 Annapoorani Muthiah
Annapoorani Muthiah Gary D. Housley
Gary D. Housley Matthias Klugmann
Matthias Klugmann Dominik Fröhlich
Dominik Fröhlich
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 26 January 2021
Sec. Cellular Neuropathology
Volume 14 - 2020 | https://doi.org/10.3389/fncel.2020.626610
This article is part of the Research Topic Myelin Repair: At the Crossing-Lines of Myelin Biology and Gene Therapy View all 10 articles
Aminoacyl-tRNA synthetases (ARSs) accurately charge tRNAs with their respective amino acids. As such, they are vital for the initiation of cytosolic and mitochondrial protein translation. These enzymes have become increasingly scrutinized in recent years for their role in neurodegenerative disorders caused by the mutations of ARS-encoding genes. This review focuses on two such genes—DARS1 and DARS2—which encode cytosolic and mitochondrial aspartyl-tRNA synthetases, and the clinical conditions associated with mutations of these genes. We also describe attempts made at modeling these conditions in mice, which have both yielded important mechanistic insights. Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL) is a disease caused by a range of mutations in the DARS2 gene, initially identified in 2003. Ten years later, hypomyelination with brainstem and spinal cord involvement and leg spasticity (HBSL), caused by mutations of cytosolic DARS1, was discovered. Multiple parallels have been drawn between the two conditions. The Magnetic Resonance Imaging (MRI) patterns are strikingly similar, but still set these two conditions apart from other leukodystrophies. Clinically, both conditions are characterized by lower limb spasticity, often associated with other pyramidal signs. However, perhaps due to earlier detection, a wider range of symptoms, including peripheral neuropathy, as well as visual and hearing changes have been described in LBSL patients. Both HBSL and LBSL are spectrum disorders lacking genotype to phenotype correlation. While the fatal phenotype of Dars1 or Dars2 single gene deletion mouse mutants revealed that the two enzymes lack functional redundancy, further pursuit of disease modeling are required to shed light onto the underlying disease mechanism, and enable examination of experimental treatments, including gene therapies.
Hypomyelination with Brainstem and Spinal Cord Involvement and Leg Spasticity (HBSL) and Leukoencephalopathy with Brainstem and Spinal Cord Involvement and Lactate Elevation (LBSL) both belong to a group of white matter disorders termed leukodystrophies. These conditions are caused by mutations of the DARS1 and DARS2 genes, respectively—two genes that encode aspartyl-tRNA synthetase enzymes responsible for the accurate charging of aspartate-specific transfer ribonucleic acid (tRNA) with aspartate (Scheper et al., 2007; Taft et al., 2013). Both enzymes are part of a larger group of enzymes known as aminoacyl-tRNA synthetases (ARSs), which are essential for protein translation. In humans, 36 ARSs exist without functional redundancy between them (Antonellis and Green, 2008).
ARSs can be divided into two subgroups based on their location of action—cytosolic and mitochondrial ARSs. In general, each ARS functions exclusively in one compartment and is responsible for the pairing of one of the 20 amino acids with its respective tRNA. Exceptions include glycyl-tRNA synthetase (GlyRS) and lysyl-tRNA synthetase (LysRS), which are capable of functioning in both compartments, and glutamyl-prolyl-tRNA synthetase (EPRS) which is able to pair both proline and glutamic acid with their respective tRNAs in the cytosolic compartment (Antonellis and Green, 2008). Interestingly, glutaminyl-tRNA synthetase (GlnRS) does not have a mitochondrial counterpart. Charging of mitochondrial tRNAGln to glutamine takes place through an indirect pathway, where “mischarging” with glutamic acid by mitochondrial glutamyl-tRNA synthetase (GluRS) is the first step (Nagao et al., 2009). While DARS1 encodes the cytosolic aspartyl-tRNA synthetase (AspRS), DARS2 encodes its mitochondrial counterpart (mt-AspRS). AspRS catalyzes the aminoacylation reaction exclusively in the cytosol, whereas mt-AspRS is transported from the cytosol to the mitochondrial matrix where it is involved in mitochondrial protein synthesis.
LBSL was first described in 2003, based on distinctive clinical features and a characteristic Magnetic Resonance Imaging (MRI) pattern (Van Der Knaap et al., 2003). Following this initial MRI-based diagnosis, a genetic basis for the condition was established in 2007, concluding mutations harbored in the nuclear-encoded DARS2 gene to be the cause (Scheper et al., 2007). This was the first mitochondrial aminoacyl-tRNA synthetase gene to be identified as disease-causing in its mutated state and marked the beginning of many more conditions being linked to this group of enzymes. Subsequently, in 2013, HBSL was first characterized by Taft et al., who established mutations in the cytosolic AspRS encoding gene DARS1 as the genetic cause for the condition (Taft et al., 2013). Both diseases are inherited in an autosomal recessive manner (Scheper et al., 2007; Taft et al., 2013). To date, more than 100 LBSL patients have been reported in the literature, while only 16 HBSL patients have been described in the past 7 years. However, the actual number of diagnosed HBSL patients is recognized to be considerably higher than the number of reported cases (pers. comm.).
Of note, neither condition features extra-neurological signs, particularly cardiac and skin symptoms, even though these tissues have high energy demand and cell turnover, respectively. This selective neurological phenotype is not unique to HBSL or LBSL but has been reported for many ARS-related diseases (Fuchs et al., 2019). The reason for the neurological preponderance of both HBSL and LBSL is the subject of ongoing studies. It is worth mentioning though that few ARS-related conditions do involve non-neural tissues; for example, some AARS2 mutations are known to cause severe cardiomyopathy (Götz et al., 2011; Taylor et al., 2014), while other AARS2 mutations do not.
The age at which symptoms are first detected is highly variable even within each condition. The onset of HBSL ranges from 4 months (Taft et al., 2013) to 18 years (Wolf et al., 2015). The average age of onset is 3.5 years; however, most patients experience first symptoms before the age of 1 year (56% of reviewed patients). For LBSL, the onset of disease occurs on average around the age of seven, but has been detected from birth (Steenweg et al., 2012) to as old as 43 years (Ikazabo et al., 2020). It is generally understood that, for both HBSL and LBSL, the earlier the onset of symptoms the more severe and rapid the progression of disease. For instance, one study found that LBSL patients who experience symptoms before the age of 1.5 years were more likely to be wheelchair-dependent than those experiencing symptoms after this age (Van Berge et al., 2014). At present, only seven LBSL patients out of 99 patients reviewed are known to be wheelchair-dependent (Serkov et al., 2004; Távora et al., 2007; Tylki-Szymanska et al., 2014; Shimojima et al., 2017; Yelam et al., 2019). These patients had onset of symptoms in infancy or early childhood. In terms of unsupported mobility, it can be argued that patients with HBSL experience more debilitating symptoms compared to patients with LBSL. All initially described HBSL patients with infantile-onset (<2 years old) were unable to mobilize independently (Taft et al., 2013). This is due to severe spasticity, a defining feature of HBSL, with lower limbs affected more severely than upper limbs. HBSL patients with childhood-onset presented with delayed motor development but were subsequently able to mobilize independently, albeit with repeated falls (Ong et al., 2020). HBSL patients with adult-onset have been shown to experience fewer and milder symptoms compared to infantile-onset patients (Wolf et al., 2015).
The age of onset also seems to predict symptomology. While there are some common clinical features that all LBSL and HBSL patients share, clinical presentation and progression seem to differ between different age groups. Infantile-onset LBSL patients typically present with hypotonia, areflexia and developmental delay or regression of achieved developmental milestones (Miyake et al., 2011; Steenweg et al., 2012; Grechanina and Zdubskaya, 2014; Tylki-Szymanska et al., 2014; Kohler et al., 2015; Navarro Vázquez et al., 2016; Yahia et al., 2018). Progression of the disease is also more rapid and typically followed by development of upper motor neuron (UMN) signs such as spasticity, hyperreflexia, and a positive Babinski sign. To date, eight deaths associated with LBSL have been reported and all of them belonged to this group of patients. Death usually occurred within 2 years of age, or ~1 year after the onset of symptoms with the exception of one patient who lived 7.5 years after onset of symptoms (Miyake et al., 2011; Orcesi et al., 2011; Steenweg et al., 2012; Navarro Vázquez et al., 2016; Rathore et al., 2017). The cause of death for these patients was either respiratory failure or respiratory tract infections. Children with disease onset in childhood or adolescence, on the other hand, typically present with gait difficulties, such as ataxia or unsteadiness and frequent falling. This is often accompanied by examination findings of spasticity, hyperreflexia, positive Babinski sign and distinct loss of proprioception and/or vibration sense. Ataxic gait in LBSL is also specified to be sensory in some reports (Labauge et al., 2007; Erturk et al., 2010; Synofzik et al., 2011; Tzoulis et al., 2012), further reinforced by findings of a positive Romberg sign (Linnankivi et al., 2004; Sharma et al., 2011; Martikainen et al., 2013; Ikazabo et al., 2020) and loss of proprioceptive and vibration sense in 11% and 49% of all clinically described LBSL cases, respectively (Table 1). In adult-onset patients, the presenting complaints also relate to difficulties in walking, however the clinical picture often includes more atypical findings. One patient, for example, experienced episodic attacks of ataxia (Synofzik et al., 2011), while another patient experienced predominantly unilateral (rather than bilateral) symptoms of spasticity and muscle weakness (Moore et al., 2012). These two patients incidentally also belonged to the few LBSL patients who did not possess the highly prevalent DARS2 intron 2 splice site mutation.
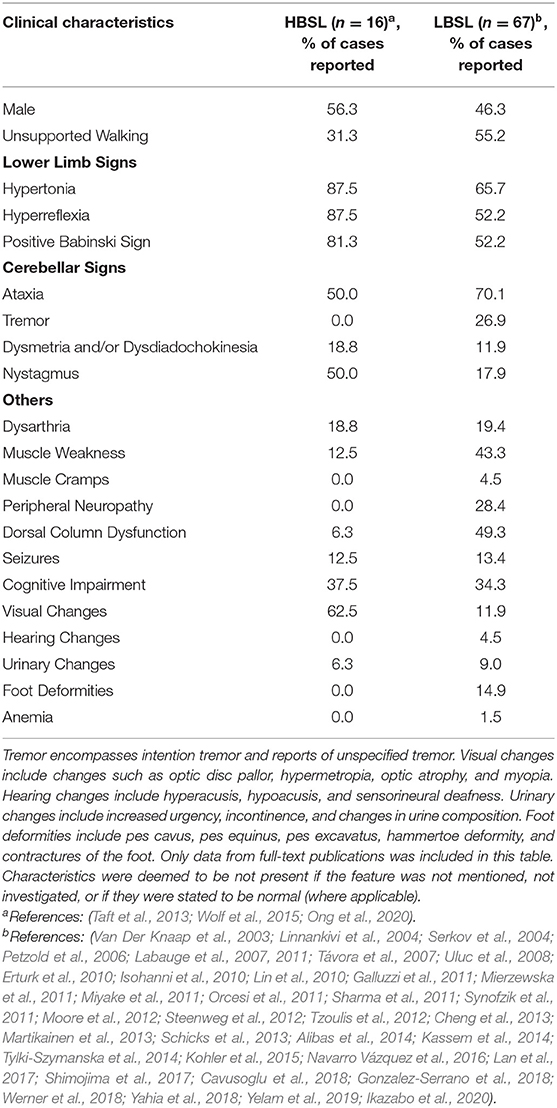
Table 1. Overview of clinical characteristics reported in HBSL and LBSL.
There are several notable exceptions to this generalized disease pattern in LBSL. The most significant is a male who experienced first symptoms at the age of 9 months (Kohler et al., 2015), where as an infant, he was irritable and had attention deficits. These worsened following a fever and respiratory infection, additionally resulting in poor eye contact and a delay in achieving motor milestones. Surprisingly, from 12.5 months of age, his condition improved to reach appropriate milestones for his age, including social smiling. His only residual symptom is mild ataxia that does not impede his ability to mobilize independently, including walking and running. It is also interesting that this patient is homozygous for DARS2 mutations (c.172C>G; p.R58G). While the majority of LBSL patients are compound heterozygous, homozygosity has only been described in five LBSL patients from three unrelated families (Miyake et al., 2011; Synofzik et al., 2011; Kohler et al., 2015). Hence, homozygosity was initially presumed to be developmentally lethal until the first LBSL patient with homozygous DARS2 mutations was described in 2011. Two of these patients (who are siblings) have experienced infantile-onset disease with severe disease course and subsequent death (Yamashita et al., 2013). Meanwhile, the third sibling experienced cognitive delay and severe motor deficits, and currently mobilizes by wheelchair.
Another patient with homozygous DARS2 mutations experienced episodic bouts of ataxia that were induced by exercise, a highly unique disease presentation that has not been described in another LBSL patient (Synofzik et al., 2011). It was suggested that the finding of episodic ataxia might be incidental rather than the result of DARS2 mutations (van der Knaap et al., 2014). Episodic ataxia, or recurring intermittent attacks of imbalance and incoordination, is a cardinal sign of a group of inherited diseases known as hereditary episodic ataxias (EA). Individuals with EA experience bouts of ataxia, of varying frequency and intensity, accompanied by other signs such as seizures and muscle fasciculation (Teive and Ashizawa, 2015). These attacks are often triggered by several environmental factors (Jen and Wan, 2018). The aforementioned patient, however, was not found to harbor any mutations for the most common EA types (Synofzik et al., 2011). Homozygous mutations, therefore, do not seem to be a reliable indicator of the prognosis in LBSL. In fact, genotype-phenotype correlations have been challenging to establish in this condition, and there are suggestions of additional genetic and non-genetic factors influencing disease presentation (Steenweg et al., 2012; Van Berge et al., 2014). This is further highlighted by the identification of a female who possessed the same two mutant DARS2 alleles as her affected sister but experienced no clinical symptoms (Labauge et al., 2011).
Ultimately, the most common sign in LBSL is cerebellar ataxia and unsteady gait. Motor impairment is mainly caused by UMN dysfunction, however lower motor neuron (LMN) signs have also been described (Miyake et al., 2011; Steenweg et al., 2012; Grechanina and Zdubskaya, 2014; Yelam et al., 2019). Some other less frequent symptoms that have been observed in LBSL patients include unspecified tremor (Isohanni et al., 2010; Galluzzi et al., 2011; Sharma et al., 2011; Yamashita et al., 2013; Werner et al., 2018) and peripheral neuropathy (Linnankivi et al., 2004; Uluc et al., 2008), among others. Cognitive deficits are usually mild, if present, and typically affect information-processing speed, concentration, and short-term memory (Martikainen et al., 2013). An overview of reported LBSL and HBSL symptoms can be found in Table 1.
HBSL is less diverse in its clinical presentation, and there has only been one known death associated with HBSL thus far (pers. comm.). This patient experienced infantile onset of symptoms and lived to 9 years of age. The most common presentation in infants is regression in motor development or developmental delay (Taft et al., 2013). Some additional congenital abnormalities including spinal cord tethering (Taft et al., 2013), vertebral malformations, trigonocephaly and Chiari malformations (Ong et al., 2020) have also been noted in a few patients. In older-onset HBSL patients, spasticity is one of the first symptoms (Wolf et al., 2015). Most reported patients have experienced leg spasticity—to varying degrees—that have impaired their ability to walk. Gait has been described as diplegic (Taft et al., 2013) or in-toeing (Ong et al., 2020) in those able to mobilize. In more severe cases, upper limb involvement is also noted. HBSL is predominantly a UMN disorder, with consistent hypertonia, hyperreflexia and positive Babinski sign noted in nearly all patients (Table 1). Nystagmus has also been a common finding. To a lesser extent, axial hypotonia (Taft et al., 2013; Wolf et al., 2015), epilepsy (Taft et al., 2013), cognitive deficits (Taft et al., 2013; Ong et al., 2020), and headaches (Ong et al., 2020) have been noted in some patients. In two adult-onset patients, symptoms of dorsal column dysfunction and urinary changes have been identified, as seen in LBSL (Wolf et al., 2015). The main differences in the clinical picture of HBSL and LBSL, are the clear lack of peripheral neuropathy and LMN signs in HBSL patients, while significant leg spasticity is the predominant feature. No hearing changes have been noted in HBSL patients. Evidence of intrafamilial variations and weak genotype-phenotype correlations, are present in HBSL as well. This is supported by a report of a family with three affected siblings carrying the same mutations but experiencing different disease courses (Ong et al., 2020).
In both conditions, a number of patients experience symptoms following clear precipitating events—for instance, fever, viral illness, or head trauma—either resulting in the first onset of symptoms (Wolf et al., 2015; Yahia et al., 2018) or further neurological deterioration (Uluc et al., 2008; Taft et al., 2013). Most patients experiencing such an exacerbation of symptoms usually recover to a new baseline with some residual deficits (Serkov et al., 2004; Isohanni et al., 2010; Wolf et al., 2015).
MRI is the tool of choice when investigating white matter (WM) diseases. Most leukodystrophies can be accurately diagnosed with this modality, as they present with a unique MRI signature (Schiffmann and van der Knaap, 2009). However, between HBSL and LBSL, MRI is not highly discriminatory, as both conditions affect similar brain structures and tracts (Taft et al., 2013; Wolf et al., 2015). While MRI remains the main imaging modality to initially identify HBSL and LBSL patients, genetic analysis is necessary to subsequently differentiate between the two conditions and is currently the only means to a definite diagnosis.
Nonetheless, prior to genetic analysis, a preliminary LBSL diagnosis can be made on an MRI basis. With the revised criteria proposed by Steenweg et al., three major criteria must be fulfilled—signal changes in the cerebral WM, involvement of the dorsal columns and lateral corticospinal tracts in the spinal cord and the involvement of either the pyramidal or medial lemniscal tracts in the medulla (Steenweg et al., 2012). These three major criteria are largely satisfied in all genetically confirmed LBSL patients (Table 2). Abnormal cerebral WM is typically observed in the periventricular region, with distinct sparing of u-fibers. A sizeable number of patients have also displayed WM changes extending into the deep cerebral WM (Kassem et al., 2014). Changes are usually bilateral and inhomogeneous, and sometimes described as spotty. Cerebral WM has also been noted to have bilateral focal lesions in some patients (Erturk et al., 2010). Rarely, additional involvement of the u-fibers has been reported (Galluzzi et al., 2011; Orcesi et al., 2011, Yamashita et al., 2013).
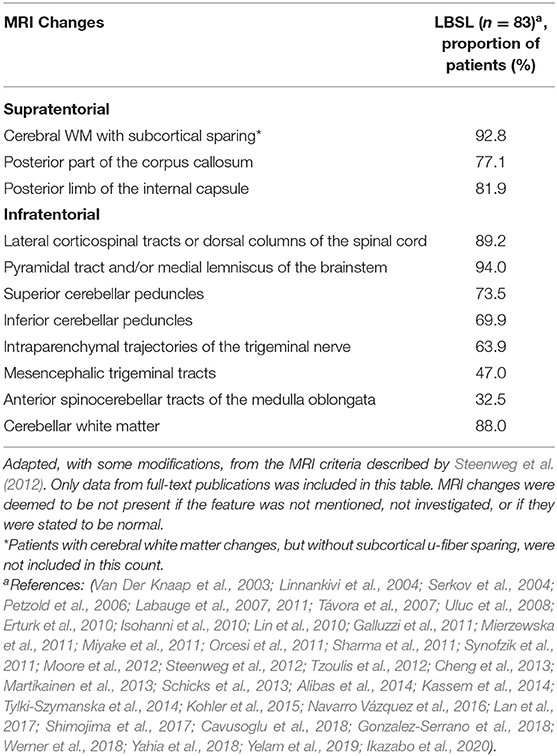
Table 2. Overview of MRI changes reported in LBSL.
In conjunction with the major criteria, at least one minor criterion should also be satisfied for diagnosis of LBSL. These minor criteria include signal changes in the cerebellar WM, posterior corpus callosum, posterior limb of the internal capsule (IC), superior and inferior cerebellar peduncles, intraparenchymal trajectories of the trigeminal nerve, mesencephalic tracts of the trigeminal nerve and anterior spinocerebellar tract in the medulla (Steenweg et al., 2012). A majority of patients display changes in the cerebellar WM, corpus callosum and posterior limb of the IC (Table 2). Involvement of the trigeminal nerve tracts and spinocerebellar tracts have been reported to a lesser extent (Table 2). Apart from these typical MRI changes, some atypical findings have been reported in genetically confirmed LBSL patients. These include the bilateral involvement of gray matter structures such as the globus pallidus (Galluzzi et al., 2011; Steenweg et al., 2012), red nucleus (Galluzzi et al., 2011), thalamus (Orcesi et al., 2011) and dentate nucleus (Galluzzi et al., 2011; Iyer and Philip, 2011; Orcesi et al., 2011; Yahia et al., 2018). Furthermore, cerebral (Tzoulis et al., 2012; Yamashita et al., 2013; Yahia et al., 2018) and cerebellar atrophy (Van Der Knaap et al., 2003; Yahia et al., 2018) has been reported, in addition to cerebellar hypoplasia (Isohanni et al., 2010). A singular case of calcifications of the cerebral WM and other regions typically affected by LBSL was identified by computed tomography (CT) scanning (Orcesi et al., 2011).
For HBSL, no such MRI criteria have been defined yet, however, some characteristic MRI features can be observed in the majority of patients (Table 3). These include homogenous cerebral WM changes (Taft et al., 2013; Wolf et al., 2015; Ong et al., 2020), abnormal posterior limb of the IC and hyperintensity of the corpus callosum on T2-weighted images (Taft et al., 2013; Wolf et al., 2015). Thinning of the corpus callosum has also been reported in some patients (Taft et al., 2013). In one HBSL patient, the additional involvement of the anterior corticospinal tract was observed on spinal MRI (Wolf et al., 2015). As seen in LBSL, the superior and inferior cerebellar peduncles, the pyramidal tracts in the medulla and spinal cord, and the dorsal columns of the spinal cord were affected in most patients. The cerebellar WM was also affected in several patients. Unlike in LBSL, medial lemniscal involvement was only observed in one patient (Taft et al., 2013), while no involvement of the trigeminal nerve tracts, spinocerebellar tracts and gray matter structures have been described for HBSL so far. The latter findings are also not common in LBSL and are therefore not reliable differentiators between the two conditions.
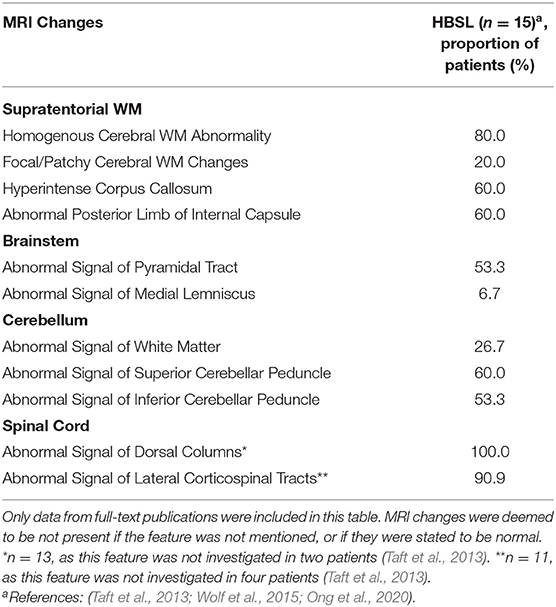
Table 3. MRI features reported in HBSL.
As the name suggests, lactate elevation detected by magnetic resonance spectroscopy (MRS) is often thought to be a defining feature of LBSL. However, elevated lactate levels were only detected in 68% of LBSL patients. Furthermore, even in patients where lactate was elevated, this elevation fluctuated over time (Van Der Knaap et al., 2003; Isohanni et al., 2010). Therefore, while raised lactate levels may be a good indicator for LBSL, an absence of lactate elevation does not necessarily exclude it, or automatically confirm HBSL. It has been suggested that lactate elevation might be more common in early-onset LBSL (Werner et al., 2018). Additional MRS analyses of affected WM regions of LBSL patients also revealed decreased N-acetylaspartate (NAA), elevated choline (Cho), elevated myoinositol (mIns) (Van Der Knaap et al., 2003) and occasionally elevated creatine (Cr) (Erturk et al., 2010). While no lactate elevation has been described in HBSL patients, one case report of HBSL showed mildly increased Cho in one patient and decreased NAA in another patient (Ong et al., 2020). A decrease in NAA, as observed in 52% of LBSL patients reviewed, implicates axonal damage or loss (Moffett et al., 2007). Increased Cho levels suggest demyelination (Van Der Knaap et al., 2003; Galluzzi et al., 2011) while an increase in mIns is a sign for gliosis (Chang et al., 2013). Finally, Cr when elevated can imply disturbance in cell metabolism (Rackayova et al., 2017). Taken together, a more detailed analysis of the metabolic changes in the brains of LBSL and HBSL patients could shed some light on the underlying mechanisms of these conditions.
Diffusion weighted imaging (DWI) has also been performed in LBSL patients, revealing diffusion restriction in parts of the affected WM (Mierzewska et al., 2011; Alibas et al., 2014; Kassem et al., 2014). DWI indicated that there are progressive changes that seem to correlate with different stages of the disease (Steenweg et al., 2011). Initially, the observed diffusion restriction is attributed to myelin-splitting and oedema within myelin. This is followed by shifting of water to the interstitial space, resulting in high T2 signals (Steenweg et al., 2011). Thereafter, the loss of water in the interstitial space indicates the final component of the disease process, leading to intermediate T2 signals (Steenweg et al., 2011). These findings have been supported by post-mortem histopathological observations in two LBSL patients, which showed intra-lamellar splitting of myelin coupled with vacuolar, spongiform degeneration of the cerebral and cerebellar WM (Yamashita et al., 2013). Similar DWI studies in HBSL patients, in addition to more detailed MRS studies, will provide valuable insight into the progression of this leukodystrophy.
Although MRI is a reliable tool for diagnosis, clinical severity of LBSL does not always reflect the MRI results. Labauge et al. described a sibling to a proband who had characteristic MRI changes typically seen in affected LBSL patients, yet did not show any symptoms (Labauge et al., 2011). Genetic testing revealed that this person was a compound heterozygous carrier of DARS2 mutations. Furthermore, there appears to be a delay between MRI changes and the onset of clinical symptoms in LBSL (Isohanni et al., 2010). These findings indicate that MRI could potentially be used to provide an early, pre-symptomatic diagnosis of the disease. An adult-onset LBSL patient was described to have solely right-sided spasticity, yet pyramidal tracts were equally affected bilaterally throughout their entire length (Moore et al., 2012), suggesting that MRI results do not always match clinical symptoms. MRI findings in HBSL patients are generally more consistent with the clinical presentation and MRI can be used to differentiate between infantile and adult-onset patients. Changes in adult-onset patients are more localized and less severe (Wolf et al., 2015). Moreover, in the least affected sibling of three reported HBSL patients, cerebral WM changes were noted to be patchy and less widespread compared to the more severely affected siblings (Ong et al., 2020).
Apart from HBSL and LBSL, there are other white matter disorders that affect similar regions of the CNS, which could potentially result in misdiagnosis. For example, one patient with mutations in the iron-sulfur cluster assembly 2 (ISCA2) gene was initially diagnosed with LBSL based on MRI and clinical features, prior to genetic testing (Toldo et al., 2018). Mutations in this gene cause multiple mitochondrial dysfunctions syndrome 4 (MMDS4), which has MRI features similar to LBSL and HBSL including signal changes in the periventricular WM, corpus callosum, posterior limb of the IC, cerebellar WM and cerebellar peduncles (Al-Hassnan et al., 2015; Alaimo et al., 2018; Toldo et al., 2018; Eidi and Garshasbi, 2019; Hartman et al., 2020). Additionally, a few MMDS4 patients also display lactate peaks on MRS (Alfadhel, 2019), as well as diffusion restriction in some affected WM regions on DWI (Alaimo et al., 2018; Toldo et al., 2018; Hartman et al., 2020). However, some MMDS4 patients show signal abnormality of the cerebral peduncles (Alaimo et al., 2018), as well as glycine elevation (Alfadhel, 2019), which have not been reported in LBSL or HBSL. Similarities have also been drawn between multiple sclerosis (MS) and HBSL and LBSL (Mierzewska et al., 2011; Wolf et al., 2015). This is likely because MS is seen to affect the same regions of the brain—the periventricular WM, brainstem, cerebellar peduncles, cerebellum and spinal cord (Filippi et al., 2019). Unlike MS however, abnormalities of WM in HBSL and LBSL are noted to be more symmetrical, and to affect specific tracts (Mierzewska et al., 2011; Filippi et al., 2019). Especially in the spinal cord, MS lesions are seen to be limited to a maximum of two vertebral levels and to be focal rather than affecting whole tracts (Wolf et al., 2015; Filippi et al., 2019). Adult-onset HBSL patients presenting with only focal MRI changes were originally misdiagnosed with MS, prior to genetic testing. As a result, treatment with steroids was initiated, resulting in transient recovery of symptoms (Wolf et al., 2015). The responsiveness to steroids paired with a relapsing-remitting course and focal white matter and spinal cord signal changes on MRI normally indicate inflammatory demyelinating diseases such as MS or neuromyelitis optica (Wolf et al., 2015). The similar presentation of adult-onset HBSL cases and MS patients indicates that the actual number of HBSL cases might be higher than originally anticipated and it has been suggested that HBSL be included in the differential diagnosis of CNS inflammatory diseases (Wolf et al., 2015).
As previously mentioned, the definite diagnosis of HBSL and LBSL requires genetic testing. The first LBSL cases reported were diagnosed using genome-wide linkage analysis to determine the disease-causing mutations (Scheper et al., 2007). The first diagnosis of HBSL was made using a novel technology at the time—full-genome trio sequencing (Taft et al., 2013). Using this method, the entire genome of the patient and both parents are sequenced and compared to limit the number of candidate genes. In a clinical setting, however, standard sequencing techniques such as next-generation sequencing (NGS) and Sanger sequencing are used to obtain a genetic diagnosis when HBSL and LBSL are already suspected based on clinical and MRI findings, which is faster and more cost-efficient than whole genome sequencing (WGS). Some LBSL patients with large deletion mutations have been reported (Lan et al., 2017). These mutations are not detected by standard sequencing techniques and it is recommended to follow up with copy number analysis to provide a diagnosis (Lan et al., 2017). Whole exon deletions have not been described for HBSL.
With the use of WGS, mutations in the nuclear genes DARS and DARS2 were identified to be the cause of HBSL and LBSL, respectively. DARS1 is located on chromosome 2 in the 2q21.3 region and encodes AspRS (Taft et al., 2013). DARS2 is located on chromosome 1 in the 1q25.1 region and encodes the mitochondrial counterpart mt-AspRS (Scheper et al., 2007). While AspRS acts exclusively in the cytosol, mt-AspRS is transported into the mitochondrial matrix and is responsible for the translation of mitochondrial transcripts.
The majority of LBSL and HBSL patients are compound heterozygous. Only very few LBSL patients are homozygous (5.6% of reported cases). In comparison, 37.5% of reported HBSL cases are homozygous. All DARS1 mutations identified thus far are missense mutations, affecting amino acids primarily located in and around the catalytic domain of AspRS (Taft et al., 2013; Wolf et al., 2015; Ong et al., 2020) (Table 4). These mutations are thought to impair aminoacylation directly through changes of the active site, or through destabilization of the side chain interactions with the active site (Taft et al., 2013). Many of these mutations occur in highly conserved amino acids of AspRS (Taft et al., 2013; Ong et al., 2020).
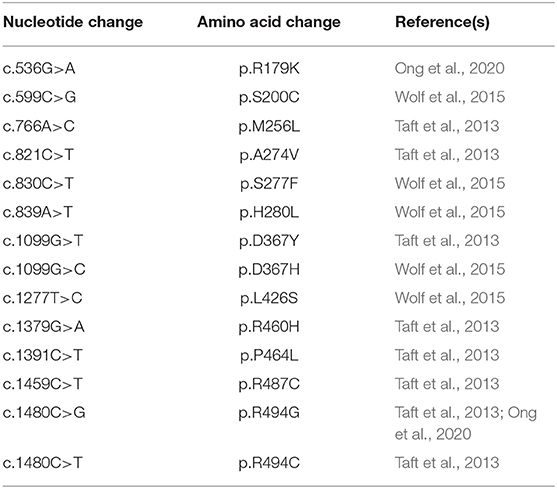
Table 4. Summary of known HBSL mutations and their corresponding amino acid change.
In contrast, LBSL-causing mutations are scattered throughout the length of the DARS2 gene (Table 5). These mutations include missense, non-sense, deletion, and splice site mutations. Most patients carry a mutation in the intron 2 splice acceptor region, several nucleotides upstream of exon 3 (Scheper et al., 2007). A mutation in this region affects about 88% of LBSL patients reviewed, and one mutation at this site (c.228-20_21delTTinsC) is estimated to have a carrier frequency of 1:95 in the Finnish population (Isohanni et al., 2010). A mutation at this splice acceptor region causes the exclusion of exon 3 in mt-AspRS transcripts. This exclusion ultimately results in a frame shift (p.R76SfsX5) and premature stop (van Berge et al., 2012), possibly producing a truncated, non-functional protein. It is important to note that this mutation is considered “leaky,” allowing low-level production of wild-type, functional mt-AspRS protein from transcripts including exon 3 (van Berge et al., 2012). Remarkably, this splicing defect is particularly profound within neural cells, and specifically in neurons, significantly lowering the amount of functional mt-AspRS produced by these cells (van Berge et al., 2012). Even in healthy individuals with two unaffected DARS2 copies, splicing of this region is less efficient in neurons compared to other cell types. When this mutation is coupled with another DARS2 mutation on the second allele, the amount of wild-type protein produced becomes insufficient (van Berge et al., 2012). The splicing differences between individuals potentially explain the large clinical variation among patients, including intrafamilial variations where affected family members possess the same DARS2 mutations (Labauge et al., 2011). Despite its high prevalence, homozygous mutations at this site have only been reported in one family with three affected siblings (Miyake et al., 2011). Additionally, one patient carrying two different mutations at this site has been reported (Orcesi et al., 2011). In HBSL, the most prevalent mutation site is in exon 9 of the DARS1 gene, causing a single amino acid change from methionine to leucine. This mutation was found in four patients from three different families (Taft et al., 2013).
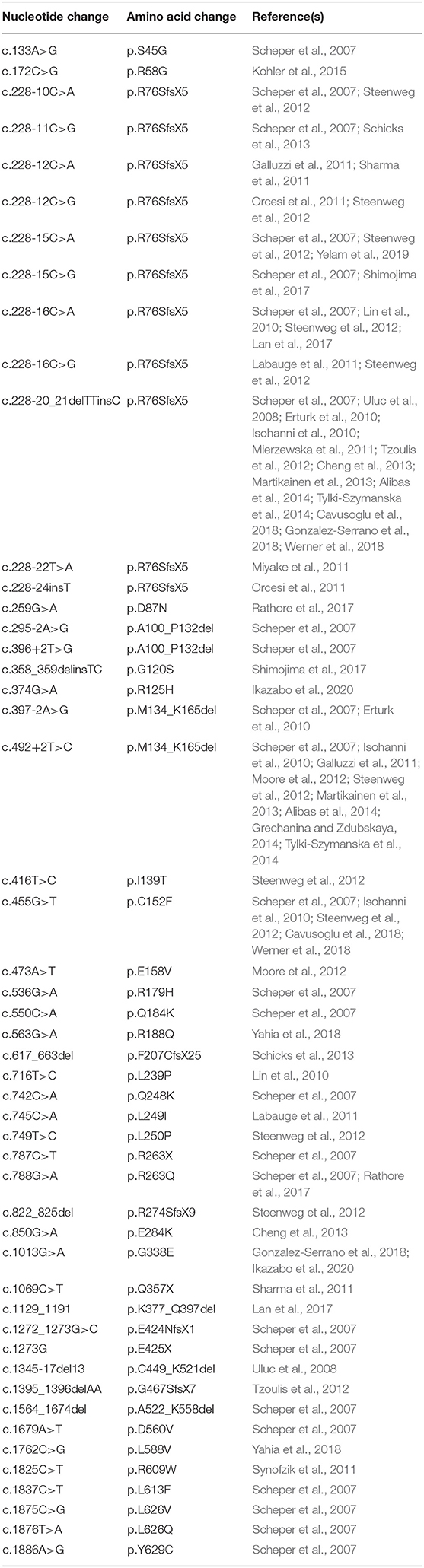
Table 5. Summary of known LBSL mutations and their corresponding amino acid change.
ARSs are primarily responsible for the accurate pairing of tRNAs to their respective amino acids, in a two-step aminoacylation reaction (Wallen and Antonellis, 2013). This is often referred to as charging. Some ARSs have additional editing capabilities to identify and correct erroneous charging (Beuning and Musier-Forsyth, 2000). However, such a capability has not been reported for AspRS or mt-AspRS. All studies investigating the mechanism of disease in HBSL and LBSL have so far been mainly targeted toward their canonical function of aminoacylation. There is a growing body of evidence, though, that the secondary ARS functions may also play a role in ARS pathologies (Guo and Schimmel, 2013). One study found missense mutations in DARS2 regions that are solely conserved in mammals, which could suggest that the evolutionary later-acquired supplementary functions of mt-AspRS may be involved in LBSL pathology (Sauter et al., 2015). These secondary functions include inflammatory and immune regulation by ARSs (Guo and Schimmel, 2013). In addition, cytosolic AspRS is known to interact with other cytosolic ARSs and aminoacyl tRNA synthetase complex interacting multifunctional proteins (AIMPs) to form a multi-synthetase complex (MSC) (Khan et al., 2020). While much of the MSC's function and role is shrouded, it is thought to regulate the secondary functions of the involved ARSs and to promote translation by ferrying tRNAs to ribosomes (Lee et al., 2004). Thus, impaired MSC function could play a role in HBSL (Figure 1).
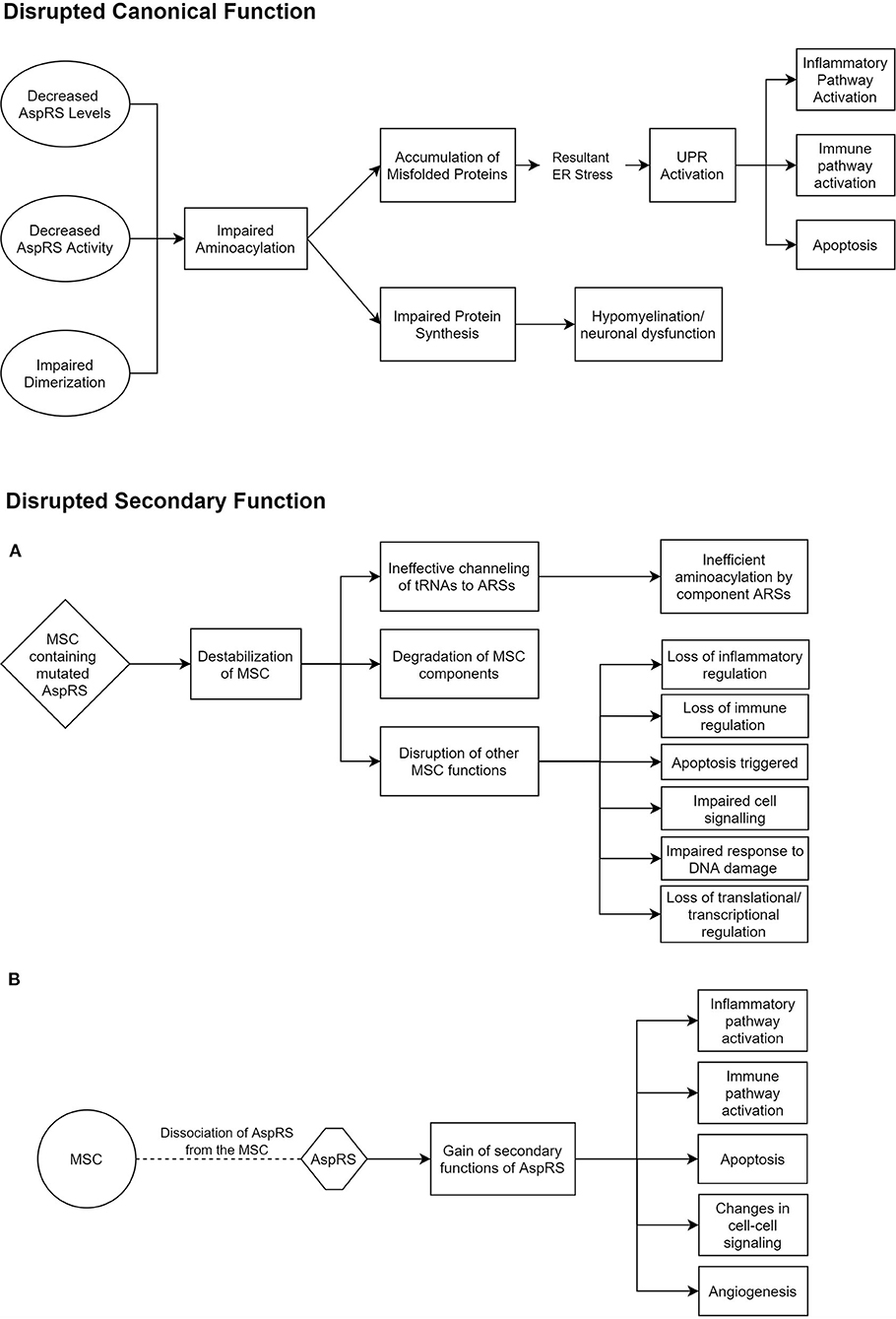
Figure 1. Potential pathomechanisms underlying HBSL. These hypothesized modes of dysfunction in HBSL are based on current knowledge of DARS1, the MSC, and secondary functions of ARSs (Lee et al., 2004; Guo et al., 2010; Pang et al., 2014; Frohlich et al., 2017). (A,B) Offer two alternative explanations for the disruption of secondary AspRS function. AspRS, Aspartyl-tRNA Synthetase; ER, Endoplasmic Reticulum; UPR, Unfolded Protein Response; MSC, Multi-tRNA Synthetase Complex; ARS, Aminoacyl-tRNA Synthetase.
Compromised aminoacylation may occur as a result of reduced enzyme production, decreased catalytic reaction or impaired dimerization. In vitro analysis of seven missense mutations of DARS2 revealed that protein levels were greatly reduced as a result of the C152F, Q184K, and D560V mutations (van Berge et al., 2013). The D560V mutation additionally caused a decrease of catalytic activity by 83.4% (van Berge et al., 2013) while another study showed a much greater reduction of up to 99.7% (Scheper et al., 2007). The R263Q and L626Q mutations also reduced catalytic activity of mt-AspRS by 99.3 and 97.7%, respectively (van Berge et al., 2013). Reduced mt-AspRS activity was also found in patient-derived lymphoblasts of a compound heterozygous (missense mutations R125H and G338E) LBSL case (Ikazabo et al., 2020), as well as in patients with the highly prevalent splicing defect and other missense mutations (Van Berge et al., 2014). It is notable that not all missense mutations studied resulted in reduced mt-AspRS expression or activity (van Berge et al., 2013). An effect of DARS1 mutations on AspRS expression levels or activity in HBSL patients has not been reported yet. However, the positions of affected amino acids in and around the catalytic domain strongly suggest that these mutations affect AspRS enzymatic activity (Taft et al., 2013).
In LBSL, impaired transport of mt-AspRS from the cytosol into mitochondria has been suggested as a potential cause of compromised aminoacylation. This disease mechanism was described for the S45G mutation, which is situated in the mitochondrial targeting sequence (MTS) of mt-AspRS (Messmer et al., 2011). The MTS enables nuclear-encoded mitochondrial precursor proteins to be recognized by and transported into mitochondria (Omura, 1998). The S45G mutation impaired the import of precursor mt-AspRS through the mitochondrial membranes, but did not affect targeting of mt-AspRS to the surface of mitochondria (Messmer et al., 2011). A change in mt-AspRS solubility has also been suggested as a potential cause of disease. One study found that the Q184K mutation significantly reduced the solubility of mt-AspRS, while the other four investigated mutations did not substantially affect solubility (Sauter et al., 2015). When combined, two mutations causing only a mild decrease in solubility might be sufficient to trigger LBSL pathology (Sauter et al., 2015). A second study replicated these findings showing that the Q184K mutation significantly decreased the matrix localization of mt-AspRS (Gonzalez-Serrano et al., 2018).
The same study also established that mt-AspRS is localized in the mitochondrial matrix and in the mitochondrial membranes, where it is anchored through electrostatic interactions (Gonzalez-Serrano et al., 2018). This raised the possibility of disrupted ARS localization as a disease mechanism for LBSL (Gonzalez-Serrano et al., 2018). Localization errors could also affect cytosolic ARSs that are part of the MSC. LysRS dissociates from the MSC in response to certain triggers (Ofir-Birin et al., 2013; Gonzalez-Serrano et al., 2018), which subsequently diverts LysRS away from translational sites (Ofir-Birin et al., 2013).
Finally, dimerization is an important aspect to consider in HBSL and LBSL, as both AspRS and mt-AspRS function as homodimers (Bonnefond et al., 2005; Kim et al., 2013). The R58G, T136S, and L626Q mt-AspRS mutations were found to impair dimerization with other mutant mt-AspRS monomers; while the C152F, Q184K, R263Q, and D560V mutations even impacted dimerization with wild-type mt-AspRS subunits (van Berge et al., 2013). None of the reported DARS1 mutations is located in the dimerization domain of AspRS. However, it has previously been demonstrated in yeast that a change in the highly conserved amino acid P273 in the dimerization domain of AspRS leads to a reduction in catalytic activity of mutant homodimers (Eriani et al., 1993).
Due to the almost identical MRI pattern and similar clinical symptoms, it has been hypothesized that the two conditions could share a common, underlying disease mechanism. Both conditions particularly affect the nervous system, despite ARSs being ubiquitously expressed enzymes. As previously mentioned, in LBSL, the splicing defects seem to be more profound in neurons compared to other cell types (van Berge et al., 2012). The characteristic presentation of LBSL patients without the intron 2 splice site mutation should be further explored. Expression pattern studies of DARS1 have revealed that neurons have a higher AspRS expression level compared to other cell types in the human brain (Frohlich et al., 2018). On a regional level, the highest AspRS expression was observed in neurons of the cerebellum, particularly in the Purkinje cell layer (Frohlich et al., 2018). To a lesser extent, the motor cortex (predominantly layers II-VI) and hippocampus (CA1, CA2, and CA3 layers) also displayed considerable DARS1 expression levels. This specific expression pattern may explain why these areas are particularly affected in HBSL (Frohlich et al., 2018). Interestingly, the white matter of the cerebellum and brainstem showed only limited DARS1 expression (Frohlich et al., 2018). This is in contrast to findings of DARS2 expression pattern analysis, which was found to be comparable across different tissues (Scheper et al., 2007). However, levels of mitochondrial tRNA levels were found to be highest in the central nervous system (CNS) (Dittmar et al., 2006; Scheper et al., 2007).
Mouse models are useful tools to explore possible disease mechanisms and are a prerequisite for trial of potential therapies. In vitro studies have also contributed valuable insight to our current understanding of the diseases, supplementing experiments in mice. Several attempts have been made to model HBSL and LBSL in mice which have provided important mechanistic insights.
The first attempt at an LBSL mouse model found that complete knockout of Dars2 is lethal; these mice died around embryonic day eight, coinciding with organogenesis (Dogan et al., 2012). Subsequent efforts were directed toward conditional knockout (cKO) of Dars2 in specific tissues. The first attempt at modeling HBSL in mice encountered the same barrier, as the complete knockout was embryologically lethal due to developmental arrest (Frohlich et al., 2017). This indicates that complete functional null mutations are not tolerated, and the resulting embryonic lethality may explain the rather low numbers of HBSL and LBSL cases diagnosed to date.
Additionally, in both HBSL and LBSL mouse models, the heterozygous null state produced largely phenotypically normal mice (Dogan et al., 2014; Frohlich et al., 2017). This is consistent with the recessive genetic trait of inheritance, that is further supported by the finding that unaffected siblings and parents of patients, who possess one wild-type copy of the DARS1 or DARS2 gene, are healthy. Altogether, these findings provide experimental evidence that some residual activity of both these ARSs is necessary for life, and that one unaffected copy of the gene is sufficient to avoid disease.
Two neuronal Dars2 cKO models have been created utilizing the Cre/LoxP system (Aradjanski et al., 2017; Nemeth et al., 2020). In both models, mice were crossed to CamKIIα Cre-driver lines, expressing Cre recombinase under control of the neuronal CamKIIα promotor. Peak CamKIIα-Cre expression, and hence recombination and Dars2 knockout, is reached at around postnatal week 4 (Aradjanski et al., 2017). Neuronal Dars2 cKO mice displayed severe morphological and behavioral changes starting around week 24 and worsening until 28 weeks of age (Aradjanski et al., 2017). These morphological changes included severe brain atrophy concentrated around the forebrain cortex and hippocampal area (Aradjanski et al., 2017; Nemeth et al., 2020) and enlargements of the third and lateral ventricles, with a notable sparing of the cerebellum (Nemeth et al., 2020). Additionally, thinning of the corpus callosum was also noted (Aradjanski et al., 2017; Nemeth et al., 2020). Microscopically, this is accompanied by neuronal cell death, as demonstrated by the presence of pyknotic cells and vacuoles in the affected regions at 28 weeks of age (Aradjanski et al., 2017). In one study, these changes occurred on the background of limited weight gain compared to wild-type mice, and a shorter lifespan. These mice also displayed self-injuries around 28 weeks of age, coinciding with the most severe morphological changes, in addition to motor abnormalities such as tremor and ataxia (Aradjanski et al., 2017). Conversely, in the other study, neuronal Dars2 cKO mice were of higher body mass than controls until 28 weeks of age, after which they were comparable to controls. In addition, neuronal Dars2 cKO mice were more active compared to controls, particularly in their explorative behavior (Nemeth et al., 2020). Both models displayed an increase in mitochondria number, however, the mitochondrial ultrastructure appeared normal in one model (Nemeth et al., 2020), while there was a loss of normal cristae in the other model indicating mitochondrial dysfunction (Aradjanski et al., 2017). It remains unclear why the two studies, despite using the same neuronal Dars2 cKO model, resulted in such different outcomes.
A third neuronal LBSL mouse model specifically targeting Purkinje cells in the cerebellum was recently reported (Rumyantseva et al., 2020). In this model, Cre recombinase was expressed under control of the Purkinje cell protein 2 promotor. Maximal recombination of Dars2 took place at postnatal weeks 2 to 3 (Rumyantseva et al., 2020). Significant Purkinje cell death was noted at 15 weeks, accompanied by motor dysfunction such as unsteady gait and lack of balance. Morphologically, no cerebellar atrophy was noted (Rumyantseva et al., 2020). Histological analysis revealed a loss of Purkinje cell layer organization at this time. Preceding these changes, mitochondrial respiratory chain (MRC) dysfunction was noted at 6 weeks of age (Rumyantseva et al., 2020). As the cerebellum is the CNS region with the highest AspRS expression in humans and mice, a similar approach to model HBSL in mice should be considered.
It is remarkable that in LBSL patients, mitochondrial dysfunction including altered MRC activity, are not a common feature and have only been reported in one patient to date (Orcesi et al., 2011). Increase in mitochondrial size in muscle cells has also been reported in another patient (Linnankivi et al., 2004). Interestingly, one in vitro study revealed a strong reduction in the number of mitochondrial DNA encoded MRC complex proteins in fibroblasts of a compound heterozygous LBSL patient, together with reduced cellular oxygen consumption and increased mitochondrial fragmentation (Lin et al., 2019).
Dars2 cKO was also performed in the myelinating cells of the central and peripheral nervous system (PNS) at the age of 4 weeks, using tamoxifen inducible Cre expression under control of the proteolipid 1 (Plp1) promoter. Similar to the neuronal Dars2 cKO model produced by the same group, strong MRC deficiencies were observed in these mice, yet no other sequelae involving demyelination, apoptosis or inflammation followed (Aradjanski et al., 2017). Morphologically and behaviorally, these mice were normal too. It was suggested that myelinating cells possess a different metabolic profile compared to neurons, which enables them to produce sufficient energy through glycolysis when mitochondrial respiration is impaired. Mitochondria in mature oligodendrocytes may be more important for myelin maintenance through fatty acid oxidation and lipid synthesis rather than ATP production (Rinholm et al., 2016; Aradjanski et al., 2017). Since Dars2 cKO only affects translation of mitochondrial DNA transcripts, and hence MRC subunit function and ATP production, myelination remained largely unaffected. Expression levels of cytosolic AspRS are much lower in oligodendrocytes compared to neurons (Frohlich et al., 2018). It has been hypothesized that the low expression levels could make these cells particularly susceptible to disturbances in protein synthesis. However, given the growing evidence of the influence of neurons on oligodendrocyte differentiation and myelination (Gibson et al., 2014; Mitew et al., 2014; Ortiz et al., 2019), it is also plausible for the primary pathology to lie in neuronal cell bodies and axonal processes. This is supported by the predominant involvement of long tracts in LBSL and HBSL (van Berge et al., 2012).
Dars2 cKO was also achieved in cardiac and skeletal muscles using the muscle creatine kinase (Ckmm) Cre-driver line. Maximal recombination of the Dars2 gene occurred at 2 weeks of age in cardiac and skeletal muscle (SkM) cells (Dogan et al., 2014). While no cardiac defects have been reported in LBSL patients, muscle tissue has a similar energy demand and therefore findings from this study could be translationally relevant for LBSL. All mice developed hypertrophic cardiomyopathy by 6 weeks of age, quickly followed by death (Dogan et al., 2014). Microscopically, the response in each muscle tissue differed. Cardiac muscle fibers were hypertrophic and disorganized progressively, while atrophy and preserved fiber organization was observed in SkM fibers. Both the cardiac and skeletal myocytes experienced substantial MRC deficiency; however, mitochondrial biogenesis, cell stress pathway activation and suppressed autophagy was only observed in cardiomyocytes (Dogan et al., 2014). It is postulated that this absence, or rather significant delay in onset of regulatory changes is owed to the skeletal myocytes' low mitochondrial transcript turnover rate and the cells' much greater capacity to regenerate (Dogan et al., 2014).
The morphological, behavioral, and microscopic changes observed in these studies have been consistently preceded by molecular changes—mainly the activation of several stress pathways accompanied by immune and inflammatory responses (Dogan et al., 2014; Aradjanski et al., 2017; Nemeth et al., 2020). In the neuronal Dars2 cKO model, these changes occur as early as 16 weeks of age and particularly impact immune pathways (Nemeth et al., 2020). The most notable change was the upregulation of Cystatin F (CST7). CST7 is a lysosomal protease inhibitor that is known to repress lysosomal proteases to prevent tissue damage (Kos et al., 2018). In the CNS, Cst7 has been shown to be expressed, and then released from activated microglia (Nemeth et al., 2020). This typically occurs in areas of active demyelination and simultaneous remyelination (Ma et al., 2011). Interestingly, when the remyelinating potential is lost (such as in chronic demyelination), CST7 levels fall (Ma et al., 2011). Therefore, the reported 200-fold increase of CST7 levels in neuronal Dars2 cKO mice (Nemeth et al., 2020) may indicate the enduring potential for remyelination. However, despite the CST7 increase, the neuronal Dars2 cKO mice continued to deteriorate. Another role of CST7 is the regulation of cathepsin C (CTSC)—a known lysosomal cysteine protease, which was also upregulated in neuronal Dars2 cKO mice (Nemeth et al., 2020). CTSC is involved in the activation of several immune cells, including T cells and neutrophils, and its absence has been shown to dampen the immune response (Colbert et al., 2009). One study found the knockout of Ctsc “rescued” the demyelinating process in multiple sclerosis (Shimizu et al., 2017). The upregulation of CTSC could potentially contribute to the progression of LBSL.
These immune changes coincided with signs of inflammation. In histological brain sections of neuronal Dars2 cKO mice, a significant increase in the number of activated microglia expressing the inflammatory marker CD68 was observed (Nemeth et al., 2020). Although the role of CD68 in inflammation is unclear, it is known to be upregulated in macrophage-mediated inflammatory processes and is hence a reliable indicator for microglia activation (Chistiakov et al., 2017). This finding is further corroborated by the second neuronal Dars2 cKO study, where increasing microglial activation was observed in the cortex until 20 weeks of age, when neuronal cells began to undergo apoptosis (Aradjanski et al., 2017). These inflammatory changes resulted in a near complete disruption of normal cellular structures in the hippocampal area close to the time of death (Aradjanski et al., 2017), and even contributed to cell-death in CamKIIα negative neurons (Nemeth et al., 2020). In the Purkinje cell Dars2 cKO mouse model, the observed microglial activation and associated neuroinflammatory changes have been attributed to degradation of Purkinje cell dendrites and their complex network (Rumyantseva et al., 2020).
In addition, molecular changes indicative of mitochondrial dysfunction and disrupted proteostasis have been observed. Following conditional knockout of Dars2 in cardiac and SkM, the activation of the mitochondrial unfolded protein response (UPRmt) in cardiomyocytes was evident by 6 weeks of age (Dogan et al., 2014). Stress pathway activators such as activation transcription factor 5 (ATF5) and C/EBP homologous protein (CHOP) were found to be upregulated at this time point (Dogan et al., 2014). These molecules stimulate the UPRmt, which is responsible for the degradation of aberrant proteins in order to maintain cell homeostasis (Haynes and Ron, 2010). The activation of the UPRmt was the first notable change in mutant cells and increased ATF5 and CHOP levels along with proteases involved in the UPRmt were identified before any detectable MRC deficits (Dogan et al., 2014). In the neuronal Dars2 cKO model, molecular changes suggestive of UPRmt activation were also observed at 20 weeks of age coinciding with severe MRC defects (Aradjanski et al., 2017). Accordingly, the activation of the unfolded protein response (UPR) as a possible disease mechanism for HBSL was suggested (Frohlich et al., 2017).
A 250-fold increase of fibroblast growth factor 21 (FGF21) within the first 2 weeks of life in the heart of Ckmm Dars2 cKO mice is further evidence of mitochondrial dysfunction preceding detectable MRC deficiency (Dogan et al., 2014). FGF21 has been shown to be elevated in mitochondrial diseases, particularly those affecting the muscles, and has even been suggested as a biomarker for mitochondrial disorders for its high sensitivity and specificity (Suomalainen et al., 2011). Notably, most of the FGF21 increase resulted from cardiomyocytes, where Fgf21 expression levels are usually low (Dogan et al., 2014). FGF21 is predominantly known for its role in modulating metabolic pathways (Potthoff et al., 2009). This is also evident in Ckmm Dars2 cKO mice through altered nutrient usage (Dogan et al., 2014). Additionally, FGF21 ensures the stability of the transcription factor Peroxisome proliferator-activated receptor-Gamma Coactivator 1-alpha (PGC-1α) (Fisher et al., 2012). PGC-1α is a key controller of mitochondrial biogenesis (Ventura-Clapier et al., 2008)—an adaptive response to increased ATP demand.
In addition to the UPR, the integrated stress response (ISR) pathway was also activated in neuronal Dars2 cKO mice. This was evident through the upregulation of Atf4, a key component of the ISR, at 16 weeks of age (Nemeth et al., 2020). ISR activation also occurred prior to any observable neuronal death. It is remarkable that only increased mitochondrial biogenesis, without any mitochondrial dysfunction, was noted in this model. In the Purkinje cell Dars2 cKO mouse model, ISR activation was noted at 8 weeks of age, as evidenced by the initiation of the one-carbon metabolic pathway. This pathway has been linked to the activation of the ISR through Atf4 upregulation (Rumyantseva et al., 2020). The ISR is a cell stress pathway intended to maintain homeostasis and is activated upon dysregulation of proteostasis, oxidative damage, loss of nutrients, and other similar events (Costa-Mattioli and Walter, 2020). When the challenge becomes unmanageable, apoptosis is triggered. Intriguingly, prolonged activation of this usually protective pathway can in itself become cytotoxic, which has been shown to be the case in several neurodegenerative disorders (Rabouw et al., 2019).
Taken together, an imbalance in protein homeostasis, which is evident through a rise in ubiquitinated proteins, appears to be the initial trigger for the disease (Dogan et al., 2014). Subsequently, this leads to the activation of stress pathways, which through prolonged activation likely contributes to neuronal cell-death and neurodegeneration (Figure 2).
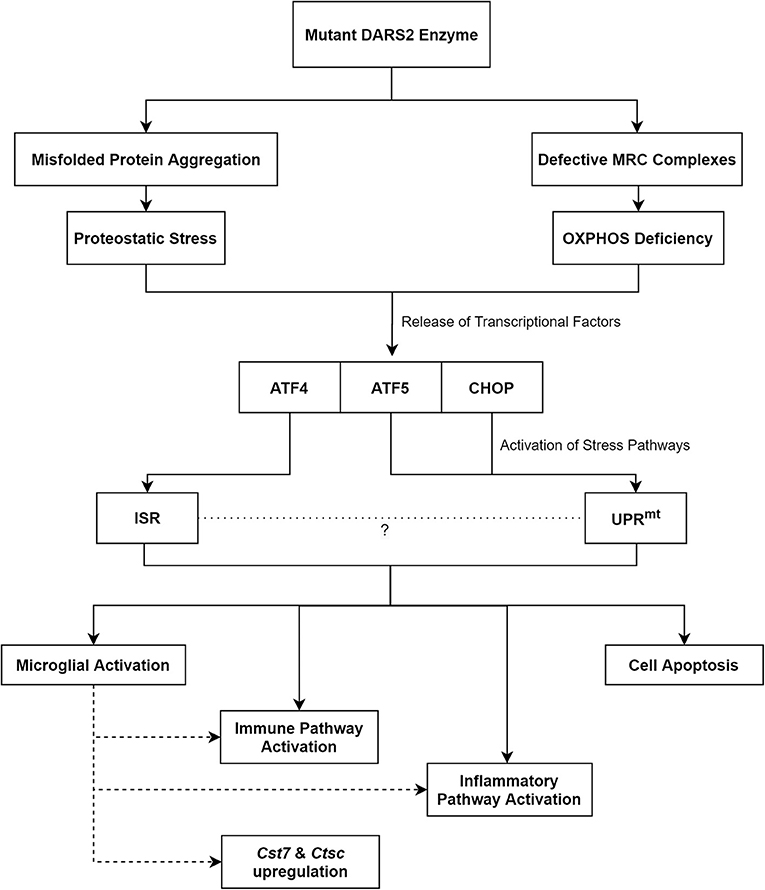
Figure 2. The proposed pathomechanisms of LBSL based on findings from the neuronal Dars2 cKO mouse models. MRC, Mitochondrial Respiratory Chain; OXPHOS, Oxidative Phosphorylation; ATF, Activation Transcription Factor; CHOP, C/EBP Homologous Protein; ISR, Integrated Stress Response; UPRmt, Mitochondrial Unfolded Protein Response; Cst7, Cystatin F; Ctsc, Cathepsin C.
The neuronal Dars2 knockout mouse model generated by Aradjanski et al. was able to capture some of the LBSL symptomatology seen in patients, such as ataxia and tremor (Aradjanski et al., 2017). Neuronal Dars2 cKO mice also displayed worsening kyphosis with age. Signs of microglial activation and cell death corroborated the histological findings in LBSL patients (Yamashita et al., 2013). Still, gross pathological changes were mainly limited to cortical and hippocampal areas, while LBSL patients displayed WM changes in the cerebellum, brainstem and spinal cord in addition to cortical changes. This discrepancy can be explained by the predominant localization of CamKIIα positive neurons within the hippocampus and cortex, while the midbrain, medulla and cerebellum lack CamKIIα positive neurons (with the exception of Purkinje cells) (Wang et al., 2013). The use of a more appropriate neuronal Cre-driver to ablate Dars2 in all neurons of the CNS might result in a more accurate LBSL model. Surprisingly, despite using the same CamKIIα Cre-driver, Nemeth et al. demonstrated increased activity of neuronal Dars2 cKO mice without any overt motor deficits (Nemeth et al., 2020). It is also important to bear in mind that regulation of mRNA translation in the human brain likely varies from the mouse brain.
For HBSL, our lab is currently working on a Dars1 cKO mouse model. In addition, we are attempting to model HBSL by introducing patient specific point mutations into the mouse Dars1 gene. The first mutation we introduced was the Dars1D367Y point mutation found in the HBSL index patient (see Froehlich et al., in this issue). While homozygous Dars1D367Y/D367Y mice failed to model HBSL, mice compound heterozygous for the Dars1D367Y and the Dars1null mutation captured some aspects of the disease.
In summary, attempts at modeling HBSL and LBSL have contributed significantly to our understanding of both conditions. Nevertheless, these studies have their limitations. In all studies, Dars2 was rendered functionally inactive postnatally. Since CNS development is accelerated in mice compared to humans, these knockouts occur relatively late during development. In contrast, DARS2 mutations are evident from birth in LBSL patients. These mutations greatly reduce, but do not completely abolish mt-AspRS function, throughout the developmental period.
In mice, complete knockout of Dars1 or Dars2 were both embryonically lethal. This indicates a lack of functional redundancy between the two enzymes and puts to rest questions on possible compensatory mechanisms between the two. However, other findings from these studies continue to offer support for a possible shared disease mechanism between LBSL and HBSL. Firstly, the finding of inflammatory processes and immune responses likely underlying neuronal dysfunction and cell-death in LBSL is reminiscent of the study by Wolf et al., who postulated that HBSL might possess a neuroinflammatory component based on clinical findings and the responsiveness of HBSL patients to steroids (Wolf et al., 2015). This should be addressed when studying the underlying disease mechanism in future HBSL mouse models.
Secondly, studies in LBSL models suggest the primary pathology to lie in neuronal or axonal dysfunction rather than in myelin-producing cells or the myelin itself. These findings are supported by the absence of noticeable phenotypic changes in mice following Dars2 cKO in myelinating cells. Moreover, one study reported findings of axonal degeneration of myelinated fibers in histopathological analysis of tissues from LBSL patients. This was accompanied by detection of myelin ovoids in the peripheral sural nerve, and the absence of segmental demyelination, hence the pattern of peripheral neuropathy in these patients was suggested to be mainly axonal (Yamashita et al., 2013). Myelin ovoids are often associated with Wallerian degeneration—a pathological process where demyelination occurs secondary to axonal pathology. Myelin ovoids result from the clearance of myelin by Schwann cells (Tricaud and Park, 2017). Segmental demyelination indicates myelin degeneration with sparing of nerve fibers, yet this was not a prominent finding in the histological analysis (Yamashita et al., 2013). Accordingly, AspRS was found to be predominantly expressed by neurons, with lower expression levels in oligodendrocytes and astrocytes. In mice, the subcellular localization of AspRS was not restricted to the cell body of neurons but also in the axons and synapses (Frohlich et al., 2017). Taken together, there is a strong rationale for a primary neuronal or axonal dysfunction underlying LBSL and HBSL pathology.
Management of HBSL and LBSL are currently solely supportive and symptomatic. While these methods can improve the quality of life, the benefits are neither sustained nor equally effective for all patients. Hence, there is a need for the development of novel treatment strategies for patients affected by these conditions. Some medications have been shown to be effective in patients and when tested in vitro for LBSL (Synofzik et al., 2011; Van Berge et al., 2014). As previously stated, for the investigation and trial of new curative treatment options accurate animal models are required to conduct pre-clinical studies. In the HBSL mouse model study, phenotypically normal heterozygous null mice possessed just 20% of AspRS protein levels compared to control mice (Frohlich et al., 2017). As such, reinstating the enzyme to this “threshold” level might be sufficient to ameliorate symptoms or prevent disease.
For the symptomatic management of LBSL, the use of allied health specialties such as speech, nutrition and physiotherapy has been reported, to support patient disability (Navarro Vázquez et al., 2016; Werner et al., 2018). In addition, medications for pain and spasticity are also used for symptom management (Werner et al., 2018). Antispastic medications have been shown to improve symptoms gradually in adult-onset LBSL patients (Petzold et al., 2006). At the same time, genetic counseling is often provided to affected families. In HBSL, management is similarly symptom-based, with chemodenervation additionally suggested to manage spasticity (Ulrick and Vanderver, 2017).
In both conditions, steroid medications have been shown to reduce the severity of symptoms experienced by patients. Particularly in HBSL, 5 out of 16 patients reported in the literature showed improvements following administration of steroids, three of whom were infantile-onset patients (Taft et al., 2013; Wolf et al., 2015). One early-onset patient was described to gain muscle strength and postural tone, although these improvements were only modest (Wolf et al., 2015). In late-onset patients, the effect of steroids was inconsistent—with one patient regaining his ability to walk independently following a single course of methylprednisolone, while another patient repeatedly experienced relapse of symptoms over a few months post-steroid treatment (Wolf et al., 2015). In LBSL, to our knowledge, the use of steroids has only been described in one patient who displayed spastic bladder symptoms, which improved following the use of glucocorticoids, although other symptoms persisted (Cheng et al., 2013). Following the uncovering of activated inflammatory and immune pathways in LBSL mouse models, the use of steroids could be beneficial for symptom management in LBSL patients in the future. Concomitantly, variable suppression of symptoms through the use of steroids in HBSL hints at inflammatory and immune activation as a component of the disease process. Further investigation into the cause of the activation of inflammatory and immune pathways—such as the UPR and ISR—will help develop more effective treatment options. An ISR inhibitor (ISRIB) is of interest, as it has delivered promising results in inhibiting the harmful effects of ISR overactivation without producing excessive side effects from inhibition of healthy, basal ISR protection (Rabouw et al., 2019).
Lately, some drugs have gained special attention for the treatment of LBSL. Firstly, the carbonic anhydrase inhibitor acetazolamide was reported to improve ataxia in one patient (Synofzik et al., 2011). This medication, however, was used in a patient with a highly atypical presentation, where ataxia was episodic and triggered by exercise. In such a patient, 125 mg bi-daily acetazolamide was shown to decrease the mean number of ataxic attacks per day by about 77%. Acetazolamide is a standard medication used in episodic ataxia syndromes, for which this patient did not harbor mutations, and is thought to exert its effect in the CNS by reducing lactate and pyruvate levels (Griggs et al., 1978). Notably, this patient displayed lactate elevation on MRS. It remains to be seen if this medication will produce similar results in more typical LBSL patients. Another drug, cantharidin, has been demonstrated to improve the efficiency of splicing at the DARS2 intron 2 splice site in vitro (Van Berge et al., 2014). There was a 5.9% increase in the inclusion of exon 3 in mRNA transcripts of DARS2 following cantharidin application. While cantharidin is toxic and unsuitable for clinical use, the use of other protein phosphatase 1 and 2A inhibitors could be explored for therapeutic use. Alternatively, antisense oligonucleotides have also been shown to increase the efficiency of splicing at this site (van Berge et al., 2012), although their use is limited by their inability to cross the blood-brain barrier (Rinaldi and Wood, 2018). As the intron 2 splice site mutation is the most prevalent mutation reported in LBSL patients, this site is a valuable target for the treatment of LBSL (Van Berge et al., 2014). For HBSL, it has been suggested that boosting AspRS activity through nutraceutical L-ornithine-L-aspartate (LOLA) supplementation might have therapeutic effects (Das et al., 2020).
Finally, gene therapy is often considered for inherited conditions. This is a particularly viable option for HBSL and LBSL, as they are both monogenic disorders and therefore highly amenable to such a treatment approach. With the recent advances in creating animal models for HBSL and LBSL, attempts at establishing such a treatment modality will follow and hopefully provide a cure for these devastating conditions.
AM and DF led the project and the manuscript production. MK and GH contributed to the manuscript preparation. All authors read and approved the final manuscript.
MK is an employee of Boehringer Ingelheim Pharma GmbH & Co. KG.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was funded by the Medical Research Future Fund (MRFF-ARLKO), the European Leukodystrophy Association (ELA 2018-014I2), and the Mission Massimo Foundation.
Alaimo, J. T., Besse, A., Alston, C. L., Pang, K., Appadurai, V., Samanta, M., et al. (2018). Loss-of-function mutations in ISCA2 disrupt 4Fe-4S cluster machinery and cause a fatal leukodystrophy with hyperglycinemia and mtDNA depletion. Hum. Mutat. 39, 537–549. doi: 10.1002/humu.23396
Alfadhel, M. (2019). Multiple mitochondrial dysfunctions syndrome 4 due to ISCA2 gene defects: a review. Child Neurol. Open 6:2329048X19847377. doi: 10.1177/2329048X19847377
Al-Hassnan, Z. N., Al-Dosary, M., Alfadhel, M., Faqeih, E. A., Alsagob, M., Kenana, R., et al. (2015). ISCA2 mutation causes infantile neurodegenerative mitochondrial disorder. J. Med. Genet. 52, 186–194. doi: 10.1136/jmedgenet-2014-102592
Alibas, H., Koytak, P. K., Ekinci, G., and Uluc, K. (2014). A case with leukoencephalopathy with brainstem and spinal cord involvement and elevated lactate (LBSL) with its characteristic clinical and neuroimaging findings. Clin. Neuroradiol. 24, 297–300. doi: 10.1007/s00062-013-0250-x
Antonellis, A., and Green, E. D. (2008). The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet. 9, 87–107. doi: 10.1146/annurev.genom.9.081307.164204
Aradjanski, M., Dogan, S. A., Lotter, S., Wang, S., Hermans, S., Wibom, R., et al. (2017). DARS2 protects against neuroinflammation and apoptotic neuronal loss, but is dispensable for myelin producing cells. Hum. Mol. Genet. 26, 4181–4189. doi: 10.1093/hmg/ddx307
Beuning, P. J., and Musier-Forsyth, K. (2000). Hydrolytic editing by a class II aminoacyl-tRNA synthetase. Proc. Natl. Acad. Sci. U.S.A. 97, 8916–8920. doi: 10.1073/pnas.97.16.8916
Bonnefond, L., Fender, A., Rudinger-Thirion, J., Giegé, R., Florentz, C., and Sissler, M. (2005). Toward the full set of human mitochondrial aminoacyl-tRNA synthetases: characterization of AspRS and TyrRS. Biochemistry 44, 4805–4816. doi: 10.1021/bi047527z
Cavusoglu, D., Olgac-Dundar, N., Oztekin, O., Ozdemir, T. R., Arican, P., and Gencpinar, P. (2018). The first pediatric case of leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL) from Turkey. Turk. J. Pediatr. 60, 216–220. doi: 10.24953/turkjped.2018.02.018
Chang, L., Munsaka, S. M., Kraft-Terry, S., and Ernst, T. (2013). Magnetic resonance spectroscopy to assess neuroinflammation and neuropathic pain. J. Neuroimmune Pharmacol. 8, 576–593. doi: 10.1007/s11481-013-9460-x
Cheng, F. B., Shen, P. P., Zhou, H. W., Meng, H. M., Yang, Y., and Feng, J. C. (2013). Adult-onset leukoencephalopathy with brain stem and spinal cord involvement in Chinese Han population: a case report and literature review. Neurol. India 61, 161–163. doi: 10.4103/0028-3886.111123
Chistiakov, D. A., Killingsworth, M. C., Myasoedova, V. A., Orekhov, A. N., and Bobryshev, Y. V. (2017). CD68/macrosialin: not just a histochemical marker. Lab. Invest. 97, 4–13. doi: 10.1038/labinvest.2016.116
Colbert, J. D., Matthews, S. P., Miller, G., and Watts, C. (2009). Diverse regulatory roles for lysosomal proteases in the immune response. Eur. J. Immunol. 39, 2955–2965. doi: 10.1002/eji.200939650
Costa-Mattioli, M., and Walter, P. (2020). The integrated stress response: from mechanism to disease. Science 368:eaat5314. doi: 10.1126/science.aat5314
Das, A., Fröhlich, D., Achanta, L. B., Rowlands, B. D., Housley, G. D., Klugmann, M., et al. (2020). L-Aspartate, L-Ornithine and L-Ornithine-L-Aspartate (LOLA) and their impact on brain energy metabolism. Neurochem. Res. 45, 1438–1450. doi: 10.1007/s11064-020-03044-9
Dittmar, K. A., Goodenbour, J. M., and Pan, T. (2006). Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2:e221. doi: 10.1371/journal.pgen.0020221
Dogan Sukru, A., Pujol, C., Maiti, P., Kukat, A., Wang, S., Hermans, S., et al. (2014). Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 19, 458–469. doi: 10.1016/j.cmet.2014.02.004
Dogan, S. A., Pujol, C., and Trifunovic, A. (2012). Mitochondrial aspartyl-tRNA synthetase (DARS2) deficiency in mice. Free Radic. Biol. Med. 1:S183. doi: 10.1016/j.freeradbiomed.2012.08.384
Eidi, M., and Garshasbi, M. (2019). A novel ISCA2 variant responsible for an early-onset neurodegenerative mitochondrial disorder: a case report of multiple mitochondrial dysfunctions syndrome 4. BMC Neurol. 19:153. doi: 10.1186/s12883-019-1387-2
Eriani, G., Cavarelli, J., Martin, F., Dirheimer, G., Moras, D., and Gangloff, J. (1993). Role of dimerization in yeast aspartyl-tRNA synthetase and importance of the class II invariant proline. Proc. Natl. Acad. Sci. U.S.A. 90, 10816–10820. doi: 10.1073/pnas.90.22.10816
Erturk, O., Yalcinkaya, C., Siva, A., and Van Der Knaap, M. S. (2010). Adult-onset leukoencephalopathy with brain stem and spinal cord involvement and normal lactate: case report. Turk Noroloji Dergisi. 16, 106–109. Available online at: https://tjn.org.tr/jvi.aspx?pdir=tjn&plng=eng&un=TJN-07752
Filippi, M., Preziosa, P., Banwell, B. L., Barkhof, F., Ciccarelli, O., De Stefano, N., et al. (2019). Assessment of lesions on magnetic resonance imaging in multiple sclerosis: practical guidelines. Brain 142, 1858–1875. doi: 10.1093/brain/awz144
Fisher, F. M., Kleiner, S., Douris, N., Fox, E. C., Mepani, R. J., Verdeguer, F., et al. (2012). FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 26, 271–281. doi: 10.1101/gad.177857.111
Frohlich, D., Suchowerska, A. K., Spencer, Z. H. T., von Jonquieres, G., Klugmann, C. B., Bongers, A., et al. (2017). In vivo characterization of the aspartyl-tRNA synthetase DARS: homing in on the leukodystrophy HBSL. Neurobiol Dis. Part A 97, 24–35. doi: 10.1016/j.nbd.2016.10.008
Frohlich, D., Suchowerska, A. K., Voss, C., He, R., Wolvetang, E., von Jonquieres, G., et al. (2018). Expression pattern of the aspartyl-tRNA Synthetase DARS in the human brain. Front. Mol. Neurosci. 11:81. doi: 10.3389/fnmol.2018.00081
Fuchs, S. A., Schene, I. F., Kok, G., Jansen, J. M., Nikkels, P. G. J., van Gassen, K. L. I., et al. (2019). Aminoacyl-tRNA synthetase deficiencies in search of common themes. Genet. Med. 21, 319–330. doi: 10.1038/s41436-018-0048-y
Galluzzi, P., Sacchini, M., Bartalini, G., Monti, L., Cerase, A., Lamantea, E., et al. (2011). LBSL (leukoencephalopathy with brain stem and spinal cord involvement and high lactate) without sparing of the u-fibers and globi pallidi: a case report. Eur. J. Radiol Extra 79, e73–e76. doi: 10.1016/j.ejrex.2011.06.002
Gibson, E. M., Purger, D., Mount, C. W., Goldstein, A. K., Lin, G. L., Wood, L. S., et al. (2014). Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344:1252304. doi: 10.1126/science.1252304
Gonzalez-Serrano, L. E., Karim, L., Pierre, F., Schwenzer, H., Rotig, A., Munnich, A., et al. (2018). Three human aminoacyl-tRNA synthetases have distinct sub-mitochondrial localizations that are unaffected by disease-associated mutations. J. Biol. Chem. 293, 13604–13615. doi: 10.1074/jbc.RA118.003400
Götz, A., Tyynismaa, H., Euro, L., Ellonen, P., Hyötyläinen, T., Ojala, T., et al. (2011). Exome sequencing identifies mitochondrial alanyl-trna synthetase mutations in infantile mitochondrial cardiomyopathy. Am. J. Hum. Genet. 88, 635–642. doi: 10.1016/j.ajhg.2011.04.006
Grechanina, E. Y., and Zdubskaya, E. P. (2014). An early manifestation of LBSL (leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation) syndrome, case description. J. Inherit. Metab. Dis. 37:114. doi: 10.1007/s10545-014-9740-5
Griggs, R. C., Moxley, R. T., Lafrance, R. A., and McQuillen, J. J. N. (1978). Hereditary paroxysmal ataxia: response to acetazolamide. Neurology 28:1259. doi: 10.1212/WNL.28.12.1259
Guo, M., and Schimmel, P. (2013). Essential nontranslational functions of tRNA synthetases. Nat. Chem. Biol. 9, 145–153. doi: 10.1038/nchembio.1158
Guo, M., Yang, X.-L., and Schimmel, P. (2010). New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell Biol. 11, 668–674. doi: 10.1038/nrm2956
Hartman, T. G., Yosovich, K., Michaeli, H. G., Blumkin, L., Ben-Sira, L., Lev, D., et al. (2020). Expanding the genotype-phenotype spectrum of ISCA2-related multiple mitochondrial dysfunction syndrome-cavitating leukoencephalopathy and prolonged survival. Neurogenetics 21, 243–249. doi: 10.1007/s10048-020-00611-8
Haynes, C. M., and Ron, D. (2010). The mitochondrial UPR – protecting organelle protein homeostasis. J. Cell Sci. 123, 3849–3855. doi: 10.1242/jcs.075119
Ikazabo, R. N. N., Mostosi, C., Jissendi, P., Labaisse, M. A., and Vandernoot, I. (2020). A new DARS2 mutation discovered in an adult patient. Case Rep. Neurol. 12, 107–113. doi: 10.1159/000506190
Isohanni, P., Linnankivi, T., Buzkova, J., Lonnqvist, T., Pihko, H., Valanne, L., et al. (2010). DARS2 mutations in mitochondrial leucoencephalopathy and multiple sclerosis. J. Med. Genet. 47, 66–70. doi: 10.1136/jmg.2009.068221
Iyer, A., and Philip, S. (2011). Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation in a 2-year-old male child. Dev. Med. Child Neurol. 53:30. doi: 10.1111/j.1469-8749.2010.03866.x
Jen, J. C., and Wan, J. (2018). “Chapter 13 - episodic ataxias,” in Handbook of Clinical Neurology, Vol. 155, eds M. Manto and T. A. G. M. Huisman (Amsterdam: Elsevier), 205–215.
Kassem, H., Wafaie, A., Abdelfattah, S., and Farid, T. (2014). Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL): assessment of the involved white matter tracts by MRI. Eur. J. Radiol. 83, 191–196. doi: 10.1016/j.ejrad.2013.09.023
Khan, K., Baleanu-Gogonea, C., Willard, B., Gogonea, V., and Fox, P. L. (2020). 3-Dimensional architecture of the human multi-tRNA synthetase complex. Nucleic Acids Res. 48, 8740–8754. doi: 10.1093/nar/gkaa569
Kim, K. R., Park, S. H., Kim, H. S., Rhee, K. H., Kim, B.-G., Kim, D. G., et al. (2013). Crystal structure of human cytosolic aspartyl-tRNA synthetase, a component of multi-tRNA synthetase complex. Proteins 81, 1840–1846. doi: 10.1002/prot.24306
Kohler, C., Heyer, C., Hoffjan, S., Stemmler, S., Lucke, T., Thiels, C., et al. (2015). Early-onset leukoencephalopathy due to a homozygous missense mutation in the DARS2 gene. Mol. Cell. Probes 29, 319–322. doi: 10.1016/j.mcp.2015.06.005
Kos, J., Nanut, M. P., Prunk, M., Sabotič, J., Dautović, E., and Jewett, A. (2018). Cystatin F as a regulator of immune cell cytotoxicity. Cancer Immunol. Immunother. 67, 1931–1938. doi: 10.1007/s00262-018-2165-5
Labauge, P., Dorboz, I., Eymard-Pierre, E., Dereeper, O., and Boespflug-Tanguy, O. (2011). Clinically asymptomatic adult patient with extensive LBSL MRI pattern and DARS2 mutations. J. Neurol. 258, 335–337. doi: 10.1007/s00415-010-5755-5
Labauge, P., Roullet, E., Boespflug-Tanguy, O., Nicoli, F., Le Fur, Y., Cozzone, P. J., et al. (2007). Familial, adult onset form of leukoencephalopathy with brain stem and spinal cord involvement: inconstant high brain lactate and very slow disease progression. Eur. Neurol. 58, 59–61. doi: 10.1159/000102171
Lan, M. Y., Chang, Y. Y., Yeh, T. H., Lin, T. K., and Lu, C. S. (2017). Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL) with a novel DARS2 mutation and isolated progressive spastic paraparesis. J. Neurol. Sci. 372, 229–231. doi: 10.1016/j.jns.2016.11.058
Lee, S. W., Cho, B. H., Park, S. G., and Kim, S. (2004). Aminoacyl-tRNA synthetase complexes: beyond translation. Cell. Mol. Life Sci. 117, 3725–3734. doi: 10.1242/jcs.01342
Lin, J., Chiconelli Faria, E., Da Rocha, A. J., Rodrigues Masruha, M., Pereira Vilanova, L. C., Scheper, G. C., et al. (2010). Leukoencephalopathy with brainstem and spinal cord involvement and normal lactate: a new mutation in the DARS2 gene. J. Child Neurol. 25, 1425–1428. doi: 10.1177/0883073810370897
Lin, T. K., Chang, Y. Y., Lin, H. Y., Liou, C. W., Wang, P. W., Chuang, J. H., et al. (2019). Mitochondrial dysfunctions in leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL). PLoS ONE 14:e0224173. doi: 10.1371/journal.pone.0224173
Linnankivi, T., Lundbom, N., Autti, T., Häkkinen, A.-M., Koillinen, H., Kuusi, T., et al. (2004). Five new cases of a recently described leukoencephalopathy with high brain lactate. Neurology 63, 688–692. doi: 10.1212/01.WNL.0000134658.35601.41
Ma, J., Tanaka, K. F., Shimizu, T., Bernard, C. C. A., Kakita, A., Takahashi, H., et al. (2011). Microglial cystatin F expression is a sensitive indicator for ongoing demyelination with concurrent remyelination. J. Neurosci. Res. 89, 639–649. doi: 10.1002/jnr.22567
Martikainen, M. H., Ellfolk, U., and Majamaa, K. (2013). Impaired information-processing speed and working memory in leukoencephalopathy with brainstem and spinal cord involvement and elevated lactate (LBSL) and DARS2 mutations: a report of three adult patients. J. Neurol. 260, 2078–2083. doi: 10.1007/s00415-013-6940-0
Messmer, M., Florentz, C., Schwenzer, H., Scheper, G. C., van der Knaap, M. S., Marechal-Drouard, L., et al. (2011). A human pathology-related mutation prevents import of an aminoacyl-tRNA synthetase into mitochondria. Biochem. J. 433, 441–446. doi: 10.1042/BJ20101902
Mierzewska, H., van der Knaap, M. S., Scheper, G. C., Bekiesinska-Figatowska, M., Szczepanik, E., and Jurkiewicz, E. (2011). Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation in the first Polish patient. Brain Dev. 33, 713–717. doi: 10.1016/j.braindev.2010.12.005
Mitew, S., Hay, C. M., Peckham, H., Xiao, J., Koenning, M., and Emery, B. (2014). Mechanisms regulating the development of oligodendrocytes and central nervous system myelin. Neuroscience 276:29–47. doi: 10.1016/j.neuroscience.2013.11.029
Miyake, N., Yamashita, S., Kurosawa, K., Miyatake, S., Tsurusaki, Y., Doi, H., et al. (2011). A novel homozygous mutation of DARS2 may cause a severe LBSL variant. Clin. Genet. 80, 293–296. doi: 10.1111/j.1399-0004.2011.01644.x
Moffett, J. R., Ross, B., Arun, P., Madhavarao, C. N., and Namboodiri, A. M. A. (2007). N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog Neurobiol. 81, 89–131. doi: 10.1016/j.pneurobio.2006.12.003
Moore, S. A., Kumar, N., and Gavrilova, R. H. (2012). Leukoencephalopathy with brain stem and spinal cord involvement (and high lactate): raising the bar for diagnosis. J. Neurol. 259, 2494–2497. doi: 10.1007/s00415-012-6596-1
Nagao, A., Suzuki, T., Katoh, T., Sakaguchi, Y., and Suzuki, T. (2009). Biogenesis of glutaminyl-mt tRNAGln in human mitochondria. Proc. Natl. Acad. Sci. U.S.A. 106, 16209–16214. doi: 10.1073/pnas.0907602106
Navarro Vázquez, I., Maestre Martínez, L., Lozano Setién, E., and Menor Serrano, F. (2016). Leucoencephalopathy with brain stem and spinal cord involvement and lactate elevation: report of two new cases. An Pediatr. 84, 291–293. doi: 10.1016/j.anpede.2015.07.034
Nemeth, C. L., Tomlinson, S. N., Rosen, M., O'Brien, B. M., Larraza, O., Jain, M., et al. (2020). Neuronal ablation of mt-AspRS in mice induces immune pathway activation prior to severe and progressive cortical and behavioral disruption. Exp. Neurol. 326:113164. doi: 10.1016/j.expneurol.2019.113164
Ofir-Birin, Y., Fang, P., Bennett, S. P., Zhang, H.-M., Wang, J., Rachmin, I., et al. (2013). Structural switch of lysyl-tRNA synthetase between translation and transcription. Mol. Cell. 49, 30–42. doi: 10.1016/j.molcel.2012.10.010
Omura, T. (1998). Mitochondria-targeting sequence, a multi-role sorting sequence recognized at all steps of protein import into mitochondria. J. Biochem. 123, 1010–1016. doi: 10.1093/oxfordjournals.jbchem.a022036
Ong, M. T., Willoughby, J., Connolly, D. J. A., Mordekar, S., Study, D. D. D., and Johnson, D. (2020). Genotype–phenotype variability of DARS mutation-case reports of a trio of siblings. Eur. J. Med. Case Rep. 4, 110–115. doi: 10.24911/ejmcr/1731551044010
Orcesi, S., La Piana, R., Uggetti, C., Tonduti, D., Pichiecchio, A., Pasin, M., et al. (2011). Spinal cord calcification in an early-onset progressive leukoencephalopathy. J. Child Neurol. 26, 876–880. doi: 10.1177/0883073810390038
Ortiz, F. C., Habermacher, C., Graciarena, M., Houry, P.-Y., Nishiyama, A., Nait Oumesmar, B., et al. (2019). Neuronal activity in vivo enhances functional myelin repair. JCI Insight 5:e123434. doi: 10.1172/jci.insight.123434
Pang, Y. L., Poruri, K., and Martinis, S. A. (2014). tRNA synthetase: tRNA aminoacylation and beyond. Wiley Interdiscip. Rev. RNA 5, 461–480. doi: 10.1002/wrna.1224
Petzold, G. C., Bohner, G., Klingebiel, R., Amberger, N., van der Knaap, M. S., and Zschenderlein, R. (2006). Adult onset leucoencephalopathy with brain stem and spinal cord involvement and normal lactate. J. Neurol. Neurosurg. Psychiatr. 77, 889–891. doi: 10.1136/jnnp.2005.078568
Potthoff, M. J., Inagaki, T., Satapati, S., Ding, X., He, T., Goetz, R., et al. (2009). FGF21 induces PGC-1α and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc. Natl. Acad. Sci. U.S.A. 106, 10853–10858. doi: 10.1073/pnas.0904187106
Rabouw, H. H., Langereis, M. A., Anand, A. A., Visser, L. J., de Groot, R. J., Walter, P., et al. (2019). Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc. Natl. Acad. Sci. U.S.A. 116, 2097–2102. doi: 10.1073/pnas.1815767116
Rackayova, V., Cudalbu, C., Pouwels, P. J. W., and Braissant, O. (2017). Creatine in the central nervous system: from magnetic resonance spectroscopy to creatine deficiencies. Anal. Biochem. 529:144–157. doi: 10.1016/j.ab.2016.11.007
Rathore, G., Star, L., Larsen, P., and Rizzo, W. (2017). “Novel mutation of DARS2 gene leads to a rare and aggressive presentation of leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL),” in Neurology Conference: 69th American Academy of Neurology Annual Meeting, AAN (Boston, MA).
Rinaldi, C., and Wood, M. J. A. (2018). Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 14, 9–21. doi: 10.1038/nrneurol.2017.148
Rinholm, J. E., Vervaeke, K., Tadross, M. R., Tkachuk, A. N., Kopek, B. G., Brown, T. A., et al. (2016). Movement and structure of mitochondria in oligodendrocytes and their myelin sheaths. Glia 64, 810–825. doi: 10.1002/glia.22965
Rumyantseva, A., Motori, E., and Trifunovic, A. (2020). DARS2 is indispensable for Purkinje cell survival and protects against cerebellar ataxia. Hum. Mol. Genet. 29, 2845–2854. doi: 10.1093/hmg/ddaa176
Sauter, C., Lorber, B., Gaudry, A., Karim, L., Schwenzer, H., Wien, F., et al. (2015). Neurodegenerative disease-associated mutants of a human mitochondrial aminoacyl-tRNA synthetase present individual molecular signatures. Sci. Rep. 5:17332. doi: 10.1038/srep17332
Scheper, G. C., Van Der Klok, T., Van Andel, R. J., Van Berkel, C. G. M., Sissler, M., Smet, J., et al. (2007). Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 39, 534–539. doi: 10.1038/ng2013
Schicks, J., Schols, L., van der Knaap, M. S., and Synofzik, M. (2013). Teaching NeuroImages: MRI guides genetics: leukoencephalopathy with brainstem and spinal cord involvement (LBSL). Neurology 80, e176–e177. doi: 10.1212/WNL.0b013e31828cf846
Schiffmann, R., and van der Knaap, M. S. (2009). Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 72, 750–759. doi: 10.1212/01.wnl.0000343049.00540.c8
Serkov, S. V., Pronin, I. N., Bykova, O. V., Maslova, O. I., Arutyunov, N. V., Muravina, T. I., et al. (2004). Five patients with a recently described novel leukoencephalopathy with brainstem and spinal cord involvement and elevated lactate. Neuropediatrics 35, 1–5. doi: 10.1055/s-2003-43548
Sharma, S., Sankhyan, N., Kumar, A., Scheper, G. C., van der Knaap, M. S., and Gulati, S. (2011). Leukoencephalopathy with brain stem and spinal cord involvement and high lactate: a genetically proven case without elevated white matter lactate. J. Child Neurol. 26, 773–776. doi: 10.1177/0883073810390695
Shimizu, T., Wisessmith, W., Li, J., Abe, M., Sakimura, K., Chetsawang, B., et al. (2017). The balance between cathepsin C and cystatin F controls remyelination in the brain of Plp1-overexpressing mouse, a chronic demyelinating disease model. Glia 65, 917–930. doi: 10.1002/glia.23134
Shimojima, K., Higashiguchi, T., Kishimoto, K., Miyatake, S., Miyake, N., Takanashi, J. I., et al. (2017). A novel DARS2 mutation in a Japanese patient with leukoencephalopathy with brainstem and spinal cord involvement but no lactate elevation. Hum. Genome Var. 4:17051. doi: 10.1038/hgv.2017.51
Steenweg, M. E., Pouwels, P. J. W., Wolf, N. I., van Wieringen, W. N., Barkhof, F., and van der Knaap, M. S. (2011). Leucoencephalopathy with brainstem and spinal cord involvement and high lactate: quantitative magnetic resonance imaging. Brain 134, 3333–3341. doi: 10.1093/brain/awr254
Steenweg, M. E., van Berge, L., van Berkel, C. G., de Coo, I. F., Temple, I. K., Brockmann, K., et al. (2012). Early-onset LBSL: how severe does it get? Neuropediatrics 43, 332–338. doi: 10.1055/s-0032-1329395
Suomalainen, A., Elo, J. M., Pietiläinen, K. H., Hakonen, A. H., Sevastianova, K., Korpela, M., et al. (2011). FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol. 10, 806–818. doi: 10.1016/S1474-4422(11)70155-7
Synofzik, M., Schicks, J., Lindig, T., Biskup, S., Schmidt, T., Hansel, J., et al. (2011). Acetazolamide-responsive exercise-induced episodic ataxia associated with a novel homozygous DARS2 mutation. J. Med. Genet. 48, 713–715. doi: 10.1136/jmg.2011.090282
Taft, R. J., Vanderver, A., Leventer, R. J., Damiani, S. A., Simons, C., Grimmond, S. M., et al. (2013). Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am. J. Hum. Genet. 92, 774–780. doi: 10.1016/j.ajhg.2013.04.006
Távora, D. G. F., Nakayama, M., Gama, R. L., Alvim TCd, L., Portugal, D., and Comerlato, E. A. (2007). Leukoencephalopathy with brainstem and spinal cord involvement and high brain lactate: report of three brazilian patients. Arq. Neuropsiquiatr. 65, 506–511. doi: 10.1590/S0004-282X2007000300028
Taylor, R. W., Pyle, A., Griffin, H., Blakely, E. L., Duff, J., He, L., et al. (2014). Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 312, 68–77. doi: 10.1001/jama.2014.7184
Teive, H. A. G., and Ashizawa, T. (2015). Primary and secondary ataxias. Curr. Opin. Neurol. 28, 413–422. doi: 10.1097/WCO.0000000000000227
Toldo, I., Nosadini, M., Boscardin, C., Talenti, G., Manara, R., Lamantea, E., et al. (2018). Neonatal mitochondrial leukoencephalopathy with brain and spinal involvement and high lactate: expanding the phenotype of ISCA2 gene mutations. Metab. Brain Dis. 33, 805–812. doi: 10.1007/s11011-017-0181-3
Tricaud, N., and Park, H. T. (2017). Wallerian demyelination: chronicle of a cellular cataclysm. Cell. Mol. Life Sci. 74, 4049–4057. doi: 10.1007/s00018-017-2565-2
Tylki-Szymanska, A., Jurkiewicz, E., Zakharova, E. Y., and Bobek-Billewicz, B. (2014). Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation: high outcome variation between two siblings. Neuropediatrics 45, 188–191. doi: 10.1055/s-0033-1364105
Tzoulis, C., Tran, G. T., Gjerde, I. O., Aasly, J., Neckelmann, G., Rydland, J., et al. (2012). Leukoencephalopathy with brainstem and spinal cord involvement caused by a novel mutation in the DARS2 gene. J. Neurol. 259, 292–296. doi: 10.1007/s00415-011-6176-9
Ulrick, N., and Vanderver, A. (2017). “Hypomyelination with brainstem and spinal cord abnormalities and leg spasticity (HBSL),” in Pediatric Demyelinating Diseases of the Central Nervous System and Their Mimics: A Case-Based Clinical Guide, eds E. Waubant and T. E. Lotze (Cham: Springer International Publishing), 285–290.
Uluc, K., Baskan, O., Yildirim, K. A., Ozsahin, S., Koseoglu, M., Isak, B., et al. (2008). Leukoencephalopathy with brain stem and spinal cord involvement and high lactate: a genetically proven case with distinct MRI findings. J. Neurol. Sci. 273, 118–122. doi: 10.1016/j.jns.2008.06.002
van Berge, L., Dooves, S., van Berkel, C. G., Polder, E., van der Knaap, M. S., and Scheper, G. C. (2012). Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation is associated with cell-type-dependent splicing of mtAspRS mRNA. Biochem. J. 441, 955–962. doi: 10.1042/BJ20110795
Van Berge, L., Hamilton, E. M., Linnankivi, T., Uziel, G., Steenweg, M. E., Isohanni, P., et al. (2014). Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation: clinical and genetic characterization and target for therapy. Brain 137, 1019–1029. doi: 10.1093/brain/awu026
van Berge, L., Kevenaar, J., Polder, E., Gaudry, A., Florentz, C., Sissler, M., et al. (2013). Pathogenic mutations causing LBSL affect mitochondrial aspartyl-tRNA synthetase in diverse ways. Biochem. J. 450, 345–350. doi: 10.1042/BJ20121564
van der Knaap, M. S., Hamilton, E. M., and van Berge, L. (2014). Reply: DARS2 gene clinical spectrum: new ideas regarding an underdiagnosed leukoencephalopathy. Brain 137(Pt. 7):e290. doi: 10.1093/brain/awu135
Van Der Knaap, M. S., Van Der Voorn, P., Barkhof, F., Van Coster, R., Krägeloh-Mann, I., Feigenbaum, A., et al. (2003). A new leukoencephalopathy with brainstem and spinal cord involvement and high lactate. Ann. Neurol. 53, 252–258. doi: 10.1002/ana.10456
Ventura-Clapier, R., Garnier, A., and Veksler, V. (2008). Transcriptional control of mitochondrial biogenesis: the central role of PGC-1α. Cardiovasc. Res. 79, 208–217. doi: 10.1093/cvr/cvn098
Wallen, R. C., and Antonellis, A. (2013). To charge or not to charge: mechanistic insights into neuropathy-associated tRNA synthetase mutations. Curr. Opin. Genet. Dev. 23, 302–309. doi: 10.1016/j.gde.2013.02.002
Wang, X., Zhang, C., Szábo, G., and Sun, Q.-Q. (2013). Distribution of CaMKIIα expression in the brain in vivo, studied by CaMKIIα-GFP mice. Brain Res. 1518, 9–25. doi: 10.1016/j.brainres.2013.04.042
Werner, R., Daum, E., Felber, S., and Wohrle, J. C. (2018). Leukoencephalopathy with brain stem and spinal cord involvement and not always lactate elevation. Clin. Neuroradiol. 28, 451–453. doi: 10.1007/s00062-017-0647-z
Wolf, N. I., Toro, C., Kister, I., Latif, K. A., Leventer, R., Pizzino, A., et al. (2015). DARS-associated leukoencephalopathy can mimic a steroid-responsive neuroinflammatory disorder. Neurology 84, 226–230. doi: 10.1212/WNL.0000000000001157
Yahia, A., Elsayed, L., Babai, A., Salih, M. A., El-Sadig, S. M., Amin, M., et al. (2018). Intra-familial phenotypic heterogeneity in a Sudanese family with DARS2-related leukoencephalopathy, brainstem and spinal cord involvement and lactate elevation: a case report. BMC Neurol. 18:175. doi: 10.1186/s12883-018-1180-7
Yamashita, S., Miyake, N., Matsumoto, N., Osaka, H., Iai, M., Aida, N., et al. (2013). Neuropathology of leukoencephalopathy with brainstem and spinal cord involvement and high lactate caused by a homozygous mutation of DARS2. Brain Dev. 35, 312–316. doi: 10.1016/j.braindev.2012.05.007
Keywords: leukodystrophy, leukoencephalopathy, aminoacyl-tRNA synthetases, aspartyl-tRNA synthetase, DARS1, DARS2, hypomyelination with brainstem and spinal cord involvement and leg spasticity, leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation
Citation: Muthiah A, Housley GD, Klugmann M and Fröhlich D (2021) The Leukodystrophies HBSL and LBSL—Correlates and Distinctions. Front. Cell. Neurosci. 14:626610. doi: 10.3389/fncel.2020.626610
Received: 06 November 2020; Accepted: 21 December 2020;
Published: 26 January 2021.
Edited by:
Egor Dzyubenko, Essen University Hospital, GermanyReviewed by:
Henna Tyynismaa, University of Helsinki, FinlandCopyright © 2021 Muthiah, Housley, Klugmann and Fröhlich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominik Fröhlich, ZC5mcm9obGljaEB1bnN3LmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.