 Erin N. Lottes
Erin N. Lottes Daniel N. Cox
Daniel N. Cox- Neuroscience Institute, Georgia State University, Atlanta, GA, United States
Cellular protein homeostasis, or proteostasis, is indispensable to the survival and function of all cells. Distinct from other cell types, neurons are long-lived, exhibiting architecturally complex and diverse multipolar projection morphologies that can span great distances. These properties present unique demands on proteostatic machinery to dynamically regulate the neuronal proteome in both space and time. Proteostasis is regulated by a distributed network of cellular processes, the proteostasis network (PN), which ensures precise control of protein synthesis, native conformational folding and maintenance, and protein turnover and degradation, collectively safeguarding proteome integrity both under homeostatic conditions and in the contexts of cellular stress, aging, and disease. Dendrites are equipped with distributed cellular machinery for protein synthesis and turnover, including dendritically trafficked ribosomes, chaperones, and autophagosomes. The PN can be subdivided into an adaptive network of three major functional pathways that synergistically govern protein quality control through the action of (1) protein synthesis machinery; (2) maintenance mechanisms including molecular chaperones involved in protein folding; and (3) degradative pathways (e.g., Ubiquitin-Proteasome System (UPS), endolysosomal pathway, and autophagy. Perturbations in any of the three arms of proteostasis can have dramatic effects on neurons, especially on their dendrites, which require tightly controlled homeostasis for proper development and maintenance. Moreover, the critical importance of the PN as a cell surveillance system against protein dyshomeostasis has been highlighted by extensive work demonstrating that the aggregation and/or failure to clear aggregated proteins figures centrally in many neurological disorders. While these studies demonstrate the relevance of derangements in proteostasis to human neurological disease, here we mainly review recent literature on homeostatic developmental roles the PN machinery plays in the establishment, maintenance, and plasticity of stable and dynamic dendritic arbors. Beyond basic housekeeping functions, we consider roles of PN machinery in protein quality control mechanisms linked to dendritic plasticity (e.g., dendritic spine remodeling during LTP); cell-type specificity; dendritic morphogenesis; and dendritic pruning.
Introduction
Some of Ramon y Cajal’s most famous drawings are of dendrites, and though much of our fascination in his work is due to Cajal’s skill in rendering each branch in minute detail, some of the appeal is naturally due to the sheer variety in shape and size of cells. In an illustration of a single slide, he might capture three or four different types of cells, crowding around each other in the same tiny slice of tissue (Garcia-Lopez et al., 2010). Cajal described the sight as “the nerve cell, the highest caste of organic elements, [appears] with its giant arms stretched out, like the tentacles of an octopus, to the provinces on the frontiers of the external world, to watch for the constant ambushes of physico-chemical forces” (qtd. in Ramón y Cajal, 1989). Our fascination with complicated dendritic arbors is not misplaced, for the shape of neurons evinces their function: the ornate arbors of hippocampal neurons need to integrate numerous inputs, and the bipolar cells of the retina only require two processes to facilitate rapid neural communication. The unique cell-specific structures of neuronal processes are vital to the function of each cell and to the function of the brain in its entirety. The brain relies on precise and reliable relationships between cells, and the cells, in turn, rely on their specific dendritic arbors to maintain proper connections between themselves and the larger cellular community.
The cell body alone is a whirring hub of activity, and the axon can stretch for incredible distances, making synaptic connections at many points along its length. Each neuron can participate in thousands of synaptic connections, which total over 100 trillion synapses in a human neocortex alone (Hanus and Schuman, 2013; Tang et al., 2001). The spatial architecture of a dendritic arbor is key to ensuring its appropriate synaptic inputs, and, thus, its proper function. There are three major physical requirements for dendrites to function correctly: (1) the arbor must cover its receptive field; (2) the branch pattern must be suited to the type and amount of incoming signals; and (3) the dendrites must be plastic, changing with both development and experience (Jan and Jan, 2010).
In short, dendrites must be both stable and dynamic. The half-life of a synaptic protein is a few days at most, but the main branches of the arbor may need to be maintained for the course of an organism’s life (Cohen et al., 2013). Another conflict: cytoskeletal proteins are moved via “slow” transport, which can be less than eight millimeters a day, but the physical changes in shape and size of dendritic spines can begin less than an hour after induction of long term potentiation (LTP) (Ostroff et al., 2018) – and protein level fluctuation starts even earlier (Bosch et al., 2014; Maday and Holzbaur, 2014; Ostroff et al., 2018; Hafner et al., 2019). Dendrites require protein transport from the cell body, but these examples illustrate that dendrites cannot simply rely on transport to maintain proteostasis. Instead, dendritic protein quality control systems must be in place to meet the needs of stability and plasticity. These systems include free ribosomes and dendritic endoplasmic reticulum (ER) tubules that facilitate local translation, cytoplasmic chaperones that monitor protein maintenance in dendrites, and dendritic autophagosomes, endosomes, lysosomes, and proteasomes that control localized protein recycling and turnover (Figure 1).
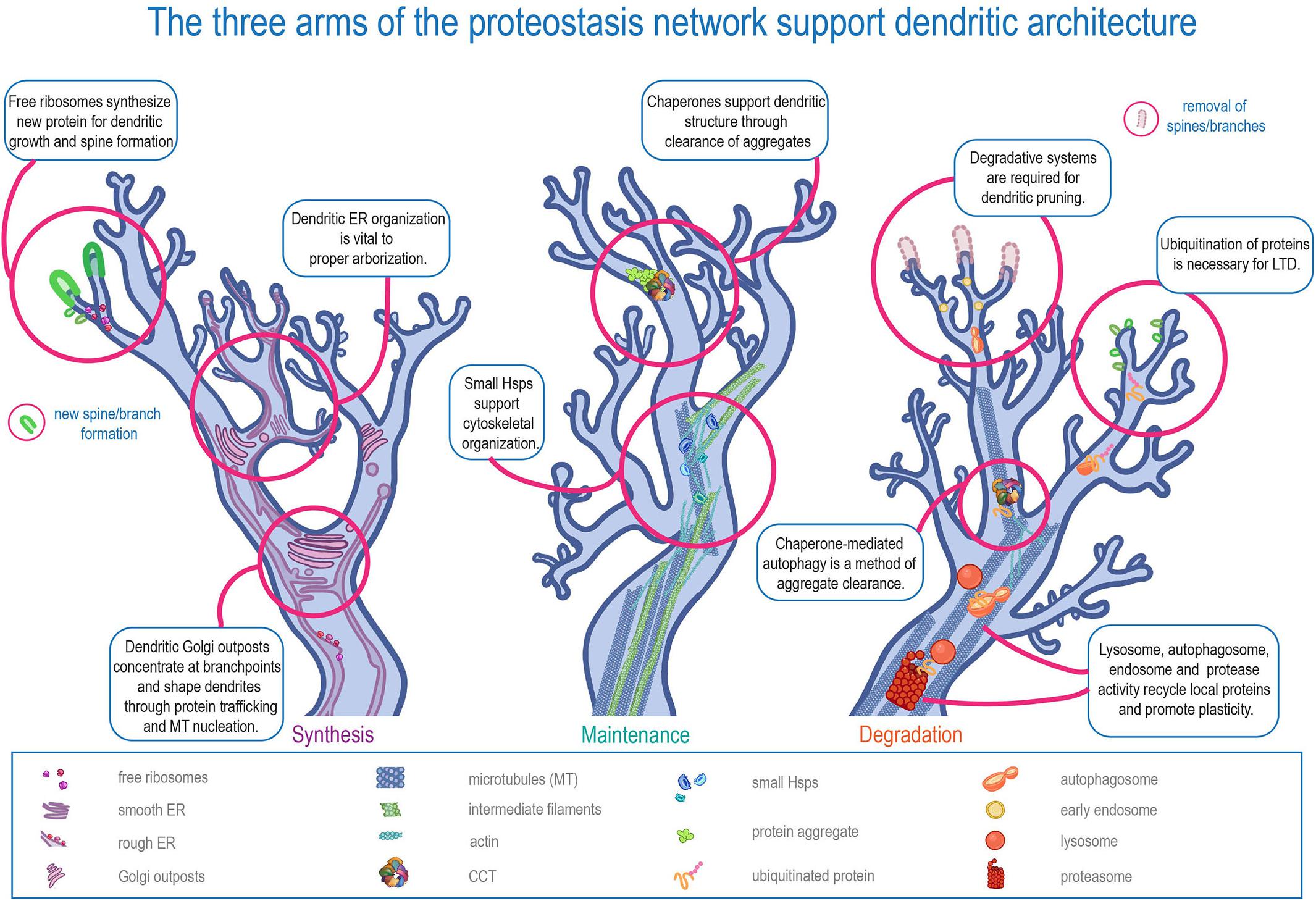
Figure 1. The three arms of the proteostasis network in supporting dendritic architecture. Schematic representation of the three major arms of proteostasis (Synthesis, Maintenance, and Degradation) in regulating distinct aspects of dendritic development and function.
Protein quality control is a sub-component of proteostasis involving protein synthesis, maintenance, and degradation (Klaips et al., 2018). This review will address the three arms of protein quality control in dendrites, from a protein’s ribosomal “birth” through its maintenance or “maturation” by chaperones, and eventually to its autophagic and ubiquitin-mediated “death.” Each proteostatic arm, when disrupted, is associated with a variety of neurological disorders, highlighting the importance of these proteostatic components to neurons, especially. Many studies have been conducted on protein quality control in cell-stress conditions (Chaari, 2019; Muranova et al., 2019; Hetz and Kaufman, 2020; Yerbury et al., 2020); however, this review will mainly address the function of each arm in homeostatic conditions. Furthermore, many of the studies discussed in this review use genetic manipulation to dissect the role of PN genes and their protein products in homeostasis. Here we discuss accumulated evidence of dendritic expression and localization of proteins and organelles that point to compartment-specific roles of PN machinery in regulating dendritic development and plasticity. With that said, an important technical caveat of the molecular genetic manipulations is that while PN genes can be genetically disrupted in a cell-type specific manner in some organisms, these manipulations are not necessarily targeted to a specific compartment and instead effect the entire cell – axon, dendrite and soma. As such, it can be technically challenging to fully disentangle putative contributions of the somatic PN from the dendritic PN. Nevertheless, that PN machinery is differentially trafficked onto dendrites, and supports biological processes such as local translation, indicates that at least somatodendritic PN machinery mechanistically functions in protein quality control linked to dendritic morphogenesis, cell-type specificity, and plasticity.
Protein Synthesis
The first arm of the protein quality control system, protein synthesis, controls translation of mRNA into protein. Ribosomes, located both in the rough ER and freely in the cytosol, shepherd the transition of mRNA to protein (Ainsley et al., 2014; Depaoli et al., 2018). Often organized in complexes comprised of many individual ribosomal subunits (Genuth and Barna, 2018), ribosomes interact with a variety of other proteins, including Ribosomal Associated Proteins (RAPs), kinases, and phosphatases, which facilitate the production of all proteins in the cell (Heise et al., 2014; Genuth and Barna, 2018).
It has long been known that free, or cytosolic, ribosomes are present in dendrites (Tiedge and Brosius, 1996; Hanus and Schuman, 2013; Genuth and Barna, 2018). In fact, ribosomal proteins have often been tagged with fluorescent markers in order to visualize dendritic arbors (Hill et al., 2012). Selectively disrupting ribosomal function has been found to affect many aspects of axonal and dendritic morphology (Perry and Fainzilber, 2014; Slomnicki et al., 2016; Genuth and Barna, 2018). This is not surprising: both axons and dendrites extend for great distances, forming intricate, complicated connections – and these polarized structures must be maintained for much longer than the lives of other cell types, requiring continual protein synthesis.
Protein Synthesis and Trafficking in Development and Maintenance of Arbors
Due to the special proteostatic demands of dendrites, it is logical that both cytosolic and ER protein translation would occur on-site. Solely depending on vesicular trafficking of essential proteins would be slow and energetically costly (Hanus and Schuman, 2013; Perry and Fainzilber, 2014). In rat brain slices, it was found that in a five-minute period – absent of any exogenous stimulation – approximately 60% of observed dendritic spines underwent active translation (Hafner et al., 2019).
The levels of some ribosomal subunits have dramatic effects on neurite formation, and some ribosomal transcription factors – transcription factors that regulate the expression of ribosomal subunits – have been specifically examined for their pro-neuritic function (Gomes et al., 2011; Das et al., 2017; Hetman and Slomnicki, 2019; Baral et al., 2020). Ribosomal subunits have been found to be required for correct dendritic arbor formation, as the knockdown of RpS3 and RpL22, 40S and 60S ribosomal subunit proteins, respectively, shrinks and simplifies arbors in Class IV (CIV) nociceptive dendritic arborization sensory neurons in Drosophila melanogaster (Olesnicky et al., 2014; Lin et al., 2015). Knockdown of many other ribosomal subunits have been found to alter CIV dendritic morphology in Drosophila larvae, such as RpL7, RpL36A, RpS2, RpS13, and RpS17 (Das et al., 2017; Nanda et al., 2018; Table 1). Mutations of RpL7 and RpL36A also resulted in reductions in dendritic F-actin and microtubule levels as well as redistribution of F-actin towards the soma, which may contribute to the observed gross morphological defects (Das et al., 2017).
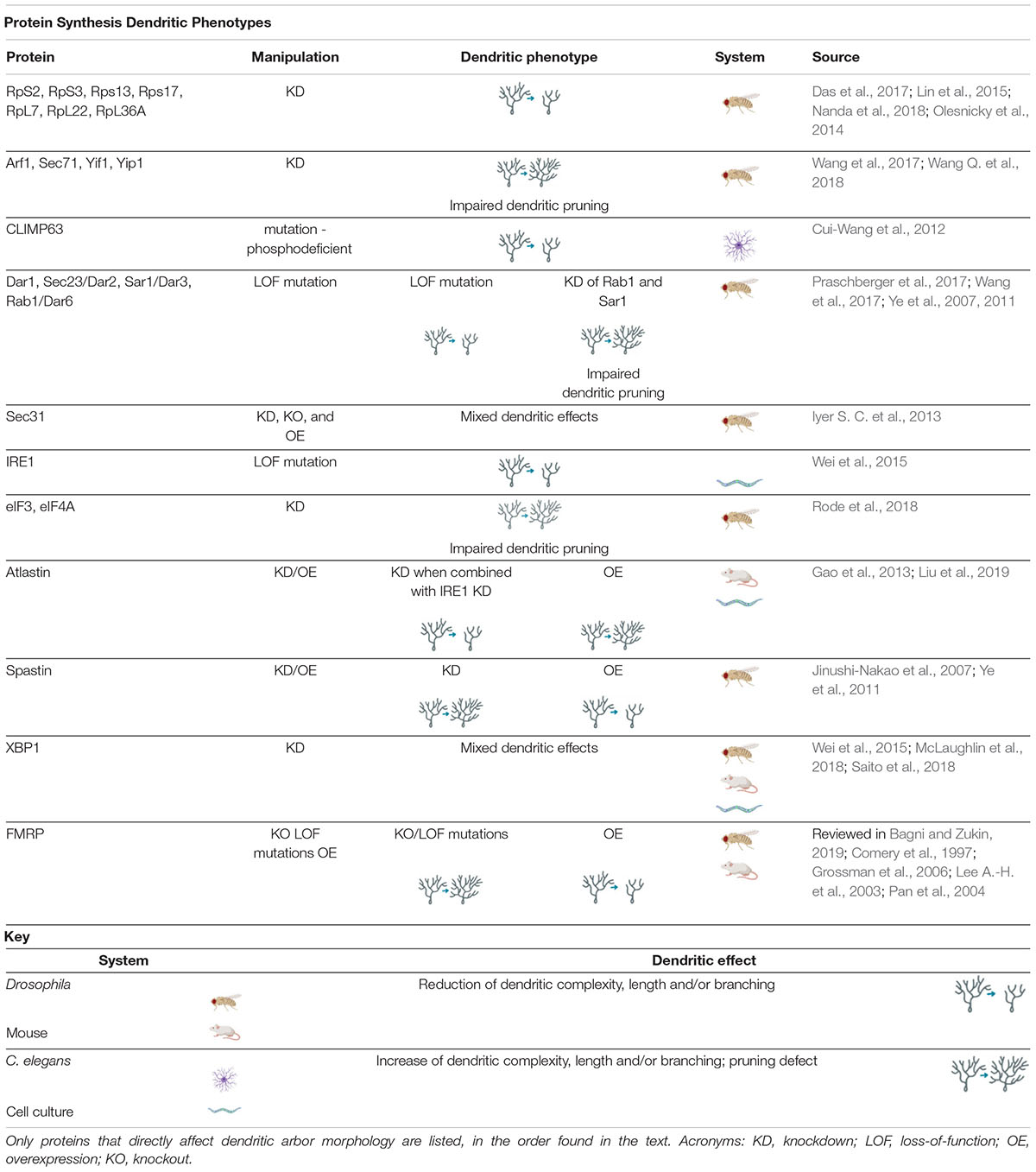
Table 1. Protein Synthesis Dendritic Phenotypes proteins involved in regulating protein synthesis cause a variety of dendritic phenotypes when manipulated.
Though some studies indicate that dendritic translation is more heavily dependent on free ribosomes than the rough ER (Koltun et al., 2020), it has been found that a healthy ER is necessary for correct dendritic arbor formation (Cui-Wang et al., 2012). Neurons have evolved a unique spatial organization of the secretory system, with satellite ER and Golgi outposts found outside of the soma (Horton and Ehlers, 2003; Aridor et al., 2004; Ye et al., 2007; Iyer S. C. et al., 2013). This specialized neuronal secretory system is necessary for neuronal polarization and the asymmetric growth and branching that distinguishes axons from dendrites (Horton and Ehlers, 2003; Horton et al., 2005; Ye et al., 2007). The ER runs in a “tubular” form in axons and the straightaways of dendrites but creates more complicated satellite networks at dendritic branch points (Liu et al., 2019). Proper ER formation may underlie correct dendritic arbor formation (Cui-Wang et al., 2012). For example, CLIMP63, an integral ER membrane protein, guides the elongation of ER tubules to the distal ends of dendritic processes when “activated” by phosphorylation. When an “inactivated” phosphodeficient version of CLIMP63 was introduced into rat hippocampal neurons, the neurons produced fewer proximal branches. Conversely, when a phosphomimetic version of CLIMP63 was introduced, there was an increase in branch number (Cui-Wang et al., 2012). These results are similar to those of experiments manipulating the protein Atlastin – an ER tubule-binding protein which, when mutated is a cause of Hereditary Spastic Paraplegia, discussed in sections “Protein Synthesis Linked to Neurological Disease” and “Protein Maintenance in Disease” (Fink, 2013; Ozdowski et al., 2015). In Drosophila, knockdown of atlastin orthologs leads to ER network fragmentation in dendrites, though dendritic arborization defects only resulted when the knockdown was combined with a knockdown of inositol-requiring enzyme-1 (IRE1) (see section “Protein Synthesis Linked to Neurological Disease”) (Liu et al., 2019; Summerville et al., 2016). Furthermore, overexpression of Atlastin in mouse cortical neurons led to increased dendritic growth both in vivo and in vitro (Gao et al., 2013).
Other components of the secretory pathway, such as Golgi outposts – specialized Golgi compartments in dendrites – have also been proven to be crucial for dendritic growth (Ye et al., 2007; Zhou et al., 2014). Dendritic Golgi outposts, which are distinct from the Golgi apparatus in the soma, depend on a RhoA-Rho kinase (Rock) signaling pathway for formation and deployment into dendrites (Quassollo et al., 2015). Satellite Golgi outposts supply the plasma membrane needed to support growth in distal dendrites, transported via vesicle trafficking. The COPII machinery needed to facilitate dendritic vesicle trafficking is also implicated in dendritic growth and branching. Mutations in COPII components such as the coat proteins Sec13, Sec23, Sec24, and Sec31, as well as GTPases Rab1 and Sar1 cause reductions in dendritic growth and branching in Drosophila CIV neurons (Ye et al., 2007; Iyer S. C. et al., 2013). Interestingly, genes involved in the secretory pathway have also been found to be necessary for the developmentally timed dendritic pruning, a regressive process, that occurs in Drosophila pupae during metamorphosis. Defects in the function of Arf1, Sec71, Yif1 and Yip1, as well as Rab1 and Sar1, have all been found to severely disrupt dendritic pruning via dysregulation of the ER-to-Golgi network (Wang et al., 2017; Wang Q. et al., 2018). It has also been suggested that Golgi outposts may play a role in supporting local translation and protein trafficking (Steward and Schuman, 2003; Ye et al., 2007). Finally, Golgi outposts have also been found to act as microtubule-organizing centers in dendrites, a role which is essential for the formation and maintenance of the dendritic arbor (Ori-McKenney et al., 2012; Yang and Wildonger, 2020). For excellent, recent reviews on the role of the secretory pathway in neurons and the role of the Golgi complex in neurological disorders see Kennedy and Hanus (2019) and Caracci et al. (2019).
Protein Synthesis and Cell-Type Specificity
Mutations of some secretory pathway proteins – (e.g., Sec23, Sar1 and Rab1) have been found to cause developmental dendritic defects (Table 1), yet mutations in similar secretory proteins (e.g., Sec23A, Sec23B, Sec24D, and Sar1b) have been linked to non-neuronal effects in humans (Ye et al., 2007; Praschberger et al., 2017). Instead of impacting neuronal morphology, the latter group of secretory proteins are found to disrupt bone formation and cause lipid absorption disorders and anemia when mutated (Praschberger et al., 2017). These findings indicate that there may be tissue-specific dependence on different components of the synthesis and secretory systems, tailored to the unique needs of each tissue. In development, certain ribosome biogenesis factors – which are crucial to ribosomal complex assembly – are specifically expressed in stem cells, and cellular ribosomal content is thought to change as select ribosomes are recruited depending on cell fate (Gabut et al., 2020).
Ribosomal distribution and cell reliance are more than just tissue-specific. The differences in ribosomal reliance are even brain-region specific. It has been discovered that Drosophila larval neuroblasts show differential responses to CRISPR-mediated knockdown of ribosome biogenesis factors. Neuroblasts of the mushroom body proliferated much longer after loss of two ribosome biogenesis factors than did other neuroblast types (Baral et al., 2020). Recent discoveries have also revealed that ribosomal protein paralogues eRpL22 and eRpL22-like show cell-type specific patterning in the Drosophila eye in addition to developmental-dependent fluctuations in their expression (Gershman et al., 2020).
The cell-type specific patterning of protein synthesis machinery may also contribute to the diversity of dendritic morphologies. As in the Drosophila eye, sensory neurons in the Drosophila larval body wall show differing expression and dependence on ribosomal expression (Iyer E. P. R. et al., 2013; Das et al., 2017). As alluded to above, a Sec31 loss-of-function mutation caused decreased dendritic length and branching in Drosophila CIV sensory neurons, however, there was no effect of Sec31 mutation in classes of sensory neurons with simpler dendritic arbors. Furthermore, Sec31 overexpression also resulted in decreased dendritic growth and branching in complex CIV neurons, whereas Sec31 overexpression in the morphologically simpler Class I (CI) sensory neurons resulted in enhanced dendritic growth and branching, revealing cell-type specific differences in dendritic development and homeostasis (Iyer S. C. et al., 2013). This phenomenon has also been observed in C. elegans, where knockdown of IRE1, which encodes a protein monitoring ER content (discussed later in section “Protein Synthesis Linked to Neurological Disease”), causes severe reductions in dendritic branching in neurons with complex dendritic arbors, but not those with simpler arbors (Wei et al., 2015). In Drosophila, ribosomal genes were found to be more highly enriched in the dendritically complex CIV neurons relative to the dendritically simpler CI neurons (Iyer E. P. R. et al., 2013). This could indicate that more complex neurons require higher levels of protein synthesis in general, or, alternatively, are more sensitive to perturbations in protein synthesis and secretory systems. It could also be the case that specific ribosomal proteins are required for complex neurons because those neurons depend heavily on “specialized ribosomes” (discussed in section “Protein Synthesis and Plasticity”) to translate the select subset of proteins required to develop such complex arbors.
Protein Synthesis and Plasticity
The formation of the ER and presence of free ribosomes is necessary for the development and stability of the dendritic arbor in many types of neurons (Martínez et al., 2018), but ribosomes also play an important role in dendritic plasticity. Free ribosomes in the cytoplasm allow compartments like the dendrites and axon to independently respond to experiences, and ribosomes themselves may have more discretion than previously imagined. Instead of identical machines that non-discriminately transcribe any strand of mRNA that comes their way, ribosomes may selectively transcribe mRNA depending on cellular conditions or post-translational modifications (Genuth and Barna, 2018; Ferretti and Karbstein, 2019).
The term “specialized ribosomes”, also referred to as the “ribosome filter hypothesis” is currently used to refer to the idea that ribosomes, able to translate many different strands of mRNA, may have selective control over the prioritization of competing strands (Mauro and Edelman, 2007; Shi et al., 2017; Genuth and Barna, 2018). This selective control springs from the ribosomal subunit composition and/or stoichiometry of the ribosomal complex itself – selectivity may arise through slight architectural distinctions between subunit isoforms or paralogs, or dynamically through changes in cell conditions like temperature or post-translational modifications like acetylation (Gerst, 2018; Guo, 2018; Li and Wang, 2020). Alternatively, ribosomal subunits and their paralogs may be tuned to translate specific sets of proteins, and the translation of these proteins may be up- or down-regulated based on the production and degradation of the ribosomal subunits themselves (Komili et al., 2007; Ferretti and Karbstein, 2019).
Ribosome specialization is an issue complicated by the sheer variety of proteins involved in translation. RAPs (Ribosomal Associated Proteins), kinases, and phosphatases are all a part of the “ribo-interactome” (Albert et al., 2019; Genuth and Barna, 2018; Heise et al., 2014). mRNA translated by ribosomes is capped at the 5′ end, and the cap requires recruitment of eukaryotic initiation factors (eIFs) such as eIF4E and eIF4G before they, in turn, recruit the ribosome (Ostroff et al., 2017; Choi et al., 2018; Das Sharma et al., 2019). Some eIFs are even preferentially involved in different stages of dendritic growth and maintenance, such as eIF4A and eIF3, which have been found to be required for dendritic pruning in Drosophila pupae (Rode et al., 2018). Additionally, some ribosomes can directly bind with mRNA using Internal Ribosome Entry Sites (IRES); however, even while skipping the “middle man”, they still depend on a whole host of other proteins that assist with initiation, elongation, and termination of the mRNA (Sutton and Schuman, 2005; Genuth and Barna, 2018).
Specialized ribosomes may be necessary for the differences in protein expression between cell types, and thus for the formation of unique dendritic arbors (Kennedy and Hanus, 2019). Interestingly, differences in the rate of mRNA translation have even been uncovered between proximal and distal dendritic branches of the same arbor (Ouwenga et al., 2017; Wu et al., 2016; Xue and Barna, 2012), implicating ribosomal specialization in the distinction and dynamics of cell compartments.
It is well known that dendritic spines physically change in response to activity as part of LTP, and ribosomal activity is thought to be an integral part of that change (Chidambaram et al., 2019; Chirillo et al., 2019; Harris, 2020). Ribosomal mRNAs have been found to be enriched in dendrites (Ohashi and Shiina, 2020), and dendritic levels of mRNA fluctuate with synaptic activity, especially those of immediate early genes such as Activity-regulated cytoskeletal associated protein (Arc) (Sutton and Schuman, 2005; Jakkamsetti et al., 2013; Na et al., 2016; Ostroff et al., 2017; Choi et al., 2018). Using a specially designed fluorescent reporter, Arc was visualized in real time in dendrites as it was translated, appearing only fifteen seconds after synaptic stimulation with glutamate (Na et al., 2016), too quickly for traditional transport mechanisms. Surprisingly, it appeared in the dendrites but not the spines themselves, lending credence to the idea that synapses might share resources (Hanus and Schuman, 2013). Local translation may occur at the dendrite level, with proteins transported short distances to activated synapses: this is aligned with the synaptic tagging model, in which synapses share pools of resources, and allocated proteins are somehow “tagged” in order to recruit them to specific synapses (Frey and Morris, 1997; Ainsley et al., 2014; Rogerson et al., 2014). Supporting this theory, ribosome numbers have been found to be elevated in dendritic shafts following LTP induction during the persistent phase of LTP, which is dependent on protein synthesis, and mRNA translation is up-regulated in both the dendrites and soma following LTP induction (Ostroff et al., 2018; Chirillo et al., 2019; Koltun et al., 2020).
Protein Synthesis Linked to Neurological Disease
Dyshomeostasis of protein synthesis can be lethal. Ribosomopathies, caused by loss-of-function mutations of ribosomes or ribosome biogenesis factors, clearly exemplify the importance of individual ribosome subunits in organismal health. Ribosomopathies, such as Diamond-Blackfan anemia, often cause congenital birth defects and heightened cancer risk (Shi et al., 2017). For reviews of ribosomopathies, see Farley-Barnes et al. (2019) and Kampen et al. (2020).
Though ribosomopathies have consequences throughout the body, some ribosomal subunit mutations are connected specifically to neurological disorders, such as those linked to cases of Autism Spectrum Disorder (ASD) and intellectual disability (ID) (Hetman and Slomnicki, 2019; Choe and Cho, 2020). Defects in ribosome function have also been connected to neurodegenerative diseases such as Alzheimer’s disease (AD) (Ding et al., 2005; Hernández-Ortega et al., 2016) and Parkinson’s disease (PD) (Taymans et al., 2015), and ribosomal frameshifting is implicated in repeat expansion disorders such as Huntington’s disease (HD) and Amyotrophic Lateral Sclerosis (ALS) (Gao et al., 2017).
Disruption of ER organization can also underlie neurological disease, though this may be due to the loss of its organizational organelle contacts rather than its peripheral protein synthesis functions (Fowler et al., 2019). Mutations in the protein Atlastin – an ER-tubule binding protein previously discussed – are responsible for 10% of autosomal dominant cases of Hereditary Spastic Paraplegia, a disease characterized by progressive weakness and loss of motor control in the lower limbs (Fink, 2013; Ozdowski et al., 2015). Changes in ER organization are characteristic of several mutations underlying forms of Hereditary Spastic Paraplegia, including changes to atlastin, spastin, reticulon 2, REEP1 and 2, and protrudin, among others (Blackstone, 2018; Fowler et al., 2019). For a recent review of the role of the ER in axons and neurodegeneration, see Öztürk et al. (2020). The neurological disease is thought to stem mainly from axon degeneration, and disruptions in axonal regeneration and bouton number have been reported with mutations in Hereditary Spastic Paraplegia-associated genes (Rao et al., 2016; Summerville et al., 2016). However, loss-of-function experiments with atlastin and other molecules have also been found to severely disrupt gross dendritic morphology and dendritic spine formation (Fink, 2013; Liu et al., 2019; Shih and Hsueh, 2018). Moreover, mutations in spastin, which encodes a microtubule-severing AAA ATPase, are known to be the most frequent cause of autosomal dominant spastic paraplegia, and have also been shown to lead to reductions in dendritic arbor complexity (Jinushi-Nakao et al., 2007; Ye et al., 2011). There is accumulating evidence that disruption of the dendritic arbor may also contribute to this disease, and may be an interesting angle at which to study Hereditary Spastic Paraplegia in the future.
The ER does not need to be physically malformed to contribute to neurological disease, as ER stress has been linked to several neurodegenerative conditions (Hetz and Mollereau, 2014; Plate and Wiseman, 2017; Martínez et al., 2018; McLaughlin et al., 2018; Morris et al., 2018). ER stress occurs when the amount of unfolded proteins in the ER reaches an unmanageable level, triggering the unfolded protein response (UPR) (Martínez et al., 2018). Many proteins in the UPR pathway have been linked to neurodegenerative diseases. For example, x-box protein 1 (XBP1) has been found to be neuroprotective in Drosophila expressing amyloid-β42 in neurons (Marcora et al., 2017). XBP1 is a downstream effector of IRE1, one of the triggers that initiates part of the UPR cascade upon sensing unfolded protein buildup in the ER (Wei et al., 2015). Interestingly, IRE1, which can initiate both cytoprotective and apoptotic cascades (Sano and Reed, 2013), has also been found in C. elegans to be required for proper dendritic arborization – implicating the UPR in not only neurodegeneration but neurodevelopment as well (Wei et al., 2015). This is supported by a recent finding that showed activation of XBP1 through IRE1 may promote developmental dendritic outgrowth through the transcriptional activation of BDNF (Saito et al., 2018). For a recent review on the molecular details of the UPR and its role in disease, see (Hetz and Kaufman, 2020).
Fragile X syndrome (FXS) is a clear example of how a neurological disorder could arise from dysregulation of protein synthesis. FXS results from a mutation in the fmr1 gene and is the leading inherited cause of ASD (Greenblatt and Spradling, 2018). The FXS mutation and subsequent silencing of the fmr1 gene prevents the production of FMRP (Fragile X Mental Retardation Protein), which normally regulates initiation of translation (Das Sharma et al., 2019; Liu et al., 2018). FMRP creates a complex with initiation proteins, including the aforementioned eIF4E, and then binds ribosomes to control translation of nascent protein (Das Sharma et al., 2019). Without FMRP, Arc becomes constitutively expressed, and overall translation is disinhibited (Park et al., 2008; Na et al., 2016). These molecular changes are thought to underlie disruptions in normal developmental dendritic pruning, as adult brains with FXS contain longer dendritic spines and more synaptic connections than average in both humans and mouse models (Grossman et al., 2006; Patel et al., 2014). In multiple animal models, loss-of-function mutations of FMRP cause an increase in terminal dendritic branches, and in Drosophila, overexpression of FMRP has been found to decrease dendritic branch number (Comery et al., 1997; Lee A. et al., 2003; Pan et al., 2004; Zhang and Broadie, 2005; Xu et al., 2008; Dahlhaus, 2018; Khayachi et al., 2018; Wang X. et al., 2018).
Disinhibition of Arc, causing over-active translation, may underlie the excess of synaptic connections found in fmr1 knock out brains. In rat pyramidal neurons, ∼13% of proteins enriched in “excitatory” synaptic terminals (those containing metabotropic glutamate receptors [mGluRs]) were definite targets of FMRP (Hafner et al., 2019). However, mGluRs are also involved in long term depression (LTD) of synapses. LTD requires well-timed protein synthesis in order to occur. Arc must be translated within five to ten minutes of experience for the excitatory AMPA receptors to be endocytosed and LTD to occur, but if Arc is constantly translated, its ability to signal LTD is extinguished (Park et al., 2008). Homeostasis is therefore attacked on two fronts: by the increased translation of proteins that encourage dendritic growth and synapse formation, and by the inability of the LTD process to naturally remove excess synapses. It is still unknown how the increase in branching and synaptic connections leads to the cognitive symptoms of FXS or what other, less visible consequences the overactive ribosomes may have on the cell. The widespread impact of the loss of FMRP – one ribosomal-associated protein – is a testament to the importance of protein synthesis in dendrites, only the first arm of the proteostasis system.
Protein Maintenance
Once proteins are synthesized, they commute to their work sites via the secretory pathway or are simply freed into the cytoplasm to carry out their duties. Post-translational modifications are largely responsible for adaptive protein responses to cell conditions and include acetylation, phosphorylation, methylation, SUMOylation, ubiquitination, ISGylation, nitrosylation, and ROS generation (Ren et al., 2014; Sambataro and Pennuto, 2017). All of these post-translational modifications are integral to dynamic control of protein activity, but are outside of the scope of this review. The protein maintenance section will chiefly focus on the role of chaperones in dendrites.
Protein structure is integral to function, thus a key component of protein maintenance is the modulation of protein folding through the action of chaperones. Chaperones are proteins that help other proteins to fold correctly. They can be found in the cytosol as well as organelles like the ER and mitochondria (Benitez et al., 2014; Brehme et al., 2014). Many chaperones are also heat shock proteins (Hsps) because of their upregulation during times of heat stress, and are commonly categorized by weight – an Hsp60 chaperone is approximately 60 kiloDaltons (kDa) (Garrido et al., 2012). A small subset of chaperones require ATP to function and are called chaperonins (Buxbaum, 2015). Chaperonins are further split into Group I – found in bacteria and mitochondria – and Group II – found in eukarya and archaea (Balchin et al., 2018; Figure 2). Chaperonins are capable of encompassing misfolded proteins and encouraging unfolding and refolding through chaperonin conformational cycling and hydrophobic residue interactions, whereas ATP-independent chaperones cannot themselves refold a protein, but can bind it to prevent it from damaging interactions with other proteins (Narberhaus, 2002). Because of this difference, chaperonins and ATP-independent chaperones are occasionally referred to as “foldases” and “holdases,” respectively (Garrido et al., 2012; Bakthisaran et al., 2015; Ali et al., 2016; Penke et al., 2018; Hipp et al., 2019).
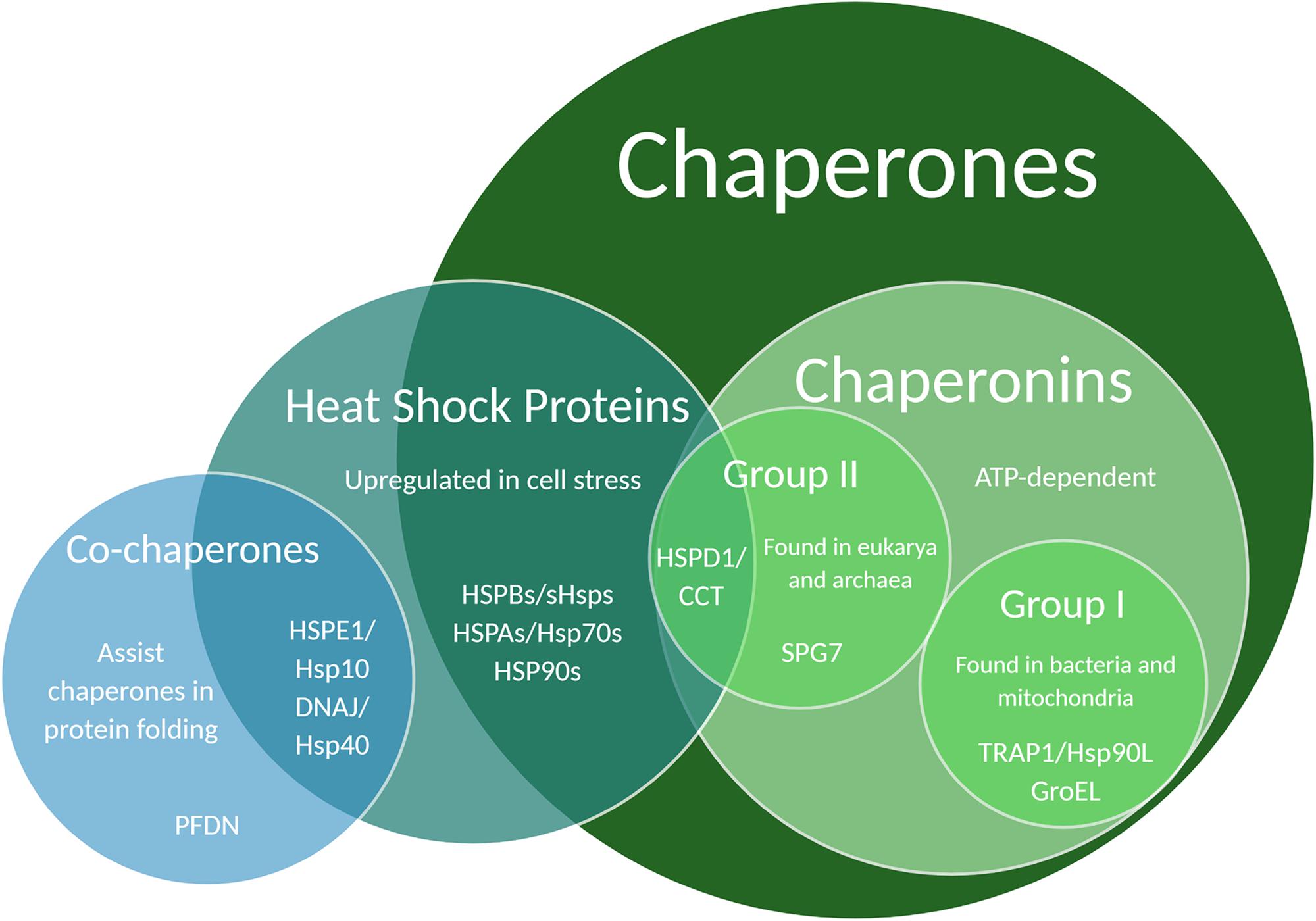
Figure 2. Venn diagram illustrating chaperone and co-chaperone families. Specific chaperones listed in this Venn diagram have been associated with regulatory effects on dendritic development and function.
Protein Maintenance in Dendritic Arborization
Protein maintenance is critical for neuronal processes: misfolded proteins must be cleared quickly before they cause damage and are either refolded or replaced. Chaperones are required from the beginning of dendritic development, as can be seen, for example, by disruptions in neurite differentiation under Hsp70 or Hsp90 knockdown conditions (Benitez et al., 2014; Miller and Fort, 2018). Hsc70, a non-heat-inducible form of Hsp70 may also be necessary for neurite differentiation. Mutations in the BAG2-Hsc70 chaperone complex was found to cause synaptic vesicles, normally located in the axon, to appear in the spines and processes of dendrites (Fukuzono et al., 2016). Chaperones contribute a great deal to cytoskeletal stability, which is especially important for neurons to sustain their complex processes (Table 2; Bakthisaran et al., 2015; Nefedova et al., 2015; Nefedova et al., 2017; Kelliher et al., 2019; Muranova et al., 2019; Vallin and Grantham, 2019; Muranova et al., 2020). Many chaperones are implicated specifically in axonal morphology: manipulation of several small heat shock proteins as well as Hsp70 and 90 was found to significantly decrease synapse number in Drosophila neuromuscular junctions (Santana et al., 2020), and an Hsp70 orthologue has been found to assist in polarized trafficking of synaptic vesicle proteins to axons (Fukuzono et al., 2016).
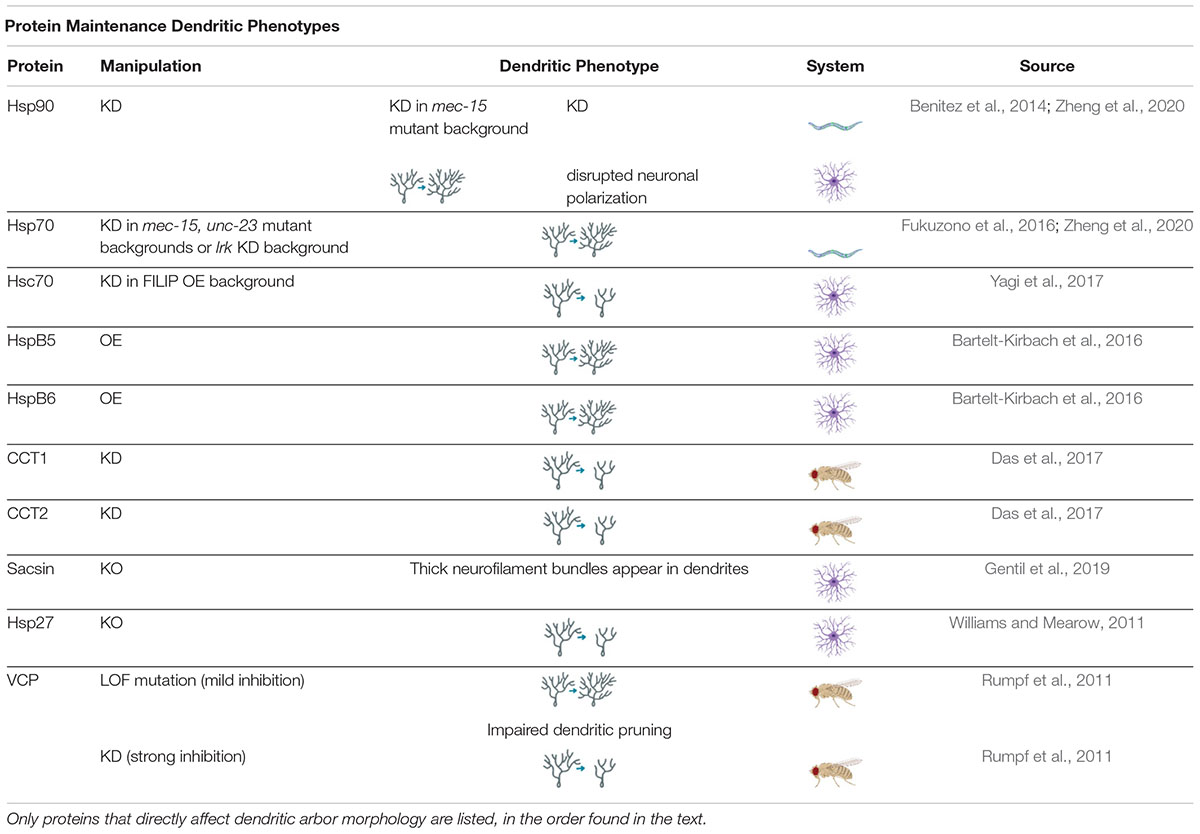
Table 2. Protein Maintenance Dendritic Phenotypes proteins involved in regulating protein maintenance cause a variety of dendritic phenotypes when manipulated.
While some chaperones have only been reported to affect axon morphology, others have been reported to affect only dendrites (Bartelt-Kirbach et al., 2016). The small heat shock proteins, Hspb5 and Hspb6, were found to increase dendritic complexity without altering axonal morphology when overexpressed in rat hippocampal neuron culture (Bartelt-Kirbach et al., 2016). Hspb5 and Hspb4, better known as αB-crystallin and αA-crystallin, make up the majority of lens protein in vertebrate eyes, and have both been found to stabilize the cytoskeleton – though Hspb4 has only been found to associate with intermediate filaments in rat retinal glia but not neurons (Bakthisaran et al., 2015; Zayas-Santiago et al., 2018).
The differing function between two such closely related chaperones is exemplary of the diversity of the small Hsp family. The small Hsp family is, ironically, one of the largest families of chaperones, with ten members in humans – HSPB1-10 – that all have diverse protein clients and are expressed in an array of tissues throughout the body, though most are expressed in the brain as well (Carra et al., 2012; Mymrikov et al., 2020). Traditionally thought of as chaperones that act as holdases to prevent protein aggregation in cell stress conditions, it has been discovered that several small Hsps contribute to proper dendritic arborization in homeostatic conditions (Narberhaus, 2002; Bakthisaran et al., 2015). Several small Hsps were found to physically interact with neurofilaments in vitro (Nefedova et al., 2017), and Hspb3 was found to distribute along neurofilaments in axons in mouse and chicken spinal motoneurons in vivo (La Padula et al., 2016). The phosphorylated forms of Hspb1 and Hspb5 associate with filamentous structures in both axons and dendrites, and mutations of Hspb1 have been found to cause cytoskeletal abnormalities, both in vitro and in vivo in mouse neurons (Schmidt et al., 2012; Lee et al., 2015; Sarparanta et al., 2020), though traditionally Hspb1 is associated with the axonal abnormalities underlying Charcot-Marie-Tooth disease (Miller and Fort, 2018; Muranova et al., 2019). More work needs to be done to uncover the role of small heat shock proteins in cytoskeletal maintenance and dendritic arborization.
Many other chaperones support dendritic development through maintenance of the cytoskeleton (Zheng et al., 2020). For example, the Hsp60 Chaperone Containing Tailless complex polypeptide-1 (CCT), is required to fold β-tubulin and actin (Brackley and Grantham, 2009; Sergeeva et al., 2014). In Drosophila CIV neurons, knockdown of CCT subunits CCT1 and 2 caused dendritic arbors to develop abnormally, with simplified, smaller arbors that contained significantly less F-actin and microtubules than controls (Das et al., 2017). Similarly, knockdown of Prefoldin 5 (Pfdn5), a component of the prefoldin co-chaperone complex, which is known to assist CCT in folding, also leads to reduction in microtubule density in CIV neurons (Tang et al., 2020). Hsp70 is also required for the development of lasting, stable dendritic arbors because it folds free tau, which is important for the stabilization of microtubules (Abisambra et al., 2013). Finally, sacsin, an enormous 520 kDa protein, has just been classified in the past decade as a chaperone because of its Hsp90 and DNAJ (Hsp40)-like domains which may function as chaperone and co-chaperone, respectively (Anderson et al., 2011). Sacsin is required for proper organization of neurofilaments in several types of neurons, including Purkinje and pyramidal neurons, and when mutated – as in the case of the hereditary disorder autosomal recessive spastic ataxia of the Charlevoix-Saguenay – leads to abnormal somatodendritic bundles of neurofilaments (Gentil et al., 2019; Larivière et al., 2019). The localization and mutation studies clearly indicate an important role of chaperones in forming and maintaining dendritic arbors.
Protein Maintenance in Cell-Type Specificity
Levels of chaperones fluctuate from tissue to tissue, as best exemplified through the small Hsp family. Hspb4 and 5 are found in high levels in the vertebrate eye lens, for example, whereas Hspb9 is only found in the testis (Mymrikov et al., 2011; Garrido et al., 2012). In a C. elegans screen of chaperones, Hsp70 showed broad expression, but a mitochondrial Hsp70 showed high expression in the intestine (Guisbert et al., 2013). Furthermore, chaperone level differences exist between cell types within the brain. In rat brain and spinal cord slices, Hsc70, a non-heat-dependent form of Hsp70, was found at higher rates in dopaminergic and motor neurons than in entorhinal cortical or hippocampal neurons (Chen and Brown, 2007). The variable levels of chaperones might mean that different cell types have variable reliance on their protein folding activities. For example, sacsin deficits have been found to cause more distinct neurofilament accumulations in certain cell types, and sacsin knockout mice displayed progressive cell loss of Purkinje neurons in the anterior cerebellar lobules at significantly greater levels than in Purkinje neurons in other cerebellar regions (Ady et al., 2018; Larivière et al., 2019). The observed differences could be due to higher cytoskeletal demands from neurons with complex arbors and thus greater sensitivity to cytoskeletal disorganization, and it has been speculated that the region-specific vulnerability of Purkinje neurons could be due to intrinsic cell qualities that differ from region to region, such as average firing rate (Ady et al., 2018; Larivière et al., 2019). However, cell-dependent reliance on chaperones could also be explained by chaperone-chaperone interactions or non-canonical homeostatic functions of chaperones.
Chaperone-chaperone interactions may contribute to cell-type differentiation. Chaperones can overlap in their “clientele,” as in the case of Hsp70 and CCT, which share seventy client proteins in common (Aswathy et al., 2016). Hsp70 has also been found to deliver clients to CCT and the two chaperonins have been shown to coordinate in folding large, multidomain proteins (Kim et al., 2013). In rat hippocampal cell culture, inhibition of Hsp90 led to Hsc70 re-localization from its usual subcellular location in proximal processes to the soma and distal axonal processes, where Hsp90 is normally found (Benitez et al., 2014), indicating that Hsc70 may change its location to compensate for the absence of Hsp90.
Small heat shock proteins are known to group together in hetero- and homo-oligomers, although the function of these oligomers is not yet known (Nefedova et al., 2015; Mymrikov et al., 2020). The interaction of chaperones may be the key to regulation of neuronal processes. Hspb6 has been found to act as a modulator of activity for other small Hsps in human cell culture (Weeks et al., 2018; Mymrikov et al., 2020; Santana et al., 2020). Furthermore, individual overexpression of Hsp23 or Hsp26 led to decreased number of synapses in the Drosophila neuromuscular junction, but combined overexpression of Hsp23 and Hsp26 together resulted in an increase in synapse numbers. From these surprising results and additional interactions with the novel kinase Pinkman, the authors hypothesize that Hsp23 and Hsp26 form a complex promoting synaptic formation and that the imbalance of the two chaperones may be the cause of synaptic dysregulation, rather than loss or gain of each individually (Santana et al., 2020).
Chaperones may also help regulate dendritic arborization in a manner apart from their canonical protein folding function. Many heat shock proteins have been found to have “moonlighting” roles in addition to their canonical protein folding duties (Jeffery, 2018). Some of these occur in separate environments, such as in the case of Hsp60 which acts as a mitochondrial chaperonin inside the cell, but as an Apo-lipoprotein A receptor on the membranes of human cultured hepatocytes (Jeffery, 2018; Bocharov et al., 2000). Some Hsps are even thought to be secreted from cells as anti-inflammatory agents (Edkins et al., 2018). Crystallins, long known for their chaperone activities in the lens of the eye, have also been found to have enzymatic functions (Fares, 2014; Jeffery, 2018).
Hsc70 performs both its canonical and moonlighting functions within the cytoplasm. In addition to its protein folding activities, Hsc70 has been found to bind to filamin-A interacting protein, which in turn binds to myosin IIb, an actin-binding protein which helps to regulate the shape a dendritic spines – in effect, Hsc70 promotes dendritic spine elongation through this pathway (Yagi et al., 2017). Additionally, the chaperone function of Hsp27 has been found to be dependent on its phosphorylation state which impacts its ability to form large homo-oligomers made up of other Hsp27 proteins. In Hsp27 knockout and phosphomimetic conditions, cultured rat neurons grew significantly fewer neuritic processes, implicating the phosphorylation state of Hsp27 in axonal and dendritic arbor formation (Williams and Mearow, 2011). Phosphorylation also affects the subcellular localization of Hspb1 and Hspb5, which moved from the soma to the dendrites and neuronal processes, respectively, when phosphorylated (Schmidt et al., 2012). This is especially interesting given that phosphorylated forms of small heat shock proteins have been shown to be less effective as chaperones in in vitro studies (Schmidt et al., 2012), supporting the idea that chaperone moonlighting may contribute to cell-specific support of diverse dendritic arbors.
Protein Maintenance in Cell Stress
Because of the dependence on cytoskeletal proteins for the maintenance of their dendritic arbors, there are some indications that neurons with large dendritic arbors may be particularly susceptible to the effects of cell stress, particularly heat shock (Dalle-Donne et al., 2001; Klose and Robertson, 2004). Without properly organized microtubules, actin, and neurofilaments, the dendritic and axonal arbors cannot be maintained (Kelliher et al., 2019). Cell stress has been shown to prevent proper maintenance of dendritic arbor and spine morphologies (Klose and Robertson, 2004; Nie et al., 2015), and in C. elegans, heat shock in adolescence has been shown to alter neuronal morphology (Hart, 2019). Therefore, chaperone-mediated maintenance of the dendritic arbor is especially important in times of neuronal stress.
Many, but not all chaperones are Hsps, named for their upregulation during heat stress. The increased numbers of Hsps is thought to combat higher levels of misfolded proteins during cell stress, and promote cell health (Miller and Fort, 2018). Hsps can be neuroprotective against cell death, as in the case of members of the Hsp70 family (HSPA1A and HSPA6), which when knocked down decreased cell viability in differentiated human neuronal cells undergoing heat shock (Deane and Brown, 2018). Upregulation of Hsps can also rescue morphology, as in the case of small Hsp23. When Hsp23 was overexpressed Drosophila muscle cells, it was able to prevent heat shock-induced axonal degeneration of the connected motor neurons (Kawasaki et al., 2016). The presence of Hsps during heat shock can even protect against dysregulation of cell dynamics. For example, boutons in the Drosophila neuromuscular junction fail to release neurotransmitter during heat shock, but overexpression of Hsp70 can rescue boutons and enable neurotransmitter release to continue (Klose and Robertson, 2004). It must be noted that these examples are all involving axons and uncategorized neurites, and the neuroprotective effects of Hsps in dendrites is in need of further research.
Heat shock is not the only type of cellular stress that can upregulate Hsp expression (Mymrikov et al., 2011; Schmidt et al., 2012; Chaari, 2019; Hu et al., 2019). Hsp70, for example, has been used as a marker of cell stress in epilepsy (Hu et al., 2019), and the upregulation of Hsp70 in these conditions is not without good reason: high expression of Hsp70 has been found to be neuroprotective in rat motoneurons experiencing excitotoxicity (Shabbir et al., 2015). Other neuronal stressors leading to the activation of heat shock proteins can include oxidative stress (Mymrikov et al., 2011; Fukui et al., 2019), hypoxia (Schmidt et al., 2012), and ER stress (Ryoo et al., 2007; Xu et al., 2011).
ER stress can induce apoptosis, and even under mild conditions of ER stress, neurite differentiation and dendritic length are disrupted (Kawada et al., 2014). As mentioned in section “Protein Synthesis Linked to Neurological Disease,” the UPR is an adaptive cell response to ER stress, which in part involves the triggering of IRE1 and subsequent activation of XBP1. XBP1 activation was found to upregulate ER DnaJ/Hsp40 (a co-chaperone to Hsp70) expression in mouse fibroblasts undergoing ER stress, and another UPR factor, ATF6, is thought to recruit BiP (an ER-specific Hsp70) in times of ER stress (Lee A.-H. et al., 2003). These ER chaperones and co-chaperones are necessary to stave off the high levels of ER stress which can lead to apoptosis. Another ER chaperone, valosin-containing protein (VCP), was found to be necessary for dendritic pruning in Drosophila (Rumpf et al., 2011). Mild inhibition of VCP caused ER stress and suppressed developmental dendritic pruning, while strong VCP inhibition caused severe dendritic morphology defects and cell death (Rumpf et al., 2011). It is possible that the connections between ER stress and chaperone response are not only neuroprotective defenses against apoptosis, but perhaps part of a larger molecular cascade regulating the dendritic arbor (Martínez et al., 2018).
Protein Maintenance in Disease
The ability of chaperones to refold misfolded proteins and disassemble protein aggregations implicates them in most proteinopathic diseases, as well as neuropathies. In select cases, mutations in the chaperone proteins themselves appear to contribute to disease etiology. For example, mutations in HSPB1, 3, and 8 are associated with Charcot-Marie-Tooth Disease (Mymrikov et al., 2011) and mutations in HSPB8 are also associated with hereditary spastic paraplegic neuropathy (Mymrikov et al., 2011), two diseases associated with axonal degeneration. Subunits four and five of the CCT complex have also been causatively linked to hereditary spastic paraplegic neuropathy; mutations in subunit five (CCT5), specifically, are the most likely candidates in causing mutilating hereditary sensory neuropathy with spastic paraplegia in a Moroccan family with the condition (Bouhouche et al., 2006; Sergeeva et al., 2014; Pavel et al., 2016). It is interesting to note that although the CCT mutations are body-wide, the effects specifically manifest in motor neurons – this, despite the fact that CCT is ubiquitously expressed in all tissues, not concentrated in neural tissue (Mymrikov et al., 2011; Guisbert et al., 2013; Hu et al., 2019). Although these examples are of mutations which cause symptoms to arise because of degeneration of the axon, there are less obvious disease links to dendritic arbor malformation. For example, in a genetic screen of mutations associated with schizophrenia and ASD, 16% of the genes screened were found to be required for proper dendritic morphology in C. elegans (Aguirre-Chen et al., 2020). Additionally, though the symptoms of autosomal recessive spastic ataxia of the Charlevoix-Saguenay are mainly due to axonal deformities, the underlying mutation of the sacsin chaperone also results in neurofilament bundling in dendrites (Anderson et al., 2011; Gentil et al., 2019; Larivière et al., 2019). The effects of dendritic arborization deficits in these rare diseases may have been previously overshadowed by the dramatic axonal effects, and should be examined in future studies.
Disruptions in protein maintenance also seem to predispose brains to neurodegenerative disease. For example, protein maintenance machinery has been found to decrease in aged brains (Brehme et al., 2014), and has thus been implicated in age-related proteinopathies such as AD and PD. CCT levels are depressed in AD patients and select subunits have also been found to be under-expressed in brains with Down syndrome, a condition known to be highly correlated with early onset AD (Aswathy et al., 2016). CCT has also been shown in vitro to inhibit the assembly of α-synuclein – as have Hsp70 and some Hsp40 co-chaperones (Aprile et al., 2017; Sot et al., 2017). α-synuclein is the aggregating protein found in the Lewy bodies of some neurodegenerative diseases such as PD (Sot et al., 2017). Though traditionally reported to aggregate in the soma and axons of neurons, it was recently found that expression of human α-synuclein in mouse cortical neurons localized to the soma and dendrites of Layer V cortical neurons and caused them to show increased dendritic spine density (Lim et al., 2020; Wagner et al., 2020). This finding runs counter to previous work stating that overexpression of α-synuclein in the cortex causes dendritic spine loss and dendritic arbor malformation in Layer V neurons (Blumenstock et al., 2017). However, since these studies were performed in younger and older mice, respectively, the findings together could indicate age-dependent dendritic effects of α-synuclein expression, perhaps underlying some of the symptoms of PD. Hsc70, Hsp27 and the mitochondrial chaperone, TRAP1, are also implicated in different hereditary forms of PD (Fukuzono et al., 2016; Brunelli et al., 2020; Vicente Miranda et al., 2020).
Chaperones are of particular interest to those studying proteinopathies and their devastating neurological effects (Smith et al., 2015). Protein aggregations are thought to be particularly for neurons because of the demands of maintaining axonal and dendritic arbors (Lim and Yue, 2015), and neurodegenerative diseases usually cause changes in dendritic morphology (Penke et al., 2018). Hsp70 has been found to co-localize with Ab plaques (Broer et al., 2011). Co-chaperones have also been explored for their contribution to proteostasis in disease. DNAJB6, an Hsp40 co-chaperone, was recently found to help regulate ataxin poly Q aggregates (Molzahn and Mayor, 2020; Thiruvalluvan et al., 2020).
CCT is widely reputed to associate with, disaggregate, or otherwise reduce toxicity of mutant huntingtin protein aggregates (Tam et al., 2006; Brackley and Grantham, 2009; Aswathy et al., 2016; Chen et al., 2018). A recent study has confirmed that CCT works to prevent formation of mutant huntingtin aggregates in adult mouse neural progenitor and stem cells (NPSCs), but more excitingly, the research implicates a balance of chaperones that work to attenuate the damages of aggregations in multiple ways over the course of cell differentiation and development. It was discovered that CCT and small Hsp levels are inversely regulated – CCT more highly expressed in NPSCs and Hspb5 more highly expressed in differentiated neurons. CCT works in the prevention of aggregates in NPSCs, but Hspb5 works in the sequestration of aggregates in differentiated neurons: all of this indicating that neurons may have developmentally regulated responses to protein aggregation, delineated by “foldase” and “holdase” chaperone properties (Molzahn and Mayor, 2020; Vonk et al., 2020).
Given their associations, upregulation of chaperones could prove to be neuroprotective, and chaperones have been targeted as potential neuroprotective agents for therapeutic interventions. There has been a recent boom in the literature of chaperone-as-medicine techniques – in 2018 the Philosophical Transactions of the Royal Society B devoted an entire themed issue to “heat shock proteins as modulators and therapeutic targets of chronic disease” (Edkins et al., 2018). Although generally the upregulation of chaperones has been found to be beneficial in fighting neurodegenerative diseases (Smith et al., 2015), counterintuitively, loss-of-function experiments with CCT subunits as well as an Hsp40 member was found to be neuroprotective in a C. elegans model of Aβ toxicity (Khabirova et al., 2014). Similarly, application of 17-AAG, which inhibits Hsp90, rescued dendritic spine loss from Aβ-induced degradation in mice (Chen et al., 2014; Smith et al., 2015). Despite these findings, the literature generally points towards a neuroprotective effect of chaperone overexpression, so several avenues have been explored in using chaperones to alleviate neurodegenerative disease (Dar et al., 2020). In primary neuronal mouse cultures with human tau mutations, application of YM-01 was used to chemically induce Hsc70 affinity for free (non-microtubule-bound) tau, leading to lower levels of the free tau, which can cause tangles within the cytoplasm (Abisambra et al., 2013). Applications of 1,4-dihydropyridine derivatives have also been used in attempts to increase levels of heat shock proteins by inducing cellular stress in mouse models of Alzheimer’s (Kasza et al., 2016). Unfortunately, the side effects of pharmacologically stimulating chaperones en masse in the brain are likely to be numerous. Hsp90 antagonists like geldanamycin have been used in clinical trials for treatment of cancer, with subjects reporting side-effects such as pain and fatigue, which may be explained by work showing that geldanamycin impairs neurite growth and cytoskeletal maintenance (Migita et al., 2019). So far, no “chaperonotherapies” have been approved for treatment of neurodegenerative diseases. However, Arimoclomol, a drug which stimulates expression of Hsp70, recently came through Phase II human clinical trials for Amyotrophic Lateral Sclerosis (ALS) with encouraging results, and is on schedule to finish Phase III trials by 2021 (Lanka et al., 2009; Elliott et al., 2020; Orphazyme’s arimoclomol receives US Fast Track Designation in Amyotrophic Lateral Sclerosis, 2020).
Protein Degradation
The final arm of the protein quality control system is protein degradation. Cellular homeostasis is achieved in part through the Ubiquitin Proteasome System (UPS), the endosome-lysosome (endolysosomal) degradation pathway, and autophagy, which are involved in protein degradation and are important not only for ridding the cell of unsalvageable misfolded proteins, but also for producing new materials by recycling protein components (Clark et al., 2018; Gerónimo-Olvera and Massieu, 2019). There is high crossover and cooperation between these systems: all three rely on ubiquitination, and all three conventionally end in fusion with a lysosome, though autophagosomes and endosomes can also fuse during trafficking (Tooze et al., 2014; Jin et al., 2018).
The UPS is used by cells to clear soluble proteins from the cytoplasm; these can be misfolded, malfunctioning proteins or even correct conformations that are simply not needed in the cytosol at the time (Ciechanover and Kwon, 2017; Kocaturk and Gozuacik, 2018). The unwanted proteins are ubiquitinated, or tagged with small ubiquitin molecules, through a process involving several enzymes (E1 through E3) (Hamilton and Zito, 2013; Kocaturk and Gozuacik, 2018). E1 is known as a ubiquitin activating enzyme, and is the enzyme that uses ATP to “activate” a ubiquitin tag. E1 then passes the ubiquitin onto E2. E2 transfers the ubiquitin to the E3 ligase – already bound to the protein target – and finally the E3 ligase transfers the ubiquitin tag to the target protein itself (Hamilton and Zito, 2013; George et al., 2018). This cycle may be repeated and more ubiquitin tags added to the first. The ubiquitinated proteins are then degraded by lysosomes or proteasomes or otherwise regulated depending on the organization of the polyubiquitination (Hamilton and Zito, 2013). Endolysosomal degradation also begins with ubiquitination, though normally of membrane proteins, which are then endocytosed and fused with early endosomes. Early endosomes then mature into multivesicular bodies, then finally to late endosomes which fuse to lysosomes (Jin et al., 2018; Zhang et al., 2014).
Autophagy, as in the other two degradative pathways, often begins in ubiquitination. However, the final location of proteins varies between UPS, endolysosomal degradation, and autophagy: UPS-mediated degradation results in proteasomes breaking down proteins, while in autophagy and endolysosomal degradation, lysosomes are often responsible for recycling the waste (Kocaturk and Gozuacik, 2018). Additionally, UPS is a highly specific system that primarily degrades single cytosolic proteins, whereas autophagy can degrade a wider variety of cytosolic substances as well as membrane proteins, which, as previously mentioned, are also degraded via endolysosomal pathways (Jin et al., 2018; Kocaturk and Gozuacik, 2018; Gerónimo-Olvera and Massieu, 2019). Since this can involve breakdown of organelles, proteins, or parasitic invaders, there are many selective forms of autophagy named for their targets, such as mitophagy (mitochondrial autophagy) and even proteaphagy (proteasome autophagy) (Yang and Klionsky, 2009; Yang et al., 2013; Kocaturk and Gozuacik, 2018; Fang et al., 2019).
There are three main types of autophagy: macro-, micro-, and chaperone-mediated autophagy (Yue et al., 2009; Gerónimo-Olvera and Massieu, 2019). In general, autophagy is the process by which cytosolic matter is transported to lysosomes to be broken down. Macroautophagy involves the creation of autophagosomes, which engulf the targets in membrane and contain them during transport to the lysosome (Maruzs et al., 2019).
Chaperones can also capture and transport misfolded proteins to the lysosome in a process called chaperone-mediated autophagy (Ciechanover and Kwon, 2017). Hsp70 is known to mediate disassembly of protein aggregates in this manner (Fukuzono et al., 2016; Ciechanover and Kwon, 2017). Chaperones not only can correct misfolded proteins but can disassemble aggregations and initiate UPS and autophagic pathways (Mymrikov et al., 2011; Fukuzono et al., 2016; Kasza et al., 2016; Pavel et al., 2016; Ciechanover and Kwon, 2017; Chaari, 2019). While they may or may not participate in physically transporting misfolded proteins to lysosomes, other chaperones - including CCT, Hspb6, and Hspb8 - are implicated in positively regulating autophagy (Mymrikov et al., 2011; Pavel et al., 2016).
Finally, microautophagy occurs when the lysosomes directly capture and break down cytosolic content (Yue et al., 2009). This wide variety of autophagic sub-systems is fascinating, and each component is individually important for proteostasis. However, generally, the word autophagy refers to macroautophagy, and that is how the term will be used for the remainder of this review.
Protein Degradation in Development and Maintenance of Dendritic Arbors
Protein degradation, like the other arms of protein quality control, is crucial for dendritic arbor formation and maintenance. Neurons are post-mitotic and thus cannot clear cellular waste through division (Son et al., 2012; Maday et al., 2014; Ciechanover and Kwon, 2017). Additionally, autophagy is especially important for degradation of long-lived proteins (Kocaturk and Gozuacik, 2018), which are often found in the brain (Alvarez-Castelao and Schuman, 2015) and membrane proteins, which are integral to the function of neuronal synapses (Hamilton and Zito, 2013).
Loss-of-function studies of components of the UPS have been found to disrupt dendritic development. Knockdown of Ube3A, an E3 ligase, was found to reduce dendritic arborization in Drosophila neurons, and mutations of Ube3A are associated with Angelman Syndrome (AS), discussed in section “Protein Degradation and Disease” (Lu et al., 2009; Hamilton and Zito, 2013). Knockdown of SkpA, an adaptor protein of the SCF (Skp-1-Cullin-F-box) E3 ubiquitin ligase complex, causes increased branching in Drosophila larval CIV neurons, and is later implicated in CIV neurons for regulating dendritic pruning at the pupal stage, discussed more in section “Protein Degradation in Neuronal Dynamics” (Wong et al., 2013; Das et al., 2017; Nanda et al., 2018).
Distinctions have been noted in the mechanisms and activity of protein degradation between axons and dendrites, and these differences may be important for developing and maintaining compartmentalization in the neuron. For example, the Anaphase Promoting Complex (APC) is a large E3 complex that ubiquitinates target proteins for UPS, and has been found to negatively regulate axonal growth through activation of downstream transcription factors; however, APC was also found to positively regulate dendritic growth and arborization (Konishi et al., 2004; Kim et al., 2009; Hamilton and Zito, 2013). Highwire, another E3 ubiquitin ligase has been found to have similar regulatory capabilities (Table 3; Wang et al., 2013). There are also differences in autophagosome transport in axons and dendrites that indicate compartmentalization of autophagosome activity. Autophagosomes are often formed in axons before being transported to the soma (Yue et al., 2009; Hernandez et al., 2012; Maday and Holzbaur, 2016). In primary mouse hippocampal cell culture, 80% of all counted autophagosomes were formed in the distal tip of the axon before moving retrograde to the cell body, where they were held before forming autolysosomes. Meanwhile, dendritic autophagosomes primarily remained stationary, and the few that moved did so bidirectionally on the dendritic arbor (Maday and Holzbaur, 2014). Based on these patterns of movement, and common mixed polarity microtubule organization in mammals, it appears that autophagosome transport is dependent on microtubules and retrograde motors, such as dynein (Yang et al., 2013; Maday and Holzbaur, 2014, 2016). Endosomes, which also appear to depend on dynein for transport, have been found to traffic bidirectionally in dendrites, as have lysosomes (Satoh et al., 2008; Jin et al., 2018; Yap et al., 2018). The movement of endosomes is also dependent on the small GTPase Rab11 (Park et al., 2006). Furthermore, locations of lysosomes, endosomes, and autophagosomes throughout the dendritic arbor differ. For example, early endosomes were found to be abundant in the medial and distal dendrites of rat hippocampal neurons, whereas lysosomes tended to concentrate closer to the soma in the proximal dendrites (Yap et al., 2018). The small GTPase Rab5, a component of the early endocytic pathway, has been shown to associate with cytoplasmic Dynein to promote dendritic branching (Satoh et al., 2008). Furthermore, components of the endosomal sorting complex required for transport (ESCRT) machinery have been implicated in regulating dendritic growth (Sweeney et al., 2006; Firkowska et al., 2019). Finally, the recycling endosome marker Rab11 has been implicated in proper dendritic arbor formation in Drosophila larvae and rat hippocampal neurons (Krämer et al., 2019; Siri et al., 2020).
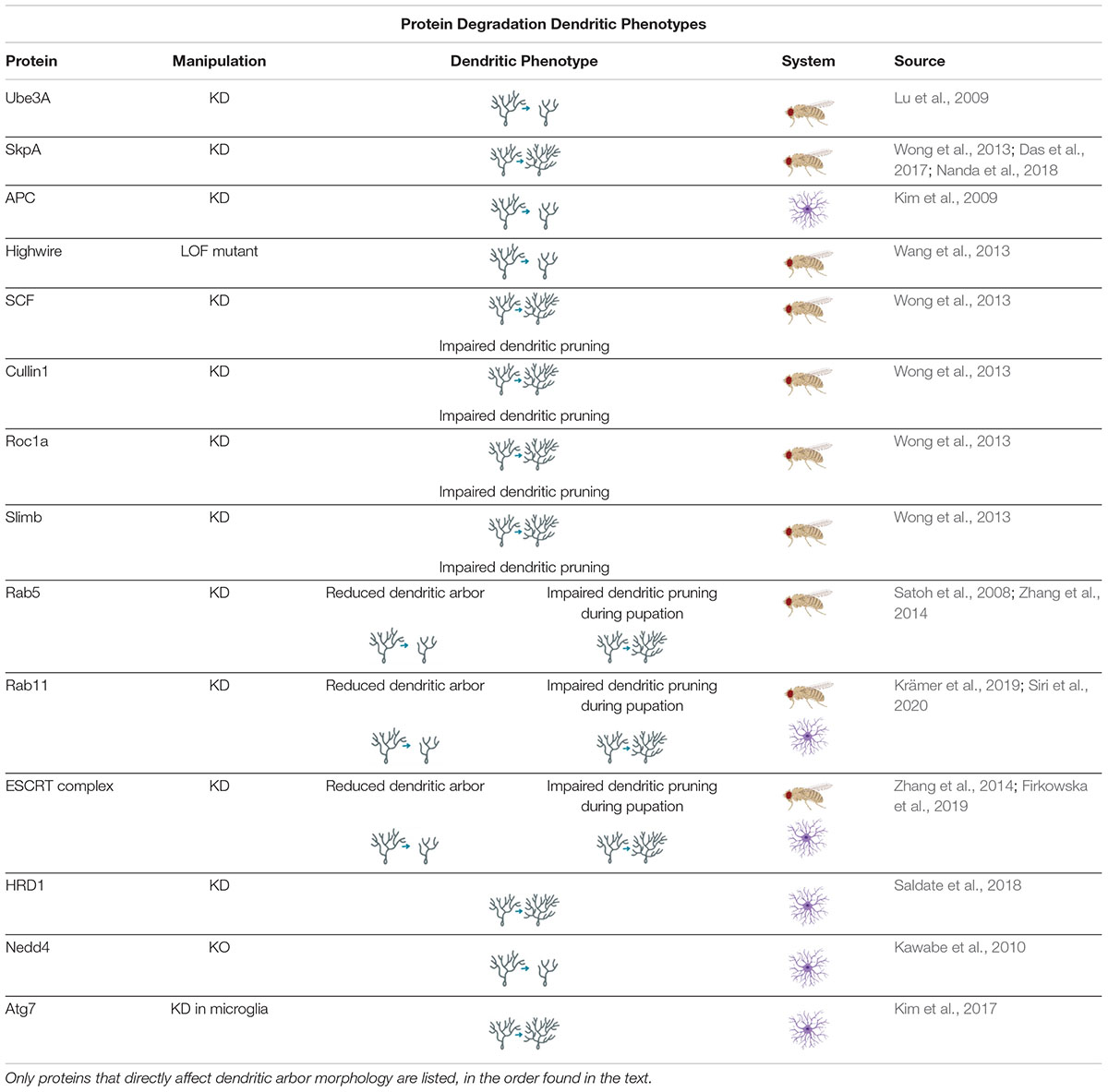
Table 3. Protein Degradation Dendritic Phenotypes proteins involved in regulating protein degradation cause a variety of dendritic phenotypes when manipulated.
There is also a lack of consensus as to whether an increase in autophagic activity positively or negatively regulates dendritic growth (Son et al., 2012). In Drosophila, knockdown or overexpression of autophagic components leads to simplification or increased branching, respectively, in the axonal neuromuscular junction (Shen and Ganetzky, 2009; Hernandez et al., 2012). However, in Drosophila sensory neurons, both knockdown and overexpression of autophagy (Atg) genes results in simplified arbors, which indicates that proper dendritic development requires carefully modulated levels of basal autophagy in homeostasis (Clark et al., 2018). In mammals, decreased autophagy can lead to increased spine density but a decrease in total dendritic length via shortening of the apical dendrite in mouse dentate gyrus neurons (Schäffner et al., 2018) – whether that is an overall increase or decrease in arbor complexity is unclear. When the autophagy of mitochondria – known as mitophagy – is manipulated in neurons, it can have effects on dendritic growth, though in what way depends on the stage of neuronal development (Brot et al., 2014).
The mixed findings may be partially due to experimental methods of autophagic induction. A common method of inducing autophagy is rapamycin, which inhibits mTOR (mammalian target of rapamycin) activity, thus disinhibiting autophagy and increasing the number of autophagosomes (Hernandez et al., 2012; Yang et al., 2013). However, the mTOR complex phosphorylates multiple kinases as well as transcription and growth factors, and inhibition of this complex may have off-target effects (Mitra et al., 2009; Lamming, 2016). Additionally, there are many studies which use starvation or other methods of induction that are not specific to just autophagy in order to examine the effects of autophagy (Brot et al., 2014; Gerónimo-Olvera and Massieu, 2019; Zhou et al., 2019). For example, tunicamycin was used to induce ER stress and, indirectly, autophagy in an experiment designed to examine the effects of autophagic flux on dendritic arbors in primary rat neuronal culture (Zhou et al., 2019). While these methods do increase autophagic action, it may be that there is no consensus on the effects of autophagic disruption in axonal and dendritic arbors because of the diversity of autophagic induction methods and the varying off-target effects of those methods (Son et al., 2012; Maday et al., 2014; Clark et al., 2018; Gerónimo-Olvera and Massieu, 2019).
Protein Degradation in Neuronal Dynamics
Protein degradation systems are required in neurons to form proper dendritic arbors. One particular way in which UPS contributes to the dynamic dendritic arbor is through pruning (Yu and Schuldiner, 2014). Pruning is a developmentally timed removal of dendritic and axonal branches, such as in humans during childhood or in Drosophila in preparation for metamorphosis (Tang et al., 2014; Yu and Schuldiner, 2014); however, similar processes can also facilitate the removal of dendritic processes and spines as a response to LTD (Piochon et al., 2016).
UPS, autophagy, and the endolysosomal pathway are required for developmental dendritic pruning, and thus for final dendritic arbor shape (Hamilton and Zito, 2013; Yu and Schuldiner, 2014). UPS inhibition leads to defects in dendritic severing, and both DIAP1 (a Drosophila E3 ubiquitin ligase) and SCF (another E3 ubiquitin ligase) are required for developmental dendritic pruning in Drosophila (Rumpf et al., 2011; Wong et al., 2013; Yu and Schuldiner, 2014). Autophagy, too, is required for developmental dendritic pruning: knockout of Atg7 – which has also been reported to selectively disrupt axonal formation – reduced spine elimination without affecting spine formation in mouse hippocampal cell culture (Tang et al., 2014). The SCF E3 ubiquitin ligase complex, comprised of cullin1, Roc1a, SkpA, and slimb is required for dendritic pruning of Drosophila CIV neurons at the pupal stage whereas knockdown of SkpA results in supernumerary growth and branching of CIV dendritic arbors at the larval stage (Wong et al., 2013; Kanamori et al., 2015a; Das et al., 2017; Nanda et al., 2018). Finally, the endolysosomal pathway also affects dendritic pruning and arbor shape. Loss of Rab5 and ESCRT proteins cause dendritic pruning defects through disrupting the endocytosis of the cell adhesion molecule, neuroglian (Zhang et al., 2014). It has also been found that Rab5 and dynamin – two more GTPases involved in the endolysosomal pathway – are required not only for the dendritic thinning that precedes detachment during developmental pruning in Drosophila CIV neurons, but also for the calcium transient currents that appear during the pruning process. The mechanism by which this occurs is still unknown, but the finding indicates that the endolysosomal pathway is not simply clearing dendritic membrane to promote pruning, but may also be actively involved in the signaling pathway that determines which processes are removed (Kanamori et al., 2015b).
Ubiquitin-proteasome system and autophagy have been well-established as an integral part of LTD (Hamilton and Zito, 2013; George et al., 2018; Mabb and Ehlers, 2018), but the mechanisms are still unknown. There are some studies which indicate that dendritic autophagy and UPS are required for NMDA-mediated LTP through removal of AMPA receptors (Lin et al., 2011; Shehata et al., 2012; Donovan and Poronnik, 2013; Tang et al., 2014; Goo et al., 2017; Gerónimo-Olvera and Massieu, 2019; Yang, 2020). Nedd4, an E3 ubiquitin ligase, was found to ubiquitinate AMPA receptors and facilitate their endocytosis (Lin et al., 2011). In cultured rat hippocampal neurons, chemical induction of LTD with low-dose NMDA led to a concurrent increase of autophagosomes and degradation of AMPA receptor subunit, mGluR1 (Shehata et al., 2012).
Other studies have indicated that degradative pathways play an important and perhaps unexpected, role in LTP as well as LTD. Recycling endosomes, which break down membrane proteins in preparation for re-use by the cell, were found to be required for LTP in rat hippocampal slices, in a Rab11-dependent manner (Park et al., 2006). This study – which also found that recycling endosomes were exocytosed at higher rates following LTP stimulation – is perhaps explained by research which has shown that recycling endosomes function in non-canonical anterograde trafficking of AMPA receptor subunit GluA1 as part of the secretory system (Park et al., 2006; Tang, 2008; Bowen et al., 2017). Rab11 knockdown has also been separately confirmed to cause reduction in dendritic spine number as well as attenuated response to LTP in rat hippocampal slices (Siri et al., 2020). UPS is also implicated in LTP: inhibition of proteasomes caused a decrease in stimulus-evoked dendritic spine growth in hippocampal slice cells (Hamilton et al., 2012; Hamilton and Zito, 2013). Other work in rat hippocampal cultures has indicated that inhibiting lysosomes causes loss of excitatory synapses (Goo et al., 2017). Furthermore, hippocampal spine density increased after knockdown of E3 ubiquitin ligase HRD1 in rat neuronal cell cultures (Saldate et al., 2018). Though perhaps paradoxical on first glance, it is likely that UPS facilitates both LTD and LTP through complex ubiquitin signals (Mabb and Ehlers, 2018; Yun et al., 2018). Interestingly, the ubiquitination and degradation of Arc, the immediate early gene discussed in section “Protein Synthesis and Plasticity,” may inhibit AMPA receptor endocytosis; the net effect being that the UPS ceases LTD through Arc inhibition (Mabb and Ehlers, 2018). Though the exact mechanisms are still incompletely understood, the contribution of protein degradation systems to dendritic pruning and dynamic spine regulation are undeniable.
Protein Degradation and Disease
Apart from dynamic changes of dendritic spine morphology, protein degradation systems are also involved in response to injury. Axonal Wallerian degeneration relies on autophagy, and although dendrites can be rapidly dismantled, it does not appear to occur through the same mechanisms as in axons, so autophagy may not be used in dendritic degeneration in the same way (Tao and Rolls, 2011; Yang et al., 2013). In mouse models with induced traumatic brain injuries, proteasomal activity decreased in the general area of injury, while autophagic markers increased; however, there is still some debate as to whether autophagy and UPS are neuroprotective or neurotoxic in cases of dendritic and axonal injury or ischemia (Yang et al., 2013; Feldmann et al., 2019; Gerónimo-Olvera and Massieu, 2019). Though it is still unclear how the two degradative subsystems interact, research indicates that they cooperate with one another in select situations (Yue et al., 2009; Feldmann et al., 2019). For comprehensive reviews on UPS-autophagy interactions see Lilienbaum (2013) and Kocaturk and Gozuacik (2018).
Like the other two arms of proteostasis, protein degradation is widely studied in neurodegenerative disease. E3 ubiquitin ligases, for example, have been linked to a wide range of neurodegenerative diseases; for a thorough review see George et al. (2018). Alterations to protein degradation have been implicated in a broad spectrum of neurodegenerative conditions including PD, HD, Multiple Sclerosis and ALS (Mitra et al., 2009; Koch et al., 2015; Albert et al., 2017; Mitsui et al., 2018). UPS and autophagy have become increasingly pursued areas of study in conjunction with neurodegenerative and protein aggregation diseases (George et al., 2018; Kabir et al., 2020; Kumar et al., 2020; Lambert-Smith et al., 2020; Lim et al., 2020; Papanikolopoulou and Skoulakis, 2020; Tundo et al., 2020).
The majority of research indicates that autophagic processes are neuroprotective, and disease-related dysfunction of autophagy contributes to or even causes neuronal degeneration (Yue et al., 2009; Yang et al., 2013; Deng et al., 2015; Koch et al., 2015; Albert et al., 2017; Beltran et al., 2019; Duan et al., 2019; Renaud et al., 2019). In ALS, for example, increased autophagy may help to clear mutant SOD1 protein and contribute to stalled degeneration of neurites in a human motor neuron cell culture model of familial ALS (Wu et al., 2019). Another instance of the neuroprotective function of autophagy is in PD models, with a recent study concluding that blockage of autophagic function causes increased levels of misfolded protein aggregation in human cell culture with induced human α-synuclein aggregates (Gerónimo-Olvera and Massieu, 2019). For recent reviews of the role of protein degradation systems in neurodegenerative disease see Kabir et al. (2020), Kumar et al. (2020), Lim et al. (2020).
The role of dendritic and spine morphology in the etiology of neuropsychiatric disorders is certainly not clear, but dysfunction of UPS and autophagy mechanisms have been linked to such disorders and are known to facilitate correct dendritic architecture. Nedd4, previously discussed for its role in AMPA receptor endocytosis, is an E3 ubiquitin ligases which facilitates proper formation of the dendritic arbor through ubiquitination of Rap2A (Donovan and Poronnik, 2013; Hamilton and Zito, 2013). Recently, single-nucleotide polymorphisms of the Nedd4 gene have been linked to schizophrenia (Han et al., 2019). For an excellent summary of neurological disorders associated with dendritic spine abnormalities see (Chidambaram et al., 2019).
Many single-nucleotide polymorphisms associated with ASD symptoms are connected to failures in the autophagic and UPS systems (Bowling and Klann, 2014; Tang et al., 2014; Devitt et al., 2015; Louros and Osterweil, 2016). ASD symptomology is linked with dendritic overgrowth and increased density of spines (Hamilton and Zito, 2013; Louros and Osterweil, 2016). ASD has also been connected to instances when the E3 ubiquitin ligase UBE3A gene is duplicated or triplicated, which leads to increases in spine density (Yi et al., 2015). Dysregulations of autophagy are also associated with ASD, as knockdown of Atg7 in microglia increased the number of somatosensory dendritic spines and caused ASD-like behavioral phenotypes to appear in mice (Kim et al., 2017). Conversely, deficiency of Ube3A is etiologically linked to AS with characteristic decreases in dendritic spine density thought to be caused by disinhibition of the PP2A phosphatase (Wang J. et al., 2019; Wang T. et al., 2019).
Though earlier discussed as a result of protein synthesis dysfunction, research implicates autophagy in Fragile X forms of ASD as well: hippocampal mouse neurons with no FMRP showed overactivation of mTORC1 – an autophagy inhibitor – causing the Fragile X model neurons to over-translate and under-degrade protein (Yan et al., 2018). These findings highlight both independent and interacting roles of proteostasis arms as checkpoints that must coordinately function in order to maintain a healthy cellular environment. It is an excellent example of the ways in which each arm of proteostasis is not only independently required, but work as a check on the other arms in order to maintain a healthy cellular environment.
Discussion
Each arm of proteostasis – translation, maintenance, and degradation – is essential to maintaining the “status quo” of homeostasis in dendrites. However, these important molecular components should not be relegated to the category of “housekeeping” proteins. They participate in neuronal cell dynamics like dendritic spine remodeling and LTP/LTD as well as developmentally vital processes such as neurite outgrowth and cell-type specification. Ribosomes, chaperones, and the ubiquitin proteasome and autophagy systems are all heavily studied in relation to neurodegenerative disease. There is a newly revived interest in using endogenous proteostatic mechanisms to combat the neurobiological signs of disease; researchers are eager to overexpress or stimulate proteins that show decreased levels with age or disease. However, in many cases of neurodegeneration, the lowered expression of a protein may not be a cause of protein aggregation, but rather a symptom of the same molecular cascade which created the root problem. Protein aggregation itself, of course, can be a symptom of a problem higher in the chain, and one can see from reports of plaques and tangles in “normal” aging brains that aggregation itself is not the single cause of cognitive decline (Guillozet et al., 2003). The necessity of discovering the roles and interactions of the many players of protein quality control in healthy cells cannot be overstated. While molecular research of pathological states is incredibly important, and may lead us more quickly to discoveries that alleviate symptoms and slow progression, true understanding of the homeostatic mechanisms of these vital proteostatic processes may be the only route we have to a true molecular solution.
Author Contributions
EL drafted the original manuscript. DC conceived the scope of the review and did the supervision. EL and DC critically analyzed, edited, and drafted final manuscript and did the funding acquisition. Both authors contributed to the article and approved the submitted version.
Funding
This work is supported by NIH R01 NS086082 to DC. EL was supported by a GSU 2CI Neurogenomics Fellowship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge Alice Van Derveer and Cox Lab member Nathaniel J. Himmel for constructive critiques on drafts of the manuscript. Select illustrations in the manuscript were created with BioRender.com.
References
Abisambra, J., Jinwal, U. K., Miyata, Y., Rogers, J., Blair, L., Li, X., et al. (2013). Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol. Psychiatry 74, 367–374. doi: 10.1016/j.biopsych.2013.02.027
Ady, V., Toscano-Márquez, B., Nath, M., Chang, P. K., Hui, J., Cook, A., et al. (2018). Altered synaptic and firing properties of cerebellar Purkinje cells in a mouse model of ARSACS. J. Physiol. 596, 4253–4267. doi: 10.1113/JP275902
Aguirre-Chen, C., Stec, N., Ramos, O. M., Kim, N., Kramer, M., McCarthy, S., et al. (2020). A Caenorhabditis elegans model for integrating the functions of neuropsychiatric risk genes identifies components required for normal dendritic morphology. G3 10, 1617–1628. doi: 10.1534/g3.119.400925
Ainsley, J. A., Drane, L., Jacobs, J., Kittelberger, K. A., and Reijmers, L. G. (2014). Functionally diverse dendritic mRNAs rapidly associate with ribosomes following a novel experience. Nat. Commun. 5:4510. doi: 10.1038/ncomms5510
Albert, B., Kos-Braun, I. C., Henras, A. K., Dez, C., Rueda, M. P., Zhang, X., et al. (2019). A ribosome assembly stress response regulates transcription to maintain proteome homeostasis. eLife 8:e45002. doi: 10.7554/eLife.45002
Albert, M., Barrantes-Freer, A., Lohrberg, M., Antel, J. P., Prineas, J. W., Palkovits, M., et al. (2017). Synaptic pathology in the cerebellar dentate nucleus in chronic multiple sclerosis. Brain Pathol. 27, 737–747. doi: 10.1111/bpa.12450
Ali, Y. O., Allen, H. M., Yu, L., Li-Kroeger, D., Bakhshizadehmahmoudi, D., Hatcher, A., et al. (2016). NMNAT2:HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 14:e1002472. doi: 10.1371/journal.pbio.1002472
Alvarez-Castelao, B., and Schuman, E. M. (2015). The regulation of synaptic protein turnover. J. Biol. Chem. 290, 28623–28630. doi: 10.1074/jbc.R115.657130
Anderson, J. F., Siller, E., and Barral, J. M. (2011). The neurodegenerative-disease-related protein sacsin is a molecular chaperone. J. Mol. Biol. 411, 870–880. doi: 10.1016/j.jmb.2011.06.016
Aprile, F. A., Källstig, E., Limorenko, G., Vendruscolo, M., Ron, D., and Hansen, C. (2017). The molecular chaperones DNAJB6 and Hsp70 cooperate to suppress α-synuclein aggregation. Sci. Rep. 7:9039. doi: 10.1038/s41598-017-08324-z
Aridor, M., Guzik, A. K., Bielli, A., and Fish, K. N. (2004). Endoplasmic reticulum export site formation and function in dendrites. J. Neurosci. 24, 3770–3776. doi: 10.1523/JNEUROSCI.4775-03.2004
Aswathy, N., Pullepu, D., and Kabir, M. A. (2016). The interactome of CCT complex – A computational analysis. Comput. Biol. Chem. 64, 396–402. doi: 10.1016/j.compbiolchem.2016.09.002
Bagni, C., and Zukin, R. S. (2019). A synaptic perspective of Fragile X Syndrome and Autism Spectrum Disorders. Neuron 101, 1070–1088. doi: 10.1016/j.neuron.2019.02.041
Bakthisaran, R., Tangirala, R., and Rao, C. M. (2015). Small heat shock proteins: role in cellular functions and pathology. Biochim. Biophys. Acta 1854, 291–319. doi: 10.1016/j.bbapap.2014.12.019
Balchin, D., Milièiæ, G., Strauss, M., Hayer-Hartl, M., and Hartl, F. U. (2018). Pathway of actin folding directed by the eukaryotic chaperonin TRiC. Cell 174, 1507–1521.e16. doi: 10.1016/J.CELL.2018.07.006
Baral, S. S., Lieux, M. E., and DiMario, P. J. (2020). Nucleolar stress in Drosophila neuroblasts, a model for human ribosomopathies. Biol. Open 9:bio046565. doi: 10.1242/bio.046565
Bartelt-Kirbach, B., Moron, M., Glomb, M., Beck, C. M., Weller, M. P., and Golenhofen, N. (2016). HspB5/αB-crystallin increases dendritic complexity and protects the dendritic arbor during heat shock in cultured rat hippocampal neurons. Cell. Mol. Life Sci. 73, 3761–3775. doi: 10.1007/s00018-016-2219-9
Beltran, S., Nassif, M., Vicencio, E., Arcos, J., Labrador, L., Cortes, B. I., et al. (2019). Network approach identifies Pacer as an autophagy protein involved in ALS pathogenesis. Mol. Neurodegener. 14:14. doi: 10.1186/s13024-019-0313-9
Benitez, M. J., Sanchez-Ponce, D., Garrido, J. J., and Wandosell, F. (2014). Hsp90 activity is necessary to acquire a proper neuronal polarization. Biochim. Biophys. Acta 1843, 245–252. doi: 10.1016/j.bbamcr.2013.11.013
Blackstone, C. (2018). “Hereditary spastic paraplegia. Handb. Clin. Neurol. 148, 633–652. doi: 10.1016/B978-0-444-64076-5.00041-7
Blumenstock, S., Rodrigues, E. F., Peters, F., Blazquez-Llorca, L., Schmidt, F., Giese, A., et al. (2017). Seeding and transgenic overexpression of alpha-synuclein triggers dendritic spine pathology in the neocortex. EMBO Mol. Med. 9, 716–731. doi: 10.15252/emmm.201607305
Bocharov, A. V., Vishnyakova, T. G., Baranova, I. N., Remaley, A. T., Patterson, A. P., and Eggerman, T. L. (2000). Heat shock protein 60 is a high-affinity high-density lipoprotein binding protein. Biochem. Biophys. Res. Commun. 277, 228–235. doi: 10.1006/bbrc.2000.3663
Bosch, M., Castro, J., Saneyoshi, T., Matsuno, H., Sur, M., and Hayashi, Y. (2014). Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 82, 444–459. doi: 10.1016/j.neuron.2014.03.021
Bouhouche, A., Benomar, A., Bouslam, N., Chkili, T., and Yahyaoui, M. (2006). Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J. Med. Genet. 43, 441–443. doi: 10.1136/jmg.2005.039230
Bowen, A. B., Bourke, A. M., Hiester, B. G., Hanus, C., and Kennedy, M. J. (2017). Golgi-Independent secretory trafficking through recycling endosomes in neuronal dendrites and spines. eLife 6:e27362. doi: 10.7554/eLife.27362
Bowling, H., and Klann, E. (2014). Shaping dendritic spines in Autism Spectrum Disorder: mTORC1-dependent macroautophagy. Neuron 83, 994–996. doi: 10.1016/j.neuron.2014.08.021
Brackley, K. I., and Grantham, J. (2009). Activities of the chaperonin containing TCP-1 (CCT): implications for cell cycle progression and cytoskeletal organisation. Cell Stress Chaperones 14, 23–31. doi: 10.1007/s12192-008-0057-x
Brehme, M., Voisine, C., Rolland, T., Wachi, S., Soper, J. H., Zhu, Y., et al. (2014). A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 9, 1135–1150. doi: 10.1016/j.celrep.2014.09.042
Broer, L., Ikram, M. A., Schuur, M., Destefano, A. L., Bis, J. C., Liu, F., et al. (2011). Association of HSP70 and its co-chaperones with Alzheimer’s Disease. J. Alzheimers Dis. 25, 93–102. doi: 10.3233/JAD-2011-101560
Brot, S., Auger, C., Bentata, R., Rogemond, V., Ménigoz, S., Chounlamountri, N., et al. (2014). Collapsin response mediator protein 5 (CRMP5) induces mitophagy, thereby regulating mitochondrion numbers in dendrites. J. Biol. Chem. 289, 2261–2276. doi: 10.1074/jbc.M113.490862
Brunelli, F., Valente, E. M., and Arena, G. (2020). Mechanisms of neurodegeneration in Parkinson’s disease: keep neurons in the PINK1. Mech. Ageing Dev. 189:111277. doi: 10.1016/j.mad.2020.111277
Buxbaum, E. (2015). “Aiding in folding: molecular chaperones and chaperonins,” in Fundamentals of Protein Structure and Function (Cham: Springer), 343–359. doi: 10.1007/978-3-319-19920-7_15
Caracci, M. O., Fuentealba, L. M., and Marzolo, M. P. (2019). Golgi complex dynamics and its implication in prevalent neurological disorders. Front. Cell Dev. Biol. 7:75. doi: 10.3389/fcell.2019.00075
Carra, S., Crippa, V., Rusmini, P., Boncoraglio, A., Minoia, M., Giorgetti, E., et al. (2012). Alteration of protein folding and degradation in motor neuron diseases: implications and protective functions of small heat shock proteins. Prog. Neurobiol. 97, 83–100. doi: 10.1016/j.pneurobio.2011.09.009
Chaari, A. (2019). Molecular chaperones biochemistry and role in neurodegenerative diseases. Int. J. Biol. Macromol. 131, 396–411. doi: 10.1016/j.ijbiomac.2019.02.148
Chen, J. Y., Parekh, M., Seliman, H., Bakshinskaya, D., Dai, W., Kwan, K., et al. (2018). Heat shock promotes inclusion body formation of mutant huntingtin (mHtt) and alleviates mHtt-induced transcription factor dysfunction. J. Biol. Chem. 293, 15581–15593. doi: 10.1074/jbc.RA118.002933
Chen, S., and Brown, I. R. (2007). Neuronal expression of constitutive heat shock proteins: implications for neurodegenerative diseases. Cell Stress Chaperones 12, 51–58. doi: 10.1379/CSC-236R.1
Chen, Y., Wang, B., Liu, D., Li, J. J., Xue, Y., Sakata, K., et al. (2014). Hsp90 chaperone inhibitor 17-AAG Attenuates Aβ-induced synaptic toxicity and memory impairment. J. Neurosci. 34, 2464–2470. doi: 10.1523/JNEUROSCI.0151-13.2014
Chidambaram, S. B., Rathipriya, A. G., Bolla, S. R., Bhat, A., Ray, B., Mahalakshmi, A. M., et al. (2019). Dendritic spines: revisiting the physiological role. Prog. Neuropsychopharmacol. Biol. Psychiatry 92, 161–193. doi: 10.1016/j.pnpbp.2019.01.005
Chirillo, M. A., Waters, M. S., Lindsey, L. F., Bourne, J. N., and Harris, K. M. (2019). Local resources of polyribosomes and SER promote synapse enlargement and spine clustering after long-term potentiation in adult rat hippocampus. Sci. Rep. 9:3861. doi: 10.1038/s41598-019-40520-x
Choe, H. K., and Cho, J. (2020). Comprehensive genome-wide approaches to activity-dependent translational control in neurons. Int. J. Mol. Sci. 21:1592. doi: 10.3390/ijms21051592
Choi, J.-H., Wang, W., Park, D., Kim, S., Kim, K.-T., and Min, K.-T. (2018). IRES -mediated translation of cofilin regulates axonal growth cone extension and turning. EMBO J. 37:e95266. doi: 10.15252/embj.201695266
Ciechanover, A., and Kwon, Y. T. (2017). Protein quality control by molecular chaperones in neurodegeneration. Front. Neurosci. 11:185. doi: 10.3389/fnins.2017.00185
Clark, S. G., Graybeal, L. L., Bhattacharjee, S., Thomas, C., Bhattacharya, S., and Cox, D. N. (2018). Basal autophagy is required for promoting dendritic terminal branching in Drosophila sensory neurons. PLoS One 13:e0206743. doi: 10.1371/journal.pone.0206743
Cohen, L. D., Zuchman, R., Sorokina, O., Müller, A., Dieterich, D. C., Armstrong, J. D., et al. (2013). Metabolic turnover of synaptic proteins: kinetics, interdependencies and implications for synaptic maintenance. PLoS One 8:e63191. doi: 10.1371/journal.pone.0063191
Comery, T. A., Harris, J. B., Willems, P. J., Oostra, B. A., Irwin, S. A., Weiler, I. J., et al. (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl. Acad. Sci. U.S.A. 94, 5401–5404. doi: 10.1073/pnas.94.10.5401
Cui-Wang, T., Hanus, C., Cui, T., Helton, T., Bourne, J., Watson, D., et al. (2012). Local zones of endoplasmic reticulum complexity confine cargo in neuronal dendrites. Cell 148, 309–321. doi: 10.1016/j.cell.2011.11.056
Dahlhaus, R. (2018). Of men and mice: modeling the fragile X syndrome. Front. Mol. Neurosci. 11:41. doi: 10.3389/fnmol.2018.00041
Dalle-Donne, I., Rossi, R., Milzani, A., Di Simplicio, P., and Colombo, R. (2001). The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic. Biol. Med. 31, 1624–1632. doi: 10.1016/S0891-5849(01)00749-3
Dar, K. B., Bhat, A. H., Amin, S., Reshi, B. A., Zargar, M. A., Masood, A., et al. (2020). Elucidating critical proteinopathic mechanisms and potential drug targets in neurodegeneration. Cell. Mol. Neurobiol. 40, 313–345. doi: 10.1007/s10571-019-00741-0
Das, R., Bhattacharjee, S., Patel, A. A., Harris, J. M., Bhattacharya, S., Letcher, J. M., et al. (2017). Dendritic cytoskeletal architecture is modulated by combinatorial transcriptional regulation in Drosophila melanogaster. Genetics 207, 1401–1421. doi: 10.1534/genetics.117.300393
Das Sharma, S., Metz, J. B., Li, H., Hobson, B. D., Hornstein, N., Sulzer, D., et al. (2019). Widespread alterations in translation elongation in the brain of juvenile Fmr1 knockout mice. Cell Rep. 26, 3313–3322.e5. doi: 10.1016/j.celrep.2019.02.086
Deane, C. A. S., and Brown, I. R. (2018). Knockdown of heat shock proteins HSPA6 (Hsp70B’) and HSPA1A (Hsp70-1) sensitizes differentiated human neuronal cells to cellular stress. Neurochem. Res. 43, 340–350. doi: 10.1007/s11064-017-2429-z
Deng, J., Yang, M., Chen, Y., Chen, X., Liu, J., Sun, S., et al. (2015). FUS interacts with HSP60 to promote mitochondrial damage. PLoS Genet. 11:e1005357. doi: 10.1371/journal.pgen.1005357
Depaoli, M. R., Hay, J. C., Graier, W. F., and Malli, R. (2018). The enigmatic ATP supply of the endoplasmic reticulum. Biol. Rev. Camb. Philos. Soc. 94, 610–628. doi: 10.1111/brv.12469
Devitt, N., Gallagher, L., and Reilly, R. (2015). Autism Spectrum Disorder (ASD) and Fragile X Syndrome (FXS): two overlapping disorders reviewed through electroencephalography—what can be interpreted from the available information? Brain Sci. 5, 92–117. doi: 10.3390/brainsci5020092
Ding, Q., Markesbery, W. R., Chen, Q., Li, F., and Keller, J. N. (2005). Ribosome dysfunction is an early event in Alzheimer’s disease. J. Neurosci. 25, 9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005
Donovan, P., and Poronnik, P. (2013). Nedd4 and Nedd4-2: ubiquitin ligases at work in the neuron. Int. J. Biochem. Cell Biol. 45, 706–710. doi: 10.1016/j.biocel.2012.12.006
Duan, W., Guo, M., Yi, L., Zhang, J., Bi, Y., Liu, Y., et al. (2019). Deletion of Tbk1 disrupts autophagy and reproduces behavioral and locomotor symptoms of FTD-ALS in mice. Aging 11, 2457–2476. doi: 10.18632/aging.101936
Edkins, A. L., Price, J. T., Graham Pockley, A., and Blatch, G. L. (2018). Heat shock proteins as modulators and therapeutic targets of chronic disease: an integrated perspective. Philos. Trans. R. Soc. B Biol. Sci. 373:20160521. doi: 10.1098/rstb.2016.0521
Elliott, E., Bailey, O., Waldron, F. M., Hardingham, G. E., Chandran, S., and Gregory, J. M. (2020). Therapeutic targeting of proteostasis in Amyotrophic Lateral Sclerosis—a systematic review and meta-analysis of preclinical research. Front. Neurosci. 14:511. doi: 10.3389/fnins.2020.00511
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412. doi: 10.1038/s41593-018-0332-9
Fares, M. A. (2014). The evolution of protein moonlighting: adaptive traps and promiscuity in the chaperonins. Biochem. Soc. Trans. 42, 1709–1714. doi: 10.1042/BST20140225
Farley-Barnes, K. I., Ogawa, L. M., and Baserga, S. J. (2019). Ribosomopathies: old concepts, new controversies. Trends Genet. 35, 754–767. doi: 10.1016/j.tig.2019.07.004
Feldmann, L. K., Le Prieult, F., Felzen, V., Thal, S. C., Engelhard, K., Behl, C., et al. (2019). Proteasome and autophagy-mediated impairment of late long-term potentiation (L-ltp) after traumatic brain injury in the somatosensory cortex of mice. Int. J. Mol. Sci. 20:3048. doi: 10.3390/ijms20123048
Ferretti, M. B., and Karbstein, K. (2019). Does functional specialization of ribosomes really exist? RNA 25, 521–538. doi: 10.1261/rna.069823.118
Fink, J. K. (2013). Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 126, 307–328. doi: 10.1007/s00401-013-1115-8
Firkowska, M., Macias, M., and Jaworski, J. (2019). ESCRT proteins control the dendritic morphology of developing and mature hippocampal neurons. Mol. Neurobiol. 56, 4866–4879. doi: 10.1007/s12035-018-1418-9
Fowler, P. C., Garcia-Pardo, M. E., Simpson, J. C., and O’Sullivan, N. C. (2019). NeurodegenERation: the central role for ER contacts in neuronal function and axonopathy, lessons from hereditary spastic paraplegias and related diseases. Front. Neurosci. 13:1051. doi: 10.3389/fnins.2019.01051
Frey, U., and Morris, R. G. M. (1997). Synaptic tagging and long-term potentiation. Nature 385, 533–536. doi: 10.1038/385533a0
Fukui, K., Okihiro, S., Ohfuchi, Y., Hashimoto, M., Kato, Y., Yoshida, N., et al. (2019). Proteomic study on neurite responses to oxidative stress: search for differentially expressed proteins in isolated neurites of N1E-115 cells. J. Clin. Biochem. Nutr. 64, 36–44. doi: 10.3164/jcbn.18-31
Fukuzono, T., Pastuhov, S. I., Fukushima, O., Li, C., Hattori, A., Iemura, S.-I., et al. (2016). Chaperone complex BAG2-HSC70 regulates localization of Caenorhabditis elegans leucine-rich repeat kinase LRK-1 to the Golgi. Genes Cells 21, 311–324. doi: 10.1111/gtc.12338
Gabut, M., Bourdelais, F., and Durand, S. (2020). Ribosome and translational control in stem cells. Cells 9:497. doi: 10.3390/cells9020497
Gao, F. B., Richter, J. D., and Cleveland, D. W. (2017). Rethinking unconventional translation in neurodegeneration. Cell 171, 994–1000. doi: 10.1016/j.cell.2017.10.042
Gao, Y., Jiang, T., Qu, C., Tao, H., Cao, H., Zhao, Y., et al. (2013). Atlastin-1 regulates dendritic morphogenesis in mouse cerebral cortex. Neurosci. Res. 77, 137–142. doi: 10.1016/j.neures.2013.08.007
Garcia-Lopez, P., Garcia-Marin, V., and Freire, M. (2010). The histological slides and drawings of Cajal. Front. Neuroanat. 4:9. doi: 10.3389/neuro.05.009.2010
Garrido, C., Paul, C., Seigneuric, R., and Kampinga, H. H. (2012). The small heat shock proteins family: the long forgotten chaperones. Int. J. Biochem. Cell Biol. 44, 1588–1592. doi: 10.1016/j.biocel.2012.02.022
Gentil, B. J., Lai, G. T., Menade, M., Larivière, R., Minotti, S., Gehring, K., et al. (2019). Sacsin, mutated in the ataxia ARSACS, regulates intermediate filament assembly and dynamics. FASEB J. 33, 2982–2994. doi: 10.1096/fj.201801556R
Genuth, N. R., and Barna, M. (2018). The discovery of ribosome heterogeneity and its implications for gene regulation and organismal life. Mol. Cell 71, 364–374. doi: 10.1016/j.molcel.2018.07.018
George, A. J., Hoffiz, Y. C., Charles, A. J., Zhu, Y., and Mabb, A. M. (2018). A comprehensive atlas of E3 ubiquitin ligase mutations in neurological disorders. Front. Genet. 9:29. doi: 10.3389/fgene.2018.00029
Gerónimo-Olvera, C., and Massieu, L. (2019). Autophagy as a homeostatic mechanism in response to stress conditions in the central nervous system. Mol. Neurobiol. 56, 6594–6608. doi: 10.1007/s12035-019-1546-x
Gershman, B. W., Pritchard, C. E., Chaney, K. P., and Ware, V. C. (2020). Tissue-specific expression of ribosomal protein paralogue eRpL22 - like in Drosophila melanogaster eye development. Dev. Dyn. doi: 10.1002/dvdy.185 [Epub ahead of print].
Gerst, J. E. (2018). Pimp my ribosome: ribosomal protein paralogs specify translational control. Trends Genet. 34, 832–845. doi: 10.1016/j.tig.2018.08.004
Gomes, C., Smith, S. C., Youssef, M. N., Zheng, J. J., Hagg, T., and Hetman, M. (2011). RNA polymerase 1-driven transcription as a mediator of BDNF-induced neurite outgrowth. J. Biol. Chem. 286, 4357–4363. doi: 10.1074/jbc.M110.170134
Goo, M. S., Sancho, L., Slepak, N., Boassa, D., Deerinck, T. J., Ellisman, M. H., et al. (2017). Activity-dependent trafficking of lysosomes in dendrites and dendritic spines. J. Cell Biol. 216, 2499–2513. doi: 10.1083/jcb.201704068
Greenblatt, E. J., and Spradling, A. C. (2018). Fragile X mental retardation 1 gene enhances the translation of large autism-related proteins. Science 361, 709–712. doi: 10.1126/science.aas9963
Grossman, A. W., Elisseou, N. M., McKinney, B. C., and Greenough, W. T. (2006). Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res. 1084, 158–164. doi: 10.1016/j.brainres.2006.02.044
Guillozet, A. L., Weintraub, S., Mash, D. C., and Marsel Mesulam, M. (2003). Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch. Neurol. 60, 729–736. doi: 10.1001/archneur.60.5.729
Guisbert, E., Czyz, D. M., Richter, K., McMullen, P. D., and Morimoto, R. I. (2013). Identification of a tissue-selective heat shock response regulatory network. PLoS Genet. 9:e1003466. doi: 10.1371/journal.pgen.1003466
Guo, H. (2018). Specialized ribosomes and the control of translation. Biochem. Soc. Trans. 46, 855–869. doi: 10.1042/BST20160426
Hafner, A. S., Donlin-Asp, P. G., Leitch, B., Herzog, E., and Schuman, E. M. (2019). Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science 364:eaau3644. doi: 10.1126/science.aau3644
Hamilton, A. M., Oh, W. C., Vega-Ramirez, H., Stein, I. S., Hell, J. W., Patrick, G. N., et al. (2012). Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron 74, 1023–1030. doi: 10.1016/j.neuron.2012.04.031
Hamilton, A. M., and Zito, K. (2013). Breaking it down: the ubiquitin proteasome system in neuronal morphogenesis. Neural Plast. 2013:196848. doi: 10.1155/2013/196848
Han, C., Cui, K., Bi, X., Wang, L., Sun, M., Yang, L., et al. (2019). Association between polymorphism of the NEDD4 gene and cognitive dysfunction of schizophrenia patients in Chinese Han population. BMC Psychiatry 19:405. doi: 10.1186/s12888-019-2386-y
Hanus, C., and Schuman, E. M. (2013). Proteostasis in complex dendrites. Nat. Rev. Neurosci. 14, 638–648. doi: 10.1038/nrn3546
Hart, M. P. (2019). Stress-induced neuron remodeling reveals differential interplay between neurexin and environmental factors in caenorhabditis elegans. Genetics 213, 1415–1430. doi: 10.1534/genetics.119.302415
Heise, C., Gardoni, F., Culotta, L., Di Luca, M., Verpelli, C., Sala, C., et al. (2014). Elongation factor-2 phosphorylation in dendrites and the regulation of dendritic mRNA translation in neurons. Front. Cell. Neurosci. 8:35. doi: 10.3389/fncel.2014.00035
Hernandez, D., Torres, C. A., Setlik, W., Cebrián, C., Mosharov, E. V., Tang, G., et al. (2012). Regulation of presynaptic neurotransmission by macroautophagy. Neuron 74, 277–284. doi: 10.1016/j.neuron.2012.02.020
Hernández-Ortega, K., Garcia-Esparcia, P., Gil, L., Lucas, J. J., and Ferrer, I. (2016). Altered machinery of protein synthesis in Alzheimer’s: from the nucleolus to the ribosome. Brain Pathol. 26, 593–605. doi: 10.1111/bpa.12335
Hetman, M., and Slomnicki, L. P. (2019). Ribosomal biogenesis as an emerging target of neurodevelopmental pathologies. J. Neurochem. 148, 325–347. doi: 10.1111/jnc.14576
Hetz, C., and Kaufman, R. J. (2020). Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 21, 421–438. doi: 10.1038/s41580-020-0250-z
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. doi: 10.1038/nrn3689
Hill, S. E., Parmar, M., Gheres, K. W., Guignet, M. A., Huang, Y., Jackson, F. R., et al. (2012). Development of dendrite polarity in Drosophila neurons. Neural Dev. 7:34. doi: 10.1186/1749-8104-7-34
Hipp, M. S., Kasturi, P., and Hartl, F. U. (2019). The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435. doi: 10.1038/s41580-019-0101-y
Horton, A. C., and Ehlers, M. D. (2003). Dual modes of endoplasmic reticulum-to-Golgi transport in dendrites revealed by live-cell imaging. J. Neurosci. 23, 6188–6199. doi: 10.1523/jneurosci.23-15-06188.2003
Horton, A. C., Rácz, B., Monson, E. E., Lin, A. L., Weinberg, R. J., and Ehlers, M. D. (2005). Polarized secretory trafficking directs cargo for asymmetric dendrite growth and morphogenesis. Neuron 48, 757–771. doi: 10.1016/j.neuron.2005.11.005
Hu, F., Zhou, J., Lu, Y., Guan, L., Wei, N.-N., Tang, Y.-Q., et al. (2019). Inhibition of Hsp70 suppresses neuronal hyperexcitability and attenuates epilepsy by enhancing A-type potassium current. Cell Rep. 26, 168–181.e4. doi: 10.1016/J.CELREP.2018.12.032
Iyer, E. P. R., Iyer, S. C., Sullivan, L., Wang, D., Meduri, R., Graybeal, L. L., et al. (2013). Functional genomic analyses of two morphologically distinct classes of Drosophila sensory neurons: post-mitotic roles of transcription factors in dendritic patterning. PLoS One 8:e72434. doi: 10.1371/journal.pone.0072434
Iyer, S. C., Iyer, E. P. R., Meduri, R., Rubaharan, M., Kuntimaddi, A., Karamsetty, M., et al. (2013). Cut, via CrebA, transcriptionally regulates the COPII secretory pathway to direct dendrite development in Drosophila. J. Cell Sci. 126, 4732–4745. doi: 10.1242/jcs.131144
Jakkamsetti, V., Tsai, N. P., Gross, C., Molinaro, G., Collins, K. A., Nicoletti, F., et al. (2013). Experience-induced Arc/Arg3.1 primes CA1 pyramidal neurons for metabotropic glutamate receptor-dependent long-term synaptic depression. Neuron 80, 72–79. doi: 10.1016/j.neuron.2013.07.020
Jan, Y. N., and Jan, L. Y. (2010). Branching out: mechanisms of dendritic arborization. Nat. Rev. Neurosci. 11, 316–328. doi: 10.1038/nrn2836
Jeffery, C. J. (2018). Protein moonlighting: What is it, and why is it important? Philos. Trans. R. Soc. B Biol. Sci. 373:20160523. doi: 10.1098/rstb.2016.0523
Jin, E. J., Kiral, F. R., and Hiesinger, P. R. (2018). The where, what, and when of membrane protein degradation in neurons. Dev. Neurobiol. 78, 283–297. doi: 10.1002/dneu.22534
Jinushi-Nakao, S., Arvind, R., Amikura, R., Kinameri, E., Liu, A. W., and Moore, A. W. (2007). Knot/Collier and Cut control different aspects of dendrite cytoskeleton and synergize to define final arbor shape. Neuron 56, 963–978. doi: 10.1016/j.neuron.2007.10.031
Kabir, M. T., Uddin, M. S., Abdeen, A., Ashraf, G. M., Perveen, A., Hafeez, A., et al. (2020). Evidence linking protein misfolding to quality control in progressive neurodegenerative diseases. Curr. Top. Med. Chem. 20. doi: 10.2174/1568026620666200618114924 [Epub ahead of print].
Kampen, K. R., Sulima, S. O., Vereecke, S., and De Keersmaecker, K. (2020). Hallmarks of ribosomopathies. Nucleic Acids Res. 48, 1013–1028. doi: 10.1093/nar/gkz637
Kanamori, T., Togashi, K., Koizumi, H., and Emoto, K. (2015a). Dendritic remodeling: lessons from invertebrate model systems. Int. Rev. Cell Mol. Biol. 318, 1–25. doi: 10.1016/bs.ircmb.2015.05.001
Kanamori, T., Yoshino, J., Yasunaga, K. I., Dairyo, Y., and Emoto, K. (2015b). Local endocytosis triggers dendritic thinning and pruning in Drosophila sensory neurons. Nat. Commun. 6:6515. doi: 10.1038/ncomms7515
Kasza, Á., Hunya, Á., Frank, Z., Fülöp, F., Török, Z., Balogh, G., et al. (2016). Dihydropyridine derivatives modulate heat shock responses and have a neuroprotective effect in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 53, 557–571. doi: 10.3233/JAD-150860
Kawabe, H., Neeb, A., Dimova, K., Young, S. M., Takeda, M., Katsurabayashi, S., et al. (2010). Regulation of Rap2A by the ubiquitin ligase Nedd4-1 controls neurite development. Neuron 65, 358–372. doi: 10.1016/j.neuron.2010.01.007
Kawada, K., Iekumo, T., Saito, R., Kaneko, M., Mimori, S., Nomura, Y., et al. (2014). Aberrant neuronal differentiation and inhibition of dendrite outgrowth resulting from endoplasmic reticulum stress. J. Neurosci. Res. 92, 1122–1133. doi: 10.1002/jnr.23389
Kawasaki, F., Koonce, N. L., Guo, L., Fatima, S., Qiu, C., Moon, M. T., et al. (2016). Small heat shock proteins mediate cell-autonomous and -nonautonomous protection in a Drosophila model for environmental-stress-induced degeneration. Dis. Model. Mech. 9, 953–964. doi: 10.1242/dmm.026385
Kelliher, M. T., Saunders, H. A., and Wildonger, J. (2019). Microtubule control of functional architecture in neurons. Curr. Opin. Neurobiol. 57, 39–45. doi: 10.1016/j.conb.2019.01.003
Kennedy, M. J., and Hanus, C. (2019). Architecture and dynamics of the neuronal secretory network. Annu. Rev. Cell Dev. Biol. 35, 543–566. doi: 10.1146/annurev-cellbio-100818-125418
Khabirova, E., Moloney, A., Marciniak, S. J., Williams, J., Lomas, D. A., Oliver, S. G., et al. (2014). The TRiC/CCT chaperone is implicated in Alzheimer’s disease based on patient GWAS and an RNAi screen in Aβ-expressing Caenorhabditis elegans. PLoS One 9:e102985. doi: 10.1371/journal.pone.0102985
Khayachi, A., Gwizdek, C., Poupon, G., Alcor, D., Chafai, M., Cassé, F., et al. (2018). Sumoylation regulates FMRP-mediated dendritic spine elimination and maturation. Nat. Commun. 9:757. doi: 10.1038/s41467-018-03222-y
Kim, A. H., Puram, S. V., Bilimoria, P. M., Ikeuchi, Y., Keough, S., Wong, M., et al. (2009). A centrosomal Cdc20-APC pathway controls dendrite morphogenesis in postmitotic neurons. Cell 136, 322–336. doi: 10.1016/j.cell.2008.11.050
Kim, H. J., Cho, M. H., Shim, W. H., Kim, J. K., Jeon, E. Y., Kim, D. H., et al. (2017). Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 22, 1576–1584. doi: 10.1038/mp.2016.103
Kim, Y. E., Hipp, M. S., Bracher, A., Hayer-Hartl, M., and Ulrich Hartl, F. (2013). Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355. doi: 10.1146/annurev-biochem-060208-092442
Klaips, C. L., Jayaraj, G. G., and Hartl, F. U. (2018). Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 217, 51–63. doi: 10.1083/JCB.201709072
Klose, M. K., and Robertson, R. M. (2004). Stress-induced thermoprotection of neuromuscular transmission. Integr. Comp. Biol. 44, 14–20. doi: 10.1093/icb/44.1.14
Kocaturk, N. M., and Gozuacik, D. (2018). Crosstalk between mammalian autophagy and the ubiquitin-proteasome system. Front. Cell Dev. Biol. 6:128. doi: 10.3389/fcell.2018.00128
Koch, J. C., Bitow, F., Haack, J., Hedouville, Z., Zhang, J. N., Tönges, L., et al. (2015). Alpha-synuclein affects neurite morphology, autophagy, vesicle transport and axonal degeneration in CNS neurons. Cell Death Dis. 6:e1811. doi: 10.1038/cddis.2015.169
Koltun, B., Ironi, S., Gershoni-Emek, N., Barrera, I., Hleihil, M., Nanguneri, S., et al. (2020). Measuring mRNA translation in neuronal processes and somata by tRNA-FRET. Nucleic Acids Res. 48:e32. doi: 10.1093/nar/gkaa042
Komili, S., Farny, N. G., Roth, F. P., and Silver, P. A. (2007). Functional specificity among ribosomal proteins regulates gene expression. Cell 131, 557–571. doi: 10.1016/j.cell.2007.08.037
Konishi, Y., Stegmüller, J., Matsuda, T., Bonni, S., and Bonni, A. (2004). Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 303, 1026–1030. doi: 10.1126/science.1093712
Krämer, R., Rode, S., and Rumpf, S. (2019). Rab11 is required for neurite pruning and developmental membrane protein degradation in Drosophila sensory neurons. Dev. Biol. 451, 68–78. doi: 10.1016/j.ydbio.2019.03.003
Kumar, D., Ambasta, R. K., and Kumar, P. (2020). Ubiquitin biology in neurodegenerative disorders: from impairment to therapeutic strategies. Ageing Res. Rev. 61:101078. doi: 10.1016/j.arr.2020.101078
La Padula, V., Staszewski, O., Nestel, S., Busch, H., Boerries, M., Roussa, E., et al. (2016). HSPB3 protein is expressed in motoneurons and induces their survival after lesion-induced degeneration. Exp. Neurol. 286, 40–49. doi: 10.1016/j.expneurol.2016.08.014
Lambert-Smith, I. A., Saunders, D. N., and Yerbury, J. J. (2020). The pivotal role of ubiquitin-activating enzyme E1 (UBA1) in neuronal health and neurodegeneration. Int. J. Biochem. Cell Biol. 123:105746. doi: 10.1016/j.biocel.2020.105746
Lamming, D. W. (2016). Inhibition of the mechanistic target of rapamycin (mTOR)–Rapamycin and beyond. Cold Spring Harb. Perspect. Med. 6:a025924. doi: 10.1101/cshperspect.a025924
Lanka, V., Wieland, S., Barber, J., and Cudkowicz, M. (2009). Arimoclomol: a potential therapy under development for ALS. Expert Opin. Investig. Drugs 18, 1907–1918. doi: 10.1517/13543780903357486
Larivière, R., Sgarioto, N., Márquez, B. T., Gaudet, R., Choquet, K., McKinney, R. A., et al. (2019). Sacs R272C missense homozygous mice develop an ataxia phenotype. Mol. Brain 12:19. doi: 10.1186/s13041-019-0438-3
Lee, A., Li, W., Xu, K., Bogert, B. A., Su, K., and Gao, F. B. (2003). Control of dendritic development by the Drosophila fragile X-related gene involves the small GTPase Rac1. Development 130, 5543–5552. doi: 10.1242/dev.00792
Lee, A.-H., Iwakoshi, N. N., and Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459. doi: 10.1128/mcb.23.21.7448-7459.2003
Lee, J., Jung, S. C., Joo, J., Choi, Y. R., Moon, H. W., Kwak, G., et al. (2015). Overexpression of mutant HSP27 causes axonal neuropathy in mice. J. Biomed. Sci. 22:43. doi: 10.1186/s12929-015-0154-y
Li, D., and Wang, J. (2020). Ribosome heterogeneity in stem cells and development. J. Cell Biol. 219:e202001108. doi: 10.1083/jcb.202001108
Lilienbaum, A. (2013). Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 4, 1–26.
Lim, J., and Yue, Z. (2015). Neuronal aggregates: formation, clearance, and spreading. Dev. Cell 32, 491–501. doi: 10.1016/j.devcel.2015.02.002
Lim, K. H., Joo, J. Y., and Baek, K. H. (2020). The potential roles of deubiquitinating enzymes in brain diseases. Ageing Res. Rev. 61:101088. doi: 10.1016/j.arr.2020.101088
Lin, A., Hou, Q., Jarzylo, L., Amato, S., Gilbert, J., Shang, F., et al. (2011). Nedd4-mediated AMPA receptor ubiquitination regulates receptor turnover and trafficking. J. Neurochem. 119, 27–39. doi: 10.1111/j.1471-4159.2011.07221.x
Lin, W. Y., Williams, C., Yan, C., Koledachkina, T., Luedke, K., Dalton, J., et al. (2015). The SLC36 transporter pathetic is required for extreme dendrite growth in Drosophila sensory neurons. Genes Dev. 29, 1120–1135. doi: 10.1101/gad.259119.115
Liu, B., Li, Y., Stackpole, E. E., Novak, A., Gao, Y., Zhao, Y., et al. (2018). Regulatory discrimination of mRNAs by FMRP controls mouse adult neural stem cell differentiation. Proc. Natl. Acad. Sci. U.S.A. 115, E11397–E11405. doi: 10.1073/pnas.1809588115
Liu, X., Guo, X., Niu, L., Li, X., Sun, F., Hu, J., et al. (2019). Atlastin-1 regulates morphology and function of endoplasmic reticulum in dendrites. Nat. Commun. 10:568. doi: 10.1038/s41467-019-08478-6
Louros, S. R., and Osterweil, E. K. (2016). Perturbed proteostasis in autism spectrum disorders. J. Neurochem. 139, 1081–1092. doi: 10.1111/jnc.13723
Lu, Y., Wang, F., Li, Y., Ferris, J., Lee, J. A., and Gao, F. B. (2009). The Drosophila homologue of the Angelman syndrome ubiquitin ligase regulates the formation of terminal dendritic branches. Hum. Mol. Genet. 18, 454–462. doi: 10.1093/hmg/ddn373
Mabb, A. M., and Ehlers, M. D. (2018). Arc ubiquitination in synaptic plasticity. Semin. Cell Dev. Biol. 77, 10–16. doi: 10.1016/j.semcdb.2017.09.009
Maday, S., and Holzbaur, E. L. F. (2014). Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell 30, 71–85. doi: 10.1016/j.devcel.2014.06.001
Maday, S., and Holzbaur, E. L. F. (2016). Compartment-specific regulation of autophagy in primary neurons. J. Neurosci. 36, 5933–5945. doi: 10.1523/JNEUROSCI.4401-15.2016
Maday, S., Twelvetrees, A. E., Moughamian, A. J., and Holzbaur, E. L. F. (2014). Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84, 292–309. doi: 10.1016/j.neuron.2014.10.019
Marcora, M. S., Belfiori-Carrasco, L. F., Bocai, N. I., Morelli, L., and Castaño, E. M. (2017). Amyloid-β42 clearance and neuroprotection mediated by X-box binding protein 1 signaling decline with aging in the Drosophila brain. Neurobiol. Aging 60, 57–70. doi: 10.1016/J.NEUROBIOLAGING.2017.08.012
Martínez, G., Khatiwada, S., Costa-Mattioli, M., and Hetz, C. (2018). ER proteostasis control of neuronal physiology and synaptic function. Trends Neurosci. 41, 610–624. doi: 10.1016/j.tins.2018.05.009
Maruzs, T., Simon-Vecsei, Z., Kiss, V., Csizmadia, T., and Juhász, G. (2019). On the fly: recent progress on autophagy and aging in Drosophila. Front. Cell Dev. Biol. 7:140. doi: 10.3389/fcell.2019.00140
Mauro, V. P., and Edelman, G. M. (2007). The ribosome filter redux. Cell Cycle 6, 2246–2251. doi: 10.4161/cc.6.18.4739
McLaughlin, T., Falkowski, M., Park, J. W., Keegan, S., Elliott, M., Wang, J. J., et al. (2018). Loss of XBP1 accelerates age-related decline in retinal function and neurodegeneration. Mol. Neurodegener. 13:16. doi: 10.1186/s13024-018-0250-z
Migita, K., Matsumoto, T., Terada, K., Ono, K., and Hara, S. (2019). Effects of geldanamycin on neurite outgrowth-related proteins and kinases in nerve growth factor-differentiated pheochromocytoma 12 cells. J. Pharmacol. Sci. 140, 255–262. doi: 10.1016/j.jphs.2019.07.011
Miller, D. J., and Fort, P. E. (2018). Heat shock proteins regulatory role in neurodevelopment. Front. Neurosci. 12:821. doi: 10.3389/fnins.2018.00821
Mitra, S., Tsvetkov, A. S., and Finkbeiner, S. (2009). Protein turnover and inclusion body formation. Autophagy 5, 1037–1038. doi: 10.4161/auto.5.7.9291
Mitsui, S., Otomo, A., Nozaki, M., Ono, S., Sato, K., Shirakawa, R., et al. (2018). Systemic overexpression of SQSTM1/p62 accelerates disease onset in a SOD1H46R-expressing ALS mouse model. Mol. Brain 11:30. doi: 10.1186/s13041-018-0373-8
Molzahn, C. M., and Mayor, T. (2020). Protein aggregation triggered by rewired protein homeostasis during neuronal differentiation. Mol. Cell 78, 195–196. doi: 10.1016/j.molcel.2020.03.028
Morris, G., Puri, B. K., Walder, K., Berk, M., Stubbs, B., Maes, M., et al. (2018). The endoplasmic reticulum stress response in neuroprogressive diseases: emerging pathophysiological role and translational implications. Mol. Neurobiol. 55, 8765–8787. doi: 10.1007/s12035-018-1028-6
Muranova, L. K., Ryzhavskaya, A. S., Sudnitsyna, M. V., Shatov, V. M., and Gusev, N. B. (2019). Small heat shock proteins and human neurodegenerative diseases. Biochemistry 84, 1256–1267. doi: 10.1134/S000629791911004X
Muranova, L. K., Sudnitsyna, M. V., Strelkov, S. V., and Gusev, N. B. (2020). Mutations in HspB1 and hereditary neuropathies. Cell Stress Chaperones 25, 655–665. doi: 10.1007/s12192-020-01099-9
Mymrikov, E. V., Riedl, M., Peters, C., Weinkauf, S., Haslbeck, M., and Buchner, J. (2020). Regulation of small heat-shock proteins by hetero-oligomer formation. J. Biol. Chem. 295, 158–169. doi: 10.1074/jbc.RA119.011143
Mymrikov, E. V., Seit-Nebi, A. S., and Gusev, N. B. (2011). Large potentials of small heat shock Proteins. Physiol. Rev. 91, 1123–1159. doi: 10.1152/physrev.00023.2010
Na, Y., Park, S., Lee, C., Kim, D. K., Park, J. M., Sockanathan, S., et al. (2016). Real-time imaging reveals properties of glutamate-induced Arc/Arg 3.1 translation in neuronal dendrites. Neuron 91, 561–573. doi: 10.1016/j.neuron.2016.06.017
Nanda, S., Das, R., Bhattacharjee, S., Cox, D. N., and Ascoli, G. A. (2018). Morphological determinants of dendritic arborization neurons in Drosophila larva. Brain Struct. Funct. 223, 1107–1120. doi: 10.1007/s00429-017-1541-9
Narberhaus, F. (2002). α-Crystallin-type heat shock proteins: socializing minichaperones in the context of a multichaperone network. Microbiol. Mol. Biol. Rev. 66, 64–93. doi: 10.1128/mmbr.66.1.64-93.2002
Nefedova, V. V., Muranova, L. K., Sudnitsyna, M. V., Ryzhavskaya, A. S., and Gusev, N. B. (2015). Small heat shock proteins and distal hereditary neuropathies. Biochemistry 80, 1734–1747. doi: 10.1134/S000629791513009X
Nefedova, V. V., Sudnitsyna, M. V., and Gusev, N. B. (2017). Interaction of small heat shock proteins with light component of neurofilaments (NFL). Cell Stress Chaperones 22, 467–479. doi: 10.1007/s12192-016-0757-6
Nie, D., Chen, Z., Ebrahimi-Fakhari, D., Di Nardo, A., Julich, K., Robson, V. K., et al. (2015). The stress-induced Atf3-gelsolin cascade underlies dendritic spine deficits in neuronal models of tuberous sclerosis complex. J. Neurosci. 35, 10762–10772. doi: 10.1523/JNEUROSCI.4796-14.2015
Ohashi, R., and Shiina, N. (2020). Cataloguing and selection of mRNAs localized to dendrites in neurons and regulated by RNA-binding proteins in RNA granules. Biomolecules 10:167. doi: 10.3390/biom10020167
Olesnicky, E. C., Killian, D. J., Garcia, E., Morton, M. C., Rathjen, A. R., Sola, I. E., et al. (2014). Extensive use of RNA-binding proteins in Drosophila sensory neuron dendrite morphogenesis. G3 4, 297–306. doi: 10.1534/g3.113.009795
Ori-McKenney, K. M., Jan, L. Y., and Jan, Y. N. (2012). Golgi outposts shape dendrite morphology by functioning as sites of acentrosomal microtubule nucleation in neurons. Neuron 76, 921–930. doi: 10.1016/j.neuron.2012.10.008
Orphazyme’s arimoclomol receives US Fast Track Designation in Amyotrophic Lateral Sclerosis (2020). Available at: https://www.globenewswire.com/news-release/2020/05/22/2037409/0/en/Orphazyme-s-arimoclomol-receives-US-Fast-Track-Designation-in-Amyotrophic-Lateral-Sclerosis.html (accessed June 19, 2020).
Ostroff, L. E., Botsford, B., Gindina, S., Cowansage, K. K., Ledoux, J. E., Klann, E., et al. (2017). Accumulation of polyribosomes in dendritic spine heads, but not bases and necks, during memory consolidation depends on cap-dependent translation initiation. J. Neurosci. 37, 1862–1872. doi: 10.1523/JNEUROSCI.3301-16.2017
Ostroff, L. E., Watson, D. J., Cao, G., Parker, P. H., Smith, H., and Harris, K. M. (2018). Shifting patterns of polyribosome accumulation at synapses over the course of hippocampal long-term potentiation. Hippocampus 28, 416–430. doi: 10.1002/hipo.22841
Ouwenga, R., Lake, A. M., O’Brien, D., Mogha, A., Dani, A., and Dougherty, J. D. (2017). Transcriptomic analysis of ribosome-bound mRNA in cortical neurites in vivo. J. Neurosci. 37, 8688–8705. doi: 10.1523/JNEUROSCI.3044-16.2017
Ozdowski, E. F., Baxter, S. L., and Sherwood, N. T. (2015). “Drosophila models of hereditary spastic paraplegia,” in Movement Disorders: Genetics and Models, 2nd Edn, ed. M. S. LeDoux, (Boston, MA: Academic Press), 1103–1122. doi: 10.1016/B978-0-12-405195-9.00073-1
Öztürk, Z., O’Kane, C. J., and Pérez-Moreno, J. J. (2020). Axonal endoplasmic reticulum dynamics and its roles in neurodegeneration. Front. Neurosci. 14:48. doi: 10.3389/fnins.2020.00048
Pan, L., Zhang, Y. Q., Woodruff, E., and Broadie, K. (2004). The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr. Biol. 14, 1863–1870. doi: 10.1016/j.cub.2004.09.085
Papanikolopoulou, K., and Skoulakis, E. M. C. (2020). “Altered proteostasis in neurodegenerative tauopathies,” in Advances in Experimental Medicine and Biology, eds R. Barrio, J. Sutherland, and M. Rodriguez, (Cham: Springer), 177–194. doi: 10.1007/978-3-030-38266-7_7
Park, M., Salgado, J. M., Ostroff, L., Helton, T. D., Robinson, C. G., Harris, K. M., et al. (2006). Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron 52, 817–830. doi: 10.1016/j.neuron.2006.09.040
Park, S., Park, J. M., Kim, S., Kim, J. A., Shepherd, J. D., Smith-Hicks, C. L., et al. (2008). Elongation factor 2 and Fragile X Mental Retardation Protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59, 70–83. doi: 10.1016/j.neuron.2008.05.023
Patel, A. B., Loerwald, K. W., Huber, K. M., and Gibson, J. R. (2014). Postsynaptic FMRP promotes the pruning of cell-to-cell connections among pyramidal neurons in the L5A neocortical network. J. Neurosci. 34, 3413–3418. doi: 10.1523/JNEUROSCI.2921-13.2014
Pavel, M., Imarisio, S., Menzies, F. M., Jimenez-Sanchez, M., Siddiqi, F. H., Wu, X., et al. (2016). CCT complex restricts neuropathogenic protein aggregation via autophagy. Nat. Commun. 7:13821. doi: 10.1038/ncomms13821
Penke, B., Bogár, F., Crul, T., Sántha, M., Tóth, M. E., and Vígh, L. (2018). Heat shock proteins and autophagy pathways in neuroprotection: from molecular bases to pharmacological interventions. Int. J. Mol. Sci. 19:325. doi: 10.3390/ijms19010325
Perry, R. B., and Fainzilber, M. (2014). Local translation in neuronal processes-in vivo tests of a “heretical hypothesis.”. Dev. Neurobiol. 74, 210–217. doi: 10.1002/dneu.22115
Piochon, C., Kano, M., and Hansel, C. (2016). LTD-like molecular pathways in developmental synaptic pruning. Nat. Neurosci. 19, 1299–1310. doi: 10.1038/nn.4389
Plate, L., and Wiseman, R. L. (2017). Regulating secretory proteostasis through the unfolded protein response: from function to therapy. Trends Cell Biol. 27, 722–737. doi: 10.1016/j.tcb.2017.05.006
Praschberger, R., Lowe, S. A., Malintan, N. T., Giachello, C. N. G., Patel, N., Houlden, H., et al. (2017). Mutations in Membrin/GOSR2 reveal stringent secretory pathway demands of dendritic growth and synaptic integrity. Cell Rep. 21, 97–109. doi: 10.1016/j.celrep.2017.09.004
Quassollo, G., Wojnacki, J., Salas, D. A., Gastaldi, L., Marzolo, M. P., Conde, C., et al. (2015). A RhoA signaling pathway regulates dendritic Golgi outpost formation. Curr. Biol. 25, 971–982. doi: 10.1016/j.cub.2015.01.075
Ramón y Cajal, S. (1989). Recollections of My Life, eds E. H. Craigie, and J. Cano, (Cambridge, MA: The MIT Press). doi: 10.7551/mitpress/5817.001.0001
Rao, K., Stone, M. C., Weiner, A. T., Gheres, K. W., Zhou, C., Deitcher, D. L., et al. (2016). Spastin, atlastin, and ER relocalization are involved in axon but not dendrite regeneration. Mol. Biol. Cell 27, 3245–3256. doi: 10.1091/mbc.E16-05-0287
Ren, R. J., Dammer, E. B., Wang, G., Seyfried, N. T., and Levey, A. I. (2014). Proteomics of protein post-translational modifications implicated in neurodegeneration. Transl. Neurodegener. 3:23. doi: 10.1186/2047-9158-3-23
Renaud, L., Picher-Martel, V., Codron, P., and Julien, J. P. (2019). Key role of UBQLN2 in pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol. Commun. 7:103. doi: 10.1186/s40478-019-0758-7
Rode, S., Ohm, H., Anhäuser, L., Wagner, M., Rosing, M., Deng, X., et al. (2018). Differential requirement for translation initiation factor pathways during ecdysone-dependent neuronal remodeling in Drosophila. Cell Rep. 24:2287. doi: 10.1016/j.celrep.2018.07.074
Rogerson, T., Cai, D. J., Frank, A., Sano, Y., Shobe, J., Lopez-Aranda, M. F., et al. (2014). Synaptic tagging during memory allocation. Nat. Rev. Neurosci. 15, 157–169. doi: 10.1038/nrn3667
Rumpf, S., Lee, S. B., Jan, L. Y., and Jan, Y. N. (2011). Neuronal remodeling and apoptosis require VCP-dependent degradation of the apoptosis inhibitor DIAP1. Development 138, 1153–1160. doi: 10.1242/dev.062703
Ryoo, H. D., Domingos, P. M., Kang, M.-J., and Steller, H. (2007). Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J. 26, 242–252. doi: 10.1038/sj.emboj.7601477
Saito, A., Cai, L., Matsuhisa, K., Ohtake, Y., Kaneko, M., Kanemoto, S., et al. (2018). Neuronal activity-dependent local activation of dendritic unfolded protein response promotes expression of brain-derived neurotrophic factor in cell soma. J. Neurochem. 144, 35–49. doi: 10.1111/jnc.14221
Saldate, J. J., Shiau, J., Cazares, V. A., and Stuenkel, E. L. (2018). The ubiquitin-proteasome system functionally links neuronal Tomosyn-1 to dendritic morphology. J. Biol. Chem. 293, 2232–2246. doi: 10.1074/jbc.M117.815514
Sambataro, F., and Pennuto, M. (2017). Post-translational modifications and protein quality control in motor neuron and polyglutamine diseases. Front. Mol. Neurosci. 10:82. doi: 10.3389/fnmol.2017.00082
Sano, R., and Reed, J. C. (2013). ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 1833, 3460–3470. doi: 10.1016/j.bbamcr.2013.06.028
Santana, E., de Los Reyes, T., and Casas-Tintó, S. (2020). Small heat shock proteins determine synapse number and neuronal activity during development. PLoS One 15:e0233231. doi: 10.1371/journal.pone.0233231
Sarparanta, J., Jonson, P. H., Kawan, S., and Udd, B. (2020). Neuromuscular diseases due to chaperone mutations: a review and some new results. Int. J. Mol. Sci. 21:1409. doi: 10.3390/ijms21041409
Satoh, D., Sato, D., Tsuyama, T., Saito, M., Ohkura, H., Rolls, M. M., et al. (2008). Spatial control of branching within dendritic arbors by dynein-dependent transport of Rab5-endosomes. Nat. Cell Biol. 10, 1164–1171. doi: 10.1038/ncb1776
Schäffner, I., Minakaki, G., Khan, M. A., Balta, E. A., Schlötzer-Schrehardt, U., Schwarz, T. J., et al. (2018). FoxO function is essential for maintenance of autophagic flux and neuronal morphogenesis in adult neurogenesis. Neuron 99, 1188–1203.e6. doi: 10.1016/j.neuron.2018.08.017
Schmidt, T., Bartelt-Kirbach, B., and Golenhofen, N. (2012). Phosphorylation-dependent subcellular localization of the small heat shock proteins HspB1/Hsp25 and HspB5/aB-crystallin in cultured hippocampal neurons. Histochem. Cell Biol. 138, 407–418. doi: 10.1007/s00418-012-0964-x
Sergeeva, O. A., Tran, M. T., Haase-Pettingell, C., and King, J. A. (2014). Biochemical characterization of mutants in chaperonin proteins CCT4 and CCT5 associated with hereditary sensory neuropathy. J. Biol. Chem. 289, 27470–27480. doi: 10.1074/JBC.M114.576033
Shabbir, A., Bianchetti, E., Cargonja, R., Petrovic, A., Mladinic, M., Pilipoviæ, K., et al. (2015). Role of HSP70 in motoneuron survival after excitotoxic stress in a rat spinal cord injury model in vitro. Eur. J. Neurosci. 42, 3054–3065. doi: 10.1111/ejn.13108
Shehata, M., Matsumura, H., Okubo-Suzuki, R., Ohkawa, N., and Inokuchi, K. (2012). Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J. Neurosci. 32, 10413–10422. doi: 10.1523/JNEUROSCI.4533-11.2012
Shen, W., and Ganetzky, B. (2009). Autophagy promotes synapse development in Drosophila. J. Cell Biol. 187, 71–79. doi: 10.1083/jcb.200907109
Shi, Z., Fujii, K., Kovary, K. M., Genuth, N. R., Röst, H. L., Teruel, M. N., et al. (2017). Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol. Cell 67, 71–83.e7. doi: 10.1016/j.molcel.2017.05.021
Shih, Y. T., and Hsueh, Y. P. (2018). The involvement of endoplasmic reticulum formation and protein synthesis efficiency in VCP- and ATL1-related neurological disorders. J. Biomed. Sci. 25:2. doi: 10.1186/s12929-017-0403-3
Siri, S. O., Rozés-Salvador, V., de la Villarmois, E. A., Ghersi, M. S., Quassollo, G., Pérez, M. F., et al. (2020). Decrease of Rab11 prevents the correct dendritic arborization, synaptic plasticity and spatial memory formation. Biochim. Biophys. Acta 1867:118735. doi: 10.1016/j.bbamcr.2020.118735
Slomnicki, L. P., Pietrzak, M., Vashishta, A., Jones, J., Lynch, N., Elliot, S., et al. (2016). Requirement of neuronal ribosome synthesis for growth and maintenance of the dendritic tree. J. Biol. Chem. 291, 5721–5739. doi: 10.1074/JBC.M115.682161
Smith, H. L., Li, W., and Cheetham, M. E. (2015). Molecular chaperones and neuronal proteostasis. Semin. Cell Dev. Biol. 40, 142–152. doi: 10.1016/j.semcdb.2015.03.003
Son, J. H., Shim, J. H., Kim, K.-H., Ha, J.-Y., and Han, J. Y. (2012). Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 44, 89–98. doi: 10.3858/emm.2012.44.2.031
Sot, B., Rubio-Muñoz, A., Leal-Quintero, A., Martínez-Sabando, J., Marcilla, M., Roodveldt, C., et al. (2017). The chaperonin CCT inhibits assembly of α-synuclein amyloid fibrils by a specific, conformation-dependent interaction. Sci. Rep. 7:40859. doi: 10.1038/srep40859
Steward, O., and Schuman, E. M. (2003). Compartmentalized synthesis and degradation of proteins in neurons. Neuron 40, 347–359. doi: 10.1016/S0896-6273(03)00635-4
Summerville, J. B., Faust, J. F., Fan, E., Pendin, D., Daga, A., Formella, J., et al. (2016). The effects of ER morphology on synaptic structure and function in Drosophila melanogaster. J. Cell Sci. 129, 1635–1648. doi: 10.1242/jcs.184929
Sutton, M. A., and Schuman, E. M. (2005). Local translational control in dendrites and its role in long-term synaptic plasticity. J. Neurobiol. 64, 116–131. doi: 10.1002/neu.20152
Sweeney, N. T., Brenman, J. E., Jan, Y. N., and Gao, F. B. (2006). The coiled-coil protein Shrub controls neuronal morphogenesis in Drosophila. Curr. Biol. 16, 1006–1011. doi: 10.1016/j.cub.2006.03.067
Tam, S., Geller, R., Spiess, C., and Frydman, J. (2006). The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 8, 1155–1162. doi: 10.1038/ncb1477
Tang, B. L. (2008). Emerging aspects of membrane traffic in neuronal dendrite growth. Biochim. Biophys. Acta 1783, 169–176. doi: 10.1016/j.bbamcr.2007.11.011
Tang, G., Gudsnuk, K., Kuo, S. H., Cotrina, M. L., Rosoklija, G., Sosunov, A., et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. doi: 10.1016/j.neuron.2014.07.040
Tang, Q., Rui, M., Bu, S., Wang, Y., Chew, L. Y., and Yu, F. (2020). A microtubule polymerase is required for microtubule orientation and dendrite pruning in Drosophila. EMBO J 39:e103549. doi: 10.15252/embj.2019103549
Tang, Y., Nyengaard, J. R., De Groot, D. M. G., and Gundersen, H. J. G. (2001). Total regional and global number of synapses in the human brain neocortex. Synapse 41, 258–273. doi: 10.1002/syn.1083
Tao, J., and Rolls, M. M. (2011). Dendrites have a rapid program of injury-induced degeneration that is molecularly distinct from developmental pruning. J. Neurosci. 31, 5398–5405. doi: 10.1523/JNEUROSCI.3826-10.2011
Taymans, J. M., Nkiliza, A., and Chartier-Harlin, M. C. (2015). Deregulation of protein translation control, a potential game-changing hypothesis for Parkinson’s disease pathogenesis. Trends Mol. Med. 21, 466–472. doi: 10.1016/j.molmed.2015.05.004
Thiruvalluvan, A., de Mattos, E. P., Brunsting, J. F., Bakels, R., Serlidaki, D., Barazzuol, L., et al. (2020). DNAJB6, a key factor in neuronal sensitivity to amyloidogenesis. Mol. Cell 78, 346–358.e9. doi: 10.1016/j.molcel.2020.02.022
Tiedge, H., and Brosius, J. (1996). Translational machinery in dendrites of hippocampal neurons in culture. J. Neurosci. 16, 7171–7181. doi: 10.1523/jneurosci.16-22-07171.1996
Tooze, S. A., Abada, A., and Elazar, Z. (2014). Endocytosis and autophagy: Exploitation or cooperation? Cold Spring Harb. Perspect. Biol. 6, 18358–18359. doi: 10.1101/cshperspect.a018358
Tundo, G. R., Sbardella, D., Santoro, A. M., Coletta, A., Oddone, F., Grasso, G., et al. (2020). The proteasome as a druggable target with multiple therapeutic potentialities: cutting and non-cutting edges. Pharmacol. Ther. 213:107579. doi: 10.1016/j.pharmthera.2020.107579
Vallin, J., and Grantham, J. (2019). The role of the molecular chaperone CCT in protein folding and mediation of cytoskeleton-associated processes: implications for cancer cell biology. Cell Stress Chaperones 24, 17–27. doi: 10.1007/s12192-018-0949-3
Vicente Miranda, H., Chegão, A., Oliveira, M. S., Fernandes Gomes, B., Enguita, F. J., and Outeiro, T. F. (2020). Hsp27 reduces glycation-induced toxicity and aggregation of alpha-synuclein. FASEB J. 34, 6718–6728. doi: 10.1096/fj.201902936R
Vonk, W. I. M., Rainbolt, T. K., Dolan, P. T., Webb, A. E., Brunet, A., and Frydman, J. (2020). Differentiation drives widespread rewiring of the neural stem cell chaperone network. Mol. Cell 78, 329–345.e9. doi: 10.1016/j.molcel.2020.03.009
Wagner, L. M., Nathwani, S. M., Ten Eyck, P. P., and Aldridge, G. M. (2020). Local cortical overexpression of human wild-type alpha-synuclein leads to increased dendritic spine density in mouse. Neurosci. Lett. 733:135051. doi: 10.1016/j.neulet.2020.135051
Wang, J., Sen Lou, S., Wang, T., Wu, R. J., Li, G., Zhao, M., et al. (2019). UBE3A-mediated PTPA ubiquitination and degradation regulate PP2A activity and dendritic spine morphology. Proc. Natl. Acad. Sci. U.S.A. 116, 12500–12505. doi: 10.1073/pnas.1820131116
Wang, Q., Wang, Y., and Fengwei, Y. (2018). Yif1 associates with yip1 on golgi and regulates dendrite pruning in sensory neurons during Drosophila metamorphosis. Development 145:dev164475. doi: 10.1242/dev.164475
Wang, T., Wang, J., Wang, J., Mao, L., Tang, B., Vanderklish, P. W., et al. (2019). HAP1 is an in vivo UBE3A target that augments autophagy in a mouse model of Angelman syndrome. Neurobiol. Dis. 132:104585. doi: 10.1016/j.nbd.2019.104585
Wang, X., Kim, J. H., Bazzi, M., Robinson, S., Collins, C. A., and Ye, B. (2013). Bimodal control of dendritic and axonal growth by the dual leucine zipper kinase pathway. PLoS Biol. 11:e1001572. doi: 10.1371/journal.pbio.1001572
Wang, X., Zorio, D. A. R., Schecterson, L., Lu, Y., and Wang, Y. (2018). Postsynaptic FMRP regulates synaptogenesis in vivo in the developing cochlear nucleus. J. Neurosci. 38, 6445–6460. doi: 10.1523/JNEUROSCI.0665-18.2018
Wang, Y., Zhang, H., Shi, M., Liou, Y. C., Lu, L., and Yu, F. (2017). Sec71 functions as a GEF for the small GTPase Arf1 to govern dendrite pruning of Drosophila sensory neurons. J. Cell Sci. 130, 1851–1862. doi: 10.1242/dev.146175
Weeks, S. D., Muranova, L. K., Heirbaut, M., Beelen, S., Strelkov, S. V., and Gusev, N. B. (2018). Characterization of human small heat shock protein HSPB1 α-crystallin domain localized mutants associated with hereditary motor neuron diseases. Sci. Rep. 8:688. doi: 10.1038/s41598-017-18874-x
Wei, X., Howell, A. S., Dong, X., Taylor, C. A., Cooper, R. C., Zhang, J., et al. (2015). The unfolded protein response is required for dendrite morphogenesis. eLife 4:e06963. doi: 10.7554/eLife.06963
Williams, K. L., and Mearow, K. M. (2011). Phosphorylation status of heat shock protein 27 influences neurite growth in adult dorsal root ganglion sensory neurons in vitro. J. Neurosci. Res. 89, 1160–1172. doi: 10.1002/jnr.22634
Wong, J. J. L., Li, S., Lim, E. K. H., Wang, Y., Wang, C., Zhang, H., et al. (2013). A Cullin1-based SCF E3 ubiquitin ligase targets the InR/PI3K/TOR pathway to regulate neuronal pruning. PLoS Biol. 11:e1001657. doi: 10.1371/journal.pbio.1001657
Wu, B., Eliscovich, C., Yoon, Y. J., and Singer, R. H. (2016). Translation dynamics of single mRNAs in live cells and neurons. Science 352, 1430–1435. doi: 10.1126/science.aaf1084
Wu, C., Watts, M. E., and Rubin, L. L. (2019). MAP4K4 activation mediates motor neuron degeneration in amyotrophic lateral sclerosis. Cell Rep. 26, 1143–1156. doi: 10.1016/j.celrep.2019.01.019
Xu, X., Gupta, S., Hu, W., McGrath, B. C., and Cavener, D. R. (2011). Hyperthermia induces the ER stress pathway. PLoS One 6:e23740. doi: 10.1371/journal.pone.0023740
Xu, X. L., Li, Y., Wang, F., and Gao, F. B. (2008). The steady-state level of the nervous-system-specific microRNA-124a is regulated by dFMR1 in Drosophila. J. Neurosci. 28, 11883–11889. doi: 10.1523/JNEUROSCI.4114-08.2008
Xue, S., and Barna, M. (2012). Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol. 13, 355–369. doi: 10.1038/nrm3359
Yagi, H., Takabayashi, T., Xie, M. J., Kuroda, K., and Sato, M. (2017). Subcellular distribution of non-muscle myosin IIb is controlled by FILIP through Hsc70. PLoS One 12:e0172257. doi: 10.1371/journal.pone.0172257
Yan, J., Porch, M. W., Court-Vazquez, B., Bennett, M. V. L., and Zukin, R. S. (2018). Activation of autophagy rescues synaptic and cognitive deficits in fragile X mice. Proc. Natl. Acad. Sci. U.S.A. 115, E9707–E9716. doi: 10.1073/pnas.1808247115
Yang, S. Z., and Wildonger, J. (2020). Golgi outposts locally regulate microtubule orientation in neurons but are not required for the overall polarity of the dendritic cytoskeleton. Genetics 215, 435–447. doi: 10.1534/genetics.119.302979
Yang, X. (2020). Characterizing spine issues: If offers novel therapeutics to Angelman syndrome. Dev. Neurobiol. doi: 10.1002/dneu.22757 [Epub ahead of print].
Yang, Y., Coleman, M., Zhang, L., Zheng, X., and Yue, Z. (2013). Autophagy in axonal and dendritic degeneration. Trends Neurosci. 36, 418–428. doi: 10.1016/j.tins.2013.04.001
Yang, Z., and Klionsky, D. J. (2009). “Autophagy in Infection and Immunity,” in Current Topics in Microbiology and Immunology, eds B. Levine, T. Yoshimori, and V. Deretic, (Berlin: Springer). doi: 10.1007/978-3-642-00302-8_1
Yap, C. C., Digilio, L., McMahon, L. P., Garcia, A. D. R., and Winckler, B. (2018). Degradation of dendritic cargos requires Rab7-dependent transport to somatic lysosomes. J. Cell Biol. 217, 3141–3159. doi: 10.1083/jcb.201711039
Ye, B., Kim, J. H., Yang, L., McLachlan, I., Younger, S., Jan, L. Y., et al. (2011). Differential regulation of dendritic and axonal development by the novel Krüppel-like factor Dar1. J. Neurosci. 31, 3309–3319. doi: 10.1523/JNEUROSCI.6307-10.2011
Ye, B., Zhang, Y., Song, W., Younger, S. H., Jan, L. Y., and Jan, Y. N. (2007). Growing dendrites and axons differ in their reliance on the secretory pathway. Cell 130, 717–729. doi: 10.1016/j.cell.2007.06.032
Yerbury, J. J., Farrawell, N. E., and Mcalary, L. (2020). Proteome homeostasis dysfunction: a unifying principle in ALS pathogenesis. Trends Neurosci. 43, 274–284. doi: 10.1016/j.tins.2020.03.002
Yi, J. J., Berrios, J., Newbern, J. M., Snider, W. D., Philpot, B. D., Hahn, K. M., et al. (2015). An Autism-linked mutation disables phosphorylation control of UBE3A. Cell 162, 795–807. doi: 10.1016/j.cell.2015.06.045
Yu, F., and Schuldiner, O. (2014). Axon and dendrite pruning in Drosophila. Curr. Opin. Neurobiol. 27, 192–198. doi: 10.1016/j.conb.2014.04.005
Yue, Z., Friedman, L., Komatsu, M., and Tanaka, K. (2009). The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim. Biophys. Acta 1793, 1496–1507. doi: 10.1016/j.bbamcr.2009.01.016
Yun, D., Zhuang, Y., Kreutz, M. R., and Behnisch, T. (2018). The role of 19S proteasome associated deubiquitinases in activity-dependent hippocampal synaptic plasticity. Neuropharmacology 133, 354–365. doi: 10.1016/j.neuropharm.2018.01.043
Zayas-Santiago, A., Ríos, D. S., Zueva, L. V., and Inyushin, M. Y. (2018). Localization of αa-Crystallin in rat retinal müller glial cells and photoreceptors. Microsc. Microanal. 24, 545–552. doi: 10.1017/S1431927618015118
Zhang, H., Wang, Y., Wong, J. J. L., Lim, K. L., Liou, Y. C., Wang, H., et al. (2014). Endocytic pathways downregulate the L1-type cell adhesion molecule Neuroglian to promote dendrite pruning in Drosophila. Dev. Cell 30, 463–478. doi: 10.1016/j.devcel.2014.06.014
Zhang, Y. Q., and Broadie, K. (2005). Fathoming fragile X in fruit flies. Trends Genet. 21, 37–45. doi: 10.1016/j.tig.2004.11.003
Zheng, C., Atlas, E., Lee, H. M. T., Jao, S. L. J., Nguyen, K. C. Q., Hall, D. H., et al. (2020). Opposing effects of an F-box protein and the HSP90 chaperone network on microtubule stability and neurite growth in Caenorhabditis elegans. Development 147:dev189886. doi: 10.1242/dev.189886
Zhou, G., Lane, G., Cooper, S. L., Kahnt, T., and Zelano, C. (2019). Characterizing functional pathways of the human olfactory system. eLife 8:e47177. doi: 10.7554/eLife.47177
Keywords: dendrite, proteostasis network, ribosome, chaperone, autophagy, ubiquitin-proteasome system (UPS), developmental homeostasis, neurological disease
Citation: Lottes EN and Cox DN (2020) Homeostatic Roles of the Proteostasis Network in Dendrites. Front. Cell. Neurosci. 14:264. doi: 10.3389/fncel.2020.00264
Received: 29 June 2020; Accepted: 28 July 2020;
Published: 14 August 2020.
Edited by:
Quan Yuan, National Institute of Neurological Disorders and Stroke (NINDS), United StatesReviewed by:
Fengwei Yu, Temasek Life Sciences Laboratory, SingaporeSung Bae Lee, Daegu Gyeongbuk Institute of Science and Technology (DGIST), South Korea
Copyright © 2020 Lottes and Cox. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel N. Cox, ZGNveDE4QGdzdS5lZHU=