular permeability in cerebral ischemia: comparison between ischemic neuronal damage and infarction. J. Neuropathol. Exp. Neurol. 41, 423–436. doi: 10.1097/00005072-198207000-00005
Poone, G. K., Hasseldam, H., Munkholm, N., Rasmussen, R. S., Grønberg, N. V., and Johansen, F. F. (2015). The hypothermic influence on CHOP and Ero1-α in an endoplasmic reticulum stress model of cerebral ischemia. Brain Sci. 5, 178–187. doi: 10.3390/brainsci5020178
Putcha, G. V., Le, S., Frank, S., Besirli, C. G., Clark, K., Chu, B., et al. (2003). JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron 38, 899–914. doi: 10.1016/s0896-6273(03)00355-6
Putney, J. W. (1986). A model for receptor-regulated calcium entry. Cell Calcium 7, 1–12. doi: 10.1016/0143-4160(86)90026-6
Rao, R. V., Hermel, E., Castro-Obregon, S., del Rio, G., Ellerby, L. M., Ellerby, H. M., et al. (2001). Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J. Biol. Chem. 276, 33869–33874.
Rissanen, A., Sivenius, J., and Jolkkonen, J. (2006). Prolonged bihemispheric alterations in unfolded protein response related gene expression after experimental stroke. Brain Res. 1087, 60–66. doi: 10.1016/j.brainres.2006.02.095
Ruiz, A., Quintela-López, T., Sánchez-Gómez, M. V., Gaminde-Blasco, A., Alberdi, E., and Matute, C. (2020). Mitochondrial division inhibitor 1 disrupts oligodendrocyte Ca homeostasis and mitochondrial function. Glia 68, 1743–1756. doi: 10.1002/glia.23802
Sanderson, T. H., Gallaway, M., and Kumar, R. (2015). Unfolding the unfolded protein response: unique insights into brain ischemia. Int. J. Mol. Sci. 16, 7133–7142. doi: 10.3390/ijms16047133
Santulli, G., Nakashima, R., Yuan, Q., and Marks, A. R. (2017). Intracellular calcium release channels: an update. J. Physiol. 595, 3041–3051. doi: 10.1113/JP272781
Schinzel, A. C., Takeuchi, O., Huang, Z., Fisher, J. K., Zhou, Z., Rubens, J., et al. (2005). Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A. 102, 12005–12010. doi: 10.1073/pnas.0505294102
Serwach, K., and Gruszczynska-Biegala, J. (2019). STIM proteins and glutamate receptors in neurons: role in neuronal physiology and neurodegenerative diseases. Int. J. Mol. Sci. 20:2289. doi: 10.3390/ijms20092289
Shaulian, E., and Karin, M. (2002). AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–E136.
Shin, S. W., Park, J. W., Suh, M. H., Suh, S. I., and Choe, B. K. (1998). Persistent expression of Fas/FasL mRNA in the mouse hippocampus after a single NMDA injection. J. Neurochem. 71, 1773–1776. doi: 10.1046/j.1471-4159.1998.71041773.x
Singh, V., Mishra, V. N., Chaurasia, R. N., Joshi, D., and Pandey, V. (2019). Modes of calcium regulation in ischemic neuron. Indian J. Clin. Biochem. 34, 246–253. doi: 10.1007/s12291-019-00838-9
Smyth, J. T., Hwang, S.-Y., Tomita, T., DeHaven, W. I., Mercer, J. C., and Putney, J. W. (2010). Activation and regulation of store-operated calcium entry. J. Cell. Mol. Med. 14, 2337–2349. doi: 10.1111/j.1582-4934.2010.01168.x
Southwood, C. M., Garbern, J., Jiang, W., and Gow, A. (2002). The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron 36, 585–596. doi: 10.1016/s0896-6273(02)01045-0
Sprenkle, N. T., Sims, S. G., Sánchez, C. L., and Meares, G. P. (2017). Endoplasmic reticulum stress and inflammation in the central nervous system. Mol. Neurodegener. 12:42. doi: 10.1186/s13024-017-0183-y
Sun, J., and Nan, G. (2016). The mitogen-activated protein kinase (MAPK) signaling pathway as a discovery target in stroke. J. Mol. Neurosci. 59, 90–98. doi: 10.1007/s12031-016-0717-8
Sun, S., Shi, G., Sha, H., Ji, Y., Han, X., Shu, X., et al. (2015). IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 17, 1546–1555. doi: 10.1038/ncb3266
Sun, X., Liu, H., Sun, Z., Zhang, B., Wang, X., Liu, T., et al. (2020). Acupuncture protects against cerebral ischemia-reperfusion injury via suppressing endoplasmic reticulum stress-mediated autophagy and apoptosis. Mol. Med. 26:105. doi: 10.1186/s10020-020-00236-5
Szydlowska, K., and Tymianski, M. (2010). Calcium, ischemia and excitotoxicity. Cell Calcium 47, 122–129. doi: 10.1016/j.ceca.2010.01.003
Tajiri, S., Oyadomari, S., Yano, S., Morioka, M., Gotoh, T., Hamada, J. I., et al. (2004). Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 11, 403–415. doi: 10.1038/sj.cdd.4401365
Takenaka, H., Adler, P. N., and Katz, A. M. (1982). Calcium fluxes across the membrane of sarcoplasmic reticulum vesicles. J. Biol. Chem. 257, 12649–12656. doi: 10.1016/s0021-9258(18)33560-9
Tang, G., Minemoto, Y., Dibling, B., Purcell, N. H., Li, Z., Karin, M., et al. (2001). Inhibition of JNK activation through NF-kappaB target genes. Nature 414, 313–317. doi: 10.1038/35104568
Thiebaut, A. M., Hedou, E., Marciniak, S. J., Vivien, D., and Roussel, B. D. (2019). Proteostasis during cerebral ischemia. Front. Neurosci. 13:637. doi: 10.3389/fnins.2019.00637
Tirasophon, W., Welihinda, A. A., and Kaufman, R. J. (1998). A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824. doi: 10.1101/gad.12.12.1812
Tsaytler, P., Harding, H. P., Ron, D., and Bertolotti, A. (2011). Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332, 91–94. doi: 10.1126/science.1201396
Tuzlak, S., Kaufmann, T., and Villunger, A. (2016). Interrogating the relevance of mitochondrial apoptosis for vertebrate development and postnatal tissue homeostasis. Genes Dev. 30, 2133–2151. doi: 10.1101/gad.289298.116
Urban, P., Pavlíková, M., Sivonová, M., Kaplán, P., Tatarková, Z., Kaminska, B., et al. (2009). Molecular analysis of endoplasmic reticulum stress response after global forebrain ischemia/reperfusion in rats: effect of neuroprotectant simvastatin. Cell. Mol. Neurobiol. 29, 181–192. doi: 10.1007/s10571-008-9309-7
Van Petegem, F. (2012). Ryanodine receptors: structure and function. J. Biol. Chem. 287, 31624–31632. doi: 10.1074/jbc.R112.349068
Verma, G., Bhatia, H., and Datta, M. (2013). JNK1/2 regulates ER-mitochondrial Ca2+ cross-talk during IL-1β-mediated cell death in RINm5F and human primary β-cells. Mol. Biol. Cell 24, 2058–2071. doi: 10.1091/mbc.E12-12-0885
Vosler, P. S., Brennan, C. S., and Chen, J. (2008). Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol. Neurobiol. 38, 78–100. doi: 10.1007/s12035-008-8036-x
Waldherr, S. M., Strovas, T. J., Vadset, T. A., Liachko, N. F., and Kraemer, B. C. (2019). Constitutive XBP-1s-mediated activation of the endoplasmic reticulum unfolded protein response protects against pathological tau. Nat. Commun. 10:4443. doi: 10.1038/s41467-019-12070-3
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wang, W.-Y., Xie, L., Zou, X.-S., Li, N., Yang, Y.-G., Wu, Z.-J., et al. (2021). Inhibition of extracellular signal-regulated kinase/calpain-2 pathway reduces neuroinflammation and necroptosis after cerebral ischemia-reperfusion injury in a rat model of cardiac arrest. Int. Immunopharmacol. 93:107377. doi: 10.1016/j.intimp.2021.107377
Wang, Y., Zhang, J. H., Sheng, J., and Shao, A. (2019). Immunoreactive cells after cerebral ischemia. Front. Immunol. 10:2781. doi: 10.3389/fimmu.2019.02781
Wang, Y.-C., Li, X., Shen, Y., Lyu, J., Sheng, H., Paschen, W., et al. (2020). PERK (protein kinase RNA-Like ER kinase) branch of the unfolded protein response confers neuroprotection in ischemic stroke by suppressing protein synthesis. Stroke 51, 1570–1577. doi: 10.1161/STROKEAHA.120.029071
White, F., McCaig, D., Brown, S. M., Graham, D. I., Harland, J., and Macrae, I. M. (2004). Up-regulation of a growth arrest and DNA damage protein (GADD34) in the ischaemic human brain: implications for protein synthesis regulation and DNA repair. Neuropathol. Appl. Neurobiol. 30, 683–691. doi: 10.1111/j.1365-2990.2004.00584.x
Xiao, P., Liu, X. W., Zhao, N. N., Fang, R., Wen, Q., Zeng, K. X., et al. (2018). Correlations of neuronal apoptosis with expressions of c-Fos and c-Jun in rats with post-ischemic reconditioning damage. Eur. Rev. Med. Pharmacol. Sci. 22, 2832–2838. doi: 10.26355/eurrev_201805_14984
Xue, X., Piao, J.-H., Nakajima, A., Sakon-Komazawa, S., Kojima, Y., Mori, K., et al. (2005). Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J. Biol. Chem. 280, 33917–33925. doi: 10.1074/jbc.m505818200
Yamamoto, K., Sato, T., Matsui, T., Sato, M., Okada, T., Yoshida, H., et al. (2007). Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 13, 365–376. doi: 10.1016/j.devcel.2007.07.018
Yamazaki, H., Hiramatsu, N., Hayakawa, K., Tagawa, Y., Okamura, M., Ogata, R., et al. (2009). Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J. Immunol. 183, 1480–1487. doi: 10.4049/jimmunol.0900017
Ye, J., Rawson, R. B., Komuro, R., Chen, X., Davé, U. P., Prywes, R., et al. (2000). ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364. doi: 10.1016/s1097-2765(00)00133-7
Yoneda, T., Imaizumi, K., Oono, K., Yui, D., Gomi, F., Katayama, T., et al. (2001). Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 276, 13935–13940. doi: 10.1074/jbc.m010677200
You, K., Wang, L., Chou, C.-H., Liu, K., Nakata, T., Jaiswal, A., et al. (2021). QRICH1 dictates the outcome of ER stress through transcriptional control of proteostasis. Science 371:eabb6896. doi: 10.1126/science.abb6896
Zerna, C., Thomalla, G., Campbell, B. C. V., Rha, J.-H., and Hill, M. D. (2018). Current practice and future directions in the diagnosis and acute treatment of ischaemic stroke. Lancet 392, 1247–1256. doi: 10.1016/S0140-6736(18)31874-9
Zhang, M., Song, J.-N., Wu, Y., Zhao, Y.-L., Pang, H.-G., Fu, Z.-F., et al. (2014). Suppression of STIM1 in the early stage after global ischemia attenuates the injury of delayed neuronal death by inhibiting store-operated calcium entry-induced apoptosis in rats. Neuroreport 25, 507–513. doi: 10.1097/WNR.0000000000000127
Zhang, Q., Liu, J., Chen, S., Liu, J., Liu, L., Liu, G., et al. (2016). Caspase-12 is involved in stretch-induced apoptosis mediated endoplasmic reticulum stress. Apoptosis 21, 432–442. doi: 10.1007/s10495-016-1217-6
Zhang, X., Yuan, Y., Jiang, L., Zhang, J., Gao, J., Shen, Z., et al. (2014). Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: involvement of PARK2-dependent mitophagy. Autophagy 10, 1801–1813. doi: 10.4161/auto.32136
Zhao, Y., Fang, Y., Zhao, H., Li, J., Duan, Y., Shi, W., et al. (2018). Chrysophanol inhibits endoplasmic reticulum stress in cerebral ischemia and reperfusion mice. Eur. J. Pharmacol. 818, 1–9. doi: 10.1016/j.ejphar.2017.10.016
Zhou, L., Bondy, S. C., Jian, L., Wen, P., Yang, F., Luo, H., et al. (2015). Tanshinone IIA attenuates the cerebral ischemic injury-induced increase in levels of GFAP and of caspases-3 and -8. Neuroscience 288, 105–111. doi: 10.1016/j.neuroscience.2014.12.028
Zhu, H., Zhu, H., Xiao, S., Sun, H., Xie, C., and Ma, Y. (2012). Activation and crosstalk between the endoplasmic reticulum road and JNK pathway in ischemia-reperfusion brain injury. Acta Neurochir. 154, 1197–1203. doi: 10.1007/s00701-012-1396-z
Keywords: cerebral ischemia, ER stress, unfolded protein response, apoptosis, inflammation, Ca2+ homeostasis
Citation: Han Y, Yuan M, Guo Y-S, Shen X-Y, Gao Z-K and Bi X (2021) Mechanism of Endoplasmic Reticulum Stress in Cerebral Ischemia. Front. Cell. Neurosci. 15:704334. doi: 10.3389/fncel.2021.704334
Received: 06 May 2021; Accepted: 09 July 2021;
Published: 02 August 2021.
Edited by:
Sharon DeMorrow, University of Texas at Austin, United StatesReviewed by:
Bert Bosche, MediClin Klinik Reichshof, GermanyCopyright © 2021 Han, Yuan, Guo, Shen, Gao and Bi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xia Bi, Yml4aWFzaEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.