 Dominik Fröhlich1*
Dominik Fröhlich1* Marisa I. Mendes2
Marisa I. Mendes2 Andrew J. Kueh3,4
Andrew J. Kueh3,4 Andre Bongers5
Andre Bongers5 Marco J. Herold3,4Gajja S. Salomons2
Marco J. Herold3,4Gajja S. Salomons2 Gary D. Housley1
Gary D. Housley1 Matthias Klugmann1*
Matthias Klugmann1*- 1Translational Neuroscience Facility & Department of Physiology, School of Medical Sciences, UNSW Sydney, Kensington, NSW, Australia
- 2Metabolic Unit/Laboratory Genetic Metabolic Diseases, Department of Clinical Chemistry, Amsterdam Neuroscience, Amsterdam Gastroenterology & Metabolism, Amsterdam UMC, Vrije Universiteit Amsterdam, Amsterdam, Netherlands
- 3The Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia
- 4Department of Medical Biology, University of Melbourne, Parkville, VIC, Australia
- 5Biomedical Resources Imaging Laboratory, UNSW Sydney, Kensington, NSW, Australia
Hypomyelination with brain stem and spinal cord involvement and leg spasticity (HBSL) is a leukodystrophy caused by missense mutations of the aspartyl-tRNA synthetase-encoding gene DARS1. The clinical picture includes the regression of acquired motor milestones, spasticity, ataxia, seizures, nystagmus, and intellectual disabilities. Morphologically, HBSL is characterized by a distinct pattern of hypomyelination in the central nervous system including the anterior brainstem, the cerebellar peduncles and the supratentorial white matter as well as the dorsal columns and the lateral corticospinal tracts of the spinal cord. Adequate HBSL animal models are lacking. Dars1 knockout mice are embryonic lethal precluding examination of the etiology. To address this, we introduced the HBSL-causing Dars1D367Y point mutation into the mouse genome. Surprisingly, mice carrying this mutation homozygously were phenotypically normal. As hypomorphic mutations are more severe in trans to a deletion, we crossed Dars1D367Y/D367Y mice with Dars1-null carriers. The resulting Dars1D367Y/− offspring displayed a strong developmental delay compared to control Dars1D367Y/+ littermates, starting during embryogenesis. Only a small fraction of Dars1D367Y/− mice were born, and half of these mice died with hydrocephalus during the first 3 weeks of life. Of the few Dars1D367Y/− mice that were born at term, 25% displayed microphthalmia. Throughout postnatal life, Dars1D367Y/− mice remained smaller and lighter than their Dars1D367Y/+ littermates. Despite this early developmental deficit, once they made it through early adolescence Dars1D367Y/− mice were phenotypically inconspicuous for most of their adult life, until they developed late onset motor deficits as well as vacuolization and demyelination of the spinal cord white matter. Expression levels of the major myelin proteins were reduced in Dars1D367Y/− mice compared to controls. Taken together, Dars1D367Y/− mice model aspects of the clinical picture of the corresponding missense mutation in HBSL. This model will enable studies of late onset deficits, which is precluded in Dars1 knockout mice, and can be leveraged to test potential HBSL therapeutics including DARS1 gene replacement therapy.
Introduction
Leukodystrophies are inherited white matter disorders often associated with an early onset, lack of treatment options and premature death. The population incidence of all leukodystrophies taken together is relatively high with one in 7,600 live births (Bonkowsky et al., 2010) underpinning the high unmet medical need. Hypomyelination with brain stem and spinal cord involvement and leg spasticity (HBSL) belongs to this group of diseases and was first described in 2013 (Taft et al., 2013). Following the initial discovery, two additional case studies, together with the original study, described a total of 16 HBSL patients (Wolf et al., 2015; Ong et al., 2020). HBSL can be seen as a spectrum disorder with a high variance in severity (mild to severe forms) and onset of the disease (4 months to 22 years) (Wolf et al., 2015). An early onset form of HBSL usually results in a more severe course of disease. The clinical symptoms typically include motor deficits, leg spasticity, regression, or delay of developmental milestones, hypertonia, hyperreflexia, positive Babinski sign, nystagmus, and gait abnormalities in patients who are able to mobilize (Taft et al., 2013; Wolf et al., 2015; Ong et al., 2020). For a comprehensive clinical review of HBSL see Muthiah et al. (2020) in this issue.
The underlying cause of HBSL are missense mutations of the aspartyl-tRNA synthetase (AspRS) gene DARS1. AspRS belongs to a group of enzymes termed aminoacyl-tRNA synthetases (ARSs) that catalyze an aminoacylation reaction in which transfer ribonucleic acids (tRNAs) are linked to their cognate amino acids. This process is known as tRNA charging and is an essential prerequisite for successful protein biosynthesis. Each ARS is specific for the charging of one tRNA with its corresponding amino acid, e.g., AspRS specifically links tRNAAsp to aspartate, and there is no redundancy amongst these enzymes. ARSs can be subdivided into two classes depending on the cell compartment in which they catalyze the aminoacylation reaction: Cytosolic ARSs and mitochondrial ARSs (mt-ARSs). AspRS functions as a homodimer (Kim et al., 2013) and is ubiquitously expressed in all cells and tissues. However, our previous studies revealed a strong prevalence in neurons of the cerebellum in the murine (Fröhlich et al., 2017) and human brain (Fröhlich et al., 2018). All DARS1 mutations identified to date are located within the catalytic domain of AspRS and are likely having a direct impact on the aminoacylation reaction and consequently on protein synthesis. Why DARS1 mutations specifically manifest in neurologic deficits remains unresolved.
Previous attempts at creating a mouse model for HBSL through complete knockout of the Dars1 gene were unsuccessful and either resulted in early embryonic lethality (homozygous Dars1-null mice), or the lack of HBSL pathology (heterozygous Dars1-null mice). A more promising strategy is to introduce HBSL-causing DARS1 point mutations into the mouse gene. The HBSL index patient was reported to be a compound heterozygous carrier of the DARS1A274V and DARS1D367Y point mutations. In this study, we employed CRISPR/Cas9-mediated gene editing to introduce the Dars1D367Y mutation into the murine Dars1 gene. Surprisingly, homozygous Dars1D367Y/D367Y mice were only mildly affected. The human DARS1 missense mutations identified so far can be classified as hypomorphic mutations, which means that they cause a partial loss of gene function either through reduced expression or through impaired activity. Hypomorphs do not usually result in a complete loss of function and are always more severe in trans to a deletion mutation. In order to enhance the phenotype of Dars1D367Y/D367Y mice, we bred them with Dars1-null carriers. The Dars1D367Y/− offspring displayed a strong developmental delay, which often resulted in early embryonic or pre-weaning death. The few surviving Dars1D367Y/− mice were smaller and lighter compared to Dars1D367Y/+ littermates and developed late onset motor deficits as well as vacuolization of the white matter of the spinal cord. In summary, Dars1D367Y/− mice recapitulate some HBSL aspects and will be instrumental for examinations of the disease etiology or therapeutic proof-of-concept studies.
Methods
Ethics
All procedures were approved by the University of New South Wales Animal Care and Ethics Committee and were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Animals
Mice were housed in ventilated cages in groups of 2 to 5 animals and fed ad libitum with standard chow. The c.1099G>T (D367Y) point mutation was introduced into the Dars1 locus using the CRISPR/Cas9 technique as previously described (Kueh et al., 2017) on a C57BL/6J background. To generate mice carrying the Dars1 c.1099G>T (D367Y) point mutation, 20 ng/μl of Cas9 mRNA, 10 ng/μl of sgRNA (CGATGAGGAAGATCTAAGGT) and 40 ng/μl of oligo donor (gagtattgcgaggctttggcaatgcttagagaagctggagttgaaatggacgatgaggaaTatctaaggttAgtctctgatattttctctttcaactttaatgcttaggtttaatgtatttcacccagctga) were injected into the cytoplasm of fertilized one-cell stage embryos. Twenty-four hours later, two-cell stage embryos were transferred into the oviducts of pseudo-pregnant female mice. Viable offspring were genotyped by next-generation sequencing. Additionally, correct targeting was also confirmed by Sanger sequencing. The region containing the c.1099G>T mutation was amplified by PCR using the following primers: Forward 5′-GCTTTTCTTGGTTCAGTCGC-3′, reverse 5′-CGGGTTACAACTAGGGGAAA-3′. The resulting 449 bp PCR fragment was purified by ethanol precipitation and subsequently sequenced using the forward primer.
Dars1-null mice were created and genotyped as previously described (Fröhlich et al., 2017). Dars1D367Y/− mice were generated by breeding homozygous Dars1D367Y/D367Y mice with heterozygous Dars1-null mice.
Behavioral Testing
Behavioral tests were either performed in Dars1D367Y/D367Y mice together with age- and sex-matched wildtype mice or in age- and sex-matched Dars1D367Y/− and Dars1D367Y/+ littermates. Locomotor behavior was assessed using the rotarod test as described (von Jonquieres et al., 2018). For habituation, mice were placed on the rotarod apparatus (Ugo Basile, Comerio, Italy) for 1 min at a constant speed of 4 revolutions per minute (rpm) before the test trials started. During the test, the speed of the rotarod constantly increased from 4 to 40 rpm over a 4-min period. Cut-off time was 5 min with the last min on full speed. Mice were tested in 3 trials per day on two consecutive days (6 trials in total). Inter-trial interval was 30 min. The latency to fall was averaged over the six trials.
The open field-test was performed as described (Fröhlich et al., 2017). Mice were placed in the center of an open field box (40 × 40 × 40 cm3) under bright light conditions (100 lux). Mice were video recorded during the 30 min trial and subsequently analyzed using the ANY-Maze™ tracking software (Stoelting, Illinnois, USA) for total distance traveled as well as the distance traveled in the inner compartment.
To determine the acoustic startle response (ASR) as well as the pre-pulse inhibition (PPI) we used the SR-LAB Startle Response System (San Diego Instruments, San Diego, USA) as described (Schneider et al., 2007; Fröhlich et al., 2017). During ASR and PPI, mice were exposed to 60 dB sound pressure level (SPL) background white noise and both the ASR and PPI, were preceded by a 5 min habituation period of 60 dB SPL white noise only. The ASR was measured as the maximum amplitude detected by the accelerometer in response to 40 ms white noise pulses with increasing intensities (60–120 dB SPL) with a 10 s interval. In order to determine the PPI, the 120 dB SPL startle pulse was preceded (100 ms) by a 20 ms pre-pulse (72, 76, or 80 dB SPL). Each trial was repeated 10 times. PPI was determined as the decrease of the ASR amplitude in response to the pre-pulse compared to the 120 dB SPL pulse alone.
Magnetic Resonance Imaging (MRI) and Body Composition Analysis
Following a lethal intraperitoneal (IP) injection of pentobarbital, mice were transcardially perfused with 10 ml phosphate buffered saline (PBS) for 5 min, followed by 10 ml 4% paraformaldehyde (PFA) for 5 min. Brains were post-fixed in 4% PFA for 2 h at room temperature (RT). Prior to imaging the fixed brains were immersed in in 9 g/l NaCl/H2O solution for 24 h at 21°C to remove fixation residues in the brain tissue. The brains were then transferred into a 1.3 mm ID, 2 ml Cryovial (Greiner, Germany) and submersed in Perfluoro-Polyether Fomblin™ 6Y for susceptibility matching. The Cryovial was then mounted on the tip of a plastic tube, which was attached to the automatic positioning system of the MRI system. MRI was performed using a 9.4T BioSpec Avance III 94/20 (Bruker, Ettlingen, Germany) magnetic resonance microimaging system equipped with BGA-12S HP gradients with maximum strength 660 mT/m and slew rate 4,570 Tm/s. A dedicated 15 mm internal diameter quadrature specimen volume coil was used for radiofrequency transmission and reception. Anatomical images were acquired using an optimized isotropic 3D multi gradient echo sequence (MGE) with 106 coronal partitions and 10 gradient echoes with the following major parameters: First TE = 2.7 ms, ΔTE = 3.45 ms, #echos = 28, TR = 100 ms, FA = 30°, FOV = 15 × 15 × 8 mm, matrix = 200 × 200× 106, image resolution = 75 μm3 (isotropic), eff. spectral BW = 78,125 Hz, total acquisition time with 2 ADC averages: 1 h and 46 min per specimen. Segmentation and surface models of the entire brain as well as the ventricles were generated using the 3D slicer software (Kikinis et al., 2014).
Body composition analysis (EchoMRI) was performed as described (von Jonquieres et al., 2018) in accordance to the manufacturer's instructions using the EchoMRI-900TM scanner equipped with A100 mouse antenna insert (Echo Medical Systems). Fat and lean mass were calculated as percentage of total mass.
Mouse Histopathology Evaluation
This study utilized the Phenomics Australia (PA; formerly known as Australian Phenomics Network) Histopathology and Organ Pathology Service (HOPS) at the University of Melbourne. This service included: Harvesting of 25 organs, fixation and embedding in paraffin blocks, sectioning, Haematoxylin and Eosin (H&E; all organs; Figures 3H,I, 5C) and Luxol Fast Blue (LFB; brain and spinal cord; Figures 5B,D) staining, slide scanning, images of histopathology, and detailed histology and pathology reports.
Immunohistochemistry
Following cardiac perfusion as described above, brains were post-fixed in 4% PFA for 2 h at RT and cryoprotected in 30% sucrose. Brains were cut in the coronal plane into 40 μm sections using a cryostat as described (von Jonquieres et al., 2013). Sections were permeabilized with 0.2% TritonX-100 in PBS (PBS-Tx) and non-specific binding was blocked with 4% goat serum in PBS-Tx. Primary antibodies were applied in 4% goat serum in PBS-Tx over night at 4°C. The following antibodies were used: rabbit anti-NF200 (neurofilament 200; 1:1000; Sigma-Aldrich no. N4142) and rat anti-PLP clone aa3 (proteolipid protein; 1:10; gift from Prof. J. Trotter, Mainz, Germany). After washing with PBS, sections were incubated with goat anti-rabbit Alexa-488 and goat anti-rat Alexa-594 secondary antibodies (1:400; Thermo Fisher Scientific no. A11012 and A11006) in 4% goat serum in PBS-Tx for 4 h at RT. After washing in PBS-Tx, sections were mounted in Mowiol (Calbiochem, Darmstadt, Germany) and imaged using an LSM710 confocal microscope (Carl Zeiss, Berlin, Germany).
RNA Isolation and qRT-PCR
Animals were euthanized at 10 months by cervical dislocation. The brain regions cortex (CX), cerebellum (CB), brainstem (BS), and basal ganglia (BG) were dissected and snap frozen. RNA extraction was performed as described before (Fröhlich et al., 2018). Briefly, brain tissue was homogenized in liquid nitrogen using mortar and pestle followed by RNA extraction according to the manufacturer's instructions (RNeasy MiniKit, Qiagen no. 74106) including on-column DNase digest (RNase-Free DNase Set, Qiagen no. 79254). RNA was transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems no. 4368813) according to the manufacturer's instructions. Quantitative real-time PCR (qRT-PCR) was performed in triplicate using the StepOnePlus™ Real-Time PCR system (Applied Biosystems). The following TaqMan probes (Applied Biosystems) were used: Dars1 (Mm00624185_m1), Dars2 (Mm01296063_m1), Aspa (Mm004808667_m1), Plp1 (Mm00456892_m1), Cnp (Mm01306640), Mbp (Mm01266402_m1), GusB (Mm01197698_m1). Data was normalized to the housekeeper GusB and the CX region using the comparative ΔΔCT method.
SDS-PAGE and Western-Blotting
Animals were euthanized at 10 months by cervical dislocation. The brain regions CX, CB, BS, and BG were dissected, snap frozen and homogenized under liquid nitrogen using mortar and pestle. 10 μl lysis buffer (50 mM Tris-Cl, pH 7.4, 1 mM EDTA pH 8.0, 250 mM NaCl, and 1% Triton-X) including a cocktail of protease inhibitors (Complete, Roche) were added per mg of brain tissue. Lysates were sonicated using a Branson 450 Digital Probe Sonifier at 10% sonication amplitude and protein concentration was determined by Bradford protein assay (Bio-Rad no. 5000006).
SDS-PAGE and Western-blotting was performed as described (Fröhlich et al., 2018). In summary, 20 μg of protein mixed with 5x Laemmli reducing sample buffer were loaded onto a 10% acrylamide gel, separated by SDS-PAGE, and transferred onto a PVDF membrane (Bio-Rad no. 162-0177). Membranes were incubated with 4% skim milk powder in PBS plus 0.1% Tween (PBS-T) to prevent unspecific binding of antibodies. Subsequently, membranes were probed with the following primary antibodies in 4% skim milk in PBS-T: mouse anti-AspRS (1:1000; SantaCruz no. sc-393275), rabbit anti-GAPDH (glyceraldehyde 3-phosphate dehydrogenase; 1:4000, Cell Signaling no. 2118S), mouse anti-CNP (2',3'-cyclic nucleotide 3' phosphodiesterase; 1:3000, Abcam no. ab6319), rat anti-MBP (myelin basic protein; 1:1000; Abcam no. ab7349), rat anti-PLP aa3 (proteolipid protein; 1:200, gift from Prof. J. Trotter, Mainz, Germany). Following 3 wash steps (15 min each) with PBS-T, membranes were probed with appropriate HRP-conjugated secondary antibodies (1:10000; Dianova, Hamburg, Germany) in 4% skim milk in PBS-T. After 3 additional wash steps with PBS-T, membranes were incubated for 1 min with 1 ml Clarity Western ECL substrate (Bio-Rad no. 170-5060) and imaged in the ChemiDoc MP system (Bio-Rad, Hercules, USA).
AspRS Enzyme Activity Assay
HEK293 cells were cultured to 70% confluency for transient expression with DARS1WT, DARS1D367Y, DARS1A274V, or an equimolar mix of DARS1A274V and DARS1D367Y. For transfection, 12 μg of plasmid DNA were mixed with 60 μl Fugene (Promega, Madison, WI) in serum–free DMEM medium (Thermo Fisher Scientific, Waltham, USA) and incubated for 15 min at room temperature. Subsequently, the mix was applied to the cells. Cells transfected with an empty vector (mock transfected) and untransfected cells were included as controls. Fluorescent microscopy was used to estimate transfection efficiency and Western blotting was performed to confirm comparable expression levels of wildtype and mutant AspRS. Cells were harvested 48 h post transfection, pelleted, snap-frozen, and stored at −80°C until further use. All transfections were performed in triplicate.
To determine aminoacylation activity, pellets were suspended in ddH2O and cells were lysed in three freeze-thaw cycles. Protein amount was quantified using the BCA assay and protein samples were adjusted to 1 mg/ml. Cell lysates were incubated in triplicate for 10 min at 37°C in reaction buffer [50 mM Tris buffer pH 7.5, 12 mM MgCl2, 25 mM KCl, 1 mg/ml bovine serum albumin, 0.5 mM spermine, 1 mM ATP, 0.2 mM E. coli total tRNA, 1 mM dithiothreitol, and 0.3 mM (13C4, 15N)-aspartate]. The reaction was stopped using trichloroacetic acid. After washing the samples with trichloroacetic acid, ammonia was added to release the labeled amino acids from the tRNAs. [D3]-aspartate was added as internal standard and the labeled amino acids were quantified by LC-MS/MS. Aminoacylation activity measured for mock transfected cells was subtracted from DARS1-transfected samples. DARS1WT transfected cells were used as a positive control and activities were expressed as percentage of DARS1WT transfected cells.
Statistics
Statistical analysis was performed with the GraphPad Prism 8 software (La Jolla, CA, USA). Following validation of normal distribution of data, one-way or two-way analysis of variance (ANOVA) with Bonferroni's multiple comparisons post-hoc test was performed as indicated. Values are displayed as mean ± SEM with p < 0.05 regarded as statistically significant.
Results
Phenotype of Homozygous Dars1D367Y/D367Y Mice
In our previous study we demonstrated that the early embryonic lethality of Dars1-null mice precluded their use as an accurate model for HBSL (Fröhlich et al., 2017). In this study we introduced the c.1099G>T (D367Y) point mutation, which was first identified in the compound heterozygous index patient, into the mouse Dars1 locus using the CRISPR/Cas9 gene editing technology. This single nucleotide change located on exon 11 of the Dars1 locus results in a TAT codon coding for tyrosine instead of GAT coding for aspartic acid (Figure 1A). To our surprise, homozygous Dars1D367Y/D367Y mice were only mildly affected. The body weight of homozygous Dars1D367Y/D367Y mice, compared to wildtype controls at 4 months of age, was unchanged (Figure 1B). Body composition analysis using EchoMRI revealed only a slight shift from fat to lean mass without reaching statistical significance (Figure 1C). We performed behavioral tests with these mice at two different timepoints, at 4 and 10 months, to detect both early as well as late onset deficits. Motor coordination was assessed using the rotarod test but revealed no differences between homozygous Dars1D367Y/D367Y mice and wildtype controls (Figure 1D). Explorative behavior, locomotor activity and anxiety were tested in the open field paradigm. Total distance traveled as well as distance traveled in the inner compartment of the apparatus were unchanged between groups (Figures 1E,F). The acoustic startle response (ASR), a reflex resulting in muscular activity in response to an acoustic stimulus, was determined to test Dars1D367Y/D367Y mice for more subtle changes in information processing speed. Homozygous mutants, exposed to short sound pulses (40 ms) with increasing intensities ranging from 60 to 120 dB SPL, showed a significantly lower ASR compared to wildtype controls from 105 dB SPL to 120 dB SPL (sound levels 620 to 740; Figure 1G). The pre-pulse inhibition (PPI) measures sensorimotor gating mechanisms, and an impairment is an indicator for attentional processing deficits (Koch, 1999). We had observed PPI deficits in heterozygous Dars1-null mice (Fröhlich et al., 2017), however, Dars1D367Y/D367Y mice were normal (Figure 1H).
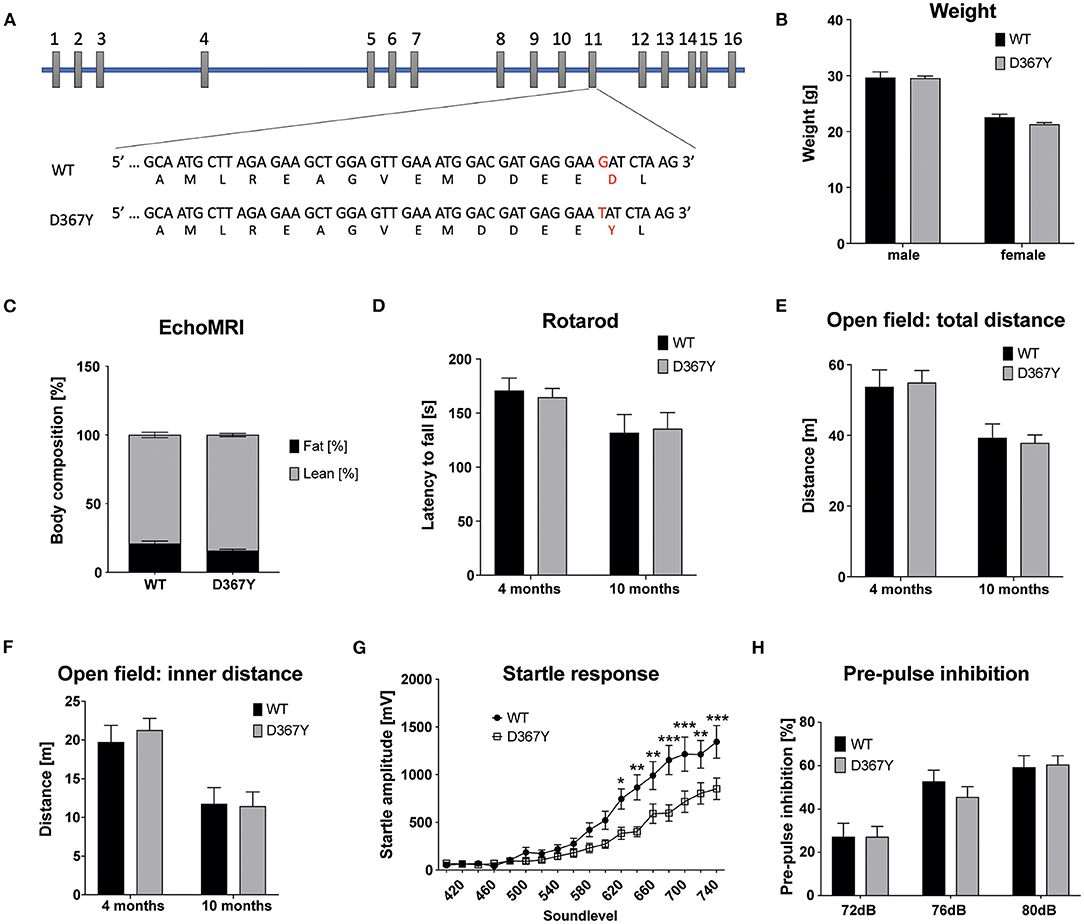
Figure 1. Characterization of homozygous Dars1D367Y mice. (A) Schematic depicting the genomic location of the D367Y mutation on exon 11 of the Dars1 gene. (B) Body weight of homozygous D367Y mice compared to wildtype controls (WT) at 4 months of age (n = 4–6). (C) EchoMRI body composition analysis revealed a slight shift from fat to lean mass in homozygous D367Y mice (n = 9). (D) Motor coordination assessed by the rotarod test was unchanged between homozygous D367Y mice and wildtype controls (n = 8–24). (E,F) Total distance traveled in an open field test apparatus [(E); n = 7–28] and distance in the inner compartment of the apparatus [(F); n = 7–28]. (G) Four-months-old homozygous D367Y mice exposed to sound pulses with increasing intensities (60–120 dB SPL, 40 ms) showed a lowered acoustic startle response compared to wildtype controls (n = 33–38). (H) Pre-pulse inhibition (72, 76, or 80 dB SPL pre-pulse 100 ms before the 120 dB SPL startle pulse) was unchanged between genotypes (n = 19–36). Data represent mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001; Two-way ANOVA).
Aminoacylation Activity of Mutant AspRS
The HBSL index patient was compound heterozygous for the A274V and D367Y DARS1 mutations. Both mutations are located in the catalytic domain of the AspRS enzyme (Taft et al., 2013) indicating a direct impact on enzymatic activity. Therefore, we measured aminoacylation activity in lysates of HEK293 cells transiently expressing DARS1WT, DARS1A274V, DARS1D367Y, or an equimolar mix of DARS1A274V and DARS1D367Y. Aminoacylation activity of mutant AspRS variants was normalized to the aminoacylation activity of wildtype AspRS (Figure 2). A higher value indicates more efficient charging of tRNAAsp with aspartate. Strikingly, the A274V mutation completely abolished aminoacylation activity of AspRS, resembling a functional null mutation. In contrast, the D367Y mutation led to a slight increase in enzymatic activity. The enzyme activity measured in lysates of HEK293 cells transiently expressing a mix of DARS1A274V and DARS1D367Y, which corresponds to the enzyme configuration of the index patient, ranged between the activities measured for DARS1A274V and DARS1D367Y alone (about 70% of wildtype AspRS activity).
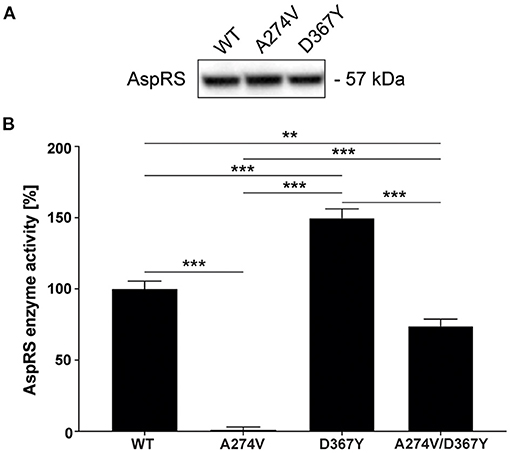
Figure 2. Aminoacylation activity of wildtype AspRS compared to AspRS carrying the A274V or D367Y mutations. (A) Western-blot depicting comparable expression levels of DARS1WT, DARS1A274V, DARS1D367Y in HEK293 cell lysates. (B) Enzyme activity was measured using lysates of HEK293 cells transiently expressing DARS1WT, DARS1A274V, DARS1D367Y, or an equimolar mix of DARS1A274V and DARS1D367Y. Cell lysates were incubated with E. coli tRNA, [13C4, 15N]-aspartate and ATP followed by quantification of [13C4,15N]-aspartate using LC-MS/MS. Aminoacylation activity of AspRS variants is displayed as percentage of WT AspRS activity. Data represent mean ± SEM (**p < 0.01, ***p < 0.001; One-way ANOVA).
Compound Heterozygous Dars1D367Y/− Mice Display Developmental Deficits
We next crossed homozygous Dars1D367Y/D367Y mice with heterozygous Dars1-null mice to create compound heterozygous Dars1D367Y/− mice. Since the A274V mutation completely abolished AspRS activity, Dars1D367Y/− mice can be considered a phenocopy of the compound heterozygous index patient (DARS1A274V/D367Y) in regard to enzyme activity.
The expected Mendelian ratio of the Dars1D367Y/− and Dars1D367Y/+ offspring in the F1 generation is 50:50. However, of 286 born mice (72 litters), only 25 mice (9%) had the genotype Dars1D367Y/−. This developmental disadvantage was sustained postnatally as 48% of Dars1D367Y/− mice (4% of all mice) developed hydrocephalus during the first 3 weeks of life and either died naturally or had to be euthanized (Figures 3A,G). 24% of Dars1D367Y/− (2% of all littermates) suffered from microphthalmia or retinal degeneration in one or both eyes (Figures 3A,H). The remainder of the born Dars1D367Y/− mice (28% of Dars1D367Y/− mice or 3% of total mice) showed growth retardation and were significantly smaller and lighter compared to Dars1D367Y/+ littermates (Figures 3A,D,E). Bodyweight of Dars1D367Y/− mice remained lower throughout life (Figure 3D) and EchoMRI body composition analysis revealed a significant reduction in body fat mass of Dars1D367Y/− mice (13.5% body fat) compared to Dars1D367Y/+ littermates (24.3% body fat; Figure 3F). Lastly, H&E staining of cross sections of the inner ear showed a reduced density of spiral ganglion cells in adult Dars1D367Y/− mice (Figure 3I).
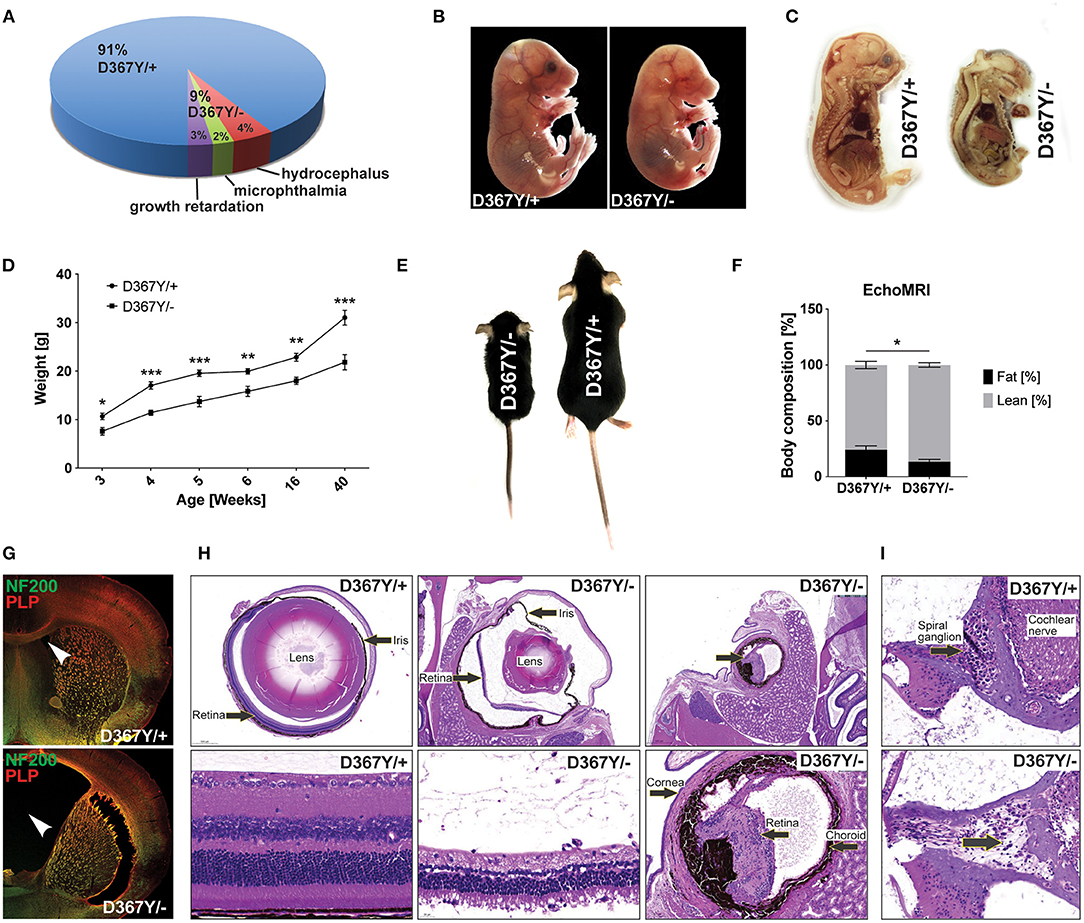
Figure 3. Compound heterozygous Dars1D367Y/− mice display developmental deficits. (A) Genotype and phenotype distribution in the F1 generation resulting from breeding of homozygous D367Y and heterozygous Dars1-null mice. Of the 286 born mice (72 litters), only 25 (9%) were compound heterozygous (D367Y/–; expected: 50%). 48% of the born D367Y/– mice (4% of total mice) developed hydrocephalus during the first weeks of their life; 24% of D367Y/– mice (2% of total mice) had microphthalmia in 1 or 2 eyes. (B) At embryonic day 18 (E18), 57% of the embryos present in utero were D367Y/– (n = 21), however, they were significantly underdeveloped at this stage. (C) Sagittal sections of D367Y/+ and D367Y/– embryos at E18 showing the severe underdevelopment at this stage. (D) Weight of D367Y/+ and D367Y/– females (n = 4–11). (E) D367Y/– mice are smaller and lighter compared to age- and sex-matched D367Y/+ littermates. (F) EchoMRI body composition analysis revealed a significant reduction of body fat in D367Y/– mice (n = 8). (G) Coronal brain sections of 3-week-old D367Y/– mice with hydrocephalus and D367Y/+ control mice. Sections were labeled with NF200 (green) and PLP (red). (H) H&E staining of cross sections of the eyes showing various degrees of microphthalmia and retinal degeneration/atrophy in D367Y/– mice compared to D367Y/+ controls. (I) H&E staining of cross sections of the inner ear indicates reduced density of spiral ganglion cells (lower panel, arrow). Data represent mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001; Two-way ANOVA).
Intriguingly, at embryonic day 18 (E18), 57% of the embryos present in utero were Dars1D367Y/− (n = 21), which is in line with the expected Mendelian ratio when crossing homozygous Dars1D367Y/D367Y mice with heterozygous Dars1-null mice. However, these embryos were severely underdeveloped at this stage (Figures 3B,C).
Compound Heterozygous Dars1D367Y/− Mice Develop Late Onset Motor Deficits
Despite the developmental delay and high mortality of Dars1D367Y/− mice, once they passed early adolescence, Dars1D367Y/− mice were phenotypically inconspicuous for most of their adult life. A hallmark of HBSL are motor deficits including severe leg spasticity and gait abnormalities. To see whether Dars1D367Y/− mice develop similar motor deficits, we performed locomotor tests at two different timepoints (four and 10 months) to distinguish between early and late onset motor deficits. Motor coordination assessed by the rotarod test was unchanged at 4 months but was impaired at 10 months in Dars1D367Y/− mice compared to Dars1D367Y/+ littermates (Figure 4A). Total distance traveled in an open field test apparatus and distance in the inner compartment of the apparatus were unchanged at 4 months of age (Figures 4B,C). At 10 months, however, the total distance and the distance in the inner compartment were significantly lower in Dars1D367Y/− mice compared to Dars1D367Y/+ littermates indicating a reduction in locomotor activity and explorative behavior (Figures 4B,C). No significant differences in the ASR were observed between Dars1D367Y/− and Dars1D367Y/+ mice (Figure 4D). PPI, on the other hand, was significantly lower in Dars1D367Y/− mice following the 72 dB SPL pre-pulse and showed a trend toward a reduction for the 76 dB SPL and 80 dB SPL pre-pulses (Figure 4E).
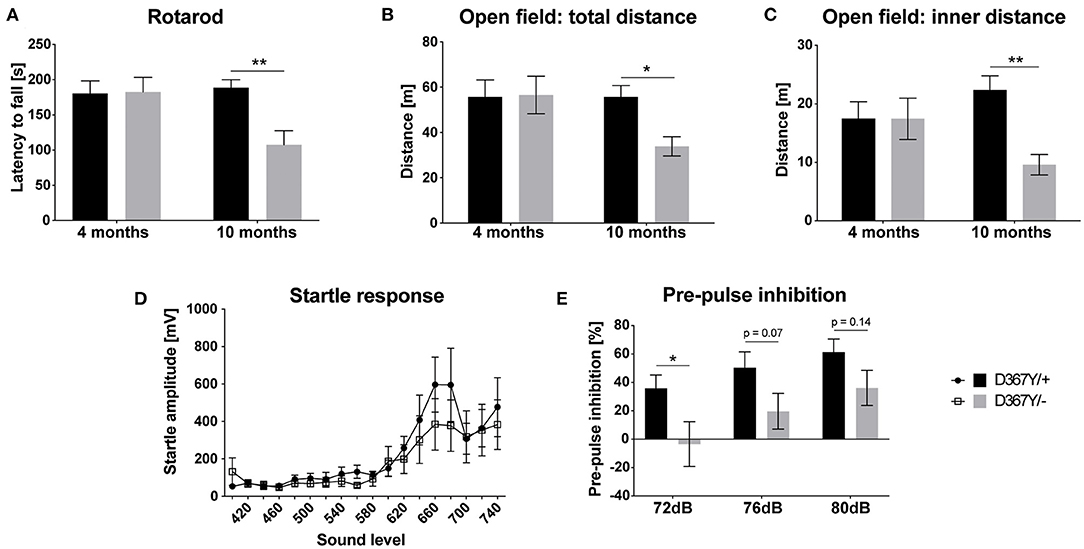
Figure 4. Compound heterozygous Dars1D367Y/− mice show impaired pre-pulse inhibition and develop late onset motor deficits. (A) Motor coordination assessed by the rotarod test was unchanged at 4 months but was impaired at 10 months in D367Y/– mice compared to D367Y/+ controls (n = 7–10). (B,C) Total distance traveled in an open field test apparatus [(B); n = 7–10] and distance in the inner compartment of the apparatus [(C); n = 7–10]. No changes were observed at 4 months but at 10 months total distance and distance in the inner compartment were significantly reduced. (D) Four-months-old mice were exposed to sound pulses with increasing intensities (60–120 dB SPL, 40 ms) and the acoustic startle response was measured. No significant differences were observed between D367Y/– and D367Y/+ mice (n = 8). (E) Pre-pulse inhibition (72, 76, or 80 dB SPL pre-pulse 100 ms before the 120 dB SPL startle pulse) was reduced in 4-months-old D367Y/– mice (n = 8). Data represent mean ± SEM (*p < 0.05, **p < 0.01; Two-way ANOVA).
Spinal Cord Abnormalities in Dars1D367Y/− Mice
In order to determine whether the behavioral deficits were accompanied by neurological changes, we analyzed central nervous system (CNS) integrity and myelination. Brain MRI revealed a smaller brain size of Dars1D367Y/− mice but did not show any other overt abnormalities or demyelination (Figure 5A). LFB staining of coronal brain sections revealed intact brain morphology and no myelination differences between genotypes (Figure 5B). In the spinal cord, however, severe vacuolization of the ventral white matter was detected in 10-months-old Dars1D367Y/− mice (Figure 5C) but not in Dars1D367Y/+ littermates. LFB staining confirmed white matter vacuolization in Dars1D367Y/− mice (Figure 5D). In addition, aged Dars1D367Y/− mice displayed severe demyelination of the lateral and dorsal white matter (Figure 5D).
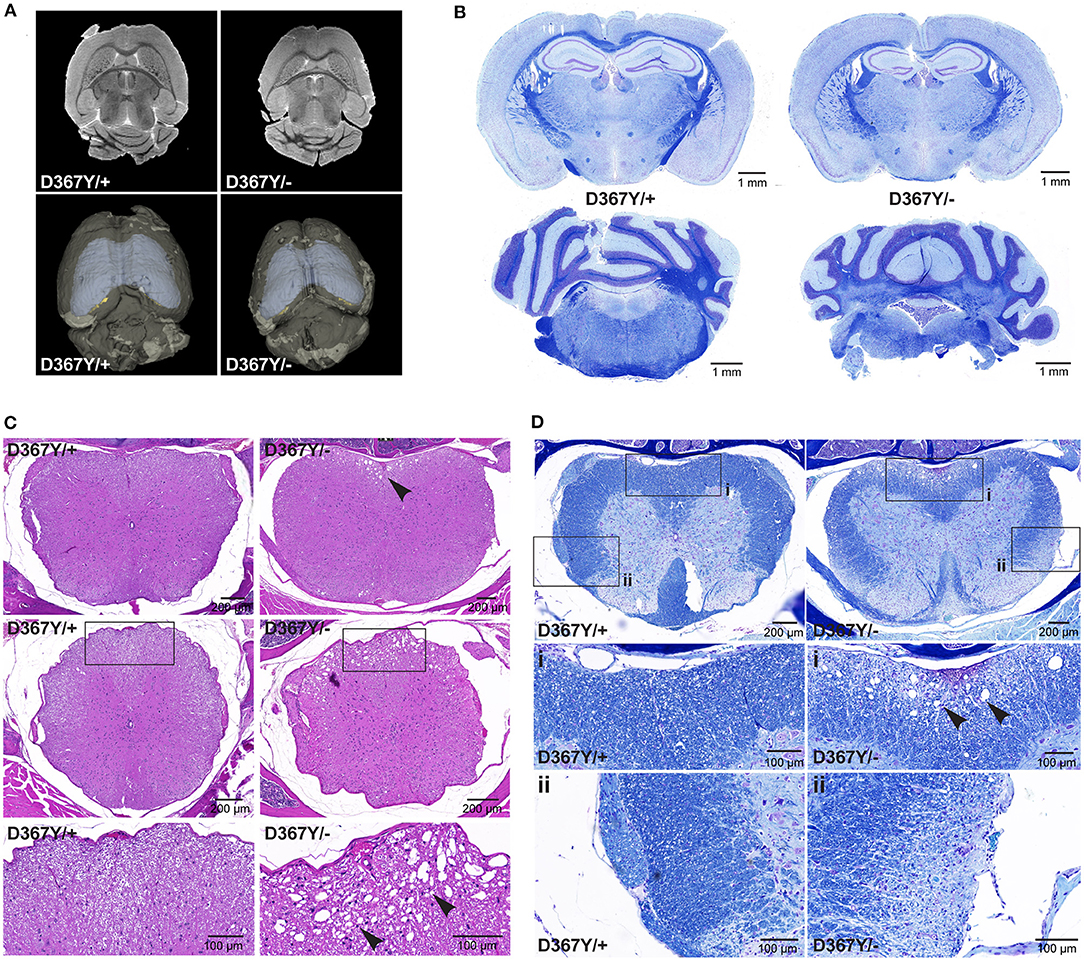
Figure 5. Compound heterozygous Dars1D367Y/− mice display spinal cord white matter abnormalities. (A) Brain magnetic resonance imaging (MRI) revealed an overall smaller brain size of D367Y/– mice but did not show any other abnormalities or demyelination. (B) Luxol fast blue (LFB) staining of coronal brain sections revealed no morphological or myelination differences between genotypes. (C) Hematoxylin and Eosin (H&E) staining of spinal cord cross sections (top row: lumbar; middle row: thoracic) revealed severe vacuolization of the ventral white matter in D367Y/– mice (arrowheads; n = 2). (D) LFB staining of lumbar spinal cord cross sections confirmed white matter vacuolization in D367Y/– mice [close-up (A), arrowheads] and revealed significant demyelination of the lateral and dorsal white matter [close-up (B); n = 2]. Analyses were performed in 10-months-old mice.
Reduced Expression of Major Myelin Proteins in Dars1D367Y/− Mice
To determine whether myelin defects could be corroborated by reduced expression levels of the major myelin proteins in Dars1D367Y/− mice, we analyzed brain tissue of 10-month-old Dars1D367Y/− and Dars1D367Y/+ mice. The brain regions analyzed included cortex, cerebellum, brainstem and basal ganglia. First, we confirmed reduction of Dars1 mRNA and AspRS protein in Dars1D367Y/− mice. As expected, Dars1 message and AspRS levels were reduced by 50% compared to Dars1D367Y/+ mice (Figure 6 top left and Figures 7A,B). mRNA levels of the mitochondrial counterpart Dars2 were unchanged ruling out potential compensatory mechanisms (Figure 6 top middle). Markers of oligodendrocytes (Aspa) or myelin (PLP, CNP and MBP) were reduced at the mRNA (Figure 6) and protein (Figure 7) level, in cerebellum and brainstem. While this trend reached statistical significance only in the brainstem, this finding suggests potential demyelination or oligodendrocyte loss in the hindbrain of Dars1D367Y/− mice.
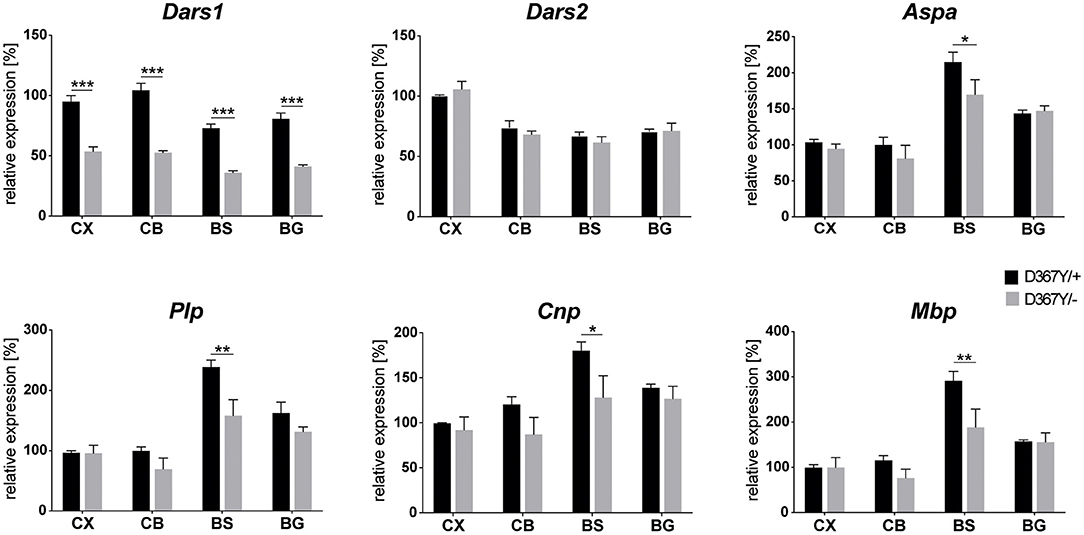
Figure 6. mRNA levels of Dars1 and the major myelin proteins are reduced in compound heterozygous Dars1D367Y/− mice. mRNA was extracted from brain tissue of 10-month-old D367Y/– and D367Y/+ mice and quantified via qRT-PCR using TaqMan probes specific for Dars1, Dars2, Aspa, Plp, Cnp, and Mbp. Brain regions analyzed included cortex (CX), cerebellum (CB), brainstem (BS), and basal ganglia (BG). Expression levels were normalized to the housekeeper GusB (n = 4). Data represent mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001; Two-way ANOVA).
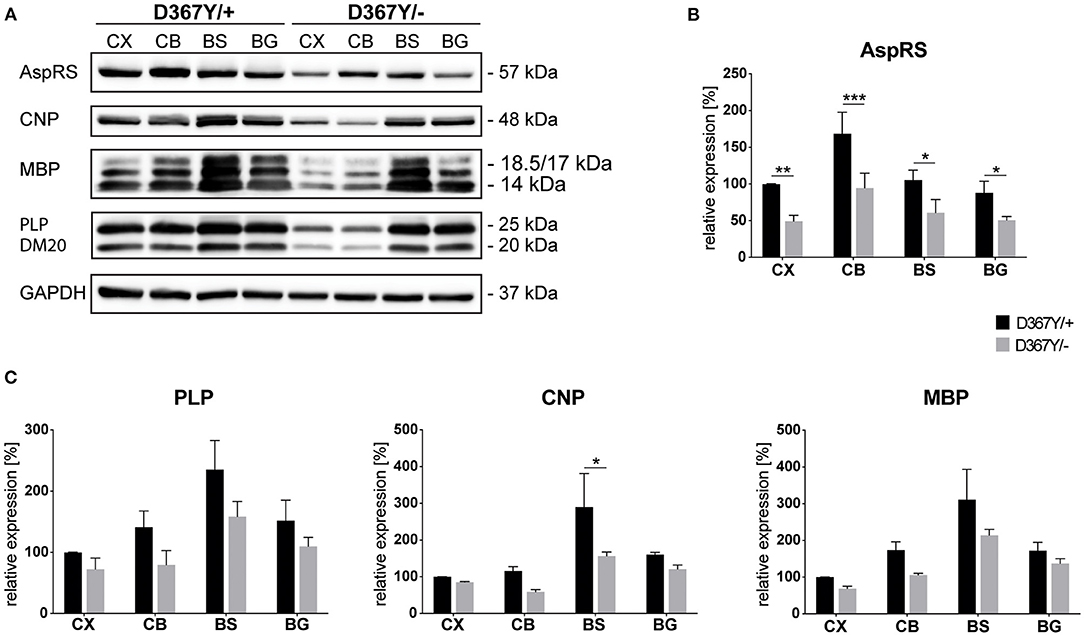
Figure 7. Expression levels of AspRS and the major myelin proteins in compound heterozygous Dars1D367Y/− mice. (A) Western-blot depicting AspRS, the myelin proteins CNP, MBP, and PLP/DM20, as well as the housekeeping protein GAPDH in different brain regions of 10-month-old D367Y/– and D367Y/+ mice. Brain regions include cortex (CX), cerebellum (CB), brainstem (BS), and basal ganglia (BG). (B) Relative AspRS expression normalized to GAPDH (n = 3). (C) Relative expression of the myelin proteins PLP, CNP, and MBP normalized to GAPDH (n = 3). Data represent mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001; Two-way ANOVA).
Discussion
To date, accurate HBSL models have been lacking, precluding examination of the underlying pathomechanism and testing of potential therapies. Here, we introduced the HBSL-causing Dars1D367Y point mutation into the mouse genome. Unexpectedly, mice carrying this mutation homozygously did not show any characteristic HBSL pathology. To enhance the phenotype of Dars1D367Y/D367Y mice, we crossed them with Dars1-null carriers. The Dars1D367Y/− offspring displayed a severe developmental delay associated with early lethality either in utero or as a result of hydrocephalus during the first 3 weeks of life. Another frequently observed feature of Dars1D367Y/− mice was microphthalmia in one or both eyes. All Dars1D367Y/− mice were underdeveloped with reduced body weight and fat mass compared to Dars1D367Y/+ littermates but remained otherwise phenotypically inconspicuous until 10 months of age when they developed late onset motor deficits as well as vacuolization and demyelination of the spinal cord white matter. These behavioral and morphological changes were accompanied by reduced expression levels of various myelin and oligodendrocyte markers, predominantly in the hindbrain of Dars1D367Y/− mice.
The A274V mutation resulted in a complete loss of aminoacylation activity, resembling a functional null mutation. In light of the lethal phenotype of homozygous null mice (Fröhlich et al., 2017) this suggests that a bi-allelic A274V mutation would not be viable in mouse or man. Intriguingly, the D367Y mutation did not decrease AspRS enzyme activity but instead led to a slight increase in activity. A potential explanation for the observed increase in enzymatic activity of Dars1D367Y could be reduced specificity of AspRS for tRNAAsp. As a consequence, other tRNAs would bind to AspRS and subsequently be mischarged with aspartate. These mischarged tRNAs would then incorporate the wrong amino acid into the growing polypeptide chain during translation ultimately resulting in either non-functional proteins or unfolded and misfolded proteins. An accumulation of un- and misfolded proteins in the endoplasmic reticulum (ER) can trigger the unfolded protein response (UPR), an adaptive pathway that under normal circumstances protects the cell from ER stress and restores normal ER function (Lin and Popko, 2009). If the UPR fails to cope with sustained ER stress, apoptosis is triggered to eliminate malfunctioning cells (Faitova et al., 2006; Szegezdi et al., 2006). This normally protective mechanism can become pathological and has been implicated in the pathophysiology of many neurodegenerative (Szegezdi et al., 2006) and white matter diseases such as Charcot-Marie-Tooth disease, Pelizaeus-Merzbacher disease, Vanishing White Matter disease, and multiple sclerosis (Lin and Popko, 2009). A similar underlying pathophysiological mechanism has been suggested for HBSL (Fröhlich et al., 2017) and is supported by data from this study. Accordingly, activation of the mitochondrial unfolded protein response (UPRmt) has been demonstrated in conditional Dars2 knockout mice (Dogan et al., 2014; Aradjanski et al., 2017). Dars2 encodes the mitochondrial aspartyl-tRNA synthetase (mt-AspRS) and mutations in the Dars2 gene result in leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL), which was first described in 2003 (van der Knaap et al., 2003; Scheper et al., 2007).
Patients affected by HBSL or LBSL display similar symptomology and MRI patterns involving the same CNS tracts and structures. While both diseases might share a common underlying disease mechanism, such as failure of the UPR and UPRmt due to an excessive accumulation of un- and misfolded proteins, there is no functional redundancy between AspRS and mt-AspRS. This has been demonstrated in Dars1 and Dars2 null mice, both displaying early embryonic lethality (Dogan et al., 2014; Fröhlich et al., 2017). HBSL and LBSL mainly affect the nervous system, a phenomenon that has also been observed in other ARS-related conditions (Fuchs et al., 2019). The majority of reported LBSL patients possess a splice site mutation in intron two of the DARS2 gene. This splice defect particularly affects neural cells providing an explanation for the CNS predilection seen in LBSL (van Berge et al., 2012). The selective vulnerability of neural cells in HBSL can be explained with the distinct AspRS expression pattern in the CNS of mice and humans (Fröhlich et al., 2017, Fröhlich et al., 2018). Accordingly, we observed CNS defects in Dars1D367Y/− mice including vacuolization and demyelination of the spinal cord white matter. White matter vacuolization is the most common myelin pathology and has been described for many leukodystrophies including Canavan's disease and Vanishing white matter disease (Duncan and Radcliff, 2016). When vacuolization occurs in conjunction with myelin degeneration it can be referred to as spongiform degeneration, however, myelin vacuolization can also occur independent of demyelination (Duncan and Radcliff, 2016). In the case of Dars1D367Y/− mice, the vacuolization in the spinal cord was accompanied by demyelination and might reflect a separation of myelin layers (Barkovich, 2000).
Protein synthesis is often regarded as a house-keeping function of all cells. Nevertheless, many aspects of protein synthesis are differently regulated across cell types and developmental stages in order to establish differences in cell identity, function and homeostasis (Buszczak et al., 2014). These differences might explain why Dars1D367Y/− mice are particularly susceptible to changes in protein synthesis during development resulting in developmental delay and in some cases premature death. Once Dars1D367Y/− mice made it through adolescence, the remaining AspRS activity seems to be sufficient to maintain normal tissue homeostasis. However, the effects of impaired protein synthesis seem to either accumulate over the lifetime or become more profound in aged mice, resulting in the observed late onset motor deficits and spinal cord abnormalities.
Many Dars1D367Y/− mice developed hydrocephalus during the first 3 weeks of life. Despite not being a frequently observed feature of HBSL, it has been described in one HBSL patient who consequently required ventriculo-peritoneal shunting to release pressure on the brain (Ong et al., 2020). Another commonly detected symptom of Dars1D367Y/− mice was severe microphthalmia or even anophthalmia, major structural malformations or even complete absence of the eye. These abnormalities were not only present in the born Dars1D367Y/− mice but also visible in 18-day-old Dars1D367Y/− embryos indicating that they occur early during embryogenesis as a result of impaired protein synthesis. The peripheral pathologies observed in Dars1D367Y/− mice, such as reduced size, body weight and fat mass as well as microphthalmia have not been reported for HBSL patients yet. While this might be due to the low number of cases reported to date, tests for peripheral deficits should be included in the clinical context in the future.
Homozygous Dars1D367Y/D367Y mice showed a lower ASR compared to wildtype mice, whereas Dars1D367Y/− mice displayed reduced PPI of the ASR. A PPI reduction is indicative of impaired sensorimotor gating mechanisms (Koch, 1999). The genetic cause of this impairment appears to be the Dars1-null mutation since reduced PPI was also a feature of heterozygous Dars1-null mice (Fröhlich et al., 2017). The ASR is mediated by a simple neuronal circuit of the lower brainstem involving neurons of the caudal pontine reticular nucleus (PnC) (Koch, 1999). PPI, on the other hand, involves a complex interplay of many brain areas ultimately resulting in a dampening of the ASR via the PnC. PPI is reduced in a variety of neurological disorders including schizophrenia, Huntington's disease, Tourette's syndrome, and ADHD (Swerdlow and Geyer, 1998; Koch, 1999) and, based on our results, might also be a feature of HBSL. Reduced PPI was present in severely affected Dars1D367Y/− mice as well as in mildly affected heterozygous Dars1-null mice (Fröhlich et al., 2017), suggesting to test for ASR and PPI in the clinical context as they might support an early diagnosis of HBSL.
While Dars1D367Y/− mice do not model every aspect of the clinical HBSL picture, they enable studies of both, early and late onset deficits, which so far was precluded in Dars1 knockout mice due to embryonic lethality. As such, mice harboring the hypomorphic Dars1D367Y allele represent the first tool enabling therapeutic proof-of-concept studies including nutraceutical L-ornithine-L-aspartate (LOLA) supplementation to boost AspRS activity (Das et al., 2020) or DARS1 gene replacement therapy. The introduction of other HBSL point mutations might produce a more accurate disease model with an early disease onset as seen in the majority of HBSL patients. Moreover, the use of a conditional knockout system will be instrumental in dissecting the contribution of Dars1-deficiency in select organs or cell lineages similar to what has been reported for Dars2 (Aradjanski et al., 2017; Nemeth et al., 2020; Rumyantseva et al., 2020).
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by University of New South Wales Animal Care and Ethics Committee.
Author Contributions
DF and MK designed the study. DF, MIM, AJK, MJH, GSS, and AB conducted the research. DF, MIM, and MK analyzed the data. GDH contributed to the experimental design and manuscript preparation. DF and MK led the project and the manuscript production. All authors read and approved the final manuscript.
Conflict of Interest
MK is an employee of Boehringer Ingelheim Pharma GmbH & Co. KG.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by the Medical Research Future Fund (MRFF-ARLKO), the European Leukodystrophy Association (ELA 2015-016I3 and ELA 2018-014I2) and the Mission Massimo Foundation. The MAGEC and the HOPS are supported by Phenomics Australia (PA; formerly known as Australian Phenomics Network) and PA is supported by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) program. We acknowledge the facilities, and the scientific and technical assistance of the Melbourne Advanced Genome Editing Center (MAGEC) at the Walter and Eliza Hall Institute of Medical Research (WEHI) and the Histopathology and Organ Pathology Service (HOPS), University of Melbourne. We acknowledge the National Imaging Facility (NIF), Australia, for the financial support of MR imaging instrumentation, and the scientific and technical assistance at the UNSW NIF node, Mark Wainwright Analytical Center, Biological Imaging Resources Laboratory. In this context, we also thank Mr. Brendan Lee for his help with image acquisitions.
References
Aradjanski, M., Dogan, S. A., Lotter, S., Wang, S., Hermans, S., Wibom, R., et al. (2017). DARS2 protects against neuroinflammation and apoptotic neuronal loss, but is dispensable for myelin producing cells. Hum. Mol. Genet. 26, 4181–4189. doi: 10.1093/hmg/ddx307
Barkovich, A. J. (2000). Concepts of myelin and myelination in neuroradiology. AJNR Am. J. Neuroradiol. 21, 1099–1109. Available online at: http://www.ajnr.org/content/21/6/1099.long
Bonkowsky, J. L., Nelson, C., Kingston, J. L., Filloux, F. M., Mundorff, M. B., and Srivastava, R. (2010). The burden of inherited leukodystrophies in children. Neurology 75, 718–725. doi: 10.1212/WNL.0b013e3181eee46b
Buszczak, M., Signer, R. A., and Morrison, S. J. (2014). Cellular differences in protein synthesis regulate tissue homeostasis. Cell 159, 242–251. doi: 10.1016/j.cell.2014.09.016
Das, A., Frohlich, D., Achanta, L. B., Rowlands, B. D., Housley, G. D., Klugmann, M., et al. (2020). L-aspartate, L-ornithine and L-ornithine-L-aspartate (LOLA) and their impact on brain energy metabolism. Neurochem. Res. 45, 1438–1450. doi: 10.1007/s11064-020-03044-9
Dogan, S. A., Pujol, C., Maiti, P., Kukat, A., Wang, S., Hermans, S., et al. (2014). Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 19, 458–469. doi: 10.1016/j.cmet.2014.02.004
Duncan, I. D., and Radcliff, A. B. (2016). Inherited and acquired disorders of myelin: the underlying myelin pathology. Exp. Neurol. 283 (Pt. B), 452–475. doi: 10.1016/j.expneurol.2016.04.002
Faitova, J., Krekac, D., Hrstka, R., and Vojtesek, B. (2006). Endoplasmic reticulum stress and apoptosis. Cell. Mol. Biol. Lett. 11, 488–505. doi: 10.2478/s11658-006-0040-4
Fröhlich, D., Suchowerska, A. K., Spencer, Z. H., von Jonquieres, G., Klugmann, C. B., Bongers, A., et al. (2017). In vivo characterization of the aspartyl-tRNA synthetase DARS: homing in on the leukodystrophy HBSL. Neurobiol. Dis. 97 (Pt. A), 24–35. doi: 10.1016/j.nbd.2016.10.008
Fröhlich, D., Suchowerska, A. K., Voss, C., He, R., Wolvetang, E., von Jonquieres, G., et al. (2018). Expression pattern of the aspartyl-tRNA synthetase DARS in the human brain. Front. Mol. Neurosci. 11:81. doi: 10.3389/fnmol.2018.00081
Fuchs, S. A., Schene, I. F., Kok, G., Jansen, J. M., Nikkels, P. G. J., van Gassen, K. L. I., et al. (2019). Aminoacyl-tRNA synthetase deficiencies in search of common themes. Genet. Med. 21, 319–330. doi: 10.1038/s41436-018-0048-y
Kikinis, R., Pieper, S. D., and Vosburgh, K. G. (2014). “3D slicer: a platform for subject-specific image analysis, visualization, and clinical support,” in Intraoperative Imaging and Image-Guided Therapy, eds F. Jolesz (New York, NY: Springer).
Kim, K. R., Park, S. H., Kim, H. S., Rhee, K. H., Kim, B. G., Kim, D. G., et al. (2013). Crystal structure of human cytosolic aspartyl-tRNA synthetase, a component of multi-tRNA synthetase complex. Proteins 81, 1840–1846. doi: 10.1002/prot.24306
Koch, M. (1999). The neurobiology of startle. Prog. Neurobiol. 59, 107–128. doi: 10.1016/S0301-0082(98)00098-7
Kueh, A. J., Pal, M., Tai, L., Liao, Y., Smyth, G. K., Shi, W., et al. (2017). An update on using CRISPR/Cas9 in the one-cell stage mouse embryo for generating complex mutant alleles. Cell Death Differ. 24, 1821–1822. doi: 10.1038/cdd.2017.122
Lin, W., and Popko, B. (2009). Endoplasmic reticulum stress in disorders of myelinating cells. Nat. Neurosci. 12, 379–385. doi: 10.1038/nn.2273
Muthiah, A., Housley, G. D., Klugmann, M., and Fröhlich, D. (2020). The leukodystrophies HBSL and LBSL - correlates and distinction. Front. Cell. Neurosci. doi: 10.3389/fncel.2020.626610
Nemeth, C. L., Tomlinson, S. N., Rosen, M., O'Brien, B. M., Larraza, O., Jain, M., et al. (2020). Neuronal ablation of mt-AspRS in mice induces immune pathway activation prior to severe and progressive cortical and behavioral disruption. Exp. Neurol. 326:113164. doi: 10.1016/j.expneurol.2019.113164
Ong, M. T., Willoughby, J., Connolly, D. J. A., Mordekar, S., Study, D., and Johnson, D. (2020). Genotype–phenotype variability of DARS mutation—case reports of a trio of siblings. Euro. J. Med. Case Rep. 4, 110–115. doi: 10.24911/ejmcr/1731551044010
Rumyantseva, A., Motori, E., and Trifunovic, A. (2020). DARS2 is indispensable for purkinje cell survival and protects against cerebellar ataxia. Hum. Mol. Genet. 29, 2845–2854. doi: 10.1093/hmg/ddaa176
Scheper, G. C., van der Klok, T., van Andel, R. J., van Berkel, C. G., Sissler, M., Smet, J., et al. (2007). Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 39, 534–539. doi: 10.1038/ng2013
Schneider, M., Spanagel, R., Zhang, S. J., Bading, H., and Klugmann, M. (2007). Adeno-associated virus. (AAV)-mediated suppression of Ca2+/calmodulin kinase IV activity in the nucleus accumbens modulates emotional behaviour in mice. BMC Neurosci. 8:105. doi: 10.1186/1471-2202-8-105
Swerdlow, N. R., and Geyer, M. A. (1998). Using an animal model of deficient sensorimotor gating to study the pathophysiology and new treatments of schizophrenia. Schizophr. Bull. 24, 285–301. doi: 10.1093/oxfordjournals.schbul.a033326
Szegezdi, E., Logue, S. E., Gorman, A. M., and Samali, A. (2006). Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885. doi: 10.1038/sj.embor.7400779
Taft, R. J., Vanderver, A., Leventer, R. J., Damiani, S. A., Simons, C., Grimmond, S. M., et al. (2013). Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am. J. Hum. Genet. 92, 774–780. doi: 10.1016/j.ajhg.2013.04.006
van Berge, L., Dooves, S., van Berkel, C. G., Polder, E., van der Knaap, M. S., and Scheper, G. C. (2012). Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation is associated with cell-type-dependent splicing of mtAspRS mRNA. Biochem. J. 441, 955–962. doi: 10.1042/BJ20110795
van der Knaap, M. S., van der Voorn, P., Barkhof, F., Van Coster, R., Krageloh-Mann, I., Feigenbaum, A., et al. (2003). A new leukoencephalopathy with brainstem and spinal cord involvement and high lactate. Ann. Neurol. 53, 252–258. doi: 10.1002/ana.10456
von Jonquieres, G., Mersmann, N., Klugmann, C. B., Harasta, A. E., Lutz, B., Teahan, O., et al. (2013). Glial promoter selectivity following AAV-delivery to the immature brain. PLoS ONE 8:e65646. doi: 10.1371/journal.pone.0065646
von Jonquieres, G., Spencer, Z. H. T., Rowlands, B. D., Klugmann, C. B., Bongers, A., Harasta, A. E., et al. (2018). Uncoupling N-acetylaspartate from brain pathology: implications for Canavan disease gene therapy. Acta Neuropathol. 135, 95–113. doi: 10.1007/s00401-017-1784-9
Keywords: HBSL, DARS1, AspRS, aminoacyl-tRNA synthetase, aspartyl-tRNA synthetase, leukodystrophy
Citation: Fröhlich D, Mendes MI, Kueh AJ, Bongers A, Herold MJ, Salomons GS, Housley GD and Klugmann M (2021) A Hypomorphic Dars1D367Y Model Recapitulates Key Aspects of the Leukodystrophy HBSL. Front. Cell. Neurosci. 14:625879. doi: 10.3389/fncel.2020.625879
Received: 04 November 2020; Accepted: 21 December 2020;
Published: 20 January 2021.
Edited by:
Rosanna Parlato, University of Heidelberg, GermanyReviewed by:
James N. Sleigh, University College London, United KingdomBilal Ersen Kerman, Istanbul Medipol University, Turkey
Copyright © 2021 Fröhlich, Mendes, Kueh, Bongers, Herold, Salomons, Housley and Klugmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominik Fröhlich, ZC5mcm9obGljaEB1bnN3LmVkdS5hdQ==; Matthias Klugmann, bS5rbHVnbWFubkB1bnN3LmVkdS5hdQ==