
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci. , 04 March 2021
Sec. Cellular Neuropathology
Volume 15 - 2021 | https://doi.org/10.3389/fncel.2021.619777
This article is part of the Research Topic Molecular Mechanisms Underlying Synaptic and Axon Degeneration in Neurodegenerative Diseases View all 9 articles
 Rima Rebiai1
Rima Rebiai1 Maria I. Givogri1*
Maria I. Givogri1* Swetha Gowrishankar1*
Swetha Gowrishankar1* Stephania M. Cologna2*
Stephania M. Cologna2* Simon T. Alford1*
Simon T. Alford1* Ernesto R. Bongarzone1*
Ernesto R. Bongarzone1*Lysosomal storage diseases (LSDs) with neurological involvement are inherited genetic diseases of the metabolism characterized by lysosomal dysfunction and the accumulation of undegraded substrates altering glial and neuronal function. Often, patients with neurological manifestations present with damage to the gray and white matter and irreversible neuronal decline. The use of animal models of LSDs has greatly facilitated studying and identifying potential mechanisms of neuronal dysfunction, including alterations in availability and function of synaptic proteins, modifications of membrane structure, deficits in docking, exocytosis, recycling of synaptic vesicles, and inflammation-mediated remodeling of synapses. Although some extrapolations from findings in adult-onset conditions such as Alzheimer’s disease or Parkinson’s disease have been reported, the pathogenetic mechanisms underpinning cognitive deficits in LSDs are still largely unclear. Without being fully inclusive, the goal of this mini-review is to present a discussion on possible mechanisms leading to synaptic dysfunction in LSDs.
Lysosomal storage diseases (LSDs) are characterized by the accumulation of biological macromolecules in the lysosomes, resulting in the formation of large intracellular deposits that affect cellular function. LSDs are inherited diseases of the metabolism caused by genetic deficiencies of proteins and enzymes involved in the biogenesis and/or degradative function of lysosomes (Table 1; Greiner-Tollersrud and Berg, 2000–2013). Lysosomal dysfunction in LSDs may impact the degradation of lipids, proteoglycans, proteins, and also those proteins involved in lysosomal trafficking and transporters (Table 1; Greiner-Tollersrud and Berg, 2000–2013). Most often, LSDs present infantile symptoms, although juvenile and late-onset forms also exist. Manifestations in early-onset neurological LSDs may include hearing loss, seizures, neuromotor regression, demyelination, intellectual disability, and developmental delay. Late-onset cases may also develop depression, psychosis, and dementia. About two-thirds of the patients affected with LSDs experience neurological deficits, and thus, a correlation between synaptic failure and cognitive decline may apply to these LSD patients (Morrison and Baxter, 2012). Although several studies have described morphological and functional changes in neurons, synapses (Lepeta et al., 2016) and electrophysiological deficits in LSDs (Zoghbi and Bear, 2012; Calabrese et al., 2016; He et al., 2018), important questions on linking lysosomal storage defects with changes in structure and function of synapses remain largely unaddressed.

Table 1. Examples of lysosomal storage diseases (LSDs) where cognitive defects were observed.
Numerous studies indicate that synaptic plasticity underlies the neurobiological basis of higher cognitive function. Dysfunction in synaptic transmission (synaptic failure and/or synaptic death) undermines mechanisms of synaptic plasticity, leading to loss of synaptic function, which may further facilitate a neurological decline in multiple pathological conditions (Lepeta et al., 2016). Spine morphology is tightly linked to synapse function and has been used often as a readout of synaptic deterioration (Holtmaat and Svoboda, 2009; Bosch and Hayashi, 2012). Progressive atrophy in spines in Alzheimer’s disease (AD) and progressive neuronal dysfunction in Parkinson’s disease (PD) are possibly good examples of this. AD is characterized by the loss of synapses and neurons in the cerebral cortex and hippocampus, as well as the formation of Aβ plaques and neurofibrillary lesions. This progressive loss of synaptic structure is believed to impair synaptic plasticity and memory and lead to cognitive decline in AD patients (Hsieh et al., 2006). Interestingly, in addition to plaques, AD has also evidence of lysosomal storage, suggesting a potential pathogenic contribution of lysosomes to AD (Orr and Oddo, 2013). Of importance, genetic associations between LSDs and adult-onset conditions such as AD and PD have been recently identified. For example, mutations in the gene GBA (encoding for glucosylceramidase, deficient in Gaucher’s disease, GD) are genetic risk factors associated with PD (Barkhuizen et al., 2016; Gan-Or et al., 2018; Sidransky et al., 2009). Additionally, SMPD1 (acid sphingomyelinase, deficient in Niemann Pick type A, B), ASAH1 (acid ceramidase, deficient in Farber’s disease and spinal muscular atrophy with progressive myoclonic epilepsy), ARSA [arylsulfatase A, deficient in metachromatic leukodystrophy (MLD)] and GALC [lysosomal galactosylceramidase, deficient in Krabbe’s disease (KD)] are also associated with vulnerability to develop adult onset neurodegenerative disorders, particularly PD (Smolders and Van Broeckhoven, 2020). These studies underline the possibility that mechanisms involved in synaptic failure in LSDs may also be playing an active role in other neurodegenerative diseases.
Analogous structural studies have started to find abnormalities in some LSDs, providing further evidence of synaptic dysfunction. For example, abnormal spine morphology (immature and reduced dendritic spines) has been reported in animal models of LSDs including mucopolysaccharidosis type IIIC or Sanfilippo disease type C and Tay-Sachs’ disease (Dwyer et al., 2017; Sambri et al., 2017). Additionally, these studies showed an abnormal distribution of several pre- and post-synaptic proteins, disorganized microtubule networks, reduced postsynaptic densities, lower synaptic vesicles, and alterations in frequency and amplitude of miniature EPSCs and IPSCs (Dwyer et al., 2017; Sambri et al., 2017). Studies in Drosophila mutants lacking neuronal glucosylceramidase (modeling for GD) have also found major synaptic loss and neurodegeneration (Kinghorn et al., 2017). An Npc1−/− mouse model for Niemann-Pick type C disease (NPC), which results from defects in cholesterol transport, develops significant alteration in presynaptic structure and function with important neuronal loss (Wang et al., 2010; Peake et al., 2011) and impairment of long-term synaptic potentiation (LTP; D’Arcangelo et al., 2016).
Although these structural studies point to synaptic dysfunction in some LSDs, much less is known on how a given lysosomal deficiency triggers these changes. Because synapses are highly complex structures with multiple components, it is unlikely that synaptic dysfunction in LSDs involves direct genetic defects of synaptic components, which tend to be embryologically lethal, but rather, are elicited by indirect defects arising from lysosomal dysfunction. Among others, synaptic efficacy may be impacted by alterations in the architecture of pre- and/or post-synaptic membranes; slowed vesicular transport reducing delivery of mitochondria and other components needed for synaptic vesicle formation; inefficient formation of SNARE complexes; and altered lysosomal/autophagosomal activity (Virmani et al., 2005; Xu et al., 2010; Button and Luo, 2017; Button et al., 2017; Sharma and Lindau, 2017). Furthermore, the exocytosis and recycling of synaptic vesicles (Rangaraju et al., 2014) and the speed and specificity of protein sorting in presynaptic active zones (Bonanomi et al., 2006) and post-synaptic structures—critical for learning (Malinow and Malenka, 2002) and homeostasis (Pérez-Otaño and Ehlers, 2005)—impose a very heavy metabolic burden on synapses. Hence, an interplay of factors such as accumulation of undegraded substrates within the synaptic endo-lysosomal pathway, altered lysosomal pH and membrane, endoplasmic reticulum stress, unfolded protein response activation, deformation of synaptic membrane architecture (e.g., shedding and invagination), autophagy impairment, and inflammation (Kim et al., 2006; Tessitore et al., 2009; Weinert et al., 2010; Hawkins-Salsbury et al., 2013; Lim et al., 2015; Karch et al., 2017; Zucca et al., 2018; Gabandé-Rodríguez et al., 2019) are more likely to contribute to synaptic disease in neurological LSDs. Figure 1 illustrates some of the possible pathways contributing to synaptic failure in LSDs. For most LSDs, the initial triggers of synaptic dysfunction are still unknown. Below, we discuss the involvement of lipids, dysregulation of synaptic components, and the role of inflammation.
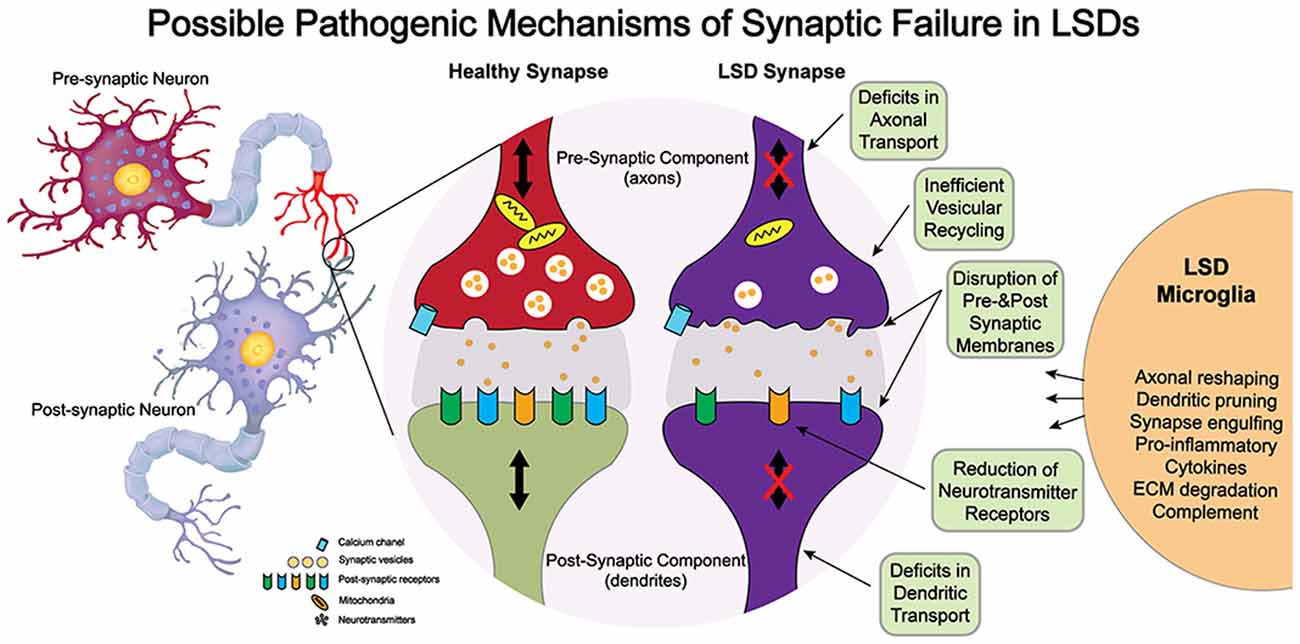
Figure 1. Potential mechanisms of neuropathogenesis affecting synaptic structure and function in lysosomal storage diseases (LSDs).
Because synapses are so closely dependent on membrane dynamics (Ceccarelli et al., 1973; Heuser and Reese, 1973), pathological conditions altering the recycling of lipid components during synaptic vesicle release and recycle are expected to impact the architecture of affected synapses. In sphingolipidoses such as KD and MLD, lysosomal defects lead to the progressive accumulation of undegraded sphingolipids in multiple membrane compartments. The build-up of psychosine (KD) and sulfatides (MLD) in lipid rafts of brain membranes provided an initial rationale to understand the link between membrane deformation and synaptic dysfunction in these LSDs (White et al., 2009; Moyano et al., 2014; Sural-Fehr et al., 2019). Psychosine is the main undegraded sphingolipid accumulated in KD caused by the deficiency of galactosylceramidase (Table 1; White et al., 2009). At high levels, psychosine becomes cytotoxic, particularly to oligodendrocytes, resulting in extensive demyelination observed in the KD brain (Suzuki, 1998). However, psychosine accumulates and disrupts the architecture of most brain membranes by altering the composition and dynamics of lipid rafts (White et al., 2009; Hawkins-Salsbury et al., 2013) and promoting membrane shedding (D’Auria et al., 2017). Lipid rafts are generally considered subdomains in the membrane where the association of sphingolipids and cholesterol facilitates the interaction with coupled raft-proteins. Some of these proteins are receptors, G-proteins, and enzymes which regulate a myriad of different signaling pathways (Bieberich, 2018). Thus, we could elaborate that the direct effect of the genetic trait in KD (i.e., the deficiency of a lysosomal enzyme in charge of degrading sphingolipids primarily impacting on myelin membranes) also leads to the accumulation of psychosine in other cell membranes, which indirectly induces an array of secondary pathogenic events. Depending on the cellular context where this occurs, distinct pathogenic mechanisms may be activated. For example, in neurons psychosine induces the dephosphorylation of neurofilaments by deregulation of two serine/threonine phosphatases (PP1 and PP2A) effecting neuronal cytoskeleton (Cantuti-Castelvetri et al., 2012). Psychosine promotes the abnormal activation of GSK3β and the reduction of fast axonal transport mechanisms (Cantuti Castelvetri et al., 2013), indirectly reducing the speed and efficiency of synaptic protein delivery. These changes are thought to be facilitated by the release of dephosphorylating activities (e.g., PP1 and PP2A) from the membrane, in response to increased local concentrations of psychosine. Because changes in pre- and post-synaptic proteins, their assembly, and axonal/dendritic transport are associated with cognitive decline (Attems and Jellinger, 2018), psychosine might increase synaptic vulnerability by inducing membrane disorganization. Additionally, psychosine binds and facilitates the abnormal aggregation of α-synuclein in neurons (Abdelkarim et al., 2018), suggesting additional and more direct involvement in neuronal pathology in KD. From our experience with psychosine, we could expect that some common mechanisms involving membrane deformation apply to other lipidoses such as MLD, GD, and NPC. While there is clear evidence of a cognitive decline in all age-related MLD variants (Hyde et al., 1992; Gieselmann et al., 1998), most reports support the hypothesis that neuronal dysfunction is primarily owed to the accumulation of sulfatides in oligodendrocytes, and the indirect damage to axon integrity and maintenance caused by demyelination (Ishibashi et al., 2002; Honke, 2013; McGonigal et al., 2019). In other lysosomal sphingolipidoses such as gangliosidoses and GD, similar mechanistic pathways may also contribute to triggering neuronal deficits (Lee et al., 2012). Thus, a growing body of evidence indicates that changes in lipid homeostasis seen in various lipidoses also are capable to trigger structural changes in synaptic membranes and function (Egawa et al., 2016; Díaz et al., 2018).
Systematic studies of pre- and post-synaptic components are largely missing and at best fragmented and incomplete for most LSDs. We can infer potential mechanisms affecting these components in LSDs from a larger and deeper understanding of synaptic components, structure, and function from formal studies in normal conditions. Presynaptic SNARE proteins (e.g., syntaxin-1, SNAP25, and VAMP2) represent the core machinery for synaptic vesicle fusion (Sutton et al., 1998) and are crucial for synaptic transmission (Hayashi et al., 1994) and are predictors of cognitive function (Ramos-Miguel et al., 2017). Analysis of these pre-synaptic proteins suggests that the level of their functional interactions is associated with better cognition, and less decline over time (Ramos-Miguel et al., 2017). Failure to properly form SNARE complexes interfere significantly with synaptic transmission. Additionally, the regulation of SNARE function is complex and offers several points where dysregulation may occur, particularly because of association with membrane lipids. Of relevance, SNAREs have various affinities for sphingolipid and sterol-rich lipid raft domains among other lipids (phospholipids, phosphoinositides), implying the alterations in lipid homeostasis in LSD also influence SNARE functionality (Salaün et al., 2005; Lam et al., 2008; Tong et al., 2009). Moreover, phosphorylation of SNARE proteins such as SNAP-25 by protein kinases plays a key role in the exocytosis of synaptic vesicles (Gao et al., 2016). The formation of functional SNARE complexes is greatly inhibited by PKA phosphorylation of SNAP-25 at Thr 138, while it is promoted by PKC phosphorylation at Ser 187 (Gao et al., 2016). Thus, abnormal signaling regulating SNARE components may impact trafficking, docking, and release of vesicles in the axonal terminal.
Although dysregulation of signaling pathways in various cellular contexts has been described in many LSDs (Seranova et al., 2017), less is known on how these changes impact SNARE function. For example, large aggregates of presynaptic proteins including VAMP2, SNAP25, and synaptophysin were observed within axonal spheroids in several brain areas in the mouse model for NPC (Pressey et al., 2012). These findings suggest that deficient transport of cholesterol, which biochemically characterizes NPC, negatively impacts the translocation/assembly of SNARE (Pressey et al., 2012) components, but other mechanisms might also be involved. For example, in mice with MPS IIIA, reduced levels of SNAP25 and VAMP2 proteins parallel decreases in synaptic vesicles, abnormal vacuoles, and enlarged mitochondria in axonal termini (Sambri et al., 2017), but surprisingly, without defects in the expression of SNAP25 and VAMP2 mRNAs. This underlines the possibility that mutant neurons might activate abnormal degradative pathways rather than impaired gene expression, impacting the availability of functional SNARE components for proper action at the presynaptic terminal (Sambri et al., 2017). This mechanism may be a consequence of dysfunctional lysosomal-autophagosomal activities, a pathogenic mechanism observed in most LSDs (Lieberman et al., 2012).
Defective transport of SNARE components in LSDs might be elicited by the loss of other synaptic components such as α-synuclein, which is implicated in neuronal loss in PD and Lewy Body dementia, and cysteine string protein α (CSPα). These two abundant presynaptic proteins act as chaperones and contribute to the SNARE complex formation at synaptic terminals (Chandra et al., 2005; Burré et al., 2010; Burgoyne and Morgan, 2011; Sharma et al., 2011). Defects in axonal transport have been described in several conditions and recently, associated with some LSDs. In KD, defects in axonal transport were first described by Cantuti Castelvetri et al. (2013). These authors found that both anterograde and retrograde transport rates are affected by psychosine. Slower transport may reduce SNARE proteins’ availability, recycling of synaptic vesicles, and translocation of mitochondria. These results were further supported by measurements of slowed retrograde transport of synaptophysin-positive vesicles in Krabbe neurons, indicating that the early steps of endocytosis and retrograde transport of endocytic and synaptic vesicles can also be impaired (Teixeira et al., 2014). Defects in the availability of functional pre-synaptic components have also been suggested for other LSDs including MPSI and IIIA (Wilkinson et al., 2012; Baldo et al., 2015), MPS IIIB (Vitry et al., 2009), MPS VII (Bayó-Puxan et al., 2018), CLN6 (Kanninen et al., 2013), and CLN5 (Amorim et al., 2015).
Likewise, defects in post-synaptic components might contribute to overall synaptic failure in LSDs. For example, several neurotransmitter receptors must be localized in lipid rafts for efficient nerve transmission (Allen et al., 2007). Regardless of their different brain localization, AMPA receptors (particularly GluR1, GluR2/3, and GluR4) along with GABA receptor A, acetylcholine receptors, and NMDA receptors (NR1, NR2A, and NR2B), are most often associated with lipid rafts. The movement between the raft and non-raft domains in the synaptic membrane is regulated by several signaling mechanisms, which can be targeted for dysregulation in LSDs. For instance, studies on KD show that psychosine facilitates the redistribution of raft-associated proteins such as Flotillin-2 and Caveolin-1 and leads to a generalized and likely irreversible disruption of membranes via shedding (White et al., 2009; D’Auria et al., 2017). Thus, we can hypothesize that in KD, psychosine-induced shedding severely changes the local concentration of key receptors and other proteins needed for optimal nerve transmission.
Similarly, we could envisage that alterations in postsynaptic raft composition and/or activity of associated signaling pathways may impact the recruitment of neurotransmitter receptors in other LSDs. For instance, the activation of CaMKII facilitates the trafficking of AMPAR to the dendritic rafts. CaMKII phosphorylates the GluA1 AMPA subunit at Ser831 (Kristensen et al., 2011) or, indirectly, through the activation of the Ras/MAPK pathway (Henley and Wilkinson, 2013), and genetic defects in the Ras-MAPK pathway are associated with deficient plasticity (Cesarini et al., 2009; Lisman et al., 2012). As in presynaptic terminals, the localization of receptors requires exocytic and endocytic processes involving SNARE complex-dependent vesicle transport, and aberrant receptors (e.g., AMPA) trafficking has been associated with impaired synaptic plasticity and cognitive deficits (Henley and Wilkinson, 2013; Chater and Goda, 2014). As stated before, little is known on neurotransmitter receptors trafficking, assembly, and regulation in the context of LSDs. Studies in a mouse model of CLN1 showed deficient developmental switch in NMDA receptors from GluN2B to GluN2A and reduced interactions between SAP-102 with GluN2B and PSD-95 with GluN2A, associated with early synaptic impairments (Koster et al., 2019). Studies in the MPS IIIA mouse have shown that accumulation of heparan sulfate in the cerebral cortex correlates with altered levels of AMPA receptor GluA2 and enhanced puncta for postsynaptic density 95 (PSD-95) in the somatosensory cortex, providing a potential link to study defects in synaptic neurotransmission (Dwyer et al., 2017). Other studies have also shown significant reductions in Homer-1, a protein enriched in the postsynaptic density of excitatory synapses in the brain of MPS I, IIIA, and IIIB (Wilkinson et al., 2012). Defects in receptor localization and interaction at the postsynaptic density likely facilitate an impaired receptor conductance state. For instance, the kinetics of the NMDA excitatory postsynaptic currents (EPSCs) showed longer activation times of GluN2B receptor (Koster et al., 2019), indicating impairment in NMDA insertion/removal from the synaptic membrane affecting both synaptic homeostasis and plasticity. Together, these few studies in LSDs underline that deregulation in several post-synaptic proteins may also be common traits affecting synaptic function in LSDs (Wilkinson et al., 2012). Undoubtedly, more studies are needed to determine whether common or disease-specific pathogenic mechanisms are present in other neurological LSDs.
During exocytosis, synaptic vesicles fuse with the plasma membrane at active zones to release neurotransmitters into the synaptic cleft. These vesicles will then be recycled to make new ones via endocytosis (Soykan et al., 2016). Endocytosis and exocytosis at the synaptic terminals is an exciting area of study, which might be of crucial relevance in many LSDs (Xu et al., 2010). For instance, evoked vesicle exocytosis was impaired in hippocampal neuronal cultures of NPC1 mice (Xu et al., 2010) and reductions in docked synaptic vesicles in cortical neurons from CLN1 mice have been described (Virmani et al., 2005). Decreased numbers of early endosomes expressing Ras-related protein 5 were also detected in dorsal root ganglion neurons from the twitcher mouse, a model for KD (Teixeira et al., 2014). Similarly, MPS IIIA neurons develop a phenotype characterized by reduced synaptic vesicles and decreased exocytosis rate (Sambri et al., 2017).
Because the rate of synaptic vesicle exocytosis/endocytosis directly controls neurotransmission release, deficits in either process may affect synaptic strength and functions. Synaptic strength is highly influenced by AMPA (Anggono and Huganir, 2012) and NMDA receptors (Bliss and Collingridge, 1993). As discussed before, localization of AMPARs within lipid rafts at the post-synaptic membrane, which is regulated by the activity of NMDA receptors (Kristensen et al., 2011) impacts synaptic strength and can elicit significant changes in LTP and LTD. A decrease in synaptic strength during LTD implies the removal of receptors by endocytosis and sorting to degradative organelles (i.e., lysosomes) to reduce the number of available AMPA receptors at the plasma membrane. Conversely, LTP involves increasing the number and recycling of AMPA receptors to the plasma membrane (Liao et al., 1995; Qin et al., 2005). During LTD, NMDA receptor-dependent internalization of AMPA receptors involves dephosphorylation at S845 of GluA1 (Parkinson and Hanley, 2018) and phosphorylation of GluA2 at S880 (Jurado, 2018), whereas the recycling of AMPA receptors during LTP requires the phosphorylation of GluA1 at S845 and S831 (Parkinson and Hanley, 2018). We speculate that alterations in membrane composition owed to altered metabolism of lipids and other lysosomal substrates impact on the recycling and availability of receptors affecting LTD/LTP and contribute to some of the neurobehavioral manifestations observed in patients with LSDs. For example, synaptic vesicles in both excitatory and inhibitory synapses are reduced in neurons from the Npc1−/− mouse, effecting the readily released pool of vesicles (Xu et al., 2010) and reducing hippocampal LTP in NPC1 mice (Võikar et al., 2002). This could help not only to understand distinct patterns of neurobehavioral symptoms in the NPC1 patients (Sévin et al., 2007) but also to illustrate possible avenues of research of cognitive function in other LSDs.
As discussed above, the efficacy of synaptic transmission can be altered at multiple levels, affecting the availability of pre- and post-synaptic components, the recycling of vesicles, and the number of neurotransmitter receptors. Activation of inflammatory responses and death-mediated pathways are also relevant mechanisms by which to modify synaptic function. For example, resting microglia are critical immune surveillance cells in charge of monitoring neuronal and glial functions in the brain and play a fundamental role in shaping synapses and synaptic plasticity during development and normal physiological activity of the nervous system (Morris et al., 2013). Expectedly, lysosomal function plays an important role in inflammation and immunity. For example, bioactive sphingolipids ceramides and sphingosine-1-phosphate are known to modulate the activation of inflammatory responses (Scheiblich et al., 2017; Sapkota et al., 2019). Glucosylsphingosine, which accumulates in GD, promotes the activation of macrophages and the release of multiple cytokines, such as chitotriosidase, TNF-α, and IL-1β (Pandey et al., 2014). During inflammatory events, microglial cells alter their function to attempt restoring homeostasis, which includes secretion of cytokines, endocytosis, and engulfment of debris. These inflammatory-related functions may drastically impact synaptic function by the secretion of factors that modify LTP/LTD (Zhou et al., 2019), degradation of extracellular matrix components inherent to the synaptic cleft (Tremblay et al., 2010; Nguyen et al., 2020), engulfment of synaptic terminals (Perry and O’Connor, 2010), and reshaping of dendrites (Cangalaya et al., 2020). An important arm of the inflammatory response in the brain is the infiltration of complement-related proteins, which have been shown to actively participate in synapse elimination (Stevens et al., 2007). This is yet another area of LSDs with minimal and fragmented understanding.
In addition to inflammation, caspases are proteins that while serving key functions in programmed cell death (Kumar, 2007), also participate in the shaping of synapses (Jonas, 2006). In particular, caspases 3, 6, and 9 appear to have key roles in dendritic pruning and axonal reshaping during learning and memory (Mattson and Duan, 1999; Forrest et al., 2013). For example, AMPA receptor subunits are known caspase substrates (Lu et al., 2002). Although the activity of caspases and other cell-related molecules is crucial during synaptic formation and maturation, their overactivation has been linked with several late-onset neurodegenerative disorders with synaptic failure (Su et al., 2001; Martin et al., 2002). Little is known about the involvement of caspases in synaptic dysfunction in LSDs. Cantuti et al reported abnormal expression of caspase 3 in presynaptic terminals of the twitcher mouse, which underlines the possibility that exacerbated degradation of synaptic components by caspase activities impact on nerve transmission (Cantuti-Castelvetri et al., 2015). Considering that inflammation of the nerve tissue is an integral part of the cellular response in most neurological LSDs (Moskot et al., 2018), the activation of microglia, inflammation, and cell death pathways are likely to influence synaptic function in these diseases and contribute to some of the neurobehavioral manifestations observed in patients.
Pathogenic mechanisms in most LSDs are multifactorial. Understanding those pathogenic mechanisms underlying synaptic failure in LSDs is critical not only to provide deeper integration and full interpretation of common vs. specific mechanisms at play in these diseases but also to enable further evaluation of short- and long-term responses of synaptic function to available treatments. Most LSDs lack a cure, but many are treated with enzyme correction approaches such as enzyme replacement therapy or hematopoietic transplantation (Wynn et al., 2009). Furthermore, gene therapy and small molecule (i.e., inhibitors, chaperones, etc.) interventions are also rapidly gaining relevance in treating some LSDs (Seregin and Amalfitano, 2011), and the concept of synergy from combining treatments is being seriously considered (Hawkins-Salsbury et al., 2015). Importantly, despite these advances, there is limited information on the extent to which synaptic function recovers or is protected in response to treatments and the extent and duration to which it can be sustained in patients undergoing a specific treatment. More in-depth studies into the triggering mechanisms modifying synaptic function and their response to different treatments are expected to contribute to a better understanding of pathogenesis and how to improve therapeutic safety and efficacy for LSDs.
All authors reviewed, edited, commented, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded with grants from the NIH (R01 NS065808) and the Legacy of Angels Foundation to ERB; NIH (R01NS111749) to STA; Together Strong NPC Foundation, Ara Parseghian Medical Research Fund, and NIH (NS114413) to SMG. The Authors acknowledge Research Open Access Publishing (ROAAP) Fund of the University of Illinois at Chicago for financial support towards the open access publishing fee for this article.
ERB is a consultant for E-Scape Bio, Bial, Affinia Therapeutics, Gain Therapeutics and Neurogene.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdelkarim, H., Marshall, M. S., Scesa, G., Smith, R. A., Rue, E., Marshall, J., et al. (2018). α-Synuclein interacts directly but reversibly with psychosine: implications for α-synucleinopathies. Sci. Rep. 8:12462. doi: 10.1038/s41598-018-30808-9
Allen, J. A., Halverson-Tamboli, R. A., and Rasenick, M. M. (2007). Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 8, 128–140. doi: 10.1038/nrn2059
Amorim, I. S., Mitchell, N. L., Palmer, D. N., Sawiak, S. J., Mason, R., Wishart, T. M., et al. (2015). Molecular neuropathology of the synapse in sheep with CLN 5 Batten disease. Brain Behav. 5:e00401. doi: 10.1002/brb3.401
Anggono, V., and Huganir, R. L. (2012). Regulation of AMPA receptor trafficking and synaptic plasticity. Curr. Opin. Neurobiol. 22, 461–469. doi: 10.1016/j.conb.2011.12.006
Attems, J., and Jellinger, K. (2018). Proteomics for synaptic markers of cognitive decline in neurodegenerative diseases. Brain 141, 329–331. doi: 10.1093/brain/awx360
Baldo, G., Lorenzini, D. M., Santos, D. S., Mayer, F. Q., Vitry, S., Bigou, S., et al. (2015). Shotgun proteomics reveals possible mechanisms for cognitive impairment in Mucopolysaccharidosis I mice. Mol. Genet. Metab. 114, 138–145. doi: 10.1016/j.ymgme.2014.12.301
Barkhuizen, M., Anderson, D. G., and Grobler, A. F. (2016). Advances in GBA-associated Parkinson’s disease-pathology, presentation and therapies. Neurochem. Int. 93, 6–25. doi: 10.1016/j.neuint.2015.12.004
Bayó-Puxan, N., Terrasso, A. P., Creyssels, S., Simão, D., Begon-Pescia, C., Lavigne, M., et al. (2018). Lysosomal and network alterations in human mucopolysaccharidosis type VII iPSC-derived neurons. Sci. Rep. 8:16644. doi: 10.1038/s41598-018-34523-3
Bieberich, E. (2018). Sphingolipids and lipid rafts: Novel concepts and methods of analysis. Chem. Phys. Lipids 216, 114–131. doi: 10.1016/j.chemphyslip.2018.08.003
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. doi: 10.1038/361031a0
Bonanomi, D., Benfenati, F., and Valtorta, F. (2006). Protein sorting in the synaptic vesicle life cycle. Prog. Neurobiol. 80, 177–217. doi: 10.1016/j.pneurobio.2006.09.002
Bosch, M., and Hayashi, Y. (2012). Structural plasticity of dendritic spines. Curr. Opin. Neurobiol. 22, 383–388. doi: 10.1016/j.conb.2011.09.002
Burgoyne, R. D., and Morgan, A. (2011). Chaperoning the SNAREs: a role in preventing neurodegeneration? Nat. Cell Biol. 13, 8–9. doi: 10.1038/ncb0111-8
Burré, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Südhof, T. C. (2010). α-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Button, R. W., and Luo, S. (2017). The formation of autophagosomes during lysosomal defect: a new source of cytotoxicity. Autophagy 13, 1797–1798. doi: 10.1080/15548627.2017.1358850
Button, R. W., Roberts, S. L., Willis, T. L., Hanemann, C. O., and Luo, S. (2017). Accumulation of autophagosomes confers cytotoxicity. J. Biol. Chem. 292, 13599–13614. doi: 10.1074/jbc.M117.782276
Calabrese, F., Riva, M. A., and Molteni, R. (2016). Synaptic alterations associated with depression and schizophrenia: potential as a therapeutic target. Expert Opin. Ther. Targets 20, 1195–1207. doi: 10.1080/14728222.2016.1188080
Cangalaya, C., Stoyanov, S., Fischer, K.-D., and Dityatev, A. (2020). Light-induced engagement of microglia to focally remodel synapses in the adult brain. eLife 9:e58435. doi: 10.7554/eLife.58435
Cantuti Castelvetri, L., Givogri, M. I., Hebert, A., Smith, B., Song, Y., Kaminska, A., et al. (2013). The sphingolipid psychosine inhibits fast axonal transport in Krabbe disease by activation of GSK3β and deregulation of molecular motors. J. Neurosci. 33, 10048–10056. doi: 10.1523/JNEUROSCI.0217-13.2013
Cantuti-Castelvetri, L., Maravilla, E., Marshall, M., Tamayo, T., D’Auria, L., Monge, J., et al. (2015). Mechanism of neuromuscular dysfunction in Krabbe disease. J. Neurosci. 35, 1606–1616. doi: 10.1523/JNEUROSCI.2431-14.2015
Cantuti-Castelvetri, L., Zhu, H., Givogri, M. I., Chidavaenzi, R. L., Lopez-Rosas, A., and Bongarzone, E. R. (2012). Psychosine induces the dephosphorylation of neurofilaments by deregulation of PP1 and PP2A phosphatases. Neurobiol. Dis. 46, 325–335. doi: 10.1016/j.nbd.2012.01.013
Ceccarelli, B., Hurlbut, W., and Mauro, A. (1973). Turnover of transmitter and synaptic vesicles at the frog neuromuscular junction. J. Cell Biol. 57, 499–524. doi: 10.1083/jcb.57.2.499
Cesarini, L., Alfieri, P., Pantaleoni, F., Vasta, I., Cerutti, M., Petrangeli, V., et al. (2009). Cognitive profile of disorders associated with dysregulation of the RAS/MAPK signaling cascade. Am. J. Med. Genet. A 149, 140–146. doi: 10.1002/ajmg.a.32488
Chandra, S., Gallardo, G., Fernández-Chacón, R., Schlüter, O. M., and Südhof, T. C. (2005). α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell 123, 383–396. doi: 10.1016/j.cell.2005.09.028
Chater, T. E., and Goda, Y. (2014). The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front. Cell. Neurosci. 8:401. doi: 10.3389/fncel.2014.00401
D’Arcangelo, G., Grossi, D., Racaniello, M., Cardinale, A., Zaratti, A., Rufini, S., et al. (2016). Miglustat reverts the impairment of synaptic plasticity in a mouse model of NPC disease. Neural Plast. 2016:3830424. doi: 10.1155/2016/3830424
D’Auria, L., Reiter, C., Ward, E., Moyano, A. L., Marshall, M. S., Nguyen, D., et al. (2017). Psychosine enhances the shedding of membrane microvesicles: implications in demyelination in Krabbe’s disease. PLoS One 12:e0178103. doi: 10.1371/journal.pone.0178103
Díaz, M., Fabelo, N., Ferrer, I., and Marín, R. (2018). “Lipid raft aging” in the human frontal cortex during nonpathological aging: gender influences and potential implications in Alzheimer’s disease. Neurobiol. Aging 67, 42–52. doi: 10.1016/j.neurobiolaging.2018.02.022
Dwyer, C. A., Scudder, S. L., Lin, Y., Dozier, L. E., Phan, D., Allen, N. J., et al. (2017). Neurodevelopmental changes in excitatory synaptic structure and function in the cerebral cortex of sanfilippo syndrome iiia mice. Sci. Rep. 7:46576. doi: 10.1038/srep46576
Egawa, J., Pearn, M. L., Lemkuil, B. P., Patel, P. M., and Head, B. P. (2016). Membrane lipid rafts and neurobiology: age-related changes in membrane lipids and loss of neuronal function. J. Physiol. 594, 4565–4579. doi: 10.1113/JP270590
Forrest, C., Darlington, L., and Stone, T. (2013). Involvement of the proteasome and caspase activation in hippocampal long-term depression induced by the serine protease subtilisin. Neuroscience 231, 233–246. doi: 10.1016/j.neuroscience.2012.11.029
Gabandé-Rodríguez, E., Pérez-Cañamás, A., Soto-Huelin, B., Mitroi, D. N., Sánchez-Redondo, S., Martínez-Sáez, E., et al. (2019). Lipid-induced lysosomal damage after demyelination corrupts microglia protective function in lysosomal storage disorders. EMBO J. 38:e99553. doi: 10.15252/embj.201899553
Gan-Or, Z., Liong, C., and Alcalay, R. N. (2018). GBA-associated Parkinson’s disease and other synucleinopathies. Curr. Neurol. Neurosci. Rep. 18:44. doi: 10.1007/s11910-018-0860-4
Gao, J., Hirata, M., Mizokami, A., Zhao, J., Takahashi, I., Takeuchi, H., et al. (2016). Differential role of SNAP-25 phosphorylation by protein kinases A and C in the regulation of SNARE complex formation and exocytosis in PC12 cells. Cell. Signal. 28, 425–437. doi: 10.1016/j.cellsig.2015.12.014
Gieselmann, V., Matzner, U., Hess, B., Lullmann-Rauch, R., Coenen, R., Hartmann, D., et al. (1998). Metachromatic leukodystrophy: molecular genetics and an animal model. J. Inherit. Metab. Dis. 21, 564–574. doi: 10.1023/a:1005471106088
Greiner-Tollersrud, O. K., and Berg, T. (2000–2013). Lysosomal Storage Disorders. Madame Curie Bioscience Database [Internet]. Austin, TX: Landes Bioscience.
Hawkins-Salsbury, J. A., Parameswar, A. R., Jiang, X., Schlesinger, P. H., Bongarzone, E., Ory, D. S., et al. (2013). Psychosine, the cytotoxic sphingolipid that accumulates in globoid cell leukodystrophy, alters membrane architecture. J. Lipid Res. 54, 3303–3311. doi: 10.1194/jlr.M039610
Hawkins-Salsbury, J. A., Shea, L., Jiang, X., Hunter, D. A., Guzman, A. M., Reddy, A. S., et al. (2015). Mechanism-based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. J. Neurosci. 35, 6495–6505. doi: 10.1523/JNEUROSCI.4199-14.2015
Hayashi, T., McMahon, H., Yamasaki, S., Binz, T., Hata, Y., Südhof, T. C., et al. (1994). Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 13, 5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x
He, H.-Y., Shen, W., Zheng, L., Guo, X., and Cline, H. T. (2018). Excitatory synaptic dysfunction cell-autonomously decreases inhibitory inputs and disrupts structural and functional plasticity. Nat. Commun. 9:2893. doi: 10.1038/s41467-018-05125-4
Henley, J. M., and Wilkinson, K. A. (2013). AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin. Neurosci. 15, 11–27. doi: 10.31887/DCNS.2013.15.1/jhenley
Heuser, J., and Reese, T. (1973). Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J. Cell Biol. 57, 315–344. doi: 10.1083/jcb.57.2.315
Holtmaat, A., and Svoboda, K. (2009). Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 10, 647–658. doi: 10.1038/nrn2699
Honke, K. (2013). Biosynthesis and biological function of sulfoglycolipids. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 89, 129–138. doi: 10.2183/pjab.89.129
Hsieh, H., Boehm, J., Sato, C., Iwatsubo, T., Tomita, T., Sisodia, S., et al. (2006). AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843. doi: 10.1016/j.neuron.2006.10.035
Hyde, T. M., Ziegler, J. C., and Weinberger, D. R. (1992). Psychiatric disturbances in metachromatic leukodystrophy: insights into the neurobiology of psychosis. Arch. Neurol. 49, 401–406. doi: 10.1001/archneur.1992.00530280095028
Ishibashi, T., Dupree, J. L., Ikenaka, K., Hirahara, Y., Honke, K., Peles, E., et al. (2002). A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. J. Neurosci. 22, 6507–6514. doi: 10.1523/JNEUROSCI.22-15-06507.2002
Jonas, E. (2006). BCL-xL regulates synaptic plasticity. Mol. Interv. 6, 208–222. doi: 10.1124/mi.6.4.7
Jurado, S. (2018). AMPA receptor trafficking in natural and pathological aging. Front. Mol. Neurosci. 10:446. doi: 10.3389/fnmol.2017.00446
Kanninen, K. M., Grubman, A., Meyerowitz, J., Duncan, C., Tan, J.-L., Parker, S. J., et al. (2013). Increased zinc and manganese in parallel with neurodegeneration, synaptic protein changes and activation of Akt/GSK3 signaling in ovine CLN6 neuronal ceroid lipofuscinosis. PLoS One 8:e58644. doi: 10.1371/journal.pone.0058644
Karch, J., Schips, T. G., Maliken, B. D., Brody, M. J., Sargent, M. A., Kanisicak, O., et al. (2017). Autophagic cell death is dependent on lysosomal membrane permeability through Bax and Bak. eLife 6:e30543. doi: 10.7554/eLife.30543
Kim, S.-J., Zhang, Z., Hitomi, E., Lee, Y.-C., and Mukherjee, A. B. (2006). Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in INCL. Hum. Mol. Genet. 15, 1826–1834. doi: 10.1093/hmg/ddl105
Kinghorn, K. J., Asghari, A. M., and Castillo-Quan, J. I. (2017). The emerging role of autophagic-lysosomal dysfunction in Gaucher disease and Parkinson’s disease. Neural Regen. Res. 12:380. doi: 10.4103/1673-5374.202934
Koster, K. P., Francesconi, W., Berton, F., Alahmadi, S., Srinivas, R., and Yoshii, A. (2019). Developmental NMDA receptor dysregulation in the infantile neuronal ceroid lipofuscinosis mouse model. eLife 8:e40316. doi: 10.7554/eLife.40316
Kristensen, A. S., Jenkins, M. A., Banke, T. G., Schousboe, A., Makino, Y., Johnson, R. C., et al. (2011). Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat. Neurosci. 14, 727–735. doi: 10.1038/nn.2804
Kumar, S. (2007). Caspase function in programmed cell death. Cell Death Differ. 14, 32–43. doi: 10.1038/sj.cdd.4402060
Lam, A. D., Tryoen-Toth, P., Tsai, B., Vitale, N., and Stuenkel, E. L. (2008). SNARE-catalyzed fusion events are regulated by syntaxin1A-lipid interactions. Mol. Biol. Cell 19, 485–497. doi: 10.1091/mbc.e07-02-0148
Lee, J., Kim, H. R., Quinley, C., Kim, J., Gonzalez-Navajas, J., Xavier, R., et al. (2012). Autophagy suppresses interleukin-1β (IL-1β) signaling by activation of p62 degradation via lysosomal and proteasomal pathways. J. Biol. Chem. 287, 4033–4040. doi: 10.1074/jbc.M111.280065
Lepeta, K., Lourenco, M. V., Schweitzer, B. C., Martino Adami, P. V., Banerjee, P., Catuara-Solarz, S., et al. (2016). Synaptopathies: synaptic dysfunction in neurological disorders-A review from students to students. J. Neurochem. 138, 785–805. doi: 10.1111/jnc.13713
Liao, D., Hessler, N. A., and Malinow, R. (1995). Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature 375, 400–404. doi: 10.1038/375400a0
Lieberman, A. P., Puertollano, R., Raben, N., Slaugenhaupt, S., Walkley, S. U., and Ballabio, A. (2012). Autophagy in lysosomal storage disorders. Autophagy 8, 719–730. doi: 10.4161/auto.19469
Lim, J.-A., Li, L., Kakhlon, O., Myerowitz, R., and Raben, N. (2015). Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy 11, 385–402. doi: 10.1080/15548627.2015.1009779
Lisman, J., Yasuda, R., and Raghavachari, S. (2012). Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 13, 169–182. doi: 10.1038/nrn3192
Lu, C., Fu, W., Salvesen, G. S., and Mattson, M. P. (2002). Direct cleavage of AMPA receptor subunit GluR1 and suppression of AMPA currents by caspase-3: implications for synaptic plasticity and excitotoxic neuronal death. Neuromolecular Med. 1, 69–79. doi: 10.1385/NMM:1:1:69
Malinow, R., and Malenka, R. C. (2002). AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126. doi: 10.1146/annurev.neuro.25.112701.142758
Martin, D. S., Lonergan, P. E., Boland, B., Fogarty, M. P., Brady, M., Horrobin, D. F., et al. (2002). Apoptotic changes in the aged brain are triggered by interleukin-1β-induced activation of p38 and reversed by treatment with eicosapentaenoic acid. J. Biol. Chem. 277, 34239–34246. doi: 10.1074/jbc.M205289200
Mattson, M. P., and Duan, W. (1999). “Apoptotic” biochemical cascades in synaptic compartments: roles in adaptive plasticity and neurodegenerative disorders. J. Neurosci. Res. 58, 152–166. doi: 10.1002/(sici)1097-4547(19991001)58:1<152::aid-jnr15>3.0.co;2-v
McGonigal, R., Barrie, J. A., Yao, D., McLaughlin, M., Cunningham, M. E., Rowan, E. G., et al. (2019). Glial sulfatides and neuronal complex gangliosides are functionally interdependent in maintaining myelinating axon integrity. J. Neurosci. 39, 63–77. doi: 10.1523/JNEUROSCI.2095-18.2018
Morris, G. P., Clark, I. A., Zinn, R., and Vissel, B. (2013). Microglia: a new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol. Learn. Mem. 105, 40–53. doi: 10.1016/j.nlm.2013.07.002
Morrison, J. H., and Baxter, M. G. (2012). The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat. Rev. Neurosci. 13, 240–250. doi: 10.1038/nrn3200
Moskot, M., Bocheńska, K., Jakóbkiewicz-Banecka, J., Banecki, B., and Gabig-Cimińska, M. (2018). Abnormal sphingolipid world in inflammation specific for lysosomal storage diseases and skin disorders. Int. J. Mol. Sci. 19:247. doi: 10.3390/ijms19010247
Moyano, A. L., Li, G., Lopez-Rosas, A., Månsson, J.-E., van Breemen, R. B., and Givogri, M. I. (2014). Distribution of C16:0, C18:0, C24:1, and C24:0 sulfatides in central nervous system lipid rafts by quantitative ultra-high-pressure liquid chromatography tandem mass spectrometry. Anal. Biochem. 467, 31–39. doi: 10.1016/j.ab.2014.08.033
Nguyen, P. T., Dorman, L. C., Pan, S., Vainchtein, I. D., Han, R. T., Nakao-Inoue, H., et al. (2020). Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell 182, 388e15–403e15. doi: 10.1016/j.cell.2020.05.050
Orr, M. E., and Oddo, S. (2013). Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res. Ther. 5:53. doi: 10.1186/alzrt217
Pérez-Otaño, I., and Ehlers, M. D. (2005). Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci. 28, 229–238. doi: 10.1016/j.tins.2005.03.004
Pandey, M. K., Jabre, N. A., Xu, Y.-H., Zhang, W., Setchell, K. D., and Grabowski, G. A. (2014). Gaucher disease: chemotactic factors and immunological cell invasion in a mouse model. Mol. Genet. Metab. 111, 163–171. doi: 10.1016/j.ymgme.2013.09.002
Parkinson, G. T., and Hanley, J. G. (2018). Mechanisms of AMPA receptor endosomal sorting. Front. Mol. Neurosci. 11:440. doi: 10.3389/fnmol.2018.00440
Peake, K. B., Campenot, R. B., Vance, D. E., and Vance, J. E. (2011). Niemann-Pick Type C1 deficiency in microglia does not cause neuron death in vitro. Biochim. Biophy. Acta 1812, 1121–1129. doi: 10.1016/j.bbadis.2011.06.003
Perry, V. H., and O’Connor, V. (2010). The role of microglia in synaptic stripping and synaptic degeneration: a revised perspective. ASN Neuro 2:e00047. doi: 10.1042/AN20100024
Pressey, S. N., Smith, D. A., Wong, A. M., Platt, F. M., and Cooper, J. D. (2012). Early glial activation, synaptic changes and axonal pathology in the thalamocortical system of Niemann-Pick type C1 mice. Neurobiol. Dis. 45, 1086–1100. doi: 10.1016/j.nbd.2011.12.027
Qin, Y., Zhu, Y., Baumgart, J. P., Stornetta, R. L., Seidenman, K., Mack, V., et al. (2005). State-dependent Ras signaling and AMPA receptor trafficking. Genes Dev. 19, 2000–2015. doi: 10.1101/gad.342205
Ramos-Miguel, A., García-Sevilla, J. A., Barr, A. M., Bayer, T. A., Falkai, P., Leurgans, S. E., et al. (2017). Decreased cortical FADD protein is associated with clinical dementia and cognitive decline in an elderly community sample. Mol. Neurodegener. 12:26. doi: 10.1186/s13024-017-0168-x
Rangaraju, V., Calloway, N., and Ryan, T. A. (2014). Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. doi: 10.1016/j.cell.2013.12.042
Sévin, M., Lesca, G., Baumann, N., Millat, G., Lyon-Caen, O., Vanier, M. T., et al. (2007). The adult form of Niemann-Pick disease type C. Brain 130, 120–133. doi: 10.1093/brain/awl260
Salaün, C., Gould, G. W., and Chamberlain, L. H. (2005). The snare proteins SNAP-25 and SNAP-23 display different affinities for lipid rafts in PC12 cells. Regulation by distinct cysteine-rich domains. J. Biol. Chem. 280, 1236–1240. doi: 10.1074/jbc.M410674200
Sambri, I., D’Alessio, R., Ezhova, Y., Giuliano, T., Sorrentino, N. C., Cacace, V., et al. (2017). Lysosomal dysfunction disrupts presynaptic maintenance and restoration of presynaptic function prevents neurodegeneration in lysosomal storage diseases. EMBO Mol. Med. 9, 112–132. doi: 10.15252/emmm.201606965
Sapkota, A., Gaire, B. P., Kang, M.-G., and Choi, J. W. (2019). S1P2 contributes to microglial activation and M1 polarization following cerebral ischemia through ERK1/2 and JNK. Sci. Rep. 9:12106. doi: 10.1038/s41598-019-48609-z
Scheiblich, H., Schlütter, A., Golenbock, D. T., Latz, E., Martinez-Martinez, P., and Heneka, M. T. (2017). Activation of the NLRP3 inflammasome in microglia: the role of ceramide. J. Neurochem. 143, 534–550. doi: 10.1111/jnc.14225
Seranova, E., Connolly, K. J., Zatyka, M., Rosenstock, T. R., Barrett, T., Tuxworth, R. I., et al. (2017). Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 61, 733–749. doi: 10.1042/EBC20170055
Seregin, S. S., and Amalfitano, A. (2011). Gene therapy for lysosomal storage diseases: progress, challenges and future prospects. Curr. Pharm. Des. 17, 2558–2574. doi: 10.2174/138161211797247578
Sharma, M., Burré, J., and Südhof, T. C. (2011). CSPα promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 13, 30–39. doi: 10.1038/ncb2131
Sharma, S., and Lindau, M. (2017). t-SNARE transmembrane domain clustering modulates lipid organization and membrane curvature. J. Am. Chem. Soc. 139, 18440–18443. doi: 10.1021/jacs.7b10677
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Smolders, S., and Van Broeckhoven, C. (2020). Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 8:63. doi: 10.1186/s40478-020-00935-4
Soykan, T., Maritzen, T., and Haucke, V. (2016). Modes and mechanisms of synaptic vesicle recycling. Curr. Opin. Neurobiol. 39, 17–23. doi: 10.1016/j.conb.2016.03.005
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Su, J. H., Zhao, M., Anderson, A. J., Srinivasan, A., and Cotman, C. W. (2001). Activated caspase-3 expression in Alzheimer’s and aged control brain: correlation with Alzheimer pathology. Brain Res. 898, 350–357. doi: 10.1016/s0006-8993(01)02018-2
Sural-Fehr, T., Singh, H., Cantuti-Catelvetri, L., Zhu, H., Marshall, M. S., Rebiai, R., et al. (2019). Inhibition of the IGF-1-PI3K-Akt-mTORC2 pathway in lipid rafts increases neuronal vulnerability in a genetic lysosomal glycosphingolipidosis. Dis. Models Mech. 12:dmm036590. doi: 10.1242/dmm.036590
Sutton, R. B., Fasshauer, D., Jahn, R., and Brunger, A. T. (1998). Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature 395, 347–353. doi: 10.1038/26412
Suzuki, K. (1998). Twenty five years of the “psychosine hypothesis”: a personal perspective of its history and present status. Neurochem. Res. 23, 251–259. doi: 10.1023/a:1022436928925
Teixeira, C. A., Miranda, C. O., Sousa, V. F., Santos, T. E., Malheiro, A. R., Solomon, M., et al. (2014). Early axonal loss accompanied by impaired endocytosis, abnormal axonal transport, and decreased microtubule stability occur in the model of Krabbe’s disease. Neurobiol. Dis. 66, 92–103. doi: 10.1016/j.nbd.2014.02.012
Tessitore, A., Pirozzi, M., and Auricchio, A. (2009). Abnormal autophagy, ubiquitination, inflammation and apoptosis are dependent upon lysosomal storage and are useful biomarkers of mucopolysaccharidosis VI. Pathogenetics 2:4. doi: 10.1186/1755-8417-2-4
Tong, J., Borbat, P. P., Freed, J. H., and Shin, Y.-K. (2009). A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol. Proc. Natl. Acad. Sci. U S A 106, 5141–5146. doi: 10.1073/pnas.0813138106
Tremblay, M.-È., Lowery, R. L., and Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8:e1000527. doi: 10.1371/journal.pbio.1000527
Võikar, V., Rauvala, H., and Ikonen, E. (2002). Cognitive deficit and development of motor impairment in a mouse model of Niemann-Pick type C disease. Behav. Brain Res. 132, 1–10. doi: 10.1016/s0166-4328(01)00380-1
Virmani, T., Gupta, P., Liu, X., Kavalali, E. T., and Hofmann, S. L. (2005). Progressively reduced synaptic vesicle pool size in cultured neurons derived from neuronal ceroid lipofuscinosis-1 knockout mice. Neurobiol. Dis. 20, 314–323. doi: 10.1016/j.nbd.2005.03.012
Vitry, S., Ausseil, J., Hocquemiller, M., Bigou, S., dos Santos Coura, R., and Heard, J. M. (2009). Enhanced degradation of synaptophysin by the proteasome in mucopolysaccharidosis type IIIB. Mol. Cell. Neurosci. 41, 8–18. doi: 10.1016/j.mcn.2009.01.001
Wang, M. L., Motamed, M., Infante, R. E., Abi-Mosleh, L., Kwon, H. J., Brown, M. S., et al. (2010). Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 12, 166–173. doi: 10.1016/j.cmet.2010.05.016
Weinert, S., Jabs, S., Supanchart, C., Schweizer, M., Gimber, N., Richter, M., et al. (2010). Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl− accumulation. Science 328, 1401–1403. doi: 10.1126/science.1188072
White, A. B., Givogri, M. I., Lopez-Rosas, A., Cao, H., van Breemen, R., Thinakaran, G., et al. (2009). Psychosine accumulates in membrane microdomains in the brain of Krabbe patients, disrupting the raft architecture. J. Neurosci. 29, 6068–6077. doi: 10.1523/JNEUROSCI.5597-08.2009
Wilkinson, F. L., Holley, R. J., Langford-Smith, K. J., Badrinath, S., Liao, A., Langford-Smith, A., et al. (2012). Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS One 7:e35787. doi: 10.1371/journal.pone.0035787
Wynn, R. F., Wraith, J. E., Mercer, J., O’Meara, A., Tylee, K., Thornley, M., et al. (2009). Improved metabolic correction in patients with lysosomal storage disease treated with hematopoietic stem cell transplant compared with enzyme replacement therapy. J. Pediatr. 154, 609–611. doi: 10.1016/j.jpeds.2008.11.005
Xu, S., Zhou, S., Xia, D., Xia, J., Chen, G., Duan, S., et al. (2010). Defects of synaptic vesicle turnover at excitatory and inhibitory synapses in Niemann-Pick C1-deficient neurons. Neuroscience 167, 608–620. doi: 10.1016/j.neuroscience.2010.02.033
Zhou, L.-J., Peng, J., Xu, Y.-N., Zeng, W.-J., Zhang, J., Wei, X., et al. (2019). Microglia are indispensable for synaptic plasticity in the spinal dorsal horn and chronic pain. Cell Rep. 27, 3844.e6–3859.e6. doi: 10.1016/j.celrep.2019.05.087
Zoghbi, H. Y., and Bear, M. F. (2012). Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 4:a009886:a009886. doi: 10.1101/cshperspect.a009886
Zucca, F. A., Vanna, R., Cupaioli, F. A., Bellei, C., De Palma, A., Di Silvestre, D., et al. (2018). Neuromelanin organelles are specialized autolysosomes that accumulate undegraded proteins and lipids in aging human brain and are likely involved in Parkinson’s disease. NPJ Parkinsons Dis. 4:17. doi: 10.1038/s41531-018-0050-8
Keywords: lysosomes, synapses, glutamate receptors, GABA, sphingolipids, cholesterol, LTP, LTD
Citation: Rebiai R, Givogri MI, Gowrishankar S, Cologna SM, Alford ST and Bongarzone ER (2021) Synaptic Function and Dysfunction in Lysosomal Storage Diseases. Front. Cell. Neurosci. 15:619777. doi: 10.3389/fncel.2021.619777
Received: 21 October 2020; Accepted: 12 February 2021;
Published: 04 March 2021.
Edited by:
Nicolas Unsain, Medical Research Institute Mercedes and Martín Ferreyra (INIMEC), ArgentinaReviewed by:
Natalia L. Kononenko, University of Cologne, GermanyCopyright © 2021 Rebiai, Givogri, Gowrishankar, Cologna, Alford and Bongarzone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria I. Givogri, bWdpdm9ncmlAdWljLmVkdQ==; Swetha Gowrishankar, c3dldGhhZ0B1aWMuZWR1; Stephanie M. Cologna, Y29sb2duYUB1aWMuZWR1; Simon T. Alford, c3RhQHVpYy5lZHU=; Ernesto R. Bongarzone, ZWJvbmdhcnpAdWljLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.