 Ruqayya Afridi
Ruqayya Afridi Kyoungho Suk
Kyoungho Suk
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci., 02 July 2021
Sec. Cellular Neuropathology
Volume 15 - 2021 | https://doi.org/10.3389/fncel.2021.691067
This article is part of the Research TopicMultifaceted interactions between immunity and the diseased brainView all 9 articles
The neuroinflammatory basis of depression encompasses the detrimental role of otherwise supportive non-neuronal cells and neuroinflammation in hampering neuronal function, leading to depressive behavior. Animals subjected to different stress paradigms show glial cell activation and a surge in proinflammatory cytokines in various brain regions. The concept of sterile inflammation observed in animal models of depression has intrigued many researchers to determine the possible triggers of central immune cell activation. Notably, microglial activation and subsequent phenotypic polarization in depression have been strongly advocated by the wealth of recent preclinical studies; however, findings from human studies have shown contradictory results. Despite intensive investigation, many research gaps still exist to elucidate the molecular mechanisms of neuroinflammatory cascades underlying the pathophysiology of depression. In this mini-review, recent progress in understanding neuroinflammatory mechanisms in light of experimental models of depression will be thoroughly discussed. The challenges of mirroring depression in animal and in vitro models will also be highlighted. Furthermore, prospects of targeting neuroinflammation to treat depressive disorder will be covered.
Major depressive disorder (MDD) is the most prevalent debilitating psychiatric disorder affecting individuals during some part of their life, resulting in a substantial health and economic burden worldwide (Konig et al., 2019; Konnopka and Konig, 2020). Neurochemical and structural alterations in mesolimbic and corticolimbic neural circuitry that regulate mood and behavior, including the prefrontal cortex (PFC), amygdala, nucleus accumbens, and hippocampus, are reported to be the cause of depression symptoms (Duman et al., 2016). Dysregulation of monoaminergic neurotransmission, including serotonin and dopamine, is a widely accepted theory of depression pathology, and various perturbations in monoamine signaling and metabolism have been identified (Jesulola et al., 2018). Moreover, deficits in synaptic plasticity induced by altered glutamatergic neurotransmission are involved in depression pathology, constituting the “neuroplasticity” hypothesis of depression (Pittenger and Duman, 2008). Various drugs interfering with glutamatergic neurotransmission have been reported to exert antidepressant actions in clinical and preclinical studies (Pittenger and Duman, 2008). Brain-derived neurotrophic factor (BDNF) is known to play an important role in neuroplasticity, and decreased BDNF expression has been reported in various brain regions of depressed patients (Dwivedi, 2009). All of these mechanisms play a crucial role in the pathology of depression; however, the inefficacy of antidepressant drugs in a subpopulation of patients with MDD and decreased remission rates highlight the involvement of diverse mechanisms in addition to these neurocentric theories.
Emerging evidence provides ample support for the involvement of non-neuronal cells leading to a neuroin-flammatory milieu in depression neurobiology (Koo and Duman, 2008; Steiner et al., 2011; Strawbridge et al., 2015). Glial cells constitute a major proportion of brain tissue and play a significant role in maintaining brain homeostasis by supporting neurons in dynamic ways. Increased microglial inflammatory activation, astrocytic atrophy, and decreased myelin basic protein immunoreactivity and fewer mature oligodendrocytes have been documented in MDD subjects and animal models of depression (Cotter et al., 2001; Tynan et al., 2013; Yang et al., 2015). The inflammatory activation of microglial cells has been reported to alter glutamatergic neurotransmission, impair monoamine synthesis, and interfere with BDNF signaling, culminating in altered synaptic plasticity and neurogenesis, and precipitating depression (Weber et al., 2019).
Neuroinflammatory perturbations identified in animal models of depression provide a strong basis for non-neuronal cell involvement in MDD pathology (Weber et al., 2017; Wang et al., 2018). Several in vitro experimental models of MDD, which provide cell-level information, have been developed to enhance the usefulness of in vivo models (Zunszain et al., 2012; Zhang et al., 2020b). All these models are of value for deciphering the fundamental mechanisms underlying MDD pathology and testing novel therapies targeted against this disease. In this review, recent literature documenting neuroinflammatory alterations observed in experimental models of depression is discussed. Subsequently, plausible reasons behind discrepancies between data from human studies and preclinical data are highlighted. Additionally, the therapeutic significance of targeting neuroinflammation in depressive disorders will be discussed.
Environmental and genetic factors have been identified as crucial drivers of depression pathology in both human and rodent models (Lesch, 2004; Levinson, 2006). As these factors are highly variable, epistatic, and complex, they are thought to regulate vulnerability to depression development and responsiveness to antidepressant therapy. Various polymorphisms have been reported in genes regulating the hypothalamic–pituitary–adrenal (HPA) axis, serotonin recycling, and immune responses, including corticotropin-releasing hormone receptor 1, the sodium-dependent serotonin transporter gene (SLC6A4), and interleukin-1β (IL-1β) (Baune et al., 2010; Schiele et al., 2021). Moreover, environmental stressors are associated with epigenetic modification of BDNF, its receptor tropomyosin-related kinase B gene, glucocorticoid receptor gene (NR3C1), and glutamate ionotropic receptor NMDA type subunit 2B (GRIN2B) (Ernst et al., 2009; Jiang et al., 2010; Sun et al., 2013; Efstathopoulos et al., 2018).
Accumulating evidence suggests the involvement of multiple biological systems, including the neuroendocrine system, immune system, and neural circuitry, in the pathophysiology of depression (de Kloet et al., 2005). Activation of the HPA axis results in increased cortisol secretion in the blood, which in turn activates peripheral immune cells (Otte et al., 2016). Inflammatory signals from peripheral immune cells are propagated through various humoral, neural, and cellular pathways and results in the activation of brain resident immune cells that interfere with neurotransmitters and directly affect neuronal integrity through excitotoxicity.
The activation of the HPA axis and sympathetic system is an adaptive response of an organism toward any psychological or environmental stimuli perceived as a threat, resulting in the release of glucocorticoids (GC) and norepinephrine (NE) in the blood (Selye, 1976; McEwen et al., 2016). Increased GC and sympathetic signaling exert proinflammatory effects by mobilizing immune cells from the bone marrow, lymph node, and spleen and increasing their inflammatory activation (Engler et al., 2004; Dhabhar et al., 2012; Powell et al., 2013). Inflammatory activation of monocytes and macrophages leads to increased secretion of proinflammatory mediators, including tumor necrosis factor-α (TNF-α), IL-1β, and interleukin-6 (IL-6) (Serrats et al., 2010). Increased proinflammatory cytokines in circulation also have the propensity to repress the expression of several tight-junction proteins of the blood–brain barrier (BBB), including claudin-5 (Dudek et al., 2020). Mice exposed to chronic social defeat stress (CSDS) exhibited decreased expression of claudin-5, positively correlating with heightened peripheral TNF-α in circulation (Dudek et al., 2020). Chronic stress-induced BBB leakage in an animal model of depression allows the passage of proinflammatory mediators (Menard et al., 2017).
Recent literature also highlights the potential role of gut microbiome in precipitating inflammatory signals in depression pathology. Among divergent pathways through which gut microbiota can alter behavior, leading to depressive-like outcomes, is an inflammation-to-brain mechanism (Guo et al., 2019). A study in rodents demonstrated the activation of the master regulator of the inflammatory pathway nuclear factor-κB (NF-κB) when gut microbiota was altered by chronic restraint stress (CRS). The depletion of Lactobacillus was accompanied by increased inflammatory cytokines as well as increased microglial activation in the hippocampus (Guo et al., 2019). Although the mechanism of immune cell activation was not investigated, it is quite plausible that inflammatory signaling is involved, as Lactobacillus treatment reduced inflammation and alleviated depressive symptoms in mice subjected to CRS, highlighting the potential role of the gut-inflammatory pathway in exerting behavioral consequences.
Astrocytes, microglia, and oligodendrocytes are the major types of the glial population, each having a distinct role in healthy and diseased states. Accumulating evidence suggests that gliosis and inflammation lead to increased levels of proinflammatory cytokines and reactive oxygen species (ROS) in various brain regions, thereby contributing to neuronal damage and leading to altered mood and behavior. Astrocytes are the most abundant glial cells that provide metabolic and trophic support to neurons. Atrophy and reduction in number of astrocytes as well as a reduction in various astrocytic proteins have been documented in MDD pathology (Fatemi et al., 2004; Zhao et al., 2018). Reduced astrocyte numbers in the hippocampus, amygdala, and prefrontal cortex of MDD patients have been reported (Altshuler et al., 2010; Cobb et al., 2016). The excitotoxicity observed in MDD can be correlated with astrocytic dysfunction. The inability of astrocytes to uptake glutamate from the synaptic cleft leads to prolonged synaptic activation, which in turn leads to excitotoxicity (Choudary et al., 2005).
Microglia are highly plastic, brain resident macrophages, that mainly guard the brain parenchyma in addition to playing other physiological roles. Microglia display extensive phenotypic plasticity dependent on surrounding cues. Recent studies suggest the important role of stress-induced damage-associated molecular patterns as a primary signal in activating microglia. The primed state of microglia that is characterized by an increased expression of proinflammatory cytokines increases the propensity for the development of severe depressive symptoms (Wohleb et al., 2014). In the CSDS model, it has been reported that microglia-secreted proinflammatory cytokines are crucial for the recruitment of peripheral immune cells in stress-responsive brain regions, and these cells remain sensitized for a longer period after cessation of the acute stressful stimuli (Wohleb et al., 2014). In addition, the study provided useful insights into the temporal effects of stress on the neuroimmune axis (Wohleb et al., 2014). A surge in proinflammatory cytokines due to microglial activation and peripheral immune cell infiltration leads to the upregulation of microglial indolamine 2,3 dioxygenase (IDO) activity (Corona et al., 2013). Increased microglial IDO activity diverts tryptophan metabolism from serotonin to quinolinic acid (QUIN), which is a N-methyl-D aspartate receptor agonist, serving as a link between immune and neurotransmitter changes in depression. Increased inflammatory cytokines, including TNF-α, in microglial cells can also influence the neuronal re-uptake of monoamine neurotransmitters by regulating neuronal mitogen-activated protein kinase (MAPK), leading to an increased surface expression of monoamine transporters on neurons (Zhu et al., 2010). Cytokine-mediated increases in microglial QUIN and reduction in astrocytic glutamate uptake can lead to excessive glutamate levels and actions, thereby altering synaptic plasticity.
With advances in our understanding of molecular mechanisms of depression, efforts have been made to establish in vivo and in vitro models that can be used efficiently for a better understanding of the enigmatic pathophysiology of depression. Although still not completely achieved, few experimental models, including in vivo and in vitro, have been used frequently in neuroscience research.
Considering stress as a major factor in predisposing humans to the development of depression, most animal models used in preclinical studies are based on stress. Though many of these models lack etiological relevance, the hyperactive HPA axis, impaired neuroplasticity and neurogenesis, and altered neurotransmitters are consistent features of these models that can be paralleled with human depression disease. Thus, the contribution of these models in providing novel insights into depression pathology cannot be underscored. Specifically, the role of neuroinflammation in the pathophysiology of depression has been well-established in these models and explains the antidepressant action of certain anti-inflammatory drugs (Table 1).
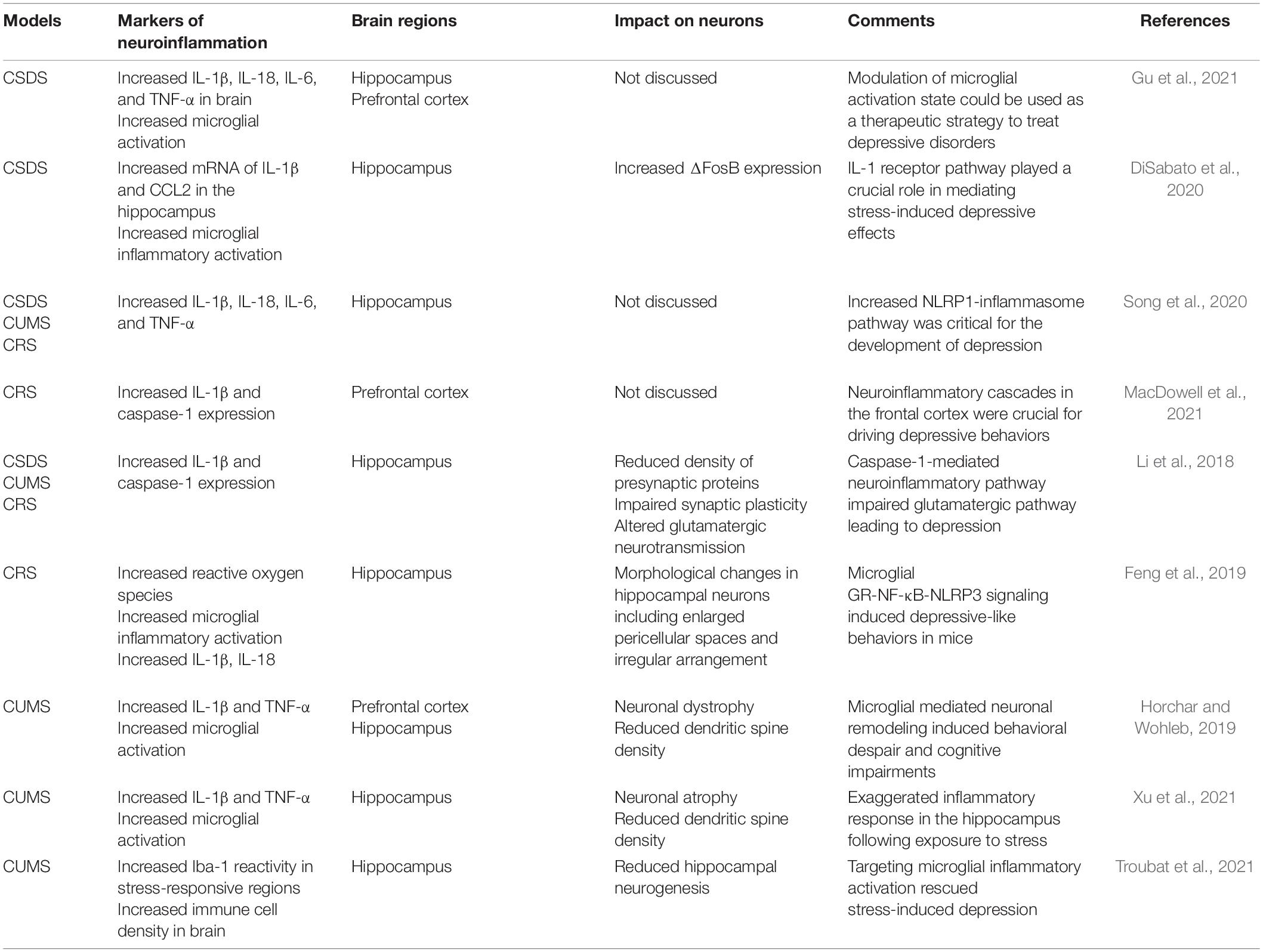
Table 1. Neuroinflammatory markers in animal models of depression.
CSDS, chronic unpredictable mild stress (CUMS), and CRS are the most widely employed animal models to decipher the neuropathological basis of depression. Increased neuroinflammatory profile characterized by elevated cytokines and the C-C Motif Chemokine ligand 2 (CCL2), and reduced anti-inflammatory regulation of neuronal-derived fractalkine ligand (CX3CL1) and microglial receptor (CX3CR1) are shared features in these models (Wohleb et al., 2014; Ramirez et al., 2016). Increased proliferation of inflammatory microglia with concomitant increase of Iba-1 immunoreactivity in the hippocampal tissue of mice subjected to CRS and CUMS has been reported (Feng et al., 2019; Horchar and Wohleb, 2019). Mice subjected to CSDS stress also exhibited microglial activation, recruitment of peripheral macrophages into the brain, and anxiety-like behavior (Tang et al., 2018). Inflammatory activation of microglia, stimulation of the microglial NLRP [NLR (nucleotide-binding domain and leucine-rich repeat) family, pyrin domain containing] inflammasome, and increased IL-1β production in the PFC were demonstrated in CUMS (Pan et al., 2014). Microglia isolated from mice subjected to CSDS have gene ontology profiles signifying increased inflammation, phagocytosis, and ROS production (Lehmann et al., 2018). The behavioral phenotypes observed in these models, including anhedonia, decreased sociability, and despair, are positively correlated with inflammatory activation of microglia.
Deficits in synaptic plasticity, altered dendritic spine density, and impaired neurogenesis because of heightened neuroinflammation have been reported in CSDS, CRS, and CUMS. Increased caspase-1 signaling in hippocampal region of mice after CSDS, CRS, and CUMS leads to dysregulated glutamatergic neurotransmission accompanied by altered dendritic spine density and reduced synaptic plasticity (Li et al., 2018). The genetic and pharmacological targeting of the IL-1β-caspase-1 pathway rescued the development of depressive behaviors in mice, highlighting the crucial role of neuroinflammatory pathways in impairing neuronal integrity (Li et al., 2018). Microglia-derived IL-1 can exert its detrimental effects on neurogenesis indirectly by stimulating the HPA axis as well as directly by activating IL-1 receptors expressed on hippocampal neural progenitor cells, resulting in decreased cell proliferation that is mediated by the NF-κB signaling pathway (Koo and Duman, 2008). Hence, microglial inflammatory activation as well as the neuroinflammatory milieu in animal models of depression may play key roles in the pathophysiology of depression.
Depression research is hampered by the absence of in vitro models that can recapitulate all molecular mechanisms of the disease. Attempts to model depression in vitro using hippocampal progenitor cell lines (HPCs) to study the pathways causing impaired neurogenesis are emerging. Thus far, studies have focused on isolated cell types in culture or occasionally two cell types in co-culture, which cannot fully model the important contributions of various cell types in disease. Various depressogenic stimuli identified in clinical and preclinical studies are used to study the mechanism or unravel pharmacological targets in neuronal cells and glial cell cultures (Table 2). Neurogenesis in the hippocampus regulates the HPA axis via a negative feedback mechanism; hence, the mechanisms underlying impairments in adult hippocampal neurogenesis have been explored in vitro (Schloesser et al., 2009). Microglia isolated from the hippocampus of cytokine-induced depressed mice suppressed neural stem/precursor cell proliferation and stimulated apoptosis of immature neurons, highlighting the role of microglia in impairing neurogenesis in depression pathology (Zhang et al., 2020a). IL-1β inhibited neurogenesis in HPCs by activating the neurotoxic branch of the kynurenine pathway, which has been postulated to be involved in the development of depressive disorders (Zunszain et al., 2012).
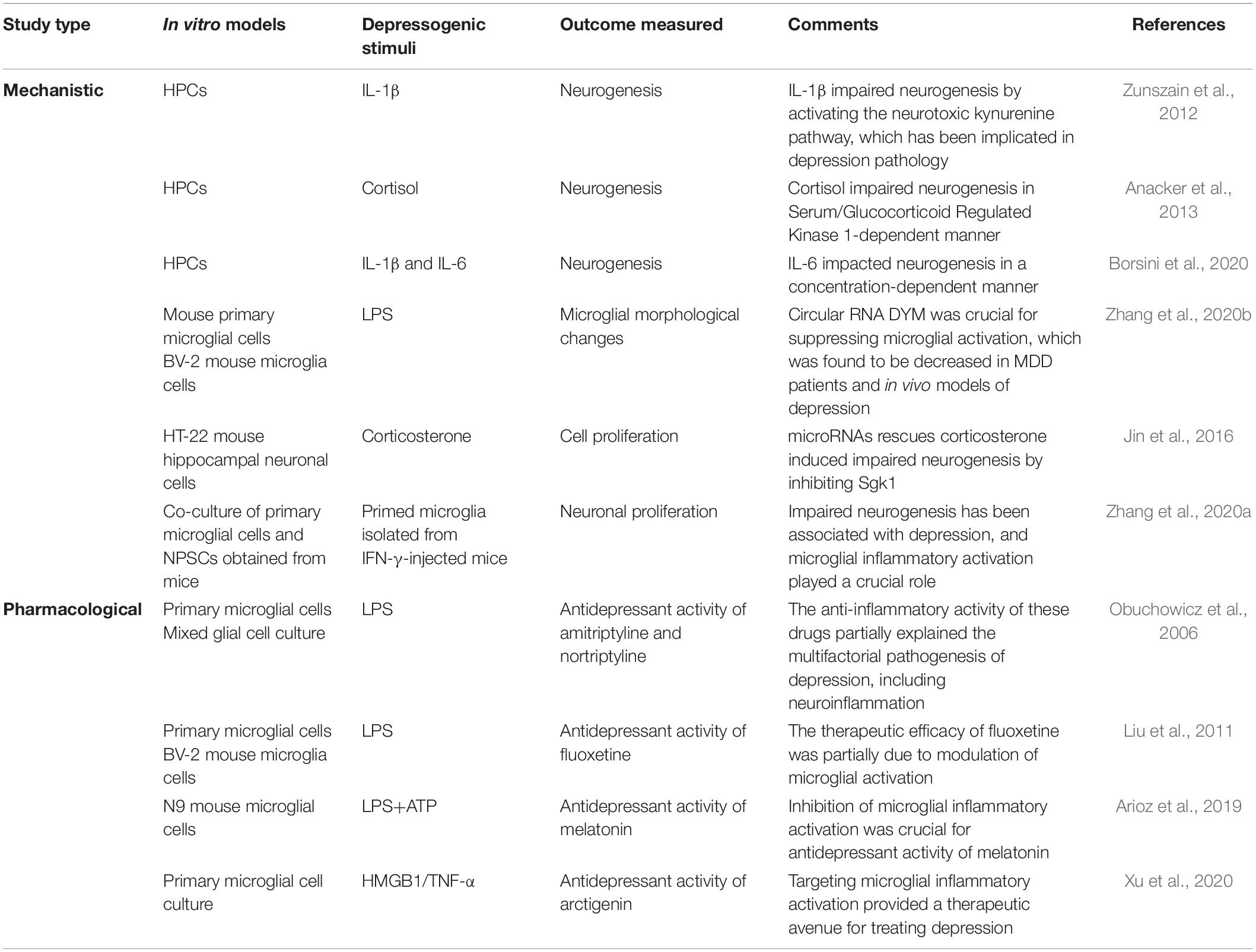
Table 2. Experimental findings in in vitro model of depression.
Although increased levels of proinflammatory cytokines, including IL-1β, IL-6, and TNF-α, in the plasma and CSF of depressed patients, have been reported, there still lies a big question mark on neuroinflammatory markers and microglial activation status (Martinez et al., 2012; Himmerich et al., 2019). The elevated translocator protein (TSPO) binding assessed by positron emission tomography studies in various brain regions of depressed patients backs microglial activation (Setiawan et al., 2015; Owen et al., 2017). Also, studies using other markers of microglial activation, such as Iba-1 or quinolinic acid, have found increased microglial reactivity in depression, whereas no difference between the density of major histocompatibility complex (HLA)-immunoreactive microglia in post-mortem brain samples of depressed subjects (Hamidi et al., 2004; Snijders et al., 2020). A recent study using single-cell high-dimensional mass cytometry (CyTOF) examined microglia from post-mortem MDD samples from different brain regions and found increased markers of homeostatic microglia, including transmembrane protein (TMEM)119 and P2Y12 in MDD compared to controls, which is in clear contrast with what has been found in animal studies (Bottcher et al., 2020). Gene expression analysis of microglia isolated from animal models of depression clearly showed enhanced inflammatory markers, including CD11b, CD45, and TLR4 (Wohleb et al., 2011; Lehmann et al., 2016). Moreover, gene expression profiling of post-mortem frontal lobe tissue from patients with MDD did not show any difference in the expression of IL-6 or TNF-α (Shelton et al., 2011). Furthermore, no differential expression of IL-6, IL-1β, or TNF-α mRNAs was found in post-mortem brain tissue of MDD cases (Bottcher et al., 2020).
It is not possible to mimic all the pathological features of human depression in animal models, owing to its multifactorial pathology involving epigenetic and genetic factors, multiple body systems working in conjunction, and subjectivity of symptoms. Yet, these models have provided useful insights into the neuroinflammatory mechanism of depression. Given that the role of neuroinflammation in human depression is yet not clear, the results of in vivo depression studies appear to be missing pieces of the puzzle of depression pathology (Nettis et al., 2021).
Recent literature highlights the crucial role of brain immune cells in depression pathology and any modality that can modulate the activity of these cells or reduce neuroinflammation, thereby bearing the potential to treat depressive symptoms. Supporting this notion, beneficial effects of anti-inflammatory drugs have been observed in MDD patients (Muller et al., 2006; Abbasi et al., 2012; Kobayashi et al., 2013; Majd et al., 2015; Cao et al., 2020; Nettis et al., 2021). Clinical trials using non-steroidal anti-inflammatory drugs in depressed patients have reported promising results, with increased remission rates in patients when used in combination with conventional antidepressant drugs (Abbasi et al., 2012; Cao et al., 2020). A tetracycline antibiotic, minocycline, an inhibitor of microglial inflammatory activation, has also shown promising antidepressant activity in treatment-resistant depression patients (Kobayashi et al., 2013; Nettis et al., 2021). The antidepressant effects of minocycline were independent of changes in peripheral inflammatory biomarkers, reflecting the possible decrease in central inflammation (Nettis et al., 2021).
Mounting evidence also suggests the protective role of targeting neuroinflammation in in vivo models of depression. Pharmacological inhibition of caspase-1, which converts IL-1β to its mature form, alleviated the depressive phenotype in preclinical models by modulating neuroinflammatory pathways and stabilizing the surface expression of glutamate receptors (Li et al., 2018). Microglial activation was inhibited by pharmacological treatment with minocycline in a variety of stress models, reducing the increase of proinflammatory cytokines (Hinwood et al., 2012; Kreisel et al., 2014). Furthermore, minocycline attenuated stress-associated deficits in cognitive memory tasks, including the Morris water maze and Barnes maze, as well as depressive-like and anxiety-like behaviors, such as reduced social interaction, sucrose preference, and open field exploration (Hinwood et al., 2012). Moreover, pharmacological inhibition of microglial ATP-gated purinergic P2X7 receptor, activation of which leads to the maturation of IL-1β, also suppressed the development of depressive behavior in rodents subjected to CUMS (Bhattacharya et al., 2018). Adipose-derived mesenchymal stem cells also produced antidepressant effects by modulating microglial phenotype, suppressing TLR4/NF-κB signaling pathways, and upregulating antioxidant pathways in mice subjected to CUMS (Huang et al., 2020). Anesthetic ketamine, which has antidepressant potential, is also known to suppress inflammatory pathways (Abdallah et al., 2020). It has been recently demonstrated in CUMS model that ketamine suppressed microglial activation and NLRP1 inflammasome pathway, exerting antidepressant effects (Aricioglu et al., 2020). These preclinical studies suggest that targeting neuroinflammation appears to be a promising therapeutic approach.
Owing to complex neurobiology and genetic variability, depression cannot be fully mimicked in animal models. However, many molecular insights can be gained from these models to identify therapeutic targets for depression. The heightened role of neuroinflammatory cascades observed in animal models of depression and the efficacy of anti-inflammatory treatment in decreasing depressive behavior pinpoint the role of neuroinflammation in the neurobiology of depression. Moreover, the inefficacy of classical antidepressant drugs partly explains the unappreciated role of neuroinflammation in depressive disorders and paves a path for targeting neuroinflammation to treat depression.
RA conducted the literature review, formulated, and wrote the manuscript. KS edited the manuscript and was involved in all aspects of manuscript preparation. Both authors contributed to the article and approved the submitted version.
This work was supported by a grant from the Basic Science Research Program through the National Research Foundation (NRF), which was funded by the Korean Government (Ministry of Science, ICT and Future Planning, MSIP; 2017R1A5A2015391 and 2020M3E5D9079764).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbasi, S. H., Hosseini, F., Modabbernia, A., Ashrafi, M., and Akhondzadeh, S. (2012). Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: randomized double-blind placebo-controlled study. J. Affect. Disord. 141, 308–314. doi: 10.1016/j.jad.2012.03.033
Abdallah, C. G., Averill, L. A., Gueorguieva, R., Goktas, S., Purohit, P., Ranganathan, M., et al. (2020). Modulation of the antidepressant effects of ketamine by the mTORC1 inhibitor rapamycin. Neuropsychopharmacology 45, 990–997. doi: 10.1038/s41386-020-0644-9
Altshuler, L. L., Abulseoud, O. A., Foland-Ross, L., Bartzokis, G., Chang, S., Mintz, J., et al. (2010). Amygdala astrocyte reduction in subjects with major depressive disorder but not bipolar disorder. Bipolar Disord. 12, 541–549. doi: 10.1111/j.1399-5618.2010.00838.x
Anacker, C., Cattaneo, A., Musaelyan, K., Zunszain, P. A., Horowitz, M., Molteni, R., et al. (2013). Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc. Natl. Acad. Sci. U.S.A. 110, 8708–8713. doi: 10.1073/pnas.1300886110
Aricioglu, F., Yalcinkaya, C., Ozkartal, C. S., Tuzun, E., Sirvanci, S., Kucukali, C. I., et al. (2020). NLRP1-mediated antidepressant effect of ketamine in chronic unpredictable mild stress model in rats. Psychiatry Investig. 17, 283–291. doi: 10.30773/pi.2019.0189
Arioz, B. I., Tastan, B., Tarakcioglu, E., Tufekci, K. U., Olcum, M., Ersoy, N., et al. (2019). Melatonin attenuates LPS-induced acute depressive-like behaviors and microglial NLRP3 inflammasome activation through the SIRT1/Nrf2 pathway. Front. Immunol. 10:1511. doi: 10.3389/fimmu.2019.01511
Baune, B. T., Dannlowski, U., Domschke, K., Janssen, D. G., Jordan, M. A., Ohrmann, P., et al. (2010). The interleukin 1 beta (IL1B) gene is associated with failure to achieve remission and impaired emotion processing in major depression. Biol. Psychiatry 67, 543–549. doi: 10.1016/j.biopsych.2009.11.004
Bhattacharya, A., Lord, B., Grigoleit, J. S., He, Y., Fraser, I., Campbell, S. N., et al. (2018). Neuropsychopharmacology of JNJ-55308942: evaluation of a clinical candidate targeting P2X7 ion channels in animal models of neuroinflammation and anhedonia. Neuropsychopharmacology 43, 2586–2596. doi: 10.1038/s41386-018-0141-6
Borsini, A., Di Benedetto, M. G., Giacobbe, J., and Pariante, C. M. (2020). Pro- and anti-inflammatory properties of interleukin (IL6) in vitro: relevance for major depression and for human hippocampal neurogenesis. Int. J. Neuropsychopharmacol. 23, 738–750. doi: 10.1093/ijnp/pyaa055
Bottcher, C., Fernandez-Zapata, C., Snijders, G. J. L., Schlickeiser, S., Sneeboer, M. a. M., Kunkel, D., et al. (2020). Single-cell mass cytometry of microglia in major depressive disorder reveals a non-inflammatory phenotype with increased homeostatic marker expression. Transl. Psychiatry 10:310.
Cao, Z. Y., Liu, Y. Z., Li, J. M., Ruan, Y. M., Yan, W. J., Zhong, S. Y., et al. (2020). Glycyrrhizic acid as an adjunctive treatment for depression through anti-inflammation: a randomized placebo-controlled clinical trial. J. Affect. Disord. 265, 247–254. doi: 10.1016/j.jad.2020.01.048
Choudary, P. V., Molnar, M., Evans, S. J., Tomita, H., Li, J. Z., Vawter, M. P., et al. (2005). Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc. Natl. Acad. Sci. U.S.A. 102, 15653–15658. doi: 10.1073/pnas.0507901102
Cobb, J. A., O’neill, K., Milner, J., Mahajan, G. J., Lawrence, T. J., May, W. L., et al. (2016). Density of GFAP-immunoreactive astrocytes is decreased in left hippocampi in major depressive disorder. Neuroscience 316, 209–220. doi: 10.1016/j.neuroscience.2015.12.044
Corona, A. W., Norden, D. M., Skendelas, J. P., Huang, Y., O’connor, J. C., Lawson, M., et al. (2013). Indoleamine 2,3-dioxygenase inhibition attenuates lipopolysaccharide induced persistent microglial activation and depressive-like complications in fractalkine receptor (CX(3)CR1)-deficient mice. Brain Behav. Immun. 31, 134–142. doi: 10.1016/j.bbi.2012.08.008
Cotter, D., Mackay, D., Landau, S., Kerwin, R., and Everall, I. (2001). Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch. Gen. Psychiatry 58, 545–553. doi: 10.1001/archpsyc.58.6.545
de Kloet, E. R., Joels, M., and Holsboer, F. (2005). Stress and the brain: from adaptation to disease. Nat. Rev. Neurosci. 6, 463–475. doi: 10.1038/nrn1683
Dhabhar, F. S., Malarkey, W. B., Neri, E., and Mcewen, B. S. (2012). Stress-induced redistribution of immune cells–from barracks to boulevards to battlefields: a tale of three hormones–Curt Richter Award winner. Psychoneuroendocrinology 37, 1345–1368. doi: 10.1016/j.psyneuen.2012.05.008
DiSabato, D. J., Nemeth, D. P., Liu, X., Witcher, K. G., O’neil, S. M., Oliver, B., et al. (2020). Interleukin-1 receptor on hippocampal neurons drives social withdrawal and cognitive deficits after chronic social stress. Mol. Psychiatry 1–13.
Dudek, K. A., Dion-Albert, L., Lebel, M., Leclair, K., Labrecque, S., Tuck, E., et al. (2020). Molecular adaptations of the blood-brain barrier promote stress resilience vs. depression. Proc. Natl. Acad. Sci. U.S.A. 117, 3326–3336. doi: 10.1073/pnas.1914655117
Duman, R. S., Aghajanian, G. K., Sanacora, G., and Krystal, J. H. (2016). Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat. Med. 22, 238–249. doi: 10.1038/nm.4050
Dwivedi, Y. (2009). Brain-derived neurotrophic factor: role in depression and suicide. Neuropsychiatr. Dis. Treat. 5, 433–449. doi: 10.2147/ndt.s5700
Efstathopoulos, P., Andersson, F., Melas, P. A., Yang, L. L., Villaescusa, J. C., Ruegg, J., et al. (2018). NR3C1 hypermethylation in depressed and bullied adolescents. Transl. Psychiatry 8:121.
Engler, H., Bailey, M. T., Engler, A., and Sheridan, J. F. (2004). Effects of repeated social stress on leukocyte distribution in bone marrow, peripheral blood and spleen. J. Neuroimmunol. 148, 106–115. doi: 10.1016/j.jneuroim.2003.11.011
Ernst, C., Deleva, V., Deng, X., Sequeira, A., Pomarenski, A., Klempan, T., et al. (2009). Alternative splicing, methylation state, and expression profile of tropomyosin-related kinase B in the frontal cortex of suicide completers. Arch. Gen. Psychiatry 66, 22–32. doi: 10.1001/archpsyc.66.1.22
Fatemi, S. H., Laurence, J. A., Araghi-Niknam, M., Stary, J. M., Schulz, S. C., Lee, S., et al. (2004). Glial fibrillary acidic protein is reduced in cerebellum of subjects with major depression, but not schizophrenia. Schizophr. Res. 69, 317–323. doi: 10.1016/j.schres.2003.08.014
Feng, X., Zhao, Y., Yang, T., Song, M., Wang, C., Yao, Y., et al. (2019). Glucocorticoid-driven NLRP3 inflammasome activation in hippocampal microglia mediates chronic stress-induced depressive-like behaviors. Front. Mol. Neurosci. 12:210. doi: 10.3389/fnmol.2019.00210
Gu, Y., Ye, T., Tan, P., Tong, L., Ji, J., Gu, Y., et al. (2021). Tolerance-inducing effect and properties of innate immune stimulation on chronic stress-induced behavioral abnormalities in mice. Brain Behav. Immun. 91, 451–471. doi: 10.1016/j.bbi.2020.11.002
Guo, Y., Xie, J. P., Deng, K., Li, X., Yuan, Y., Xuan, Q., et al. (2019). Prophylactic effects of bifidobacterium adolescentis on anxiety and depression-like phenotypes after chronic stress: a role of the gut microbiota-inflammation axis. Front. Behav. Neurosci. 13:126. doi: 10.3389/fnbeh.2019.00126
Hamidi, M., Drevets, W. C., and Price, J. L. (2004). Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes. Biol. Psychiatry 55, 563–569. doi: 10.1016/j.biopsych.2003.11.006
Himmerich, H., Patsalos, O., Lichtblau, N., Ibrahim, M. a. A., and Dalton, B. (2019). Cytokine research in depression: principles, challenges, and open questions. Front. Psychiatry 10:30. doi: 10.3389/fpsyt.2019.00030
Hinwood, M., Morandini, J., Day, T. A., and Walker, F. R. (2012). Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb. Cortex 22, 1442–1454. doi: 10.1093/cercor/bhr229
Horchar, M. J., and Wohleb, E. S. (2019). Glucocorticoid receptor antagonism prevents microglia-mediated neuronal remodeling and behavioral despair following chronic unpredictable stress. Brain Behav. Immun. 81, 329–340. doi: 10.1016/j.bbi.2019.06.030
Huang, X., Fei, G. Q., Liu, W. J., Ding, J., Wang, Y., Wang, H., et al. (2020). Adipose-derived mesenchymal stem cells protect against CMS-induced depression-like behaviors in mice via regulating the Nrf2/HO-1 and TLR4/NF-kappaB signaling pathways. Acta Pharmacol. Sin. 41, 612–619. doi: 10.1038/s41401-019-0317-6
Jesulola, E., Micalos, P., and Baguley, I. J. (2018). Understanding the pathophysiology of depression: from monoamines to the neurogenesis hypothesis model– are we there yet? Behav Brain Res 341, 79–90. doi: 10.1016/j.bbr.2017.12.025
Jiang, Y., Jakovcevski, M., Bharadwaj, R., Connor, C., Schroeder, F. A., Lin, C. L., et al. (2010). Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J. Neurosci. 30, 7152–7167. doi: 10.1523/jneurosci.1314-10.2010
Jin, J., Kim, S. N., Liu, X., Zhang, H., Zhang, C., Seo, J. S., et al. (2016). miR-17-92 cluster regulates adult hippocampal neurogenesis, anxiety, and depression. Cell Rep. 16, 1653–1663. doi: 10.1016/j.celrep.2016.06.101
Kobayashi, K., Imagama, S., Ohgomori, T., Hirano, K., Uchimura, K., Sakamoto, K., et al. (2013). Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 4:e525. doi: 10.1038/cddis.2013.54
Konig, H., Konig, H. H., and Konnopka, A. (2019). The excess costs of depression: a systematic review and meta-analysis. Epidemiol. Psychiatr. Sci. 29:e30.
Konnopka, A., and Konig, H. (2020). Economic burden of anxiety disorders: a systematic review and meta-analysis. Pharmacoeconomics 38, 25–37. doi: 10.1007/s40273-019-00849-7
Koo, J. W., and Duman, R. S. (2008). IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc. Natl. Acad. Sci. U.S.A. 105, 751–756. doi: 10.1073/pnas.0708092105
Kreisel, T., Frank, M. G., Licht, T., Reshef, R., Ben-Menachem-Zidon, O., Baratta, M. V., et al. (2014). Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol. Psychiatry 19, 699–709. doi: 10.1038/mp.2013.155
Lehmann, M. L., Cooper, H. A., Maric, D., and Herkenham, M. (2016). Social defeat induces depressive-like states and microglial activation without involvement of peripheral macrophages. J. Neuroinflammation 13:224.
Lehmann, M. L., Weigel, T. K., Cooper, H. A., Elkahloun, A. G., Kigar, S. L., and Herkenham, M. (2018). Decoding microglia responses to psychosocial stress reveals blood-brain barrier breakdown that may drive stress susceptibility. Sci. Rep. 8:11240.
Lesch, K. P. (2004). Gene-environment interaction and the genetics of depression. J. Psychiatry Neurosci. 29, 174–184.
Levinson, D. F. (2006). The genetics of depression: a review. Biol. Psychiatry 60, 84–92. doi: 10.1016/j.biopsych.2005.08.024
Li, M. X., Zheng, H. L., Luo, Y., He, J. G., Wang, W., Han, J., et al. (2018). Gene deficiency and pharmacological inhibition of caspase-1 confers resilience to chronic social defeat stress via regulating the stability of surface AMPARs. Mol. Psychiatry 23, 556–568. doi: 10.1038/mp.2017.76
Liu, D., Wang, Z., Liu, S., Wang, F., Zhao, S., and Hao, A. (2011). Anti-inflammatory effects of fluoxetine in lipopolysaccharide(LPS)-stimulated microglial cells. Neuropharmacology 61, 592–599. doi: 10.1016/j.neuropharm.2011.04.033
MacDowell, K. S., Martin-Hernandez, D., Ulecia-Moron, C., Bris, A. G., Madrigal, J. L. M., Garcia-Bueno, B., et al. (2021). Paliperidone attenuates chronic stress-induced changes in the expression of inflammasomes-related protein in the frontal cortex of male rats. Int. Immunopharmacol. 90:107217. doi: 10.1016/j.intimp.2020.107217
Majd, M., Hashemian, F., Hosseini, S. M., Vahdat Shariatpanahi, M., and Sharifi, A. (2015). A randomized, double-blind, placebo-controlled trial of celecoxib augmentation of sertraline in treatment of drug-naive depressed women: a pilot study. Iran J. Pharm. Res. 14, 891–899.
Martinez, J. M., Garakani, A., Yehuda, R., and Gorman, J. M. (2012). Pro-inflammatory and “resiliency” proteins in the CSF of patients with major depression. Depress Anxiety 29, 32–38. doi: 10.1002/da.20876
McEwen, B. S., Nasca, C., and Gray, J. D. (2016). Stress effects on neuronal structure: hippocampus, amygdala, and prefrontal cortex. Neuropsychopharmacology 41, 3–23. doi: 10.1038/npp.2015.171
Menard, C., Pfau, M. L., Hodes, G. E., Kana, V., Wang, V. X., Bouchard, S., et al. (2017). Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 20, 1752–1760. doi: 10.1038/s41593-017-0010-3
Muller, N., Schwarz, M. J., Dehning, S., Douhe, A., Cerovecki, A., Goldstein-Muller, B., et al. (2006). The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol. Psychiatry 11, 680–684. doi: 10.1038/sj.mp.4001805
Nettis, M. A., Lombardo, G., Hastings, C., Zajkowska, Z., Mariani, N., Nikkheslat, N., et al. (2021). Augmentation therapy with minocycline in treatment-resistant depression patients with low-grade peripheral inflammation: results from a double-blind randomised clinical trial. Neuropsychopharmacology 46, 939–948. doi: 10.1038/s41386-020-00948-6
Obuchowicz, E., Kowalski, J., Labuzek, K., Krysiak, R., Pendzich, J., and Herman, Z. S. (2006). Amitriptyline and nortriptyline inhibit interleukin-1 release by rat mixed glial and microglial cell cultures. Int. J. Neuropsychopharmacol. 9, 27–35. doi: 10.1017/s146114570500547x
Otte, C., Gold, S. M., Penninx, B. W., Pariante, C. M., Etkin, A., Fava, M., et al. (2016). Major depressive disorder. Nat. Rev. Dis. Primers 2:16065.
Owen, D. R., Narayan, N., Wells, L., Healy, L., Smyth, E., Rabiner, E. A., et al. (2017). Pro-inflammatory activation of primary microglia and macrophages increases 18 kDa translocator protein expression in rodents but not humans. J. Cereb. Blood Flow Metab. 37, 2679–2690. doi: 10.1177/0271678x17710182
Pan, Y., Chen, X. Y., Zhang, Q. Y., and Kong, L. D. (2014). Microglial NLRP3 inflammasome activation mediates IL-1beta-related inflammation in prefrontal cortex of depressive rats. Brain Behav. Immun. 41, 90–100. doi: 10.1016/j.bbi.2014.04.007
Pittenger, C., and Duman, R. S. (2008). Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 33, 88–109. doi: 10.1038/sj.npp.1301574
Powell, N. D., Sloan, E. K., Bailey, M. T., Arevalo, J. M., Miller, G. E., Chen, E., et al. (2013). Social stress upregulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc. Natl. Acad. Sci. U.S.A. 110, 16574–16579. doi: 10.1073/pnas.1310655110
Ramirez, K., Niraula, A., and Sheridan, J. F. (2016). GABAergic modulation with classical benzodiazepines prevent stress-induced neuro-immune dysregulation and behavioral alterations. Brain Behav. Immun. 51, 154–168. doi: 10.1016/j.bbi.2015.08.011
Schiele, M. A., Zwanzger, P., Schwarte, K., Arolt, V., Baune, B. T., and Domschke, K. (2021). Serotonin transporter gene promoter hypomethylation as a predictor of antidepressant treatment response in major depression: a replication study. Int. J. Neuropsychopharmacol. 24, 191–199. doi: 10.1093/ijnp/pyaa081
Schloesser, R. J., Manji, H. K., and Martinowich, K. (2009). Suppression of adult neurogenesis leads to an increased hypothalamo-pituitary-adrenal axis response. Neuroreport 20, 553–557. doi: 10.1097/wnr.0b013e3283293e59
Serrats, J., Schiltz, J. C., Garcia-Bueno, B., Van Rooijen, N., Reyes, T. M., and Sawchenko, P. E. (2010). Dual roles for perivascular macrophages in immune-to-brain signaling. Neuron 65, 94–106. doi: 10.1016/j.neuron.2009.11.032
Setiawan, E., Wilson, A. A., Mizrahi, R., Rusjan, P. M., Miler, L., Rajkowska, G., et al. (2015). Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 72, 268–275. doi: 10.1001/jamapsychiatry.2014.2427
Shelton, R. C., Claiborne, J., Sidoryk-Wegrzynowicz, M., Reddy, R., Aschner, M., Lewis, D. A., et al. (2011). Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Mol. Psychiatry 16, 751–762. doi: 10.1038/mp.2010.52
Snijders, G. J. L. J., Sneeboer, M. a. M., Fernández-Andreu, A., Udine, E., Psychiatric donor program of the Netherlands Brain Bank (Nbb-Psy), Boks, M. P., et al. (2020). Distinct non-inflammatory signature of microglia in post-mortem brain tissue of patients with major depressive disorder. Mol. Psychiatry 1–14.
Song, A. Q., Gao, B., Fan, J. J., Zhu, Y. J., Zhou, J., Wang, Y. L., et al. (2020). NLRP1 inflammasome contributes to chronic stress-induced depressive-like behaviors in mice. J. Neuroinflammation 17:178.
Steiner, J., Walter, M., Gos, T., Guillemin, G. J., Bernstein, H. G., Sarnyai, Z., et al. (2011). Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J. Neuroinflammation 8:94. doi: 10.1186/1742-2094-8-94
Strawbridge, R., Arnone, D., Danese, A., Papadopoulos, A., Herane Vives, A., and Cleare, A. J. (2015). Inflammation and clinical response to treatment in depression: a meta-analysis. Eur. Neuropsychopharmacol. 25, 1532–1543. doi: 10.1016/j.euroneuro.2015.06.007
Sun, H., Kennedy, P. J., and Nestler, E. J. (2013). Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology 38, 124–137. doi: 10.1038/npp.2012.73
Tang, J., Yu, W., Chen, S., Gao, Z., and Xiao, B. (2018). Microglia polarization and endoplasmic reticulum stress in chronic social defeat stress induced depression mouse. Neurochem. Res. 43, 985–994. doi: 10.1007/s11064-018-2504-0
Troubat, R., Leman, S., Pinchaud, K., Surget, A., Barone, P., Roger, S., et al. (2021). Brain immune cells characterization in UCMS exposed P2X7 Knock-Out mouse. Brain Behav. Immun. 94, 159–174. doi: 10.1016/j.bbi.2021.02.012
Tynan, R. J., Beynon, S. B., Hinwood, M., Johnson, S. J., Nilsson, M., Woods, J. J., et al. (2013). Chronic stress-induced disruption of the astrocyte network is driven by structural atrophy and not loss of astrocytes. Acta Neuropathol. 126, 75–91. doi: 10.1007/s00401-013-1102-0
Wang, Y. L., Han, Q. Q., Gong, W. Q., Pan, D. H., Wang, L. Z., Hu, W., et al. (2018). Microglial activation mediates chronic mild stress-induced depressive- and anxiety-like behavior in adult rats. J. Neuroinflammation 15, 21.
Weber, M. D., Godbout, J. P., and Sheridan, J. F. (2017). Repeated social defeat, neuroinflammation, and behavior: monocytes carry the signal. Neuropsychopharmacology 42, 46–61. doi: 10.1038/npp.2016.102
Weber, M. D., Mckim, D. B., Niraula, A., Witcher, K. G., Yin, W., Sobol, C. G., et al. (2019). The influence of microglial elimination and repopulation on stress sensitization induced by repeated social defeat. Biol. Psychiatry 85, 667–678. doi: 10.1016/j.biopsych.2018.10.009
Wohleb, E. S., Hanke, M. L., Corona, A. W., Powell, N. D., Stiner, L. M., Bailey, M. T., et al. (2011). beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J. Neurosci. 31, 6277–6288. doi: 10.1523/jneurosci.0450-11.2011
Wohleb, E. S., Mckim, D. B., Shea, D. T., Powell, N. D., Tarr, A. J., Sheridan, J. F., et al. (2014). Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol. Psychiatry 75, 970–981. doi: 10.1016/j.biopsych.2013.11.029
Xu, X., Piao, H. N., Aosai, F., Zeng, X. Y., Cheng, J. H., Cui, Y. X., et al. (2020). Arctigenin protects against depression by inhibiting microglial activation and neuroinflammation via HMGB1/TLR4/NF-kappaB and TNF-alpha/TNFR1/NF-kappaB pathways. Br. J. Pharmacol. 177, 5224–5245. doi: 10.1111/bph.15261
Xu, X., Xiao, X., Yan, Y., and Zhang, T. (2021). Activation of Liver X receptors prevents emotional and cognitive dysfunction by suppressing microglial M1-polarization and restoring synaptic plasticity in the hippocampus of mice. Brain Behav. Immun. 94, 111–124. doi: 10.1016/j.bbi.2021.02.026
Yang, P., Gao, Z., Zhang, H., Fang, Z., Wu, C., Xu, H., et al. (2015). Changes in proinflammatory cytokines and white matter in chronically stressed rats. Neuropsychiatr. Dis. Treat. 11, 597–607. doi: 10.2147/ndt.s78131
Zhang, J., He, H., Qiao, Y., Zhou, T., He, H., Yi, S., et al. (2020a). Priming of microglia with IFN-gamma impairs adult hippocampal neurogenesis and leads to depression-like behaviors and cognitive defects. Glia 68, 2674–2692. doi: 10.1002/glia.23878
Zhang, Y., Du, L., Bai, Y., Han, B., He, C., Gong, L., et al. (2020b). CircDYM ameliorates depressive-like behavior by targeting miR-9 to regulate microglial activation via HSP90 ubiquitination. Mol. Psychiatry 25, 1175–1190. doi: 10.1038/s41380-018-0285-0
Zhao, Y., Lin, Z., Chen, L., Ouyang, L., Gu, L., Chen, F., et al. (2018). Hippocampal astrocyte atrophy in a mouse depression model induced by corticosterone is reversed by fluoxetine instead of benzodiazepine diazepam. Prog. Neuropsychopharmacol. Biol. Psychiatry 83, 99–109. doi: 10.1016/j.pnpbp.2018.01.011
Zhu, C. B., Lindler, K. M., Owens, A. W., Daws, L. C., Blakely, R. D., and Hewlett, W. A. (2010). Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology 35, 2510–2520. doi: 10.1038/npp.2010.116
Keywords: glia, depression, neuroinflammation, cytokines, immune cells, experimental models
Citation: Afridi R and Suk K (2021) Neuroinflammatory Basis of Depression: Learning From Experimental Models. Front. Cell. Neurosci. 15:691067. doi: 10.3389/fncel.2021.691067
Received: 05 April 2021; Accepted: 08 June 2021;
Published: 02 July 2021.
Edited by:
Axel Montagne, University of Edinburgh, United KingdomReviewed by:
Alexandre Bonnin, University of Southern California, United StatesCopyright © 2021 Afridi and Suk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kyoungho Suk, a3N1a0BrbnUuYWMua3I=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.