 Daniel C. Shippy
Daniel C. Shippy Tyler K. Ulland
Tyler K. Ulland
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci. , 18 September 2020
Sec. Non-Neuronal Cells
Volume 14 - 2020 | https://doi.org/10.3389/fncel.2020.563446
This article is part of the Research Topic The Metabolism of the Neuron-Glia Unit View all 8 articles
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by amyloid-β (Aβ) plaques and the formation of neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau. In response to Aβ and tau aggregates, microglia, the primary innate immune cells of the central nervous system (CNS), facilitate Aβ and tau clearance and contribute to neuroinflammation that damages neurons. Microglia also perform a wide range of other functions, e.g., synaptic pruning, within the CNS that require a large amount of energy. Glucose appears to be the primary energy source, but microglia can utilize several other substrates for energy production including other sugars and ketone bodies. Recent studies have demonstrated that changes in the metabolic profiles of immune cells, including macrophages, are important in controlling their activation and effector functions. Additional studies have focused on the role of metabolism in neuron and astrocyte function while until recently microglia metabolism has been considerably less well understood. Considering many neurological disorders, such as neurodegeneration associated with AD, are associated with chronic inflammation and alterations in brain energy metabolism, it is hypothesized that microglial metabolism plays a significant role in the inflammatory responses of microglia during neurodegeneration. Here, we review the role of microglial immunometabolism in AD.
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder associated with memory loss and impaired cognitive abilities. AD is a major cause of disability and dependency in the United States and worldwide, causing a significant impact on not only the individual patient, but also their family, community, and the healthcare system (Collaborators, 2019). To date, no effective treatment for AD exists, so advances in our understanding of AD neuropathology, and the associated immune responses, are necessary to develop therapeutic strategies to combat AD.
AD neuropathology is a complex process with several key features. Macroscopically, the AD brain displays cortical atrophy mostly affecting the medial temporal lobes (Serrano-Pozo et al., 2011). On the cellular level, AD is characterized by the accumulation of extracellular amyloid-β (Aβ) plaques followed by the formation of intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau (p-tau) resulting in synapse loss (Holtzman et al., 2011). In response to the accumulation of Aβ plaques and NFTs, microglia are activated and facilitate Aβ and tau clearance in addition to inducing a neuroinflammatory response that damages neurons suggesting a delicate balance between a beneficial, detrimental, or mixed microglia reaction to AD progression (Lue et al., 2010; Leyns and Holtzman, 2017).
Early studies of immunometabolic functions focused on the requirements of certain metabolites to provide energy and support biosynthesis in activated macrophages (Oren et al., 1963; Newsholme et al., 1986; Fukuzumi et al., 1996). Numerous recent studies suggest changes in intracellular metabolic pathways in immune cells can alter their functions (Chang et al., 2013; Huang et al., 2014; O’Neill et al., 2016; Zhao et al., 2020). The role of metabolism in neuron and astrocyte function (Pfrieger and Ungerer, 2011; Turner and Adamson, 2011; Jha and Morrison, 2018), has been studied while until recently the role of cellular metabolism in microglia has been less well understood. Considering many neurological disorders are associated with inflammation and alterations in energy metabolism in the brain, it is hypothesized that microglial metabolism plays a significant role in the inflammatory responses of microglia during neurodegeneration associated with AD. Here, we review the role of microglial immunometabolism in AD. We discuss our understanding of the overall role of microglia in AD, metabolism in the brain, and the importance of glucose and ketone body metabolism in AD.
Microglia, the innate immune cells of the central nervous system (CNS), account for 10–15% of the adult glial cell population in the brain (Nayak et al., 2014). Microglia develop in the yolk sac and migrate to the developing CNS during embryogenesis where they can continuously self-renew without support from bone marrow-derived precursor cells (Ginhoux and Prinz, 2015). Microglial activation in AD was first described over 100 years ago by Alois Alzheimer (English Translation: Alzheimer et al., 1995). Recently, significant progress has been made in our understanding of how microglia develop, function, and participate in AD (Lue et al., 2010; Ulrich et al., 2014, 2017; Condello et al., 2015; Vincenti et al., 2015; Wang Y. et al., 2015; Wang et al., 2016; Ulrich and Holtzman, 2016; Yuan et al., 2016; Keren-Shaul et al., 2017; Ulland et al., 2017; Gotzl et al., 2019; Mathys et al., 2019; Schlepckow et al., 2020; Zhou et al., 2020).
Despite a great deal of progress, the precise role of microglia in AD is not completely understood. Several reports suggest microglial activation in the early stages of AD delays disease progression through clearance of soluble and oligomeric Aβ (Frackowiak et al., 1992; Qiu et al., 1997; Frautschy et al., 1998). Activated microglia are hypothesized to reduce Aβ accumulation through phagocytosis mediated clearance (Qiu et al., 1997, 1998; Frautschy et al., 1998; Figure 1). Electron microscopy data show that microglia rapidly respond to Aβ deposition, extending their processes, and engulfing Aβ (Frackowiak et al., 1992). Additionally, microglia can act as a barrier to decrease the neurotoxic effects of plaque contact on adjacent neurons (Condello et al., 2015; Wang et al., 2016; Yuan et al., 2016; Figure 1). Overall, there is an abundance of data to suggest that proper microglia function protects against pathology early in AD development, however, in contrast to these findings, plentiful evidence also exists that microglia can be neurotoxic and contribute to neurodegeneration in AD. Microglia are directly linked to synapse loss (Wu et al., 2015; Spangenberg and Green, 2017) and provoke tauopathy-mediated pathology (Leyns and Holtzman, 2017; Leyns et al., 2017). Furthermore, evidence suggests tau pathology itself can stimulate microglial activation (Morales et al., 2013).
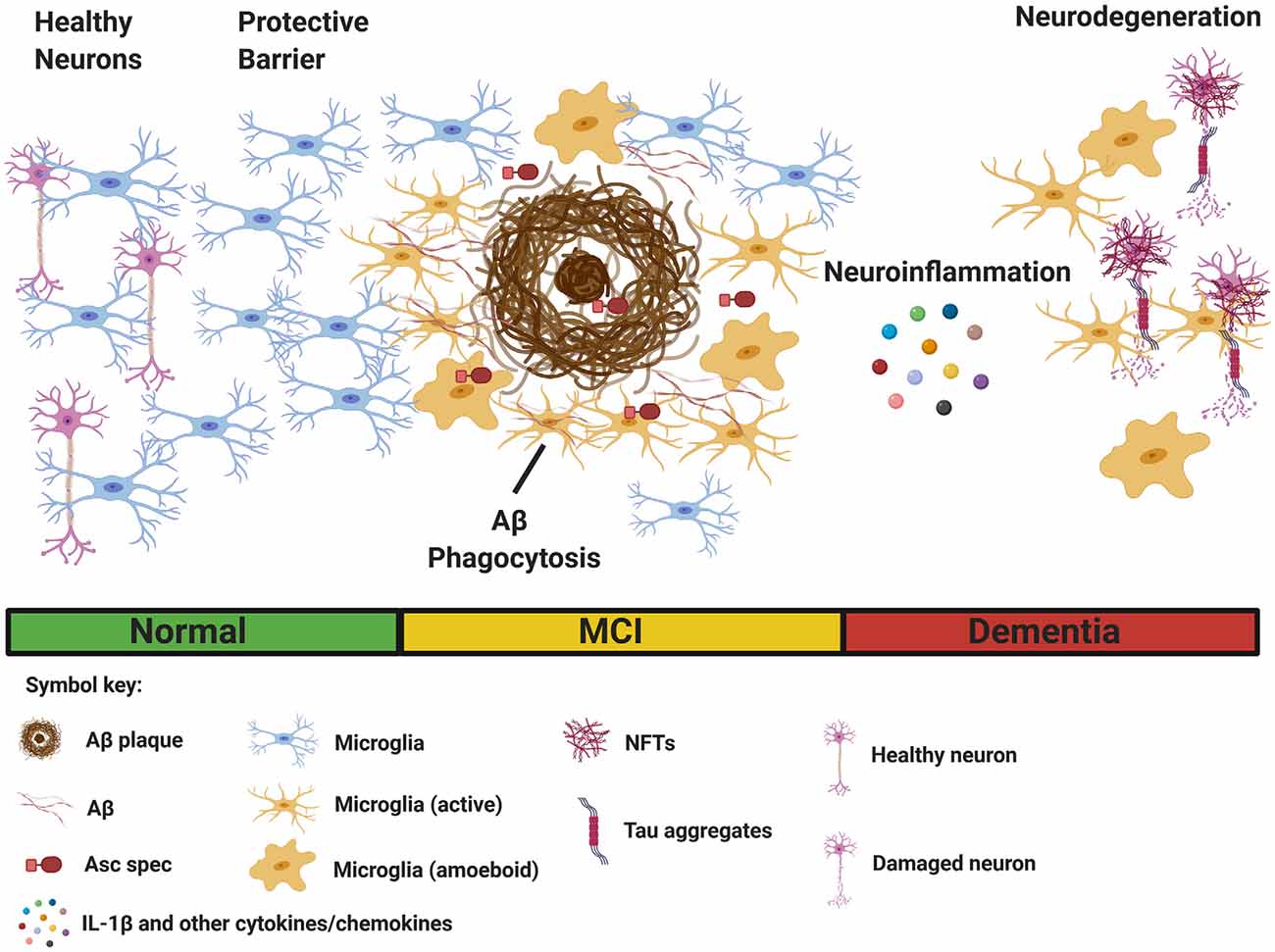
Figure 1. Multiple functions of microglia in Alzheimer’s disease (AD). Microglia sense pathological Aβ accumulation in the brain, and rapidly respond to the site of injury. (Left) Initially, microglia provide protective functions by facilitating Aβ clearance through phagocytosis to restore tissue homeostasis. Microglia also acts as a protective barrier to inhibit plaque expansion and contact with adjacent, healthy neurons. (Right) In contrast, sustained microglial activation promotes detrimental effects such as inflammasome activation and the secretion of IL-1β and other inflammatory cytokines and chemokines, leading to neuroinflammation. Asc specs, formed following NLRP3 inflammasome activation, and released by microglia, also seed new Aβ plaques. Finally, chronic neuroinflammation enhances the aggregation of hyperphosphorylated tau in neurofibrillary tangles (NFTs), resulting in neurodegeneration. (Bottom) Clinical stages of AD [normal, mild cognitive impairment (MCI), and dementia]. The figure was created with BioRender.com.
Microglia are hypothesized to exhibit functional plasticity within the context of neurodegenerative diseases (Jha et al., 2016). Many factors, such as complement, can influence microglial polarization (Bohlson et al., 2014). Activated microglia can somewhat resemble classically activated macrophages, formerly referred to as M1 macrophages, release pro-inflammatory cytokines (IL-1β, IL-18, TNF-α, IFN-γ, and IL-6), produce reactive oxygen species, and other pro-inflammatory molecules implicated in neurodegeneration in AD (Nayak et al., 2014; Wang W. Y. et al., 2015; Spangenberg and Green, 2017). In contrast, microglia may assume a phenotype similar to alternatively activated macrophages resulting in enhanced phagocytosis and anti-inflammatory responses (Park et al., 2016; Figure 1). The M1/M2 phenotype hypothesis, remains very controversial, as distinct microglial polarization has not been properly supported by research findings (Ransohoff, 2016).
Regardless of whether microglia provide a protective, pathogenic, or mixed contribution, in AD it is clear microglia is a key player in AD progression. Therefore, understanding characteristics of microglia during AD progression, like metabolism, may lead to novel approaches to treat and/or prevent AD.
While the brain only makes up about 2% of the total human body mass it accounts for approximately 25% of the glucose and 20% of the oxygen consumed by the body (Attwell and Laughlin, 2001; Alle et al., 2009). The underlying reasons for the high energy demand of the brain include neurotransmitter reuptake, action potential generation, and the generation and renewal of ion gradients (Attwell and Laughlin, 2001). The brain is therefore highly sensitive to changes in energy supply, with minor alterations in energy processing linked to hindered brain function and neurodegenerative disorders, like AD (Edison et al., 2013; Andersen et al., 2017; Skotte et al., 2018).
A growing body of evidence suggests AD pathology is driven by metabolic dysfunction (de la Monte and Tong, 2014). Diabetes appears to be an important risk factor for developing AD (Baumgart et al., 2015) with several studies linking diabetes and impaired insulin signaling in the brain to AD pathogenesis (Biessels et al., 2006; Bomfim et al., 2012; Talbot et al., 2012; Zhao et al., 2017; Kim et al., 2019). Overall, diabetics are at a greater risk for AD and the brains of individuals with AD have higher levels of insulin, insulin receptor, and insulin signaling (Hoyer, 2004; Steen et al., 2005; Craft et al., 2013). These studies highlight the importance of brain energy metabolism in AD development and provide a solid basis for the hypothesis that microglial immunometabolism is a critical component of the inflammatory responses of microglia in AD development.
Glucose is the main energy source for microglia, and they express several glucose transporters (Maher et al., 1994; Duelli and Kuschinsky, 2001), with GLUT1 (SLC2A1) and GLUT3 (SLC2A3) being the major isoforms (Kalsbeek et al., 2016; Wang et al., 2019). A study of genes associated with energy metabolism in mouse microglia, astrocytes, and neurons indicates microglia express the required genes for both glycolytic and oxidative energy metabolism (Zhang et al., 2014). A large scale proteomic study also identified several proteins expressed by activated microglia linked to sugar metabolism, highlighting the importance of microglial sugar metabolism in AD (Johnson et al., 2020). Additional studies show non-activated microglia depend on oxidative phosphorylation for ATP production while activated microglia rely on glycolysis (Bernhart et al., 2010). Further studies validated these findings showing that LPS induced significant metabolic changes in BV2 cells resulting in decreased mitochondrial function and increased glycolysis (Voloboueva et al., 2013; Gimeno-Bayon et al., 2014). Additionally, microglial response to LPS is enhanced under high glucose conditions as indicated by significantly increased release of IL-6 and TNF-α (Zhang et al., 2015).
Of further interest are the number of recent studies which suggest the nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome is activated by glycolytic enzymes (Hughes and O’Neill, 2018); activation of the NLRP3 inflammasome has been demonstrated to contribute to AD pathology (Heneka et al., 2013; Venegas et al., 2017; Ising et al., 2019). For example, inhibition of the mammalian target of rapamycin complex 1 (mTORC1) suppressed hexokinase 1-dependent glycolysis and caspase-1 activation, implicating NLRP3 inflammasome activation in macrophage metabolism (Moon et al., 2015). A further study showed hyperglycolysis and hexokinase induction activates microglia and is essential for neuroinflammation by microglia under hypoxic conditions (Li et al., 2018). Furthermore, microglia that have activated the NLRP3 inflammasome switch their metabolism towards glycolysis which has the potential to impact energy-requiring processes, like phagocytosis (Rubio-Araiz et al., 2018). Interestingly, the addition of anti-TLR2 increased microglial phagocytosis of Aβ with decreased expression of an important glycolysis enzyme, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase. These findings were indirectly linked to the inhibition of inflammasome activation by the anti-TLR2 antibody (Rubio-Araiz et al., 2018).
Recently, a link between defective microglial function associated with triggering receptor on myeloid cells (TREM2) and glucose metabolism in neurodegeneration has been identified (Kleinberger et al., 2017; Ulland et al., 2017). Mutations in TREM2 are associated with an increased risk for the development of AD (Guerreiro et al., 2013; Jonsson et al., 2013; Song et al., 2017). TREM2 T66M knock-in mice displayed an age-dependent decline in microglial activity along with a significant decrease in cerebral blood flow and brain glucose metabolism suggesting a potential microglial function in managing brain glucose metabolism (Kleinberger et al., 2017). TREM2 was also found to play a major role in microglial metabolic fitness (Ulland et al., 2017). Trem2−/− 5XFAD mice were less metabolically competent as they exhibited large decreases in glycolytic and mammalian target of rapamycin (mTOR) activity compared to wild-type cells; decreases in mTOR signaling were associated with increased autophagy. Additionally, the metabolic deficiency, and lack of microglial responsiveness, was restored in Trem2−/− 5XFAD mice by increasing microglial energy capacity with cyclocreatine (Ulland et al., 2017). Together, these studies highlight the importance of microglial metabolism of glucose in AD. Further investigation is needed to determine the precise mechanisms in which microglial metabolism of glucose influences AD pathology.
Microglia can use ketone bodies as an alternative energy source to glucose. The three main ketone body components are acetate, β-hydroxybutyrate (BHB), and acetoacetate (Laffel, 1999). Levels of ketone bodies increase during periods of extended exercise, starvation, caloric restriction, or in individuals on low carbohydrate diets, e.g., the ketogenic diet. Dietary regimens, like a ketogenic diet, have been shown to reduce inflammation and suppress microglial activation; therefore, there is interest in using the ketogenic diet as a potential therapeutic option for AD. Ketones are known to have a protective effect in AD by improving synaptic plasticity and reducing oxidative stress (Yin et al., 2016). BHB activates G-protein-coupled receptor 109A (GPR109A), also called hydroxycarboxylic acid receptor 2 (HCA2), which attenuates NF-κB signaling, pro-inflammatory enzyme (Cox-2 and iNOS), and cytokine (IL-6, TNF-α, and IL-1β) production in both macrophages and microglia (Rahman et al., 2014; Fu et al., 2015; Huang et al., 2018). Although it appears ketone body metabolism by microglia has a significant role in AD, much work is needed to elucidate the mechanistic insights into how this metabolism modulates microglial activity and function.
To date AD drug discovery research has focused on tauopathy or Aβ reduction. As discussed above, glycolysis is a major factor in maintaining activated microglia, while non-activated microglia rely more on oxidative metabolism (Bernhart et al., 2010). Based on this data, it is reasonable to suggest that reprogramming microglia towards oxidative metabolism may be a useful therapeutic strategy to reduce neuroinflammation in AD. The study by Gu et al. (2017) shows a reduced expression of Clock (clk)1, a mitochondrial hydroxylase, enhanced inflammation, and aerobic glycolysis in microglia by an NF-κB-dependent mechanism. Additionally, their study showed that inhibition of glycolytic metabolism abolished the enhanced inflammatory phenotype seen in Clk1-deficient BV2 cells (Gu et al., 2017). Based on this observation, several molecules could be potential therapeutic candidates, including dimethyl fumarate and its metabolite, monomethyl fumarate, as they have been shown to inhibit NF-κB activity (Gillard et al., 2015; Al-Jaderi and Maghazachi, 2016; Kornberg et al., 2018). Also, short-chain fatty acids, like dichloroacetate and butyrate, could potentially be used as therapeutics, as both have been shown to promote metabolic shifts away from glycolysis towards oxidative metabolic pathways (Blouin et al., 2011; Matt et al., 2018).
Microglia shift to an anti-inflammatory phenotype in response to BHB (Huang et al., 2018). Additionally, studies in macrophages (Youm et al., 2015) and primary microglia (Deora et al., 2017) indicate BHB blocks NLRP3 inflammasome activation. In macrophages, BHB can block NLRP3 inflammasome activation by preventing potassium efflux, which in turn reduces apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) oligomerization and ASC speck formation (Youm et al., 2015). Whether this mechanism is similar in microglia remains unclear. These findings suggest BHB treatment or dietary regimens that promote elevated BHB levels, could be a promising therapy for AD. Indeed, several studies of mice on a ketogenic diet have shown reduced tau and amyloid pathologies (Van der Auwera et al., 2005; Kashiwaya et al., 2013). BHB also protects against AD pathology by targeting multiple aspects of AD pathogenesis (Wu et al., 2020). BHB administration to 5XFAD mice improved cognitive functions, decreased microgliosis, and reduced Aβ accumulation (Wu et al., 2020). Furthermore, several studies in humans demonstrate a ketogenic diet may improve cognitive abilities in patients with neurodegenerative disorders, with higher ketone levels correlating with improved cognitive functioning (Reger et al., 2004; Henderson et al., 2009; Krikorian et al., 2012; Taylor et al., 2018; Ota et al., 2019). In contrast, five-month-old mice on a ketogenic diet for three months did not improve cognition in the amyloid or tau mouse model of AD (Brownlow et al., 2013). This study, however, showed improvement in the motor performance of mice, suggesting BHB may enhance existing neuron function without modifying the rate of neuropathy in AD (Brownlow et al., 2013). Additionally, BHB did not inhibit synuclein fibril mediated inflammasome activation in microglia, suggesting that activation of NLRP3 by synuclein fibrils acts through different mechanisms compared to adenosine triphosphate (ATP) and monosodium urate (MSU) activation (Deora et al., 2017). Based on these contrasting findings, further work is needed to determine if BHB is a viable treatment option for AD.
Medium-chain triglyceride diets have been developed to provide a more palatable alternative to the ketogenic diet (Huttenlocher et al., 1971). Caprylic triglyceride (CT) is a medium-chain triglyceride, which is metabolized into ketone bodies that can be used as an alternative energy source for neuronal metabolism (Bach and Babayan, 1982) and has been developed as a medical food therapy to promote mitochondrial metabolism in AD (Roman, 2010). Several studies suggest an increase in brain ketone metabolism can increase overall brain energy supply to improve mild cognitive impairment (MCI; Croteau et al., 2018; Fortier et al., 2019; Neth et al., 2020). Apolipoprotein E (ApoE) appears to be an important factor in the efficacy of this therapy, as carriers of the APOE4 allele do not see the improvement in cognitive function as subjects administered CT who are not carriers of the APOE4 allele (Reger et al., 2004; Henderson et al., 2009; Farah, 2014; Yamazaki et al., 2019). While CT administration is generally thought to function through the generation of ketones to provide an alternative energy source for brain cells, including microglia, the underlying mechanisms, however, are still largely unknown. Further characterization of all forms of the ketogenic diet might improve and increase their use as a therapeutic regime for AD.
Another approach to AD treatment is to target microglial genes important in microglial metabolism. As previously discussed, TREM2 is vital to microglial metabolic fitness (Ulland et al., 2017). Therapeutic strategies that promote TREM2 expression and function may have beneficial effects in AD patients (Ulland and Colonna, 2018). For example, TREM2 signaling could potentially be increased by using small molecule inhibitors or agonistic antibodies to phospholipid ligands, and inhibiting protease-mediated cleavage could increase TREM2 expression. Furthermore, since TREM2 maintains microglial mTOR metabolism and signaling, the use of metabolic agents that promote microglial metabolic fitness may also be a viable option (Ulland and Colonna, 2018). However, the use of these potential therapies is questionable as there is conflicting evidence about the impact of modifying TREM2 signaling in a tau model of AD. Leyns et al. suggest microglial TREM2 signaling is detrimental during disease progression, as TREM2 deficiency results in decreased neuroinflammation and protects against neurodegeneration (Leyns et al., 2017). In contrast, the study by Bemiller et al. demonstrated that deficiency of microglial TREM2 increases tau pathology (Bemiller et al., 2017). Further investigation is needed, as much is to be learned before therapeutic agents targeting TREM2 signaling, microglia, and metabolism in AD can be developed and implemented clinically.
The emerging field of immunometabolism has provided significant progress in our understanding of how cellular and systemic metabolism affects immune responses. More importantly, these data suggest that targeting immune cell metabolism may be a valuable strategy for the development of advanced therapeutics to treat human disease (Bettencourt and Powell, 2017; Matsushita and Pearce, 2018). Little is known, however, about microglial immunometabolism in the context of neurodegeneration and AD. A major challenge in targeting microglia-specific metabolism as a therapeutic strategy is to determine the possible conflicting functions microglia may have in AD progression. As discussed above, there is evidence to suggest microglia may have a beneficial and/or detrimental effect during AD pathogenesis depending on several factors, including the stage of AD progression. Further work is necessary to address these concerns and design microglia-targeted therapeutic strategies for AD intervention.
DS drafted the manuscript. TU reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by start-up funds to TU from the Department of Pathology and Laboratory Medicine, Graduate School, and School of Medicine and Public Health at the University of Wisconsin.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The figure was created with BioRender.com.
Al-Jaderi, Z., and Maghazachi, A. A. (2016). Utilization of dimethyl fumarate and related molecules for treatment of multiple sclerosis, cancer and other diseases. Front. Immunol. 7:278. doi: 10.3389/fimmu.2016.00278
Alle, H., Roth, A., and Geiger, J. R. (2009). Energy-efficient action potentials in hippocampal mossy fibers. Science 325, 1405–1408. doi: 10.1126/science.1174331
Alzheimer, A., Stelzmann, R. A., Schnitzlein, H. N., and Murtagh, F. R. (1995). An English translation of Alzheimer’s 1907 paper, Uber eine eigenartige Erkankung der Hirnrinde. Clin. Anat. 8, 429–431. doi: 10.1002/ca.980080612
Andersen, J. V., Christensen, S. K., Aldana, B. I., Nissen, J. D., Tanila, H., and Waagepetersen, H. S. (2017). Alterations in cerebral cortical glucose and glutamine metabolism precedes amyloid plaques in the APPswe/PSEN1dE9 mouse model of Alzheimer’s disease. Neurochem. Res. 42, 1589–1598. doi: 10.1007/s11064-016-2070-2
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Bach, A. C., and Babayan, V. K. (1982). Medium-chain triglycerides: an update. Am. J. Clin. Nutr. 36, 950–962. doi: 10.1093/ajcn/36.5.950
Baumgart, M., Snyder, H. M., Carrillo, M. C., Fazio, S., Kim, H., and Johns, H. (2015). Summary of the evidence on modifiable risk factors for cognitive decline and dementia: a population-based perspective. Alzheimers Dement. 11, 718–726. doi: 10.1016/j.jalz.2015.05.016
Bemiller, S. M., McCray, T. J., Allan, K., Formica, S. V., Xu, G., Wilson, G., et al. (2017). TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 12:74. doi: 10.1186/s13024-017-0216-6
Bernhart, E., Kollroser, M., Rechberger, G., Reicher, H., Heinemann, A., Schratl, P., et al. (2010). Lysophosphatidic acid receptor activation affects the C13NJ microglia cell line proteome leading to alterations in glycolysis, motility and cytoskeletal architecture. Proteomics 10, 141–158. doi: 10.1002/pmic.200900195
Bettencourt, I. A., and Powell, J. D. (2017). Targeting metabolism as a novel therapeutic approach to autoimmunity, inflammation and transplantation. J. Immunol. 198, 999–1005. doi: 10.4049/jimmunol.1601318
Biessels, G. J., Staekenborg, S., Brunner, E., Brayne, C., and Scheltens, P. (2006). Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 5, 64–74. doi: 10.1016/S1474-4422(05)70284-2
Blouin, J. M., Penot, G., Collinet, M., Nacfer, M., Forest, C., Laurent-Puig, P., et al. (2011). Butyrate elicits a metabolic switch in human colon cancer cells by targeting the pyruvate dehydrogenase complex. Int. J. Cancer 128, 2591–2601. doi: 10.1002/ijc.25599
Bohlson, S. S., O’Conner, S. D., Hulsebus, H. J., Ho, M. M., and Fraser, D. A. (2014). Complement, c1q and c1q-related molecules regulate macrophage polarization. Front. Immunol. 5:402. doi: 10.3389/fimmu.2014.00402
Bomfim, T. R., Forny-Germano, L., Sathler, L. B., Brito-Moreira, J., Houzel, J. C., Decker, H., et al. (2012). An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Aβ oligomers. J. Clin. Invest. 122, 1339–1353. doi: 10.1172/jci57256
Brownlow, M. L., Benner, L., D’Agostino, D., Gordon, M. N., and Morgan, D. (2013). Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PLoS One 8:e75713. doi: 10.1371/journal.pone.0075713
Chang, C. H., Curtis, J. D., Maggi, L. B. Jr., Faubert, B., Villarino, A. V., O’Sullivan, D., et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. doi: 10.1016/j.cell.2013.05.016
Collaborators, G. B. D. D. (2019). Global, regional and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18, 88–106. doi: 10.1016/S1474-4422(18)30403-4
Condello, C., Yuan, P., Schain, A., and Grutzendler, J. (2015). Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 6:6176. doi: 10.1038/ncomms7176
Craft, S., Cholerton, B., and Baker, L. D. (2013). Insulin and Alzheimer’s disease: untangling the web. J. Alzheimers Dis. 33, S263–S275. doi: 10.3233/JAD-2012-129042
Croteau, E., Castellano, C. A., Richard, M. A., Fortier, M., Nugent, S., Lepage, M., et al. (2018). Ketogenic medium chain triglycerides increase brain energy metabolism in Alzheimer’s disease. J. Alzheimers Dis. 64, 551–561. doi: 10.3233/jad-180202
de la Monte, S. M., and Tong, M. (2014). Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol. 88, 548–559. doi: 10.1016/j.bcp.2013.12.012
Deora, V., Albornoz, E. A., Zhu, K., Woodruff, T. M., and Gordon, R. (2017). The ketone body β-hydroxybutyrate does not inhibit synuclein mediated inflammasome activation in microglia. J. Neuroimmune Pharmacol. 12, 568–574. doi: 10.1007/s11481-017-9754-5
Duelli, R., and Kuschinsky, W. (2001). Brain glucose transporters: relationship to local energy demand. News Physiol. Sci. 16, 71–76. doi: 10.1152/physiologyonline.2001.16.2.71
Edison, P., Ahmed, I., Fan, Z., Hinz, R., Gelosa, G., Ray Chaudhuri, K., et al. (2013). Microglia, amyloid and glucose metabolism in Parkinson’s disease with and without dementia. Neuropsychopharmacology 38, 938–949. doi: 10.1038/npp.2012.255
Farah, B. A. (2014). Effects of caprylic triglyceride on cognitive performance and cerebral glucose metabolism in mild Alzheimer’s disease: a single-case observation. Front. Aging Neurosci. 6:133. doi: 10.3389/fnagi.2014.00133
Fortier, M., Castellano, C. A., Croteau, E., Langlois, F., Bocti, C., St-Pierre, V., et al. (2019). A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimers Dement. 15, 625–634. doi: 10.1016/j.jalz.2018.12.017
Frackowiak, J., Wisniewski, H. M., Wegiel, J., Merz, G. S., Iqbal, K., and Wang, K. C. (1992). Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce β-amyloid fibrils. Acta Neuropathol. 84, 225–233. doi: 10.1007/bf00227813
Frautschy, S. A., Yang, F., Irrizarry, M., Hyman, B., Saido, T. C., Hsiao, K., et al. (1998). Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol. 152, 307–317.
Fu, S. P., Wang, J. F., Xue, W. J., Liu, H. M., Liu, B. R., Zeng, Y. L., et al. (2015). Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflammation 12:9. doi: 10.1186/s12974-014-0230-3
Fukuzumi, M., Shinomiya, H., Shimizu, Y., Ohishi, K., and Utsumi, S. (1996). Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1. Infect. Immun. 64, 108–112. doi: 10.1128/iai.64.1.108-112.1996
Gillard, G. O., Collette, B., Anderson, J., Chao, J., Scannevin, R. H., Huss, D. J., et al. (2015). DMF, but not other fumarates, inhibits NF-κB activity in vitro in an Nrf2-independent manner. J. Neuroimmunol. 283, 74–85. doi: 10.1016/j.jneuroim.2015.04.006
Gimeno-Bayon, J., Lopez-Lopez, A., Rodriguez, M. J., and Mahy, N. (2014). Glucose pathways adaptation supports acquisition of activated microglia phenotype. J. Neurosci. Res. 92, 723–731. doi: 10.1002/jnr.23356
Ginhoux, F., and Prinz, M. (2015). Origin of microglia: current concepts and past controversies. Cold Spring Harb. Perspect. Biol. 7:a020537. doi: 10.1101/cshperspect.a020537
Gotzl, J. K., Brendel, M., Werner, G., Parhizkar, S., Sebastian Monasor, L., Kleinberger, G., et al. (2019). Opposite microglial activation stages upon loss of PGRN or TREM2 result in reduced cerebral glucose metabolism. EMBO Mol. Med. 11:e9711. doi: 10.15252/emmm.201809711
Gu, R., Zhang, F., Chen, G., Han, C., Liu, J., Ren, Z., et al. (2017). Clk1 deficiency promotes neuroinflammation and subsequent dopaminergic cell death through regulation of microglial metabolic reprogramming. Brain Behav. Immun. 60, 206–219. doi: 10.1016/j.bbi.2016.10.018
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Henderson, S. T., Vogel, J. L., Barr, L. J., Garvin, F., Jones, J. J., and Costantini, L. C. (2009). Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 6:31. doi: 10.1186/1743-7075-6-31
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Holtzman, D. M., Morris, J. C., and Goate, A. M. (2011). Alzheimer’s disease: the challenge of the second century. Sci. Transl. Med. 3:77sr71. doi: 10.1126/scitranslmed.3002369
Hoyer, S. (2004). Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur. J. Pharmacol. 490, 115–125. doi: 10.1016/j.ejphar.2004.02.049
Huang, S. C., Everts, B., Ivanova, Y., O’Sullivan, D., Nascimento, M., Smith, A. M., et al. (2014). Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 15, 846–855. doi: 10.1038/ni.2956
Huang, C., Wang, P., Xu, X., Zhang, Y., Gong, Y., Hu, W., et al. (2018). The ketone body metabolite β-hydroxybutyrate induces an antidepression-associated ramification of microglia via HDACs inhibition-triggered Akt-small RhoGTPase activation. Glia 66, 256–278. doi: 10.1002/glia.23241
Hughes, M. M., and O’Neill, L. A. J. (2018). Metabolic regulation of NLRP3. Immunol. Rev. 281, 88–98. doi: 10.1111/imr.12608
Huttenlocher, P. R., Wilbourn, A. J., and Signore, J. M. (1971). Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology 21, 1097–1103. doi: 10.1212/wnl.21.11.1097
Ising, C., Venegas, C., Zhang, S., Scheiblich, H., Schmidt, S. V., Vieira-Saecker, A., et al. (2019). NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. doi: 10.1038/s41586-019-1769-z
Jha, M. K., Lee, W. H., and Suk, K. (2016). Functional polarization of neuroglia: implications in neuroinflammation and neurological disorders. Biochem. Pharmacol. 103, 1–16. doi: 10.1016/j.bcp.2015.11.003
Jha, M. K., and Morrison, B. M. (2018). Glia-neuron energy metabolism in health and diseases: new insights into the role of nervous system metabolic transporters. Exp. Neurol. 309, 23–31. doi: 10.1016/j.expneurol.2018.07.009
Johnson, E. C. B., Dammer, E. B., Duong, D. M., Ping, L., Zhou, M., Yin, L., et al. (2020). Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 26, 769–780. doi: 10.1038/s41591-020-0815-6
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Kalsbeek, M. J., Mulder, L., and Yi, C. X. (2016). Microglia energy metabolism in metabolic disorder. Mol. Cell. Endocrinol. 438, 27–35. doi: 10.1016/j.mce.2016.09.028
Kashiwaya, Y., Bergman, C., Lee, J. H., Wan, R., King, M. T., Mughal, M. R., et al. (2013). A ketone ester diet exhibits anxiolytic and cognition-sparing properties and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 1530–1539. doi: 10.1016/j.neurobiolaging.2012.11.023
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276.e17–1290.e17. doi: 10.1016/j.cell.2017.05.018
Kim, B., Elzinga, S. E., Henn, R. E., McGinley, L. M., and Feldman, E. L. (2019). The effects of insulin and insulin-like growth factor I on amyloid precursor protein phosphorylation in in vitro and in vivo models of Alzheimer’s disease. Neurobiol. Dis. 132:104541. doi: 10.1016/j.nbd.2019.104541
Kleinberger, G., Brendel, M., Mracsko, E., Wefers, B., Groeneweg, L., Xiang, X., et al. (2017). The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion and glucose metabolism. EMBO J. 36, 1837–1853. doi: 10.15252/embj.201796516
Kornberg, M. D., Bhargava, P., Kim, P. M., Putluri, V., Snowman, A. M., Putluri, N., et al. (2018). Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 360, 449–453. doi: 10.1126/science.aan4665
Krikorian, R., Shidler, M. D., Dangelo, K., Couch, S. C., Benoit, S. C., and Clegg, D. J. (2012). Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 33, e419–427. doi: 10.1016/j.neurobiolaging.2010.10.006
Laffel, L. (1999). Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 15, 412–426. doi: 10.1002/(sici)1520-7560(199911/12)15:6<412::aid-dmrr72>3.0.co;2-8
Leyns, C. E. G., and Holtzman, D. M. (2017). Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegener. 12:50. doi: 10.1186/s13024-017-0192-x
Leyns, C. E. G., Ulrich, J. D., Finn, M. B., Stewart, F. R., Koscal, L. J., Remolina Serrano, J., et al. (2017). TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. U S A 114, 11524–11529. doi: 10.1073/pnas.1710311114
Li, Y., Lu, B., Sheng, L., Zhu, Z., Sun, H., Zhou, Y., et al. (2018). Hexokinase 2-dependent hyperglycolysis driving microglial activation contributes to ischemic brain injury. J. Neurochem. 144, 186–200. doi: 10.1111/jnc.14267
Lue, L. F., Kuo, Y. M., Beach, T., and Walker, D. G. (2010). Microglia activation and anti-inflammatory regulation in Alzheimer’s disease. Mol. Neurobiol. 41, 115–128. doi: 10.1007/s12035-010-8106-8
Maher, F., Vannucci, S. J., and Simpson, I. A. (1994). Glucose transporter proteins in brain. FASEB J. 8, 1003–1011. doi: 10.1096/fasebj.8.13.7926364
Mathys, H., Davila-Velderrain, J., Peng, Z., Gao, F., Mohammadi, S., Young, J. Z., et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. doi: 10.1038/s41586-019-1195-2
Matsushita, M., and Pearce, E. J. (2018). Disrupting metabolism to treat autoimmunity. Science 360, 377–378. doi: 10.1126/science.aat4984
Matt, S. M., Allen, J. M., Lawson, M. A., Mailing, L. J., Woods, J. A., and Johnson, R. W. (2018). Butyrate and dietary soluble fiber improve neuroinflammation associated with aging in mice. Front. Immunol. 9:1832. doi: 10.3389/fimmu.2018.01832
Moon, J. S., Hisata, S., Park, M. A., DeNicola, G. M., Ryter, S. W., Nakahira, K., et al. (2015). mTORC1-induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep. 12, 102–115. doi: 10.1016/j.celrep.2015.05.046
Morales, I., Jimenez, J. M., Mancilla, M., and Maccioni, R. B. (2013). Tau oligomers and fibrils induce activation of microglial cells. J. Alzheimers Dis. 37, 849–856. doi: 10.3233/jad-131843
Nayak, D., Roth, T. L., and McGavern, D. B. (2014). Microglia development and function. Annu. Rev. Immunol. 32, 367–402. doi: 10.1146/annurev-immunol-032713-120240
Neth, B. J., Mintz, A., Whitlow, C., Jung, Y., Solingapuram Sai, K., Register, T. C., et al. (2020). Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: a pilot study. Neurobiol. Aging 86, 54–63. doi: 10.1016/j.neurobiolaging.2019.09.015
Newsholme, P., Curi, R., Gordon, S., and Newsholme, E. A. (1986). Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem. J. 239, 121–125. doi: 10.1042/bj2390121
O’Neill, L. A., Kishton, R. J., and Rathmell, J. (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565. doi: 10.1038/nri.2016.70
Oren, R., Farnham, A. E., Saito, K., Milofsky, E., and Karnovsky, M. L. (1963). Metabolic patterns in three types of phagocytizing cells. J. Cell Biol. 17, 487–501. doi: 10.1083/jcb.17.3.487
Ota, M., Matsuo, J., Ishida, I., Takano, H., Yokoi, Y., Hori, H., et al. (2019). Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 690, 232–236. doi: 10.1016/j.neulet.2018.10.048
Park, H. J., Oh, S. H., Kim, H. N., Jung, Y. J., and Lee, P. H. (2016). Mesenchymal stem cells enhance α-synuclein clearance via M2 microglia polarization in experimental and human parkinsonian disorder. Acta Neuropathol. 132, 685–701. doi: 10.1007/s00401-016-1605-6
Pfrieger, F. W., and Ungerer, N. (2011). Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 50, 357–371. doi: 10.1016/j.plipres.2011.06.002
Qiu, W. Q., Walsh, D. M., Ye, Z., Vekrellis, K., Zhang, J., Podlisny, M. B., et al. (1998). Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J. Biol. Chem. 273, 32730–32738. doi: 10.1074/jbc.273.49.32730
Qiu, W. Q., Ye, Z., Kholodenko, D., Seubert, P., and Selkoe, D. J. (1997). Degradation of amyloid β-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J. Biol. Chem. 272, 6641–6646. doi: 10.1074/jbc.272.10.6641
Rahman, M., Muhammad, S., Khan, M. A., Chen, H., Ridder, D. A., Muller-Fielitz, H., et al. (2014). The β-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 5:3944. doi: 10.1038/ncomms4944
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Reger, M. A., Henderson, S. T., Hale, C., Cholerton, B., Baker, L. D., Watson, G. S., et al. (2004). Effects of β-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 25, 311–314. doi: 10.1016/S0197-4580(03)00087-3
Roman, M. W. (2010). Axona (Accera, Inc.): a new medical food therapy for persons with Alzheimer’s disease. Issues Ment. Health Nurs. 31, 435–436. doi: 10.3109/01612841003768231
Rubio-Araiz, A., Finucane, O. M., Keogh, S., and Lynch, M. A. (2018). Anti-TLR2 antibody triggers oxidative phosphorylation in microglia and increases phagocytosis of β-amyloid. J. Neuroinflammation 15:247. doi: 10.1186/s12974-018-1281-7
Schlepckow, K., Monroe, K. M., Kleinberger, G., Cantuti-Castelvetri, L., Parhizkar, S., Xia, D., et al. (2020). Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol. Med. 12:e11227. doi: 10.15252/emmm.201911227
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Skotte, N. H., Andersen, J. V., Santos, A., Aldana, B. I., Willert, C. W., Norremolle, A., et al. (2018). Integrative characterization of the R6/2 mouse model of Huntington’s disease reveals dysfunctional astrocyte metabolism. Cell Rep. 23, 2211–2224. doi: 10.1016/j.celrep.2018.04.052
Song, W., Hooli, B., Mullin, K., Jin, S. C., Cella, M., Ulland, T. K., et al. (2017). Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. 13, 381–387. doi: 10.1016/j.jalz.2016.07.004
Spangenberg, E. E., and Green, K. N. (2017). Inflammation in Alzheimer’s disease: lessons learned from microglia-depletion models. Brain Behav. Immun. 61, 1–11. doi: 10.1016/j.bbi.2016.07.003
Steen, E., Terry, B. M., Rivera, E. J., Cannon, J. L., Neely, T. R., Tavares, R., et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes? J. Alzheimers Dis. 7, 63–80. doi: 10.3233/jad-2005-7107
Talbot, K., Wang, H. Y., Kazi, H., Han, L. Y., Bakshi, K. P., Stucky, A., et al. (2012). Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation and cognitive decline. J. Clin. Invest. 122, 1316–1338. doi: 10.1172/jci59903
Taylor, M. K., Sullivan, D. K., Mahnken, J. D., Burns, J. M., and Swerdlow, R. H. (2018). Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement. 4, 28–36. doi: 10.1016/j.trci.2017.11.002
Turner, D. A., and Adamson, D. C. (2011). Neuronal-astrocyte metabolic interactions: understanding the transition into abnormal astrocytoma metabolism. J. Neuropathol. Exp. Neurol. 70, 167–176. doi: 10.1097/nen.0b013e31820e1152
Ulland, T. K., and Colonna, M. (2018). TREM2—a key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 14, 667–675. doi: 10.1038/s41582-018-0072-1
Ulland, T. K., Song, W. M., Huang, S. C., Ulrich, J. D., Sergushichev, A., Beatty, W. L., et al. (2017). TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170, 649.e13–663.e13. doi: 10.1016/j.cell.2017.07.023
Ulrich, J. D., Finn, M. B., Wang, Y., Shen, A., Mahan, T. E., Jiang, H., et al. (2014). Altered microglial response to Aβ plaques in APPPS1–21 mice heterozygous for TREM2. Mol. Neurodegener. 9:20. doi: 10.1186/1750-1326-9-20
Ulrich, J. D., and Holtzman, D. M. (2016). TREM2 function in Alzheimer’s disease and neurodegeneration. ACS Chem. Neurosci. 7, 420–427. doi: 10.1021/acschemneuro.5b00313
Ulrich, J. D., Ulland, T. K., Colonna, M., and Holtzman, D. M. (2017). Elucidating the role of TREM2 in Alzheimer’s disease. Neuron 94, 237–248. doi: 10.1016/j.neuron.2017.02.042
Van der Auwera, I., Wera, S., Van Leuven, F., and Henderson, S. T. (2005). A ketogenic diet reduces amyloid β 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. 2:28. doi: 10.1186/1743-7075-2-28
Venegas, C., Kumar, S., Franklin, B. S., Dierkes, T., Brinkschulte, R., Tejera, D., et al. (2017). Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552, 355–361. doi: 10.1038/nature25158
Vincenti, J. E., Murphy, L., Grabert, K., McColl, B. W., Cancellotti, E., Freeman, T. C., et al. (2015). Defining the microglia response during the time course of chronic neurodegeneration. J. Virol. 90, 3003–3017. doi: 10.1128/jvi.02613-15
Voloboueva, L. A., Emery, J. F., Sun, X., and Giffard, R. G. (2013). Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett. 587, 756–762. doi: 10.1016/j.febslet.2013.01.067
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Wang, L., Pavlou, S., Du, X., Bhuckory, M., Xu, H., and Chen, M. (2019). Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol. Neurodegener. 14:2. doi: 10.1186/s13024-019-0305-9
Wang, W. Y., Tan, M. S., Yu, J. T., and Tan, L. (2015). Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 3:136. doi: 10.3978/j.issn.2305-5839.2015.03.49
Wang, Y., Ulland, T. K., Ulrich, J. D., Song, W., Tzaferis, J. A., Hole, J. T., et al. (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 213, 667–675. doi: 10.1084/jem.20151948
Wu, Y., Dissing-Olesen, L., MacVicar, B. A., and Stevens, B. (2015). Microglia: dynamic mediators of synapse development and plasticity. Trends Immunol. 36, 605–613. doi: 10.1016/j.it.2015.08.008
Wu, Y., Gong, Y., Luan, Y., Li, Y., Liu, J., Yue, Z., et al. (2020). BHBA treatment improves cognitive function by targeting pleiotropic mechanisms in transgenic mouse model of Alzheimer’s disease. FASEB J. 34, 1412–1429. doi: 10.1096/fj.201901984R
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C. C., and Bu, G. (2019). Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15, 501–518. doi: 10.1038/s41582-019-0228-7
Yin, J. X., Maalouf, M., Han, P., Zhao, M., Gao, M., Dharshaun, T., et al. (2016). Ketones block amyloid entry and improve cognition in an Alzheimer’s model. Neurobiol. Aging 39, 25–37. doi: 10.1016/j.neurobiolaging.2015.11.018
Youm, Y. H., Nguyen, K. Y., Grant, R. W., Goldberg, E. L., Bodogai, M., Kim, D., et al. (2015). The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269. doi: 10.1038/nm.3804
Yuan, P., Condello, C., Keene, C. D., Wang, Y., Bird, T. D., Paul, S. M., et al. (2016). TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739. doi: 10.1016/j.neuron.2016.05.003
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O’Keeffe, S., et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014
Zhang, X., Dong, H., Zhang, S., Lu, S., Sun, J., and Qian, Y. (2015). Enhancement of LPS-induced microglial inflammation response via TLR4 under high glucose conditions. Cell Physiol. Biochem. 35, 1571–1581. doi: 10.1159/000373972
Zhao, N., Liu, C. C., Van Ingelgom, A. J., Martens, Y. A., Linares, C., Knight, J. A., et al. (2017). Apolipoprotein E4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron 96, 115.e5–129.e5. doi: 10.1016/j.neuron.2017.09.003
Zhao, H., Raines, L. N., and Huang, S. C. (2020). Carbohydrate and amino acid metabolism as hallmarks for innate immune cell activation and function. Cells 9:562. doi: 10.3390/cells9030562
Keywords: Alzheimer’s disease, microglia, immunometabolism, neuroinflammation, neurodegeneration, glucose, ketone bodies
Citation: Shippy DC and Ulland TK (2020) Microglial Immunometabolism in Alzheimer’s Disease. Front. Cell. Neurosci. 14:563446. doi: 10.3389/fncel.2020.563446
Received: 18 May 2020; Accepted: 28 August 2020;
Published: 18 September 2020.
Edited by:
Marie-Eve Tremblay, University of Victoria, CanadaReviewed by:
Eric B. Dammer, Emory University, United StatesCopyright © 2020 Shippy and Ulland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tyler K. Ulland, dHVsbGFuZEB3aXNjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.