 Rebecca Stevenson
Rebecca Stevenson Evgeniia Samokhina
Evgeniia Samokhina Ilaria Rossetti
Ilaria Rossetti John W. Morley
John W. Morley Yossi Buskila
Yossi Buskila
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 21 August 2020
Sec. Non-Neuronal Cells
Volume 14 - 2020 | https://doi.org/10.3389/fncel.2020.00278
This article is part of the Research Topic Neuronal Pathways Affecting Glial Function View all 4 articles
Glia, a non-excitable cell type once considered merely as the connective tissue between neurons, is nowadays acknowledged for its essential contribution to multiple physiological processes including learning, memory formation, excitability, synaptic plasticity, ion homeostasis, and energy metabolism. Moreover, as glia are key players in the brain immune system and provide structural and nutritional support for neurons, they are intimately involved in multiple neurological disorders. Recent advances have demonstrated that glial cells, specifically microglia and astroglia, are involved in several neurodegenerative diseases including Amyotrophic lateral sclerosis (ALS), Epilepsy, Parkinson’s disease (PD), Alzheimer’s disease (AD), and frontotemporal dementia (FTD). While there is compelling evidence for glial modulation of synaptic formation and regulation that affect neuronal signal processing and activity, in this manuscript we will review recent findings on neuronal activity that affect glial function, specifically during neurodegenerative disorders. We will discuss the nature of each glial malfunction, its specificity to each disorder, overall contribution to the disease progression and assess its potential as a future therapeutic target.
Glia are non-neuronal cells of the nervous system which do not generate electrical impulses yet communicate via other means such as calcium signals. Due to their lack of electrical activity, it was previously assumed that glial cells primarily functioned as “nerve-glue” (Virchow, 1860) and performed house-keeping functions for neurons; however, this concept has shifted due to recent findings showing glia are key components in many neuronal functions that go far beyond housekeeping (Araque et al., 1999; Buskila et al., 2019a).
Glial cells are categorized into two main groups; macroglia, which includes astrocytes, oligodendrocytes, NG2-glia and ependymal cells, and microglia which are the resident phagocytes of the central nervous system (CNS). Each population of glial cells is specialized for a particular function in the central or peripheral nervous system (García-Cabezas et al., 2016), and normal brain function depends on the interplay between neurons and the various types of glial cells. In this review, we will focus on astrocytes and microglia.
Glia are involved in many fundamental processes in the CNS including regulation of microcirculation (Zonta et al., 2003), formation and pruning of synapses (Allen et al., 2012), synaptic plasticity (Buskila and Amitai, 2010), and learning and memory (Ben Menachem-Zidon et al., 2011). Glia are also essential for the regulation of the blood-brain barrier (BBB; Lippmann et al., 2012), clearance of metabolic waste during sleep (Xie et al., 2013) and maintenance of synaptic transmission via ion homeostasis which includes K+ clearance (Capuani et al., 2016) and uptake of neurotransmitters (Lehre and Danbolt, 1998). Moreover, glia plays a key role during neuroinflammation (Kékesi et al., 2019) and their activity determines its severity and overall neurotoxic effects (Liddelow et al., 2017).
Glia play a key role in synapse formation and function during development and adulthood (John Lin et al., 2017). Synapse development can be divided into synaptogenesis, pruning, and stabilization (Koeppen et al., 2018). Microglia and astroglia are ideally positioned to affect these activities as they are in close association with synapses, axons, and dendrites, which allows them to monitor and alter synaptic functions (Perez-Alvarez et al., 2014). Evidence suggests synapses can be formed without glial support (Ullian et al., 2001); however, these synapses are functionally premature. Hence, interactions between neurons and glia are essential for synapse generation and function (Ullian et al., 2004; Stogsdill and Eroglu, 2017).
During synaptogenesis, astrocytes play a pivotal role in the formation and regulation of the number of synapses. This was first recognized in retinal ganglion cells cultured in the absence of astroglia, which formed 10-fold fewer synapses compared to cultures containing astrocytes (Ullian et al., 2001). Moreover, astrocyte-conditioned medium originated from astrocytes from different brain regions, including the midbrain, cortex, hippocampus, and cerebellum, found to differ in their gene expression profiles, which effect their synaptogenesis potential (Buosi et al., 2018). The formation of synapses requires the assembly of presynaptic axon terminals and postsynaptic dendritic processes into a functioning unit that allows the transmission of electrochemical signals (Priller et al., 2006; Zeng et al., 2018). This transformation is induced by astrocytic secreted factors including EphrinA3 (Iadecola and Nedergaard, 2007; Nishida and Okabe, 2007; Schiweck et al., 2018), thrombospondin (Christopherson et al., 2005; Xu et al., 2010), neuroligin (Stogsdill and Eroglu, 2017), and D-Serine (Sultan et al., 2015) which are involved in the formation of excitatory synapses. Moreover, astrocytes are involved in the regulation of molecules that can modulate synaptic plasticity and long-term potentiation (Ben Menachem-Zidon et al., 2011), including glutamate, ATP, adenosine and lactate (Araque et al., 1998; Coco et al., 2003; Martín et al., 2007; Sotelo-Hitschfeld et al., 2015; Lepannetier et al., 2018; Gonçalves et al., 2019). Astrocytes also seem to regulate the stability of synapses via direct contact, as they are essential for the morphological maturation of dendritic protrusions and promote their conversion into spines (Nishida and Okabe, 2007).
In addition to their role during synaptogenesis, plasticity, and maintenance, glial cells can remove synapses in a process termed synaptic pruning, further supporting their role in memory and learning processes (Jo et al., 2014). Microglial processes are constantly scavenging the extracellular space for pathogens and cellular debris. Throughout this surveillance process, in which they are in contact with axons, synapses, and dendritic spines, they phagocyte disused synapses (Lauterbach and Klein, 2006; Paolicelli et al., 2011; Hong et al., 2016b; Diaz-Aparicio et al., 2020; Milior et al., 2020), and therefore prevent energy and nutrient loss that is much needed at more active synapses. Hence, this synaptic pruning optimizes the effectiveness of neuronal transmission and functional plasticity in the brain and has been postulated to improve learning and memory capability (Millán et al., 2019). Indeed, abnormalities in synaptic pruning by microglia have been associated with the cognitive decline that accompanies many neurodegenerative disorders such as Alzheimer’s disease (AD; Hong et al., 2016a).
Astrocytes have also been implicated in synapse loss and regulation of long-term memory (Ben Menachem-Zidon et al., 2011). A recent study showed that over-expression of the protein Ephrin-B1 in astrocytes (that is typically linked to synapse formation and stabilization) led to synaptic loss and a decrease of glutamatergic signals (Koeppen et al., 2018). It is thought that during physiological activity, Ephrin-B1 expressed in astrocytes regulates synaptic remodeling by competing with neuronal Ephrin-B1 and thus restricting new synapse formation (Nikolakopoulou et al., 2016; Koeppen et al., 2018). Moreover, Nikolakopoulou et al. (2016) suggested that astrocytic Ephrin-B1 is activated via the astrocytic STAT-3 mediated signaling and affects EphB receptors in microglia which then triggers microglia-mediated synaptic pruning. These recent studies provide new insights about astrocyte-microglia communication that can impact synaptic pruning and therefore should be investigated as a new therapeutic target to prevent synapse loss in neurodegenerative diseases.
A crucial glial function is the maintenance of the concentration of ions in the interstitial space to allow proper synaptic transmission (Capuani et al., 2016). Neuronal activity causes a transitory rise in extracellular K+ concentration, which must be cleared to avoid hyperactivity of the neuron (Chever et al., 2010). K+ clearance takes place primarily by active transport through astrocytic transporters and channels (termed K+ uptake) which then transport K+ via gap junctions to other astrocytes and back to the extracellular space, in a process known as spatial buffering (Orkand et al., 1966; Buskila et al., 2019a). This is an essential astrocytic function that prevents the harmful build-up of K+ levels in the extracellular space (Bellot-Saez et al., 2017). Impairments in astrocytic K+ clearance are thought to contribute to hypersynchronous oscillations (Bellot-Saez et al., 2018) and initiation of seizure activity in various neurological disorders as recently reviewed by Bellot-Saez et al. (2017).
In addition to K+, glial cells are responsible for the regulation of several other ions in the extracellular space including sodium (via the activity of the Na+, K+-ATPase together with a plethora of voltage-gated sodium channels; Wang et al., 2012), chloride (Ratté and Prescott, 2011) and hydrogen (via the activity of the Na+-HCO3 and Na+/H+ exchanger (Ghandour et al., 2000). Moreover, glia form the glymphatic system, which is responsible for the removal of metabolic waste products in the brain, facilitated by Aquaporin-4 (AQP4) channels expressed in astrocytic end-feet (Iliff et al., 2012). Astrocytic AQP4 channels facilitate CSF-ISF exchange and thus modulate water flux (and shrinkage) from the astrocyte to optimize the interstitial space during neuronal activity (Nagelhus et al., 2004) and the sleep-wake cycle (Ding et al., 2016).
For proper synaptic transmission, neurotransmitters (e.g., GABA, glycine, and glutamate) must be transported away from the synaptic cleft immediately after release. Glutamate is the most common excitatory neurotransmitter in the brain, however, excess glutamate residing in the extracellular space can be neurotoxic (Ye et al., 2013). Many cells in the CNS are involved in glutamate reuptake, but astrocytes are responsible for the majority of this uptake (Bergles and Jahr, 1997; Lutgen et al., 2016) with some estimates as high as 60–80% of total glutamate uptake (Rothstein et al., 1996) via the excitatory amino acid transporters EAAT1 and EAAT2 (Bergles and Jahr, 1998). Hence, changes in astrocytic function to regulate neurotransmitter uptake are prone to affect synaptic function as well as cytotoxicity. Indeed, glutamate excitotoxicity is implicated in some neurodegenerative diseases, for a detailed review see Dong et al. (2009).
The BBB is a highly selective border around the cerebral vasculature that prevents pathogens, toxic substances, red blood cells, and leukocytes from the circulating blood to cross into the interstitial fluid (Kanmogne et al., 2007). Astrocytes form a crucial part of the BBB by supporting the integrity of the tight junctions between endothelial cells and facilitating the provision of nutrients from blood vessels (Lippmann et al., 2012). Moreover, astrocytes regulate the constriction and dilation of microvessels (Iadecola and Nedergaard, 2007) and thus form the bridge in the neurovascular unit that couples the activity of neurons, astrocytes and BBB endothelial cells (Viggars et al., 2011). These features allow astrocytes to regulate the cerebral blood flow (CBF) in regions of high neuronal activity that requires more glucose and oxygen (Logothetis et al., 2001; Metea and Newman, 2006).
Previous studies indicated a close association between aging and the breakdown of the BBB, suggesting that impairment of neurovascular coupling is responsible for the cognitive decline seen in the elderly (Fabiani et al., 2014; Balbi et al., 2015) and during neurodegenerative diseases such as AD (Fabiani et al., 2014; Tarantini et al., 2017) and MS (Sivakolundu et al., 2020).
The brain has a high metabolic rate (Attwell and Laughlin, 2001), yet does not have a traditional lymphatic system to clear waste products; instead, the glymphatic system, a term coined by the Nedergaard group (Xie et al., 2013), is utilized as the functional waste clearance system of the CNS. This glial-dependant system facilitates the removal of metabolic by-products and soluble proteins from the brain parenchyma via the exchange of solutes between the interstitial fluid and the cerebrospinal fluid (Xie et al., 2013; Wang et al., 2017). The exchange of fluids is driven by arterial pulsation and activity of AQP4 channels expressed in astrocytic endfeet (Iliff et al., 2012), suggesting that impairment of the BBB function can impact waste clearance mechanisms, induction of neuroinflammation and thus neurodegeneration, as seen in BBB associated pathology (Yang et al., 2013). Indeed, AQP4 knockout mice experienced a 55–65% reduction in Aβ clearance compared to WT mice (Iliff et al., 2012). Furthermore, a 40% reduction has been reported in aged mice compared to young mice (Kress et al., 2014), which is consistent with the notion that the glymphatic system is compromised with age, while age is also the biggest risk factor for dementia and several neurodegenerative conditions such as in late-onset AD (Alzheimer’s disease facts and figures; Alzheimer’s Association, 2019). Sleep has also been shown to affect the glymphatic system, with the clearance of toxic proteins during sleep occurring twice as fast as during awake periods in mice (Xie et al., 2013).
Neuroinflammation is an immune response to harmful stimuli in the nervous system, triggered by infection or injury that leads to microglia and astrocyte activation, aiming to defend the neuronal tissue by clearing and controlling the harmful stimuli (Davalos et al., 2005). Microglia are the resident immune cells of the CNS and are known to actively investigate their microenvironment using their processes (Nimmerjahn et al., 2005). During an injury, infection, or disease state, microglia become “activated,” whereby they migrate towards sites of injury or infection and initiate an inflammatory response by producing cytokines and chemokines (Brockhaus et al., 1996). However, recent studies showed that microglial activation can span between the canonical pro-inflammatory response (termed M1 microglia) to an alternative activation (termed M2), in which microglia release anti-inflammatory cytokines (e.g., BDNF, IGF-1, and TGF-β), and facilitate phagocytosis, tissue repair, and regeneration (Park et al., 2016; Zhou et al., 2017). Moreover, the M2 phenotype is associated with increased Aβ clearance in AD (Kawahara et al., 2012), neurogenesis and higher survival rate of progenitor stem cells (Yuan et al., 2017), indicating that shifting microglia into the M2 phenotype is a plausible treatment in neurodegenerative disorders, as recently suggested by Zhang et al. (2018).
However, some have argued the M1/M2 polarization model is a conceptual deficit and needs to be reconsidered as the categorization into “classical” or “alternative” state is an over simplification that does not capture the heterogeneity of microglia (for extensive reviews see Martinez and Gordon, 2014; Ransohoff, 2016). Indeed, transcriptomics has uncovered gene expression changes much beyond what can be appreciated with a handful of markers. One such study looked at gene expression profiles of mice that underwent traumatic brain injury (TBI). Individual cells were analyzed using RNA-sequencing to investigate if M1-like or M2-like macrophages could be detected (Kim et al., 2016). The study found that expression of a polarization marker was no more likely than random to be co-expressed within the same cell, concluding that microglia are not made up of polarized groups, but proposing that individual macrophages undergo a wide range of activation (Kim et al., 2016).
Like microglia, astrocytic activation is also hypothesized to range between two poles of a spectrum. On the one side of the spectrum, A1 activated astrocytes promote cell death, the release of neurotoxins (i.e., IL-1β, IL-6, TNF-α, IFN-γ) and activation of catabolic processes (Liddelow et al., 2017). On the other side of the spectrum, A2 astrocytes are beneficial as they provide neuroprotection by releasing antioxidants and neurotrophic factors via activation of the JAK/STAT3 pathway (Zamanian et al., 2012; Liddelow et al., 2017). Evidence suggests that A1 reactive astrocytes are induced by cytokines released from activated microglia (i.e., Il-1α, TNFα, and C1q; Zhang et al., 2014). These activated A1 astrocytes lose the capacity to support neuronal survival, induce the death of oligodendrocytes and neurons and accompany many neurodegenerative diseases such as Huntington’s disease (HD), AD and Amyotrophic lateral sclerosis (ALS; Liddelow et al., 2017). Hence, the prevention of A1 astrocytic formation and transformation of A1 astrocytes towards the A2 phenotype can serve as a therapeutic target. Although this classification provides insight towards astrocytic heterogeneity, the categorization of activated astrocytes into the A1/A2 model has been recently challenged and concerns have been raised that this classification would be used as a shortcut to imply a mechanistic understanding (Vainchtein and Molofsky, 2020). Cunningham et al. (2019) recently suggested that this classification is oversimplified which puts into risk the complexity of the factors shaping astrocyte phenotype, and thus astrocytic heterogeneity. Evidence in the field is yet to establish whether the A1/A2 classification is suitable, with recent opinion questioning if the grouping may be a simplification of the various cellular states that astroglia assumes, both in diseased and healthy states (Arranz and De Strooper, 2019).
The tripartite synapse is a functional unit comprised of a neuronal component (pre- and post-synaptic membranes) along with an ensheathing astrocyte (Araque et al., 1999). Evidence shows that the number of tripartite synapses varies between brain regions, suggesting different levels of neuron-glial communication. In the rodent brain, synapses ensheathed by astrocytes account for 60–90% of total synapses in the cerebellum (Xu-Friedman et al., 2001), 77% in the striatum (Chai et al., 2017), 86% in the hippocampus (Chai et al., 2017) and more than 90% of synapses in the barrel cortex (Genoud et al., 2006). Although it remains unclear if tripartite synapse numbers in rodents are comparable to humans, a previous study shows that human astrocytes are significantly larger and structurally more complex when compared to rodents (Oberheim et al., 2009). However, this number may be harder to estimate as tripartite synapses are dynamic and can change following neuronal activity or sensory stimulation (Schipke et al., 2008). It is not well documented how these synapses and their associated mechanisms are affected in pathology, however, evidence suggests that an imbalance in the tripartite synapse activity is implicated in epilepsy and neuropsychiatric disorders such as schizophrenia (Mitterauer, 2018).
Astrocytes are functionally coupled via gap junctions comprised of a pair of connexons which connect across intracellular space (Nagy et al., 2001; Johnson et al., 2018) and link the cytoplasm of two cells (Hofer and Dermietzel, 1998; Wei et al., 2004; Wilson and Mongin, 2019). Astrocytic gap junctions have several functions, including facilitation of electrical and chemical communication between cells (Kettenmann and Ranson, 1988), affecting neuronal rhythmicity in the brainstem (Kadala et al., 2015; Condamine et al., 2018), distribution of excess K+ from high concentration regions to low concentration areas, and allowing transmembrane transport of small molecules to share metabolic demands across cells (Huguet et al., 2016). This communication method allows for astrocytes to work as a syncytium and propagate signals through the astrocytic network (Halassa et al., 2007). The failure of such connectivity and dysfunction of signal propagation results in a malfunctioning nervous system and can be a factor in the progression of neurodegenerative disorders.
Astrocytic connectivity is also paramount for neuronal—astrocytic communication. Previous studies showed that neuronal activity triggers glial responses, resulting in changes in the astrocytic Ca2+ concentration (Poskanzer and Yuste, 2011, 2016). Astrocytes respond to neurotransmitters and neuromodulators via activation of G-protein coupled receptors (GPCRs) which leads to the release of 1,4,5-triphosphate that induces Ca2+ release (Lev-Ram and Ellisman, 1995). These glial Ca2+ signals are not restricted to a single cell and can propagate as a calcium wave within the glial syncytium (Cornell-Bell et al., 1990; Schipke et al., 2002) and thus amplify the original neuronal signal, as recently proposed by Chen et al. (2019).
Neurons can modulate glial function via activation of a wide variety of receptors for different neuromodulators, including acetylcholine (Ach, nicotinic α/β and metabotropic M1–4; Oikawa et al., 2005; Amar et al., 2010), histamine (H1–3; Inagaki et al., 1989), serotonin (5-HT1,2,5,6,7; Hirst et al., 1998), Noradrenaline (NA, α1,2-adrenoreceptors and α1,2-adrenoreceptors; Bekar et al., 2008; Morin et al., 1997) and dopamine (DA, D1–5; Khan et al., 2001; Wei et al., 2018). In most cases, the neuromodulatory agent will lead to either change in calcium signaling in astrocytes (see Figure 1) and microglia or alterations of the coupling ratio, affecting glia to work as a syncitium. Indeed, recently, Ma et al. (2016) showed that neuromodulators can signal through astrocytes by affecting their Ca2+ oscillations to alter neuronal circuitry and consequently behavioral output. In line with these observations, Nedergaard’s group further demonstrated that bath application of a cocktail of neuromodulators to cortical brain slices increased [K+]o regardless of synaptic activity (Ding et al., 2016), suggesting that increased [K+]o could serve as a mechanism to maximize the impact of neuromodulators on synchronous activity and recruitment of neurons into networks (Bellot-Saez et al., 2018; Zupanc, 2020). Moreover, Bellot-Saez et al. (2019) recently showed that different neuromodulators, such as 5-HT, NA, DA, and histamine affect the astrocytic K+ clearance rate via different pathways, establishing a synergistic mechanism between neurons and astrocytes that promote excitability and network oscillations. Also, Chen et al. (2019) reported that in the hypothalamus, norepinephrine activates corticotropin-releasing hormone (CRH) neurons to engage astrocytes via dendritic retrograde signaling. This activation leads to astrocytic release of ATP in upstream neurons, thus allowing CRH neurons to amplify their dendritic signaling and control distal presynaptic neurons.
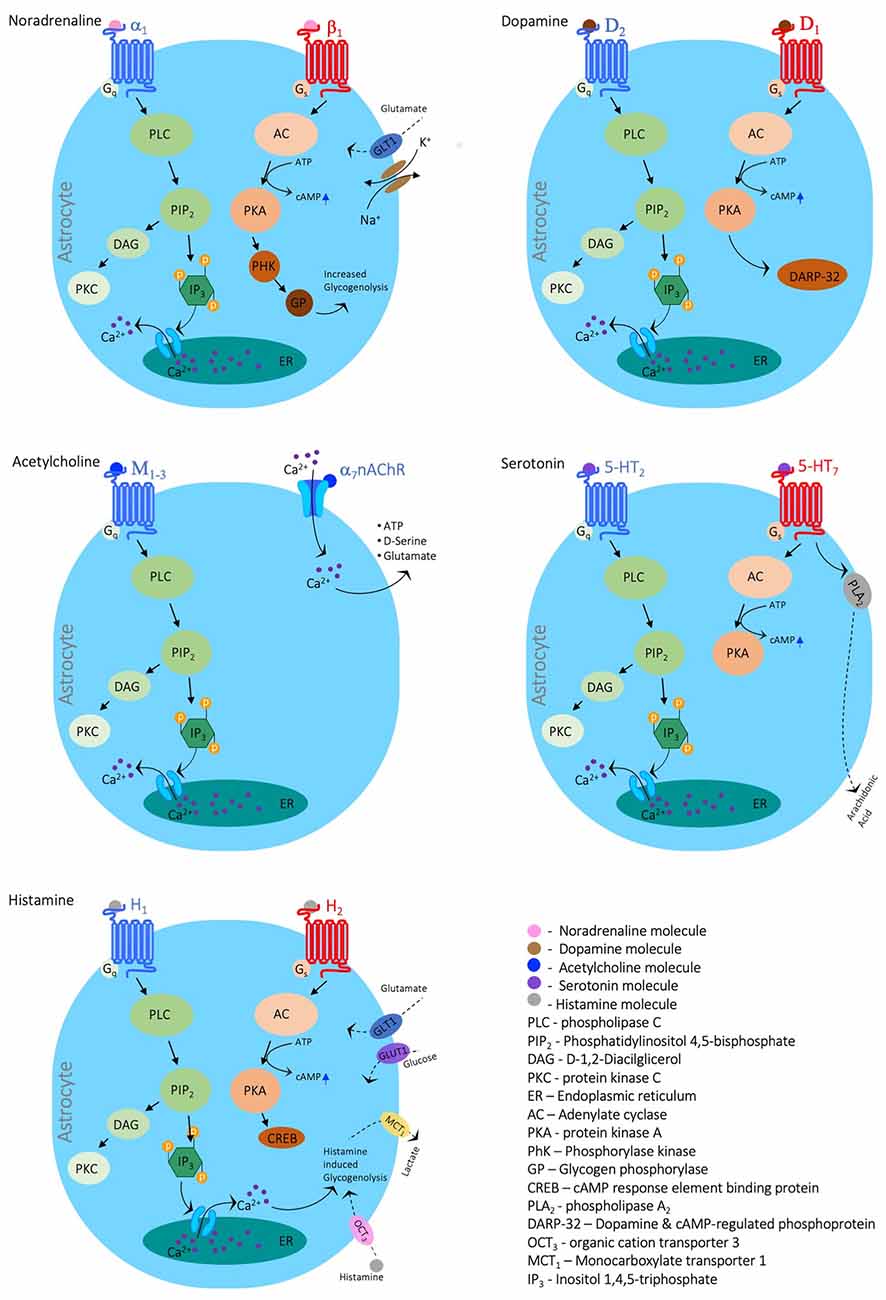
Figure 1. Neuromodulators’ impact on astrocytic calcium signaling. Schematic diagrams depicting the main signaling cascades in which the neuromodulators Noradrenaline, Dopamine, Acetylcholine, Serotonin, and Histamine evoke calcium signals. The figure was created with the Motifolio toolkit (Motifolio Inc., Ellicott City, MD, USA).
Both noradrenaline and serotonin exhibit profound effects on astrocytic metabolism (Cambray-Deakin et al., 1988). Adrenergic agonists cause a disruption in astrocytic metabolism in the form of increased ATPase activity (Kimelberg et al., 1978) and have been shown to prime astrocytes for local circuity alterations and changes in behavioral state (Paukert et al., 2014). Whereas serotonin has the effect of altering K+ permeability in glial cells of rodent astrocytes (Hösli et al., 1993). It is hypothesized that a breakdown of communication in the adrenergic pathway in the locus coeruleus and the serotonergic pathway in the raphe nuclei leads to a break down in metabolic and functional neuron-glia communication causing the degradation of neurons in those regions, contributing to Alzheimer’s progression (Hertz, 1989).
However, the impact of neuromodulators on glial activity is complex and highly dependent on the receptor identity. For example, activation of α7 nAChRs led to a decrease in the release of pro-inflammatory cytokines such as IL-6 and TNF-α (Liu et al., 2012; Li et al., 2013; Kalashnyk et al., 2014), an increased expression of glial cell-derived neurotrophic factor (GDNF; Takarada et al., 2012) and upregulation of glutamate uptake via the glutamate/aspartate transporter (GLAST; Mashimo et al., 2010; Morioka et al., 2014), suggesting acetylcholine promotes neuroprotection via inhibition of microglia. On the other hand, ACh was found to cause an increase in intracellular Ca2+ which stimulates the release of glutamate (Wang et al., 2013), triggering GABAergic transmission in astrocytes (Banerjee et al., 2012; Beggiato et al., 2013) and thus affecting the activity of network oscillations. Moreover, cholinergic transmission affects satellite glia to enwrap and support sensory peripheral neurons that were previously deprived of nerve growth factor (Enes et al., 2020).
Recently, Bosson et al. (2015) showed that blockade of dopaminergic transmission in the substantia nigra increased astrocytic calcium activity and gap junction coupling, resembling morphological alterations of Parkinson’s disease (PD; Bosson et al., 2015), and further indicating that neuron-glia dopaminergic communication may play a part in the etiology of PD. While histamine plays a critical role in maintaining the energy balance of the CNS by regulating glucose allocation/distribution (Jurič et al., 2011), it is also purported to increase the excitability of neurons and increase astrocytic uptake of Ca2+ in vitro (Jung et al., 2000). Further exploration of the effect of neuromodulators on astrocytic processes will enrich our understanding of astrocyte functionality and the roles they play in healthy and pathological states.
While our understanding of the neuroinflammatory process is constantly developing it is clear that it is a common characteristic which is preserved across several neurodegenerative diseases (Thameem Dheen et al., 2007) including PD (Ghadery et al., 2017), AD (Schuitemaker et al., 2013), ALS (Sargsyan et al., 2005) and multiple sclerosis (MS; Kozela et al., 2011). A plausible explanation for this phenotype is that the inflammatory profile of microglia changes with age (DiSabato et al., 2016). It is thought that microglia in the aged brain is primed, as demonstrated by their elevated expression of inflammatory mediators, with a reduced activation threshold and higher levels of inflammation following activation. Moreover, once activated, aged microglia are more resistant to regulation (Norden and Godbout, 2013), which can be a contributing factor to the progression of neurodegenerative diseases, as active microglia synthesize and release neurotoxic substances that cause further neuronal damage (Lull and Block, 2010). In a healthy environment, activation of microglia serves as a protective mechanism, however, due to the complex and multifactorial nature of microglial activation, subtle changes may lead to amplified or dysfunctional activation which is thought to enhance neurodegenerative pathology, with cytokines presenting at almost all disease phases (Brambilla, 2019).
Previous studies showed that glial activation is controlled by neuronal activity, which can “turn-on” or “turn-off” signals that either keep microglia in their resting quiescent state or activate them into a proinflammatory state, for a detailed review see Biber et al. (2007). One of the main agents through which neurons affect glial activation is the CD200 glycoprotein, which inhibits microglial priming and holds microglia in a quiescent state (Shrivastava et al., 2012). Indeed, alterations in the distribution of the CD200 or its glial receptor CD200R can lead to overactivation of microglia and neuroinflammation (Hernangómez et al., 2012) that accompany neurodegeneration (Oria et al., 2018). Other neuronal agents that affect the course of neuroinflammation are chemokines, such as CX3CL1, CXCR4 and CCL2 which promotes microglial scavenging (Meucci et al., 2000; Hatori et al., 2002; Paolicelli et al., 2014), regulation of the BBB integrity (Rabinovich-Nikitin et al., 2016) and chemotaxis (Coughlan et al., 2000; Conductier et al., 2010) respectively. These chemokines activate diverse intracellular signaling pathways, including PI3K, AKT, and NF-κB to regulate phagocytosis, proliferation, and migration.
The Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) pathway is a common mediator of astrocyte reactivity in neurodegenerative diseases (ND) and is highly conserved between disease states (Ben Haim et al., 2015). Previous studies showed that this pathway shifts astrocytes towards the more beneficial A2 phenotype, which reduces inflammation and astrogliosis (Okada et al., 2006). Indeed, recently Ben Haim et al. (2015) showed that inhibition of the (JAK/STAT3) pathway increased the number of pathological huntingtin aggregates in animal models for HD. On the other hand, activation of the NF-κB pathway in glial cells during inflammation was found to promote neurotoxicity and neurodegeneration, reviewed by Dresselhaus and Meffert (2019).
Some molecules released by neurons are found to have a dual effect on the development and progression of neuroinflammation. For example, the brain-derived neurotrophic factor (BDNF), which is released from neurons, can either activate the JAK/STAT3 pathway, which shifts microglia and astrocytes towards a more beneficial phenotype that reduces inflammation and astrogliosis (Okada et al., 2006; Lund et al., 2008; Hixson et al., 2019), or activate the receptor TrkB which leads to activation of the NF-κB detrimental pathways (Colombo et al., 2012). These alterations might stem from the mode of activation, as continuous administration of BDNF or its overexpression led to attenuation of microglial activation in a mouse model of HD (Lund et al., 2008; Xie et al., 2010; Giampà et al., 2013; Hixson et al., 2019), while acute administration of BDNF increased microglial activity (Jiang et al., 2010) and accelerated neurodegeneration (Colombo et al., 2012).
Glial recruitment and activation during neuroinflammation are also modulated by neuronal release of neurotransmitters (e.g., glutamate and GABA) and extracellular nucleotides, in particular ATP and UDP (Haynes et al., 2006; Ulmann et al., 2013). While GABA and glycine lead to a decrease in the LPS-induced secretion of several cytokines (Kuhn et al., 2004) and attenuation of the phagocytic activity of microglia (Carmans et al., 2010), the neuronal release of glutamate can either inhibit microglia activation via metabotropic receptors (Taylor et al., 2002, 2003) or mediate glial activation via AMPA and kainate ionotropic receptors (Hagino et al., 2004). Moreover, the release mechanisms of ATP (non-specific release as occurs during necrosis vs. vesicular exocytosis), were found to be key effectors on the inflammatory pathway and are capable of fine-tuning the glial response throughout neuroinflammation, reviewed by Dosch et al. (2018). Given the importance of the above signaling pathways on the progression of inflammation, in which, even minor changes are sufficient to cause fatal dysregulation of glial function and neurodegeneration, they are increasingly becoming the focus in the development of new therapeutics.
Neurodegenerative diseases are conventionally regarded as a progressive neurological disorder associated with the loss of specific populations of neurons. As a result, neurodegenerative diseases such as Parkinson disease, AD, epilepsy, and ALS have traditionally been associated with neurodegeneration of dopaminergic, cholinergic, GABAergic and motor neurons respectively (Davies and Maloney, 1976; Leenders et al., 1990; Xu-Friedman et al., 2001; Buskila et al., 2019b). However, increasing evidence suggests that glial dysfunction may be a significant contributor to the pathology of these diseases, as indicated in Supplementary Table S1. In this chapter, we will summarize specific contributions of glial dysfunction to different neurodegenerative diseases (see also Figure 2).
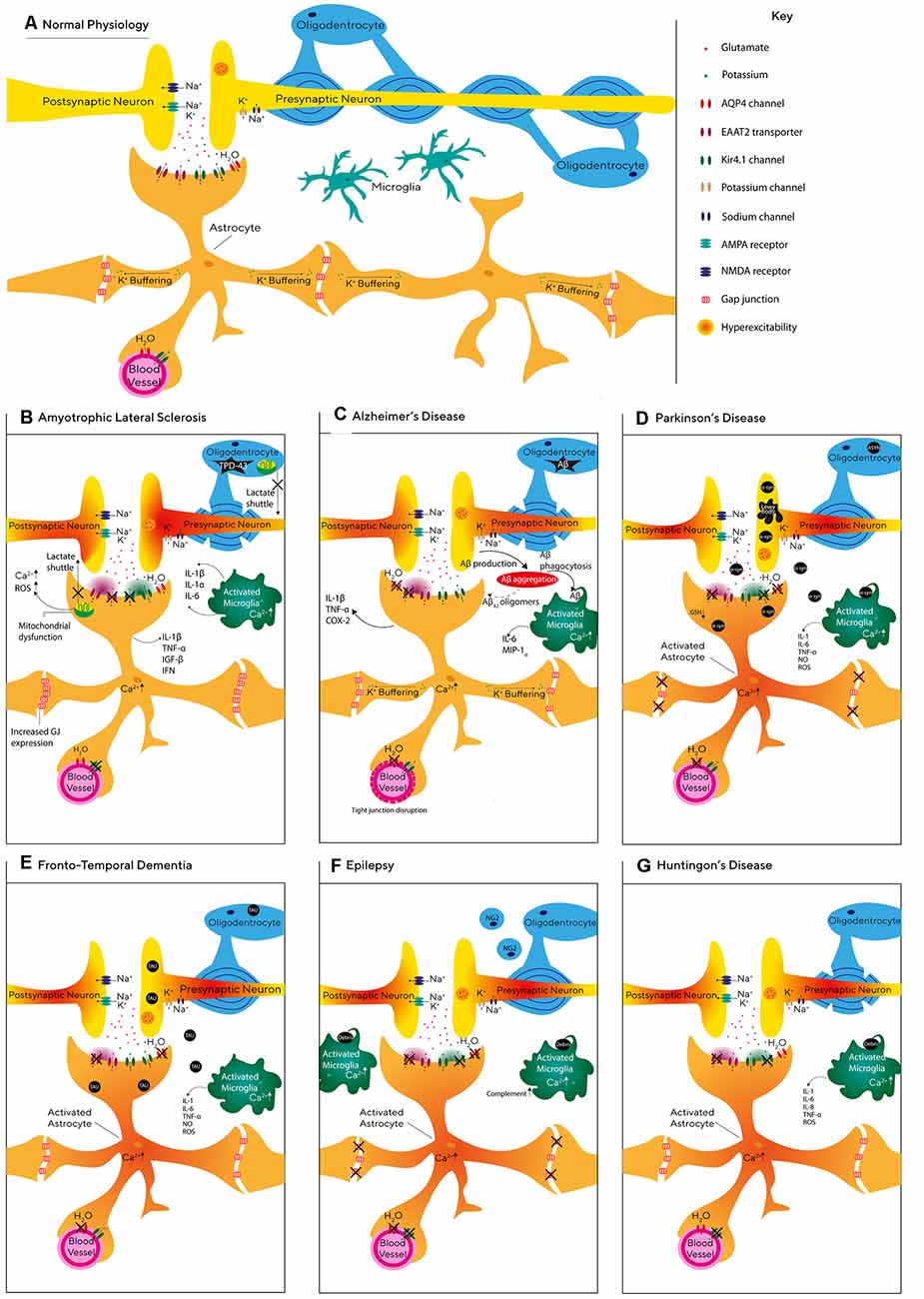
Figure 2. Glial pathophysiology during neurodegeneration. (A) Schematic representation of the relationship between neurons and glial cells, including astrocytes, oligodendrocytes, and microglia. (B–G) Graphic illustration of the molecular and cellular processes affecting astrocytes, microglia, and oligodendrocytes during Alzheimer’s disease (AD; C), Epilepsy (F), Frontotemporal dementia (FTD; E), Parkinson’s disease (PD; D), Huntington disease (G), and amyotrophic lateral sclerosis (ALS; B).
Epilepsy is a complex neurodegenerative disorder characterized by recurrent seizures and includes both focal and generalized seizures (Devinsky et al., 2013). Epilepsy can develop for several reasons including genetic disposition (Ottman, 2005), TBI, infections (Löscher et al., 2015), prenatal injury (Scher, 2003), and developmental disorders (Lehéricy et al., 1995). Seizures are thought to be caused by the synchronized firing of groups of neurons which results from an imbalance between inhibitory and excitatory currents (Scharfman, 2007). Therefore, previous epilepsy research focused on neurons, however, recent evidence indicates that glial cells are also involved in the pathology of epilepsy.
One of the hallmarks of acquired epilepsy is glial scarring formed by hypertrophic astrocytes (Das et al., 2012; Do Nascimento et al., 2012), in which the gap-junction connectivity between astrocytes is reduced (Wallraff et al., 2006). Indeed, recently Bedner et al. (2015) showed that impairments in astrocytic gap-junction coupling start within 4 h following status epilepticus. In line with this study, acute brain slices prepared from transgenic mice with coupling-deficient astrocytes expressed spontaneous epileptiform activity and increased synaptic transmission (Wallraff et al., 2006; Pannasch et al., 2011). Dysfunctional astrocytes may be pivotal to the generation and propagation of epileptic seizures (Seifert et al., 2006) via their inability to sufficiently clear K+, as increasing extracellular K+ concentration has been shown to increase synchronous oscillatory activity (Bellot-Saez et al., 2018) and induce epileptiform activity in brain slices (Gabriel et al., 2004). K+ buffering in astrocytes is also regulated by Kir4.1 channels which are found in astrocytic endfeet (Zurolo et al., 2012). Indeed, the variation of the KCNJ10 gene that encodes Kir4.1 channels has been linked to seizure susceptibility (Ferraro et al., 2004; Wallraff et al., 2006). Moreover, dysfunctional astrocytes with decreased expression of Kir4.1 channels displayed impaired clearance of extracellular K+ and glutamate which led to neuronal hyperexcitability (Patel et al., 2019). Although there is an ongoing debate as to whether gliosis is consequential or a causative effect of seizures (Patel et al., 2019), evidence in support of the latter has shown that astrogliosis alone can cause spontaneous seizures in the absence of other pathologies (Robel et al., 2015).
It has been hypothesized for some time that astrocytic processes grow towards synapses which release high levels of glutamate as a mechanism to offset the spill-over and prevent hyperexcitability (Ventura and Harris, 1999). Indeed, previous studies found that patients with epilepsy expressed lower levels of glutamine synthetase (GS) in the hippocampus (Petroff et al., 2002; Eid et al., 2004), which results in accumulation of glutamate in astrocytes and the extracellular space. Similarly, animal models for epilepsy showed a significant reduction of GS activity during the chronic epileptogenic phase (Kang et al., 2006; Bidmon et al., 2008; Hammer et al., 2008). Hence, it has been suggested that reduction in GS activity is the main reason for increased extracellular glutamate levels and seizure generation.
Research has established that astrocytes alter their morphology and domain organization in neurodegenerative disease, to the extent that there is a 10-fold increase in overlap of astrocytic processes in mouse models of epilepsy (Golarai et al., 2008). A recent study suggested that during the progression of epilepsy, the tripartite synapse undergoes molecular, structural, and functional changes in the hippocampus (Clarkson et al., 2020). The study reported that in addition to decreased density in GLT-1 expression, astrocyte processes increased ensheathment at synapses in an attempt to decrease excitability by raising the levels of glutamate uptake (Clarkson et al., 2020). Another astrocytic mechanism that can lead to epilepsy involves the dislocation of AQP4 water channels and reduced expression of Kir channels in astrocytes, which affect K+ clearance and enhance the propagation of seizures (Lee et al., 2004; Binder et al., 2006; Seifert et al., 2006; Szu et al., 2020) These findings demonstrate the importance of neuron-glia communication mechanisms when considering new therapeutic targets for epilepsy.
HD is an inherited brain disorder characterized by the progressive loss of selected neuronal populations within the corticostriatal circuit and glial cell malfunction (Tong et al., 2014; Jiang et al., 2017). One of the earliest astrocytic dysfunctions in HD is a decrease in the expression of glutamate transporters (Faideau et al., 2010) and a decrease in extracellular glutamate uptake in both the striatum and cerebral cortex (Przedborski et al., 2000; Liévens et al., 2001; Behrens et al., 2002; Shin et al., 2005; Hassel et al., 2008; Baljit et al., 2017). Moreover, the mutated huntingtin protein is reported to lead to a decrease in the expression of Kir4.1 channels and impairment of astrocytic K+ clearance capabilities (Tong et al., 2014), leading to hyperexcitability of local striatal neurons (Tong et al., 2014; Benraiss et al., 2016). Indeed, restoring the expression of astrocytic Kir4.1 channels in an HD mouse model rescued glutamate signaling, emphasizing the importance of K+ homeostasis in the development of HD (Jiang et al., 2016).
Recently, the Khakh group reported that astrocyte morphology and spatial territory at the striatum are significantly lower and precede synaptic dysfunction in an HD mouse model (Octeau et al., 2018). Dysfunctional glial cells have been shown to play a critical role in HD progression, as mutant huntingtin expressing human glial progenitor cells engrafted in healthy mice were found to impart the disease phenotypes (Benraiss et al., 2016). Moreover, the introduction of healthy human glial cells into transgenic HD mice significantly increased survival rates and reduced motor deterioration, along with the reinstatement of K+ homeostasis (Benraiss et al., 2016). Furthermore, Garcia et al. (2019) used patient-derived HD astrocytes to demonstrate that the dysfunction of these cells leads to less astrocytic support and decreased protection from glutamate toxicity, suggesting astrocytic malfunction should be regarded as a major therapeutic target in HD.
ALS is the most common type of motor neuron disease, affecting the brain and spinal cord and causing progressive loss of the upper and lower motor neurons controlling muscle movement, breathing, and speech (Martin et al., 2017). Astrocytes are thought to contribute to the progression of ALS through the abnormal function of glutamate transporters and extracellular K+ concentration, leading to elevated levels of extracellular glutamate and excitotoxicity (Rothstein et al., 1995; Do-Ha et al., 2018). Indeed, SOD1 mutations that are thought to facilitate oxidative stress (Barber et al., 2006) and are typically found in patients who suffer from the familial form of ALS (Tang et al., 2019) were accompanied with reduced expression of EAAT2 transporters in astrocytes (Pedersen et al., 1998). Moreover, Gong et al. (2000) showed that the expression of mutated SOD1 must exist in both neurons and astrocytes for ALS to progress, and expression of mutated SOD1 in neurons alone was not sufficient to trigger ALS (Pramatarova et al., 2001). Although, other in vivo studies do indicate that transplanted glial-restricted precursor cells in wild-type rats are sufficient to induce motor neuron death (Papadeas et al., 2011). This is evidence that ALS is not a disease of astrocytes or neurons alone, and that both cell types are involved in the pathogenesis of ALS (Rothstein et al., 1995; Do-Ha et al., 2018).
Frontotemporal dementia (FTD) is the most common type of dementia for those aged 65 and under (Hogan et al., 2016) and is characterized by atrophy of the frontal anterior temporal lobes (Broe et al., 2004). Although the role astrocytes play in FTD is not fully understood, a meta-analysis of FTD cases revealed that astrocytic cell death increased as the disease progressed, while neuronal apoptosis remained constant (Broe et al., 2004). These studies indicate that the neurodegeneration seen in FTD may be facilitated by the loss of astrocytic support. Indeed, a recent study conducted by Hallmann et al. (2017) showed that FTD astrocytes demonstrated changes in whole-genome expression including upregulation of genes linked with synapse organization and transmission (CHRNA1). Moreover, decreased expression of EAAT2 has also been observed in the frontal cortex of FTD patients compared with controls (Umoh et al., 2018), suggesting that deficiencies in astrocytic glutamate uptake may contribute to excitotoxicity and neuronal degeneration (see Figure 2).
FTD progression is accompanied by protein alterations, neuroinflammation, and vascular dysfunction (Martinac et al., 2001; Garbuzova-Davis et al., 2012), which profoundly involve microglia and astrocytes (Philips and Robberecht, 2011). Indeed, microglial activation has been associated with neuronal loss and TAU deposition suggesting that such microglial activation occurs in the early stages of the disease (Schofield et al., 2003). Moreover, autopsy samples from FTD patients expressed high levels of reactive astrocytes showing signs of degradation which would likely contribute to neuronal cell death (Su et al., 2000).
One of the hallmarks in FTD patients is a reduction of the resting CBF in the frontal, temporal and parietal cortices (Dopper et al., 2016), which is correlated with astrocytic damage and astrogliosis (Martinac et al., 2001). As evidence suggests that altered cerebral perfusion may be an early biomarker of FTD and may even show distinct perfusion patterns (Hu et al., 2010), therapeutics aimed at maintaining cerebral circulation, to retain astrocytic and neuronal health, maybe a beneficial future target.
PD is the second most common neurodegenerative disease (de Lau and Breteler, 2006) with symptoms including resting tremor and bradykinesia (Salarian et al., 2007). PD is characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the formation of α-syn deposits known as Lewy bodies (Kordower et al., 2013; Diniz et al., 2019). Recently, Bosson et al. (2015) reported an increase in the number of glutamatergic synapses, which was accompanied by alterations in astrocytic morphology in the SNr of a rat model to PD. Moreover, intracerebroventricular injection of α-syn increased the number of astrocytes, the density of excitatory synapses, and the levels of TGF-b1 in the striatum of injected animals (Diniz et al., 2019). Similarly, astrocytic spatial territories were found to be larger in the striatum of parkinsonian monkeys (Villalba and Smith, 2011), suggesting that astrocytic remodeling is a compensatory mechanism aiming to decrease the high extracellular glutamate levels and overall neuronal hyperactivity observed in PD.
Neurodegeneration of dopaminergic neurons has also been associated with alteration in the glial antioxidative system. Solano et al. (2008) showed that dysfunction of the PD-associated protein Parkin can lead to a decrease in the synthesis of the antioxidant glutathione (GSH) by astrocytes, underlying the lower levels of GSH found in PD patients (Baillet et al., 2010). Indeed, previous studies indicate that the prevention of oxidative stress protected dopaminergic neurons in an animal model of PD (Chen et al., 2009; Kitamura et al., 2011), further supporting the role of glial malfunction in the degeneration of dopaminergic neurons.
It is well established that glial cells of post-mortem PD patients show high levels of α-syn accumulation (Wakabayashi et al., 2000). Direct neuron-glia transmission plays an essential role in the progression of PD via the release of α-syn at axon terminal sites where the α-syn is taken-up into astrocytes, triggering an inflammatory response (Lee et al., 2010). It is generally thought that astrocytes remove α-syn from the synapse as a protective mechanism (Booth et al., 2017), however, a recent study postulates that astrocytes amplify PD pathology by accumulating and spreading α-syn to neighboring dopamine-producing neurons and thus intensifying neurodegeneration (di Domenico et al., 2019). Indeed, cultured neurons originated from healthy individuals were more susceptible to neuronal death once they were in contact with iPSC-derived astrocytes from a PD patient. However, when healthy astrocytes were cultured with PD patient neurons, α-syn did not accumulate and neuronal function was restored (di Domenico et al., 2019). These studies highlight the role that malfunctioning astrocytes play in the spread of α-syn, accumulating abnormal levels due to impaired protein degradation which may enhance the spread of PD in the brain. As the SNpc region has the lowest astrocyte to neuron ratio in the CNS (Damier et al., 1993), this may explain why dopaminergic neurons are susceptible to PD as they have less astrocytic support than neurons in other regions (de Majo et al., 2020).
AD, the most prevalent form of dementia worldwide (Ferri et al., 2005), is a progressive neurodegenerative disorder characterized by the build-up of β-amyloid (Aβ) plaques, intracellular neurofibrillary tangles (NFTs) with hyper-phosphorylated tau and activated glial cells surrounding senile plaques (Morley et al., 2018). Aβ deposits and tau are primarily derived from neurons, however, the genes associated with the development of late-onset Alzheimer’s, including apolipoprotein (APOE), apolipoproteinJ (APOJ) and sortilin-related-receptor-1 (SORL) are mainly expressed by glial cells (Arranz and De Strooper, 2019), suggesting that both glia and neurons are involved in the pathogenesis of the disease. Moreover, a recent report by Habib et al. (2020) identified a population of disease-associated astrocytes in an AD mouse model, which appeared at early disease stages and increased with disease progression.
Aβ deposits in the brain lead to early loss of synapses and neurons (Buskila et al., 2013), abnormal activation of microglia, and oxidative stress (Leyns et al., 2019). Recently, Beth Stevens’ group reported that phagocytic microglia mediate synaptic loss in the early stages of AD via the complement system, indicating an abnormal activation of the synaptic pruning pathway (Hong et al., 2016b). Moreover, there is growing evidence that glutamatergic communication between neurons and astrocytes at the tripartite synapse is affected during AD progression, as Aβ leads to increased glutamate release from neurons and astrocytes (Pirttimaki et al., 2013; Talantova et al., 2013). Previous studies in an animal model for AD demonstrated that astrocytes in the entorhinal cortex, prefrontal cortex, and hippocampus undergo morphological and functional alterations. These changes include a decrease in the number of astrocytes expressing the enzyme glutamine synthase, atrophy, and a reduction in the surface area, volume, and complexity of astrocytic processes (Olabarria et al., 2011; Beauquis et al., 2013, 2014; Kulijewicz-Nawrot et al., 2013). Moreover, astrocytes at the pre-symptomatic AD phase exhibited a decrease in vesicle trafficking (Stenovec et al., 2016), as well as lower expression of AQP4 and GLT-1, which results in the disruption of water and glutamate homeostasis (Hoshino et al., 2017) and an increase of glutamate release from astrocytes, leading to hyperexcitability and cell death (Buskila et al., 2005; Pirttimaki et al., 2013; Talantova et al., 2013).
Multiple sclerosis (MS) is a disease in which the myelin sheath of axons degenerates, resulting in a wide range of symptoms and functional impairments including pain, muscle weakness, and tremors (Trapp et al., 1998). The canonical view of this disease progression is that CD4+ T-lymphocytes cross the BBB into the parenchyma which causes a cascade of several events leading to inflammatory lesion and demyelination (Laplaud et al., 2004; Bahbouhi et al., 2009). However, a secondary hypothesis states that the disease is neurally initiated and begins in the CNS, caused by metabolic changes affecting neurons and glia which leads to BBB breakdown (Chaudhuri and Behan, 2004). Although somewhat controversial, evidence supporting this theory suggests that neuronal loss and inflammation are present at the very early stages of MS (Filippi et al., 2003). If it is the case that MS begins in the CNS, neuroprotective interventions may be effective as future MS treatments. However, the autoimmune vs. CNS-derived theories of MS are still the source of an ongoing debate in the literature.
The classification of different neurodegenerative diseases is based on certain neurological symptoms expressed in each disorder, however, there is considerable clinical and pathological comorbidity across neurodegenerative disorders. Patients with FTD may present with ALS or parkinsonian syndromes and vice versa (Lomen-Hoerth et al., 2002; Padovani et al., 2007). Also, major risk factors that lead to glial malfunction, such as cardiovascular dysfunction, TBI, and stroke are common in different neurodegenerative diseases, suggesting that similarities between different disorders stem from common glial dysfunction.
The brain has a disproportionate energy demand relative to its size. While it constitutes only 2% of the total body weight, it requires 20% of the oxygen and glucose metabolism (Sokoloff, 1991). Hence, regulation of energy metabolism is essential to maintain proper brain function, see review by Watts et al. (2018). In the adult brain, both neurons and glia use glucose as an energy substrate, however, a small portion of glucose molecules in the brain is stored as glycogen chains preferentially within astrocytes (Obel et al., 2012).
Glycogen storage in the adult brain is highly compartmentalized, with the highest levels in the hippocampus, striatum, and superficial layers of the cortex (Oe et al., 2016). Immunohistochemical studies showed that glycogen has a punctate distribution localized predominantly in astrocytic processes (Oe et al., 2016; Hirase et al., 2019). These studies further indicated an astrocytic heterogeneity, whereas some astrocytes were “glycogen-rich” while others found to be glycogen-poor. Further investigation of the functional compartmentalization of glycogen stores in astrocytes showed that glycogen granules are preferentially located in astrocytic processes around synapses and blood vessels, rather than randomly distributed in the astrocytic cytosol (Calì et al., 2016; Mohammed et al., 2018). This distribution suggests that astrocytic glycogen plays a role in synaptic processes.
Historically, astrocytic glycogen was considered to be an emergency fuel, which is mobilized only once there is discontinuation in the cerebral glucose supply. However, glycogen content in the CNS is low (8–12 μmol/g tissue in the rat brain; Cruz and Dienel, 2002), and can support brain function only for a few minutes under ischemic conditions (Swanson et al., 1989). Recent studies suggest that glycogen metabolism is associated with different brain functions including the regulation of awake-sleep cycles and high cognitive processes such as learning and memory consolidation (Kong et al., 2002; Gibbs et al., 2007; Gibbs and Hutchinson, 2012; Gibbs, 2016; Bellesi et al., 2018). Moreover, glycogen is a precursor for glutamate and glutamine synthesis (Sickmann et al., 2005; Gibbs et al., 2007) and is found to play a neuroprotective role, as it protects neurons from stress conditions including hypoglycemia or hypoxic stress (Obel et al., 2012; Öz et al., 2015; Adeva-Andany et al., 2016).
Glycogen metabolism in astrocytes is actively regulated by neuromodulators, which activates adenyl cyclase and increases intracellular calcium levels (Sorg and Magistretti, 1992; Cardinaux and Magistretti, 1996; Sickmann et al., 2009; Obel et al., 2012; Dinuzzo et al., 2013; Xu et al., 2014). Classical studies by Pierre Magistretti’s group found that neuromodulators such as vasoactive intestinal peptide (VIP), histamine and NA, induce glycogenolysis (the breakdown of glycogen) in astrocytes and lead to a local increase of ATP within the stimulated networks in a concentration-dependent manner (Magistretti et al., 1981; Magistretti and Schorderet, 1984; Magistretti and Morrison, 1988). In line with this work, in vivo studies involving photostimulation of the retina, confirmed that sensory stimulation is associated with glial glycogenolysis (Tsacopoulos et al., 1988; Poitry-Yamate and Tsacopoulos, 1991). Moreover, glycogenolysis was detected in the somatosensory cortex of awake rats immediately after sensory stimulation of the vibrissae, further supporting the impact of neuromodulators on energy metabolism (Poitry-Yamate and Tsacopoulos, 1991; Swanson et al., 1992; Dienel et al., 2007). In contrast, under conditions of low glutamatergic and monoaminergic activity (e.g., anesthesia; slow-wave sleep), glycogen levels increase (Folbergrová et al., 1970; Watanabe and Passonneau, 1973; Karnovsky et al., 1983; Morgenthaler et al., 2006; Bellesi et al., 2018), suggesting that glycogen is mostly used during intense neuronal activity. Consistent with this hypothesis, Gibbs and Hutchinson (2012) showed that NA affects glycogen turnover to enhance memory consolidation and glycogen-derived lactate was found to be central to higher cognitive function and memory formation (Newman et al., 2011; Suzuki et al., 2011).
However, the impact of neuromodulators on astrocytic glycogen metabolism is complex and highly dependent on the identity of the receptors. For example, while activation of the hippocampal β1-adrenergic receptor was found to induce glycogenolysis during the transition between the first and second phases of intermediate memory, after 30 min it caused α2-adrenoreceptors to induce glycogen synthesis (Gibbs et al., 2007; Hutchinson et al., 2011; Gibbs and Hutchinson, 2012). Similar to noradrenaline, vasoactive intestinal polypeptide (VIP) in addition to its glycogenolytic effect, was also found to elicit temporally-delayed resynthesis of glycogen (Sorg and Magistretti, 1991).
Given the impact of neuromodulators on glycogen metabolism, any impairment in their activity is primed to affect the energy metabolism in the brain, as seen in different neurodegenerative diseases. Indeed, noradrenergic dysfunction during Alzheimer’s disease (Mann et al., 1987) has been linked to disruptions in glycogen metabolism (Hertz, 1989; Marien et al., 2004; Weinshenker, 2008). Other studies showed that epileptic patients had increased levels of glycogen in the hippocampus, and sustained seizures led to the synthesis and accumulation of abnormal glycogen molecules that are resistant to degradation (Dalsgaard et al., 2007; Dinuzzo et al., 2015). Accumulation of glycogen was also found in the spinal cord of animal models for ALS (Li et al., 2019) and epilepsy (Rubio-Villena et al., 2018), which was due to deteriorated glycogenolysis.
Energy metabolism in astrocytes is not only modulated by neurons but also affects neuronal activity. Specifically, glycogen-derived energy supports the clearance of extracellular potassium and glutamate following neuronal activation (Choi et al., 2012). Following the astrocyte-neuron lactate shuttle hypothesis, glucose-derived lactate from astrocytes is transferred to neurons to maintain neuronal metabolism (Pellerin et al., 2007), including oxidative phosphorylation and restoration of a Na+ gradient following glutamatergic neurotransmission (Sickmann et al., 2005; Gibbs et al., 2007). Inhibition of astrocyte energy turnover induces deterioration of both neuronal energy metabolism and extracellular clearance, which can cause neuronal hyperexcitability (Kreisman and Smith, 1993; Xu et al., 2013; Kilic et al., 2018). Hence, an initial disturbance in neuron-astrocyte interactions may create a vicious cycle of disease development through positive feedback loops, resulting in augmented neuronal disfunction.
Glial cells make up an integral part of the CNS architecture and are critical for the maintenance of healthy brain function. In recent years the field has shifted from viewing glial cells as passive “housekeeping” cells to becoming active participants in brain function and modulation of neuronal output. In addition to their functions in the healthy brain, a growing body of evidence suggests glia are implicated in the progression of neurodegeneration, either through the loss of normal function or the introduction of abnormal function (Sofroniew and Vinters, 2010). Moreover, there is cumulative experimental evidence suggesting that functional and genetic alterations in glia are driving disease pathogenesis, see De Strooper and Karran (2016) and Gleichman and Carmichael (2020) for detailed reviews. For these reasons, neuron-glia transmission can no longer be viewed as a passive method of communication and must be seen as an active and important signaling thread that joins many neurodegenerative disorders.
Current therapeutic strategies for treating neurodegeneration include targeting the ApoE4 signaling pathway, which is the greatest risk factor for AD and primarily expressed in glial cells, and nicotinamide adenine dinucleotide (NAD), which is thought to regulate inflammatory repair processes (Blacher et al., 2015; Roboon et al., 2019). Indeed, Sawmiller et al. (2019) reported that disrupting the ApoE signaling cascade ameliorated cerebral Aβ and tau pathologies, including neuronal apoptosis and synaptic loss in a mouse model of AD. Moreover, the deletion of CD38, the enzyme that hydrolyzes NAD and controls its bioavailability, was recently found to suppress glial activation and neuroinflammation in a mouse model for demyelination (Roboon et al., 2019). As levels of NAD reduce with age, this strategy represents a promising target for MS, AD, and other age-related neurodegenerative disorders.
Another therapeutic strategy includes astrocyte-targeted production of interleukin-6, which has been shown to reduce the activation of both astrocytes and microglia and delay demyelination of neurons in a mouse model of MS (Petković et al., 2016). Moreover, targeting the histaminergic system with anti-histamine proved as a strategy that can preserve myelin and initiate myelination in animal models of MS (Mei et al., 2014, 2016; Liu et al., 2016), and this has now been tested in a randomized control trial of 50 MS patients with promising results (Green et al., 2017).
Recently, strategies directed towards targeting the astrocytic glutamate transporter EAAT2 proved to be beneficial in alleviating the hyperexcitability symptoms associated with some neurodegenerative diseases and decrease the level of neurodegeneration in animal models of epilepsy (Kong et al., 2012, 2014; Goodrich et al., 2013; Zaitsev et al., 2019), ALS (Kong et al., 2014), PD (Chotibut et al., 2014; Hsu et al., 2015) and HD (Sari et al., 2010). Moreover, modulation of astrocytic energy metabolism via inhibition of lactate dehydrogenase (Sada et al., 2015; Rosati et al., 2018) or via ketonic diet (Valdebenito et al., 2016) had a neuroprotective effect in both animal models of PD (Shaafi et al., 2016), and patients with HD (Adanyeguh et al., 2015) and epilepsy (van Berkel et al., 2018). Also, treatments that targeted the suppression of the inflammatory NF-κB pathway by increasing the expression of neurotrophic growth factor IGF-1 (Hu et al., 2018), or decreasing microglial proliferation (Martínez-Muriana et al., 2016), postponed the onset and slowed down the progression of motoneuron death in a mouse model of ALS.
The etiology of neurodegeneration is still unclear, however recent studies established that lack of glial support and dysregulation in neuron-glia interactions are central for disease initiation and progression. More research is needed to address key questions on neuronal-glia interactions, such as why do astrocytes contact some synapses and not others, and how does this communication change over developmental stages or with the progression of the disease?
We know neuron-glia interactions are dynamic, can be altered by neuronal activity, and can change throughout life (Bernardinelli et al., 2014). However, during neurodegeneration, alterations in neuron-glia interactions take place slowly and their source is largely unknown. While glial modulation of synaptic morphology and function has been heavily investigated in the past decade, the neuron-glia communication processes that affect glial function are still unclear. Molecular, cellular, and genetic approaches can be employed to investigate these communication mechanisms, as addressing this gap will provide novel targets for much needed therapeutic intervention.
All authors contributed to the article and approved the submitted version.
This work was supported by the Motor Neurone Disease Research Institute of Australia (MNDRIA) awarded to YB and Ainsworth medical research innovation fund awarded to YB and JM. RS, ES, and IR are supported by the Ainsworth MRIF scholarship program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2020.00278/full#supplementary-material.
Adanyeguh, I. M., Rinaldi, D., Henry, P. G., Caillet, S., Valabregue, R., Durr, A., et al. (2015). Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 84, 490–495. doi: 10.1212/wnl.0000000000001214
Adeva-Andany, M. M., González-Lucán, M., Donapetry-García, C., Fernández-Fernández, C., and Ameneiros-Rodríguez, E. (2016). Glycogen metabolism in humans. BBA Clin. 5, 85–100. doi: 10.1016/j.bbacli.2016.02.001
Allen, N. J., Bennett, M. L., Foo, L. C., Wang, G. X., Chakraborty, C., Smith, S. J., et al. (2012). Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486, 410–414. doi: 10.1038/nature11059
Alzheimer’s Association. (2019). 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 15, 321–387. doi: 10.1016/j.jalz.2019.01.010
Amar, M., Lucas-Meunier, E., Baux, G., and Fossier, P. (2010). Blockade of different muscarinic receptor subtypes changes the equilibrium between excitation and inhibition in rat visual cortex. Neuroscience 169, 1610–1620. doi: 10.1016/j.neuroscience.2010.06.019
Araque, A., Parpura, V., Sanzgiri, R. P., and Haydon, P. G. (1998). Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur. J. Neurosci. 10, 2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x
Araque, A., Parpura, V., Sanzgiri, R. P., and Haydon, P. G. (1999). Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 22, 208–215. doi: 10.1016/s0166-2236(98)01349-6
Aronica, E., Boer, K., van Vliet, E. A., Redeker, S., Baayen, J. C., Spliet, W. G. M., et al. (2007). Complement activation in experimental and human temporal lobe epilepsy. Neurobiol. Dis. 26, 497–511. doi: 10.1016/j.nbd.2007.01.015
Arranz, A. M., and De Strooper, B. (2019). The role of astroglia in Alzheimer’s disease: pathophysiology and clinical implications. Lancet Neurol. 18, 406–414. doi: 10.1016/S1474-4422(18)30490-3
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Bahbouhi, B., Berthelot, L., Pettré, S., Michel, L., Wiertlewski, S., Weksler, B., et al. (2009). Peripheral blood CD4+ T lymphocytes from multiple sclerosis patients are characterized by higher PSGL-1 expression and transmigration capacity across a human blood-brain barrier-derived endothelial cell line. J. Leukoc. Biol. 86, 1049–1063. doi: 10.1189/jlb.1008666
Baillet, A., Chanteperdrix, V., Trocmé, C., Casez, P., Garrel, C., and Besson, G. (2010). The role of oxidative stress in amyotrophic lateral sclerosis and Parkinson’s disease. Neurochem. Res. 35, 1530–1537. doi: 10.1007/s11064-010-0212-5
Balbi, M., Ghosh, M., Longden, T. A., Jativa Vega, M., Gesierich, B., Hellal, F., et al. (2015). Dysfunction of mouse cerebral arteries during early aging. J. Cereb. Blood Flow Metab. 35, 1445–1453. doi: 10.1038/jcbfm.2015.107
Baljit, S. K., Beaumont, V., Cachope, R., Munoz-Sanjuan, I., Goldman, S. A., and Rosemarie, G. (2017). Unravelling and exploiting astrocyte dysfunction in Huntington’s disease. Trends Neurosci. 40, 422–437. doi: 10.1016/j.tins.2017.05.002
Banerjee, J., Alkondon, M., Pereira, E. F. R., and Albuquerque, E. X. (2012). Regulation of GABAergic inputs to CA1 pyramidal neurons by nicotinic receptors and kynurenic acid. J. Pharmacol. Exp. Ther. 341, 500–509. doi: 10.1124/jpet.111.189860
Barber, S. C., Mead, R. J., and Shaw, P. J. (2006). Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 1762, 1051–1067. doi: 10.1016/j.bbadis.2006.03.008
Barriola, S., Pérez-Cerdá, F., Matute, C., Bribián, A., and López-Mascaraque, L. (2020). A clonal NG2-glia cell response in a mouse model of multiple sclerosis. Cells 9:1279. doi: 10.3390/cells9051279
Bataveljić, D., Nikolić, L., Milosević, M., Todorović, N., and Andjus, P. R. (2012). Changes in the astrocytic aquaporin-4 and inwardly rectifying potassium channel expression in the brain of the amyotrophic lateral sclerosis SOD1G93A rat model. Glia 60, 1991–2003. doi: 10.1002/glia.22414
Beauquis, J., Pavía, P., Pomilio, C., Vinuesa, A., Podlutskaya, N., Galvan, V., et al. (2013). Environmental enrichment prevents astroglial pathological changes in the hippocampus of APP transgenic mice, model of Alzheimer’s disease. Exp. Neurol. 239, 28–37. doi: 10.1016/j.expneurol.2012.09.009
Beauquis, J., Vinuesa, A., Pomilio, C., Pavía, P., Galván, V., and Saravia, F. (2014). Neuronal and glial alterations, increased anxiety, and cognitive impairment before hippocampal amyloid deposition in PDAPP mice, model of Alzheimer’s disease. Hippocampus 24, 257–269. doi: 10.1002/hipo.22219
Bedner, P., Dupper, A., Hüttmann, K., Müller, J., Herde, M. K., Dublin, P., et al. (2015). Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 138, 1208–1222. doi: 10.1093/brain/awv067
Beggiato, S., Antonelli, T., Tomasini, M. C., Tanganelli, S., Fuxe, K., Schwarcz, R., et al. (2013). Kynurenic acid, by targeting α7 nicotinic acetylcholine receptors, modulates extracellular GABA levels in the rat striatum in vivo. Eur. J. Neurosci. 37, 1470–1477. doi: 10.1111/ejn.12160
Behrendt, G., Baer, K., Buffo, A., Curtis, M. A., Faull, R. L., Rees, M. I., et al. (2013). Dynamic changes in myelin aberrations and oligodendrocyte generation in chronic amyloidosis in mice and men. Glia 61, 273–286. doi: 10.1002/glia.22432
Behrens, P. F., Franz, P., Woodman, B., Lindenberg, K. S., and Landwehrmeyer, G. B. (2002). Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain 125, 1908–1922. doi: 10.1093/brain/awf180
Bekar, L. K., He, W., and Nedergaard, M. (2008). Locus coeruleus α-adrenergic-mediated activation of cortical astrocytes in vivo. Cereb. Cortex 18, 2789–2795. doi: 10.1093/cercor/bhn040
Bellesi, M., de Vivo, L., Koebe, S., Tononi, G., and Cirelli, C. (2018). Sleep and wake affect glycogen content and turnover at perisynaptic astrocytic processes. Front. Cell. Neurosci. 12:308. doi: 10.3389/fncel.2018.00308
Bellot-Saez, A., Cohen, G., van Schaik, A., Ooi, L., Morley, J.W., and Buskila, Y. (2018). Astrocytic modulation of cortical oscillations. Sci. Rep. 8:11565. doi: 10.1038/s41598-018-30003-w
Bellot-Saez, A., Kékesi, O., Ben-Abu, Y., Morley, J. W., and Buskila, Y. (2019). Neuromodulation of astrocytic K+ clearance. bioRxiv [Preprint]. doi: 10.1101/841643
Bellot-Saez, A., Kékesi, O., Morley, J. W., and Buskila, Y. (2017). Astrocytic modulation of neuronal excitability through K+ spatial buffering. Neurosci. Biobehav. Rev. 77, 87–97. doi: 10.1016/j.neubiorev.2017.03.002
Ben Haim, L., Ceyzériat, K., Sauvage, M. A. C., Aubry, F., Auregan, G., Guillermier, M., et al. (2015). The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci. 35, 2817–2829. doi: 10.1523/JNEUROSCI.3516-14.2015
Ben Menachem-Zidon, O., Avital, A., Ben-Menahem, Y., Goshen, I., Kreisel, T., Shmueli, E. M., et al. (2011). Astrocytes support hippocampal-dependent memory and long-term potentiation via interleukin-1 signaling. Brain. Behav. Immun. 25, 1008–1016. doi: 10.3410/f.12585956.13821055
Benraiss, A., Wang, S., Herrlinger, S., Li, X., Chandler-Militello, D., Mauceri, J., et al. (2016). Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat. Commun. 7:11758. doi: 10.1038/ncomms11758
Bergles, D. E., and Jahr, C. E. (1997). Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron 19, 1297–1308. doi: 10.1016/s0896-6273(00)80420-1
Bergles, D. E., and Jahr, C. E. (1998). Glial contribution to glutamate uptake at Schaffer collateral- commissural synapses in the hippocampus. J. Neurosci. 18, 7709–7716. doi: 10.1523/JNEUROSCI.18-19-07709.1998
Bernardinelli, Y., Randall, J., Janett, E., Nikonenko, I., König, S., Jones, E. V., et al. (2014). Activity-dependent structural plasticity of perisynaptic astrocytic domains promotes excitatory synapse stability. Curr. Biol. 24, 1679–1688. doi: 10.1016/j.cub.2014.06.025
Biber, K., Neumann, H., Inoue, K., and Boddeke, H. W. G. M. (2007). Neuronal “On” and “Off” signals control microglia. Trends Neurosci. 30, 596–602. doi: 10.1016/j.tins.2007.08.007
Bidmon, H. J., Görg, B., Palomero-Gallagher, N., Schleicher, A., Häussinger, D., Speckmann, E. J., et al. (2008). Glutamine synthetase becomes nitrated and its activity is reduced during repetitive seizure activity in the pentylentetrazole model of epilepsy. Epilepsia 49, 1733–1748. doi: 10.1111/j.1528-1167.2008.01642.x
Binder, D. K., Yao, X., Zador, Z., Sick, T. J., Verkman, A. S., and Manley, G. T. (2006). Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 53, 631–636. doi: 10.1002/glia.20318
Birger, A., Ben-Dor, I., Ottolenghi, M., Turetsky, T., Gil, Y., Sweetat, S., et al. (2019). Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 50, 274–289. doi: 10.1016/j.ebiom.2019.11.026
Blacher, E., Dadali, T., Bespalko, A., Haupenthal, V. J., Grimm, M. O. W., Hartmann, T., et al. (2015). Alzheimer’s disease pathology is attenuated in a CD38-deficient mouse model. Ann. Neurol. 78, 88–103. doi: 10.1002/ana.24425
Booth, H. D. E., Hirst, W. D., and Wade-Martins, R. (2017). The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci. 40, 358–370. doi: 10.1016/j.tins.2017.04.001
Bosson, A., Boisseau, S., Buisson, A., Savasta, M., and Albrieux, M. (2015). Disruption of dopaminergic transmission remodels tripartite synapse morphology and astrocytic calcium activity within substantia nigra pars reticulata. Glia 63, 673–683. doi: 10.1002/glia.22777
Brambilla, R. (2019). Neuroinflammation, the thread connecting neurological disease. Acta Neuropathol. 137, 689–691. doi: 10.1007/s00401-019-02009-9
Brawek, B., Schwendele, B., Riester, K., Kohsaka, S., Lerdkrai, C., Liang, Y., et al. (2014). Impairment of in vivo calcium signaling in amyloid plaque-associated microglia. Acta Neuropathol. 127, 495–505. doi: 10.1007/s00401-013-1242-2
Brockhaus, J., Möller, T., and Kettenmann, H. (1996). Phagocytozing ameboid microglial cells studied in a mouse corpus callosum slice preparation. Glia 16, 81–90. doi: 10.1002/(sici)1098-1136(199601)16:1<81::aid-glia9>3.0.co;2-e
Broe, M., Kril, J., and Halliday, G. M. (2004). Astrocytic degeneration relates to the severity of disease in frontotemporal dementia. Brain 127, 2214–2220. doi: 10.1093/brain/awh250
Buosi, A. S., Matias, I., Araujo, A. P. B., Batista, C., and Gomes, F. C. A. (2018). Heterogeneity in synaptogenic profile of astrocytes from different brain regions. Mol. Neurobiol. 55, 751–752. doi: 10.1007/s12035-016-0343-z
Buschmann, J. P., Berger, K., Awad, H., Clarner, T., Beyer, C., and Kipp, M. (2012). Inflammatory response and chemokine expression in the white matter corpus callosum and gray matter cortex region during cuprizone-induced demyelination. J. Mol. Neurosci. 48, 66–76. doi: 10.1007/s12031-012-9773-x
Buskila, Y., and Amitai, Y. (2010). Astrocytic iNOS-dependent enhancement of synaptic release in mouse neocortex. J. Neurophysiol. 103, 1322–1328. doi: 10.1152/jn.00676.2009
Buskila, Y., Bellot-Saez, A., and Morley, J. W. (2019a). Generating brain waves, the power of astrocytes. Front. Neurosci. 13:1125. doi: 10.3389/fnins.2019.01125
Buskila, Y., Kékesi, O., Bellot-Saez, A., Seah, W., Berg, T., Trpceski, M., et al. (2019b). Dynamic interplay between H-current and M-current controls motoneuron hyperexcitability in amyotrophic lateral sclerosis. Cell Death Dis. 10:310. doi: 10.1038/s41419-019-1538-9
Buskila, Y., Crowe, S. E., and Ellis-Davies, G. C. R. (2013). Synaptic deficits in layer 5 neurons precede overt structural decay in 5xFAD mice. Neuroscience 254, 152–159. doi: 10.1016/j.neuroscience.2013.09.016
Buskila, Y., Farkash, S., Hershfinkel, M., and Amitai, Y. (2005). Rapid and reactive nitric oxide production by astrocytes in mouse neocortical slices. Glia 52, 169–176. doi: 10.1002/glia.20217
Calì, C., Baghabra, J., Boges, D. J., Holst, G. R., Kreshuk, A., Hamprecht, F. A., et al. (2016). Three-dimensional immersive virtual reality for studying cellular compartments in 3D models from EM preparations of neural tissues. J. Comp. Neurol. 524, 23–38. doi: 10.1002/cne.23852
Cambray-Deakin, M., Pearce, B., Morrow, C., and Murphy, S. (1988). Effects of neurotransmitters on astrocyte glycogen stores in vitro. J. Neurochem. 51, 1852–1857. doi: 10.1111/j.1471-4159.1988.tb01168.x
Capuani, C., Melone, M., Tottene, A., Bragina, L., Crivellaro, G., Santello, M., et al. (2016). Defective glutamate and K+ clearance by cortical astrocytes in familial hemiplegic migraine type 2. EMBO Mol. Med. 8, 967–986. doi: 10.15252/emmm.201505944
Cardinaux, J. R., and Magistretti, P. J. (1996). Vasoactive intestinal peptide, pituitary adenylate cyclase-activating peptide and noradrenaline induce the transcription factors CCAAT/enhancer binding protein (C/EBP)-β and C/EBPδ in mouse cortical astrocytes: involvement in cAMP-regulated glycogen meta. J. Neurosci. 16, 919–929. doi: 10.1523/JNEUROSCI.16-03-00919.1996
Carmans, S., Hendriks, J. J. A., Thewissen, K., van den Eynden, J., Stinissen, P., Rigo, J. M., et al. (2010). The inhibitory neurotransmitter glycine modulates macrophage activity by activation of neutral amino acid transporters. J. Neurosci. Res. 88, 2420–2430. doi: 10.1002/jnr.22395
Chai, H., Diaz-Castro, B., Shigetomi, E., Monte, E., Octeau, J. C., Yu, X., et al. (2017). Neural circuit-specialized astrocytes: transcriptomic, proteomic, morphological, and functional evidence. Neuron 95, 531.e9–549.e9. doi: 10.1016/j.neuron.2017.06.029
Chaudhuri, A., and Behan, P. O. (2004). Multiple sclerosis is not an autoimmune disease. Arch. Neurol. 61, 1610–1612. doi: 10.1001/archneur.61.10.1610
Chen, C., Jiang, Z. Y., Fu, X., Yu, D., Huang, H., and Tasker, J. G. (2019). Astrocytes amplify neuronal dendritic volume transmission stimulated by norepinephrine. Cell Rep. 29, 4349.e4–4361.e4. doi: 10.1016/j.celrep.2019.11.092
Chen, P. C., Vargas, M. R., Pani, A. K., Smeyne, R. J., Johnson, D. A., Kan, Y. W., et al. (2009). Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: critical role for the astrocyte. Proc. Natl. Acad. Sci. U S A 106, 2933–2938. doi: 10.1073/pnas.0813361106
Chever, O., Djukic, B., McCarthy, K. D., and Amzica, F. (2010). Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J. Neurosci. 30, 15769–15777. doi: 10.1523/JNEUROSCI.2078-10.2010
Choi, H. B., Gordon, G. R. J., Zhou, N., Tai, C., Rungta, R. L., Martinez, J., et al. (2012). Metabolic communication between astrocytes and neurons via bicarbonate-responsive soluble adenylyl cyclase. Neuron 75, 1094–1104. doi: 10.1016/j.neuron.2012.08.032
Chotibut, T., Davis, R. W., Arnold, J. C., Frenchek, Z., Gurwara, S., Bondada, V., et al. (2014). Ceftriaxone increases glutamate uptake and reduces striatal tyrosine hydroxylase loss in 6-OHDA Parkinson’s model. Mol. Neurobiol. 49, 1282–1292. doi: 10.1007/s12035-013-8598-0
Christopherson, K. S., Ullian, E. M., Stokes, C. C. A., Mullowney, C. E., Hell, J. W., Agah, A., et al. (2005). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. doi: 10.1016/j.cell.2004.12.020
Clarkson, C., Smeal, R. M., Hasenoehrl, M. G., White, J. A., Rubio, M. E., and Wilcox, K. S. (2020). Ultrastructural and functional changes at the tripartite synapse during epileptogenesis in a model of temporal lobe epilepsy. Exp. Neurol. 326:113196. doi: 10.1016/j.expneurol.2020.113196
Coco, S., Calegari, F., Pravettoni, E., Pozzi, D., Taverna, E., Rosa, P., et al. (2003). Storage and release of ATP from astrocytes in culture. J. Biol. Chem. 278, 1354–1362. doi: 10.1074/jbc.M209454200
Colombo, E., Cordiglieri, C., Melli, G., Newcombe, J., Krumbholz, M., Parada, L. F., et al. (2012). Stimulation of the neurotrophin receptor trkb on astrocytes drives nitric oxide production and neurodegeneration. J. Exp. Med. 209, 521–535. doi: 10.1084/jem.20110698
Condamine, S., Lavoie, R., Verdier, D., and Kolta, A. (2018). Functional rhythmogenic domains defined by astrocytic networks in the trigeminal main sensory nucleus. Glia 66, 311–326. doi: 10.1002/glia.23244
Conductier, G., Blondeau, N., Guyon, A., Nahon, J. L., and Rovère, C. (2010). The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J. Neuroimmunol. 224, 93–100. doi: 10.1016/j.jneuroim.2010.05.010
Cornell-Bell, A. H., Finkbeiner, S. M., Cooper, M. S., and Smith, S. J. (1990). Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science 247, 470–473. doi: 10.1126/science.1967852
Coughlan, C. M., McManus, C. M., Sharron, M., Gao, Z. Y., Murphy, D., Jaffer, S., et al. (2000). Expression of multiple functional chemokine receptors and monocyte chemoattractant protein-1 in human neurons. Neuroscience 97, 591–600. doi: 10.1016/s0306-4522(00)00024-5
Cruz, N. F., and Dienel, G. A. (2002). High glycogen levels in brains of rats with minimal environmental stimuli: implications for metabolic contributions of working astrocytes. J. Cereb. Blood Flow Metab. 22, 1476–1489. doi: 10.1097/01.WCB.0000034362.37277.C0
Cunningham, C., Dunne, A., and Lopez-Rodriguez, A. B. (2019). Astrocytes: heterogeneous and dynamic phenotypes in neurodegeneration and innate immunity. Neuroscientist 25, 455–474. doi: 10.1177/1073858418809941
Dalsgaard, M. K., Madsen, F. F., Secher, N. H., Laursen, H., and Quistorff, B. (2007). High glycogen levels in the hippocampus of patients with epilepsy. J. Cereb. Blood Flow Metab. 27, 1137–1141. doi: 10.1038/sj.jcbfm.9600426
Damier, P., Hirsch, E. C., Zhang, P., Agid, Y., and Javoy-Agid, F. (1993). Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 52, 1–6. doi: 10.1016/0306-4522(93)90175-f
Das, A., Wallace, G. C. IV., Holmes, C., McDowell, M. L., Smith, J. A., Marshall, J. D., et al. (2012). Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 220, 237–246. doi: 10.1016/j.neuroscience.2012.06.002
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
David, Y., Cacheaux, L. P., Ivens, S., Lapilover, E., Heinemann, U., Kaufer, D., et al. (2009). Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J. Neurosci. 29, 10588–10599. doi: 10.1523/JNEUROSCI.2323-09.2009
Davies, P., and Maloney, A. J. F. (1976). Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 308:1403. doi: 10.1016/s0140-6736(76)91936-x
de Lau, L. M. L., and Breteler, M. M. B. (2006). Epidemiology of Parkinson’s disease. Lancet Neurol. 5, 525–535. doi: 10.1016/S1474-4422(06)70471-9
de Majo, M., Koontz, M., Rowitch, D., and Ullian, E. M. (2020). An update on human astrocytes and their role in development and disease. Glia 68, 685–704. doi: 10.1002/glia.23771
Desai, M. K., Mastrangelo, M. A., Ryan, D. A., Sudol, K. L., Narrow, W. C., and Bowers, W. J. (2010). Early oligodendrocyte/myelin pathology in Alzheimer’s disease mice constitutes a novel therapeutic target. Am. J. Pathol. 177, 1422–1435. doi: 10.2353/ajpath.2010.100087
De Strooper, B., and Karran, E. (2016). The cellular phase of Alzheimer’s disease. Cell 164, 603–615. doi: 10.1016/j.cell.2015.12.056
Devinsky, O., Vezzani, A., Najjar, S., De Lanerolle, N. C., and Rogawski, M. A. (2013). Glia and epilepsy: excitability and inflammation. Trends Neurosci. 36, 174–184. doi: 10.1016/j.tins.2012.11.008
Diaz-Aparicio, I., Paris, I., Sierra-Torre, V., Plaza-Zabala, A., Rodríguez-Iglesias, N., Márquez-Ropero, M., et al. (2020). Microglia actively remodel adult hippocampal neurogenesis through the phagocytosis secretome. J. Neurosci. 40, 1453–1482. doi: 10.1523/JNEUROSCI.0993-19.2019
di Domenico, A., Carola, G., Calatayud, C., Pons-Espinal, M., Muñoz, J. P., Richaud-Patin, Y., et al. (2019). Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Reports 12, 213–229. doi: 10.1016/j.stemcr.2018.12.011
Dienel, G. A., Ball, K. K., and Cruz, N. F. (2007). A glycogen phosphorylase inhibitor selectively enhances local rates of glucose utilization in brain during sensory stimulation of conscious rats: implications for glycogen turnover. J. Neurochem. 102, 466–478. doi: 10.1111/j.1471-4159.2007.04595.x
Di Giorgio, F. P., Boulting, G. L., Bobrowicz, S., and Eggan, K. C. (2008). Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 3, 637–648. doi: 10.1016/j.stem.2008.09.017
Ding, F., O’Donnell, J., Xu, Q., Kang, N., Goldman, N., and Nedergaard, M. (2016). Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science 352, 550–555. doi: 10.1126/science.aad4821
Diniz, L. P., Matias, I., Araujo, A. P. B., Garcia, M. N., Barros-Aragão, F. G. Q., Alves-Leon, S. V., et al. (2019). α-synuclein oligomers enhance astrocyte-induced synapse formation through TGF-β1 signaling in a Parkinson’s disease model. J. Neurochem. 150, 138–157. doi: 10.1111/jnc.14710
Dinuzzo, M., Mangia, S., Maraviglia, B., and Giove, F. (2013). Regulatory mechanisms for glycogenolysis and K+ uptake in brain astrocytes. Neurochem. Int. 63, 458–464. doi: 10.1016/j.neuint.2013.08.004
Dinuzzo, M., Mangia, S., Maraviglia, B., and Giove, F. (2015). Does abnormal glycogen structure contribute to increased susceptibility to seizures in epilepsy? Metab. Brain Dis. 30, 307–316. doi: 10.1007/s11011-014-9524-5
DiSabato, D. J., Quan, N., and Godbout, J. P. (2016). Neuroinflammation: the devil is in the details. J. Neurochem. 139, 136–153. doi: 10.1111/jnc.13607
Do-Ha, D., Buskila, Y., and Ooi, L. (2018). Impairments in motor neurons, interneurons and astrocytes contribute to hyperexcitability in ALS: underlying mechanisms and paths to therapy. Mol. Neurobiol. 55, 1410–1418. doi: 10.1007/s12035-017-0392-y
Do Nascimento, A. L., Dos Santos, N. F., Campos Pelágio, F., Aparecida Teixeira, S., De Moraes Ferrari, E. A., and Langone, F. (2012). Neuronal degeneration and gliosis time-course in the mouse hippocampal formation after pilocarpine-induced status epilepticus. Brain Res. 1470, 98–110. doi: 10.1016/j.brainres.2012.06.008
Dong, X. X., Wang, Y., and Qin, Z. H. (2009). Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 30, 379–387. doi: 10.1038/aps.2009.24
Dopper, E. G. P., Chalos, V., Ghariq, E., den Heijer, T., Hafkemeijer, A., Jiskoot, L. C., et al. (2016). Cerebral blood flow in presymptomatic MAPT and GRN mutation carriers: a longitudinal arterial spin labeling study. NeuroImage Clin. 12, 460–465. doi: 10.1016/j.nicl.2016.08.001
Dosch, M., Gerber, J., Jebbawi, F., and Beldi, G. (2018). Mechanisms of ATP release by inflammatory cells. Int. J. Mol. Sci. 19:1222. doi: 10.3390/ijms19041222
Dresselhaus, E. C., and Meffert, M. K. (2019). Cellular specificity of NF-κB function in the nervous system. Front. Immunol. 10:1043. doi: 10.3389/fimmu.2019.01043
Eid, T., Thomas, M. J., Spencer, D. D., Rundén-Pran, E., Lai, J. C. K., Malthankar, G. V., et al. (2004). Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363, 28–37. doi: 10.1016/s0140-6736(03)15166-5
Enes, J., Haburcák, M., Sona, S., Gerard, N., Mitchell, A. C., Fu, W., et al. (2020). Satellite glial cells modulate cholinergic transmission between sympathetic neurons. PLoS One 15:e0218643. doi: 10.1371/journal.pone.0218643
Fabiani, M., Gordon, B. A., Maclin, E. L., Pearson, M. A., Brumback-Peltz, C. R., Low, K. A., et al. (2014). Neurovascular coupling in normal aging: a combined optical, ERP and fMRI study. NeuroImage 85, 592–607. doi: 10.1016/j.neuroimage.2013.04.113
Faideau, M., Kim, J., Cormier, K., Gilmore, R., Welch, M., Auregan, G., et al. (2010). in vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington’s disease subjects. Hum. Mol. Genet. 19, 3053–3067. doi: 10.1093/hmg/ddq212
Ferraiuolo, L., Higginbottom, A., Heath, P. R., Barber, S., Greenald, D., Kirby, J., et al. (2011). Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134, 2627–2641. doi: 10.1093/brain/awr193
Ferraro, T. N., Golden, G. T., Smith, G. G., Martin, J. F., Lohoff, F. W., Gieringer, T. A., et al. (2004). Fine mapping of a seizure susceptibility locus on mouse Chromosome 1: nomination of Kcnj10 as a causative gene. Mamm. Genome 15, 239–251. doi: 10.1007/s00335-003-2270-3
Ferri, C. P., Prince, M., Brayne, C., Brodaty, H., Fratiglioni, L., Ganguli, M., et al. (2005). Global prevalence of dementia: a Delphi consensus study. Lancet 366, 2112–2117. doi: 10.1016/S0140-6736(05)67889-0
Filippi, M., Bozzali, M., Rovaris, M., Gonen, O., Kesavadas, C., Ghezzi, A., et al. (2003). Evidence for widespread axonal damage at the earliest clinical stage of multiple sclerosis. Brain 126, 433–437. doi: 10.1093/brain/awg038
Folbergrová, J., Lowry, O. H., and Passonneau, J. V. (1970). Changes in metabolites of the energy reserves in individual layers of mouse cerebral cortex and subjacent white matter during ischaemia and anaesthesia. J. Neurochem. 17, 1155–1162. doi: 10.1111/j.1471-4159.1970.tb03363.x
Gabriel, S., Njunting, M., Pomper, J. K., Merschhemke, M., Sanabria, E. R. G., Eilers, A., et al. (2004). Stimulus and potassium-induced epileptiform activity in the human dentate gyrus from patients with and without hippocampal sclerosis. J. Neurosci. 24, 10416–10430. doi: 10.1523/JNEUROSCI.2074-04.2004
Garbuzova-Davis, S., Hernandez-Ontiveros, D. G., Rodrigues, M. C. O., Haller, E., Frisina-Deyo, A., Mirtyl, S., et al. (2012). Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 1469, 114–128. doi: 10.1016/j.brainres.2012.05.056
Garcia, V. J., Rushton, D. J., Tom, C. M., Allen, N. D., Kemp, P. J., Svendsen, C. N., et al. (2019). Huntington’s disease patient-derived astrocytes display electrophysiological impairments and reduced neuronal support. Front. Neurosci. 13:669. doi: 10.3389/fnins.2019.00669
García-Cabezas, M., John, Y. J., Barbas, H., and Zikopoulos, B. (2016). Distinction of neurons, glia and endothelial cells in the cerebral cortex: an algorithm based on cytological features. Front. Neuroanat. 10:107. doi: 10.3389/fnana.2016.00107
Genoud, C., Quairiaux, C., Steiner, P., Hirling, H., Welker, E., and Knott, G. W. (2006). Plasticity of astrocytic coverage and glutamate transporter expression in adult mouse cortex. PLoS Biol. 4:e343. doi: 10.1371/journal.pbio.0040343
Ghadery, C., Koshimori, Y., Coakeley, S., Harris, M., Rusjan, P., Kim, J., et al. (2017). Microglial activation in Parkinson’s disease using [18F]-FEPPA. J. Neuroinflammation 14:8. doi: 10.1186/s12974-016-0778-1
Ghandour, M. S., Parkkila, A. K., Parkkila, S., Waheed, A., and Sly, W. S. (2000). Mitochondrial carbonic anhydrase in the nervous system: expression in neuronal and glial cells. J. Neurochem. 75, 2212–2220. doi: 10.1046/j.1471-4159.2000.0752212.x
Giampà, C., Montagna, E., Dato, C., Melone, M. A. B., Bernardi, G., and Fusco, F. R. (2013). Systemic delivery of recombinant brain derived neurotrophic factor (BDNF) in the R6/2 mouse model of Huntington’s disease. PLoS One 8:e64037. doi: 10.1371/journal.pone.0064037
Gibbs, M. E. (2016). Role of glycogenolysis in memory and learning: regulation by noradrenaline, serotonin and ATP. Front. Integr. Neurosci. 9:70. doi: 10.3389/fnint.2015.00070
Gibbs, M. E., and Hutchinson, D. S. (2012). Rapid turnover of glycogen in memory formation. Neurochem. Res. 37, 2456–2463. doi: 10.1007/s11064-012-0805-2
Gibbs, M. E., Lloyd, H. G. E., Santa, T., and Hertz, L. (2007). Glycogen is a preferred glutamate precursor during learning in 1-day-old chick: biochemical and behavioral evidence. in. J. Neurosci. Res. 85, 3326–3333. doi: 10.1002/jnr.21307
Gleichman, A. J., and Carmichael, S. T. (2020). Glia in neurodegeneration: drivers of disease or along for the ride? Neurobiol. Dis. 142:104957. doi: 10.1016/j.nbd.2020.104957
Golarai, G., Greenwood, A., Feeney, D., and Connor, J. (2008). Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J. Neurosci. 21, 8523–8537. doi: 10.1523/JNEUROSCI.21-21-08523.2001
Gonçalves, F. Q., Lopes, J. P., Silva, H. B., Lemos, C., Silva, A. C., Gonçalves, N., et al. (2019). Synaptic and memory dysfunction in a β-amyloid model of early Alzheimer’s disease depends on increased formation of ATP-derived extracellular adenosine. Neurobiol. Dis. 132:104570. doi: 10.1016/j.nbd.2019.104570
Gong, Y. H., Parsadanian, A. S., Andreeva, A., Snider, W. D., and Elliott, J. L. (2000). Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration 20, 660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000
Goodrich, G. S., Kabakov, A. Y., Hameed, M. Q., Dhamne, S. C., Rosenberg, P. A., and Rotenberg, A. (2013). Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J. Neurotrauma 30, 1434–1441. doi: 10.1089/neu.2012.2712
Green, A. J., Gelfand, J. M., Cree, B. A., Bevan, C., Boscardin, W. J., Mei, F., et al. (2017). Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): a randomised, controlled, double-blind, crossover trial. Lancet 390, 2481–2489. doi: 10.1016/s0140-6736(17)32346-2
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer’s disease risk gene cd33 inhibits microglial uptake of amyloid β. Neuron 78, 631–643. doi: 10.1016/j.neuron.2013.04.014
Habib, N., McCabe, C., Medina, S., Varshavsky, M., Kitsberg, D., Dvir-Szternfeld, R., et al. (2020). Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 23, 701–706. doi: 10.1097/00002093-198802030-00114
Hagino, Y., Kariura, Y., Manago, Y., Amano, T., Wang, B., Sekiguchi, M., et al. (2004). Heterogeneity and potentiation of AMPA type of glutamate receptors in rat cultured microglia. Glia 47, 68–77. doi: 10.1002/glia.20034
Haindl, M. T., Köck, U., Zeitelhofer-Adzemovic, M., Fazekas, F., and Hochmeister, S. (2019). The formation of a glial scar does not prohibit remyelination in an animal model of multiple sclerosis. Glia 67, 467–481. doi: 10.1002/glia.23556
Halassa, M. M., Fellin, T., Takano, H., Dong, J.-H., and Haydon, P. G. (2007). Synaptic islands defined by the territory of a single astrocyte. J. Neurosci. 27, 6473–6477. doi: 10.1523/JNEUROSCI.1419-07.2007
Hallmann, A. L., Araúzo-Bravo, M. J., Mavrommatis, L., Ehrlich, M., Röpke, A., Brockhaus, J., et al. (2017). Astrocyte pathology in a human neural stem cell model of frontotemporal dementia caused by mutant TAU protein. Sci. Rep. 7:42991. doi: 10.1038/srep42991
Hammer, J., Alvestad, S., Osen, K. K., Skare, Ö., Sonnewald, U., and Ottersen, O. P. (2008). Expression of glutamine synthetase and glutamate dehydrogenase in the latent phase and chronic phase in the kainate model of temporal lobe epilepsy. Glia 56, 856–868. doi: 10.1002/glia.20659
Hassel, B., Tessler, S., Faull, R. L. M., and Emson, P. C. (2008). Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem. Res. 33, 232–237. doi: 10.1007/s11064-007-9463-1
Hatori, K., Nagai, A., Heisel, R., Ryu, J. K., and Kim, S. U. (2002). Fractalkine and fractalkine receptors in human neurons and glial cells. J. Neurosci. Res. 69, 418–426. doi: 10.1002/jnr.10304
Haynes, S. E., Hollopeter, G., Yang, G., Kurpius, D., Dailey, M. E., Gan, W. B., et al. (2006). The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 9, 1512–1519. doi: 10.1038/nn1805
Hernangómez, M., Mestre, L., Correa, F. G., Loría, F., Mecha, M., Iñigo, P. M., et al. (2012). CD200-CD200R1 interaction contributes to neuroprotective effects of anandamide on experimentally induced inflammation. Glia 60, 1437–1450. doi: 10.1002/glia.22366
Hertz, L. (1989). Is Alzheimer’s disease an anterograde degeneration, originating in the brainstem and disrupting metabolic and functional interactions between neurons and glial cells? Brain Res. Rev. 14, 335–353. doi: 10.1016/0165-0173(89)90017-9
Hirase, H., Akther, S., Wang, X., and Oe, Y. (2019). Glycogen distribution in mouse hippocampus. J. Neurosci. Res. 97, 923–932. doi: 10.1002/jnr.24386
Hirst, W. D., Cheung, N. Y., Rattray, M., Price, G. W., and Wilkin, G. P. (1998). Cultured astrocytes express messenger RNA for multiple serotonin receptor subtypes, without functional coupling of 5-HT1 receptor subtypes to adenylyl cyclase. Mol. Brain Res. 61, 90–99. doi: 10.1016/s0169-328x(98)00206-x
Hixson, K. M., Cogswell, M., Brooks-Kayal, A. R., and Russek, S. J. (2019). Evidence for a non-canonical JAK/STAT signaling pathway in the synthesis of the brain’s major ion channels and neurotransmitter receptors. BMC Genomics 20:677. doi: 10.1186/s12864-019-6033-2
Hofer, A., and Dermietzel, R. (1998). Visualization and functional blocking of gap junction hemichannels (connexons) with antibodies against external loop domains in astrocytes. Glia 24, 141–154. doi: 10.1002/(sici)1098-1136(199809)24:1<141::aid-glia13>3.0.co;2-r
Hogan, D. B., Jetté, N., Fiest, K. M., Roberts, J. I., Pearson, D., Smith, E. E., et al. (2016). The prevalence and incidence of frontotemporal dementia: a systematic review. Can. J. Neurol. Sci. 43, S96–S109. doi: 10.1017/cjn.2016.25
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016a). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hong, S., Dissing-Olesen, L., and Stevens, B. (2016b). New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 36, 128–134. doi: 10.1016/j.conb.2015.12.004
Hoshino, K., Hayakawa, M., and Morimoto, Y. (2017). Minocycline prevents the impairment of hippocampal long-term potentiation in the septic mouse. Shock 48, 209–214. doi: 10.1097/shk.0000000000000847
Hösli, L., Hösli, E., Winter, T., and Käser, H. (1993). Electrophysiological evidence for the presence of receptors for cholecystokinin and bombesin on cultured astrocytes of rat central nervous system. Neurosci. Lett. 163, 145–147. doi: 10.1016/0304-3940(93)90367-t
Howland, D. S., Liu, J., She, Y., Goad, B., Maragakis, N. J., Kim, B., et al. (2002). Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. U S A 99, 1604–1609. doi: 10.1073/pnas.032539299
Hsu, C. Y., Hung, C. S., Chang, H. M., Liao, W. C., Ho, S. C., and Ho, Y. J. (2015). Ceftriaxone prevents and reverses behavioral and neuronal deficits in an MPTP-induced animal model of Parkinson’s disease dementia. Neuropharmacology 91, 43–56. doi: 10.1016/j.neuropharm.2014.11.023
Hu, H. J., Lin, H. Q., Duan, W. S., Cui, C., Li, Z. Y., Liu, Y. K., et al. (2018). Intrathecal injection of scAAV9-hIGF1 prolongs the survival of ALS model mice by inhibiting the NF-κB pathway. Neuroscience 381, 1–10. doi: 10.1016/j.neuroscience.2018.02.004
Hu, W. T., Wang, Z., Lee, V. M. Y., Trojanowski, J. Q., Detre, J. A., and Grossman, M. (2010). Distinct cerebral perfusion patterns in FTLD and AD. Neurology 75, 881–888. doi: 10.1212/wnl.0b013e3181f11e35
Huang, B., Wei, W. J., Wang, G., Gaertig, M. A., Feng, Y., Wang, W., et al. (2015). Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron 85, 1212–1226. doi: 10.1016/j.neuron.2015.02.026
Huguet, G., Joglekar, A., Messi, L. M., Buckalew, R., Wong, S., and Terman, D. (2016). Neuroprotective role of gap junctions in a neuron astrocyte network model. Biophys. J. 111, 452–462. doi: 10.1016/j.bpj.2016.05.051
Hutchinson, D. S., Catus, S. L., Merlin, J., Summers, R. J., and Gibbs, M. E. (2011). α2-Adrenoceptors activate noradrenaline-mediated glycogen turnover in chick astrocytes. J. Neurochem. 117, 915–926. doi: 10.1111/j.1471-4159.2011.07261.x
Iadecola, C., and Nedergaard, M. (2007). Glial regulation of the cerebral microvasculature. Nat. Neurosci. 10, 1369–1376. doi: 10.1038/nn2003
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Goldman, S. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 4:147ra111. doi: 10.1126/scitranslmed.3003748
Inagaki, N., Fukui, H., Taguchi, Y., Wang, N. P., Yamatodani, A., and Wada, H. (1989). Characterization of histamine H1-receptors on astrocytes in primary culture: [3H]mepyramine binding studies. Eur. J. Pharmacol. 173, 43–51. doi: 10.1016/0014-2999(89)90007-1
Jiang, R., Diaz-Castro, B., Looger, L. L., and Khakh, B. S. (2016). Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J. Neurosci. 36, 3453–3470. doi: 10.1523/JNEUROSCI.3693-15.2016
Jiang, Y., Wei, N., Zhu, J., Lu, T., Chen, Z., Xu, G., et al. (2010). Effects of brain-derived neurotrophic factor on local inflammation in experimental stroke of rat. Mediators Inflamm. 2010:372423. doi: 10.1155/2010/372423
Jiang, X., Zhang, H., Duan, F., and Quan, X. (2017). Identify Huntington’s disease associated genes based on restricted Boltzmann machine with RNA-seq data. BMC Bioinformatics 18:447. doi: 10.1186/s12859-017-1859-6
Jo, S., Yarishkin, O., Hwang, Y. J., Chun, Y. E., Park, M., Woo, D. H., et al. (2014). GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 20, 886–896. doi: 10.1038/nm.3639
Johann, S., Heitzer, M., Kanagaratnam, M., Goswami, A., Rizo, T., Weis, J., et al. (2015). NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63, 2260–2273. doi: 10.1002/glia.22891
John Lin, C. C., Yu, K., Hatcher, A., Huang, T. W., Lee, H. K., Carlson, J., et al. (2017). Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 20, 396–405. doi: 10.1038/nn.4493
Johnson, A. M., Roach, J. P., Hu, A., Stamatovic, S. M., Zochowski, M. R., Keep, R. F., et al. (2018). Connexin 43 gap junctions contribute to brain endothelial barrier hyperpermeability in familial cerebral cavernous malformations type III by modulating tight junction structure. FASEB J. 32, 2615–2629. doi: 10.1096/fj.201700699r
Jones, V. C., Atkinson-Dell, R., Verkhratsky, A., and Mohamet, L. (2017). Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis. 8:e2696. doi: 10.1038/cddis.2017.89
Jung, S., Pfeiffer, F., and Deitmer, J. W. (2000). Histamine-induced calcium entry in rat cerebellar astrocytes: evidence for capacitative and non-capacitative mechanisms. J. Physiol. 527, 549–561. doi: 10.1111/j.1469-7793.2000.00549.x
Jurič, D. M., Mele, T., and Čarman-Kržan, M. (2011). Involvement of histaminergic receptor mechanisms in the stimulation of NT-3 synthesis in astrocytes. Neuropharmacology 60, 1309–1317. doi: 10.1016/j.neuropharm.2011.01.019
Kadala, A., Verdier, D., Morquette, P., and Kolta, A. (2015). Ion homeostasis in rhythmogenesis: the interplay between neurons and astroglia. Physiology 30, 371–388. doi: 10.1152/physiol.00023.2014
Kalashnyk, O., Lykhmus, O., Oliinyk, O., Komisarenko, S., and Skok, M. (2014). α7 Nicotinic acetylcholine receptor-specific antibody stimulates interleukin-6 production in human astrocytes through p38-dependent pathway. Int. Immunopharmacol. 23, 475–479. doi: 10.1016/j.intimp.2014.09.022
Kang, T. C., Kim, D. S., Kwak, S. E., Kim, J. E., Won, M. H., Kim, D. W., et al. (2006). Epileptogenic roles of astroglial death and regeneration in the dentate gyrus of experimental temporal lobe epilepsy. Glia 54, 258–271. doi: 10.1002/glia.20380
Kanmogne, G. D., Schall, K., Leibhart, J., Knipe, B., Gendelman, H. E., and Persidsky, Y. (2007). HIV-1 gp120 compromises blood-brain barrier integrity and enhance monocyte migration across blood-brain barrier: implication for viral neuropathogenesis. J. Cereb. Blood Flow Metab. 27, 123–134. doi: 10.1038/sj.jcbfm.9600330
Karnovsky, M. L., Reich, P., Anchors, J. M., and Burrows, B. L. (1983). Changes in brain glycogen during slow-wave sleep in the rat. J. Neurochem. 41, 1498–1501. doi: 10.1111/j.1471-4159.1983.tb00853.x
Kawahara, K., Suenobu, M., Yoshida, A., Koga, K., Hyodo, A., Ohtsuka, H., et al. (2012). Intracerebral microinjection of interleukin-4/interleukin-13 reduces β-amyloid accumulation in the ipsilateral side and improves cognitive deficits in young amyloid precursor protein 23 mice. Neuroscience 207, 243–260. doi: 10.1016/j.neuroscience.2012.01.049
Kékesi, O., Liang, H., Münch, G., Morley, J. W., Gyengesi, E., and Buskila, Y. (2019). The differential impact of acute microglia activation on the excitability of cholinergic neurons in the mouse medial septum. Brain Struct. Funct. 224, 2297–2309. doi: 10.1007/s00429-019-01905-w
Kettenmann, H., and Ranson, B. R. (1988). Electrical coupling between astrocytes and between oligodendrocytes studied in mammalian cell cultures. Glia 1, 64–73. doi: 10.1002/glia.440010108
Khan, Z. U., Koulen, P., Rubinstein, M., Grandy, D. K., and Goldman-Rakic, P. S. (2001). An astroglia-linked dopamine D2-receptor action in prefrontal cortex. Proc. Natl. Acad. Sci. U S A 98, 1964–1969. doi: 10.1073/pnas.98.4.1964
Kilic, K., Karatas, H., Dönmez-Demir, B., Eren-Kocak, E., Gursoy-Ozdemir, Y., Can, A., et al. (2018). Inadequate brain glycogen or sleep increases spreading depression susceptibility. Ann. Neurol. 83, 61–73. doi: 10.1002/ana.25122
Kim, C. C., Nakamura, M. C., and Hsieh, C. L. (2016). Brain trauma elicits non-canonical macrophage activation states. J. Neuroinflammation 13:117. doi: 10.1186/s12974-016-0581-z
Kimelberg, H. K., Narumi, S., and Bourke, R. S. (1978). Enzymatic and morphological properties of primary rat brain astrocyte cultures and enzyme development in vivo. Brain Res. 153, 55–77. doi: 10.1016/0006-8993(78)91128-9
Kitamura, Y., Watanabe, S., Taguchi, M., Takagi, K., Kawata, T., Takahashi-Niki, K., et al. (2011). Neuroprotective effect of a new DJ-1-binding compound against neurodegeneration in Parkinson’s disease and stroke model rats. Mol. Neurodegener. 6:48. doi: 10.1186/1750-1326-6-48
Koeppen, J., Nguyen, A. Q., Nikolakopoulou, A. M., Garcia, M., Hanna, S., Woodruff, S., et al. (2018). Functional consequences of synapse remodeling following astrocyte-specific regulation of Ephrin-B1 in the adult hippocampus. J. Neurosci. 38, 5710–5726. doi: 10.1523/JNEUROSCI.3618-17.2018
Kong, J., Shepel, P. N., Holden, C. P., Mackiewicz, M., Pack, A. I., and Geiger, J. D. (2002). Brain glycogen decreases with increased periods of wakefulness: implications for homeostatic drive to sleep. J. Neurosci. 22, 5581–5587. doi: 10.1523/JNEUROSCI.22-13-05581.2002
Kong, Q., Glicksman, M. A., Lin, G., Kong, Q., Chang, L., Takahashi, K., et al. (2014). Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection Find the latest version: small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J. Clin. Invest. 124, 1255–1267. doi: 10.1172/jci66163
Kong, Q., Takahashi, K., Schulte, D., Stouffer, N., Lin, Y., and Lin, C. L. G. (2012). Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol. Dis. 47, 145–154. doi: 10.1016/j.nbd.2012.03.032
Kordower, J. H., Olanow, C. W., Dodiya, H. B., Chu, Y., Beach, T. G., Adler, C. H., et al. (2013). Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136, 2419–2431. doi: 10.1093/brain/awt192
Kozela, E., Lev, N., Kaushansky, N., Eilam, R., Rimmerman, N., Levy, R., et al. (2011). Cannabidiol inhibits pathogenic T cells, decreases spinal microglial activation and ameliorates multiple sclerosis-like disease in C57BL/6 mice. Br. J. Pharmacol. 163, 1507–1519. doi: 10.1111/j.1476-5381.2011.01379.x
Kreisman, N. R., and Smith, M. L. (1993). Potassium-induced changes in excitability in the hippocampal CA1 region of immature and adult rats. Dev. Brain Res. 76, 67–73. doi: 10.1016/0165-3806(93)90123-r
Kress, B. T., Iliff, J. J., Xia, M., Wang, M., Wei Bs, H. S., Zeppenfeld, D., et al. (2014). Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 76, 845–861. doi: 10.1002/ana.24271
Kuhn, S. A., Van Landeghem, F. K. H., Zacharias, R., Färber, K., Rappert, A., Pavlovic, S., et al. (2004). Microglia express GABAB receptors to modulate interleukin release. Mol. Cell. Neurosci. 25, 312–322. doi: 10.1016/j.mcn.2003.10.023
Kulijewicz-Nawrot, M., Syková, E., Chvátal, A., Verkhratsky, A., and Rodríguez, J. J. (2013). Astrocytes and glutamate homoeostasis in Alzheimer’s disease: a decrease in glutamine synthetase, but not in glutamate transporter-1, in the prefrontal cortex. ASN Neuro 5, 273–282. doi: 10.1042/an20130017
Laplaud, D. A., Ruiz, C., Wiertlewski, S., Brouard, S., Berthelot, L., Guillet, M., et al. (2004). Blood T-cell receptor β chain transcriptome in multiple sclerosis. Characterization of the T cells with altered CDR3 length distribution. Brain 127, 981–995. doi: 10.1093/brain/awh119
Lastres-Becker, I., Ulusoy, A., Innamorato, N. G., Sahin, G., Rábano, A., Kirik, D., et al. (2012). α-synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 21, 3173–3192. doi: 10.1093/hmg/dds143
Lauterbach, J., and Klein, R. (2006). Release of full-length EphB2 receptors from hippocampal neurons to cocultured glial cells. J. Neurosci. 26, 11575–11581. doi: 10.1523/JNEUROSCI.2697-06.2006
Lee, T. S., Eid, T., Mane, S., Kim, J. H., Spencer, D. D., Ottersen, O. P., et al. (2004). Aquaporin-4 is increased in the sclerotic hippocampus in human temporal lobe epilepsy. Acta Neuropathol. 108, 493–502. doi: 10.1007/s00401-004-0910-7
Lee, Y., Morrison, B. M., Li, Y., Lengacher, S., Farah, M. H., Hoffman, P. N., et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. doi: 10.1038/nature11314
Lee, H. J., Suk, J. E., Patrick, C., Bae, E. J., Cho, J. H., Rho, S., et al. (2010). Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 285, 9262–9272. doi: 10.1074/jbc.M109.081125
Leenders, K. L., Salmon, E. P., Tyrrell, P., Perani, D., Brooks, D. J., Sager, H., et al. (1990). The nigrostriatal dopaminergic system assessed in vivo by positron emission tomography in healthy volunteer subjects and patients with Parkinson’s disease. Arch. Neurol. 47, 1290–1298. doi: 10.1001/archneur.1990.00530120034007
Lehéricy, S., Dormont, D., Sémah, F., Clémenceau, S., Granat, O., Marsault, C., et al. (1995). Developmental abnormalities of the medial temporal lobe in patients with temporal lobe epilepsy. Am. J. Neuroradiol. 16, 617–626.
Lehre, K. P., and Danbolt, N. C. (1998). The number of glutamate transport subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J. Neurosci. 18, 8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998
Lepannetier, S., Gualdani, R., Tempesta, S., Schakman, O., Seghers, F., Kreis, A., et al. (2018). Activation of TRPC1 channel by metabotropic glutamate receptor mGluR5 modulates synaptic plasticity and spatial working memory. Front. Cell. Neurosci. 12:318. doi: 10.3389/fncel.2018.00318
Lev-Ram, V., and Ellisman, M. H. (1995). Axonal activation-induced calcium transients in myelinating Schwann cells, sources, and mechanisms. J. Neurosci. 15, 2628–2637. doi: 10.1523/JNEUROSCI.15-04-02628.1995
Leyns, C. E. G., Gratuze, M., Narasimhan, S., Jain, N., Koscal, L. J., Jiang, H., et al. (2019). TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 22, 1217–1222. doi: 10.1038/s41593-019-0433-0
Li, Z., Cao, J., Ren, X., Zhao, Q., Zhou, M., and Zang, W. (2013). Nicotine protects dopaminergic neurons against lipopolysaccharide-induced damage through a7 nAChRs in microglia. Life Sci. J. 10(3):1198–1203.
Li, C., Wei, Q., Gu, X., Chen, Y., Chen, X., Cao, B., et al. (2019). Decreased glycogenolysis by miR-338–3p promotes regional glycogen accumulation within the spinal cord of amyotrophic lateral sclerosis mice. Front. Mol. Neurosci. 12:114. doi: 10.3389/fnmol.2019.00114
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Liévens, J. C., Woodman, B., Mahal, A., Spasic-Boscovic, O., Samuel, D., Kerkerian-Le Goff, L., et al. (2001). Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol. Dis. 8, 807–821. doi: 10.1006/nbdi.2001.0430
Lippmann, E. S., Azarin, S. M., Kay, J. E., Nessler, R. A., Wilson, H. K., Al-Ahmad, A., et al. (2012). Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 30, 783–791. doi: 10.1038/nbt.2247
Liu, J., Dupree, J. L., Gacias, M., Frawley, R., Sikder, T., Naik, P., et al. (2016). Clemastine enhances myelination in the prefrontal cortex and rescues behavioral changes in socially isolated mice. J. Neurosci. 36, 957–962. doi: 10.1523/JNEUROSCI.3608-15.2016
Liu, Y., Hu, J., Wu, J., Zhu, C., Hui, Y., Han, Y., et al. (2012). α7 nicotinic acetylcholine receptor-mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. J. Neuroinflammation 9:98. doi: 10.1186/1742-2094-9-98
Logothetis, N. K., Pauls, J., Augath, M., Trinath, T., and Oeltermann, A. (2001). Neurophysiological investigation of the basis of the fMRI signal. Nature 412, 150–157. doi: 10.1038/35084005
Lomen-Hoerth, C., Anderson, T., and Miller, B. (2002). The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59, 1077–1079. doi: 10.1212/wnl.59.7.1077
Löscher, W., Hirsch, L. J., and Schmidt, D. (2015). The enigma of the latent period in the development of symptomatic acquired epilepsy—traditional view versus new concepts. Epilepsy Behav. 52, 78–92. doi: 10.1016/j.yebeh.2015.08.037
Lull, M. E., and Block, M. L. (2010). Microglial activation and chronic neurodegeneration. Neurotherapeutics 7, 354–365. doi: 10.1016/j.nurt.2010.05.014
Lund, I. V., Hu, Y., Raol, Y. H., Benham, R. S., Russek, S. J., and Brooks-Kayal, A. R. (2008). BDNF selectively regulates gabaa receptor transcription. Sci. Signal. 1:ra9. doi: 10.1126/scisignal.1162396
Lutgen, V., Narasipura, S. D., Sharma, A., Min, S., and Al-Harthi, L. (2016). β-catenin signaling positively regulates glutamate uptake and metabolism in astrocytes. J. Neuroinflammation 13:242. doi: 10.1186/s12974-016-0691-7
Ma, Z., Stork, T., Bergles, D. E., and Freeman, M. R. (2016). Neuromodulators signal through astrocytes to alter neural circuit activity and behavior. Nature 539, 428–432. doi: 10.1038/nature20145
Magistretti, P. J., and Morrison, J. H. (1988). Noradrenaline- and vasoactive intestinal peptide-containing neuronal systems in neocortex: functional convergence with contrasting morphology. Neuroscience 24, 367–378. doi: 10.1016/0306-4522(88)90338-7
Magistretti, P. J., and Schorderet, M. (1984). VIP and noradrenaline act synergistically to increase cyclic AMP in cerebral cortex. Nature 308, 280–282. doi: 10.1038/308280a0
Magistretti, P. J., Morrison, J. H., Shoemaker, W. J., Sapin, V., and Bloom, F. E. (1981). Vasoactive intestinal polypeptide induces glycogenolysis in mouse cortical slices: a possible regulatory mechanism for the local control of energy metabolism. Proc. Natl. Acad. Sci. U S A 78, 6535–6539. doi: 10.1073/pnas.78.10.6535
Mann, D. M., Sumpter, P. Q., Davies, C. A., and Yates, P. O. (1987). Glycogen accumulations in the cerebral cortex in Alzheimer’s disease. Acta Neuropathol. 73, 181–184. doi: 10.1007/bf00693786
Marien, M. R., Colpaert, F. C., and Rosenquist, A. C. (2004). Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res. Rev. 45, 38–78. doi: 10.1016/j.brainresrev.2004.02.002
Martín, E. D., Fernández, M., Perea, G., Pascual, O., Haydon, P. G., Araque, A., et al. (2007). Adenosine released by astrocytes contributes to hypoxia-induced modulation of synaptic transmission. Glia 55, 36–45. doi: 10.1002/glia.20431
Martin, S., Al Khleifat, A., and Al-Chalabi, A. (2017). What causes amyotrophic lateral sclerosis? F1000Research 6:371. doi: 10.12688/f1000research.10476.1
Martinac, J. A., Craft, D. K., Su, J. H., Kim, R. C., and Cotman, C. W. (2001). Astrocytes degenerate in frontotemporal dementia: possible relation to hypoperfusion. Neurobiol. Aging 22, 195–207. doi: 10.1016/s0197-4580(00)00231-1
Martinez, F. O., and Gordon, S. (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6:13. doi: 10.12703/p6-13
Martínez-Muriana, A., Mancuso, R., Francos-Quijorna, I., Olmos-Alonso, A., Osta, R., Perry, V. H., et al. (2016). CSF1R blockade slows the progression of amyotrophic lateral sclerosis by reducing microgliosis and invasion of macrophages into peripheral nerves. Sci. Rep. 6:25663. doi: 10.1038/srep25663
Mashimo, M., Okubo, Y., Yamazawa, T., Yamasaki, M., Watanabe, M., Murayama, T., et al. (2010). Inositol 1,4,5-trisphosphate signaling maintains the activity of glutamate uptake in Bergmann glia. Eur. J. Neurosci. 32, 1668–1677. doi: 10.1111/j.1460-9568.2010.07452.x
Mason, J. L., Toews, A., Hostettler, J. D., Morell, P., Suzuki, K., Goldman, J. E., et al. (2004). Oligodendrocytes and progenitors become progressively depleted within chronically demyelinated lesions. Am. J. Pathol. 164, 1673–1682. doi: 10.1016/s0002-9440(10)63726-1
Mei, F., Fancy, S. P. J., Shen, Y. A. A., Niu, J., Zhao, C., Presley, B., et al. (2014). Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 20, 954–960. doi: 10.1038/nm.3618
Mei, F., Lehmann-Horn, K., Shen, Y. A. A., Rankin, K. A., Stebbins, K. J., Lorrain, D. S., et al. (2016). Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. Elife 5:e18246. doi: 10.7554/elife.18246
Metea, M. R., and Newman, E. A. (2006). Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J. Neurosci. 26, 2862–2870. doi: 10.1523/jneurosci.4048-05.2006
Meucci, O., Fatatis, A., Simen, A. A., and Miller, R. J. (2000). Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc. Natl. Acad. Sci. U S A 97, 8075–8080. doi: 10.1073/pnas.090017497
Milior, G., Morin-Brureau, M., Chali, F., Le Duigou, C., Savary, E., Huberfeld, G., et al. (2020). Distinct P2Y receptors mediate extension and retraction of microglial processes in epileptic and peritumoral human tissue. J. Neurosci. 40, 1373–1388. doi: 10.1523/jneurosci.0218-19.2019
Millán, A. P., Torres, J. J., and Marro, J. (2019). How memory conforms to brain development. Front. Comput. Neurosci. 13:22. doi: 10.3389/fncom.2019.00022
Mitterauer, B. (2018). Imbalances of tripartite synapses responsible for the pathophysiology of mental disorders and epilepsy. J. Neurol. Neuromedicine 3, 57–63. doi: 10.29245/2572.942x/2018/5.1216
Mohammed, H., Al-Awami, A. K., Beyer, J., Cali, C., Magistretti, P., Pfister, H., et al. (2018). Abstractocyte: a visual tool for exploring nanoscale astroglial cells. IEEE Trans. Vis. Comput. Graph. 24, 853–861. doi: 10.1109/tvcg.2017.2744278
Morales, I., Sanchez, A., Rodriguez-Sabate, C., and Rodriguez, M. (2017). Striatal astrocytes engulf dopaminergic debris in Parkinson’s disease: a study in an animal model. PLoS One 12:e0185989. doi: 10.1371/journal.pone.0185989
Morgenthaler, F. D., Koski, D. M., Kraftsik, R., Henry, P. G., and Gruetter, R. (2006). Biochemical quantification of total brain glycogen concentration in rats under different glycemic states. Neurochem. Int. 48, 616–622. doi: 10.1016/j.neuint.2005.12.034
Morin, D., Sapena, R., Zini, R., Onteniente, B., and Tillement, J. P. (1997). Characterization of β-adrenergic receptors of freshly isolated astrocytes and neurons from rat brain. Life Sci. 60, 315–324. doi: 10.1016/s0024-3205(96)00632-7
Morioka, N., Tokuhara, M., Nakamura, Y., Idenoshita, Y., Harano, S., Zhang, F. F., et al. (2014). Primary cultures of rat cortical microglia treated with nicotine increases in the expression of excitatory amino acid transporter 1 (GLAST) via the activation of the α7 nicotinic acetylcholine receptor. Neuroscience 258, 374–384. doi: 10.1016/j.neuroscience.2013.11.044
Morley, J. E., Farr, S. A., and Nguyen, A. D. (2018). Alzheimer disease. Clin. Geriatr. Med. 34, 591–601. doi: 10.1016/j.cger.2018.06.006
Nagelhus, E. A., Mathiisen, T. M., and Ottersen, O. P. (2004). Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with Kir4.1. Neuroscience 129, 905–913. doi: 10.1016/j.neuroscience.2004.08.053
Nagy, J. I., Li, X., Rempel, J., Stelmack, G., Patel, D., Staines, W. A., et al. (2001). Connexin26 in adult rodent central nervous system: demonstration at astrocytic gap junctions and colocalization with connexin30 and connexin43. J. Comp. Neurol. 441, 302–323. doi: 10.1002/cne.1414
Newman, L. A., Korol, D. L., and Gold, P. E. (2011). Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One 6:e28427. doi: 10.1371/journal.pone.0028427
Nikolakopoulou, A. M., Koeppen, J., Garcia, M., Leish, J., Obenaus, A., and Ethell, I. M. (2016). Astrocytic Ephrin-B1 regulates synapse remodeling following traumatic brain injury. Am. Soc. Neurochem. 8, 1–18. doi: 10.1177/1759091416630220
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Neuroscience: resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Nishida, H., and Okabe, S. (2007). Direct astrocytic contacts regulate local maturation of dendritic spines. J. Neurosci. 27, 331–340. doi: 10.1523/JNEUROSCI.4466-06.2007
Norden, D. M., and Godbout, J. P. (2013). Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 39, 19–34. doi: 10.1111/j.1365-2990.2012.01306.x
Obel, L. F., Müller, M. S., Walls, A. B., Sickmann, H. M., Bak, L. K., Waagepetersen, H. S., et al. (2012). Brain glycogen—new perspectives on its metabolic function and regulation at the subcellular level. Front. Neuroenergetics 4:3. doi: 10.3389/fnene.2012.00003
Oberheim, N. A., Takano, T., Han, X., He, W., Lin, J. H. C., Wang, F., et al. (2009). Uniquely hominid features of adult human astrocytes. J. Neurosci. 29, 3276–3287. doi: 10.1523/jneurosci.4707-08.2009
Octeau, J. C., Chai, H., Jiang, R., Bonanno, S. L., Martin, K. C., and Khakh, B. S. (2018). An optical neuron-astrocyte proximity assay at synaptic distance scales. Neuron 98, 49.e9–66.e9. doi: 10.1016/j.neuron.2018.03.003
Oe, Y., Baba, O., Ashida, H., Nakamura, K. C., and Hirase, H. (2016). Glycogen distribution in the microwave-fixed mouse brain reveals heterogeneous astrocytic patterns. Glia 64, 1532–1545. doi: 10.1002/glia.23020
Oikawa, H., Nakamichi, N., Kambe, Y., Ogura, M., and Yoneda, Y. (2005). An increase in intracellular free calcium ions by nicotinic acetylcholine receptors in a single cultured rat cortical astrocyte. J. Neurosci. Res. 79, 535–544. doi: 10.1002/jnr.20398
Okada, S., Nakamura, M., Katoh, H., Miyao, T., Shimazaki, T., Ishii, K., et al. (2006). Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med. 12, 829–834. doi: 10.1038/nm1425
Oksanen, M., Petersen, A. J., Naumenko, N., Puttonen, K., Lehtonen, Š., Gubert Olivé, M., et al. (2017). PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Reports 9, 1885–1897. doi: 10.1016/j.stemcr.2017.10.016
Olabarria, M., Noristani, H. N., Verkhratsky, A., and Rodríguez, J. J. (2011). Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: mechanism for deficient glutamatergic transmission? Mol. Neurodegener. 6:55. doi: 10.1186/1750-1326-6-55
Oria, M., Figueira, R. L., Scorletti, F., Sbragia, L., Owens, K., Li, Z., et al. (2018). CD200-CD200R imbalance correlates with microglia and pro-inflammatory activation in rat spinal cords exposed to amniotic fluid in retinoic acid-induced spina bifida. Sci. Rep. 8:10638. doi: 10.1038/s41598-018-28829-5
Orkand, R. K., Nicholls, J. G., and Kuffler, S. W. (1966). Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J. Neurophysiol. 29, 788–806. doi: 10.1152/jn.1966.29.4.788
Ottman, R. (2005). Analysis of genetically complex epilepsies. Epilepsia 10, 7–14. doi: 10.1111/j.1528-1167.2005.00350.x
Öz, G., DiNuzzo, M., Kumar, A., Moheet, A., and Seaquist, E. R. (2015). Revisiting glycogen content in the human brain. Neurochem. Res. 40, 2473–2481. doi: 10.1007/s11064-015-1664-4
Padovani, A., Agosti, C., Premi, E., Bellelli, G., and Borroni, B. (2007). Extrapyramidal symptoms in frontotemporal dementia: prevalence and clinical correlations. Neurosci. Lett. 422, 39–42. doi: 10.1016/j.neulet.2007.05.049
Pannasch, U., Vargová, L., Reingruber, J., Ezan, P., Holcman, D., and Giaume, C. (2011). Astroglial networks scale synaptic activity and plasticity. Proc. Natl. Acad. Sci. U S A 108, 8467–8472. doi: 10.1073/pnas.1016650108
Paolicelli, R. C., Bisht, K., and Tremblay, M. È. (2014). Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front. Cell. Neurosci. 8:129. doi: 10.3389/fncel.2014.00129
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Papadeas, S. T., Kraig, S. E., O’Banion, C., Lepore, A. C., and Maragakis, N. J. (2011). Astrocytes carrying the superoxide dismutase 1 (SOD1 G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc. Natl. Acad. Sci. U S A 108, 17803–17808. doi: 10.1073/pnas.1103141108
Park, H. J., Oh, S. H., Kim, H. N., Jung, Y. J., and Lee, P. H. (2016). Mesenchymal stem cells enhance α-synuclein clearance via M2 microglia polarization in experimental and human parkinsonian disorder. Acta Neuropathol. 132, 685–701. doi: 10.1007/s00401-016-1605-6
Patel, D. C., Tewari, B. P., Chaunsali, L., and Sontheimer, H. (2019). Neuron-glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 20, 282–297. doi: 10.1038/s41583-019-0126-4
Paukert, M., Agarwal, A., Cha, J., Doze, V. A., Kang, J. U., and Bergles, D. E. (2014). Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 82, 1236–1270. doi: 10.1016/j.neuron.2014.04.038
Pedersen, W. A., Fu, W., Keller, J. N., Markesbery, W. R., Appel, S., Smith, R. G., et al. (1998). Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann. Neurol. 44, 819–824. doi: 10.1002/ana.410440518
Pellerin, L., Bouzier-Sore, A.-K., Aubert, A., Serres, S., Merle, M., Costalat, R., et al. (2007). Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55, 1251–1262. doi: 10.1002/glia.20528
Perez-Alvarez, A., Navarrete, M., Covelo, A., Martin, E. D., and Araque, A. (2014). Structural and functional plasticity of astrocyte processes and dendritic spine interactions. J. Neurosci. 34, 12738–12744. doi: 10.1523/jneurosci.3913-14.2014
Petković, F., Campbell, I. L., Gonzalez, B., and Castellano, B. (2016). Astrocyte-targeted production of interleukin-6 reduces astroglial and microglial activation in the cuprizone demyelination model: implications for myelin clearance and oligodendrocyte maturation. Glia 64, 2104–2119. doi: 10.1002/glia.23043
Petroff, O. A. C., Errante, L. D., Rothman, D. L., Kim, J. H., and Spencer, D. D. (2002). Glutamate-glutamine cycling in the epileptic human hippocampus. Epilepsia 43, 703–710. doi: 10.1046/j.1528-1157.2002.38901.x
Philips, T., Bento-Abreu, A., Nonneman, A., Haeck, W., Staats, K., Geelen, V., et al. (2013). Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 136, 471–482. doi: 10.1093/brain/aws339
Philips, T., and Robberecht, W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 10, 253–263. doi: 10.1016/s1474-4422(11)70015-1
Pirttimaki, T. M., Codadu, N. K., Awni, A., Pratik, P., Nagel, D. A., Hill, E. J., et al. (2013). α7 nicotinic receptor-mediated astrocytic gliotransmitter release: Aβ effects in a preclinical Alzheimer’s mouse model. PLoS One 8:e81828. doi: 10.1371/journal.pone.0081828
Poitry-Yamate, C., and Tsacopoulos, M. (1991). Glial (Müller) cells take up and phosphorylate [3H]2-deoxy-D-glucose in mammalian retina. Neurosci. Lett. 122, 241–244. doi: 10.1016/0304-3940(91)90868-t
Politis, M., Lahiri, N., Niccolini, F., Su, P., Wu, K., Giannetti, P., et al. (2015). Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol. Dis. 83, 115–121. doi: 10.1016/j.nbd.2015.08.011
Poskanzer, K. E., and Yuste, R. (2011). Astrocytic regulation of cortical UP states. Proc. Natl. Acad. Sci. U S A 108, 18453–18458. doi: 10.1073/pnas.1112378108
Poskanzer, K. E., and Yuste, R. (2016). Astrocytes regulate cortical state switching in vivo. Proc. Natl. Acad. Sci. U S A 113, E2675–E2684. doi: 10.1073/pnas.1520759113
Pramatarova, A., Laganière, J., Roussel, J., Brisebois, K., and Rouleau, G. A. (2001). Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci. 21, 3369–3374. doi: 10.1523/jneurosci.21-10-03369.2001
Priller, C., Bauer, T., Mitteregger, G., Krebs, B., Kretzschmar, H. A., and Herms, J. (2006). Synapse formation and function is modulated by the amyloid precursor protein. J. Neurosci. 26, 7212–7221. doi: 10.1523/JNEUROSCI.1450-06.2006
Przedborski, S., Jackson-Lewis, V., Djaldetti, R., Liberatore, G., Vila, M., Vukosavic, S., et al. (2000). The parkinsonian toxin MPTP: action and mechanism. Restor. Neurol. Neurosci. 16, 135–142.
Rabinovich-Nikitin, I., Ezra, A., Barbiro, B., Rabinovich-Toidman, P., and Solomon, B. (2016). Chronic administration of AMD3100 increases survival and alleviates pathology in SOD1G93A mice model of ALS. J. Neuroinflammation 13:123. doi: 10.1186/s12974-016-0587-6
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist. Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Ratté, S., and Prescott, S. A. (2011). CLC-2 channels regulate neuronal excitability, not intracellular chloride levels. J. Neurosci. 31, 15838–15843. doi: 10.1523/jneurosci.2748-11.2011
Robel, S., Buckingham, S. C., Boni, J. L., Campbell, S. L., Danbolt, N. C., Riedemann, T., et al. (2015). Reactive astrogliosis causes the development of spontaneous seizures. J. Neurosci. 35, 3330–3345. doi: 10.1523/JNEUROSCI.1574-14.2015
Roboon, J., Hattori, T., Ishii, H., Takarada-Iemata, M., Le, T. M., Shiraishi, Y., et al. (2019). Deletion of CD38 suppresses glial activation and neuroinflammation in a mouse model of demyelination. Front. Cell. Neurosci. 13:258. doi: 10.3389/fncel.2019.00258
Rosati, A., Ilvento, L., Rizzi, R., Doccini, V., Leo, M. C., Pugi, A., et al. (2018). Long-term efficacy of add-on lacosamide treatment in children and adolescents with refractory epilepsies: a single-center observational study. Epilepsia 59, 1004–1010. doi: 10.1111/epi.14071
Rothstein, J. D., Dykes-Hoberg, M., Pardo, C. A., Bristol, L. A., Jin, L., Kuncl, R. W., et al. (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16, 675–686. doi: 10.1016/s0896-6273(00)80086-0
Rothstein, J. D., Van Kammen, M., Levey, A. I., Martin, L. J., and Kuncl, R. W. (1995). Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 38, 73–84. doi: 10.1002/ana.410380114
Rubio-Villena, C., Viana, R., Bonet, J., Garcia-Gimeno, M. A., Casado, M., Heredia, M., et al. (2018). Astrocytes: new players in progressive myoclonus epilepsy of Lafora type. Hum. Mol. Genet. 27, 1290–1300. doi: 10.1093/hmg/ddy044
Sada, N., Lee, S., Katsu, T., Otsuki, T., and Inoue, T. (2015). Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 347, 1362–1367. doi: 10.1126/science.aaa1299
Salarian, A., Russmann, H., Wider, C., Burkhard, P. R., Vingerhoets, F. J. G., and Aminian, K. (2007). Quantification of tremor and bradykinesia in Parkinson’s disease using a novel ambulatory monitoring system. IEEE Trans. Biomed. Eng. 54, 313–322. doi: 10.1109/TBME.2006.886670
Sargsyan, S. A., Monk, P. N., and Shaw, P. J. (2005). Microglia as potential contributors to motor neuron injury in amyotrophic lateral sclerosis. Glia 51, 241–253. doi: 10.1002/glia.20210
Sari, Y., Prieto, A. L., Barton, S. J., Miller, B. R., and Rebec, G. V. (2010). Ceftriaxone-induced up-regulation of cortical and striatal GLT1 in the R6/2 model of Huntington’s disease. J. Biomed. Sci. 17:62. doi: 10.1186/1423-0127-17-62
Savage, J. C., St-Pierre, M. K., Carrier, M., El Hajj, H., Novak, S. W., Sanchez, M. G., et al. (2020). Microglial physiological properties and interactions with synapses are altered at presymptomatic stages in a mouse model of Huntington’s disease pathology. J. Neuroinflammation 17:98. doi: 10.1186/s12974-020-01782-9
Sawmiller, D., Habib, A., Hou, H., Mori, T., Fan, A., Tian, J., et al. (2019). A novel apolipoprotein E antagonist functionally blocks apolipoprotein E interaction with N-terminal amyloid precursor protein, reduces β-amyloid-associated pathology and improves cognition. Biol. Psychiatry 86, 208–220. doi: 10.1016/j.biopsych.2019.04.026
Scharfman, H. E. (2007). The neurobiology of epilepsy. Curr. Neurol. Neurosci. Rep. 7, 348–354. doi: 10.1007/s11910-007-0053-z
Scher, M. S. (2003). Prenatal contributions to epilepsy: lessons from the bedside. Epileptic Disord. 5, 77–91.
Schipke, C. G., Boucsein, C., Ohlemeyer, C., Kirchhoff, F., and Kettenmann, H. (2002). Astrocyte Ca2+ waves trigger responses in microglial cells in brain slices. FASEB J. 16, 255–257. doi: 10.1096/fj.01-0514fje
Schipke, C. G., Haas, B., and Kettenmann, H. (2008). Astrocytes discriminate and selectively respond to the activity of a aubpopulation of neurons within the barrel cortex. Cereb. Cortex 18, 2450–2459. doi: 10.1093/cercor/bhn009
Schiweck, J., Eickholt, B. J., and Murk, K. (2018). Important shapeshifter: mechanisms allowing astrocytes to respond to the changing nervous system during development, injury and disease. Front. Cell. Neurosci. 12:261. doi: 10.3389/fncel.2018.00261
Schofield, E., Kersaitis, C., Shepherd, C. E., Kril, J. J., and Halliday, G. M. (2003). Severity of gliosis in Pick’s disease and frontotemporal lobar degeneration: tau-positive glia differentiate these disorders. Brain 126, 827–840. doi: 10.1093/brain/awg085
Schuitemaker, A., Kropholler, M. A., Boellaard, R., van der Flier, W. M., Kloet, R. W., van der Doef, T. F., et al. (2013). Microglial activation in Alzheimer’s disease: an (R)-[11C]PK11195 positron emission tomography study. Neurobiol. Aging 34, 128–136. doi: 10.1016/j.neurobiolaging.2012.04.021
Seifert, G., Schilling, K., and Steinhäuser, C. (2006). Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat. Rev. Neurosci. 7, 194–206. doi: 10.1038/nrn1870
Shaafi, S., Najmi, S., Aliasgharpour, H., Mahmoudi, J., Sadigh-Etemad, S., Farhoudi, M., et al. (2016). The efficacy of the ketogenic diet on motor functions in Parkinson’s disease: a rat model. Iran. J. Neurol. 15, 63–69.
Shin, J. Y., Fang, Z. H., Yu, Z. X., Wang, C. E., Li, S. H., and Li, X. J. (2005). Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 171, 1001–1012. doi: 10.1083/jcb.200508072
Shrivastava, K., Gonzalez, P., and Acarin, L. (2012). The immune inhibitory complex CD200/CD200R is developmentally regulated in the mouse brain. J. Comp. Neurol. 520, 2657–2675. doi: 10.1002/cne.23062
Sickmann, H. M., Schousboe, A., Fosgerau, K., and Waagepetersen, H. S. (2005). Compartmentation of lactate originating from glycogen and glucose in cultured astrocytes. Neurochem. Res. 30, 1295–1304. doi: 10.1007/s11064-005-8801-4
Sickmann, H. M., Walls, A. B., Schousboe, A., Bouman, S. D., and Waagepetersen, H. S. (2009). Functional significance of brain glycogen in sustaining glutamatergic neurotransmission. J. Neurochem. 109, 80–86. doi: 10.1111/j.1471-4159.2009.05915.x
Sivakolundu, D. K., West, K. L., Zuppichini, M., Turner, M. P., Abdelkarim, D., Zhao, Y., et al. (2020). The neurovascular basis of processing speed differences in humans: a model-systems approach using multiple sclerosis. NeuroImage 215:116812. doi: 10.1016/j.neuroimage.2020.116812
Sjögren, M., Folkesson, S., Blennow, K., and Tarkowski, E. (2004). Increased intrathecal inflammatory activity in frontotemporal dementia: pathophysiological implications. J. Neurol. Neurosurg. Psychiatry 75, 1107–1111. doi: 10.1136/jnnp.2003.019422
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Sokoloff, L. (1991). “Measurement of local cerebral glucose utilization and its relation to local functional activity in the brain,” in Advances in Experimental Medicine and Biology, eds M. Vranic, S. Efendic and C. H. Hollenberg (Boston, MA: Springer), 21–42.
Solano, R. M., Casarejos, M. J., Menéndez-Cuervo, J., Rodriguez-Navarro, J. A., De Yébenes, J. G., and Mena, M. A. (2008). Glial dysfunction in parkin null mice: effects of aging. J. Neurosci. 28, 598–611. doi: 10.1523/jneurosci.4609-07.2008
Sorg, O., and Magistretti, P. J. (1991). Characterization of the glycogenolysis elicited by vasoactive intestinal peptide, noradrenaline and adenosine in primary cultures of mouse cerebral cortical astrocytes. Brain Res. 563, 227–233. doi: 10.1016/0006-8993(91)91538-c
Sorg, O., and Magistretti, P. J. (1992). Vasoactive intestinal peptide and noradrenaline exert long-term control on glycogen levels in astrocytes: blockade by protein synthesis inhibition. J. Neurosci. 12, 4923–4931. doi: 10.1523/jneurosci.12-12-04923.1992
Sotelo-Hitschfeld, T., Niemeyer, M. I., Mächler, P., Ruminot, I., Lerchundi, R., Wyss, M. T., et al. (2015). Channel-mediated lactate release by K+ -stimulated astrocytes. J. Neurosci. 35, 4168–4178. doi: 10.1523/JNEUROSCI.5036-14.2015
Stenovec, M., Trkov, S., Lasič, E., Terzieva, S., Kreft, M., Rodríguez Arellano, J. J., et al. (2016). Expression of familial Alzheimer disease presenilin 1 gene attenuates vesicle traffic and reduces peptide secretion in cultured astrocytes devoid of pathologic tissue environment. Glia 64, 317–329. doi: 10.1002/glia.22931
Stogsdill, J. A., and Eroglu, C. (2017). The interplay between neurons and glia in synapse development and plasticity. Curr. Opin. Neurobiol. 42, 1–8. doi: 10.1016/j.conb.2016.09.016
Su, J. H., Nichol, K. E., Sitch, T., Sheu, P., Chubb, C., Miller, B. L., et al. (2000). DNA damage and activated caspase-3 expression in neurons and astrocytes: evidence for apoptosis in frontotemporal dementia. Exp. Neurol. 163, 9–19. doi: 10.1006/exnr.2000.7340
Sultan, S., Li, L., Moss, J., Petrelli, F., Cassé, F., Gebara, E., et al. (2015). Synaptic integration of adult-born hippocampal neurons is locally controlled by astrocytes. Neuron 88, 957–972. doi: 10.1016/j.neuron.2015.10.037
Suzuki, A., Stern, S. A., Bozdagi, O., Huntley, G. W., Walker, R. H., Magistretti, P. J., et al. (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823. doi: 10.1016/j.cell.2011.02.018
Swanson, R. A., Morton, M. M., Sagar, S. M., and Sharp, F. R. (1992). Sensory stimulation induces local cerebral glycogenolysis: demonstration by autoradiography. Neuroscience 51, 451–461. doi: 10.1016/0306-4522(92)90329-z
Swanson, R. A., Sagar, S. M., and Sharp, F. R. (1989). Regional brain glycogen stores and metabolism during complete global ischaemia. Neurol. Res. 11, 24–28. doi: 10.1080/01616412.1989.11739856
Szu, J. I., Chaturvedi, S., Patel, D. D., and Binder, D. K. (2020). Aquaporin-4 dysregulation in a controlled cortical impact injury model of posttraumatic epilepsy. Neuroscience 428, 140–153. doi: 10.1016/j.neuroscience.2019.12.006
Takarada, T., Nakamichi, N., Kawagoe, H., Ogura, M., Fukumori, R., Nakazato, R., et al. (2012). Possible neuroprotective property of nicotinic acetylcholine receptors in association with predominant upregulation of glial cell line-derived neurotrophic factor in astrocytes. J. Neurosci. Res. 90, 2074–2085. doi: 10.1002/jnr.23101
Talantova, M., Sanz-Blasco, S., Zhang, X., Xia, P., Akhtar, M. W., Okamoto, S. I., et al. (2013). Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation and synaptic loss. Proc. Natl. Acad. Sci. U S A 110, E2518–E2527. doi: 10.1073/pnas.1306832110
Tang, L., Ma, Y., Liu, X. L., Chen, L., and Fan, D. S. (2019). Better survival in female SOD1-mutant patients with ALS: a study of SOD1-related natural history. Transl. Neurodegener. 8:2. doi: 10.1186/s40035-018-0142-8
Tarantini, S., Fulop, G. A., Kiss, T., Farkas, E., Zölei-Szénási, D., Galvan, V., et al. (2017). Demonstration of impaired neurovascular coupling responses in TG2576 mouse model of Alzheimer’s disease using functional laser speckle contrast imaging. GeroScience 39, 465–473. doi: 10.1007/s11357-017-9980-z
Taylor, D. L., Diemel, L. T., Cuzner, M. L., and Pocock, J. M. (2002). Activation of group II metabotropic glutamate receptors underlies microglial reactivity and neurotoxicity following stimulation with chromogranin A, a peptide up-regulated in Alzheimer’s disease. J. Neurochem. 82, 1179–1191. doi: 10.1046/j.1471-4159.2002.01062.x
Taylor, D. L., Diemel, L. T., and Pocock, J. M. (2003). Activation of microglial group III metabotropic glutamate receptors protects neurons against microglial neurotoxicity. J. Neurosci. 23, 2150–2160. doi: 10.1523/jneurosci.23-06-02150.2003
Thameem Dheen, S., Kaur, C., and Ling, E.-A. (2007). Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 14, 1189–1197. doi: 10.2174/092986707780597961
Tong, X., Ao, Y., Faas, G. C., Nwaobi, S. E., Xu, J., Haustein, M. D., et al. (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci. 17, 694–703. doi: 10.1038/nn.3691
Trapp, B. D., Peterson, J., Ransohoff, R. M., Rudick, R., Mörk, S., and Bö, L. (1998). Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 338, 278–285. doi: 10.1056/NEJM199801293380502
Tsacopoulos, M., Evêquoz-Mercier, V., Perrottet, P., and Buchner, E. (1988). Honeybee retinal glial cells transform glucose and supply the neurons with metabolic substrate. Proc. Natl. Acad. Sci. U S A 85, 8727–8731. doi: 10.1073/pnas.85.22.8727
Ullian, E. M., Harris, B. T., Wu, A., Chan, J. R., and Barres, B. A. (2004). Schwann cells and astrocytes induce synapse formation by spinal motor neurons in culture. Mol. Cell. Neurosci. 25, 241–251. doi: 10.1016/j.mcn.2003.10.011
Ullian, E. M., Sapperstein, S. K., Christopherson, K. S., and Barres, B. A. (2001). Control of synapse number by glia. Science 291, 657–661. doi: 10.1126/science.291.5504.657
Ulmann, L., Levavasseur, F., Avignone, E., Peyroutou, R., Hirbec, H., Audinat, E., et al. (2013). Involvement of P2X4 receptors in hippocampal microglial activation after status epilepticus. Glia 61, 1306’1319. doi: 10.1002/glia.22516
Umoh, M. E., Dammer, E. B., Dai, J., Duong, D. M., Lah, J. J., Levey, A. I., et al. (2018). A proteomic network approach across the ALS - FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain . EMBO Mol. Med. 10, 48–62. doi: 10.15252/emmm.201708202
Vainchtein, I. D., and Molofsky, A. V. (2020). Astrocytes and microglia: in sickness and in health. Trends Neurosci. 43, 144–154. doi: 10.1016/j.tins.2020.01.003
Valdebenito, R., Ruminot, I., Garrido-Gerter, P., Fernández-Moncada, I., Forero-Quintero, L., Alegría, K., et al. (2016). Targeting of astrocytic glucose metabolism by β-hydroxybutyrate. J. Cereb. Blood Flow Metab. 36, 1813–1822. doi: 10.1177/0271678x15613955
van Berkel, A. A., IJff, D. M., and Verkuyl, J. M. (2018). Cognitive benefits of the ketogenic diet in patients with epilepsy: a systematic overview. Epilepsy Behav. 87, 69–77. doi: 10.1016/j.yebeh.2018.06.004
Ventura, R., and Harris, K. M. (1999). Three-dimensional relationships between hippocampal synapses and astrocytes. J. Neurosci. 19, 6897–6906. doi: 10.1523/jneurosci.19-16-06897.1999
Viggars, A. P., Wharton, S. B., Simpson, J. E., Matthews, F. E., Brayne, C., Savva, G. M., et al. (2011). Alterations in the blood brain barrier in ageing cerebral cortex in relationship to Alzheimer-type pathology: a study in the MRC-CFAS population neuropathology cohort. Neurosci. Lett. 505, 25–30. doi: 10.1016/j.neulet.2011.09.049
Villalba, R. M., and Smith, Y. (2011). Neuroglial plasticity at striatal glutamatergic synapses in Parkinson’s disease. Front. Syst. Neurosci. 5:68. doi: 10.3389/fnsys.2011.00068
Virchow, R. (1860). Cellular Pathology as Based Upon Physiological and Pathological Histology. London: John Churchill.
Voß, E. V., Škuljec, J., Gudi, V., Skripuletz, T., Pul, R., Trebst, C., et al. (2012). Characterisation of microglia during de- and remyelination: can they create a repair promoting environment? Neurobiol. Dis. 45, 519–528. doi: 10.1016/j.nbd.2011.09.008
Wakabayashi, K., Hayashi, S., Yoshimoto, M., Kudo, H., and Takahash, H. (2000). NACP αsynuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 99, 14–20. doi: 10.1007/pl00007400
Wallraff, A., Köhling, R., Heinemann, U., Theis, M., Willecke, K., and Steinhäuser, C. (2006). The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. 26, 5438–5447. doi: 10.1523/jneurosci.0037-06.2006
Wang, M., Ding, F., Deng, S. Y., Guo, X., Wang, W., Iliff, J. J., et al. (2017). Focal solute trapping and global glymphatic pathway impairment in a murine model of multiple microinfarcts. J. Neurosci. 37, 2870–2877. doi: 10.1523/jneurosci.2112-16.2017
Wang, X., Lippi, G., Carlson, D. M., and Berg, D. K. (2013). Activation of α7-containing nicotinic receptors on astrocytes triggers AMPA receptor recruitment to glutamatergic synapses. J. Neurochem. 127, 632–643. doi: 10.1111/jnc.12436
Wang, F., Smith, N. A., Xu, Q., Fujita, T., Baba, A., Matsuda, T., et al. (2012). Astrocytes modulate neural network activity by Ca2+ dependent uptake of extracellular K+. Sci. Signal. 5:ra26. doi: 10.1126/scisignal.2002334
Watanabe, H., and Passonneau, J. V. (1973). Factors affecting the turnover of cerebral glycogen and limit dextrin in vivo. J. Neurochem. 20, 1543–1554. doi: 10.1111/j.1471-4159.1973.tb00272.x
Watts, M. E., Pocock, R., and Claudianos, C. (2018). Brain energy and oxygen metabolism: emerging role in normal function and disease. Front. Mol. Neurosci. 11:216. doi: 10.3389/fnmol.2018.00216
Wei, X., Ma, T., Cheng, Y., Huang, C. C. Y., Wang, X., Lu, J., et al. (2018). Dopamine D1 or D2 receptor-expressing neurons in the central nervous system. Addict. Biol. 23, 569–584. doi: 10.1111/adb.12512
Wei, C.-J., Xu, X., and Lo, C. W. (2004). Connexins and cell signaling in development and disease. Annu. Rev. Cell Dev. Biol. 20, 811–838. doi: 10.1146/annurev.cellbio.19.111301.144309
Weinshenker, D. (2008). Functional consequences of locus coeruleus degeneration in Alzheimers disease. Curr. Alzheimer Res. 5, 342–345. doi: 10.2174/156720508784533286
Wilcock, D. M., Vitek, M. P., and Colton, C. A. (2009). Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 159, 1055–1069. doi: 10.1016/j.neuroscience.2009.01.023
Wilson, C. S., and Mongin, A. A. (2019). The signaling role for chloride in the bidirectional communication between neurons and astrocytes. Neurosci. Lett. 689, 33’44. doi: 10.1016/j.neulet.2018.01.012
Wu, X. L., Zhou, J. S., Wang, L. H., Liu, J. X., Hu, H. B., Zhang, X. T., et al. (2019). Proliferation of NG2 cells in the epileptic hippocampus. Epilepsy Res. 152, 62–72. doi: 10.1016/j.eplepsyres.2019.03.006
Xie, Y., Hayden, M. R., and Xu, B. (2010). BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. J. Neurosci. 30, 14708–14718. doi: 10.1523/jneurosci.1637-10.2010
Xie, L., Kang, H., Xu, Q., Chen, M. J., Liao, Y., Thiyagarajan, M., et al. (2013). Sleep drives metabolite clearance from the adult brain. Science 342, 373–377. doi: 10.1126/science.1241224
Xu, J., Song, D., Bai, Q., Cai, L., Hertz, L., and Peng, L. (2014). Basic mechanism leading to stimulation of glycogenolysis by isoproterenol, EGF, elevated extracellular K+ concentrations, or GABA. Neurochem. Res. 39, 661–667. doi: 10.1007/s11064-014-1244-z
Xu, J., Song, D., Xue, Z., Gu, L., Hertz, L., and Peng, L. (2013). Requirement of glycogenolysis for uptake of increased extracellular K− in astrocytes: potential implications for K+ homeostasis and glycogen usage in brain. Neurochem. Res. 38, 472–485. doi: 10.1007/s11064-012-0938-3
Xu, J., Xiao, N., and Xia, J. (2010). Thrombospondin 1 accelerates synaptogenesis in hippocampal neurons through neuroligin 1. Nat. Neurosci. 13, 22–24. doi: 10.1038/nn.2459
Xu-Friedman, M. A., Harris, K. M., and Regehr, W. G. (2001). Three-dimensional comparison of ultrastructural characteristics at depressing and facilitating synapses onto cerebellar Purkinje cells. J. Neurosci. 21, 6666–6672. doi: 10.1523/jneurosci.21-17-06666.2001
Yang, L., Kress, B. T., Weber, H. J., Thiyagarajan, M., Wang, B., Deane, R., et al. (2013). Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J. Transl. Med. 11:107. doi: 10.1186/1479-5876-11-107
Ye, L., Huang, Y., Zhao, L., Li, Y., Sun, L., Zhou, Y., et al. (2013). IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J. Neurochem. 125, 897–908. doi: 10.1111/jnc.12263
Yuan, J., Ge, H., Liu, W., Zhu, H., Chen, Y., Zhang, X., et al. (2017). M2 microglia promotes neurogenesis and oligodendrogenesis from neural stem/progenitor cells via the PPARγ signaling pathway. Oncotarget 8, 19855–19865. doi: 10.18632/oncotarget.15774
Zaitsev, A. V., Malkin, S. L., Postnikova, T. Y., Smolensky, I. V., Zubareva, O. E., Romanova, I. V., et al. (2019). Ceftriaxone treatment affects EAAT2 expression and glutamatergic neurotransmission and exerts a weak anticonvulsant effect in young rats. Int. J. Mol. Sci. 20:5852. doi: 10.3390/ijms20235852
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012
Zeng, M., Chen, X., Guan, D., Xu, J., Wu, H., Tong, P., et al. (2018). Reconstituted postsynaptic density as a molecular platform for understanding synapse formation and plasticity. Cell 174, 1172.e16–1187.e16. doi: 10.1016/j.cell.2018.06.047
Zeng, X. N., Sun, X. L., Gao, L., Fan, Y., Ding, J. H., and Hu, G. (2007). Aquaporin-4 deficiency down-regulates glutamate uptake and GLT-1 expression in astrocytes. Mol. Cell. Neurosci. 34, 34–39. doi: 10.1016/j.mcn.2006.09.008
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O’Keeffe, S., et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014
Zhang, L., Zhang, J., and You, Z. (2018). Switching of the microglial activation phenotype is a possible treatment for depression disorder. Front. Cell. Neurosci. 12:306. doi: 10.3389/fncel.2018.00306
Zhao, X., Liao, Y., Morgan, S., Mathur, R., Feustel, P., Mazurkiewicz, J., et al. (2018). Noninflammatory changes of microglia are sufficient to cause epilepsy. Cell Rep. 22, 2080–2093. doi: 10.1016/j.celrep.2018.02.004
Zhao, Y., Wu, X., Li, X., Jiang, L. L., Gui, X., Liu, Y., et al. (2018). TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron 97, 1023.e7–1031.e7. doi: 10.1016/j.neuron.2018.01.031
Zhao, W., Xie, W., Le, W., Beers, D. R., He, Y., Henkel, J. S., et al. (2004). Activated microglia initiate motor neuron injury by a nitric oxide and glutamate-mediated mechanism. J. Neuropathol. Exp. Neurol. 63, 964–977. doi: 10.1093/jnen/63.9.964
Zhou, T., Huang, Z., Sun, X., Zhu, X., Zhou, L., Li, M., et al. (2017). Microglia polarization with M1/M2 phenotype changes in rd1 mouse model of retinal degeneration. Front. Neuroanat. 11:77. doi: 10.3389/fnana.2017.00077
Zhukareva, V., Mann, D., Pickering-Brown, S., Uryu, K., Shuck, T., Shah, K., et al. (2002). Sporadic Pick’s disease: a tauopathy characterized by a spectrum of pathological τ isoforms in gray and white matter. Ann. Neurol. 51, 730–739. doi: 10.1002/ana.10222
Zonta, M., Angulo, M. C., Gobbo, S., Rosengarten, B., Hossmann, K. A., Pozzan, T., et al. (2003). Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 6, 43–50. doi: 10.1038/nn980
Zupanc, G. K. H. (2020). Development of a sexual dimorphism in a central pattern generator driving a rhythmic behavior: the role of glia-mediated potassium buffering in the pacemaker nucleus of the weakly electric fish Apteronotus leptorhynchus. Dev. Neurobiol. 80, 6–15. doi: 10.1002/dneu.22736
Keywords: glia, neurodegeneration, astrocytes, spatial buffering, K+ clearance
Citation: Stevenson R, Samokhina E, Rossetti I, Morley JW and Buskila Y (2020) Neuromodulation of Glial Function During Neurodegeneration. Front. Cell. Neurosci. 14:278. doi: 10.3389/fncel.2020.00278
Received: 24 May 2020; Accepted: 05 August 2020;
Published: 21 August 2020.
Edited by:
Arturo Ortega, Instituto Politécnico Nacional de México (CINVESTAV), MexicoReviewed by:
Marina Guizzetti, Oregon Health and Science University, United StatesCopyright © 2020 Stevenson, Samokhina, Rossetti, Morley and Buskila. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yossi Buskila, eS5idXNraWxhQHdlc3Rlcm5zeWRuZXkuZWR1LmF1
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.