 Bronac Flanagan
Bronac Flanagan Liam McDaid
Liam McDaid John Joseph Wade
John Joseph Wade Marinus Toman
Marinus Toman KongFatt Wong-Lin
KongFatt Wong-Lin Jim Harkin
Jim Harkin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 12 July 2021
Sec. Non-Neuronal Cells
Volume 15 - 2021 | https://doi.org/10.3389/fncel.2021.682460
This article is part of the Research Topic Neurotransmitter transporters: from the biophysics of the transport process to its implications for synapse and circuit function View all 14 articles
Neurotransmitter dynamics within neuronal synapses can be controlled by astrocytes and reflect key contributors to neuronal activity. In particular, Glutamate (Glu) released by activated neurons is predominantly removed from the synaptic space by perisynaptic astrocytic transporters EAAT-2 (GLT-1). In previous work, we showed that the time course of Glu transport is affected by ionic concentration gradients either side of the astrocytic membrane and has the propensity for influencing postsynaptic neuronal excitability. Experimental findings co-localize GABA transporters GAT-3 with EAAT-2 on the perisynaptic astrocytic membrane. While these transporters are unlikely to facilitate the uptake of synaptic GABA, this paper presents simulation results which demonstrate the coupling of EAAT-2 and GAT-3, giving rise to the ionic-dependent reversed transport of GAT-3. The resulting efflux of GABA from the astrocyte to the synaptic space reflects an important astrocytic mechanism for modulation of hyperexcitability. Key results also illustrate an astrocytic-mediated modulation of synaptic neuronal excitation by released GABA at the glutamatergic synapse.
Glutamate (Glu) and γ-aminobutyric acid (GABA) are the brain’s most prevalent excitatory and inhibitory neurotransmitters, respectively (Petroff, 2002). Exposure of neurons expressing appropriate excitatory ionotropic receptors, including N-methyl-D-aspartate receptors (NMDA-Rs) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPA-Rs), to Glu can result in an influx of cations elevating the neuronal membrane potential toward the firing threshold (Meldrum, 2000). Conversely, the exposure of neurons expressing GABA ionotropic receptors (GABAA-Rs) to GABA can result in a hyperpolarising current, decreasing the neuronal membrane potential away from the firing threshold (Sigel and Steinmann, 2012). As the metabolism of Glu and GABA is an intracellular process, these neurotransmitters must be rapidly removed from the extracellular space (ECS) by their corresponding transporters, to avoid the over-exposure of the agonist to the excitatory and inhibitory ionotropic receptors (Petroff, 2002). Broadly speaking on a network level, a balance between excitation and inhibition is necessary for normal brain activity (Fellin et al., 2006). Moreover, imbalance of excitatory/inhibitory transmission is believed to underlie such conditions as epilepsy (Clasadonte and Haydon, 2012; Coulter and Steinhäuser, 2015), autism spectrum disorders (Pizzarelli and Cherubini, 2011), schizophrenia (Coyle, 2004) and Alzheimer’s disease (Robinson, 2000). In particular, studies have indicated altered Glu and GABA concentrations within focal seizure sites (During and Spencer, 1993; Petroff, 2002), favoring excitatory neuronal behavior over inhibition.
In previous work (Flanagan et al., 2018) the effects of synaptic glutamatergic dynamics on postsynaptic firing were explored, where it was found that an increased astrocytic Glu content was sufficient to slow synaptic Glu clearance due to reduced driving force across the excitatory amino-acid transporter 2 (EAAT-2, homologue of GLT-1). Altered sodium (Na+) and calcium (Ca2+) dynamics within the astrocyte were also found and are attributed to variations in the EAAT-2 currents.
A strong extracellular-to-intracellular Na+ concentration gradient is imperative for a range of homeostatic functions, including neurotransmitter transport (Kirischuk et al., 2012; Verkhratsky and Nedergaard, 2018). The influx of Glu across the astrocytic membrane, against a large (∼106 times) concentration gradient, by EAAT-2 requires the concerted transport of 3Na+ and 1H+ and counter-transport of 1K+ for each Glu ion (Zerangue and Kavanaugh, 1996). Due to its Na+-dependence, the reversal potential of the EAAT-2 lies well above the astrocytic resting membrane potential of ∼−80 mV (Verkhratsky and Nedergaard, 2018), ensuring the astrocytic influx of Glu upon synaptic Glu release.
In contrast, the reversal potential of GABA transporter type-3 (GAT-3) approximates to the astrocytic membrane potential and is also dependent on co-transport substrate (Na+ and Cl–) concentrations at equilibrium. Where the clearance of synaptic-released Glu appears as a predominantly astrocytic function (Danbolt, 2001), synaptic-released GABA is mostly retaken by the releasing neuron and subsequently recycled into vesicles (Hertz et al., 1999; Schousboe et al., 2014). As the GABA concentration in the ECS close to the astrocyte would be unlikely to increase based on this synaptic self-recovery of neurotransmitter, the direction of GABA transport by GAT-3 transporter is highly sensitive to fluctuations in astrocytic and extracellular ionic concentrations. In other words, the development of an astrocytic [Na+] microdomain (Breslin et al., 2018) may be sufficient to prompt the release of GABA into the ECS by disturbing the electrochemical potential of the transporter and eliciting an efflux of transporter substrates from the astrocyte. In particular, EAAT-2 activation has been observed experimentally to initiate the GAT-3-mediated release of GABA (Héja et al., 2012), believed to modulate tonic neuronal inhibition through the action of GABAA-Rs (Rossi et al., 2003; Farrant and Nusser, 2005; Héja et al., 2012). Considering this observation, the co-localization of the major Glu and GABA transporters, EAAT-2 and GAT-3, respectively, on the astrocytic membrane (Minelli et al., 1996; Héja et al., 2012; Kirischuk et al., 2012) may indicate a finely balanced excitatory-inhibitory mechanism: the uptake of Glu coupled to the astrocytic release of cytoplasmic GABA (Héja et al., 2012).
Traditionally, the classification of neuronal synapses was determined by the presynaptic neuron-released neurotransmitter (O’Rourke et al., 2012), for example, the presynaptic neuron of a glutamatergic synapse would release Glu by activity-induced exocytosis (Kandel et al., 2012). Consequently, computational models of neuron-astrocyte synapses consider neuronal and astrocytic activity as a function of neuronal-released neurotransmitter (Bentzen et al., 2009; Allam et al., 2012; Tewari and Majumdar, 2012; Li et al., 2016; Hübel et al., 2017). The neurotransmitter almost exclusively modeled at the tripartite synapse is Glu (Manninen et al., 2018), and the possibility of a secondary neurotransmitter at the same synapse has hitherto been ignored. The significance and novelty of this paper lies in the fact that it explores the astrocyte-mediated symbiosis of two neurotransmitters, Glu and GABA, at the glutamatergic synapse with a view to creating a more complete view of ionic dynamics at the tripartite synapses.
Furthermore, this paper considers the electrochemical potentials of both EAAT-2 and GAT-3 proteins with a view to (a) explain experimentally observed phenomena, (b) explore the effectiveness of this balance where astrocytic Glu concentrations are elevated and (c) predict the effects of this balance for postsynaptic neuron activity.
To consider the effects of synaptic neurotransmitter fluxes, namely the presynaptic and postsynaptic neuronal synaptic-driven dynamics, the model in Figure 1 was considered. The model extends a previously developed framework (Flanagan et al., 2018) to characterize tripartite synapse signaling, to include GABAergic signaling as well. A pulsed depolarizing current of 5 μA/cm2 was applied for 50 s to the presynaptic neuronal membrane, generating a presynaptic neuronal 10 Hz firing rate. Consideration was also given to the influence of astrocytic Glu content, in line with previous simulation (Flanagan et al., 2018), specifically in the transport of Glu across the astrocytic membrane. To achieve this, the basal astrocytic [Glu] of 1.5 mM and 5 mM were chosen to represent the bounds of the physiological range (Attwell et al., 1993) and the basal astrocytic [Glu] of 10 mM hypothesized pathological state (Flanagan et al., 2018), following glutamine synthetase downregulation (Eid et al., 2008; Perez et al., 2012). At each neuronal spike, Glu and K+ were released from the presynaptic neuron into the synaptic cleft. The model was also simulated with the exclusion of GAT-3 to act as a control in determining the role of EAAT-2-induced GAT-3 transport at the neuronal synapse. The key ions considered were Na+, K+, Glu, Ca2+ and GABA.
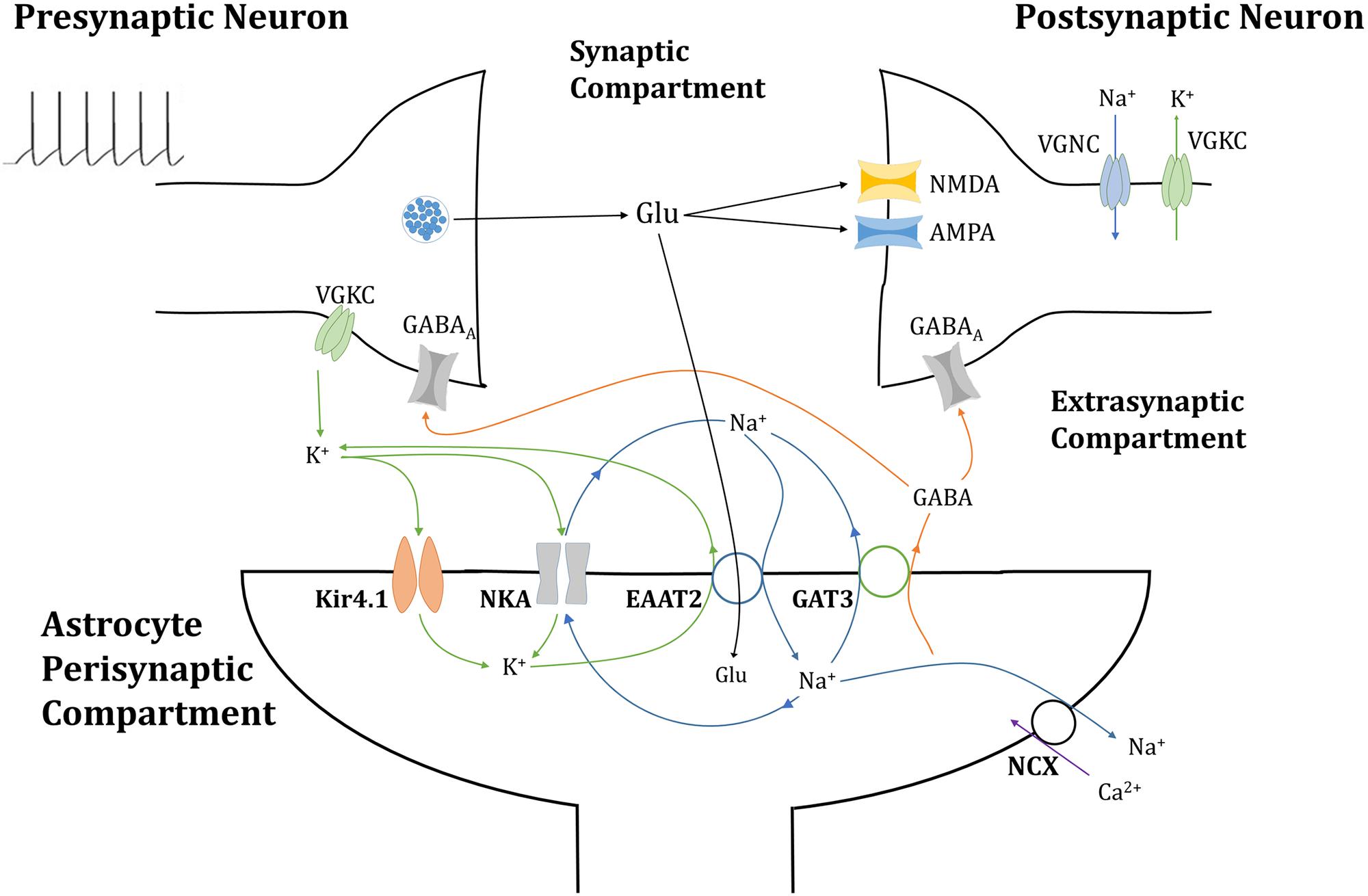
Figure 1. Tripartite glutamatergic synapse compartment model: consisting of neuronal, synaptic, and astrocytic compartments, within which ionic concentrations are dynamic and neuronal membrane currents respond to neurotransmitter-mediated ionotropic currents (not to scale). Glutamate (Glu) neurotransmitter is released into the synapse by the presynaptic neuron in a membrane potential-dependent mechanism, where it activates neurotransmitter-gated ionotropic receptors NMDA and AMPA channels located on the postsynaptic neuron. This postsynaptic membrane activity triggers the activation of voltage-gated sodium (Na+) and potassium (K+) channels (VGNC and VGKC, respectively), also located on the postsynaptic neuronal membrane. Synaptic Glu is exclusively taken up by astrocytic membrane-bound excitatory amino acid transporter 2 (EAAT2), along with synaptic Na+ and the counter-transport of astrocytic K+. The influx of Na+ into the astrocytic compartment triggers the release of GABA (and Na+) through GABA transporter type 3 (GAT3), into the synaptic compartment, where it activated inhibitory ionotropic GABAA receptors located both on the pre- and postsynaptic neuronal membranes. Synaptic K+ transport is completed with the inclusion of inwardly rectifying K+ channels (Kir4.1) and the Na+/K+ ATPase transporter (NKA). Na+ dynamics are completed by the NKA and reversible Na+/Ca2+ exchanger (NCX).
The presynaptic neuron is modeled using a Hodgkin-Huxley-based (Hodgkin and Huxley, 1952; Golomb et al., 2006) description for voltage-gated Na+ and K+ dynamics. The presynaptic membrane potential uses the formalism.
where INa,Preneuron, IK,Preneuron and IL,Preneuron reflect voltage-gated Na+, K+ and leak presynaptic currents, respectively, and are described in Table 1 with parameters enumerated in Table 2. IPreGABAA is the GABAA mediated current in response to synaptic astrocyte-released GABA, see below, and Iapp is an applied stimulus.

Table 1. Neuron membrane dynamics.
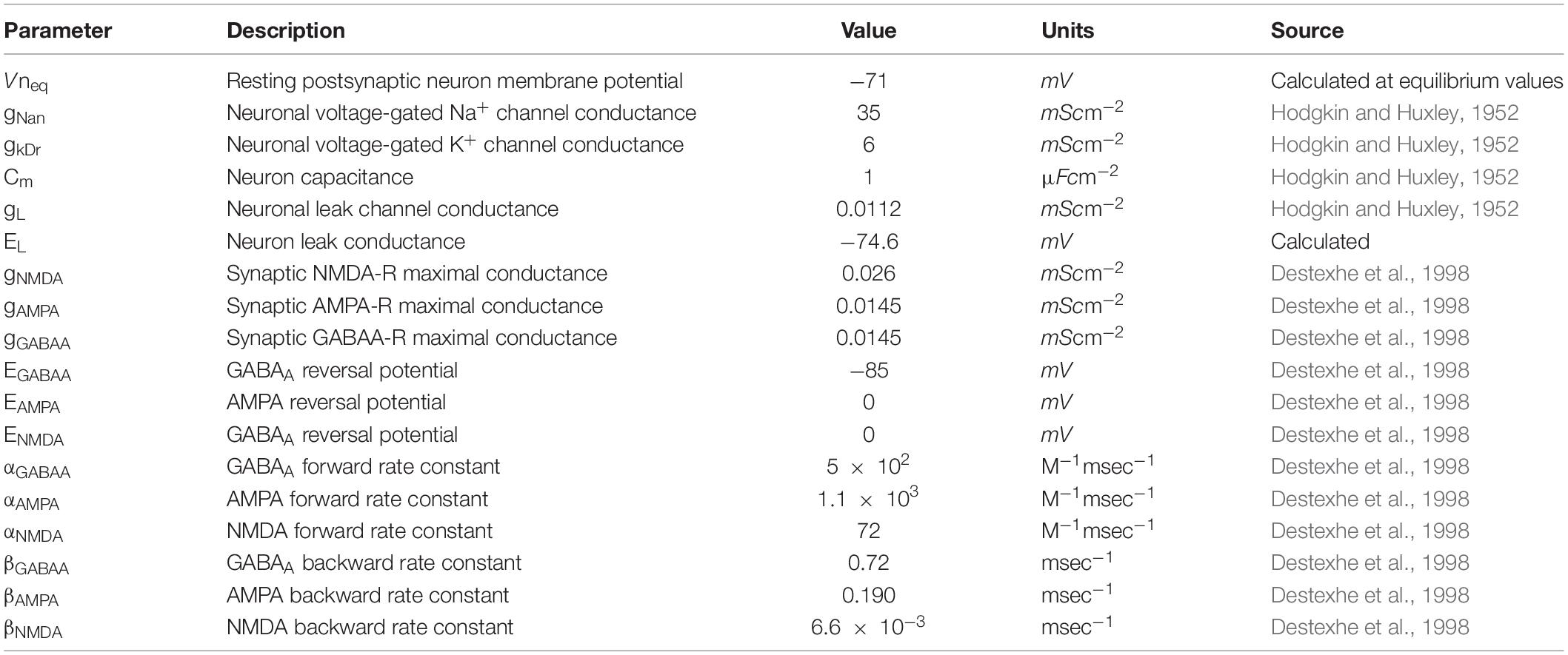
Table 2. Neuronal membrane model parameters.
To improve upon previous work (Flanagan et al., 2018), a more realistic description of synaptic resources was accounted for to preserve biological realism. To this end, the model adopts the Tsodyks-Markram model (Tsodyks et al., 1998) description for the use of synaptic resources within a facilitating synapse, so that any alteration of the neuronal activity would be directly influenced by the GABA-induced inhibition rather than the nature of the synapse model. This model describes the fraction of recovered resources (x), active resources (y) and inactive resources (z) using the model scheme
using parameters detailed in Table 3.
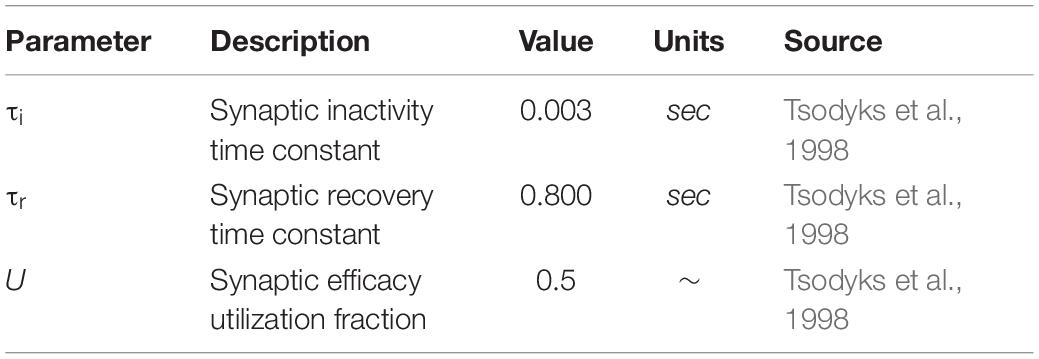
Table 3. Presynaptic resource model parameters.
In this model the amount of Glu released by the presynaptic neuron is proposed to be proportional to the fraction of active resources (Eq. 3) is scaled by constant parameter of 0.1 mM, chosen to sufficiently perturb the system under all cases, namely to excite the postsynaptic membrane to its firing threshold. At each presynaptic neuronal spike, Glu is released by the presynaptic neuron into the synaptic compartment, along with a small amount of K+ representing the input to the system.
Astrocytic membrane ionic currents, subject to changes in ionic concentrations (Glu, K+, Na+, Ca2+ and GABA), are then calculated using the following equations
within this scheme IEAAT, INKA, INCX, IGAT and IKir denote EAAT-2, NKA, Na+-Ca2+ exchanger (NCX), GAT-3 and inwardly rectifying K+ channel (Kir4.1) generated currents, respectively. The inclusion of Na+, K+, Glu, and Ca2+ leak currents, denoted INa,L, IK,L, IGlu,L, and ICaL, L respectively, provide model stability. Each transport current is calculated using the existing concentration of its corresponding substrate(s), the equations of which are found in Table 4 and parameters used in the model are contained in Table 5. To study the effects of these currents on concentrations alone, the membrane potential of the astrocyte is set to be constant.
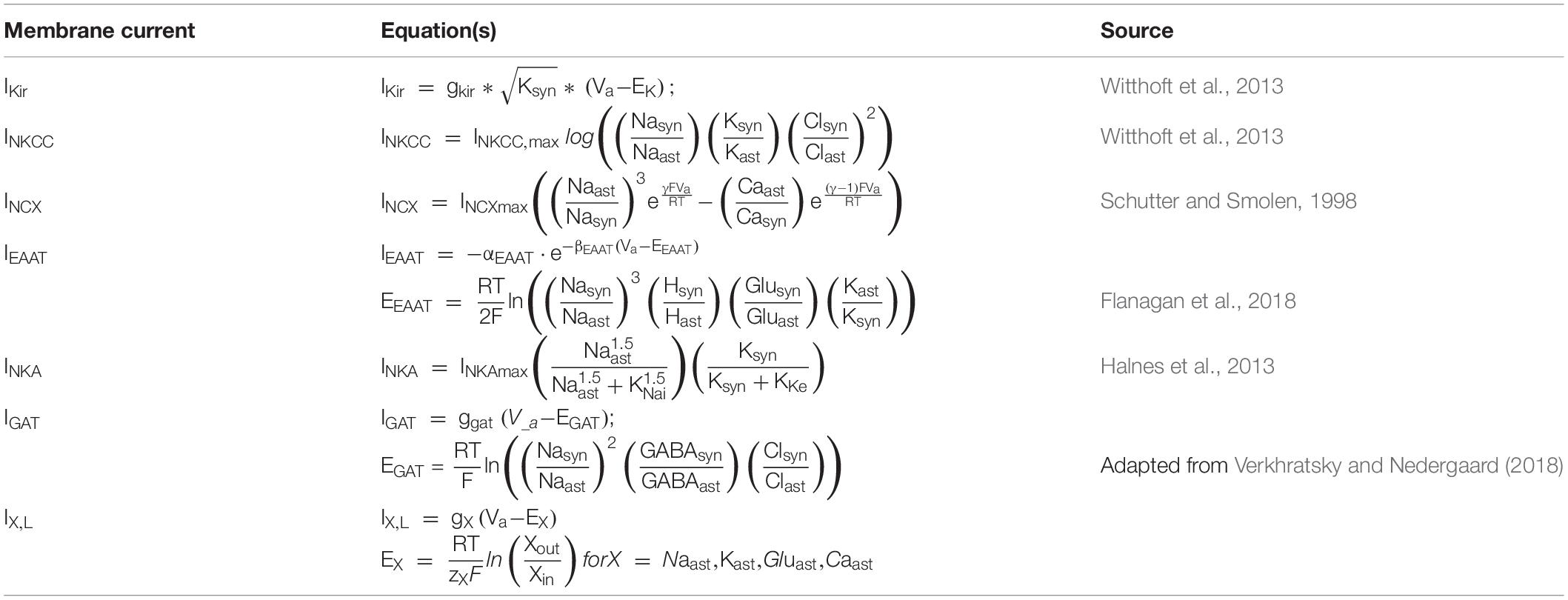
Table 4. Astrocyte membrane transporter equations.
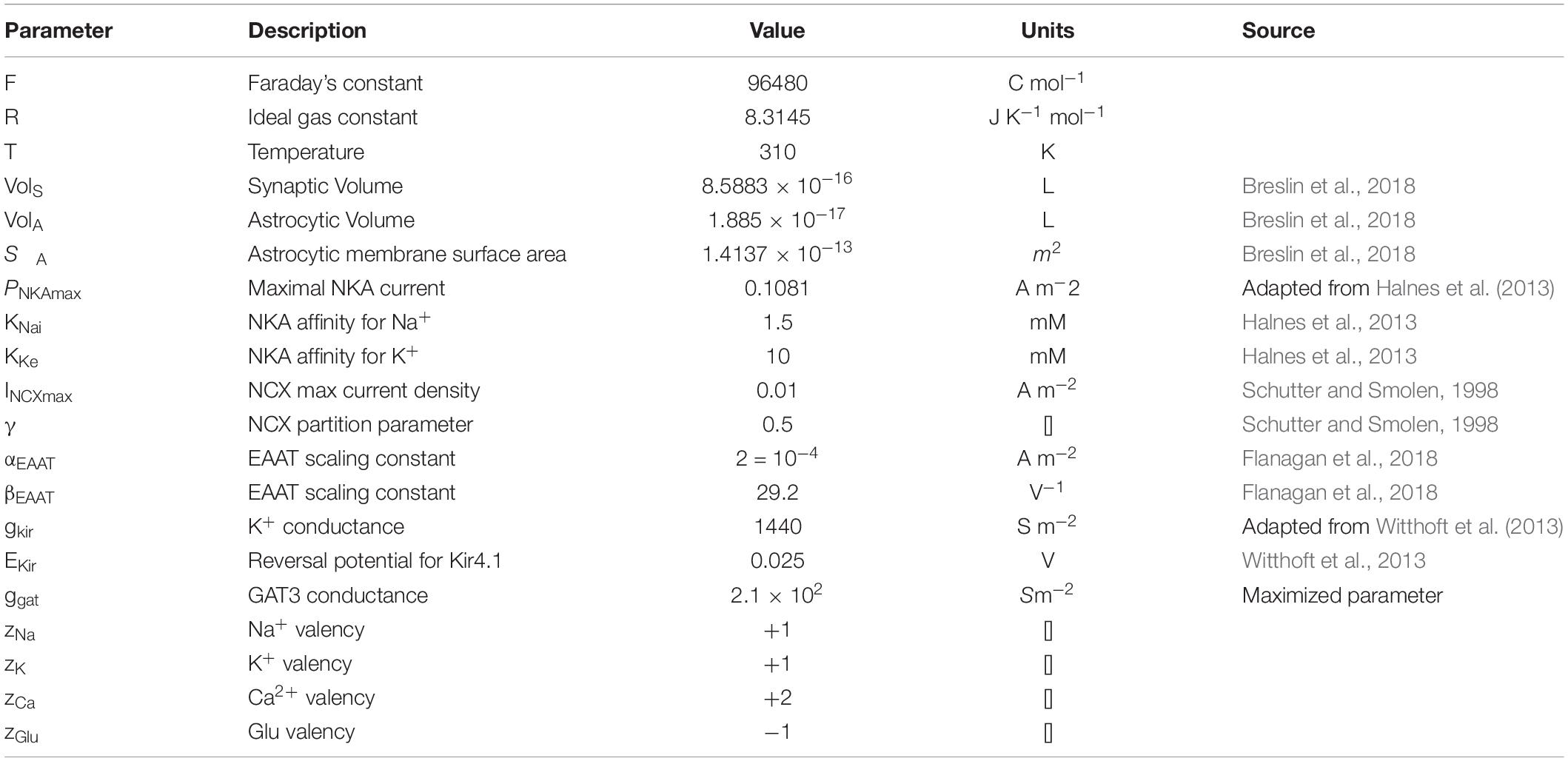
Table 5. Astrocytic membrane transporter parameters.
Currents are converted to ionic fluxes by Faraday’s law, where the change in the astrocytic concentration of ion X is given by
and corresponding change in synaptic concentration given by
using the surface area of the peri-synaptic astrocytic membrane (SA) and volume of astrocyte (VolA) and synaptic compartments (VolS) as parameters.
Synaptic Glu and GABA concentrations are also used to calculated local postsynaptic neuronal membrane dynamics, the neurotransmitters activating corresponding receptors on the postsynaptic terminal, the equations for which are found in Table 1 and the parameters used in Table 2.
The localized effect of these neurotransmitter-driven currents on the postsynaptic neuron are also calculated using a Hodgkin-Huxley-based (Hodgkin and Huxley, 1952; Golomb et al., 2006) description, with the change in the postsynaptic neuron membrane potential (Vm) is thus calculated as the (negative) sum of intrinsic voltage-gated Na+ and K+ currents, a leak current and synaptic NMDA, AMPA and GABAA mediated currents (Destexhe et al., 1998) as given by
The simulation uses the forward Euler numerical integration scheme with 0.01 ms time step using MATLAB R2017b. Each time step of the model was considered in three separate settings, where only the initial astrocytic Glu concentration differs, i.e., [Glu]ast,eq = 1.5, 5, and 10 mM and leak conductances are adjusted accordingly so that equilibrium conditions are met initially.
Results are split into two sections: the first considers the resulting ionic changes due to the simulation while the second considers the effects of the neurotransmitter dynamics on the pre- and postsynaptic neuronal membrane. As astrocytes are considered the main controller of ionic homeostasis, this model only considers changes in ionic concentrations due to astrocytic membrane-mediated currents.
In agreement with previously reported findings (Breslin et al., 2018; Flanagan et al., 2018), the activation of EAAT-2 transporter by neuronal-released Glu was sufficient to generate an astrocytic influx of Na+ within the model containing GAT-3 (Figure 2A). This resulted in a decrease in synaptic [Na+] (Figure 2A.i) and a corresponding increase in astrocytic [Na+] (Figure 2A.ii). The simulation was repeated without GAT-3, as a control, and found similar, yet exaggerated results (Figures 2B.i,ii). This is expected from the dual effect of decreased EAAT-2-mediated influx, resulting from reduced presynaptic neuronal activity, and the Na+-dependent efflux of Na+ through GAT-3. [K+] increased in the synaptic compartment (Figures 2A.iii,B.iii) with neuronal activity and correspondingly decreased in the astrocytic compartment (Figures 2A.iv,B.iv). Considering this inverse behavior between intracellular and extracellular concentration change, these results suggest that the astrocytic efflux of K+ by EAAT-2 dominates over the influx of K+ by the Na+/K+ ATPase (NKA), with a net increase of [K+] in the cleft. However, the rate of change of [K+] in both compartments was higher where the model did not include GAT-3 (Figures 2B.iii,iv) because the NKA is sensitive to astrocytic [Na+] and therefore in the absence of GAT-3 a heightened NKA activity results. This generated a more pronounced change in [K+] in both compartments. Due to the fact this is not a closed system, as K+ has been injected due to the presynaptic activity, the concentration levels do not return to baseline following the simulation.
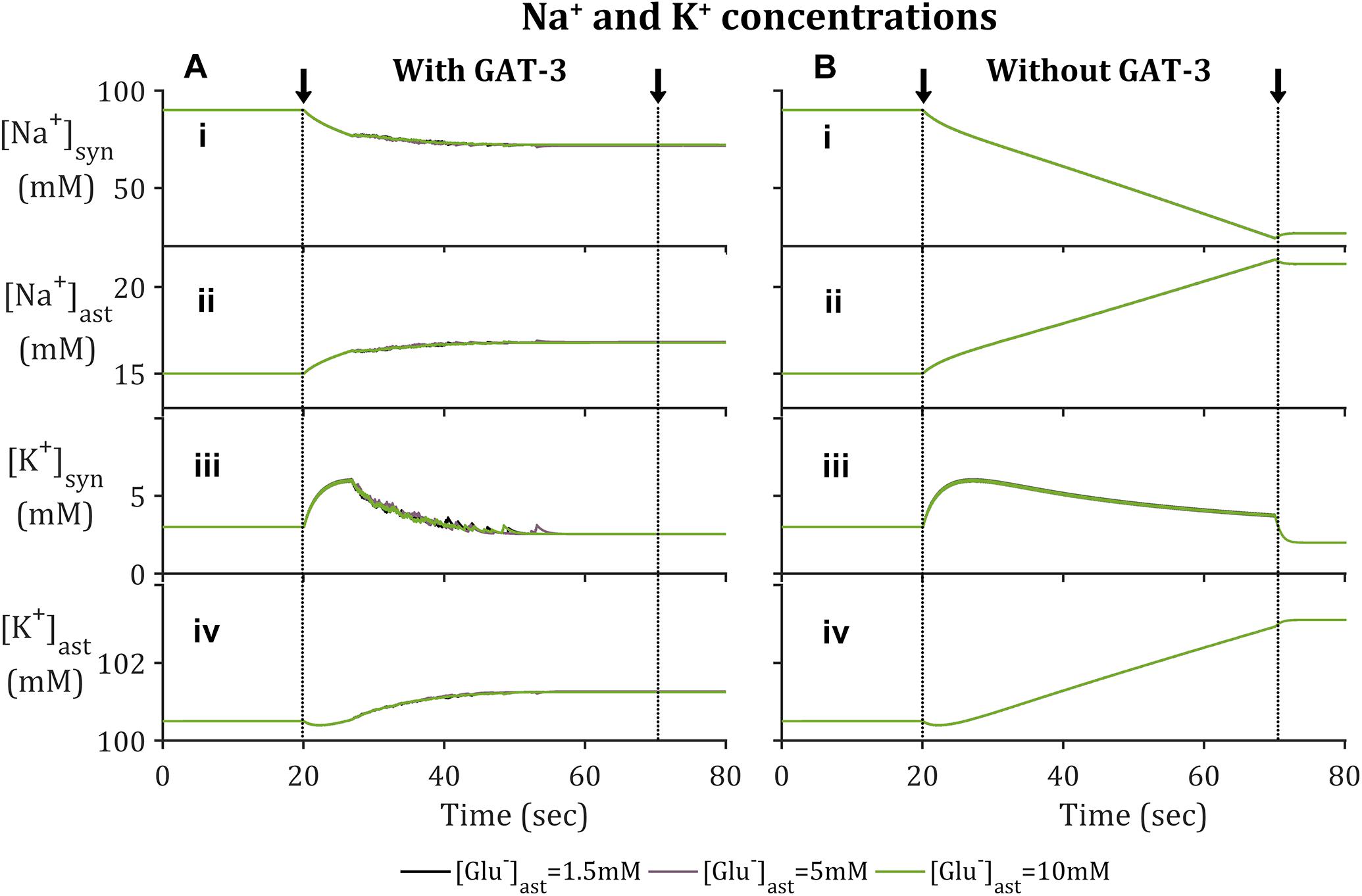
Figure 2. Astrocytic and synaptic concentrations of Na+ and K+ where the model (A) includes GAT-3 activity and (B) where GAT-3 activity is not included; (i–iv) describes (respectively) synaptic Na+, astrocytic [Na+], synaptic [K+] and astrocytic [K+]. The results show the ionic concentrations of a simulation in which the presynaptic neuron is subject to an applied current, pulsed at a frequency of 10 Hz over a 50 s window starting at 20 s (window of applied current given by arrows and dashed vertical lines).
The reversal potential of GAT-3 (EGAT) is heavily dependent on the [Na+] gradient across the astrocytic membrane and, at equilibrium conditions, is close to parity with the astrocytic membrane potential (Verkhratsky and Nedergaard, 2018). This indicates that the direction of its mediated ionic fluxes is highly sensitive to any change in [Na+]. The reversible nature of the transporter is demonstrated in Figure 4B, where a reduction in the transmembrane [Na+] gradient is sufficient to reduce EGAT to below the astrocytic membrane potential, facilitating the release of its substrates, GABA, Na+ and Cl– (not included in this model). Note that the recovery rate of EGAT following activation is markedly faster where astrocytic Glu is lower, which can be attributed to the correlation between EAAT-2 activity and astrocytic [Glu] (Flanagan et al., 2018). EAAT-2 activity increased as astrocytic Glu decreased and thus EAAT-2- mediated influx of Na+ resulted in heightened NKA activity. This increase in transmembrane currents resulted in a faster recovery of EGAT.
As expected, the inclusion of a GAT-3 transporter restricted astrocytic [Na+] as the concentration dependent GAT-3 reversal potential dropped below the astrocytic membrane potential, resulting in the net efflux of Na+ through this transporter. Little difference was recorded in comparison between simulation setups regarding basal astrocytic [Glu], due to the relative size of the Glu-mediated fluxes compared to the magnitude of the ionic concentrations.
As with GAT-3 transport, the rate of Glu transport by astrocytic EAAT-2 is largely dependent on the transmembrane [Na+] gradient (Zerangue and Kavanaugh, 1996; Levy et al., 1998) in addition to the Glu concentration gradient across the astrocytic membrane (Flanagan et al., 2018). In support of previously presented results (Flanagan et al., 2018), increased astrocytic Glu content promotes a longer rate of clearance and higher concentration attainment of synaptic Glu (Figure 3A.i), despite the fraction of active synaptic resources (Figure 3A.ii) being identical in all cases. This rate of clearance is increased further if GAT-3 is not included (Figure 3B) as a result of the heightened shift in [Na+] and [K+] transmembrane gradients (Figure 2).
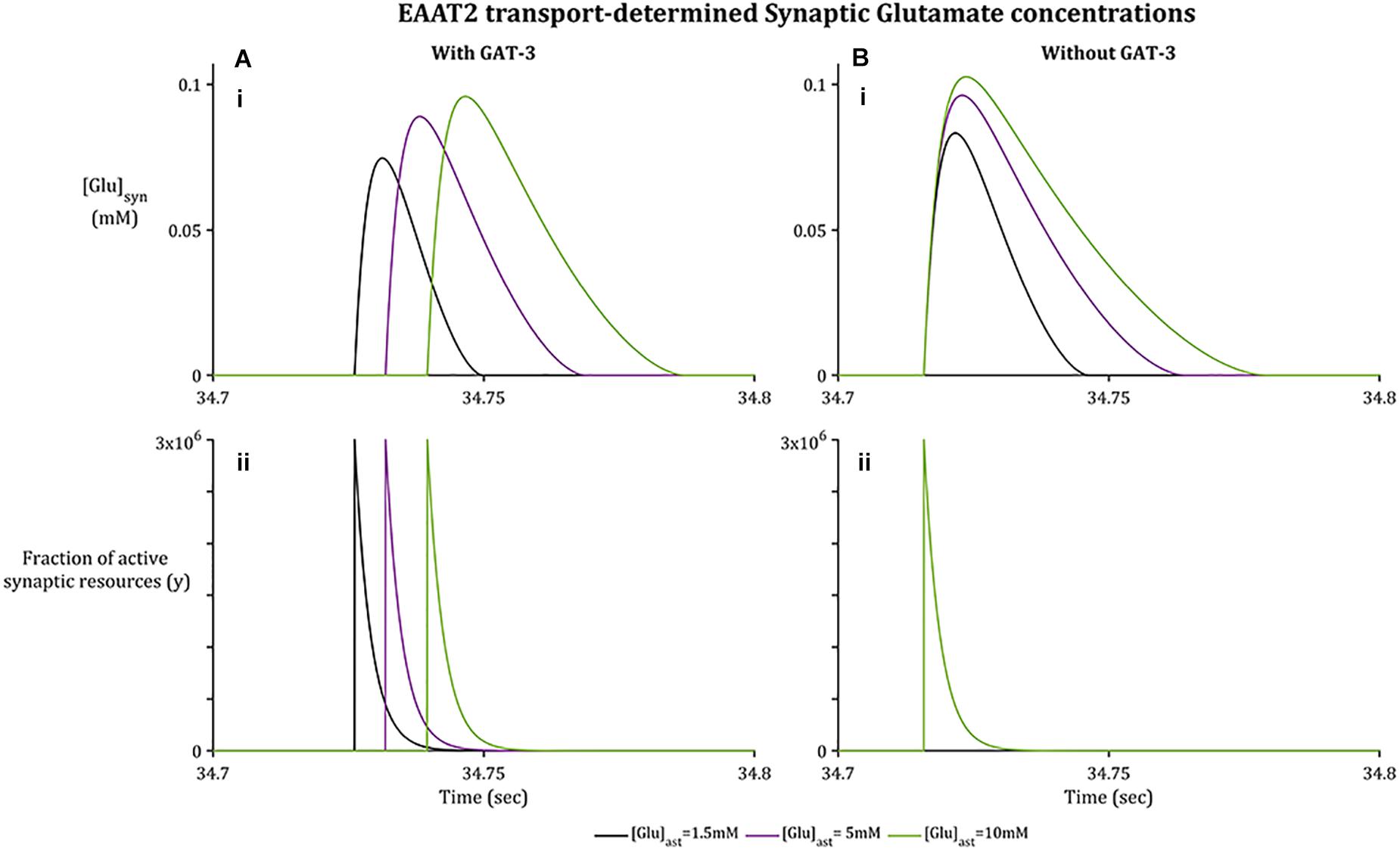
Figure 3. Synaptic [Glu] dynamics for a 0.1 s window within the simulation associated with basal [Glu]ast.(A.i) shows [Glu]syn increasing to a higher concentration where the basal [Glu]ast is higher, despite the fraction of active synaptic resources (A.ii) being identical in all cases. Within (B.i), [Glu]syn increases to a higher concentration again for all [Glu]ast where the GAT3-mediated transport is not included, again despite an identical fraction of active resources (B.ii) being released by the presynaptic neuron.
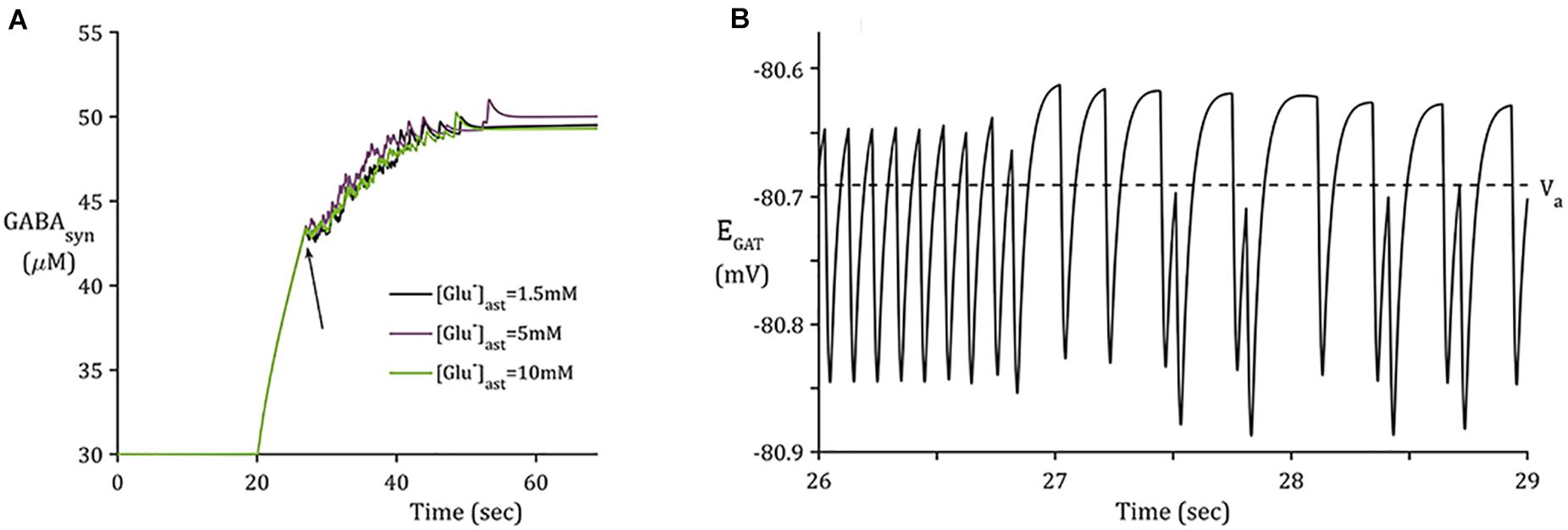
Figure 4. Synaptic GABA dynamics: (A) GABA concentration due to GAT-3 activity (B) 3-s window of GAT-3 reversal potential for [Glu]ast = 1.5 mM (initial time of window indicated by arrow in panel (A).
In contrast to the sharp increase of [Glu] (Figure 3A), as implicitly described by neuronal exocytosis, the rate of GABA release by reversed GAT-3 transport is much slower (Figure 4), increasing in line with neuronal activity and decreasing slowly as the reversal potential increases above the astrocytic membrane potential. The slow time course of GAT-3 mediated GABA release describes the tonic inhibition described by Rossi et al. (2003); Farrant and Nusser (2005), and Héja et al. (2012). As GAT-3 is predominantly controlled by [Na+] gradients, and from Figures 2A.i,ii, 4, it can be seen than these differ little due to basal astrocytic [Glu], little difference can be seen in the GAT-3-mediated synaptic [GABA] (Figure 4A). Note that the flux of GABA fluctuates a little in line with astrocytic [Na+], at ∼26 s in the simulation these fluctuations become more pronounced due to the slowing of presynaptic neuronal activity (Figure 5) as the GAT-3-mediated efflux of Na+ attempts to correct the equilibrium concentration gradient (Figure 4B). Following the termination of presynaptic firing and Glu-mediated ionic currents, the transmembrane [Na+] gradient stabilizes (Figures 2A.i,ii), resulting in no net release of GABA (Figure 4A).
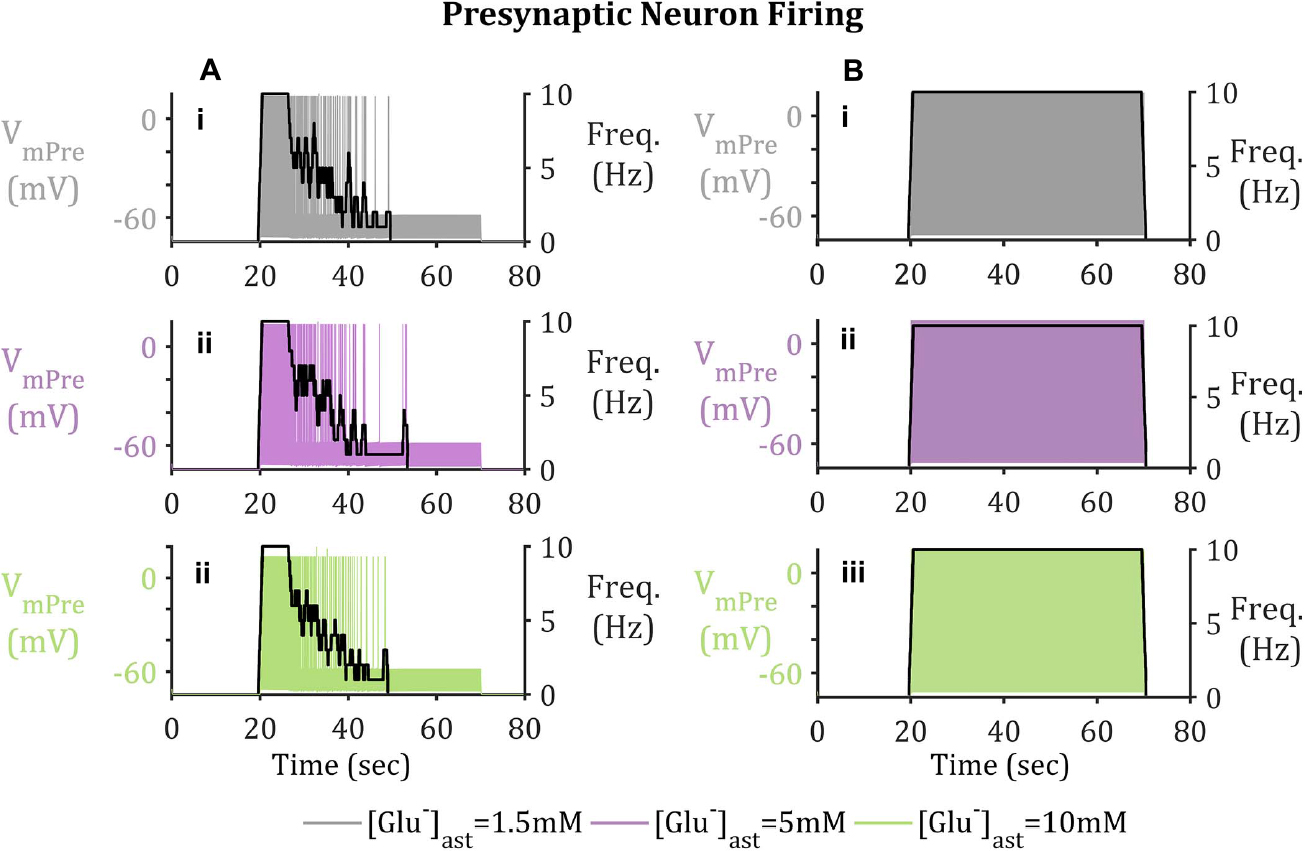
Figure 5. Presynaptic Neuron Membrane Activity (A) with and (B) without GAT-3-mediated GABA release. Upon activation of the presynaptic neuron by means of a pulsed applied current, Glu released into the synapse triggers the activation of EAAT-2 currents, located on the astrocytic membrane. EAAT-2 transports both Glu and Na+ into the astrocyte disturbing the equilibrium state of GAT-3, resulting in the release of GABA back to the presynaptic neuron, where upon binding to presynaptic membrane-bound GABAA-Rs inhibits the neuron, even in the presence of a pulsed applied current. (i–iii) shows presynaptic neuron membrane potential, overlaid with presynaptic neuron firing frequency (given in black with the right-hand y-axis). Results describe basal (i) [Glu]ast = 1.5 mM, (ii) [Glu]ast = 5 mM, and (iii) [Glu]ast = 10 mM.
In order to model the longer-term neuronal effects of EAAT-2-GAT-3 coupling, a similar simulation to a previously published study (Flanagan et al., 2018) was performed. The major differences between the former and latter models being the inclusion of GAT-3 transport and more realistic presynaptic firing activity. The presynaptic neuronal membrane dynamics were modeled using a Hodgkin-Huxley formalism. Within this model a pulsed periodic current of 5 μA/cm2 was applied, sufficient to initiate a 10 Hz presynaptic neuronal firing for 50 s. In addition to the applied current, the presynaptic neuron is exposed to inhibitory currents mediated by synaptic GABA-activating GABAA receptors.
From Figure 5, the current applied to the presynaptic neuron results in an initial firing frequency of ∼10 Hz. Where synaptic GABA is released by the astrocytic GAT-3 (Figure 4) and subsequently activates presynaptic GABAA-Rs; GABAA-mediated currents then compete with the simulated applied current to generate subthreshold presynaptic potentials (Figure 5A.i), reducing the presynaptic firing frequency (Figure 5B.i). In contrast, if no GABA is released by astrocytic GAT-3, the presynaptic neuronal firing persists (Figure 5.ii).
Besides GABAA mediated currents, the postsynaptic terminal is subject to Glu-mediated activation of NMDA and AMPA receptors. Higher frequency firing was observed in the postsynaptic neuron (Figures 6B.i,ii) where the model does not include GAT-3 activity in comparison to the model containing GAT-3 activity (Figures 6A.i,ii). We attribute this to the dual effect of a prolonged synaptic Glu time course and over-activation of NMDA and AMPA-Rs (Flanagan et al., 2018), coupled with the exclusion of GABA-mediated inhibitory currents. Postsynaptic neuronal excitability has been shown to be correlated to time course of synaptic Glu (Flanagan et al., 2018) through activation of ionotropic NMDA and AMPA-Rs. Thus, as basal astrocytic [Glu] increased, time course of synaptic Glu increased (Figure 4) increasing the postsynaptic firing frequency (Figure 6) in both cases (with and without GAT-3).
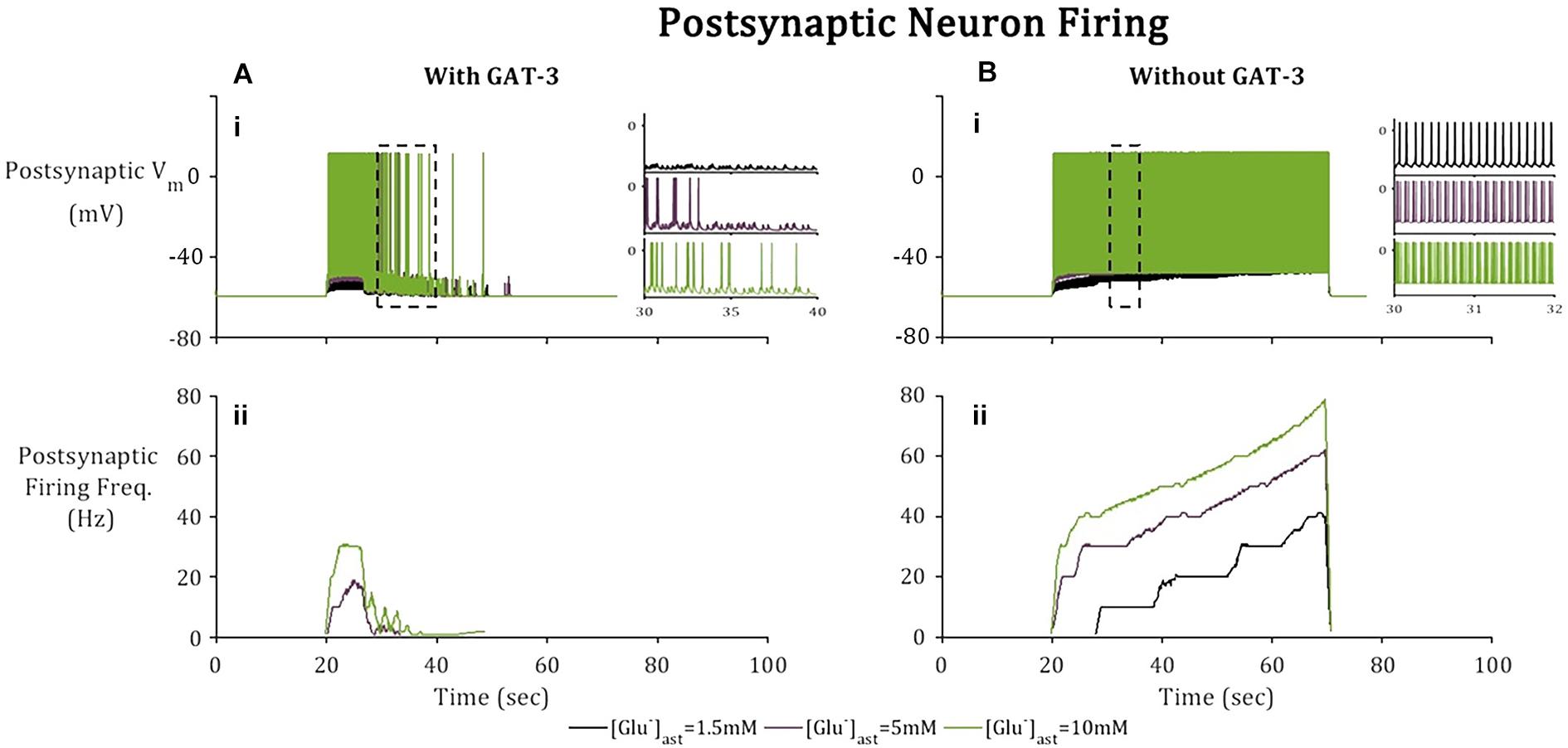
Figure 6. Postsynaptic neuronal activity in model simulation across 3 paradigms, [Glu]ast = 1.5, 5, and 10 mM. Postsynaptic neuronal membrane is subject to (A) NMDA, AMPA, and GABAA mediated currents with inclusion of astrocytic GAT-3-mediated GABA transport and (B) where the GAT-3-mediated GABA release has been omitted. (i) Postsynaptic neuron membrane potential and (ii) firing frequency as a result of an applied current (pulsed at a frequency of 10 Hz over a 50 s window starting at 20 s). All graphs depict firing frequency calculated using a moving average using window size 1 s, 10% overlap). Inset Figs describe 10 s window (left) for each of the basal [Glu]ast and 2 s window (right) for each basal [Glu]ast, where the time window was selected according to extent of neuronal activity.
To consider the effects of astrocytic GAT-3 mediated release of GABA for postsynaptic neuronal activity, three cases were considered; (i) where GABA activates GABAA-mediated inhibitory currents located on both pre- and postsynaptic neuronal membranes (as above) (ii) where GABA activates GABAA mediated inhibitory currents located on the postsynaptic neuronal membrane alone and (iii) where GAT-3 is not included in the model, as a control. Figure 7 illustrates the simulation results using the setup described for each of the basal astrocytic [Glu] of 1.5, 5, and 10 mM. Where astrocytic [Glu] is low, the inclusion of GABA dynamics results in sub- threshold postsynaptic potentials only and thus the firing is completely suppressed (Figure 7A) in comparison to where GAT-3 is excluded from the model and thus GABA dynamics and GABA-mediated inhibition are ignored.
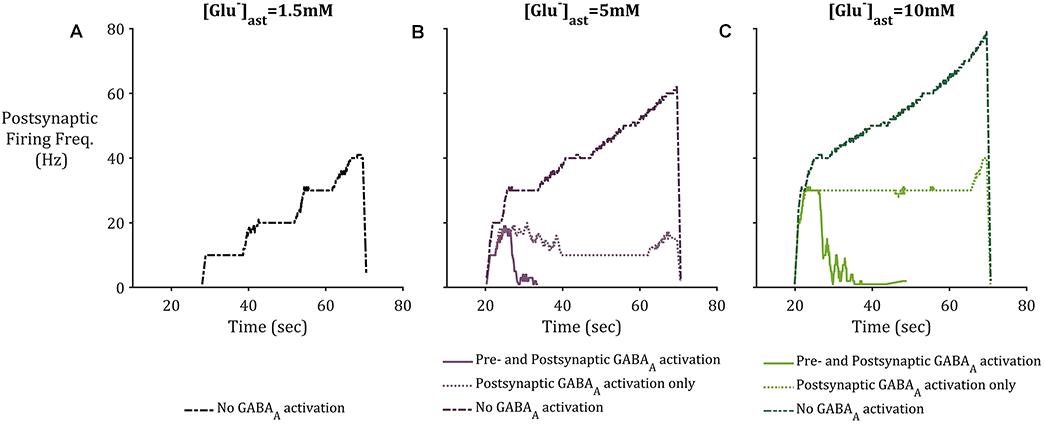
Figure 7. Postsynaptic neuronal firing frequency where (A) basal [Glu]ast = 1.5 mM, (B) basal [Glu]ast = 5 mM, and (C) basal [Glu]ast = 10 mM. Within each plot results from the modeling simulation described above were recorded, where inclusion of astrocytic GAT-3 resulted in GABA-mediated activation of GABAA-Rs are either located on both presynaptic and postsynaptic neuronal membrane (solid line), or the postsynaptic neuronal membrane only (dotted line). Exclusion of GAT-3 and thus no GABAA-R mediated currents are included as control (dash-dotted line). In all cases, the inclusion of GABA dynamics resulted in diminished postsynaptic activity, specifically where [Glu]ast = 1.5 mM, which resulted in no postsynaptic activity where GABAA-R activation was considered.
In the cases of higher astrocytic [Glu], the inclusion of GABA dynamics in both cases (i) and (ii) is sufficient to not only significantly reduce the firing frequency of the postsynaptic neuron, but also curtail the runaway Glu-mediated excitation of the neuronal membrane, as seen in the dash-dotted lines in Figures 7B,C.
The effects of astrocytic function and dysfunction on synaptic activity are widely researched areas in both experimental and computational fields. In this study, we focused on one particular recognized function of astrocytes: the control of extracellular Glu and GABA neurotransmitter concentrations by astrocytic transporters.
The experimental observation that astrocytic GABA transporters, GAT-3, respond to Glu mediated EAAT-2 activation (Héja et al., 2009) appears to indicate a synaptic feedback determined by the influx of shared substrate, Na+. Governed by the reversal potential of GAT-3, this activation mediates the release of GABA from the astrocyte and may modulate a long-lasting tonic inhibition of nearby neurons (Farrant and Nusser, 2005; Héja et al., 2009; Kersanté et al., 2013) as opposed to transient, or phasic, inhibition typically resulting from exocytotic release (Farrant and Nusser, 2005). A model to describe the interaction between EAAT-2 and GAT-3 activity was developed, exploring their effects for synaptic Glu and GABA concentrations and consequential perturbation of both the pre- and postsynaptic neuronal membrane potential.
Within this paper, a simulation sought to explore and explain the interplay of EAAT-2 and GAT-3 transport. Within the simulated time, it was noted that astrocytic GAT-3 released inhibitory neurotransmitter GABA in addition to acting as a non-ATP dependent mechanism for regulating intracellular Na+: a role traditionally assigned to the NKA which relies directly on ATP availability. Based on the findings of this paper, the proposition of previous experimental study (Héja et al., 2012) is supported, that GAT-3 acts to provide a modulatory effect when faced with excessive synaptic excitation. In addition, the modulatory effect is diminished where astrocytic [Glu] is elevated, as the time course of synaptic Glu is prolonged (Flanagan et al., 2018), thus locally exciting the postsynaptic neuron for longer. Results indicate that astrocyte-released GABA through GAT-3 acting on the postsynaptic neuron alone is also sufficient to suppress postsynaptic neuronal activity. It was noted that presynaptic neuronal inhibition decreases where astrocytic [Glu] is elevated.
As in Flanagan et al. (2018), consideration was taken of the astrocytic [Glu], reflecting the hypothesized effects of astrocytic glutamine synthetase downregulation (Perez et al., 2012) as observed in the focal sites of some epilepsies, particularly mesial temporal lobe epilepsy (MTLE) (Petroff et al., 2002; Eid et al., 2004). Previously reported results (Flanagan et al., 2018) indicate the slowing of synaptic Glu clearance in line with increasing astrocytic [Glu]. This resulted in the over-activation of postsynaptic NMDA and AMPA receptors and thus heightened local postsynaptic neuronal firing frequencies. Within this paper, the inclusion of astrocytic GAT-3 modulates the postsynaptic firing frequencies despite increased astrocytic Glu content, however, the strength of astrocytic GABA-mediated neuronal inhibition decreases where astrocytic [Glu] is elevated. The implications of this within the context of MTLE would be an ineffective astrocytic GABA-induced synaptic modulation, which would be further impaired by chronic GS downregulation (Hammer et al., 2008), resulting in increased neuronal hyperexcitability and seizure generation.
Although astrocytic-released GABA may act on an extrasynaptic location and the corresponding GABAA-Rs have been seen to have a higher affinity to GABA than their synaptic counterparts (Farrant and Nusser, 2005), this has not been accounted for in this model and remains a direction for future work. In addition, this study took account of neuronal inhibition mediated by the ubiquitous GABAA-Rs due to experimental results demonstrating their fast activating inhibitory effect of neuronal hyperexcitability and epileptiform activity (Mann et al., 2009). These receptors were modeled to counteract the fast-excitatory behavior of NMDA-R and AMPA-R mediated currents. Further developments of this model would also take account of slower GABAB receptor-activation and their longer-term effects on neuronal hyperexcitability.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
BF, LM, JW, MT, KW-L, and JH contributed to conception and design of the study. BF wrote the code. BF, LM, and JW wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work acknowledges funding support from Northern Ireland’s Department for the Economy.
Allam, S. L., Ghaderi, V. S., Bouteiller, J.-M. C., Legendre, A., Ambert, N., Greget, R., et al. (2012). A computational model to investigate astrocytic glutamate uptake influence on synaptic transmission and neuronal spiking. Front. Comput. Neurosci. 6:70. doi: 10.3389/fncom.2012.00070
Attwell, D., Barbour, B., and Szatkowski, M. (1993). Nonvesicular release of neurotransmitter. Neuron 11, 401–407. doi: 10.1016/0896-6273(93)90145-h
Bentzen, N. C. K., Zhabotinsky, A. M., and Laugesen, J. L. (2009). Modeling of glutamate-induced dynamical patterns. Int. J. Neural Syst. 19, 395–407. doi: 10.1142/s0129065709002105
Breslin, K., Joseph Wade, J., Wong-Lin, K. F., Harkin, J., Flanagan, B., Van Zalinge, H., et al. (2018). Potassium and sodium microdomains in thin astroglial processes: a computational model study. PLoS Comput. Biol. 14:e1006151. doi: 10.1371/journal.pcbi.1006151
Clasadonte, J., and Haydon, P. G. (2012). “Astrocytes and epilepsy,” in Jasper’s Basic Mechanisms of the Epilepsies, 4th Edn, eds J. Noebels, M. Avoli, M. Rogawski, R. Olsen, and A. Delgado-Escueta (New York, NY: Oxford University Press).
Coulter, D. A., and Steinhäuser, C. (2015). Role of astrocytes in epilepsy. Cold Spring Harb. Perspect. Med. 5:a022434.
Coyle, J. T. (2004). The GABA-glutamate connection in schizophrenia: which is the proximate cause? Biochem. Pharmacol. 68, 1507–1514. doi: 10.1016/j.bcp.2004.07.034
Destexhe, A., Mainen, Z. F., and Sejnowski, T. J. (1998). “Kinetic models of synaptic transmission,” in Methods in Neuronal Modeling: From Ions to Networks, 2nd Edn, eds C. Koch and I. Segev (Cambridge, MA: Massachusetts Intitute of Technology), 1–25.
During, M. J., and Spencer, D. D. (1993). Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 341, 1607–1610. doi: 10.1016/0140-6736(93)90754-5
Eid, T., Ghosh, A., Wang, Y., Beckström, H., Zaveri, H. P., Lee, T. S. W., et al. (2008). Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain 131, 2061–2070. doi: 10.1093/brain/awn133
Eid, T., Thomas, M. J., Spencer, D. D., Rundén-Pran, E., Lai, J. C. K., Malthankar, G. V., et al. (2004). Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363, 28–37. doi: 10.1016/s0140-6736(03)15166-5
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat. Rev. Neurosci. 6, 215–229. doi: 10.1038/nrn1625
Fellin, T., Pascual, O., and Haydon, P. G. (2006). Astrocytes coordinate synaptic networks: balanced excitation and inhibition. Physiology 21, 208–215. doi: 10.1152/physiol.00161.2005
Flanagan, B., McDaid, L., Wade, J., Wong-Lin, K., and Harkin, J. (2018). A computational study of astrocytic glutamate influence on post-synaptic neuronal excitability. PLoS Comput. Biol. 14:e1006040. doi: 10.1371/journal.pcbi.1006040
Golomb, D., Yue, C., and Yaari, Y. (2006). Contribution of persistent Na+ current and M-type K+ current to somatic bursting in CA1 pyramidal cells: combined experimental and modeling study. J. Neurophysiol. 96, 1912–1926. doi: 10.1152/jn.00205.2006
Halnes, G., Østby, I., Pettersen, K. H., Omholt, S. W., and Einevoll, G. T. (2013). Electrodiffusive model for astrocytic and neuronal ion concentration dynamics. PLoS Comput. Biol. 9:e1003386. doi: 10.1371/journal.pcbi.1003386
Hammer, J., Alvestad, S., Osen, K. K., Skare, Ø, Sonnewald, U., Ottersen, O. P., et al. (2008). Expression of glutamine synthetase and glutamate dehydrogenase in the latent phase and chronic phase in the kainate model of temporal lobe epilepsy. Glia 56, 856–868. doi: 10.1002/glia.20659
Héja, L., Barabás, P., Nyitrai, G., Kékesi, K. A., Lasztóczi, B., Toke, O., et al. (2009). Glutamate uptake triggers transporter-mediated GABA release from astrocytes. PLoS One 4:e7153. doi: 10.1371/journal.pone.0007153
Héja, L., Nyitrai, G., Kékesi, O., Dobolyi, Á, Szabó, P., Fiáth, R., et al. (2012). Astrocytes convert network excitation to tonic inhibition of neurons. BMC Biol. 10:26. doi: 10.1186/1741-7007-10-26
Hertz, L., Dringen, R., Schousboe, A., and Robinson, S. R. (1999). Astrocytes: glutamate producers for neurons. J. Neurosci. Res. 57, 417–428. doi: 10.1002/(sici)1097-4547(19990815)57:4<417::aid-jnr1>3.0.co;2-n
Hodgkin, A. L., and Huxley, A. F. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117, 500–544. doi: 10.1113/jphysiol.1952.sp004764
Hübel, N., Hosseini-Zare, M. S., Žiburkus, J., and Ullah, G. (2017). The role of glutamate in neuronal ion homeostasis: a case study of spreading depolarization. PLoS Comput. Biol. 13:e1005804. doi: 10.1371/journal.pcbi.1005804
Kandel, E. R., Schwartz, J. H., Jessell, T. M., Siegelbaum, S. A., Hudspeth, A. J., and Mack, S. (2012). Principles of Neural Science, 5th Edn, eds A. Sydor and H. Lebowitz (New York: McGraw Hill Professional).
Kersanté, F., Rowley, S. C. S., Pavlov, I., Gutièrrez-Mecinas, M., Semyanov, A., Reul, J. M. H. M., et al. (2013). A functional role for both -aminobutyric acid (GABA) transporter-1 and GABA transporter-3 in the modulation of extracellular GABA and GABAergic tonic conductances in the rat hippocampus. J. Physiol. 591, 2429–2441. doi: 10.1113/jphysiol.2012.246298
Kirischuk, S., Parpura, V., and Verkhratsky, A. (2012). Sodium dynamics: another key to astroglial excitability? Trends Neurosci. 35, 497–506. doi: 10.1016/j.tins.2012.04.003
Levy, L. M., Warr, O., and Attwell, D. (1998). Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J. Neurosci. 18, 9620–9628. doi: 10.1523/jneurosci.18-23-09620.1998
Li, J., Tang, J., Ma, J., Du, M., Wang, R., and Wu, Y. (2016). Dynamic transition of neuronal firing induced by abnormal astrocytic glutamate oscillation. Sci. Rep. 6:32343.
Mann, E. O., Kohl, M. M., and Paulsen, O. (2009). Distinct roles of GABAA and GABAB receptors in balancing and terminating persistent cortical activity. J. Neurosci. 29, 7513–7518. doi: 10.1523/jneurosci.6162-08.2009
Manninen, T., Havela, R., and Linne, M.-L. (2018). Computational models for calcium-mediated astrocyte functions. Front. Comput. Neurosci. 12:14. doi: 10.3389/fncom.2018.00014
Meldrum, B. S. (2000). Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J. Nutr. 130(4 Suppl.), 1007S–1015S.
Minelli, A., DeBiasi, S., Brecha, N. C., Zuccarello, L. V., and Conti, F. (1996). GAT-3, a high-affinity GABA plasma membrane transporter, is localized to astrocytic processes, and it is not confined to the vicinity of GABAergic synapses in the cerebral cortex. J. Neurosci. 16, 6255–6264. doi: 10.1523/jneurosci.16-19-06255.1996
O’Rourke, N. A., Weiler, N. C., Micheva, K. D., and Smith, S. J. (2012). Deep molecular diversity of mammalian synapses: why it matters and how to measure it. Nat. Rev. Neurosci. 13, 365–379. doi: 10.1038/nrn3170
Perez, E. L., Lauritzen, F., Wang, Y., Lee, T. S. W., Kang, D., Zaveri, H. P., et al. (2012). Evidence for astrocytes as a potential source of the glutamate excess in temporal lobe epilepsy. Neurobiol. Dis. 47, 331–337. doi: 10.1016/j.nbd.2012.05.010
Petroff, O. A. C. (2002). GABA and glutamate in the human brain. Neuroscientist 8, 562–573. doi: 10.1177/1073858402238515
Petroff, O. A. C., Errante, L. D., Rothman, D. L., Kim, J. H., and Spencer, D. D. (2002). Glutamate-glutamine cycling in the epileptic human hippocampus. Epilepsia 43, 703–710. doi: 10.1046/j.1528-1157.2002.38901.x
Pizzarelli, R., and Cherubini, E. (2011). Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast. 2011:297153.
Robinson, S. R. (2000). Neuronal expression of glutamine synthetase in Alzheimer’s disease indicates a profound impairment of metabolic interactions with astrocytes. Neurochem. Int. 36, 471–482. doi: 10.1016/s0197-0186(99)00150-3
Rossi, D. J., Hamann, M., and Attwell, D. (2003). Multiple modes of GABAergic inhibition of rat cerebellar granule cells. J. Physiol. 548(Pt 1), 97–110.
Schousboe, A., Scafidi, S., Bak, L. K., Waagepetersen, H. S., and McKenna, M. C. (2014). “Glutamate metabolism in the brain focusing on astrocytes,” in Glutamate and {ATP} at the Interface of Metabolism and Signaling in the Brain, eds V. Parpura, A. Schousboe, and A. Verkhratsky (Cham: Springer International Publishing), 13–30.
Schutter, E. D. E., and Smolen, P. (1998). “Calcium dynamics in large neuronal models,” in Methods in Neuronal Modeling: From Ions to Networks, 2nd Edn, eds C. Koch and I. Segev (Cambridge, MA: Massachusetts Intitute of Technology), 211–250.
Sigel, E., and Steinmann, M. E. (2012). Structure, function, and modulation of GABA(A) receptors. J. Biol. Chem. 287, 40224–40231.
Tewari, S. G., and Majumdar, K. K. (2012). A mathematical model of the tripartite synapse: astrocyte-induced synaptic plasticity. J. Biol. Phys. 38, 232–236.
Tsodyks, M., Pawelzik, K., and Markram, H. (1998). Neural networks with dynamic synapses. Neural Comput. 10, 821–835.
Witthoft, A., Filosa, J. A., and Karniadakis, G. E. (2013). Potassium buffering in the neurovascular unit: models and sensitivity analysis. Biophys. J. 105, 2046–2054.
Keywords: astrocyte, sodium-signaling, neurotransmission, synapse, glutamate, GABA
Citation: Flanagan B, McDaid L, Wade JJ, Toman M, Wong-Lin K and Harkin J (2021) A Computational Study of Astrocytic GABA Release at the Glutamatergic Synapse: EAAT-2 and GAT-3 Coupled Dynamics. Front. Cell. Neurosci. 15:682460. doi: 10.3389/fncel.2021.682460
Received: 18 March 2021; Accepted: 15 June 2021;
Published: 12 July 2021.
Edited by:
Annalisa Scimemi, University at Albany, United StatesReviewed by:
Maurizio De Pittà, Basque Center for Applied Mathematics, SpainCopyright © 2021 Flanagan, McDaid, Wade, Toman, Wong-Lin and Harkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bronac Flanagan, Yi5mbGFuYWdhbkB1bHN0ZXIuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.