 Ifeoluwa O. Awogbindin1,2*
Ifeoluwa O. Awogbindin1,2* Benneth Ben-Azu1,3†
Benneth Ben-Azu1,3† Babatunde A. Olusola4†
Babatunde A. Olusola4† Elizabeth T. Akinluyi1,5
Elizabeth T. Akinluyi1,5 Philip A. Adeniyi6
Philip A. Adeniyi6 Therese Di Paolo7,8
Therese Di Paolo7,8 Marie-Ève Tremblay1,7,9,10,11*
Marie-Ève Tremblay1,7,9,10,11*- 1Division of Medical Sciences, University of Victoria, Victoria, BC, Canada
- 2Neuroimmunology Group, Molecular Drug Metabolism and Toxicology Laboratory, Department of Biochemistry, College of Medicine, University of Ibadan, Ibadan, Nigeria
- 3Neuropharmacology Unit, Department of Pharmacology and Therapeutics, Faculty of Basic Medical Sciences, College of Health Sciences, Delta State University, Abraka, Nigeria
- 4Department of Virology, College of Medicine, University of Ibadan, Ibadan, Nigeria
- 5Department of Pharmacology and Therapeutics, College of Medicine and Health Sciences, Afe Babalola University, Ado-Ekiti, Nigeria
- 6Department of Comparative Biomedical Sciences, School of Veterinary Medicine, Louisiana State University, Baton Rouge, LA, United States
- 7Axe Neurosciences, Centre de Recherche du CHU de Québec-Université Laval, Québec, QC, Canada
- 8Faculté de Pharmacie, Université Laval, Québec, QC, Canada
- 9Neurology and Neurosurgery Department, McGill University, Montréal, QC, Canada
- 10Department of Molecular Medicine, Université Laval, Québec, QC, Canada
- 11Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, BC, Canada
Since December 2019, humankind has been experiencing a ravaging severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) outbreak, the second coronavirus pandemic in a decade after the Middle East respiratory syndrome coronavirus (MERS-CoV) disease in 2012. Infection with SARS-CoV-2 results in Coronavirus disease 2019 (COVID-19), which is responsible for over 3.1 million deaths worldwide. With the emergence of a second and a third wave of infection across the globe, and the rising record of multiple reinfections and relapses, SARS-CoV-2 infection shows no sign of abating. In addition, it is now evident that SARS-CoV-2 infection presents with neurological symptoms that include early hyposmia, ischemic stroke, meningitis, delirium and falls, even after viral clearance. This may suggest chronic or permanent changes to the neurons, glial cells, and/or brain vasculature in response to SARS-CoV-2 infection or COVID-19. Within the central nervous system (CNS), microglia act as the central housekeepers against altered homeostatic states, including during viral neurotropic infections. In this review, we highlight microglial responses to viral neuroinfections, especially those with a similar genetic composition and route of entry as SARS-CoV-2. As the primary sensor of viral infection in the CNS, we describe the pathogenic and neuroinvasive mechanisms of RNA viruses and SARS-CoV-2 vis-à-vis the microglial means of viral recognition. Responses of microglia which may culminate in viral clearance or immunopathology are also covered. Lastly, we further discuss the implication of SARS-CoV-2 CNS invasion on microglial plasticity and associated long-term neurodegeneration. As such, this review provides insight into some of the mechanisms by which microglia could contribute to the pathophysiology of post-COVID-19 neurological sequelae and disorders, including Parkinson’s disease, which could be pervasive in the coming years given the growing numbers of infected and re-infected individuals globally.
Introduction
Since the turn of 2019, a widespread and lingering severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic has been ravaging the humanity (Rabi et al., 2020). Coronaviruses are large, pleomorphic, enveloped, and positive-stranded RNA viruses that belong to three genera: alphacoronavirus, betacoronavirus, gammacoronavirus. These viruses are responsible for a wide range of respiratory, gastrointestinal, hepatic, and neurological diseases with varying severity levels (Jin et al., 2020; Vargas et al., 2020). SARS-CoV-2 belongs to the genus betacoronaviruses of the family (Walker et al., 2019). Examples of previously existing coronaviruses are severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) and Middle East respiratory syndrome coronavirus (MERS-CoV), which were the causes of the 2002 and 2012 epidemics, respectively (Vargas et al., 2020). Others are the human coronavirus 229E (HCoV-229E; one of the first alphacoronavirus strains to be reported), human coronavirus OC43 (HCoV-OC43; a betacoronavirus), human coronavirus NL63 (HCoV-NL63; an alphacoronavirus), and human coronavirus HKU1 (HCoV-HKUI; a betacoronavirus) (Fehr and Perlman, 2015; Chen Y. et al., 2020).
The genome of SARS-CoV-2 is 79.5% similar to other SARS-CoVs. This shows extensive sequence homology and conservation within the family (Choudhury and Mukherjee, 2020; Jin et al., 2020; Motayo et al., 2020). The major structural proteins are the spike, matrix, envelope, and nucleocapsid (Jin et al., 2020; Onofrio et al., 2020). However, a significant striking difference between SARS-CoV-2 and other coronaviruses is the longer length of the spike protein amino acid (Onofrio et al., 2020). This disparity has been suggested to confer higher transmissibility potential to SARS-CoV-2, making it possible for the virus to infect humans of different races and geographical origins (Onofrio et al., 2020). As of May 1, 2021, more than 1.9% of the world population are infected with SARS-CoV-2. The infection has been reported in over 86.85% of countries and territories globally with over 3.1% deaths (World Health Organization [WHO], 2021). Infection with SARS-CoV-2 results in Coronavirus disease 2019 (COVID-19) with a signature collection of symptoms including dyspnea, fatigue, pulmonary insufficiency, fever, dry cough, nasal congestion, myalgia, headache, and intestinal dysfunction (Wiersinga et al., 2020).
With a second surge in reported cases globally and growing numbers of multiple re-infections or relapses, SARS-CoV-2 infection shows no sign of abating (Iwasaki, 2020). Notably, re-infection or relapses, characterized by inflammatory rebound, pose more threats than the previous infection and commonly result in fatalities (Gousseff et al., 2020; Lafaie et al., 2020). Homing of SARS-CoV-2 into the cells of vulnerable organs relies on angiotensin-converting enzyme 2 (ACE2) binding (Letko et al., 2020), while neuropilin-1 (NRP-1) has recently been discovered as a receptor capable of facilitating SARS-CoV-2 entry with ACE2-independent mechanism (Cantuti-Castelvetri et al., 2020). SARS-CoV-2 can enter various organs displaying abundant ACE2 and NRP-1 expression, such as the nasopharynx, lungs, stomach, small intestine, lymph nodes, spleen, kidney, and brain (Hamming et al., 2004). The severity of COVID-19 results in complications and loss of function across these multiple organs, especially in individuals with co-morbidities (Zou et al., 2020). Also, SARS-CoV-2 infection results in neurological symptoms, including early hyposmia, reduced or total inability to detect odors, ischemic stroke, meningitis, cerebral thrombosis, delirium, and dizziness (Ahmad and Rathore, 2020). Moreover, documented and non-documented reports of neuropsychiatry symptoms and movement abnormalities post-infection are also emerging (Poyiadji et al., 2020; Thakur et al., 2021). Evidently, some reports have described the lack of coordination in movement, gait shuffling, falls, confusion, and clumsiness of thoughts, even after SARS-CoV-2 antibodies were detected and testing negative to the virus for several days (Mao et al., 2020; Piscitelli et al., 2020). This may suggest a continuous or permanent remodeling of ACE2-expressing neurons, glial cells including microglia, and/or brain vasculature in response to SARS-CoV-2 or COVID-19. However, very few pathophysiological details are currently available.
In this review, we highlight microglial response to viral neuroinfections, especially those with similar genetic composition and route of entry as SARS-CoV-2, given that microglia have been implicated in the pathophysiology of viral-mediated neurodevelopmental, neuropsychiatric, and neurodegenerative disease conditions (Rock et al., 2004). Microglia are the primary non-neuronal innate sensors of viral infections in the central nervous system (CNS) (Chen et al., 2019). They survey the CNS parenchyma, respond to viral stressors, and regulate the egress of viral particles across brain regions (Nimmerjahn et al., 2005; Fekete et al., 2018). Specifically, we describe the available literature on SARS-CoV-2 neurotropism, with a special focus on its route of entry to the CNS. Furthermore, we discuss a few neurotropic RNA viruses with similar route of entry to the CNS, providing specific details on the interaction between viral pathogen-associated molecular patterns (PAMPs) and microglia-expressed receptors. Since microglia are important mediators of neuroprotection and neurodegeneration in the CNS (Chen and Trapp, 2016; Tay et al., 2017; Bellver-Landete et al., 2019), we emphasize the outcomes of SARS-CoV-2 CNS invasion on microglial responses. Further, we provide concise explanations on the specific microglial response vis-à-vis the associated immediate and long-term implications. Consequently, this article offers insight into possible pathogenic mechanisms by which microglial reactivity during SAR-CoV-2 infection could be involved in the development of post-COVID-19 neurological sequelae and syndromes, including Parkinson’s disease.
Evidence of SARS-CoV-2 Neurotropism
Increasing number of reports describe a prevalence of neurological alterations and symptoms in COVID-19 (Brown et al., 2020; Helms et al., 2020; Mao et al., 2020; Piscitelli et al., 2020). Accordingly, the neurotropic hypothesis and capacity of SARS-CoV-2 were first proposed following the manifestation of anosmia in COVID-19 patients, as observed in other coronaviruses (Su et al., 2016; Dubé et al., 2018; Qian et al., 2020). Among the common neuropsychiatric presentations, delirium is suggested to be the most prevalent in over 30% of patient during the acute phase of SARS-CoV-2 infection (Rogers et al., 2020). Although delirium is often believed to occur as an early prodromal of mild brain dysfunction, in the case of infectious diseases, delirium can be caused by aggravated peripheral inflammatory response or be a direct effect of infectious agents on the CNS (Tsuruta and Oda, 2016).
In line with the neuropathological sequelae, mined data from brain single-nuclear RNA sequencing and spatial distribution analyses found relatively high levels of ACE2 expression in several brain regions including the choroid plexus, substantia nigra (SN), ventricles, and neurovasculature (Chen T. et al., 2020; Hu X. et al., 2020). Results from the analysis of single-cell sequencing data from human middle temporal gyrus also found that ACE2 was widely distributed in human and mouse neurons, astrocytes and oligodendrocytes, but rarely in microglia (Chen R. et al., 2020). An ultrastructural characterization of the spike protein component of the virus by high-resolution cryoelectron microscopy further revealed that ACE2 was critical for the neuroinvasiveness of SARS-CoV-2 (Wrapp et al., 2019). In comparison with previous coronaviruses, the study also reported that the spike protein octodomain of SARS-CoV-2 had a higher affinity for the ACE2 receptor (Wrapp et al., 2019), suggesting that SARS-CoV-2 may have a more potent neuroinvasive property (Natoli et al., 2020).
In some instances, specific SARS-CoV-2 antibodies and viral antigens have been confirmed in the cerebrospinal fluid (CSF) of infected individuals by genomic sequencing (Benameur et al., 2020; Moriguchi et al., 2020; Wu et al., 2020). Radiographic examinations showed traces of COVID-19-associated neuropathological features, including acute hemorrhagic necrotizing encephalitis (AHLE) and acute disseminated encephalomyelitis (ADEM) (Poyiadji et al., 2020; Reichard et al., 2020; Thakur et al., 2021). A biopsy examination has been used to confirm temporal lobe encephalitis enriched by perivascular lymphocytic infiltration, microglial nodule, and neuronal destabilization (Hu J. et al., 2020; Thakur et al., 2021). As such, it has been hypothesized that the auto-antigenicity of SARS-CoV-2 antibodies causes macrophage functional recruitment and ADEM. The immunological reactivity of SARS-CoV-2 is crossed-linked to human neuronal myelin sheath and thus may likely promote a post-infectious autoimmune demyelinated pathology of the brain (Garg et al., 2020; Parsons et al., 2020). Furthermore, evidence derived from forebrain specific human neural progenitor cells (hNPCs) generated from human-induced pluripotent stem cell (hiPSC) lines demonstrated replication of SARS-CoV-2 in 2-week old hNPCs with peak infection in less than 12 h post-infection. The study showed increased cell death with TUNEL assay (Song et al., 2020). The hiPSC-derived brain organoids showed at 9 weeks increased viral particles and infection of microtubule-associated protein 2 (MAP2)-positive mature neurons 24 h post-infection. The study suggested that SARS-CoV-2 can infect cells of neural origin and hypothesized that infected cells can cause death of nearby cells (Solomon et al., 2020). In comparison with other viruses, such as Zika virus (ZIKV), SARS-CoV-2 related brain infection has been linked to an upregulation of cell division, organelle fission, and metabolic processes, via moderate interferon-stimulated gene activation (Blanco-Melo et al., 2020). This finding suggest that the brain is a site of high replicative potential for SARS-CoV-2 (Song et al., 2020). Additionally, using transgenic mice expressing ACE2 under the K18 promoter (hACE2-K18), intranasal administration of SARS-CoV-2 (Song et al., 2020) resulted in viral titers in the brain, especially in the forebrain and cerebral cortex. However, low distribution in the dentate gyrus, globus pallidus, and cortical layer 4 was reported. Together, these findings suggest that SARS-CoV-2 can infect patients brain’s neurons via its neurotropic properties (Song et al., 2020). However, given that microglia act as the central housekeepers against altered homeostatic states, even during viral neurotropic infections, it is tempting to speculate that microglial reactivity and cell infectivity by SARS-CoV-2 might play an important role in the transmission and activity of the virus across the brain parenchyma (Vargas et al., 2020).
Route of Entry and Neuroinvasive Mechanisms of SARS-CoV-2 and RNA Viruses
Neurotropic RNA viruses gain access to the CNS via different routes. While some viruses overcome the barrier between the periphery and CNS, a few usurp the direct access between the olfactory canal and the brain (Chen G. et al., 2020; Hu X. et al., 2020). Other viruses channel the retrograde transport between peripheral and central nervous systems before achieving neuroinfection (Chen G. et al., 2020; Hu X. et al., 2020). For instance, West Nile virus (WNV), Canine distemper virus, and Mumps virus breach the blood-brain barrier (BBB), meningeal blood-barrier, and blood-CSF barrier to invade the brain after achieving a significant viremia (Rudd et al., 2006). Using this hematogenous approach, these viruses infect the CNS vasculature endothelium or “trojan horse” lymphocytes, which extravasate through the structural layers of the barrier, before spreading to the adjoining glial cells and neurons within the brain parenchyma. In the process, the virus can infect the components of the protective barriers, including pericytes, astrocytes, microglia, and endothelial cells, thereby increasing their permeability (Miner and Diamond, 2016). Various immune cells derived from the periphery, such as circulating macrophages and dendritic cells, may then crosstalk with the CNS’s neuronal and non-neuronal components (Aghagoli et al., 2020). Following brain invasion, viral proteins can act as PAMPs or induce damage-associated molecular patterns (DAMPs), which in turn trigger an innate immune response via pattern recognition receptors (PRRs) (Dantzer, 2018). Alternatively, airborne coronaviruses in the upper respiratory tract mucosa can infect the bipolar olfactory sensory neurons with unmyelinated axons that penetrate the cribriform plate into the olfactory bulb and form a direct synapse with the mitral cells (Aghagoli et al., 2020; Hu X. et al., 2020). As such, the virus is transmitted transneuronally by usurping the neuronal dynein-kinesins transportation network via anterograde axonal transport within the olfactory receptor neurons and via synaptic transfers to the olfactory bulb mitral cells. Following the linear anterograde and retrograde transport between the olfactory bulb and different bran regions, the virus can access the limbic system and other deeper nuclei (Aghagoli et al., 2020; Hu X. et al., 2020).
There are three primary routes of SARS-CoV-2 transmission: droplets, aerosol, and contact (Adhikari et al., 2020; Kabir et al., 2020; Qian et al., 2020; Zhang et al., 2020a). Cell and tissue tropism determines the host restriction (Lee et al., 2020; Saxena et al., 2020). Similar to SARS-CoV-1, SARS-CoV-2 spike protein binds to ACE2, even with a 10–20-fold higher affinity (Jin et al., 2020). However, the fusion requires a proteolytic activation by transmembrane protease serine 2 (TMPRSS2) as well as cathepsin B and L for full entry (Letko et al., 2020; Wan et al., 2020). The ubiquitous nature of ACE2 enables SARS-CoV-2 to gain access to various cell types and elicit multiorgan failure. It is proposed that SARS-CoV-2 neuroinvasion is possible essentially through the hematogenous and transneuronal routes (Chen G. et al., 2020; Hu X. et al., 2020). For instance, autopsied brain analysis by electron microscopy of COVID-19 patients showed endotheliitis, mononuclear inflammatory cells, and viral-like structures in the brain capillary endothelium of the frontal lobe (Chigr et al., 2020; Varga et al., 2020). This is suggestive of direct endothelial viral infection. Furthermore, because ACE2 is expressed in both the lymph node-associated CD68-positive (+) macrophages and tissue-based CD169+ macrophage, the trojan horse mechanism of SARS-CoV-2 neuroinvasion may involve a number of mechanisms. First, a viral infection of leukocytes or monocyte-macrophages before a paracellular and circumventricular transfer to the brain, mainly through organs that are deficient in BBB (Chen G. et al., 2020). It can also proceed via dorsal root or autonomic ganglia. A detailed mechanistic study in mice revealed that S1 subunit of SARS-CoV-2 spike protein was initially retained on the luminal side of capillary bed after which it crossed the BBB via adsorptive transcytosis with an intense tropism in different brain regions including the striatum, midbrain, hypothalamus and olfactory bulb, following intravenous injection (Rhea et al., 2020). This suggests that the S1 subunit may drive the brain uptake of SARS-CoV-2 through the BBB. Shed SARS-CoV-2 S1 subunit may also display the full pathogenic attributes of whole SARS-CoV-2, even after viral clearance, since it is very stable in the brain and can bind to ACE2 (Rhea et al., 2020). That anosmia and hypogeusia are early neurological alterations mediated by SARS-CoV-2 suggests CNS invasion through the olfactory system. In hACE2-K18 mice, SARS-CoV-1 infects the brain majorly through the olfactory nerve and subsequently spreads to other brain regions transneuronally (Netland et al., 2008). Similarly, in patients with COVID-19, recent images from fluid-attenuated inversion recovery (FLAIR) on magnetic resonance imaging (MRI) sequence revealed a bilateral hyper-intensity of the olfactory bulbs and right gyrus rectus (Politi et al., 2020). This further reinforces the transneuronal or olfactory pathway hypothesis (Hu J. et al., 2020). This finding could, in part, provide a possible mechanism underlying the cranial nerve symptoms observed in COVID-19.
Some studies have also highlighted the possibility of SARS-CoV-2 transmission through the digestive tract and the vagus nerve (Adhikari et al., 2020; Kabir et al., 2020; Qian et al., 2020; Zhang et al., 2020a). Previous findings showed that nucleus tractus solitarius and dorsal motor nucleus of the vagus nerve, which connect and activate the central autonomic network (sympathetic and parasympathetic systems) (Benarroch, 1993), express ACE2 (Doobay et al., 2007). Thus, SAR-CoV-2 is capable of infecting the terminal structures of the vagal afferents and the early parts of the vagal efferents to cause downexpression of ACE2, a mechanism that is already being implicated in disease severity and organ damage (Bonaz et al., 2020). Emerging evidence indicates that dysregulated cholinergic anti-inflammatory cascade by SAR-CoV-2 is linked to leaky gut, immune reactivity, BBB breakdown, and microglial reactivity (Changeux et al., 2020). Vagus nerve stimulation profoundly attenuated the microglial reactivity in lipopolysaccharide (LPS)-exposed mice (Meneses et al., 2016) and SARS-CoV-2-induced symptoms in COVID-19 patients (Changeux et al., 2020). Although it remains a hypothesis, two possible mechanisms may be used by SARS-CoV-2 to infect intestinal cells. The first has been described previously to occur during acute entero- and retroviral infections of the intestine. This process includes the disruption of the gut microbiota by these invading organisms, leading to the release of plasma LPS and other inflammatory biomarkers. The systemic inflammation leads to dysbiosis together with water and electrolyte imbalances causing gastrointestinal symptoms (Bergamini et al., 2018; Rao, 2020). The other mechanism has been reviewed elsewhere (de Oliveira et al., 2020). This hypothesis involves the downregulation of ACE2 on intestinal cells after SARS-CoV-2 binding. Reduction in the expression of ACE2 leads to decreased activation of the mechanistic target of rapamycin (mTOR) signaling cascade, which signals exacerbated inflammation, leaky gut, and increased autophagy (Chiappini et al., 2020; da Silva et al., 2020).
Viral PAMPs and DAMPs Following RNA Virus CNS Infection
RNA viruses, which are mostly old-world viruses (e.g., measles, mumps, rubella, rabies) or zoonotic, i.e., transmitted from other animals to humans, either through insect vectors (arthropod-borne, e.g., West Nile and flaviviruses), rodents (rodent-borne, e.g., Lassa fever viruses) or originating from bats (Lyssaviruses, SARS-CoV-2), are highly neuroinvasive and associated with CNS infections (Lund et al., 2004; Wang et al., 2006; Cui et al., 2010; Faul et al., 2010; Furr and Marriott, 2012; Jureka et al., 2015; Sallenave and Guillot, 2020). Viral CNS infections result in exacerbated inflammation responsible for several symptoms, such as fever, headache, confusion, stroke, seizure, or death (Furr and Marriott, 2012). Such infections trigger reactivity of resident immune and glial cells that serve as defenses for the CNS, namely, microglia and astrocytes. This results in the release of proinflammatory cytokines and subsequent activation of both innate and adaptive immune responses. These immune responses may limit the viral replication and spread; however, sustained inflammation can elicit damage to the function and structure of neurons directly or indirectly (Ising and Heneka, 2018). For example, early induction and secretion of interferons may protect neighboring cells from viral infection (see section “Microglial Response During RNA Viral Infections”). However, inflammatory mediators can also impact negatively on neuronal function at synapses thus affecting interneuronal networks. For instance, under an inflammatory environment, microglia may lose their homeostatic function of synaptic pruning and remodeling, notably due to process retraction (Stence et al., 2001; Schafer et al., 2012). Glial-mediated trophic support secretion also reduces under inflammatory conditions, resulting in functional and structural impairment of neurons (Parkhurst et al., 2013; Pöyhönen et al., 2019). Evidence also revealed that suppression of synaptic plasticity particularly long-term potentiation co-exists with sustained levels of tumor necrosis factor (TNF)-α (Tancredi et al., 1992), interleukin (IL)-6 (Tancredi et al., 2000), nitric oxide synthase 2 (Wang et al., 2004), and complement factor 3 (C3) (Lian et al., 2015) in the brain parenchyma. These inflammatory mediators may modulate astrocytes to trigger neurodegeneration (Liddelow et al., 2017).
Viral epitopes are recognized by pathogen recognition receptors (PRRs) or sensors on the plasma membrane, in the endosome, and within the immune cells’ cytoplasm. These PRRs interact with conserved PAMPs and nucleic acids, as well as DAMPs (Pichlmair and Reis e Sousa, 2007; Pedraza et al., 2010). The PAMPs are unique footprints of pathogens that are conserved among similar pathogens. While membrane-bound and endosomal PRRs, which target viral single-stranded (ss) and double-stranded (ds) RNA and DNA, sufficiently discriminate between self and exogenous nucleic acids, the cytoplasmic sensors, particularly those that bind to dsDNA, do not differentiate these nucleic acids (Roers et al., 2016). The PRRs (Table 1) include Toll-like receptors (TLRs), nucleotide oligomerization domain (NOD)-like receptors (NLRs), and retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), and cytosolic DNA sensors, such as cyclic GMP-AMP synthase (cGAS) (Pedraza et al., 2010; Jensen and Thomsen, 2012; Said et al., 2018; Lee et al., 2019).

Table 1. Pattern recognition receptors (PRRs) with presence in microglia: recognized viruses and ligands.
Despite their CNS homeostasis and neuronal cell functions, microglia and astrocytes are mainly involved in the protective immune responses during neuroinflammation (Facci et al., 2018; Jha et al., 2019; Tremblay et al., 2020). Following the recognition of PAMPs by PRR, glial cells express antiviral inflammatory mediators including type-1 interferons (IFN), IL-1β, TNF-α, and IL-6, which can induce peripheral immune cell infiltration across the BBB (Chauhan et al., 2008; Furr et al., 2010; Furr and Marriott, 2012; Jensen and Thomsen, 2012; Chen et al., 2017; Lee et al., 2019; Choudhury and Mukherjee, 2020). The endogenous DAMPs produced by stressed or dying neurons can also initiate PRR-mediated inflammatory responses (Nan et al., 2014; Fleshner and Crane, 2017; Lee et al., 2019). Although secreted cytokines are essential for inducing efficient and robust adaptive immune responses, excessive IFN production, as well as prolonged inflammatory responses, together elicit CNS damage, as discussed in the section “Microglial Response During RNA Viral Infections” (Bastard et al., 2020; Zhang et al., 2020d). On the other hand, the ineffective PRR signaling can increase viral infection severity (Lee et al., 2019). Hence, there is the need for a tight regulation of PRRs mediated signal transduction. Viruses have evolved several strategies (Table 2) to evade host immune detection and clearance. These strategies include interruption of viral sensors and manipulating molecules within signaling cascades (Gack et al., 2007; Jensen and Thomsen, 2012; Lee et al., 2019). A myriad of viral proteins target RIG-I because it is pivotal to the signaling cascades leading to the induction and secretion of antiviral IFN. These viral proteins interfere directly with RIG-I. For instance, the nucleocapsid of SARS-CoVs down-regulates RIG-I activity by targeting tripartite motif-containing protein 25 (TRIM25) thereby preventing RIG-I ubiquitination and subsequent IFN production (Pedraza et al., 2010; Furr and Marriott, 2012; Jensen and Thomsen, 2012; Lee et al., 2019).
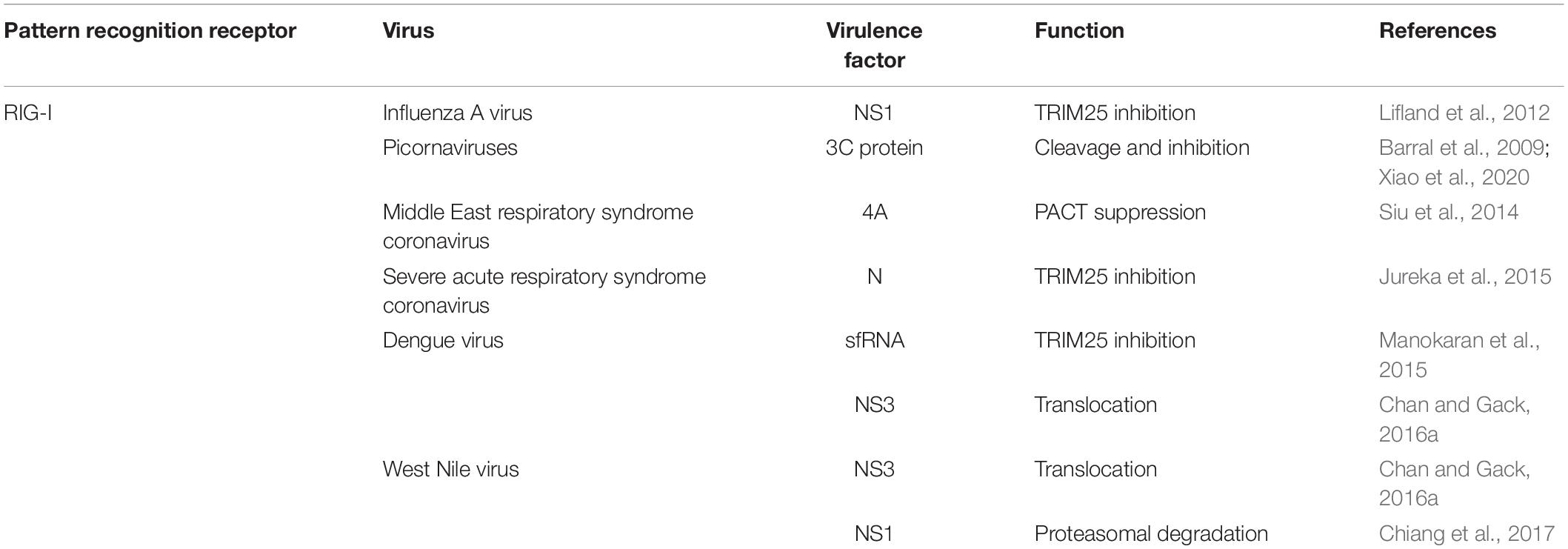
Table 2. Viral evasion mechanism for neurotropic RNA virus PRRs.
Pathogenesis of SARS-CoV-2 Infection
Following entry, there is an incubation period of about 5 days before symptoms begin to appear. It takes around 6–40 days from symptoms appearance to death, although the mean interval is 14 days (Kabir et al., 2020), whereas the mean interval is about 11.5 days for patients over 70 years of age (Jin et al., 2020). Several studies have noted that due to the acute nature of SARS-CoV-2 infection, innate immune responses may play a critical role in determining its eventual outcomes since the rapidity rather than memory of immune responses is important. This is particularly essential in novel viral infections, such as SARS-CoV-2 in which a pre-existing immunity is not possible (Pichlmair and Reis e Sousa, 2007; Ayegbusi et al., 2018; Costela-Ruiz et al., 2020). The acute nature of the infection may also account for the possibility of severe re-infection due to its deceptive imprinting (Kohler and Nara, 2020; Westerhuis et al., 2020). Deceptive imprinting refers to immune evasion mechanisms by certain viruses in which antibodies are mounted against immunodominant epitopes, thereby inducing strain-specific immunity, which offers little or no neutralization activity between serotypes and subtypes. This phenomenon has been described for Human Immunodeficiency virus (HIV), Influenza, and Dengue viruses (Tobin et al., 2008). Since coronaviruses are ubiquitous and certain strains have been identified in common seasonal colds; deceptive imprinting may account for severe COVID-19 cases with higher antibody titers against seasonal coronaviruses compared to SARS-CoV-2 (Westerhuis et al., 2020).
TLRs and RIG-1 are likely involved in viral clearance as well as in the development of severe COVID-19 disease (Jensen and Thomsen, 2012; Chen et al., 2017; Said et al., 2018). Upon recognition of dsRNA motifs, TLR3 recruits TIR-domain-containing adapter-inducing interferon-β (TRIF) adaptor proteins, which results in NF-κB signaling (Jensen and Thomsen, 2012). TLR4 is expressed although at low basal levels in bronchial, epithelial, and alveolar cells. Its expression and activation are increased upon cellular infiltration in response to viral infection (Pedraza et al., 2010; Jensen and Thomsen, 2012; Said et al., 2018). Both myeloid differentiation primary response 88 (MyD88) and TRIF sorting adaptors have been implicated in the proliferation of acute respiratory distress syndrome caused by other respiratory viruses (Jensen and Thomsen, 2012; Totura et al., 2015). Studies in mice have shown that TLR3–/–, TLR4–/–, and TRAM–/– mice are more susceptible to SARS-CoV infection, although they experience only transient weight loss with no mortality. In contrast, mice deficient in TLR4 adaptor proteins are highly susceptible to SARS-CoV, showing remarkable weight loss, mortality, impaired lung function, and lung pathology. These mice also show acute respiratory distress syndromes and deranged proinflammatory cytokines as well as interferon-stimulated gene signaling (Totura et al., 2015).
In silico studies have suggested a strong binding affinity between the spike protein and TLR4 (Choudhury and Mukherjee, 2020). Experimental mouse models of acute respiratory distress induced by multiple causes, including SARS-CoV-1, also showed the protective role of TLR4 (Totura et al., 2015). TLR activation via MyD88 and TRIF dependent pathways is involved in the pathogenesis of SARS-COV-1 (Jensen and Thomsen, 2012). These pathways lead to the production of proinflammatory cytokines (IL-1, IL-6, TNF-α) and type I IFN-α/β. Although necessary for viral clearance, unabated induction of these pathways may lead to a marked increase of a myriad of pro-inflammatory mediators up to a life-threatening level (cytokine storm), neuroinflammation, and autoimmunity, which contribute significantly to immunopathology (Hosseini et al., 2020; Varatharaj et al., 2020; Zhao H. et al., 2020). Autoimmunity may be another key mechanism of SARS-CoV-2 pathogenesis. Autoimmune responses can be induced by molecular mimicry or genetic defects (Jensen and Thomsen, 2012). Production of antiphospholipid autoantibodies may cause coagulopathy and cerebral infarction, which has been reported in patients with severe COVID-19 (Zhang et al., 2020e; Thakur et al., 2021). Studies have also documented an increased cytokine storm susceptibility in individuals with autoimmune diseases (Schwartz and Deczkowska, 2016). The antiviral role of interferons in the clearance of SARS-CoV-1 infection has been previously described (Jensen and Thomsen, 2012). Recent studies have also reported the enrichment in type I and III IFN genes (Bastard et al., 2020) as well as neutralizing autoantibodies against type I IFN-α2 and IFN-ω in patients with severe COVID-19 pneumonia (Zhang et al., 2020d).
The entry of SARS-CoV-2 into susceptible host cells eventually leads to apoptosis, pyroptosis, ACE2 downregulation, and shedding (Yang, 2020; Zhao Y. et al., 2020; Zhou et al., 2020). This, in turn, leads to primary inflammatory responses involving cytokine and chemokine release, antiviral factors expression, pulmonary cell infiltration, vascular permeability, lymphopenia, and acute respiratory distress (Nimmerjahn and Ravetch, 2007; Wang et al., 2008; Haslwanter et al., 2017). The secondary inflammatory responses involve virus-neutralizing antibody complex, which leads to Fc Receptor (FcR) and complement system activation, accompanied by antibody-mediated cellular cytotoxicity (Nimmerjahn and Ravetch, 2007; Wang et al., 2008; Haslwanter et al., 2017). These inflammatory responses lead to skewing of macrophage responses, abrogation of wound healing, monocyte chemoattractant protein 1 (MCP-1) and IL-8 production, acute lung injury, and cellular damage (Liu et al., 2019; Zhang et al., 2020a; Zhao Y. et al., 2020). Uncontrolled pulmonary inflammation and infiltration are accordingly the leading causes of death among SARS-CoV-2 infected individuals (Wang et al., 2008; Liu et al., 2019; Yang, 2020).
Microglia, the CNS-Resident Innate Immune Cells, as Sensors of CNS Viral Infection
Upon CNS viral infection, inflammatory responses have long been proposed to involve infiltrating peripheral monocytes and leukocytes, as a vast majority of resident CNS cells assumedly lacked immune functions (Sochocka et al., 2017). However, glial cells, most notably astrocytes and microglia, are now recognized as key players in protective and detrimental host responses during CNS disease states (Serramía et al., 2015; Li and Barres, 2018). Microglia are CNS-resident mononuclear phagocytes characterized by a distinctive ramified structure and specific gene expression (Hickman et al., 2013). These cells constitute 5–10% of total brain cells and are derived from embryonic yolk sac precursors (Ginhoux et al., 2010; Gomez Perdiguero et al., 2015), which seed the brain early in development (Gomez Perdiguero et al., 2015). While microglia play a vital role in the maintenance of CNS homeostasis, they are additionally known to dynamically scan the brain parenchyma, detecting the occurrence of pathologies (Schafer et al., 2012; Neniskyte and Gross, 2017). They also contribute to numerous developmental events and physiological processes, such as neurogenesis (Cunningham et al., 2013; Tremblay et al., 2015), programmed cell death (Wakselman et al., 2008), myelination (Voet et al., 2019; Sariol et al., 2020), synaptic remodeling and maturation (Paolicelli et al., 2011; Schafer et al., 2012; Tremblay et al., 2015). As myeloid cells, microglia are immunologically competent, swiftly responding to pathogenic infections within the CNS by modifying their function with a broad spectrum of reactivity states (Aguzzi et al., 2013; Ransohoff, 2016; Stratoulias et al., 2019). Accordingly, it is expected that microglial reactivity and dysfunction (i.e., altered physiological functions) are implicated in practically all CNS infections (Colonna and Butovsky, 2017; Tay et al., 2017; Wolf et al., 2017). Environmental factors including viral infections modulate microglial functions resulting in pathological synaptic remodeling, which culminates in altered cognition and behavior (Shi et al., 2003; Brown, 2012). Indeed, microglial reactivity outlasts the initial immune response with longlasting effects. This reactivity is often characterized of hypercytokinemia, altered ramification and dystrophy, leading to the formation of lysosomal inclusion proteins, up-regulation of proinflammatory cytokine genes, changes in brain neurochemistry and decreased neurogenesis, which can together result in microglial senescence, dystrophy or dysfunction within the CNS microenviroment (Colonna and Butovsky, 2017; Tay et al., 2017).
Microglia are equipped with the viral DAMPs recognition system (Jeffries and Marriott, 2017), which activates intracellular signaling cascades and promotes transcriptional activation, as well as expression of proinflammatory and antiviral cytokines (Furr and Marriott, 2012). As the principal immune sentinels of the CNS parenchyma, microglia- and astrocyte-mediated immune responses substantially contribute to antiviral immune-mediated events. Recent genomic studies of reactive astrogliosis have identified the neuroinflammatory and neuroprotective activities of astrocytes. Reactive astrocytes are commonly involved in processes of neurodegeneration and neuroinflammation via up-regulation of inflammatory cytokine genes and increased BBB permeability. However, homeostatic astrocytes have been reported to contribute to neuromodulation and neuroprotection via induction of immune tolerance genes (Liddelow and Barres, 2017; Liddelow et al., 2017). Strong pieces of evidence have shown that exposure to PAMPs and microglial-mediated proinflammatory cytokine release contribute to determining astrocyte reactivity (Liddelow et al., 2017). Since microglia are an important component of the intrinsic immune response in the CNS, it is clear that any viral infection will cause direct and indirect microglial responses, which are essential for the anti-viral mechanisms and to a large extent can determine the long-term neurological manifestations of the infection (Vargas et al., 2020). Unlike the innate peripheral cells, the functionality of viral nucleotide-sensing PRR in microglia is not well-known since the BBB is generally thought to prevent peripheral microbes from invading the brain during homeostasis. However, the contribution of microglial nucleotide-sensing mediated antiviral defense to brain pathology in contexts of neuroinvasive viral infections is being appreciated lately (Reinert et al., 2016). Subsequent to viral CNS infection, extracellular nucleotides, such as ATPs are released from neurons, astrocytes or microglia in response to noxious stimuli (Sperlágh and Illes, 2007). The released ATPs are strong chemotactic signals for microglia (Figure 1) and microglia express PRR for their detection (Kawasaki and Kawai, 2014). As infection persists, there is a sustained increase in the levels of these nucleotides, which subsequently trigger the recruitment and phagocytic action of microglia notably via the purinergic receptor P2RY12 signaling pathway (Fekete et al., 2018). This chemotactic tracing contributes to microglial recognition of compromised cells and regulation of phagocytic activity including upon viral infections (Fekete et al., 2018). Furthermore, viruses replicate in cells, accumulating massive amounts of nucleic acids, RNA, and DNA. Cytosolic mitochondrial proteins and dsDNA or ssDNA have been shown to alter microglial and astrocytic activities by triggering intracellular inflammatory pathways (Bajwa et al., 2019). The nucleotide DAMPs accumulate in the cytoplasm when phagocytosed viral particles overwhelm the lysosomal processing pathway. Under cellular stress or DNA damage, dsDNA from the nucleus or mitochondria further infiltrates the cytoplasm of neuronal and glial cells (Roers et al., 2016). DAMPs from neurotropic RNA viruses are prominently recognized by RIG-I dependent mechanisms (Furr et al., 2010; Furr and Marriott, 2012). For instance, RIG-I was markedly upregulated with the concomitant production of IL-6, TNF-α, and antiviral IFN-β when immortalized microglial cells defective in TLR4 or primary astrocyte/microglia were infected with either vesicular stomatitis virus (VSV), 5’ triphosphate double-stranded RNA (50ppp-dsRNA) or 5’-triphosphate single-stranded RNA (50ppp-ssRNA) (Crill et al., 2015). Upon RIG-I knockdown, these effects were significantly attenuated (Crill et al., 2015).
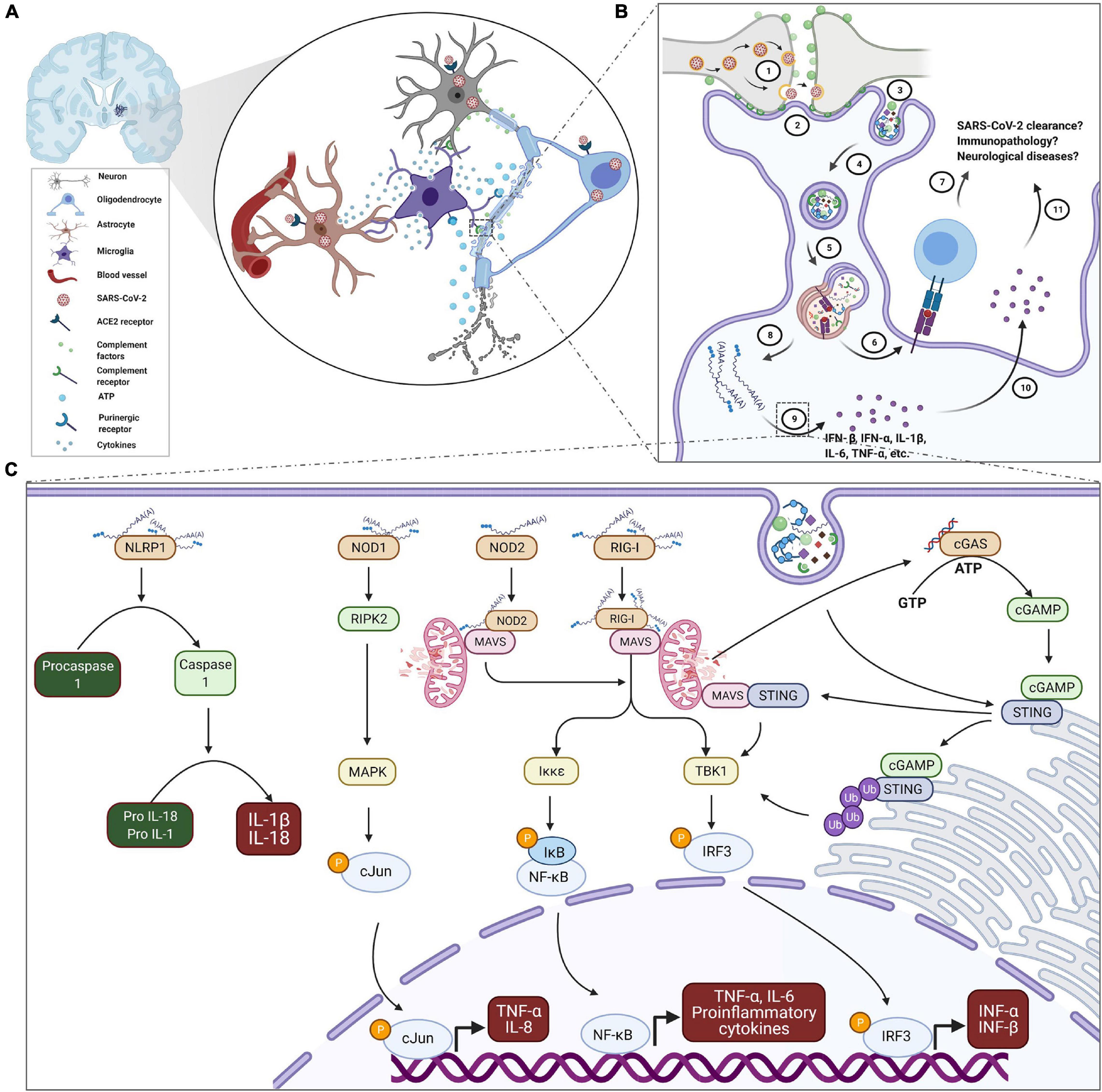
Figure 1. Proposed schematic of microglial reactivity and implications in SARS-CoV-2 infection and COVID-19. (A) COVID-19-associated focal hemorrhagic infarcts in the brain are characterized with microglia nodules, degenerating neurons and infiltrated T cells. Thus, microglia may be coordinating the inflammatory events around the infarct’s milieu in a number of ways via reactivity to signals from oligodendrocytes, neurons and astrocytes after SARS-CoV-2 infection, including ATP and complement (C1q or C3) tags, as well as secretion of cytokines. (B) For instance, complement coating of SARS-CoV-2-infected synapses (1) may trigger microglial recruitment and interaction via their complement receptors (2) culminating in encapsulation (3) and phagocytosis (4) of synaptic elements in membrane cargoes, which subsequently fuse with lysosomes for adequate processing (5). In the process, fragments of viral peptides may be presented via MHC-I and/or MHC-II to cytotoxic and/or helper T cells (6), respectively, of elicit adaptive immune response. However excessive phagocytosis of synaptic elements may overwhelm the phagolysosomal processing (8) resulting in the exposure of microglia to SARS-CoV-2 genome and functional/structural impairment of vital organelles. The exposure sensitizes microglia to produce (9) and secrete (10) both antiviral and inflammatory cytokines in significant quantity. Although microglia are equipped with a competent innate recognition system, their contribution in the context of SARS-CoV-2 infection and COVID-19 is yet unknown (7 and 11). (C) For emphasis, upon cytosolic exposure, microglia may detect SARS-CoV-2 genome through a battery of sensors. NLRP1 sensing of dsRNA and ssRNA activates inflammasome, which processes IL-1β and IL-18 production through caspase 1. NOD1 binding of dsRNA activates the translocation of cJun to the nucleus with subsequent upregulation of pro-inflammatory mediators. RIG-I-bound dsRNA and ssRNA as well as NOD-2- ssRNA complex exacerbate production of TNF-α, IL-6, and IL8 through mitochondrial adaptor protein MAVS mediated NF-κB signaling. Simultaneously, they also regulate the transcription of antiviral type 1 interferons through IRF3. In addition, DAMPs from stressed microglial organelles, such as mitochondrial DNA may trigger cGAS receptor to synthesize cGAMP, an agonist of STING. STING activation potentiates IRF3 signaling. Membrane fusion of endosomatic cargoes may also initiate cGAS-independent STING-interferons signaling through MAVS. Thus, characterization of the specific contribution of microglia in the development of neuronal damage and associated neurological sequelae, or the involvement in debris clearance, SARS-CoV-2 resolution and disease outcome is an active area of research. ATP, adenosine triphosphate; COVID-19, coronavirus disease 2019; DAMPs, damage-associated molecular patterns; cGAMP, cyclic GMP-AMP; cGAS, cyclic GMP-AMP synthase; GTP, guanosine triphosphate; IL, interleukin; IκB, inhibitor of κB; Iκκε, IκB kinase; IFN-α/β, interferon alpha/beta; IRF3, interferon Regulatory Factor 3; MAPK, mitogen activated protein kinase; MAVS, mitochondrial antiviral signaling protein; MHC-I/II, major histocompatibility complex I/II; NOD2/1, nucleotide-binding oligomerization domain 2/1; NF-κB, nuclear factor kappa light chain enhancer of activated B cells; NLRP1, NLR family pyrin domain containing 1; P, phosphate; RIG-I, retinoic acid-inducible gene I; RIPK2, receptor interacting serine/threonine protein kinase 2; dsRNA, double stranded viral RNA; ssRNA, single stranded viral RNA; SARS-CoV-2, severe acute respiratory syndrome coronavirus; STING, stimulator of type I interferon genes; TBK-1, TANK-binding kinase 1; TNF-α, tumor necrosis factor alpha; Ub, ubiquitin.
A signaling cascade involving the stimulator of type I interferon genes (STING) and absent in melanoma 2 (AIM2) is an alternative microglial sensing medium for viruses. STING serves as an adaptor molecule upon recognition of a sensor-dsDNA adduct, the best-described being cGAS-dsDNA (Sun et al., 2013; Zhang et al., 2014; Cox et al., 2015). Binding of cGAS-dsDNA to STING forms a complex, which brings in proximity the TANK-binding kinase (TBK1) and its substrate interferon regulatory factor (IRF3) via the recruitment of poly-ubiquitination apparatus. Phosphorylation of the transcription factor IRF3 stimulates the production of type 1 IFN α and β. On the other hand, AIM2 directs the production and secretion of the proinflammatory cytokine IL-1β through its precursor’s proteolytic processing by caspase-1 upon the formation of an active inflammasome complex. Although DNA viruses and retroviruses, via dsDNA, activate this pathway, emerging evidence indicates that positive-stranded RNA viruses can evade immune recognition by suppressing the STING pathway. Positive-stranded RNA viruses seem to interfere with the innate defense mechanisms by disrupting IFN production and its effects (Kabir et al., 2020; Larenas-Linnemann et al., 2020). For instance, the cleavage of STING by non-structural protein (NS) 2B and NS3 proteins was recognized as a conserved strategy used by flavivirus including dengue virus (DENV), WNV, ZIKV, and Japanese encephalitis virus (JEV) to establish infections in human (Aguirre et al., 2012; Yu et al., 2012; Ding et al., 2018). While ZIKV NS1 additionally cleaves cGAS, NS4B of Yellow Fever Virus (YFV) and DENV forms a complex with STING to achieve immune evasion (Ding et al., 2013; Nitta et al., 2013; Chan and Gack, 2016b). The protein product of human coronavirus NL6 and SARS-CoV papain-like proteases particularly disrupt antiviral cGAS-STING-mediated signaling by abolishing ubiquitin-STING conjugation (Sun et al., 2012; Xing et al., 2013; Chen et al., 2014). However, it remains to be investigated whether the inhibition of STING cascade is a significant evasion strategy during SARS-CoV-2 neuroinvasion. Moreover, characterizing the evasion strategies specifically employed by SARS-CoV-2 may present novel pharmacological targets.
Another pathway through which microglia detect CNS viral infections is the classical complement cascade. The complement system, a vital component of the innate immune pathogen defense, consists of approximately 30 proteins and membrane-bound receptors and regulators, which are also involved in pattern recognition and clearance (Ojha et al., 2014; Agrawal et al., 2017). Viruses, such as the WNV and their PAMPs induce complement activation within the CNS (Mehlhop et al., 2005; Vasek et al., 2016). In such infections, the complement system plays a key role in controlling viral propagation. This involves targeting and binding of viral particles and recruitment of proinflammatory peptides and immune cells as well as clearance of cells that express complement receptors. The complement component C3, along with its phagocytic receptor 3 (CR3/CD11b-CD18/Mac-1) expressed on the surface of microglia, recruits immune cells to the site of an injury thereby promoting internalization of synaptic structures. This was demonstrated using mice lacking C3- and its receptor CR3, which had reduced microglial engulfment of synapses (Schafer et al., 2012). Microglia also remove C1q-coated neurites through CR3-mediated internalization (Linnartz et al., 2012). Howbeit, viruses have developed a series of strategies in order to subvert complement detection mechanisms. This includes targeting of recognition molecules and key pathway enzymes, stimulation of proteases that cleave the complement proteins, and/or outright inhibition of the synthesis of complement proteins (Ojha et al., 2014; Agrawal et al., 2017). For instance, several studies report that flaviviruses including WNV, ZIKV, JEV, DENV, and YFV contain a conserved region which codes for a particular complement regulator, non-structural protein 1 (NS-1). This protein is necessary for viral RNA replication (Alcon-Lepoder et al., 2008). As a subversion strategy, the NS-1 protein antagonizes the component C4 (Conde et al., 2016) of the complement cascade, and recruits host major soluble inhibitor of the complement cascade C4 by complexing binding protein and factor H (Kyung et al., 2006). Also, NS-1 inhibits C9 polymerization (Conde et al., 2016), thus aiding flaviviruses to evade the complement detection system and thereby enhancing their survival in host cells.
Microglial Response During RNA Viral Infections
So far, about 180 species of RNA viruses with the capacity to infect humans have been recognized, and about two new species are added every year on average (Woolhouse et al., 2013). Compared to other groups of viral infection, viral RNA infections are more frightening because they are highly evolving with increased likelihood to infect a new host species due to their outstandingly shorter generation time. The high rate of replication makes the reproductive cycles more error-prone (Holmes, 2010). This gives RNA viruses a potential to quickly produce new strains within a shorter timeframe (Sanjuán and Domingo-Calap, 2016). Although an avalanche of reports have documented neurological symptoms related to COVID-19, the specific role, contributions and implications of microglia during SARS-CoV-2 infection and COVID-19 are still elusive. Thus, proper understanding of the myriads of ways microglia respond to neuroinvasive viral infections may provide insight into how to manipulate the pathogenic mediators to achieve effective viral control thereby advancing therapeutic development.
Antiviral type 1 IFN signaling cascade is pivotal to curtailing viral spread in the CNS parenchyma (Detje et al., 2009). For instance, intranasal VSV instillation at a dose that was harmless to wild-type mice resulted in death within 2–3 days in mice deficient in IFN-α/β receptor (IFNAR). However, the hemizygous mutant of IFNAR, which presented with about 100-fold high viral load, survived 5–6 days before the onset of mortality (Detje et al., 2009). Microglial functions may be pivotal to protecting the brain from neuropathological assaults mediated by viral infection and viral encephalitis (Chen et al., 2019; Hatton and Duncan, 2019). Evidence from rodent studies in the context of viral encephalitis correlated microglial ablation with reduced survival, amplified viral burden, and negative clinical outcomes, including the development of overt neurological diseases and mortalities (Sanchez-Mejias et al., 2016; Fekete et al., 2018; Seitz et al., 2018; Waltl et al., 2018; Sanchez et al., 2019). To unravel the specific contribution of microglial function against neuroinvasive infections, independent investigations in mice have shown that microglial cells can generate antiviral innate immune response (Sorgeloos et al., 2013). Using IFN-β promoter-luciferase reporter mice, Kallfass et al. (2013) observed that astrocytes and F4/80 positive cells, which could be either microglia or infiltrating macrophages, accounted for a significant IFN-β staining in the brain following intraperitoneal La Crosse virus (LACV) infection, even though LACV replicated largely in neurons. However, when mutant LACV-infected mice deficient in NS proteins were used, the IFN-β were mainly detected in astrocytes (Kallfass et al., 2013). Knowing that NS proteins of LACV are an inherent viral strategy to subvert host’s type 1 IFN antiviral responses (Blakqori et al., 2007), this may imply that the mechanism underlining IFN-β production by the F4/80-expressing cells may be non-redundant and more pivotal to the survival of the host cell, particularly when astrocytic antiviral mediators are suppressed. In similitude, the study of Wheeler et al. (2018) observed a significant mRNA expression of IFN-α4, IFN-β, and IL-6, which correlated with viral load in the olfactory bulb and brainstem when mice were intracranially injected with mouse hepatitis virus (MHV), and after microglia were pharmacological depleted with PLX5622 treatment for a week (Wheeler et al., 2018). PLX5622 is an inhibitor of microglia-expressed colony-stimulating factor 1 receptor (CSF1R) required for their survival, although some subsets of microglia are CSF1R negative and thus resistant to this depletion (Erblich et al., 2012). This further emphasizes the presence of extra-microglial antiviral and pro-inflammatory response to various neuroinvasive RNA viral infection, including flaviviruses (Seitz et al., 2018). However, this non-microglial response may be blunt against MHV considering that increasing viral titer and spread co-existed in this context with the expression of antiviral cytokines.
To differentiate the protective role of microglia, treatment of mice with PLX5622, a week before and after an intranasal instillation of neuroattenuated coronavirus MHV infection, was performed, resulting in 100% mortality. However, mice that did not receive PLX5622 survived (Wheeler et al., 2018). Enhanced mortality was also observed in PLX5622-mediated microglia-depleted JEV-infected mice (Seitz et al., 2018). When the timing of PLX5622 treatment was adjusted to day 0 or 3 post infection (p.i.), the survival of mice increased to 10 and 40%, respectively. However, administering PLX5622 on day 6 p.i. did not rescue the MHV-infected mice from mortality (Wheeler et al., 2018). This suggests that microglial activity is pivotal to surviving fatal neurotropic viral infection in mice especially at the early stage of infection. Again, this points to the involvement of microglia in early innate immune responses against neuroinvasive viral infections. A glimpse at the protective mechanism initiated by uninfected microglia against an invading RNA virus in the CNS showed that microglial antiviral type I IFN against VSV was potent at suppressing the anterograde trans-synaptic viral propagation (Drokhlyansky et al., 2017). When eGFP-labeled VSV (VSV-eGFP) or its type 1 IFN-stimulating virus-free interfering particles (DIPs) were injected into the caudate-putamen of mice, FACS-isolated microglial cells (CD11b+FCRLS+ cells) with or without VSV-eGFP infection upregulated the mRNA expression of IR7 to a level comparable to microglia from the DIP-injected brain (Drokhlyansky et al., 2017). IR7, a master regulator of type 1 IFN, and Rsad2, is an IFN stimulated gene (ISG). The DIP injection limited the lateral anterograde spread of the replication-competent rVSV-eGFP across the caudate-putamen circuitry on the contralateral side of the brain. However, microglia infected with VSV-eGFP expressed IFN-β and IL-1β, which were not upregulated in the DIP-injected brain through its antiviral IFN secretion. This suggests that viral-infected microglia are equipped with DIP-equivalent mechanism to limit transneuronal viral transmission and uninfected microglia are inductively primed via IFN-β paracrine signaling emanating from the infected cells. Meanwhile, excessive microglial reaction may result in more damage to the healthy neurons and synapses, hence resulting in further neurodegeneration (Lecours et al., 2018). Whether the additional pro-inflammatory IL-1β production is capable of contributing to neuropathology remains to be investigated.
Following adequate initiation of innate response, microglia may be presenting peptides from the invading neurotropic virus to initiate adaptive immune response and resolution of infection. Using intravital imaging, a recent study by Moseman et al. (2020) demonstrated that intranasal inoculation of mice with VSV and subsequent infection of the CNS was resolved non-cytolytically by cytotoxic T lymphocytes (CTL) through neuronal major histocompatibility complex 1 (MHC-1)-dependent microglial, but not neuronal, presentation of viral peptides. In the study, ablation of neuronal MHC-1 did not affect viral control, whereas depletion of microglia significantly interfered with viral clearance (Moseman et al., 2020). This suggests that lack of microglia may impair T cell recruitment and viral clearance. Following microglial ablation with PLX5622, which resulted in the loss of MHC-II expression in the brain, peripheral CD45hiCD11b+ macrophages with capability to initiate adaptive response were recruited to the brain. This is suggestive of a compensatory mechanism since PLX5622 does not affect their antigen presentation to CD4+ or CD8+ T cells in response to subclinical intraperitoneal MHV infection in the periphery (Wheeler et al., 2018), although a recent study demonstrated the side effect of PLX5622 on hematopoiesis and peripheral macrophages (Lei et al., 2020). Similarly, brain infiltrating cells expressing markers of microglia and invading monocytes, ionized calcium binding adaptor molecule 1 (IBA1) and Mac-3, were detected following microglial depletion with PLX5622 in a mouse model of picornavirus-mediated viral encephalitis-induced seizure development (Waltl et al., 2018). This emphasizes a microglial role in viral antigen presentation to T cells. Specifically, depletion of microglia with PLX5622 resulted in reduced frequency of CD4+ T cells and FOXP3+ Tregs in the draining lymph nodes of the brain, diminished IFN-γ expression by virus-specific CD4+ T cell response, and enhanced CD8+ T cells (Wheeler et al., 2018). However, using a lethal WNV infection, a recent study showed that 2 weeks of PLX5622 pre-treatment in mice was associated with the depletion of both microglia and infiltrating antigen presenting cells within the CNS, which resulted in limited CD8+ T cells reactivation and aberrant viral load in the CNS. As opposed to the lethal WNV, the subclinical MHV infection may model the current SARS-CoV-2 infection in human.
With respect to COVID-19, a detailed analysis of 43 post-mortem brains of patients by Matschke et al. (2020) revealed the involvement of microglia in the neuropathology of SARS-CoV-2. The study revealed via an in silico analysis of publicly available dataset that both neuronal and non-neuronal cells were vulnerable to SARS-CoV-2 infection with the neurons, oligodendrocytes, microglia, and astrocytes being the most enriched in viral entry apparatus, TMPRSS2/4, ACE2, cathepsin L, and two pore segment channel 2 (TPCN2) expression, respectively. Clinical CNS manifestation of COVID-19 was associated with spike or nucleocapsid mRNA and protein expression in the basal ganglia, cerebellum, frontal cortex, and medulla oblongata as well as pronounced leaky meninges. This is characterized by CTL infiltration clustering around microglia (defined by HLA-DR and CD68 staining) at the brainstem perivasculature (Matschke et al., 2020). Overall, this may imply significant infection and remodeling of perivascular microglia upon SARS-CoV-2 CNS entry and a potential crosstalk with CTL. Previous observations have also confirmed microgliosis in the brainstem (Deigendesch et al., 2020), but the characteristics of the remodeled microglia in response to COVID-19 or SARS-CoV-2 infection remain to be investigated.
Perspectives: Implications of Microglial Reactivity in SARS-CoV-2 Related Neuropsychiatric Disorders and Neurodegenerative Diseases
It is now generally accepted that many neuropsychiatric disorders and neurodegenerative diseases arise from the influence of environmental factors (Chin-Chan et al., 2015; Khan et al., 2019). The SARS-CoV-2 pandemic may overwhelmingly impact the mental health due to the multiple psychosocial stressors, such as self-isolation/quarantine, fear, anxiety, worry, social restriction, lockdown, and stigmatism created by the disease. Symptoms of neuropsychiatric disorders, such as obsessive-compulsive disorders, insomnia, depression, anxiety and psychoses are being reported in some cases and survivors of SARS-CoV-2 infection (Valdés-Florido et al., 2020). Psychosis is known as one of the neuropsychiatric disorders requiring special care and attention. Of note, since the days of the Spanish Flu pandemic, psychosis of influenza has been documented in many other pandemics (Kêpińska et al., 2020). Anecdotal clinical reports from mental health facilities have recorded increased paranoia amongst persons who are close to infected patients (Brown et al., 2020). Indeed, physical distancing measures have been proposed to serve as risk factor for increased vulnerability to neuropsychiatric disorder including psychosis (Brown et al., 2020). Moreover, evidence has linked both peripheral and neurotropic viral infections with neurodegenerative conditions (Karim et al., 2014). It is now clear that chronic HIV infection correlates with dementia and other neurocognitive disorders (Naghavi, 2018). Babies from ZIKV-infected mothers have microencephaly with consequences on their developmental and cognitive abilities into adulthood, while ZIKV causes neurodegenerative complications including myelitis, neuropathy and Guillain-Barre syndrome (Christian et al., 2019). Of note, different quarters are beginning to sensitize the public about the possibility of post-COVID-19 pandemic of neurodegenerative diseases (Serrano-Castro et al., 2020; Singal et al., 2020), given the neurological symptoms displayed during and after the infection (see section “Evidence of SARS-CoV-2 Neurotropism”). The question begging for answers is how SAR-CoV-2 viruses orchestrate the pathogenesis of neuropsychiatric disorders and neurodegenerative diseases, and whether microglia are involved. Experimental studies have shown that prolonged episode of chronic stress promotes dystrophic microglial phenotype with higher propensity for phagocytosis and apoptosis (Hinwood et al., 2013; Kreisel et al., 2014; Milior et al., 2016; Frank et al., 2019). Ablation of microglial C-X3-C motif chemokine receptor 1 (CX3CR1) in mice resulted in phenotypes associated with autism spectrum disorders including cognitive impairment (Kreisel et al., 2014), social withdrawal (Zhang et al., 2014) and resistance to chronic psychological stress-induced anhedonia- and anxiety-like phenotype (Wohleb et al., 2014; Milior et al., 2016). Also, coronavirus neurovirulence is associated with microglia-mediated up-regulation of proinflammatory signals for the recruitment of blood-derived inflammatory cells (Li et al., 2004; Olajide et al., 2020). Mice infected with mouse hepatitis virus (MHV)-A59 developed a meningoencephalopathy characterized by perivascular inflammation, microglial nodules, and astrocytic proliferation (Lavi et al., 1984; Das Sarma et al., 2000; Li et al., 2004). At 10-day p.i., when viral clearance was achieved in the neurons, viral RNA persisted in the astrocytes and microglia within the olfactory and limbic regions with continued chronic inflammatory demyelination as detected by in situ hybridization (Lavi et al., 1984; Das Sarma et al., 2000). Since microglia- and astrocyte-induced neuroinflammation are risk factors for the development of major depressive disorder (Brites and Fernandes, 2015; Troubat et al., 2020), as evident in individuals who committed suicide (Steiner et al., 2008; Schnieder et al., 2014), SARS-CoV-2 neurotropism may trigger or exacerbate neuropsychiatric disorders (Steardo et al., 2020). Also, recent advances in biological psychiatry have suggested that chronic psychosocial stress, such as the one generated by COVID-19 pandemic, could enhance microglial reactivity and impact significantly vulnerability of the brain to various neuropsychiatric disorders including depression, cognitive decline, and schizophrenia (Vargas et al., 2020). This suggests that microglia could contribute significantly to the changes in brain function including altered regulation of neuroendocrine, renin-angiotensin aldosterone tryptophan-kynurenine dysregulation, increased release of proinflammatory cytokines, chemokines, and neurotoxins in stress-sensitive regions (Suzuki et al., 2019; Picard et al., 2021). Moreover, stress-induced microglial remodeling has been linked to increased expression and function of catecholamine reuptake transporters or decrease catecholamine precursors; notably altering the synaptic availability of catecholamine neurotransmitters (Miller et al., 2017) all of which could be associated with the onset of neurological disorders in SAR-CoV-2 patients and/or survivors (Alharthy et al., 2020).
Evidence showed that SARS-CoV-2 infection may result in demyelination suggesting the possibility of immunopathogenic events that lead to the development of neurological disorders including multiple sclerosis (Wu and Perlman, 1999; Khateb et al., 2020). Moreover, an animal model of coronavirus (MHV-4) has been reported to induce demyelination (Fleming et al., 1987). In particular, coronavirus RNA sequences have been observed in the brain and demyelinating structures of multiple sclerosis patients using in situ hybridization (Murray et al., 1992). Some of the proposed mechanisms of coronavirus-induced demyelination include cytopathogenic properties of the virus for oligodendrocytes, which is linked to E2 sub-structure (Fleming et al., 1987) and T-cell cross-reactivity (Boucher et al., 2007). This is mainly orchestrated by T cell activation following widespread infection of the CNS parenchyma as lack of MHC I and II in β2-Macroglobulin–/– and Aβ–/– MHV-J2.2-v1 mice or deficiency of CD4 in CD4–/– mice resulted in reduced viral clearance with limited demyelination (Houtman, 1996; Lane et al., 2000). This implies that the recruitment of T cells, which is necessary for viral clearance, may also drive demyelination. Of note, Wu et al. (2000) strongly implicated CD8+ T cells, albeit in the presence of rapid viral spread (Marten et al., 2000), in the demyelination of spinal cord that accompanied MHV-JHM-infection in recombination-activating gene 1–/– (RAG1–/–) mice lacking T and B cells. A recent study by Kaddatz et al. (2020) demonstrated with a mouse cuprizone induced demyelinating model that CD8+ T cells, which predominated around the demyelinating foci, were highly proliferating with extensive cytotoxic granule. This is suggestive of antigenic-primed activated CD8+ T cells orchestrated by antigen presenting cells, possibly microglia. Meanwhile, in addition to regulating CD8+ T cells during viral neurotropic infection (Waltl et al., 2018), microglia play pivotal roles in the demyelination and remyelination within the CNS (Lampron et al., 2015; Laflamme et al., 2018). Therapeutic treatments of cuprizone-intoxicated mice with BLZ945, a pharmacological CSF1R kinase inhibitor, resulted in striatal and cortical remyelination, which correlated with reduced microglial, but enhanced oligodendroglial density (Beckmann et al., 2018). On the other hand, prophylactic BLZ945 treatment attenuated extensive demyelination in the corpus callosum with the oligodendroglial and microglial dynamics showing similar patterns to the therapeutic treatment (Beckmann et al., 2018). This suggests that microglial reactivity contributes negatively to demyelination. However, in the external capsule—which was not affected by BLZ945 prophylactic treatment—of either cuprizone-treated or triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice, oligodendrocytes were depleted, with accumulation of myelin debris and axonal damage without any impact on microglial density (Beckmann et al., 2018; Wies Mancini et al., 2019). This may be an indication of dysfunctional microglial phagocytic capacity during demyelination. Indeed, a dynamic gene expression profile with increasing mRNA upregulation of proinflammatory and phagocytic markers, which correlated with peak demyelination and was sustained even after viral clearance, was observed in a non-lethal glial-tropic MHV model of demyelination (Savarin et al., 2018). This may indicate the presence of a continuously reactive microglial phenotype during demyelination with huge implication on long-term demyelination. Nevertheless, cases of SARS-CoV-2-associated inflammatory CNS demyelinating diseases have been documented across the globe. For instance, acute multi-infarct encephalopathy was reported in a 40-year-old woman by Zhang et al. (2020b), while acute transverse myelitis (Durrani et al., 2020; Sarma and Bilello, 2020) and neuromyelitis optica were recently described in patient with SARS-CoV-2 infection (Miskin, 2020). However, it is yet to be unraveled if microglia are the villain in the pathophysiology of the CNS demyelinating diseases associated with SARS-CoV-2 infection.
Although aging has been established as one of the prominent factors responsible for the induction of neurodegenerative diseases, there is now a strong evidence that viral pathogens can precipitate or exacerbate neurodegenerative diseases including Parkinson’s disease (PD) (Matsui and Takahashi, 2009). Existing evidence showed that SARS-CoV-2 infection may worsen PD symptoms. For instance, 10 of 17 PD patients experienced severe PD symptoms and 25 of 214 experienced severe COVID-19 disease in a systematic review of 26 reports Kubota and Kuroda (2021). Also, 148 of 694 (vs. 4,074 of 74,065 in non-PD patients) in a cohort from the United States (Zhang et al., 2020c), as well as 23 of 117 PD patients followed in Spain, the United Kingdom, Iran and Italy across 21 health centers, died of COVID-19 complications (Fasano et al., 2020). Consequently, an area of future concern is whether SARS-CoV-2 infection would expand the exploding cases of PD worldwide (Otero-Losada et al., 2020). In the past, the incidence of parkinsonism was observed to increase after the Spanish flu pandemic in 1918 with people born during the pandemic having 2–3-fold risk to develop parkinsonism compared to people born before to 1888 or after 1924 (Jang et al., 2009; Eldeeb et al., 2020). Since then, association of various viruses and parkinsonism has been reported (Jang et al., 2009; Eldeeb et al., 2020). PD is the fastest growing neurodegenerative disease and movement disorder with a prevalence described to have achieved pandemic status (Dorsey et al., 2018). Among other etiological factors, aging and chronic stress are regarded as a major driver of PD (Reeve et al., 2014; Herrera et al., 2015; Dodiya et al., 2020). Available data confirmed that the severity and transmissibility of SARS-CoV-2 infection is proportional to age and aging remains a major risk factor for SARS-CoV-2 infection and severity of COVID-19 (Nanda et al., 2020). During aging, microglia within the SN take up remodeled dystrophic structural, physiologic and phenotypic features in the healthy state. The microglial “inflammaging” renders SN more vulnerable to any environmental assault which may contribute to the onset or progression of PD (Sharaf et al., 2013; Awogbindin et al., 2020). Moreover, the SN is a brain region enriched with SARS-CoV-2 receptors, including ACE2 and TMPRSS2 (Hamming et al., 2004). Also, a recent article suggested that SARS-CoV-2 may hijack the host by disrupting the mitochondrial, autophagic and lysosomal machineries, which are pivotal to microglial functions including synaptic pruning, neurogenesis, surveillance and phagocytosis (Colonna and Butovsky, 2017; Tay et al., 2017), via direct binding (Gordon et al., 2020). Moreover, infection with Influenza A virus (IAV) and SARS−CoV, which infiltrate the CNS via the olfactory canal like SARS-CoV-2, modulates cellular aging pathways (López-Otín et al., 2013). In addition, the virulent IAV, H1N1, infects dopaminergic neurons resulting in α-synuclein aggregation, the intraneuronal hallmark of PD, via a mechanism related to the inhibition of autophagy (Marreiros et al., 2020). Recently, intracytoplasmic SARS-CoV-2 was detected in the brain of COVID-19 patients but gliosis or microgliosis was not determined (Gomez-Pinedo et al., 2020). This is suggestive of a vacuolation which may template unfolded proteins associated with PD. In the long term, this speculates that SARS-CoV-2, through microglial dysfunction, may have potential to contribute to the progression and onset of PD in the aging population. Viruses, and possibly SARS-CoV-2, can be a precipitating factor in the development of PD. SARS-CoV-2, could be the first “hit” in a two-hit hypothesis, that could sensitize the brain to a later assault. Experimental evidence of a multi-hit PD hypothesis was shown in mice with a synergy between influenza and 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP) toxicity that was eliminated with influenza therapeutics (Sadasivan et al., 2017).
Concluding Remark
Taken together, microglia could play the role of a double-edged sword during viral neuroinfections. By extension, microglial reactivity, if initiated effectively, could orchestrate the clearance of SARS-CoV-2 in the CNS or trigger neuroinflammation and contribute to the severity of the sequelae associated with SARS-CoV-2 neurotropism. For instance, Olajide et al. (2020) recently demonstrated that SARS-CoV-2 spike S1 elicited a robust NF-kB/NLRP3 inflammasome-mediated pro-inflammatory response in BV-2 microglial cells, although the implication and the receptor(s) mediating the stimulatory effect of S1 glycoprotein were not investigated. Given that the adverse effect of microglial reactivity in the CNS exceptionally outlasts the direct damaging effect of viral neurotropism (Thakur et al., 2021), the present SARS-CoV-2 pandemic provides a global opportunity of proactive research to establish the predicted implications of microglia to avert potential incidence of neuropsychiatric disorders and neurodegenerative diseases, which could be pervasive in the coming years as a result of the growing numbers of cases, survivors and re-current waves.
Author Contributions
IA and M-ÈT conceived the review manuscript. IA prepared the figure. All the authors contributed to the design, writing, and revision.
Funding
IA was supported by an International Brain Research Organization African Regional Committee (IBRO-ARC) 2019 Fellowship at the Université Laval, Québec, QC, Canada. BB-A was a recipient of IBRO-ARC 2020 Fellowship. EA was a recipient of the Committee for Aid and Education in Neurochemistry (CAEN) 1A grant from the International Society for Neurochemistry (ISN). PA was a recipient of 2019 Young IBRO Regions Connecting Award. BO and IA were recipients of the University of Ibadan MEPI Junior Faculty Research Training Program (UI-MEPI-J) mentored research award through the National Institute of Health (NIH), United States grant funded by Fogarty International Centre, the office of AIDS Research and National Human Genome Research Institute of NIH, the Health Resources and Services Administration (HRSA), and the Office of the U.S. Global AIDS Coordinator under award number D43TW01014. MÈ-T holds a Canada Research Chair (Tier 2) in Neurobiology of Aging and Cognition. TD and M-ÈT hold an operating grant from the Canadian Institutes of Health Research (#341846) for their work on microglia and Parkinson’s disease.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge that the University of Victoria is located on the territory of the Lekwungen peoples and that the Songhees, Esquimalt and WSÁNEĆ peoples have relationships to this land.
Glossary
| ACE2, | angiotensin-converting enzyme 2 |
| ADEM, | acute disseminated encephalomyelitis |
| AHLE, | acute hemorrhagic necrotizing encephalitis |
| AIM2, | absent in melanoma 2 |
| BBB, | blood-brain barrier |
| cGAS, | cyclic GMP-AMP synthase |
| COVID-19, | coronavirus disease 2019 |
| CSF, | cerebrospinal fluid |
| CSF1R, | colony-stimulating factor 1 receptor |
| CTL, | cytotoxic T lymphocytes |
| DAMPs, | damage-associated molecular patterns |
| DENV, | dengue virus |
| FcR, | Fc receptor |
| FLAIR, | fluid-attenuated inversion recovery |
| HCoV, | human coronavirus |
| hiPSC, | human-induced pluripotent stem cells |
| hNPCs, | human neural progenitor cells |
| IFN-α/β, | interferon α/β |
| IL-1/6, | interleukin 1/6 |
| IRF3, | interferon Regulatory Factor 3 |
| ISG, | Interferon Stimulated Gene |
| JEV, | Japanese encephalitis virus |
| LACV, | La crosse virus |
| MERS-CoV, | middle east respiratory syndrome coronavirus |
| MHV, | mouse hepatitis virus |
| MRI, | magnetic resonance imaging |
| mTOR, | rapamycin |
| MyD88, | myeloid differentiation primary response 88 |
| NF-κB, | nuclear factor kappa light chain enhancer of activated B cells |
| NLRs, | nucleotide oligomerization domain (NOD)-like receptors |
| NOD, | nucleotide-binding oligomerization domain |
| NRP-1, | neuropilin-1 |
| PAMPs, | pathogen-associated molecular patterns |
| PRRs, | pattern recognition receptors |
| RIG-1, | retinoic acid-inducible gene I |
| RLRs, | retinoic acid-inducible gene (RIG)-I-like receptors |
| RNA, | ribonucleic acid |
| SARS-CoV, | severe acute respiratory syndrome coronavirus |
| STING, | stimulator of type I interferon genes |
| TBK1, | TANK-binding kinase 1 |
| TLR, | toll-like receptors |
| TMPRSS2, | rans membrane protease serine 2 |
| TNF-α, | tumor necrosis factor alpha |
| TRAM, | TRIF-related adaptor molecule |
| TRIF TIR, | domain-containing adaptor protein-inducing interferon β |
| WNV, | West Nile virus |
| YFV, | Yellow fever virus |
| ZIKV, | Zika virus |
References
Adhikari, S. P., Meng, S., Wu, Y. J., Mao, Y. P., Ye, R. X., Wang, Q. Z., et al. (2020). Epidemiology, causes, clinical manifestation and diagnosis, prevention and control of coronavirus disease (COVID-19) during the early outbreak period: a scoping review. Infect. Dis. Poverty 9:29. doi: 10.1186/s40249-020-00646-x
Aghagoli, G., Gallo Marin, B., Katchur, N. J., Chaves-Sell, F., Asaad, W. F., and Murphy, S. A. (2020). Neurological involvement in COVID-19 and potential mechanisms: a review. Neurocrit. Care [Epub ahead of print]. doi: 10.1007/s12028-020-01049-4
Agrawal, P., Nawadkar, R., Ojha, H., Kumar, J., and Sahu, A. (2017). Complement evasion strategies of viruses: an overview. Front. Microbiol. 8:1117. doi: 10.3389/fmicb.2017.01117
Aguirre, S., Maestre, A. M., Pagni, S., Patel, J. R., Savage, T., Gutman, D., et al. (2012). DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 8:e1002934. doi: 10.1371/journal.ppat.1002934
Aguzzi, A., Barres, B. A., and Bennett, M. L. (2013). Microglia: scapegoat, saboteur, or something else? Science 339, 156–161. doi: 10.1126/science.1227901
Ahmad, I., and Rathore, F. A. (2020). Neurological manifestations and complications of COVID-19: a literature review. J. Clin. Neurosci. 77, 8–12. doi: 10.1016/j.jocn.2020.05.017
Alcon-Lepoder, S., Sivard, P., Drouet, M. T., Talarmin, A., Rice, C., and Flamand, M. (2008). “Secretion of flaviviral non-structural protein NS1: from diagnosis to pathogenesis,” in New Treatment Strategies for Dengue and Other Flaviviral Diseases, eds G. R. Bock and J. A. Goode (Hoboken, NJ: Wiley), 233–250.
Alharthy, A., Faqihi, F., Memish, Z. A., and Karakitsos, D. (2020). Fragile endothelium and brain dysregulated neurochemical activity in COVID-19. ACS Chem. Neurosci. 11, 2159–2162. doi: 10.1021/acschemneuro.0c00437
Awogbindin, I. O., Ishola, I. O., St-Pierre, M. K., Carrier, M., Savage, J. C., Di Paolo, T., et al. (2020). Remodeling microglia to a protective phenotype in Parkinson’s disease? Neurosci. Lett. 735:135164. doi: 10.1016/j.neulet.2020.135164
Ayegbusi, O. T., Ajagbe, O. A., Afowowe, T. O., Aransi, A. T., Olusola, B. A., Awogbindin, I. O., et al. (2018). Virus genes and host correlates of pathology are markedly reduced during respiratory syncytial and in fl uenza virus co-infection in BALB / c mice. Heliyon 5:e01094. doi: 10.1016/j.heliyon.2018.e01094
Bajwa, E., Pointer, C. B., and Klegeris, A. (2019). The role of mitochondrial damage-associated molecular patterns in chronic neuroinflammation. Mediat. Inflamm. 2019:4050796. doi: 10.1155/2019/4050796
Bastard, P., Rosen, L. B., Zhang, Q., Michailidis, E., Hoffmann, H. H., Zhang, Y., et al. (2020). Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370:eabd4585. doi: 10.1126/science.abd4585
Barral, P. M., Sarkar, D., Fisher, P. B., and Racaniello, V. R. (2009). RIG-I is cleaved during picornavirus infection. Virology 391, 171–176. doi: 10.1016/j.virol.2009.06.045
Beckmann, N., Giorgetti, E., Neuhaus, A., Zurbruegg, S., Accart, N., Smith, P., et al. (2018). Brain region-specific enhancement of remyelination and prevention of demyelination by the CSF1R kinase inhibitor BLZ945. Acta Neuropathol. Commun. 6:9. doi: 10.1186/s40478-018-0510-8
Beignon, A. S., McKenna, K., Skoberne, M., Manches, O., DaSilva, I., Kavanagh, D. G., et al. (2005). Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Invest. 115, 3265–3275. doi: 10.1172/JCI26032
Bellver-Landete, V., Bretheau, F., Mailhot, B., Vallières, N., Lessard, M., Janelle, M. E., et al. (2019). Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat. Commun. 10:518. doi: 10.1038/s41467-019-08446-0
Benameur, K., Agarwal, A., Auld, S. C., Butters, M. P., Webster, A. S., Ozturk, T., et al. (2020). Encephalopathy and encephalitis associated with cerebrospinal fluid cytokine alterations and coronavirus disease, atlanta, georgia, usa, 2020. Emerg. Infect. Dis. 26, 2016–2021. doi: 10.3201/eid2609.202122
Benarroch, E. E. (1993). The central autonomic network: functional organization, dysfunction, and perspective. Mayo Clin. Proc. 68, 988–1001.
Bergamini, A., Faggioli, E., Bolacchi, F., Gessani, S., Cappannoli, L., Uccella, I., et al. (2018). Enhanced production of tumor necrosis factor- a and interleukin-6 due to prolonged response to lipopolysaccharide in human macrophages infected in vitro with human immunodeficiency virus type 1. J. Infect. Dis. 179, 832–842.
Bieback, K., Lien, E., Klagge, I. M., Avota, E., Schneider-Schaulies, J., Duprex, W. P., et al. (2002). Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J. Virol. 76, 8729–8736. doi: 10.1128/jvi.76.17.8729-8736.2002
Blakqori, G., Delhaye, S., Habjan, M., Blair, C. D., Sánchez-Vargas, I., Olson, K. E., et al. (2007). La crosse bunyavirus nonstructural protein NSs serves to suppress the type I interferon system of mammalian hosts. J. Virol. 81, 4991–4999. doi: 10.1128/jvi.01933-06
Blanco-Melo, D., Nilsson-Payant, B. E., Liu, W. C., Uhl, S., Hoagland, D., Møller, R., et al. (2020). Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181, 1036.e9–1045.e9. doi: 10.1016/j.cell.2020.04.026
Bonaz, B., Sinniger, V., and Pellissier, S. (2020). Targeting the cholinergic anti-inflammatory pathway with vagus nerve stimulation in patients with Covid-19? Bioelectron. Med. 6:15. doi: 10.1186/s42234-020-00051-7
Boucher, A., Desforges, M., Duquette, P., and Talbot, P. J. (2007). Long-term human coronavirus-myelin cross-reactive T-cell clones derived from multiple sclerosis patients. Clin. Immunol. 123, 258–267. doi: 10.1016/j.clim.2007.02.002
Brites, D., and Fernandes, A. (2015). Neuroinflammation and depression: microglia activation, extracellular microvesicles and microRNA dysregulation. Front. Cell. Neurosci. 9:476. doi: 10.3389/fncel.2015.00476
Brown, A. S. (2012). Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev. Neurobiol. 72, 1272–1276. doi: 10.1002/dneu.22024
Brown, E., Gray, R., Lo Monaco, S., O’Donoghue, B., Nelson, B., Thompson, A., et al. (2020). The potential impact of COVID-19 on psychosis: a rapid review of contemporary epidemic and pandemic research. Schizophr. Res. 222, 79–87. doi: 10.1016/j.schres.2020.05.005
Cantuti-Castelvetri, L., Ojha, R., Pedro, L. D., Djannatian, M., Franz, J., Kuivanen, S., et al. (2020). Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 370:eabd2985. doi: 10.1126/science.abd2985
Cárdenas, W. B., Loo, Y.-M., Gale, M., Hartman, A. L., Kimberlin, C. R., Martínez-Sobrido, L., et al. (2006). Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol. 80, 5168–5178. doi: 10.1128/jvi.02199-05
Chan, Y. K., and Gack, M. U. (2016a). A phosphomimetic-based mechanism of dengue virus to antagonize innate immunity. Nat. Immunol. 17, 523–530. doi: 10.1038/ni.3393
Chan, Y. K., and Gack, M. U. (2016b). Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 14, 360–373. doi: 10.1038/nrmicro.2016.45
Changeux, J. P., Amoura, Z., Rey, F. A., and Miyara, M. (2020). A nicotinic hypothesis for Covid-19 with preventive and therapeutic implications. Comptes Rendus. Biol. 343, 33–39. doi: 10.5802/crbiol.8
Chauhan, V. S., Sterka, D. G., Gray, D. L., Bost, K. L., and Marriott, I. (2008). Neurogenic exacerbation of microglial and astrocyte responses to Neisseria meningitidis and Borrelia burgdorferi. J. Immunol. 180, 8241–8249. doi: 10.4049/jimmunol.180.12.8241
Chen, G., Wu, D., Guo, W., Cao, Y., Huang, D., Wang, H., et al. (2020). Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 130, 2620–2629. doi: 10.1172/JCI137244
Chen, N., Xia, P., Li, S., Zhang, T., Wang, T. T., and Zhu, J. (2017). RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life 69, 297–304. doi: 10.1002/iub.1625
Chen, R., Wang, K., Yu, J., Howard, D., French, L., Chen, Z., et al. (2020). The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in human and mouse brain. bioRxiv [Preprint]. doi: 10.1101/2020.04.07.030650
Chen, T., Wu, D., Chen, H., Yan, W., Yang, D., Chen, G., et al. (2020). Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ 368:m1295. doi: 10.1136/bmj.m1091
Chen, X., Yang, X., Zheng, Y., Yang, Y., Xing, Y., and Chen, Z. (2014). SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell 5, 369–381. doi: 10.1007/s13238-014-0026-3
Chen, Y., Liu, Q., and Guo, D. (2020). Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 92, 418–423. doi: 10.1002/jmv.25681
Chen, Z., and Trapp, B. D. (2016). Microglia and neuroprotection. J. Neurochem. 136, 10–17. doi: 10.1111/jnc.13062
Chen, Z., Zhong, D., and Li, G. (2019). The role of microglia in viral encephalitis: a review. J. Neuroinflamm. 16, 250–258. doi: 10.1186/s12974-019-1443-2
Chiang, C., Pauli, E.-K., Biryukov, J., Feister, K. F., Meng, M., White, E. A., et al. (2017). The human papillomavirus E6 oncoprotein targets USP15 and TRIM25 To suppress RIG-I-mediated innate immune signaling. J. Virol. 92:e01737-17. doi: 10.1128/jvi.01737-17
Chiappini, E., Licari, A., Motisi, M. A., Manti, S., Marseglia, G. L., Galli, L., et al. (2020). Gastrointestinal involvement in children with SARS-COV-2 infection: an overview for the pediatrician. Pediatr. Allergy Immunol. 31, 92–95. doi: 10.1111/pai.13373
Chigr, F., Merzouki, M., and Najimi, M. (2020). Comment on “The neuroinvasive potential of SARS-CoV-2 may play a role in the respiratory failure of COVID-19 patients.”. J. Med. Virol. 92, 703–704. doi: 10.1002/jmv.25960
Chin-Chan, M., Navarro-Yepes, J., and Quintanilla-Vega, B. (2015). Environmental pollutants as risk factors for neurodegenerative disorders: alzheimer and Parkinson diseases. Front. Cell. Neurosci. 9:124. doi: 10.3389/fncel.2015.00124
Choudhury, A., and Mukherjee, S. (2020). In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 92, 2105–2113. doi: 10.1002/jmv.25987
Christian, K. M., Song, H., and Ming, G. L. (2019). Pathophysiology and mechanisms of zika virus infection in the nervous system. Annu. Rev. Neurosci. 42, 249–269. doi: 10.1146/annurev-neuro-080317-062231
Colonna, M., and Butovsky, O. (2017). Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 35, 441–468. doi: 10.1146/annurev-immunol-051116-052358
Conde, J. N., da Silva, E. M., Allonso, D., Coelho, D. R., Andrade, I., da, S., et al. (2016). Inhibition of the membrane attack complex by dengue virus NS1 through interaction with vitronectin and terminal complement proteins. J. Virol. 90, 9570–9581. doi: 10.1128/jvi.00912-16
Costela-Ruiz, V. J., Illescas-Montes, R., Puerta-Puerta, J. M., Ruiz, C., and Melguizo-Rodríguez, L. (2020). SARS-CoV-2 infection: the role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 54, 62–75. doi: 10.1016/j.cytogfr.2020.06.001
Cox, D. J., Field, R. H., Williams, D. G., Baran, M., Bowie, A. G., Cunningham, C., et al. (2015). DNA sensors are expressed in astrocytes and microglia in vitro and are upregulated during gliosis in neurodegenerative disease. Glia 63, 812–825. doi: 10.1002/glia.22786
Crill, E. K., Furr-Rogers, S. R., and Marriott, I. (2015). RIG-I is required for VSV-induced cytokine production by murine glia and acts in combination with DAI to initiate responses to HSV-1. Glia 63, 2168–2180. doi: 10.1002/glia.22883
Cui, J., Zhu, L., Xia, X., Wang, H. Y., Legras, X., Hong, J., et al. (2010). NLRC5 Negatively regulates the NF-κB and type I interferon signaling pathways. Cell 141, 483–496. doi: 10.1016/j.cell.2010.03.040
Cunningham, C. L., Martínez-Cerdeño, V., and Noctor, S. C. (2013). Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. 33, 4216–4233. doi: 10.1523/JNEUROSCI.3441-12.2013
Daffis, S., Samuel, M. A., Suthar, M. S., Gale, M., and Diamond, M. S. (2008). Toll-like receptor 3 has a protective role against west nile virus infection. J. Virol. 82, 10349–10358. doi: 10.1128/jvi.00935-08
Dantzer, R. (2018). Neuroimmune interactions: from the brain to the immune system and vice versa. Physiol. Rev. 98, 477–504. doi: 10.1152/physrev.00039.2016
Das Sarma, J., Fu, L., Tsai, J. C., Weiss, S. R., and Lavi, E. (2000). Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J. Virol. 74, 9206–9213. doi: 10.1128/jvi.74.19.9206-9213.2000
de Oliveira, A. P., Lopes, A. L. F., Pacheco, G., de Sá Guimarães Nolêto, I. R., Nicolau, L. A. D., and Medeiros, J. V. R. (2020). Premises among SARS-CoV-2, dysbiosis and diarrhea: walking through the ACE2/mTOR/autophagy route. Med. Hypotheses 144:110243. doi: 10.1016/j.mehy.2020.110243
Deigendesch, N., Sironi, L., Kutza, M., Wischnewski, S., Fuchs, V., Hench, J., et al. (2020). Correlates of critical illness-related encephalopathy predominate postmortem COVID-19 neuropathology. Acta Neuropathol. 140, 583–586. doi: 10.1007/s00401-020-02213-y
Detje, C. N., Meyer, T., Schmidt, H., Kreuz, D., Rose, J. K., Bechmann, I., et al. (2009). Local Type I IFN receptor signaling protects against virus spread within the central nervous system. J. Immunol. 182, 2297–2304. doi: 10.4049/jimmunol.0800596
Ding, Q., Cao, X., Lu, J., Huang, B., Liu, Y. J., Kato, N., et al. (2013). Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 59, 52–58. doi: 10.1016/j.jhep.2013.03.019
Ding, Q., Gaska, J. M., Douam, F., Wei, L., Kim, D., Balev, M., et al. (2018). Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc. Natl. Acad. Sci. U.S.A. 115, E6310–E6318. doi: 10.1073/pnas.1803406115
Dodiya, H. B., Forsyth, C. B., Voigt, R. M., Engen, P. A., Patel, J., Shaikh, M., et al. (2020). Chronic stress-induced gut dysfunction exacerbates Parkinson’s disease phenotype and pathology in a rotenone-induced mouse model of Parkinson’s disease. Neurobiol. Dis. 135:104352. doi: 10.1016/j.nbd.2018.12.012
Doobay, M. F., Talman, L. S., Obr, T. D., Tian, X., Davisson, R. L., and Lazartigues, E. (2007). Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am. J. Physiol. - Regul. Integr. Comp. Physiol. 292, R373–R381. doi: 10.1152/ajpregu.00292.2006
Dorsey, E. R., Sherer, T., Okun, M. S., and Bloemd, B. R. (2018). The emerging evidence of the Parkinson pandemic. J. Parkinsons. Dis. 8, S3–S8. doi: 10.3233/JPD-181474
Drokhlyansky, E., Aytürk, D. G., Soh, T. K., Chrenek, R., O’Loughlin, E., Madore, C., et al. (2017). The brain parenchyma has a type I interferon response that can limit virus spread. Proc. Natl. Acad. Sci. U. S. A. 114, E95–E104. doi: 10.1073/pnas.1618157114
Dubé, M., Le Coupanec, A., Wong, A. H. M., Rini, J. M., Desforges, M., and Talbot, P. J. (2018). Axonal transport enables neuron-to-neuron propagation of human coronavirus OC43. J. Virol. 92, e404–e418. doi: 10.1128/jvi.00404-18
Durrani, M., Kucharski, K., Smith, Z., and Fien, S. (2020). Acute transverse myelitis secondary to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): a case report. Clin. Pract. Cases Emerg. Med. 4, 344–348. doi: 10.5811/cpcem.2020.6.48462
Eldeeb, M. A., Hussain, F. S., and Siddiqi, Z. A. (2020). COVID-19 infection may increase the risk of parkinsonism – Remember the Spanish flu? Cytokine Growth Factor Rev. 54, 6–7. doi: 10.1016/j.cytogfr.2020.06.009
Erblich, B., Zhu, L., Etgen, A. M., Dobrenis, K., and Pollard, J. W. (2012). Correction: absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One 7:e0026317. doi: 10.1371/journal.pone.0026317
Facci, L., Barbierato, M., and Skaper, S. D. (2018). Astrocyte/microglia cocultures as a model to study neuroinflammation. Methods Mol. Biol. 1727, 127–137. doi: 10.1007/978-1-4939-7571-6_10
Fasano, A., Elia, A. E., Dallocchio, C., Canesi, M., Alimonti, D., Sorbera, C., et al. (2020). Predictors of COVID-19 outcome in Parkinson’s disease. Park. Relat. Disord. 78, 134–137. doi: 10.1016/j.parkreldis.2020.08.012
Faul, E. J., Wanjalla, C. N., Suthar, M. S., Gale, M., Wirblich, C., and Schnell, M. J. (2010). Rabies virus infection induces type I interferon production in an IPS-1 dependent manner while dendritic cell activation relies on IFNAR signaling. PLoS Pathog. 6:e1001016. doi: 10.1371/journal.ppat.1001016
Fehr, A. R., and Perlman, S. (2015). Coronaviruses: an overview of their replication and pathogenesis. Coronaviruses Methods Protoc. 1282, 1–23. doi: 10.1007/978-1-4939-2438-7_1
Fekete, R., Cserép, C., Lénárt, N., Tóth, K., Orsolits, B., Martinecz, B., et al. (2018). Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms. Acta Neuropathol. 136, 461–482. doi: 10.1007/s00401-018-1885-0
Fleming, J. O., Trousdale, M. D., Bradbury, J., Stohlman, S. A., and Weiner, L. P. (1987). Experimental demyelination induced by coronavirus JHM (MHV-4): molecular identification of a viral determinant of paralytic disease. Microb. Pathog. 3, 9–20. doi: 10.1016/0882-4010(87)90033-7
Fleshner, M., and Crane, C. R. (2017). Exosomes, DAMPs and miRNA: features of stress physiology and immune homeostasis. Trends Immunol. 38, 768–776. doi: 10.1016/j.it.2017.08.002
Frank, M. G., Fonken, L. K., Watkins, L. R., and Maier, S. F. (2019). Microglia: neuroimmune-sensors of stress. Semin. Cell Dev. Biol. 94, 176–185. doi: 10.1016/j.semcdb.2019.01.001
Fredericksen, B. L., Keller, B. C., Fornek, J., Katze, M. G., and Gale, M. (2008). Establishment and maintenance of the innate antiviral response to west nile virus involves both RIG-I and MDA5 signaling through IPS-1. J. Virol. 82, 609–616. doi: 10.1128/jvi.01305-07
Furr, S. R., and Marriott, I. (2012). Viral CNS infections: role of glial pattern recognition receptors in neuroinflammation. Front. Microbiol. 3:201. doi: 10.3389/fmicb.2012.00201
Furr, S. R., Moerdyk-Schauwecker, M., Grdzelishvili, V. Z., and Marriott, I. (2010). RIG-I mediates nonsegmented negative-sense RNA virus-induced inflammatory immune responses of primary human astrocytes. Glia 58, 1620–1629. doi: 10.1002/glia.21034
Gack, M. U., Shin, Y. C., Joo, C., Urano, T., Liang, C., Sun, L., et al. (2007). TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920. doi: 10.1038/nature05732
Garg, R. K., Paliwal, V. K., and Gupta, A. (2020). Encephalopathy in patients with COVID-19: a review. J. Med. Virol. 93, 206–222. doi: 10.1002/jmv.26207
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Gitlin, L., Barchet, W., Gilfillan, S., Cella, M., Beutler, B., Flavell, R. A., et al. (2006). Essential role of mda-5 in type I IFN responses to polyriboinosinic: polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. U.S.A. 103, 8459–8464. doi: 10.1073/pnas.0603082103
Gomez Perdiguero, E., Klapproth, K., Schulz, C., Busch, K., Azzoni, E., Crozet, L., et al. (2015). Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551. doi: 10.1038/nature13989
Gomez-Pinedo, U., Matias-Guiu, J., Sanclemente-Alaman, I., Moreno-Jimenez, L., Montero-Escribano, P., and Matias-Guiu, J. A. (2020). SARS-CoV-2 as a potential trigger of neurodegenerative diseases. Mov. Disord. 35, 1104–1105. doi: 10.1002/mds.28179
Gordon, D. E., Jang, G. M., Bouhaddou, M., Xu, J., Obernier, K., White, K. M., et al. (2020). A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468. doi: 10.1038/s41586-020-2286-9
Gousseff, M., Penot, P., Gallay, L., Batisse, D., Benech, N., Bouiller, K., et al. (2020). Clinical recurrences of COVID-19 symptoms after recovery: viral relapse, reinfection or inflammatory rebound? J. Infect. 81, 816–846. doi: 10.1016/j.jinf.2020.06.073
Guillot, L., Le Goffic, R., Bloch, S., Escriou, N., Akira, S., Chignard, M., et al. (2005). Involvement of Toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J. Biol. Chem. 280, 5571–5580. doi: 10.1074/jbc.M410592200
Habjan, M., Andersson, I., Klingström, J., Schümann, M., Martin, A., Zimmermann, P., et al. (2008). Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS One 3:e2032. doi: 10.1371/journal.pone.0002032
Hamming, I., Timens, W., Bulthuis, M. L. C., Lely, A. T., Navis, G. J., and van Goor, H. (2004). Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203, 631–637. doi: 10.1002/path.1570
Hardy, A. W., Graham, D. R., Shearer, G. M., and Herbeuval, J. P. (2007). HIV turns plasmacytoid dendritic cells (pDC) into TRAIL-expressing killer pDC and down-regulates HIV coreceptors by Toll-like receptor 7-induced IFN-α. Proc. Natl. Acad. Sci. U.S.A. 104, 17453–17458. doi: 10.1073/pnas.0707244104
Haslwanter, D., Blaas, D., Heinz, F. X., and Stiasny, K. (2017). A novel mechanism of antibody-mediated enhancement of flavivirus infection. PLoS Pathog. 13:e1006643. doi: 10.1371/journal.ppat.1006643
Hatton, C. F., and Duncan, C. J. A. (2019). Microglia are essential to protective antiviral immunity: lessons from mouse models of viral encephalitis. Front. Immunol. 10:2656. doi: 10.3389/fimmu.2019.02656
Helms, J., Kremer, S., Merdji, H., Clere-Jehl, R., Schenck, M., Kummerlen, C., et al. (2020). Neurologic features in severe SARS-CoV-2 infection. N. Engl. J. Med. 382, 2268–2270. doi: 10.1056/NEJMC2008597
Herrera, A. J., Espinosa-Oliva, A. M., Carrillo-Jiménez, A., Oliva-Martín, M. J., García-Revilla, J., García-Quintanilla, A., et al. (2015). Relevance of chronic stress and the two faces of microglia in Parkinson’s disease. Front. Cell. Neurosci. 9:312. doi: 10.3389/fncel.2015.00312
Hickman, S. E., Kingery, N. D., Ohsumi, T. K., Borowsky, M. L., Wang, L. C., Means, T. K., et al. (2013). The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905. doi: 10.1038/nn.3554
Hinwood, M., Tynan, R. J., Charnley, J. L., Beynon, S. B., Day, T. A., and Walker, F. R. (2013). Chronic stress induced remodeling of the prefrontal cortex: structural re-organization of microglia and the inhibitory effect of minocycline. Cereb. Cortex 23, 1784–1797. doi: 10.1093/cercor/bhs151
Holmes, E. C. (2010). The comparative genomics of viral emergence. Proc. Natl. Acad. Sci. U.S.A. 107, 1742–1746. doi: 10.1073/pnas.0906193106
Hosseini, A. A., Shetty, A. K., Sprigg, N., Auer, D. P., and Constantinescu, C. S. (2020). Delirium as a presenting feature in COVID-19: neuroinvasive infection or autoimmune encephalopathy? Brain Behav. Immun. 88, 68–70. doi: 10.1016/j.bbi.2020.06.012
Houtman, J. J. (1996). Dissociation of demyelination and viral clearance in congenitally immunodeficient mice infected with murine coronavirus JHM. J. Neurovirol. 2, 101–110. doi: 10.3109/13550289609146543
Hu, J., Jolkkonen, J., and Zhao, C. (2020). Neurotropism of SARS-CoV-2 and its neuropathological alterations: similarities with other coronaviruses. Neurosci. Biobehav. Rev. 119, 184–193. doi: 10.1016/j.neubiorev.2020.10.012
Hu, X., Cai, X., Song, X., Li, C., Zhao, J., Luo, W., et al. (2020). Possible SARS-coronavirus 2 inhibitor revealed by simulated molecular docking to viral main protease and host toll-like receptor. Future Virol. 15, 359–368. doi: 10.2217/fvl-2020-0099
Ising, C., and Heneka, M. T. (2018). Functional and structural damage of neurons by innate immune mechanisms during neurodegeneration review-Article. Cell Death Dis. 9:120. doi: 10.1038/s41419-017-0153-x
Iwasaki, A. (2020). What reinfections mean for COVID-19. Lancet Infect. Dis. 21, 3–5. doi: 10.1016/s1473-3099(20)30783-0
Jang, H., Boltz, D. A., Webster, R. G., and Smeyne, R. J. (2009). Viral parkinsonism. Biochim. Biophys. Acta Mol. Basis Dis. 1792, 714–721. doi: 10.1016/j.bbadis.2008.08.001
Jeffries, A. M., and Marriott, I. (2017). Human microglia and astrocytes express cGAS-STING viral sensing components. Neurosci. Lett. 658, 53–56. doi: 10.1016/j.neulet.2017.08.039
Jensen, S., and Thomsen, A. R. (2012). Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. 86, 2900–2910. doi: 10.1128/JVI.05738-11
Jha, M. K., Jo, M., Kim, J. H., and Suk, K. (2019). Microglia-astrocyte crosstalk: an intimate molecular conversation. Neuroscientist 25, 227–240. doi: 10.1177/1073858418783959
Jin, Y., Yang, H., Ji, W., Wu, W., Chen, S., Zhang, W., et al. (2020). Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses 12, 1–17. doi: 10.3390/v12040372
Jureka, A. S., Kleinpeter, A. B., Cornilescu, G., Cornilescu, C. C., and Petit, C. M. (2015). Structural basis for a novel interaction between the NS1 protein derived from the 1918 influenza virus and RIG-I. Structure 23, 2001–2010. doi: 10.1016/j.str.2015.08.007
Kabir, M. T., Uddin, M. S., Hossain, M. F., Abdulhakim, J. A., Alam, M. A., Ashraf, G. M., et al. (2020). nCOVID-19 pandemic: from molecular pathogenesis to potential investigational therapeutics. Front. Cell Dev. Biol. 8:616. doi: 10.3389/fcell.2020.00616
Kaddatz, H., Joost, S., Nedelcu, J., Chrzanowski, U., Schmitz, C., Gingele, S., et al. (2020). Cuprizone-induced demyelination triggers a CD8 -pronounced T cell recruitment. Glia 69, 925–942. doi: 10.1002/glia.23937
Kallfass, C., Lienenklaus, S., Weiss, S., and Staeheli, P. (2013). Visualizing the beta interferon response in mice during infection with influenza a viruses expressing or lacking nonstructural protein 1. J. Virol. 87, 6925–6930. doi: 10.1128/jvi.00283-13
Kanneganti, T. D., Body-Malapel, M., Amer, A., Park, J. H., Whitfield, J., Franchi, L., et al. (2006). Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 281, 36560–36568. doi: 10.1074/jbc.M607594200
Karim, S., Mirza, Z., Kamal, M., Abuzenadah, A., Azhar, E., Al-Qahtani, M., et al. (2014). The role of viruses in neurodegenerative and neurobehavioral diseases. CNS Neurol. Disord. Drug Targets 13, 1213–1223. doi: 10.2174/187152731307141015122638
Kato, H., Sato, S., Yoneyama, M., Yamamoto, M., Uematsu, S., Matsui, K., et al. (2005). Cell type-specific involvement of RIG-I in antiviral response. Immunity 23, 19–28. doi: 10.1016/j.immuni.2005.04.010
Kawasaki, T., and Kawai, T. (2014). Toll-like receptor signaling pathways. Front. Immunol. 5:461. doi: 10.3389/fimmu.2014.00461
Kępińska, A. P., Iyegbe, C. O., Vernon, A. C., Yolken, R., Murray, R. M., and Pollak, T. A. (2020). Schizophrenia and influenza at the centenary of the 1918-1919 spanish influenza pandemic: mechanisms of psychosis risk. Front. Psychiatry 11:72. doi: 10.3389/fpsyt.2020.00072
Khan, A., Plana-Ripoll, O., Antonsen, S., Brandt, J., Geels, C., Landecker, H., et al. (2019). Environmental pollution is associated with increased risk of psychiatric disorders in the US and Denmark. PLoS Biol. 17:e3000353. doi: 10.1371/journal.pbio.3000353
Khateb, M., Bosak, N., and Muqary, M. (2020). Coronaviruses and central nervous system manifestations. Front. Neurol. 11:715. doi: 10.3389/fneur.2020.00715
Kohler, H., and Nara, P. (2020). Perspective A Novel Hypothesis for Original Antigenic Sin in the Severe Disease of SARS-CoV-2 Infection. Monoclon. Antib. Immunodiagn. Immunother. 39, 107–111. doi: 10.1089/mab.2020.0029
Kreisel, T., Frank, M. G., Licht, T., Reshef, R., Ben-Menachem-Zidon, O., Baratta, M. V., et al. (2014). Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol. Psychiatry 19, 699–709. doi: 10.1038/mp.2013.155
Kubota, T., and Kuroda, N. (2021). Exacerbation of neurological symptoms and COVID-19 severity in patients with preexisting neurological disorders and COVID-19: a systematic review. Clin. Neurol. Neurosurg. 200:106349. doi: 10.1016/j.clineuro.2020.106349
Kyung, M. C., Liszewski, M. K., Nybakken, G., Davis, A. E., Townsend, R. R., Fremont, D. H., et al. (2006). West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc. Natl. Acad. Sci. U.S.A. 103, 19111–19116. doi: 10.1073/pnas.0605668103
Lafaie, L., Célarier, T., Goethals, L., Pozzetto, B., Grange, S., Ojardias, E., et al. (2020). Recurrence or relapse of COVID-19 in older patients: a description of three cases. J. Am. Geriatr. Soc. 68, 2179–2183. doi: 10.1111/jgs.16728
Laflamme, N., Cisbani, G., Préfontaine, P., Srour, Y., Bernier, J., St-Pierre, M. K., et al. (2018). mCSF-induced microglial activation prevents myelin loss and promotes its repair in a mouse model of multiple sclerosis. Front. Cell. Neurosci. 12:178. doi: 10.3389/fncel.2018.00178
Lampron, A., Larochelle, A., Laflamme, N., Préfontaine, P., Plante, M. M., Sánchez, M. G., et al. (2015). Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 212, 481–495. doi: 10.1084/jem.20141656
Lane, T. E., Liu, M. T., Chen, B. P., Asensio, V. C., Samawi, R. M., Paoletti, A. D., et al. (2000). A central role for CD4+ T Cells and RANTES in virus-induced central nervous system inflammation and demyelination. J. Virol. 74, 1415–1424. doi: 10.1128/jvi.74.3.1415-1424.2000
Larenas-Linnemann, D., Rodríguez-Pérez, N., Arias-Cruz, A., Blandón-Vijil, M. V., Del-Río-Navarro, B. E., Estrada-Cardona, A., et al. (2020). Enhancing innate immunity against virus in times of COVID-19: trying to untangle facts from fictions. World Allergy Organ. J. 13:100476. doi: 10.1016/j.waojou.2020.100476
Lavi, E., Gilden, D. H., Highkin, M. K., and Weiss, S. R. (1984). Persistence of mouse hepatitis virus A59 RNA in a slow virus demyelinating infection in mice as detected by in situ hybridization. J. Virol. 51, 563–566. doi: 10.1128/jvi.51.2.563-566.1984
Lecours, C., Bordeleau, M., Cantin, L., Parent, M., di Paolo, T., and Tremblay, M. È (2018). Microglial implication in Parkinson’s disease: loss of beneficial physiological roles or gain of inflammatory functions? Front. Cell. Neurosci. 12:282. doi: 10.3389/fncel.2018.00282
Lee, H., Jung, O., and Hennighausen, L. (2020). Activation of interferon-stimulated transcriptomes and ACE2 isoforms in human airway epithelium is curbed by janus kinase inhibitors. Res. Sq. [Epub ahead of print]. doi: 10.21203/RS.3.RS-119695/V1
Lee, H. C., Chathuranga, K., and Lee, J. S. (2019). Intracellular sensing of viral genomes and viral evasion. Exp. Mol. Med. 51, 1–13. doi: 10.1038/s12276-019-0299-y
Lei, F., Cui, N., Zhou, C., Chodosh, J., Vavvas, D. G., and Paschalis, E. I. (2020). CSF1R inhibition by a small-molecule inhibitor is not microglia specific; Affecting hematopoiesis and the function of macrophages. Proc. Natl. Acad. Sci. U.S.A. 117, 23336–23338. doi: 10.1073/pnas.1922788117
Letko, M., Marzi, A., and Munster, V. (2020). Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 5, 562–569. doi: 10.1038/s41564-020-0688-y
Li, Q., and Barres, B. A. (2018). Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225–242. doi: 10.1038/nri.2017.125
Li, Y., Fu, L., Gonzales, D. M., and Lavi, E. (2004). Coronavirus neurovirulence correlates with the ability of the virus to induce proinflammatory cytokine signals from astrocytes and microglia. J. Virol. 78, 3398–3406. doi: 10.1128/jvi.78.7.3398-3406.2004
Lian, H., Yang, L., Cole, A., Sun, L., Chiang, A. C. A., Fowler, S. W., et al. (2015). NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 85, 101–115. doi: 10.1016/j.neuron.2014.11.018
Liddelow, S. A., and Barres, B. A. (2017). Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967. doi: 10.1016/j.immuni.2017.06.006
Liddelow, S. A., Guttenplan, K. A., and Clarke, L. E. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lifland, A. W., Jung, J., Alonas, E., Zurla, C., Crowe, J. E., and Santangelo, P. J. (2012). Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J. Virol. 86, 8245–8258. doi: 10.1128/jvi.00215-12
Linnartz, B., Kopatz, J., Tenner, A. J., and Neumann, H. (2012). Sialic acid on the neuronal glycocalyx prevents complement c1 binding and complement receptor-3-mediated removal by microglia. J. Neurosci. 32, 946–952. doi: 10.1523/JNEUROSCI.3830-11.2012
Liu, L., Wei, Q., Lin, Q., Fang, J., Wang, H., Kwok, H., et al. (2019). Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 4:e123158. doi: 10.1172/jci.insight.123158
Loo, Y.-M., Fornek, J., Crochet, N., Bajwa, G., Perwitasari, O., Martinez-Sobrido, L., et al. (2008). Distinct RIG-I and MDA5 Signaling by RNA Viruses in Innate Immunity. J. Virol. 82, 335–345. doi: 10.1128/jvi.01080-07
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153:1194. doi: 10.1016/j.cell.2013.05.039
Lund, J. M., Alexopoulou, L., Sato, A., Karow, M., Adams, N. C., Gale, N. W., et al. (2004). Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. U.S.A. 101, 5598–5603. doi: 10.1073/pnas.0400937101
Manokaran, G., Finol, E., Wang, C., Gunaratne, J., Bahl, J., Ong, E. Z., et al. (2015). Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 350, 217–221. doi: 10.1126/science.aab3369
Mao, L., Jin, H., Wang, M., Hu, Y., Chen, S., He, Q., et al. (2020). Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 77, 683–690. doi: 10.1001/jamaneurol.2020.1127
Marreiros, R., Müller-Schiffmann, A., Trossbach, S. V., Prikulis, I., Hänsch, S., Weidtkamp-Peters, S., et al. (2020). Disruption of cellular proteostasis by H1N1 influenza A virus causes $α$-synuclein aggregation. Proc. Natl. Acad. Sci. U.S.A. 117, 6741–6751. doi: 10.1073/pnas.1906466117
Marten, N. W., Stohlman, S. A., Atkinson, R. D., Hinton, D. R., Fleming, J. O., and Bergmann, C. C. (2000). Contributions of CD8 + T cells and viral spread to demyelinating disease. J. Immunol. 164, 4080–4088. doi: 10.4049/jimmunol.164.8.4080
Matschke, J., Lütgehetmann, M., Hagel, C., Sperhake, J. P., Schröder, A. S., Edler, C., et al. (2020). Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol. 19, 919–929. doi: 10.1016/S1474-4422(20)30308-2
Matsui, H., and Takahashi, R. (2009). Pathological mechanisms of Parkinson’s disease. Brain Nerve 61, 441–446.
Mehlhop, E., Whitby, K., Oliphant, T., Marri, A., Engle, M., and Diamond, M. S. (2005). Complement activation is required for induction of a protective antibody response against west nile virus infection. J. Virol. 79, 7466–7477. doi: 10.1128/jvi.79.12.7466-7477.2005
Meneses, G., Bautista, M., Florentino, A., Díaz, G., Acero, G., Besedovsky, H., et al. (2016). Electric stimulation of the vagus nerve reduced mouse neuroinflammation induced by lipopolysaccharide. J. Inflamm. 13, 1–11. doi: 10.1186/s12950-016-0140-5
Milior, G., Lecours, C., Samson, L., Bisht, K., Poggini, S., Pagani, F., et al. (2016). Fractalkine receptor deficiency impairs microglial and neuronal responsiveness to chronic stress. Brain Behav. Immun. 55, 114–125. doi: 10.1016/j.bbi.2015.07.024
Miller, A. H., Haroon, E., and Felger, J. C. (2017). Therapeutic implications of brain-immune interactions: treatment in translation. Neuropsychopharmacology 42, 334–359. doi: 10.1038/npp.2016.167
Miner, J. J., and Diamond, M. S. (2016). Mechanisms of restriction of viral neuroinvasion at the blood-brain barrier. Curr. Opin. Immunol. 38, 18–23. doi: 10.1016/j.coi.2015.10.008
Miskin, D. (2020). COVID-19-Associated CNS Demyelinating Diseases. Philadelphia, PA: Thomas Jefferson University.
Moriguchi, T., Harii, N., Goto, J., Harada, D., Sugawara, H., Takamino, J., et al. (2020). A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int. J. Infect. Dis. 94, 55–58. doi: 10.1016/j.ijid.2020.03.062
Moseman, E. A., Blanchard, A. C., Nayak, D., and McGavern, D. B. (2020). T cell engagement of cross-presenting microglia protects the brain from a nasal virus infection. Sci. Immunol. 5:eabb1817. doi: 10.1126/SCIIMMUNOL.ABB1817
Motayo, B. O., Oluwasemowo, O. O., and Akinduti, P. A. (2020). Evolutionary dynamics and geographic dispersal of beta coronaviruses in african bats. bioRxiv [Preprint]. doi: 10.1101/2020.05.14.056085
Murray, R. S., Brown, B., Brain, D., and Cabirac, G. F. (1992). Detection of coronavirus RNA and antigen in multiple sclerosis brain. Ann. Neurol. 31, 525–533. doi: 10.1002/ana.410310511
Naghavi, M. H. (2018). “APP”reciating the complexity of HIV-induced neurodegenerative diseases. PLoS Pathog. 14:e1007309. doi: 10.1371/journal.ppat.1007309
Nan, Y., Nan, G., and Zhang, Y. J. (2014). Interferon induction by RNA viruses and antagonism by viral pathogens. Viruses 6, 4999–5027. doi: 10.3390/v6124999
Nanda, A., Vura, N. V. R. K., and Gravenstein, S. (2020). COVID-19 in older adults. Aging Clin. Exp. Res. 32, 1199–1202. doi: 10.1007/s40520-020-01581-5
Natoli, S., Oliveira, V., Calabresi, P., Maia, L. F., and Pisani, A. (2020). Does SARS-Cov-2 invade the brain? Translational lessons from animal models. Eur. J. Neurol. 27, 1764–1773. doi: 10.1111/ene.14277
Negishi, H., Osawa, T., Ogami, K., Ouyang, X., Sakaguchi, S., Koshiba, R., et al. (2008). A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc. Natl. Acad. Sci. U.S.A. 105, 20446–20451. doi: 10.1073/pnas.0810372105
Neniskyte, U., and Gross, C. T. (2017). Errant gardeners: Glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat. Rev. Neurosci. 18, 658–670. doi: 10.1038/nrn.2017.110
Netland, J., Meyerholz, D. K., Moore, S., Cassell, M., and Perlman, S. (2008). Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J. Virol. 82, 7264–7275. doi: 10.1128/jvi.00737-08
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Neuroscience: resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Nimmerjahn, F., and Ravetch, J. V. (2007). Fc-receptors as regulators of immunity. Adv. Immunol. 96, 179–204. doi: 10.1016/S0065-2776(07)96005-8
Nitta, S., Sakamoto, N., Nakagawa, M., Kakinuma, S., Mishima, K., Kusano-Kitazume, A., et al. (2013). Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology 57, 46–58. doi: 10.1002/hep.26017
Ojha, H., Panwar, H. S., Gorham, R. D., Morikis, D., and Sahu, A. (2014). Viral regulators of complement activation: structure, function and evolution. Mol. Immunol. 61, 89–99. doi: 10.1016/j.molimm.2014.06.004
Olajide, O. A., Iwuanyanwu, V. U., and Adegbola, O. D. (2020). SARS-CoV-2 spike glycoprotein S1 induces neuroinflammation in BV-2 microglia. bioRxiv [Preprint]. doi: 10.1101/2020.12.29.424619
Onofrio, L., Caraglia, M., Facchini, G., Margherita, V., De Placido, S., and Buonerba, C. (2020). Toll-like receptors and COVID-19: a two-faced story with an exciting ending. Futur. Sci. OA 6:FSO605. doi: 10.2144/fsoa-2020-0091
Oshiumi, H., Okamoto, M., Fujii, K., Kawanishi, T., Matsumoto, M., Koike, S., et al. (2011). The TLR3/TICAM-1 pathway is mandatory for innate immune responses to poliovirus infection. J. Immunol. 187, 5320–5327. doi: 10.4049/jimmunol.1101503
Otero-Losada, M., Kobiec, T., Udovin, L., Chevalier, G., Quarracino, C., Menéndez Maissonave, C., et al. (2020). Parkinson’s disease in the era of a novel respiratory virus pandemic. Front. Neurol. 11:995. doi: 10.3389/fneur.2020.00995
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Parkhurst, C. N., Yang, G., and Ninan, I. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Parsons, T., Banks, S., Bae, C., Gelber, J., Alahmadi, H., and Tichauer, M. (2020). COVID-19-associated acute disseminated encephalomyelitis (ADEM). J. Neurol. 267, 2799–2802. doi: 10.1007/s00415-020-09951-9
Pedraza, S. T., Guillermo, B., Biol, J., and Urcuqui-Inchima, S. (2010). Viral recognition by the innate immune system: the role of pattern recognition receptors. Colomb. Med. 41, 377–387.
Picard, K., St-Pierre, M. K., Vecchiarelli, H. A., Bordeleau, M., and Tremblay, M. È (2021). Neuroendocrine, neuroinflammatory and pathological outcomes of chronic stress: a story of microglial remodeling. Neurochem. Int. 145:104987. doi: 10.1016/j.neuint.2021.104987
Pichlmair, A., and Reis e Sousa, C. (2007). Innate recognition of viruses. Immunity 27, 370–383. doi: 10.1016/j.immuni.2007.08.012
Piscitelli, D., Perin, C., Tremolizzo, L., Peroni, F., Cerri, C. G., and Cornaggia, C. M. (2020). Functional movement disorders in a patient with COVID-19. Neurol. Sci. 41, 2343–2344. doi: 10.1007/s10072-020-04593-1
Plumet, S., Herschke, F., Bourhis, J.-M., Valentin, H., Longhi, S., and Gerlier, D. (2007). Cytosolic 5’-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response. PLoS One 2:e279. doi: 10.1371/journal.pone.0000279
Politi, L. S., Salsano, E., and Grimaldi, M. (2020). Magnetic resonance imaging alteration of the brain in a patient with coronavirus disease 2019 (COVID-19) and anosmia. JAMA Neurol. 77, 1028–1029. doi: 10.1001/jamaneurol.2020.2125
Pöyhönen, S., Er, S., Domanskyi, A., and Airavaara, M. (2019). Effects of neurotrophic factors in glial cells in the central nervous system: expression and properties in neurodegeneration and injury. Front. Physiol. 10:486. doi: 10.3389/fphys.2019.00486
Poyiadji, N., Shahin, G., Noujaim, D., Stone, M., Patel, S., and Griffith, B. (2020). COVID-19-associated acute hemorrhagic necrotizing encephalopathy: imaging features. Radiology 296, E119–E120. doi: 10.1148/radiol.2020201187
Qian, X., Ren, R., Wang, Y., Guo, Y., Fang, J., Wu, Z. D., et al. (2020). Fighting against the common enemy of COVID-19: a practice of building a community with a shared future for mankind. Infect. Dis. Poverty 9:34. doi: 10.1186/s40249-020-00650-1
Rabi, F. A., Al Zoubi, M. S., Al-Nasser, A. D., Kasasbeh, G. A., and Salameh, D. M. (2020). Sars-cov-2 and coronavirus disease 2019: what we know so far. Pathogens 9:231. doi: 10.3390/pathogens9030231
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist. Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Rao, C. D. (2020). Enteroviruses in gastrointestinal diseases. Rev. Med. Virol. 11:460. doi: 10.1002/rmv.2148
Reeve, A., Simcox, E., and Turnbull, D. (2014). Ageing and Parkinson’s disease: why is advancing age the biggest risk factor? Ageing Res. Rev. 14, 19–30. doi: 10.1016/j.arr.2014.01.004
Reichard, R. R., Kashani, K. B., Boire, N. A., Constantopoulos, E., Guo, Y., and Lucchinetti, C. F. (2020). Neuropathology of COVID-19: a spectrum of vascular and acute disseminated encephalomyelitis (ADEM)-like pathology. Acta Neuropathol. 140, 1–6. doi: 10.1007/s00401-020-02166-2
Reinert, L. S., Lopušná, K., Winther, H., Sun, C., Thomsen, M. K., Nandakumar, R., et al. (2016). Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 10:13348. doi: 10.1038/ncomms13348
Rhea, E. M., Logsdon, A. F., Hansen, K. M., Williams, L. M., Reed, M. J., Baumann, K. K., et al. (2020). The S1 protein of SARS-CoV-2 crosses the blood–brain barrier in mice. Nat. Neurosci. 24, 368–378. doi: 10.1038/s41593-020-00771-8
Rock, R. B., Gekker, G., Hu, S., Sheng, W. S., Cheeran, M., Lokensgard, J. R., et al. (2004). Role of microglia in central nervous system infections. Clin. Microbiol. Rev. 17, 942–964. doi: 10.1128/CMR.17.4.942-964.2004
Roers, A., Hiller, B., and Hornung, V. (2016). Recognition of endogenous nucleic acids by the innate immune system. Immunity 44, 739–754. doi: 10.1016/j.immuni.2016.04.002
Rogers, J. P., Chesney, E., Oliver, D., Pollak, T. A., McGuire, P., Fusar-Poli, P., et al. (2020). Psychiatric and neuropsychiatric presentations associated with severe coronavirus infections: a systematic review and meta-analysis with comparison to the COVID-19 pandemic. Lancet Psychiatry 7, 611–627. doi: 10.1016/S2215-0366(20)30203-0
Rudd, P. A., Cattaneo, R., and von Messling, V. (2006). Canine distemper virus uses both the anterograde and the hematogenous pathway for neuroinvasion. J. Virol. 80, 9361–9370. doi: 10.1128/jvi.01034-06
Sadasivan, S., Sharp, B., Schultz-Cherry, S., and Smeyne, R. J. (2017). Synergistic effects of influenza and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) can be eliminated by the use of influenza therapeutics: experimental evidence for the multi-hit hypothesis. Npj Park. Dis. 3:18. doi: 10.1038/s41531-017-0019-z
Said, E. A., Tremblay, N., Al-Balushi, M. S., Al-Jabri, A. A., and Lamarre, D. (2018). Viruses seen by our cells: the role of viral RNA sensors. J. Immunol. Res. 2018:9480497. doi: 10.1155/2018/9480497
Sallenave, J. M., and Guillot, L. (2020). Innate immune signaling and proteolytic pathways in the resolution or exacerbation of SARS-CoV-2 in Covid-19: key therapeutic targets? Front. Immunol. 11:1229. doi: 10.3389/fimmu.2020.01229
Sanchez, J. M. S., DePaula-Silva, A. B., Doty, D. J., Truong, A., Libbey, J. E., and Fujinami, R. S. (2019). Microglial cell depletion is fatal with low level picornavirus infection of the central nervous system. J. Neurovirol. 25, 415–421. doi: 10.1007/s13365-019-00740-3
Sanchez-Mejias, E., Navarro, V., Jimenez, S., Sanchez-Mico, M., Sanchez-Varo, R., Nuñez-Diaz, C., et al. (2016). Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol. 132, 897–916. doi: 10.1007/s00401-016-1630-5
Sanjuán, R., and Domingo-Calap, P. (2016). Mechanisms of viral mutation. Cell. Mol. Life Sci. 73, 4433–4448. doi: 10.1007/s00018-016-2299-6
Sariol, A., Mackin, S., Allred, M. G., Ma, C., Zhou, Y., Zhang, Q., et al. (2020). Microglia depletion exacerbates demyelination and impairs remyelination in a neurotropic coronavirus infection. Proc. Natl. Acad. Sci. U.S.A. 117, 24464–24474. doi: 10.1073/pnas.2007814117
Sarma, D., and Bilello, L. (2020). A case report of acute transverse myelitis following novel coronavirus infection. Clin. Pract. Cases Emerg. Med. 4, 321–323. doi: 10.5811/cpcem.2020.5.47937
Savarin, C., Dutta, R., and Bergmann, C. C. (2018). Distinct gene profiles of bone marrow-derived macrophages and microglia during neurotropic coronavirus-induced demyelination. Front. Immunol. 9:1325. doi: 10.3389/fimmu.2018.01325
Saxena, S. K., Kumar, S., Baxi, P., Srivastava, N., Puri, B., and Ratho, R. K. (2020). Chasing COVID-19 through SARS-CoV-2 spike glycoprotein. Virusdisease 31, 1–9. doi: 10.1007/s13337-020-00642-7
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Schnieder, T. P., Trencevska, I., Rosoklija, G., Stankov, A., Mann, J. J., Smiley, J., et al. (2014). Microglia of prefrontal white matter in suicide. J. Neuropathol. Exp. Neurol. 73, 880–890. doi: 10.1097/NEN.0000000000000107
Schwartz, M., and Deczkowska, A. (2016). Neurological disease as a failure of brain–immune crosstalk: the multiple faces of neuroinflammation. Trends Immunol. 37, 668–679. doi: 10.1016/j.it.2016.08.001
Seitz, S., Clarke, P., and Tyler, K. L. (2018). Pharmacologic depletion of microglia increases viral load in the brain and enhances mortality in murine models of flavivirus-induced encephalitis. J. Virol. 92:e00525-18. doi: 10.1128/jvi.00525-18
Serramía, M. J., and Muñoz-Fernández, M. Á, and Álvarez, S. (2015). HIV-1 increases TLR responses in human primary astrocytes. Sci. Rep. 5:17887. doi: 10.1038/srep17887
Serrano-Castro, P. J., Estivill-Torrús, G., Cabezudo-García, P., Reyes-Bueno, J. A., Ciano Petersen, N., Aguilar-Castillo, M. J., et al. (2020). Impact of SARS-CoV-2 infection on neurodegenerative and neuropsychiatric diseases: a delayed pandemic? Neurologia 35, 245–251. doi: 10.1016/j.nrl.2020.04.002
Sharaf, A., Krieglstein, K., and Spittau, B. (2013). Distribution of microglia in the postnatal murine nigrostriatal system. Cell Tissue Res. 351, 373–382. doi: 10.1007/s00441-012-1537-y
Shi, L., Fatemi, S. H., Sidwell, R. W., and Patterson, P. H. (2003). Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J. Neurosci. 23, 297–302. doi: 10.1523/jneurosci.23-01-00297.2003
Silva, F. A. F., de Brito, B. B., Santos, M. L. C., Marques, H. S., da Silva Júnior, R. T., de Carvalho, L. S., et al. (2020). COVID-19 gastrointestinal manifestations: a systematic review. Rev. Soc. Bras. Med. Trop. 53:e20200714. doi: 10.1590/0037-8682-0714-2020
Singal, C. M. S., Jaiswal, P., and Seth, P. (2020). SARS-CoV-2, more than a respiratory virus: its potential role in neuropathogenesis. ACS Chem. Neurosci. 11, 1887–1899. doi: 10.1021/acschemneuro.0c00251
Siu, K.-L., Yeung, M. L., Kok, K.-H., Yuen, K.-S., Kew, C., Lui, P.-Y., et al. (2014). Middle east respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J. Virol. 88, 4866–4876. doi: 10.1128/jvi.03649-13
Sochocka, M., Diniz, B. S., and Leszek, J. (2017). Inflammatory response in the CNS: friend or foe? Mol. Neurobiol. 54, 8071–8089. doi: 10.1007/s12035-016-0297-1
Solomon, I. H., Normandin, E., Bhattacharyya, S., Mukerji, S. S., Keller, K., Ali, A. S., et al. (2020). Neuropathological features of Covid-19. N. Engl. J. Med. 383, 989–992. doi: 10.1056/nejmc2019373
Song, E., Zhang, C., Israelow, B., Lu-Culligan, A., Prado, A. V., Skriabine, S., et al. (2020). Neuroinvasion of SARS-CoV-2 in human and mouse brain. bioRxiv [Preprint]. doi: 10.1101/2020.06.25.169946
Sorgeloos, F., Kreit, M., Hermant, P., Lardinois, C., and Michiels, T. (2013). Antiviral type I and type III interferon responses in the central nervous system. Viruses 5, 834–857. doi: 10.3390/v5030834
Sperlágh, B., and Illes, P. (2007). Purinergic modulation of microglial cell activation. Purinerg. Signal. 3, 117–127. doi: 10.1007/s11302-006-9043-x
Steardo, L., Steardo, L., and Verkhratsky, A. (2020). Psychiatric face of COVID-19. Transl. Psychiatry 10:261. doi: 10.1038/s41398-020-00949-5
Steiner, J., Bielau, H., Brisch, R., Danos, P., Ullrich, O., Mawrin, C., et al. (2008). Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J. Psychiatr. Res. 42, 151–157. doi: 10.1016/j.jpsychires.2006.10.013
Stence, N., Waite, M., and Dailey, M. E. (2001). Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia 33, 256–266. doi: 10.1002/1098-1136(200103)33:3<256::AID-GLIA1024<3.0.CO;2-J
Stratoulias, V., Venero, J. L., Tremblay, M., and Joseph, B. (2019). Microglial subtypes: diversity within the microglial community. EMBO J. 38:e101997. doi: 10.15252/embj.2019101997
Su, S., Wong, G., Shi, W., Liu, J., Lai, A. C. K., Zhou, J., et al. (2016). Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 24, 490–502. doi: 10.1016/j.tim.2016.03.003
Sun, L., Wu, J., Du, F., Chen, X., and Chen, Z. J. (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type-I interferon pathway. Science 339, 786–791. doi: 10.1038/jid.2014.371
Sun, L., Xing, Y., Chen, X., Zheng, Y., Yang, Y., Nichols, D. B., et al. (2012). Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One 7:e0030802. doi: 10.1371/journal.pone.0030802
Suzuki, H., Ohgidani, M., Kuwano, N., Chrétien, F., Lorin de la Grandmaison, G., Onaya, M., et al. (2019). Suicide and microglia: recent findings and future perspectives based on human studies. Front. Cell. Neurosci. 13:31. doi: 10.3389/fncel.2019.00031
Tancredi, V., D’Antuono, M., and Cafè, C. (2000). The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase ERK. J. Neurochem. 75, 634–643. doi: 10.1046/j.1471-4159.2000.0750634.x
Tancredi, V., D’Arcangelo, G., Grassi, F., Tarroni, P., Palmieri, G., Santoni, A., et al. (1992). Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci. Lett. 146, 176–178. doi: 10.1016/0304-3940(92)90071-E
Tay, T. L., Savage, J. C., Hui, C. W., Bisht, K., and Tremblay, M. È (2017). Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J. Physiol. 595, 1929–1945. doi: 10.1113/JP272134
Thakur, K. T., Miller, E. H., Glendinning, M. D., Al-Dalahmah, O., Banu, M. A., Boehme, A. K., et al. (2021). COVID-19 neuropathology at columbia university irving medical center/New York presbyterian hospital. Brain [Epub ahead of print]. doi: 10.1093/brain/awab148
Tobin, G. J., Trujillo, J. D., Bushnell, R. V., Lin, G., Chaudhuri, A. R., Long, J., et al. (2008). Deceptive imprinting and immune refocusing in vaccine design. Vaccine 26, 6189–6199. doi: 10.1016/j.vaccine.2008.09.080
Totura, A. L., Whitmore, A., Agnihothram, S., Schäfer, A., Katze, M. G., Heise, M. T., et al. (2015). Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio 6:e00638-15. doi: 10.1128/mBio.00638-15
Tremblay, M.-E., Madore, C., Bordeleau, M., Tian, L., and Verkhratsky, A. (2020). Neuropathobiology of COVID-19: the role for glia. Front. Cell. Neurosci. 14:592214. doi: 10.3389/fncel.2020.592214
Tremblay, M. È, Lecours, C., Samson, L., Sánchez-Zafra, V., and Sierra, A. (2015). From the Cajal alumni Achúcarro and Río-Hortega to the rediscovery of never-resting microglia. Front. Neuroanat. 9:45. doi: 10.3389/fnana.2015.00045
Troubat, R., Barone, P., Leman, S., Desmidt, T., Cressant, A., Atanasova, B., et al. (2020). Neuroinflammation and depression: a review. Eur. J. Neurosci. 53, 151–171. doi: 10.1111/ejn.14720
Tsuruta, R., and Oda, Y. (2016). A clinical perspective of sepsis-associated delirium. J. Intensive Care 4:18. doi: 10.1186/s40560-016-0145-4
Valdés-Florido, M. J., López-Díaz, Á, Palermo-Zeballos, F. J., Martínez-Molina, I., Martín-Gil, V. E., Crespo-Facorro, B., et al. (2020). Reactive psychoses in the context of the COVID-19 pandemic: clinical perspectives from a case series. Rev. Psiquiatr. Salud Ment. 13, 90–94. doi: 10.1016/j.rpsm.2020.04.009
Varatharaj, A., Thomas, N., Ellul, M. A., Davies, N. W. S., Pollak, T. A., Tenorio, E. L., et al. (2020). Neurological and neuropsychiatric complications of COVID-19 in 153 patients: a UK-wide surveillance study. Lancet Psychiatry 7, 875–882. doi: 10.1016/S2215-0366(20)30287-X
Varga, Z., Flammer, A. J., Steiger, P., Haberecker, M., Andermatt, R., Zinkernagel, A. S., et al. (2020). Endothelial cell infection and endotheliitis in COVID-19. Lancet 395, 1417–1418. doi: 10.1016/S0140-6736(20)30937-5
Vargas, G., Medeiros Geraldo, L. H., Gedeão Salomão, N., Viana Paes, M., Regina Souza Lima, F., and Carvalho Alcantara Gomes, F. (2020). Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and glial cells: insights and perspectives. Brain Behav. Immun. Health 7:100127. doi: 10.1016/j.bbih.2020.100127
Vasek, M. J., Garber, C., Dorsey, D., Durrant, D. M., Bollman, B., Soung, A., et al. (2016). A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534, 538–543. doi: 10.1038/nature18283
Voet, S., Prinz, M., and van Loo, G. (2019). Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol. Med. 25, 112–123. doi: 10.1016/j.molmed.2018.11.005
Wakselman, S., Béchade, C., Roumier, A., Bernard, D., Triller, A., and Bessis, A. (2008). Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J. Neurosci. 28, 8138–8143. doi: 10.1523/JNEUROSCI.1006-08.2008
Walker, P. J., Siddell, S. G., Lefkowitz, E. J., Mushegian, A. R., Dempsey, D. M., Dutilh, B. E., et al. (2019). Changes to virus taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses (2019). Arch. Virol. 164, 2417–2429. doi: 10.1007/s00705-019-04306-w
Waltl, I., Käufer, C., Gerhauser, I., Chhatbar, C., Ghita, L., Kalinke, U., et al. (2018). Microglia have a protective role in viral encephalitis-induced seizure development and hippocampal damage. Brain Behav. Immun. 74, 186–204. doi: 10.1016/j.bbi.2018.09.006
Wan, Y., Shang, J., Graham, R., Baric, R. S., and Li, F. (2020). Receptor recognition by the novel coronavirus from wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 94, 127–147. doi: 10.1128/jvi.00127-20
Wang, J. P., Liu, P., Latz, E., Golenbock, D. T., Finberg, R. W., and Libraty, D. H. (2006). Flavivirus activation of plasmacytoid dendritic cells delineates key elements of TLR7 signaling beyond endosomal recognition. J. Immunol. 177, 7114–7121. doi: 10.4049/jimmunol.177.10.7114
Wang, Q., Rowan, M. J., and Anwyl, R. (2004). β-amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J. Neurosci. 24, 6049–6056. doi: 10.1523/JNEUROSCI.0233-04.2004
Wang, S., Guo, F., Liu, K., Wang, H., Rao, S., Yang, P., et al. (2008). Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE2. Virus Res. 136, 8–15. doi: 10.1016/j.virusres.2008.03.004
Westerhuis, B. M., Aguilar-Bretones, M., Raadsen, M. P., Bruin, D., Okba, N. M., Haagmans, B. L., et al. (2020). Severe COVID-19 patients display a back boost of seasonal coronavirus-specific antibodies. medRxiv [Preprint]. doi: 10.1101/2020.10.10.20210070
Wheeler, D. L., Sariol, A., Meyerholz, D. K., and Perlman, S. (2018). Microglia are required for protection against lethal coronavirus encephalitis in mice. J. Clin. Invest. 128, 931–943. doi: 10.1172/JCI97229
Wiersinga, W. J., Rhodes, A., Cheng, A. C., Peacock, S. J., and Prescott, H. C. (2020). Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA J. Am. Med. Assoc. 324, 782–793. doi: 10.1001/jama.2020.12839
Wies Mancini, V. S. B., Pasquini, J. M., Correale, J. D., and Pasquini, L. A. (2019). Microglial modulation through colony-stimulating factor-1 receptor inhibition attenuates demyelination. Glia 67, 291–308. doi: 10.1002/glia.23540
Wohleb, E. S., McKim, D. B., Shea, D. T., Powell, N. D., Tarr, A. J., Sheridan, J. F., et al. (2014). Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking fromthe spleen to the brain. Biol. Psychiatry 75, 970–981.
Wolf, S. A., Boddeke, H. W. G. M., and Kettenmann, H. (2017). Microglia in physiology and disease. Annu. Rev. Physiol. 79, 619–643. doi: 10.1146/annurev-physiol-022516-034406
Woolhouse, M. E. J., Adair, K., and Brierley, L. (2013). RNA viruses: a case study of the biology of emerging infectious diseases. Microbiol. Spectr. 1, 83–97. doi: 10.1128/microbiolspec.oh-0001-2012
World Health Organization [WHO] (2021). WHO Coronavirus (COVID-19) Dashboard. Available online at: https://covid19.who.int/table (accessed May 01, 2021).
Wrapp, D., Wang, N., Corbett, K. S., Goldsmith, J. A., Hsieh, C.-L., Abiona, O., et al. (2019). Cryo-EM Structure of the 2019-nCoV Spike in the Prefusion Conformation. Available online at: http://science.sciencemag.org/ (accessed December 14, 2020).
Wu, G. F., Dandekar, A. A., Pewe, L., and Perlman, S. (2000). CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. J. Immunol. 165, 2278–2286. doi: 10.4049/jimmunol.165.4.2278
Wu, G. F., and Perlman, S. (1999). Macrophage infiltration, but not apoptosis, is correlated with immune-mediated demyelination following murine infection with a neurotropic coronavirus. J. Virol. 73, 8771–8780. doi: 10.1128/jvi.73.10.8771-8780.1999
Wu, Y., Xu, X., Chen, Z., Duan, J., Hashimoto, K., Yang, L., et al. (2020). Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 87, 18–22. doi: 10.1016/j.bbi.2020.03.031
Xiao, H., Li, J., Yang, X., Li, Z., Rui, Y., and Zhang, W. (2020). Picornavirus 3C proteins inhibit RIG-I-mediated innate immune responses by reducing TRIM25 expression. Res. Sq. [preprint]. doi: 10.21203/rs.3.rs-54603/v1
Xing, Y., Chen, J., Tu, J., Zhang, B., Chen, X., Shi, H., et al. (2013). The papain-like protease of porcine epidemic diarrhea virus negatively regulates type I interferon pathway by acting as a viral deubiquitinase. J. Gen. Virol. 94, 1554–1567. doi: 10.1099/vir.0.051169-0
Yang, M. (2020). Cell Pyroptosis, a Potential Pathogenic Mechanism of 2019-nCoV Infection. SSRN Electron. J. [Epub ahead of print]. doi: 10.2139/ssrn.3527420
Yu, C. Y., Chang, T. H., Liang, J. J., Chiang, R. L., Lee, Y. L., Liao, C. L., et al. (2012). Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog. 8:1002780. doi: 10.1371/journal.ppat.1002780
Zhang, H., Kang, Z., Gong, H., Xu, D., Wang, J., Li, Z., et al. (2020a). The digestive system is a potential route of 2019-nCov infection: a bioinformatics analysis based on single-cell transcriptomes. bioRxiv [Preprint]. doi: 10.1101/2020.01.30.927806
Zhang, Q., Bastard, P., Liu, Z., Le Pen, J., Moncada-Velez, M., Chen, J., et al. (2020b). Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370:eabd4570. doi: 10.1126/science.abd4570
Zhang, Q., Schultz, J. L., Aldridge, G. M., Simmering, J. E., and Narayanan, N. S. (2020c). Coronavirus disease 2019 case fatality and Parkinson’s disease. Mov. Disord. 35, 1914–1915. doi: 10.1002/mds.28325
Zhang, T., Hirsh, E., Zandieh, S., and Rodricks, M. B. (2020d). COVID-19-associated acute multi-infarct encephalopathy in an asymptomatic CADASIL patient. Neurocrit. Care [Epub ahead of print]. doi: 10.1007/s12028-020-01119-7
Zhang, X., Wu, J., Du, F., Xu, H., Sun, L., Chen, Z., et al. (2014). The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 6, 421–430. doi: 10.1016/j.celrep.2014.01.003
Zhang, Y., Xiao, M., Zhang, S., Xia, P., Cao, W., Jiang, W., et al. (2020e). Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N. Engl. J. Med. 382:e38. doi: 10.1056/nejmc2007575
Zhao, H., Shen, D., Zhou, H., Liu, J., and Chen, S. (2020). Guillain-Barré syndrome associated with SARS-CoV-2 infection: causality or coincidence? Lancet Neurol. 19, 383–384. doi: 10.1016/S1474-4422(20)30109-5
Zhao, Y., Zhao, Z., Wang, Y., Zhou, Y., Ma, Y., and Zuo, W. (2020). Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCov. bioRxiv [Preprint]. doi: 10.1101/2020.01.26.919985
Zhou, S., Cerny, A. M., Zacharia, A., Fitzgerald, K. A., Kurt-Jones, E. A., and Finberg, R. W. (2010). Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 84, 9452–9462. doi: 10.1128/jvi.00155-10
Zhou, Z., Kang, H., Li, S., and Zhao, X. (2020). Understanding the neurotropic characteristics of SARS-CoV-2: from neurological manifestations of COVID-19 to potential neurotropic mechanisms. J. Neurol. 267, 2179–2184. doi: 10.1007/s00415-020-09929-7
Keywords: microglia, SARS-CoV-2, COVID-19, brain, viral RNA neurotropism, Parkinson’s disease, neuropsychiatric disorders, neurodegenerative diseases
Citation: Awogbindin IO, Ben-Azu B, Olusola BA, Akinluyi ET, Adeniyi PA, Di Paolo T and Tremblay M-È (2021) Microglial Implications in SARS-CoV-2 Infection and COVID-19: Lessons From Viral RNA Neurotropism and Possible Relevance to Parkinson’s Disease. Front. Cell. Neurosci. 15:670298. doi: 10.3389/fncel.2021.670298
Received: 20 February 2021; Accepted: 05 May 2021;
Published: 15 June 2021.
Edited by:
Youichi Shinozaki, University of Yamanashi, JapanReviewed by:
Yukari Shigemoto-mogami, National Institute of Health Sciences (NIHS), JapanOlumayokun Adebodun Olajide, University of Huddersfield, United Kingdom
Steven Sheridan, Massachusetts General Hospital and Harvard Medical School, United States
Copyright © 2021 Awogbindin, Ben-Azu, Olusola, Akinluyi, Adeniyi, Di Paolo and Tremblay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ifeoluwa O. Awogbindin, aW8uYXdvZ2JpbmRpbkBtYWlsLnVpLmVkdS5uZw==; Marie-Ève Tremblay, ZXZldHJlbWJsYXlAdXZpYy5jYQ==
†These authors have contributed equally to this work