 Anthony Poerwoatmodjo
Anthony Poerwoatmodjo Geert J. Schenk
Geert J. Schenk Antonio Luchicchi
Antonio Luchicchi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Cell. Neurosci. , 01 December 2020
Sec. Cellular Neuropathology
Volume 14 - 2020 | https://doi.org/10.3389/fncel.2020.612383
This article is part of the Research Topic “Inside-out” vs “Outside-in” Paradigms in Multiple Sclerosis Etiopathogenesis View all 10 articles
Multiple sclerosis (MS) is a chronic neuroinflammatory disease of the central nervous system (CNS) of which the main pathological feature is the loss of white matter myelin (demyelination) (Ringold et al., 2005).
Although a solid cause to explain MS origin is still debated, a classic etiological view (named outside-in hypothesis of MS) regards this disorder as primarily mediated by a faulty CD4+ T-lymphocytic attack against the myelin (Lassmann and Ransohoff, 2004; Chitnis, 2007).
Despite its prevalence, this hypothesis has been recently questioned by an opposing view (inside-out hypothesis of MS) which posits that subtle primary cytodegenerative processes might happen in the CNS itself, leading to myelin disintegration and a secondary immune system response (Stys et al., 2012). For instance, studies have shown that MS pathological correlates, like widening of myelin lamellae and oligodendrocyte apoptosis, are often present in regions separated form inflammation foci (Barnett and Prineas, 2004; Henderson et al., 2009), and that loss of inner myelin sheath-expressed proteins (like the myelin associated glycoprotein, MAG) temporarily precedes that of outer-sheath expressed molecules in newly forming lesions (Aboul-Enein et al., 2003). Furthermore, clinical observations have recurrently reported that immunosuppressant agents, elective to treat relapsing-remitting forms of MS, are largely ineffective in halting less inflammatory progressive forms of the disease (Stys et al., 2012).
Although a solid sequence of events is still elusive, one putative condition to explain this primary myelin degeneration in MS may be the subtle imbalance at the level of the axon-myelinic synapse (AMS, Figure 1A; Micu et al., 2018). The AMS is a recently proposed form of glutamate-mediated communication between axon and myelin which regulates axonal myelination and action potential propagation via n-methyl-d-aspartate (NMDA) receptor-dependent myelin Ca2+ supply (Figure 1A, 1–3; Micu et al., 2018). Briefly, controlled myelinic Ca2+ influx takes part in glycolysis processes which lead to pyruvate and lactate production (Figure 1A, 4). Lactate is then back-transported to the axon where it boosts energy production during electric activity (Figure 1A, 5).
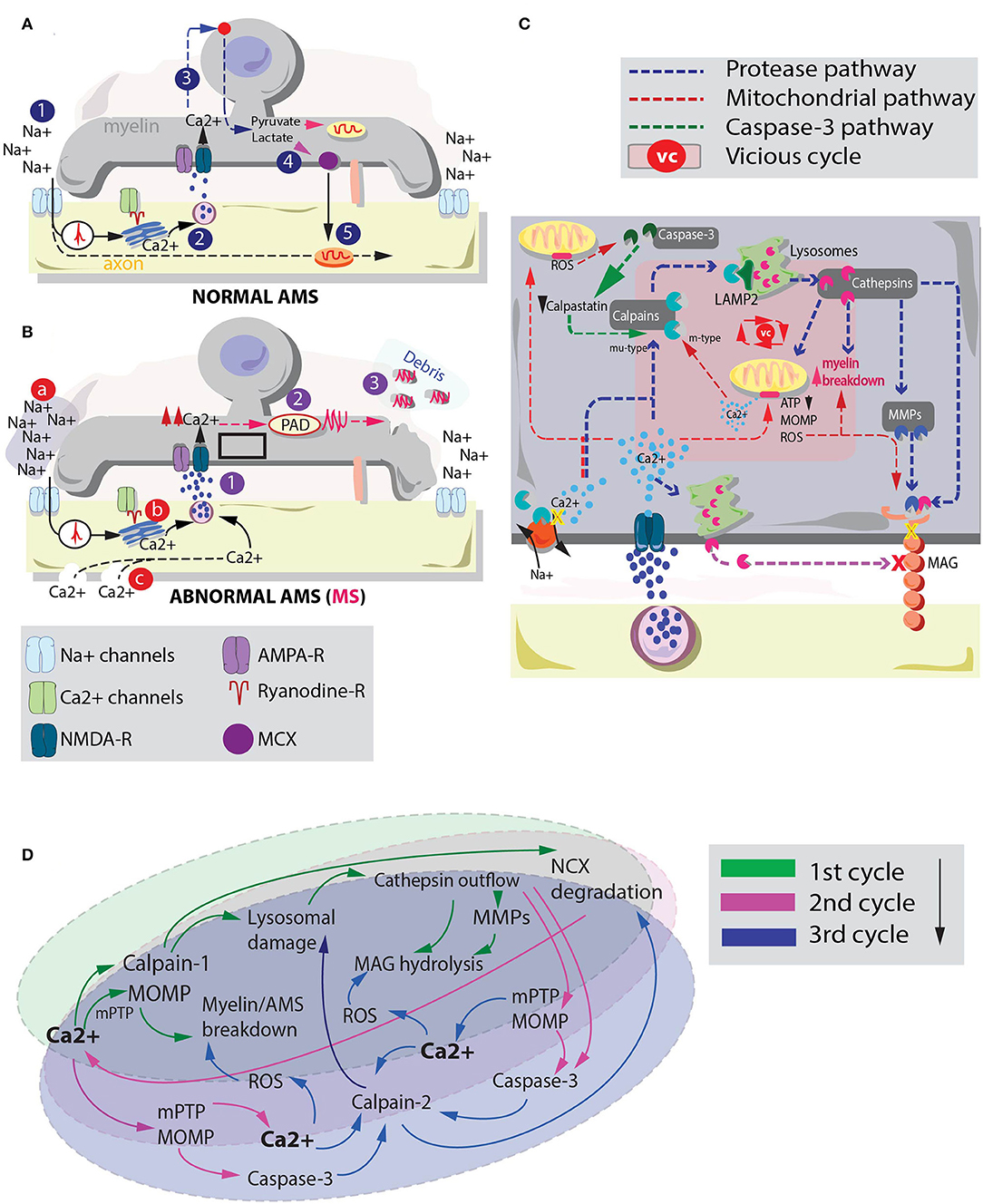
Figure 1. Interplay between cysteine protease activation and mitochondrial deteriorating processes in MS myelin degeneration. (A) graphical depiction of the AMS in physiological conditions (Micu et al., 2018). The action potential (1) is sensed by Ca2+ channels which via a ryanodine receptor-dependent mechanism mobilize the axoplasmic Ca2+ reserves. Ca2+ triggers vesicle fusion and glutamate release (2). Glutamate activates NMDA-Rs promoting myelinic Ca2+ influx and glycolysis processes. This sequence of events leads to pyruvate and lactate production (3). Lactate is ultimately transported to the axon to metabolically assist axonal mitochondria. (B) In pathological conditions, excessive Ca2+ myelinic influx is promoted via altered stimulation of NMDA receptors (Micu et al., 2018). Possible players involved in such effect are: (a) an altered Na+ homeostasis, where augmented extracellular Na+ may lead to intra-axonal aberrant processes that promote Ca2+ influx (Craner et al., 2004); (b) altered Ca2+ mobilization inside the axoplasmic reticulum (Micu et al., 2018) and (c) the presence of subtle axonal nanoruptures which make axonal cylinder permeable to Ca2+ (Witte et al., 2019). Irrespective of the mechanism by which altered Ca2+ entrance in the myelin is triggered, such phenomenon (1) may lead to a consequent activation of PADs (2). PADs citrullinate myelin proteins (e.g., MBP). Myelin degenerative processes together with citrullination of MBP promote biochemically altered (highly immunogenic) debris formation (3). (C) Summary of the principal pathways by which cysteine proteases (blue pathway) and dysregulated mitochondria (red pathway) can affect myelin and MAG stability. These processes and their interplay may be responsible of myelin and axonal breakdown in MS. Pink box inside myelin depicts the players of a possible vicious cycle between cysteine protease activation and mitochondrial instability in MS-related myelin breakdown. (D) Schematic representation of a possible vicious cycle activated by enhanced myelinic Ca2+ entrance. Primary Ca2+ entrance instigates a parallel activation of mu-type-calpains (calpain-1) and mitochondria degenerative processes. Secondary Ca2+ accumulation due to Na+/Ca2+ exchanger (NCX) degradation and Ca2+ efflux from mitochondria might take part in a tertiary activation of m-type calpains (calpain-2) which can exacerbate lysosomal and mitochondria instability leading to heightened myelin damage.
Consequently, alterations at the level of the AMS, triggered by aberrant stimuli like dysfunctional Ca2+ mobilization from the axoplasmic reticulum (Micu et al., 2018), Na+ homeostasis alterations (Inglese et al., 2010) and/or the pathological presence of axonal nanoruptures (Witte et al., 2019) (Figure 1B, a–c) may instigate an augmented myelinic Ca2+ entrance. As elegantly shown in studies employing cuprizone mouse models of MS, this condition is responsible of many aberrant biochemical processes relevant for myelin stability including the citrullination of the myelin basic protein (MBP) (Caprariello et al., 2018). Citrullination is a posttranslational modification process instigated by the activation of the enzyme protein arginine deiminase (PAD) that promotes the formation of highly immunogenic myelin (Figure 1B, 1–3).
Several parallel aberrant processes may lead citrullinated myelin to degrade. Among them, the activation of cysteine proteases like the Ca2+-dependent non-lysosomal protease calpain and the lysosomal Ca2+-independent cathepsin might very well-recapitulate MS myelin degenerative processes.
Calpain and cathepsin play a role in apoptotic processes (Leist and Jaattela, 2001; Kroemer and Jaattela, 2005), Na+/Ca2+ exchange pump/actin cytoskeleton degradation (McConkey, 1998; Zong and Thompson, 2006), and myelin retraction from the node of Ranvier (Baraban et al., 2018). Moreover, studies on ischemia and sporadic Creutzfeldt-Jakob disease have shown that, under specific circumstances (e.g., aberrant cellular Ca2+ influx), these two proteases engage into a sequential activation named “calpain-cathepsin axis” where calpain promotes a cleavage of lysosomal associated membrane 2 (LAMP2). As a result, a programmed cell death through lysosomal cathepsin leakage is activated (Miyazawa et al., 1998; Zong and Thompson, 2006; Villalpando Rodriguez and Torriglia, 2013; Llorens et al., 2017).
This axial activation, combined with the hypothesized role of cathepsin in degrading the MAG (Stebbins et al., 1997), an inner-sheath adhesion molecule important for axon-myelinic stability (Trapp and Quarles, 1982; Pronker et al., 2016), may explain the primary degenerative processes involved in MS-related AMS instability and myelin disintegration.
Despite this, whether an aberrant cysteine protease activation is relevant and alone sufficient to explain demyelination in MS is still not understood.
In this perspective article we evaluate the possibility that a disruptive activation of the calpain-cathepsin axis in MS myelin can be reinforced by the activation of parallel Ca2+-dependent aberrant events to induce primary myelin degeneration.
One that seems very relevant for MS is the cascade generated by Ca2+-dependent mitochondrial dysfunctions (Nicholls, 2009). Mitochondria instability is not a new concept in MS pathology (Witte et al., 2014). In particular, Ca2+-mediated aberrant events in the mitochondria, such as the opening of the mitochondria permeability transition pore (mPTP), trigger the release of intramembrane proteins (like cytochrome c) which, in turn, activate an apoptotic process named mitochondrial outer-membrane permeabilization (MOMP) (Ichas and Mazat, 1998).
Interestingly, cysteine protease activation and mitochondria pathological events share a number of common pathways. The activation of these pathways might instigate a “vicious cycle,” strongly contributing to explain MS-related myelin degeneration. Here we propose that in MS, following excessive myelinic Ca2+ influx, calpain-cathepsin axis activation and MOMP play a synergic role in AMS destabilization and MAG degradation.
Cysteine proteases actively promote protein catabolism inside the cellular compartment (Verma et al., 2016). Among them, calpain co-exists in two main isoforms: calpain-1 (mu-type) and calpain-2 (m-type, which requires higher Ca2+ concentrations to be activated) (Yamashima, 2004; Potz et al., 2016). Similarly, cathepsins are present in different forms (Turk et al., 2012).
Besides their role in cellular degeneration, some of these proteases seem particularly relevant for MS pathology. For instance, the L form of cathepsin is thought to play a role in MAG hydrolysis, truncating this molecule into a less functional version named dMAG (Stebbins et al., 1997). Notably, a selective MAG loss is spotted in the lesion formation in several neurodegenerative diseases including Kearns-Sayre syndrome (Lax et al., 2012) and MS (Aboul-Enein et al., 2003). Therefore, a dMAG formation operated by a calpain-cathepsin axis activation may hold important consequences for AMS stability, recapitulating the dynamic of AMS destabilization/myelin-axonal degeneration in MS.
Although the biochemical steps that lead to MAG degradation are not entirely understood (Paivalainen et al., 2003), studies have proposed that cathepsin-dependent MAG truncation may happen via different pathways. One implies a direct effect of cysteine proteases on MAG intracellular domains (Stebbins et al., 1997), while another mechanism operates via an indirect Ca2+-dependent lysosomal fusion with the plasma membrane. The latter promotes cathepsin secretion through vesicular exocytosis (Rodriguez et al., 1997; Hashimoto et al., 2015). Finally, cathepsins are also shown to activate matrix metalloproteases (MMPs) (Milward et al., 2008). This third pathway may influence MAG stability through an extracellular matrix degradation and increase in myelin motility (Kroemer and Jaattela, 2005; Gu et al., 2015). Altered myelin motility may profoundly affect the ability of axon and myelin to establish a rigidly regulated point-to-point synapse (Micu et al., 2018), hampering the metabolic coupling they share (Beirowski et al., 2014; Micu et al., 2018; Figure 1C).
Mitochondria dysfunctionalities may corroborate the effects of these proteases on MS myelin. For instance, MOMP is shown to induce a cascade of deteriorating events such as those catalyzed by caspase-9 and−3 (Parsons and Green, 2010). Additionally, due to the role of mitochondria in Ca2+ storage, instability at the level of these organelles is able to instigate a detrimental extra-mitochondrial Ca2+ release (Montero et al., 2001). The latter effect might add up to the effects caused by augmented NMDAR-dependent myelinic Ca2+ influx.
On top of these processes, mitochondria respiratory chain defects are thought to cause MAG loss in relatively non-inflammatory environments. For instance, studies have shown that MAG loss is associated with prominent nuclear expression of HIF-1α, a marker for hypoxia-like metabolic tissue injury. HIF-1α can be induced by either mitochondrial increase in intracellular reactive oxygen species (ROS) production (Aboul-Enein et al., 2003) or by impaired mitochondrial respiratory chain functions (Semenza, 2000). Finally, studies on Kearns-Sayre syndrome reported primary MAG loss and consequent demyelination (Lax et al., 2012). The onset of this disorder is thought to be due to primary mitochondrial respiratory chain defects as a result of a single mtDNA deletion. Therefore, it might be possible that also in MS mitochondrial instability instigates MAG loss with consequent induction of AMS instability/myelin degeneration.
Aberrant myelin Ca2+ influx in the AMS may be an essential trigger in MS (Micu et al., 2018). Although a clear sequence of events is still ignored, Ca2+ dysregulations are a potent source of parallel biochemical processes that may synergistically partake in degenerative conditions. This interplay might generate a vicious cycle where Ca2+-mediated events reinforce each other, explaining complex structural and biochemical alterations like those observed in MS brains. Interestingly, in the case of cysteine proteases and mitochondria dysfunctionality, both processes are crucially involved in degeneration/apoptosis (McConkey, 1998; Festjens et al., 2006; Zong and Thompson, 2006). Therefore, a combined activation of these pathways would likely explain the consequent highly immunogenic myelin fragmentation observed e.g., in cuprizone mouse models (Caprariello et al., 2018).
As one of the roles of the AMS is to supply the axonal compartment with lactate for metabolic purposes, a cysteine protease/mitochondrial-dependent myelin breakup can induce a secondary stage of axonal virtual hypoxia leading to axon disintegration (Stys et al., 2012; Micu et al., 2018).
Cysteine proteases and mitochondria might hypothetically interact in several ways to decide the fate of myelin in MS. Following an initial myelinic Ca2+ increase a primary co-activation of calpain-1 and mPTP/MOMP might occur (Figures 1C,D). These events may instigate lysosomal cathepsin outflow and primitive myelin breakdown, respectively (Figure 1D, 1st cycle). Elevated levels of cathepsins can either directly or indirectly affect MAG stability (Rodriguez et al., 1997; Stebbins et al., 1997; Milward et al., 2008; Hashimoto et al., 2015). Furthermore, simultaneously altered level of calpains can promote a cleavage of the Na+/Ca2+ exchange pump (NCX) (McConkey, 1998; Zong and Thompson, 2006). The latter phenomenon facilitates an additional intracellular Ca2+ accumulation which together with the ability of cathepsin-L and D to activate caspase-3 (Li et al., 1997; Yamashima, 2000) and MOMP (Kroemer and Jaattela, 2005) might greatly reinforce the cysteine protease/mitochondria vicious cycle (Figure 1D, 2nd cycle). In fact, caspase-3 is thought to be able to cleave calpastatin, an endogenous calpain inhibitor (Porn-Ares et al., 1998; Yamashima, 2004), instigating a protracted permanence of calpains in the myelinic compartment. Cleavage of calpastatin together with mPTP-dependent mitochondria Ca2+ release (De Marchi et al., 2014) might promote the activation and accumulation of other forms of degenerative calpains, like calpain-2, which require Ca2+ in the mM range to be activated (Baudry and Bi, 2016). Finally, parallel mitochondria disruptions caused by Ca2+ overload can induce ROS enhancement (Festjens et al., 2006) strongly adding up to the ongoing AMS instability, myelin breakdown and MAG degradation (Figure 1D, 3rd cycle and beyond).
In this opinion article we proposed a scenario where a primary altered AMS stability might explain myelin breakdown and axonal degeneration in MS via Ca2+-mediated aberrant events (Micu et al., 2018). Possible instigators of such processes are the cysteine proteases and mitochondria dysfunctionalities. Here we posit that these two players reinforce each other leading to a possible vicious cycle which may hold important consequences for myelin stability. To confirm this hypothesis rigidly controlled experiments should define whether an actual interplay between the activation of the calpain-cathepsin axis and MOMP exists and, in this case, whether one pathway temporarily precedes the other one.
AP, GS, and AL: conceived the study. AP: performed the literature research. GS and JG: provided critical revision to the manuscript. AP and AL: wrote the manuscript with input from all the authors.
This study was supported by MS Research Stichting (pilot project number 16-954 MS), and Ammodo KNAW award (2017) awarded to JG.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aboul-Enein, F., Rauschka, H., Kornek, B., Stadelmann, C., Stefferl, A., Bruck, W., et al. (2003). Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J. Neuropathol. Exp. Neurol. 62, 25–33. doi: 10.1093/jnen/62.1.25
Baraban, M., Koudelka, S., and Lyons, D. A. (2018). Ca (2+) activity signatures of myelin sheath formation and growth in vivo. Nat. Neurosci. 21, 19–23. doi: 10.1038/s41593-017-0040-x
Barnett, M. H., and Prineas, J. W. (2004). Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann. Neurol. 55, 458–468. doi: 10.1002/ana.20016
Baudry, M., and Bi, X. (2016). Calpain-1 and calpain-2: the yin and yang of synaptic plasticity and neurodegeneration. Trends Neurosci. 39, 235–245. doi: 10.1016/j.tins.2016.01.007
Beirowski, B., Babetto, E., Golden, J. P., Chen, Y. J., Yang, K., Gross, R. W., et al. (2014). Metabolic regulator LKB1 is crucial for Schwann cell-mediated axon maintenance. Nat. Neurosci. 17, 1351–1361. doi: 10.1038/nn.3809
Caprariello, A. V., Rogers, J. A., Morgan, M. L., Hoghooghi, V., Plemel, J. R., Koebel, A., et al. (2018). Biochemically altered myelin triggers autoimmune demyelination. Proc. Natl. Acad. Sci. U.S.A. 115, 5528–5533. doi: 10.1073/pnas.1721115115
Chitnis, T. (2007). The role of CD4 T cells in the pathogenesis of multiple sclerosis. Int. Rev. Neurobiol. 79, 43–72. doi: 10.1016/S0074-7742(07)79003-7
Craner, M. J., Hains, B. C., Lo, A. C., Black, J. A., and Waxman, S. G. (2004). Co-localization of sodium channel Nav1.6 and the sodium-calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain 127, 294–303. doi: 10.1093/brain/awh032
De Marchi, E., Bonora, M., Giorgi, C., and Pinton, P. (2014). The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium 56, 1–13. doi: 10.1016/j.ceca.2014.03.004
Festjens, N., Vanden Berghe, T., and Vandenabeele, P. (2006). Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta 1757, 1371–1387. doi: 10.1016/j.bbabio.2006.06.014
Gu, Y. H., Kanazawa, M., Hung, S. Y., Wang, X., Fukuda, S., Koziol, J. A., et al. (2015). Cathepsin L acutely alters microvessel integrity within the neurovascular unit during focal cerebral ischemia. J. Cereb. Blood Flow Metab. 35, 1888–1900. doi: 10.1038/jcbfm.2015.170
Hashimoto, Y., Kondo, C., and Katunuma, N. (2015). An active 32-kDa cathepsin L is secreted directly from HT 1080 fibrosarcoma cells and not via lysosomal exocytosis. PLoS ONE 10:e0145067. doi: 10.1371/journal.pone.0145067
Henderson, A. P., Barnett, M. H., Parratt, J. D., and Prineas, J. W. (2009). Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann. Neurol. 66, 739–753. doi: 10.1002/ana.21800
Ichas, F., and Mazat, J. P. (1998). From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta 1366, 33–50. doi: 10.1016/S0005-2728(98)00119-4
Inglese, M., Madelin, G., Oesingmann, N., Babb, J. S., Wu, W., Stoeckel, B., et al. (2010). Brain tissue sodium concentration in multiple sclerosis: a sodium imaging study at 3 tesla. Brain 133, 847–857. doi: 10.1093/brain/awp334
Kroemer, G., and Jaattela, M. (2005). Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 5, 886–897. doi: 10.1038/nrc1738
Lassmann, H., and Ransohoff, R. M. (2004). The CD4-Th1 model for multiple sclerosis: a critical [correction of crucial] re-appraisal. Trends Immunol. 25, 132–137. doi: 10.1016/j.it.2004.01.007
Lax, N. Z., Campbell, G. R., Reeve, A. K., Ohno, N., Zambonin, J., Blakely, E. L., et al. (2012). Loss of myelin-associated glycoprotein in kearns-sayre syndrome. Arch. Neurol. 69, 490–499. doi: 10.1001/archneurol.2011.2167
Leist, M., and Jaattela, M. (2001). Triggering of apoptosis by cathepsins. Cell Death Differ. 8, 324–326. doi: 10.1038/sj.cdd.4400859
Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S. M., Ahmad, M., Alnemri, E. S., et al. (1997). Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489. doi: 10.1016/S0092-8674(00)80434-1
Llorens, F., Thune, K., Sikorska, B., Schmitz, M., Tahir, W., Fernandez-Borges, N., et al. (2017). Altered Ca(2+) homeostasis induces calpain-cathepsin axis activation in sporadic creutzfeldt-jakob disease. Acta Neuropathol. Commun. 5:35. doi: 10.1186/s40478-017-0431-y
McConkey, D. J. (1998). Biochemical determinants of apoptosis and necrosis. Toxicol. Lett. 99, 157–168. doi: 10.1016/S0378-4274(98)00155-6
Micu, I., Plemel, J. R., Caprariello, A. V., Nave, K. A., and Stys, P. K. (2018). Axo-myelinic neurotransmission: a novel mode of cell signalling in the central nervous system. Nat. Rev. Neurosci. 19, 49–58. doi: 10.1038/nrn.2017.128
Milward, E., Kim, K. J., Szklarczyk, A., Nguyen, T., Melli, G., Nayak, M., et al. (2008). Cleavage of myelin associated glycoprotein by matrix metalloproteinases. J. Neuroimmunol. 193, 140–148. doi: 10.1016/j.jneuroim.2007.11.001
Miyazawa, T., Matsumoto, K., Ohmichi, H., Katoh, H., Yamashima, T., and Nakamura, T. (1998). Protection of hippocampal neurons from ischemia-induced delayed neuronal death by hepatocyte growth factor: a novel neurotrophic factor. J. Cereb. Blood Flow Metab. 18, 345–348. doi: 10.1097/00004647-199804000-00001
Montero, M., Alonso, M. T., Albillos, A., Garcia-Sancho, J., and Alvarez, J. (2001). Mitochondrial Ca(2+)-induced Ca(2+) release mediated by the Ca(2+) uniporter. Mol. Biol. Cell. 12, 63–71. doi: 10.1091/mbc.12.1.63
Nicholls, D. G. (2009). Mitochondrial calcium function and dysfunction in the central nervous system. Biochim. Biophys. Acta 1787, 1416–1424. doi: 10.1016/j.bbabio.2009.03.010
Paivalainen, S., Suokas, M., Lahti, O., and Heape, A. M. (2003). Degraded myelin-associated glycoprotein (dMAG) formation from pure human brain myelin-associated glycoprotein (MAG) is not mediated by calpain or cathepsin L-like activities. J. Neurochem. 84, 533–545. doi: 10.1046/j.1471-4159.2003.01539.x
Parsons, M. J., and Green, D. R. (2010). Mitochondria in cell death. Essays Biochem. 47, 99–114. doi: 10.1042/bse0470099
Porn-Ares, M. I., Samali, A., and Orrenius, S. (1998). Cleavage of the calpain inhibitor, calpastatin, during apoptosis. Cell Death Differ. 5, 1028–1033. doi: 10.1038/sj.cdd.4400424
Potz, B. A., Abid, M. R., and Sellke, F. W. (2016). Role of calpain in pathogenesis of human disease processes. J. Nat. Sci. 2:e218.
Pronker, M. F., Lemstra, S., Snijder, J., Heck, A. J., Thies-Weesie, D. M., Pasterkamp, R. J., et al. (2016). Structural basis of myelin-associated glycoprotein adhesion and signalling. Nat. Commun. 7:13584. doi: 10.1038/ncomms13584
Ringold, S., Lynm, C., and Glass, R. M. (2005). JAMA patient page. Multiple sclerosis. JAMA 293:514. doi: 10.1001/jama.293.4.514
Rodriguez, A., Webster, P., Ortego, J., and Andrews, N. W. (1997). Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell. Biol. 137, 93–104. doi: 10.1083/jcb.137.1.93
Semenza, G. L. (2000). Surviving ischemia: adaptive responses mediated by hypoxia-inducible factor 1. J. Clin. Invest. 106, 809–812. doi: 10.1172/JCI11223
Stebbins, J. W., Jaffe, H., Fales, H. M., and Moller, J. R. (1997). Determination of a native proteolytic site in myelin-associated glycoprotein. Biochemistry 36, 2221–2226. doi: 10.1021/bi962385x
Stys, P. K., Zamponi, G. W., Van Minnen, J., and Geurts, J. J. (2012). Will the real multiple sclerosis please stand up? Nat. Rev. Neurosci. 13, 507–514. doi: 10.1038/nrn3275
Trapp, B. D., and Quarles, R. H. (1982). Presence of the myelin-associated glycoprotein correlates with alterations in the periodicity of peripheral myelin. J. Cell. Biol. 92, 877–882. doi: 10.1083/jcb.92.3.877
Turk, V., Stoka, V., Vasiljeva, O., Renko, M., Sun, T., Turk, B., et al. (2012). Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim. Biophys. Acta 1824, 68–88. doi: 10.1016/j.bbapap.2011.10.002
Verma, S., Dixit, R., and Pandey, K. C. (2016). Cysteine proteases: modes of activation and future prospects as pharmacological targets. Front. Pharmacol. 7:107. doi: 10.3389/fphar.2016.00107
Villalpando Rodriguez, G. E., and Torriglia, A. (2013). Calpain 1 induce lysosomal permeabilization by cleavage of lysosomal associated membrane protein 2. Biochim. Biophys. Acta 1833, 2244–2253. doi: 10.1016/j.bbamcr.2013.05.019
Witte, M. E., Mahad, D. J., Lassmann, H., and Van Horssen, J. (2014). Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol. Med. 20, 179–187. doi: 10.1016/j.molmed.2013.11.007
Witte, M. E., Schumacher, A. M., Mahler, C. F., Bewersdorf, J. P., Lehmitz, J., Scheiter, A., et al. (2019). Calcium influx through plasma-membrane nanoruptures drives axon degeneration in a model of multiple sclerosis. Neuron 101, 615–624.e615. doi: 10.1016/j.neuron.2018.12.023
Yamashima, T. (2000). Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog. Neurobiol. 62, 273–295. doi: 10.1016/S0301-0082(00)00006-X
Yamashima, T. (2004). Ca2+-dependent proteases in ischemic neuronal death: a conserved 'calpain-cathepsin cascade' from nematodes to primates. Cell Calcium 36, 285–293. doi: 10.1016/j.ceca.2004.03.001
Keywords: multiple sclerosis, axo-myelinic synapse, cysteine proteases, mitochondria, myelin and axon breakdown, myelin associated glycoprotein
Citation: Poerwoatmodjo A, Schenk GJ, Geurts JJG and Luchicchi A (2020) Cysteine Proteases and Mitochondrial Instability: A Possible Vicious Cycle in MS Myelin? Front. Cell. Neurosci. 14:612383. doi: 10.3389/fncel.2020.612383
Received: 30 September 2020; Accepted: 12 November 2020;
Published: 01 December 2020.
Edited by:
Egor Dzyubenko, Essen University Hospital, GermanyReviewed by:
Tim Vanmierlo, University of Hasselt, BelgiumCopyright © 2020 Poerwoatmodjo, Schenk, Geurts and Luchicchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Luchicchi, YS5sdWNoaWNjaGlAYW1zdGVyZGFtdW1jLm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.