 Fumio Nakamura
Fumio Nakamura Toshio Ohshima
Toshio Ohshima Yoshio Goshima
Yoshio Goshima- 1Department of Biochemistry, Tokyo Women’s Medical University, Tokyo, Japan
- 2Department of Life Science and Medical Bio-Science, Waseda University, Tokyo, Japan
- 3Department of Molecular Pharmacology and Neurobiology, Yokohama City University Graduate School of Medicine, Yokohama, Japan
Collapsin response mediator proteins (CRMPs), which consist of five homologous cytosolic proteins, are one of the major phosphoproteins in the developing nervous system. The prominent feature of the CRMP family proteins is a new class of microtubule-associated proteins that play important roles in the whole process of developing the nervous system, such as axon guidance, synapse maturation, cell migration, and even in adult brain function. The CRMP C-terminal region is subjected to posttranslational modifications such as phosphorylation, which, in turn, regulates the interaction between the CRMPs and various kinds of proteins including receptors, ion channels, cytoskeletal proteins, and motor proteins. The gene-knockout of the CRMP family proteins produces different phenotypes, thereby showing distinct roles of all CRMP family proteins. Also, the phenotypic analysis of a non-phosphorylated form of CRMP2-knockin mouse model, and studies of pharmacological responses to CRMP-related drugs suggest that the phosphorylation/dephosphorylation process plays a pivotal role in pathophysiology in neuronal development, regeneration, and neurodegenerative disorders, thus showing CRMPs as promising target molecules for therapeutic intervention.
Introduction
Cell to cell interactions mediated by extracellular molecules drives numerous physiological processes and helps enable coordinated functioning in neuronal development and regeneration. Extracellular signals are often integrated into complex regulatory networks in which cytoskeletal rearrangement and membrane reorganization are precisely regulated. The growth cone is a characteristic structure at the distal tip of growing axons during development. Growth cones are composed of an actin-rich peripheral domain and a microtubule-rich central domain. At the distal tip of the growth cone, finger-like filopodia and sheet-like lamellipodia extend and retract rapidly as they sense the environmental axon guidance cues around the growth cone.
The first member of the Collapsin Response Mediator Proteins (CRMPs) family, was originally identified as an intracellular protein mediating the action of semaphorin-3A (Sema3A)-signaling, a repulsive axon guidance molecule (Goshima et al., 1995). The initial name of the protein was CRMP-62 because collapsin is the former name of Sema3A and 62 was the new molecule’s molecular weight. As CRMP-62 has significant homology with UNC-33, which is involved in axon guidance in C. elegans through the regulation of tubulin-cytoskeleton, CRMP-62 has been thought to mediate the intracellular signaling involved in axon guidance via its modulation of the cytoskeleton at various developmental stages. After the identification of CRMP-62, an additional four members of the CRMP family were identified by several groups, such as TOAD-64, Ulip, DRP, DPYSL (Schmidt and Strittmatter, 2007). Currently, the nomenclature has been unified by calling the family members “CRMP1” through “CRMP5” (Supplementary Table S1); CRMP-62 has been renamed as CRMP2. The CRMP family of proteins are now recognized as multifunctional proteins, not only being involved in neuronal development, regeneration and inflammation, but also in various neurological and psychiatric disease states (Tobe et al., 2017; Tsutiya et al., 2017).
In this review, we summarize the molecular aspects of the CRMPs and discuss their possible involvement in pathophysiological conditions of various disease states. Comprehensive reviews on the implication of CRMPs in Alzheimer’s disease (AD) and psychiatric disorders have been described elsewhere (Gu and Ihara, 2000; Yamashita and Goshima, 2012; Quach et al., 2015; Hensley and Kursula, 2016; Nagai et al., 2017; Tobe et al., 2017; Nakashima et al., 2018).
Structure of the CRMPs
CRMP1, 2, and 4 have long and short alternate splicing isoforms (Leung et al., 2002). Short isoforms of CRMP1, 2 and 4, CRMP3, and CRMP5 are 565–572 amino acid lengths. The apparent molecular size of these proteins on SDS-PAGE is 62–65 kDa. The long isoforms of CRMP1, 2, and 4 extend approximately 100 amino acids at their N-termini and exhibit 72–75 kDa on SDS-PAGE. We hereafter describe long and short isoforms as “L-CRMP” and “CRMP,” respectively. CRMP1 to CRMP4 share 69–76% amino acid identity while these members and CRMP5 share approximately 50% identity. The long isoforms of CRMPs are minor components in most of the cells and organs. The amino acid identity of the N-terminal regions of L-CRMP1, 2, and 4 is 35% to 54%. The N-terminal extended region has several unique functions such as distal localization of L-CRMP2 (CRMP2A) in axons (Balastik et al., 2015), L-CRMP4 (CRMP4b) and RhoA interaction in Nogo signaling (Alabed et al., 2007), and correlation of L-CRMP1 expression and cancer cell migration (Pan et al., 2011).
X-ray crystal structures of the short isoforms of CRMP1, 2, 4, and 5 have been reported (Deo et al., 2004; Stenmark et al., 2007; Ponnusamy and Lohkamp, 2013; Ponnusamy et al., 2014). Central regions of the CRMPs (8–490) forms a tetramer (Figure 1). The folded CRMP structure resembles dihydropyrimidinase (DHPase), which hydrolyzes the amide bond of pyrimidine bases (Gojkovic et al., 2003). Each CRMP monomer consists of an N-terminal β-sheet enriched region (15–69) and central α/β barrel domain (70–490). The central domain contains tetramer interfaces. The ternary structure of the entire CRMP C-terminal region (490–572) has not been determined possibly because the region stays flexible and the structure is somewhat random, altering its conformation upon posttranslational modification such as phosphorylation. However, the partial ternary structure of the C-terminal proximal region of CRMP2 has been reported (Niwa et al., 2017). The C-terminal visible residues from the α-helix19 (480–487) further extend in the same direction and several residues in (491–506) interact with the neighboring monomer (Figure 1), contributing to stabilizing the tetramer. It has been shown that CRMPs form hetero-oligomerized complexed in the brain (Wang and Strittmatter, 1996). In vitro reconstitution revealed that CRMP1, CRMP2, and CRMP3 prefer hetero oligomerization. However, the biological significance of the hetero-complexed CRMPs has not yet been fully addressed (Makihara et al., 2016).
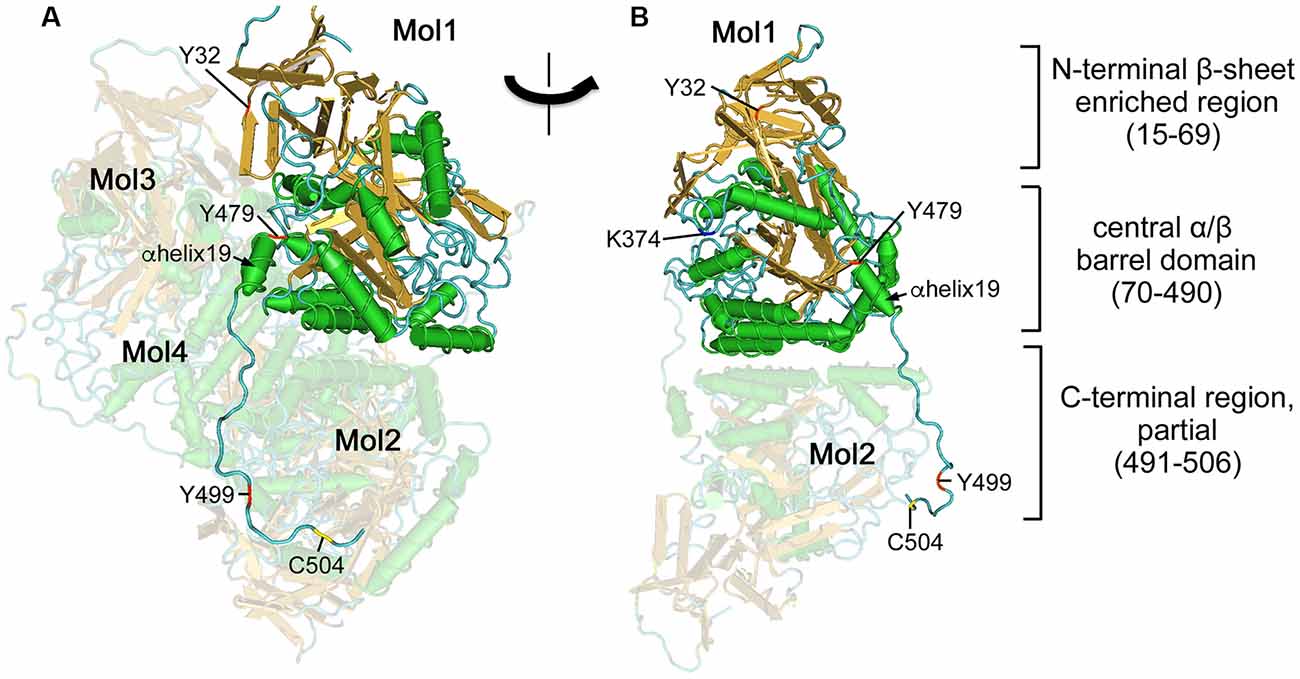
Figure 1. Ternary structure of Collapsin response mediator proteins 2 (CRMP2). (A) Crystal structure of human CRMP2 (1–525) homo-tetramer (5X1A; Niwa et al., 2017). A secondary structure-based view was rendered using Cn3D (version 4.3). One monomer (Mol1) is shown in secondary structure-based color (α-helix, green; β-sheet, gold; random coil, cyan). The structure is visible between 14 and 506 residues. Phosphorylation (Tyr32, Tyr479, and Tyr499), SUMOylation (Lys374), and oxidation (Cys504) sites are indicated by red, blue, and yellow, respectively. Other monomers (Mol2–4) are shown in faded colors. The C-terminal domain of Mol1 interacts with Mol2. (B) Rotate view. The interface of Mol1 to Mol3 is shown by omitting Mol3 and Mol4. Note that Lys374 and Tyr479 are present on the interface.
Although the CRMPs have 51–54% amino acid identity with DHPase, they have no enzymatic activity toward dihydropyrimidines (Wang and Strittmatter, 1996), as His69 and His248 residues coordinating Zn ions in the catalytic center of DHPase are not conserved in the CRMPs. Instead, CRMP3 has histone H4 deacetylase activity, which is involved in neuronal cell death after the translocation of CRMP3 to the nucleus (Hou et al., 2013). Other enzymatic activities of the CRMPs are currently unknown.
Phosphorylation of CRMPs
Posttranslational modifications play a crucial role in regulating the folding of proteins, their targeting to specific subcellular compartments, their interaction with other proteins, and their functional states, such as activation and inactivation in signal transduction pathways. Protein phosphorylation is the major molecular mechanism through which protein function is regulated in response to extracellular stimuli both inside and outside the nervous system. Many kinases phosphorylate CRMPs (Figure 2, Supplementary Table S2). Most of the phosphorylation sites are localized in their C-terminal regions. As the primary structures of their C-termini are relatively not conserved compared to their central domains, some kinases specifically phosphorylate one of the CRMPs but not the others. Such phosphorylation may contribute to the unique function of each member of the CRMP family protein. Here we summarize the phosphorylation by kinase-based classification.
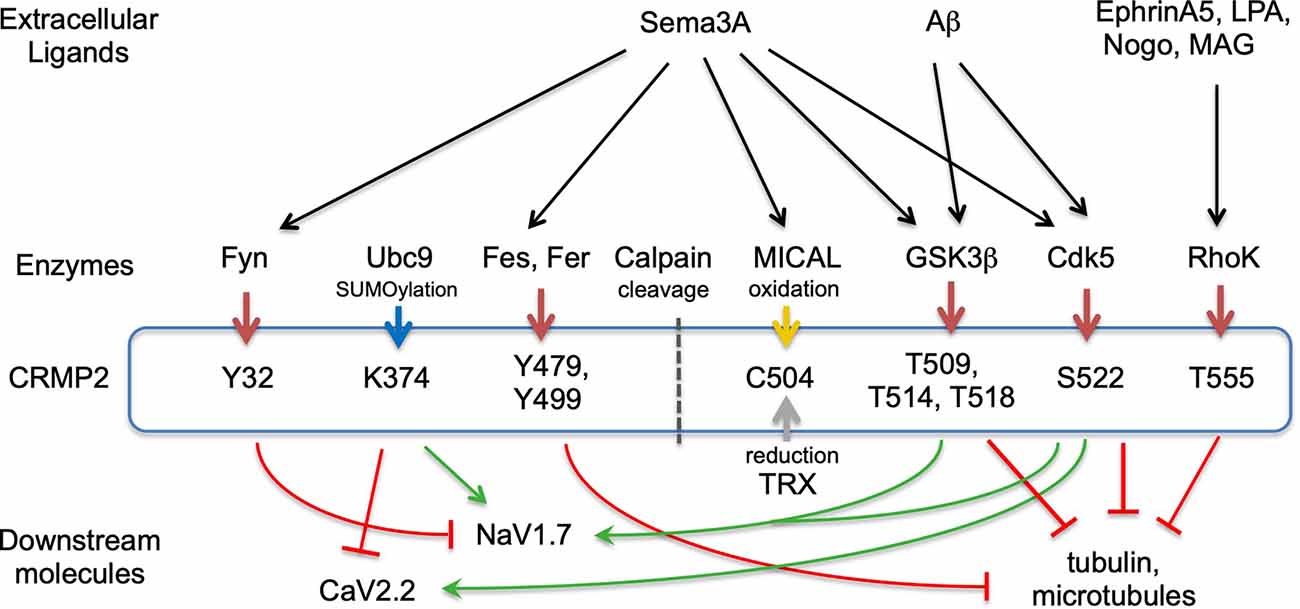
Figure 2. CRMP2-posttranslational modification and signaling. Extracellular ligands, modulating enzymes, posttranslational medication sites in CRMP2, and downstream molecules are represented. Red arrows from enzymes to CRMP2 represent phosphorylation. Blue, yellow, and gray arrows indicate SUMOylation, oxidation, and reduction, respectively. Green and red stop arrows represent facilitation and inhibition, respectively. Not all ligands or downstream molecules are shown.
Cyclin-dependent kinase 5 (Cdk5) and glycogen kinase 3β (GSK3β)—the phosphorylation of CRMP2 by Cdk5 and GSK3β is the most extensively studied among the CRMP members (Figure 2). CRMP2 is primarily phosphorylated at Ser522 by Cdk5 and subsequently phosphorylated at Ser518, Thr514, and Thr509 by GSK3β (Cole et al., 2006; Brittain et al., 2012; Yamashita and Goshima, 2012; Nagai et al., 2017). The latter phosphorylation requires Cdk5-primed phosphorylation of Ser522, therefore, the substitution of Ser522 with Ala eliminates the phosphorylation of Ser518, Thr514, and Thr509 in CRMP2. CRMP’s phosphorylation state determines its biological function. Non-phosphorylated CRMP2 binds tubulin dimers to facilitate the axonal elongation; the phosphorylation eliminates that function. Instead, the phosphorylated CRMP2 acts as an intracellular signaling mediator for inhibition of axonal guidance such as via Sema3A. Recent crystal structural analysis of CRMP2 revealed that non-phosphorylated CRMP2 monomer forms hetero-trimer between CRMP2 monomer and the GTP-tubulin hetero-dimer (Sumi et al., 2018). Using the phosphorylation-mimicking form of CRMP2, Sumi et al. (2018) revealed that the increased negative charge of the C-terminal region alters CRMP2 homo-tetramer conformation and reduces the interaction of CRMP2 and tubulin-dimers. Ser522 phosphorylation of CRMP2 augments the interaction with CaV2.2 to enhance Ca2+ influx (Brittain et al., 2012).
Phosphorylation state is not only regulated by kinase but also by phosphatases that dephosphorylate. PP2A dephosphorylates CRMP2 Thr514 and facilitates the non-phospho CRMP2 effect on neurite outgrowth (Zhu et al., 2010). L-CRMP2 is phosphorylated by Cdk5 at Ser27 in N-terminal extended region as well as at Ser623, an equivalent residue of Ser522 in the short form (Balastik et al., 2015). Prolyl Isomerase Pin1 catalyzes and stabilizes phosho-Ser32-Pro33 in L-CRMP2. This brings distal localization of L-CRMP2 in axons and attenuates Sema3A-repulsive response despite Cdk5-dependent phosphorylation.
The C-terminal phosphorylation sites, Thr509, Thr514, Ser518, and Ser522, are conserved in human CRMP1 and CRMP4. Different priming kinases, Cdk5 and dual-specificity tyrosine phosphorylation-regulated kinase 2 (DYRK2) phosphorylate Ser522 of CRMP1 and CRMP4, respectively (Cole et al., 2006). GSK3β secondarily phosphorylates human CRMP1 and CRMP4 like CRMP2. However, Cdk5 directly phosphorylates Thr509 of mouse/rat CRMP1 because Thr514 is replaced with Ala. CRMP5 is phosphorylated by GSK3β at Thr516 and this phosphorylation is essential for tubulin-binding of CRMP5 but inhibits neurite outgrowth (Brot et al., 2014).
Rho-kinase—Rho-kinase phosphorylation of Thr555 CRMP2 was initially identified by in vitro kinase assays (Arimura et al., 2000). As Rho-kinase acts as the downstream molecule of lysophosphatidic acid (LPA), the primary cultured dorsal root ganglion (DRG) neurons stimulated with LPA induced CRMP2-Thr555 phosphorylation (Figure 2). Overexpression of CRMP2-Thr555Ala in DRG neurons suppressed the LPA-induced growth cone collapse response but not the Sema3A-response. A similar mechanism is utilized in Ephrin-A5 signaling. Ephrin-A5-stimulation induced growth cone collapse response and the Thr555 phosphorylation in cultured DRG neurons. This response was abrogated by a Rho-kinase inhibitor or by the overexpression of CRMP2 Thr555Ala (Arimura et al., 2005).
Other Ser/Thr kinases—it has been shown that Protein kinase A (PKA), Protein kinase C (PKC), and Calmodulin kinase II (CaMKII) phosphorylate CRMP2 in pathological conditions. PKA-dependent phosphorylation of CRMP2 was observed in the nucleus accumbens neurons of cocaine-sensitized rats (Boudreau et al., 2009). The PKA-phosphorylation site has not been determined. PKC-βII phosphorylates CRMP2 at the Thr514 residue. This phosphorylation prevents calpain-mediated CRMP2 proteolysis during ischemic injury (Yang et al., 2016). CaMKII phosphorylates CRMP2 at Thr555, which, in turn, attenuates glutamate toxicity in ischemic brains (Hou et al., 2009).
Tyrosine kinases—it has been shown that the constitutive active form of Fyn phosphorylates all members of CRMPs (Uchida et al., 2009). The constitutive active form phosphorylates Tyr32 of CRMP2; this phosphorylation is involved in Sema3A-signaling in mouse DRG neurons (Figure 2). In contrast, wild-type Fyn selectively phosphorylates CRMP1 at Tyr504 but not other CRMPs (Buel et al., 2010). The absence of an autoinhibitory mechanism in the constitutive active form may contribute to the non-selective phosphorylation of CRMPs. Fyn-related kinase Fes and Fer phosphorylate CRMP2 and CRMP5. In vitro phosphorylation assay and mass spectrometry analysis revealed that Fer phosphorylates CRMP2 at the 32, 251, 275, 431, 479, and 499 Tyr residues (Zheng et al., 2018). Among these sites, Tyr479 and/or Tyr499 phosphorylation prevents the tetramerization of CRMP2, which, in turn, decreases the interaction of CRMP2 and microtubules (Figures 1, 2).
Other Posttranslational Modifications
SUMOylation—CRMP2 is post-translationally modulated by a small ubiquitin-like modifier (SUMO) at Lys374, and this modulation alters CRMP2-interaction with Ca2+ and Na+ channels in a different manner (Figure 2). While SUMOylated CRMP2 enhances Na+ currents through NaV1.7 surface expression (Dustrude et al., 2013), it reduces Ca2+ influx through CaV2.2 (Ju et al., 2013). De-SUMOylation and de-phosphorylation of CRMP2 at Thr514 contribute to the formation and maturation of dendritic spines (Zhang et al., 2018). As the SUMO consensus motif including Lys374 is conserved among CRMPs, SUMOylation may regulate other CRMPs.
Oxidation—genetic dissection of drosophila axon guidance revealed the involvement of flavoprotein oxidoreductase MICAL in semaphorin-signaling (Terman et al., 2002). CRMP2 binds and activates MICAL, by releasing its autoinhibition domain (Schmidt et al., 2008). CRMP2 is subsequently oxidized at Cys504 by MICAL upon Sema3A-stimulation and transiently forms disulfate bonds with thioredoxin (TRX; Figure 2). The CRMP2-TRX complex facilitates the phosphorylation of CRMP2 at Thr509 by GSK3β (Morinaka et al., 2011), indicating cross-talk between oxidation and phosphorylation in Sema3A-signaling.
Proteolysis—various brain injuries such as ischemia and glutamate toxicity induce calpain-mediated cleavage of all members of the CRMP family and produce approximately 54 kDa truncated products (Jiang et al., 2007). Calpain cleavage sites have been identified in CRMP3 and CRMP4. CRMP3 is cleaved at N-terminal Arg75-Leu76 bond and CRMP4 is at the C-terminal Arg550-Ser551 bond (Kowara et al., 2005; Hou et al., 2006). Processing of CRMP2 at the C-terminal region exposes nuclear localization signal (NLS) within residues Arg471-Lys472, which brings the truncated CRMP2 to the nucleus (Rogemond et al., 2008). Each truncated CRMP has a unique function. Truncated CRMP2 suppresses neurite outgrowth and reduces surface expression of the NR2B NMDA receptor subunit to protect neurons from glutamate toxicity (Bretin et al., 2006; Rogemond et al., 2008). Cleaved CRMP3 translocates to the nucleus and acts as a histone H4 deacetylase, which, in turn, induces neuronal cell death (Hou et al., 2006; Hou et al., 2013).
CRMP Interacting Molecules
CRMPs interact with membrane and intracellular proteins (Supplementary Table S3). Here, we classify those proteins into several categories including cytoskeletal proteins and ion channels.
Tubulin and microtubules—overexpression CRMP2 in hippocampal neurons switch neurite identity from dendrites to axons (Fukata et al., 2002; Yoshimura et al., 2005). This action is accomplished by two functions (Niwa et al., 2017). First, CRMP2 monomers bind to tubulin dimers and transport them to the distal end of axons to facilitate the neurite outgrowth. Second, CRMP2 tetramer stabilizes microtubules (Lin et al., 2011). These actions are canceled by the C-terminal phosphorylation of CRMP2 by GSK3β because phosphorylated CRMP2 loses its binding ability (Figure 2). It has been shown that Fer phosphorylates CRMP2 at Tyr479 and Tyr499. Crystal structures of the CRMP2-Tyr479Glu phospho-mimicking mutant revealed that the phosphorylation of Tyr479 interferes with the tetramerization of CRMP2 by introducing a negative charge in the hydrophobic cavity of the tetramer interface (Zheng et al., 2018). CRMP2 α-helix 19 (476–487) serves as a tubulin-dimer interface (Niwa et al., 2017). As CRMP2-tubulin dimer interaction is abrogated by the C-terminal phosphorylation of CRMP2 (Sumi et al., 2018), this modulation would also alter the higher structure of the C-terminal region and α-helix 19. However, such alteration of full-length CRMPs has not been revealed in a structural basis. CRMP3 inhibits tubulin polymerization and neurite outgrowth in cultured cerebellar granule neurons (Aylsworth et al., 2009). CRMP4 interacts with microtubules in hippocampal neurons to facilitate axon outgrowth (Khazaei et al., 2014). CRMP5 is phosphorylated by GSK3β at Thr516 (Brot et al., 2014). This phosphorylation is essential for tubulin-binding of CRMP5 but inhibits neurite outgrowth.
Actin—CRMP1 indirectly regulates actin-cytoskeleton through the interaction with actin-binding proteins including filamin-A and Ena/VASP proteins (Nakamura et al., 2014; Yu-Kemp et al., 2017). Interestingly, distinct roles were demonstrated for CRMP1 and CRMP2 in steering axonal outgrowth, using microscale-chromophore-assisted local inactivation (micro-CALI) method (Higurashi et al., 2012). CRMP1 and CRMP2 have characteristic distribution in the growth cones. CRMP1 was co-localized with actin in the peripheral domain, while CRMP2 was co-localized with tubulin in the central domain. It has been shown that CRMP4 binds F-actin and causes actin bundling (Rosslenbroich et al., 2005). This actin bundling is involved in the filopodia extension of hippocampal growth cones (Khazaei et al., 2014). CRMP5 interacts with actin and tubulin in growth cones (Gong et al., 2016). CRMP5 is localized in filopodia of growth cones and its overexpression promotes filopodial formation (Hotta et al., 2005).
Motor proteins—both Antero- and retro-grade motor proteins bind to CRMP2. Kinesin-1 binds to CRMP2 through the interaction of the kinesin light chain and the C-terminal region of CRMP2 (Kimura et al., 2005). In contrast, the N-terminus of CRMP2 binds to the dynein heavy chain but the interaction interferes with the retrograde transporting activity (Arimura et al., 2009). Combining these actions may facilitate the transport of the CRMP2-tubulin dimer complex to the distal end of the axons. The CRMP2 and Kinesin-1 complex also transports TrkB-containing vesicles to axonal plasma membranes (Arimura et al., 2009).
Na+ channel—CRMP1 interacts with NaV1.7 and modulates the Na+ currents by lowering the threshold of the channel (Yamane et al., 2017). This action is probably augmented by C-terminal phosphorylation because the overexpression of phosphomimetic CRMP1-Thr509Asp/Ser522Asp mutant enhanced the current. CRMP2 also interacts with NaV1.7 and phosphorylation of Thr509 and Ser522 of CRMP2 augments the action (Dustrude et al., 2013). This interaction is modulated by SUMOylation and phosphorylation of CRMP2. While Ser522 phosphorylation and Lys374 SUMOylation prevent NaV1.7 internalization, Tyr32-phosphorylation facilitates the endocytosis of NaV1.7 (Dustrude et al., 2016).
Ca2+ channel—CRMP2 binds to CaV2.2 and increases Ca2+ current by cell surface expression of the channel (Brittain et al., 2009). Cdk5-phosphorylation at Ser522 augments the interaction with and Ca2+ influx through CaV2.2 (Brittain et al., 2012). However, in contrast to NaV1.7 modulation, Lys374 SUMOylation of CRMP2 attenuates the Ca2+ current (Ju et al., 2013). Three domains in CRMP2, CBD1(94–166), CBD2(212–297), and CBD3(479–500), are involved in CaV2.2-binding (Brittain et al., 2009). The peptide fuses TAT (transduction domain of HIV), and CBD3 interferes with CRMP2 and CaV2.2 coupling to suppress inflammatory and neuropathic pain in model mice (Brittain et al., 2011). This effect is due to the reduction of membrane surface expression of CaV2.2. As the CBD3 sequence partially overlaps with α-helix 19 (476–487), a tubulin-dimer binding domain, TAT-CBD3 may also affect CRMP2-tubulin interaction. Also, CRMP2 interacts with NMDAR and Na/Ca Exchanger 3 to facilitate glutamate-induced Ca2+ dysregulation in hippocampal neurons (Brustovetsky et al., 2014). CRMP3 facilitates the depolarization-evoked Ca2+ response of L- and N-type Ca channels to promote dendrite morphogenesis of hippocampal neurons (Quach et al., 2013).
Other membrane and intracellular proteins—CRMP1 interacts with actin-binding protein Filamin-A. This interaction is augmented by the Cdk5-phosphorylation of CRMP1 (Nakamura et al., 2014). CRMP1-binding to Filamin-A decreases the F-actin gelation cross-linked by Filamin-A, which, in turn, facilitates remodeling of the actin cytoskeleton. CRMP1 also interacts with EVL, one of Ena/VASP proteins, to facilitate actin filament elongation at the barbed end (Yu-Kemp et al., 2017). CRMP2 interacts with Numb, one of the endocytosis adaptor proteins, to regulate endocytosis and recycling of axonal growth cone membranes (Nishimura et al., 2003). Long-form CRMP4, but not short-form, interacts with RhoA and suppresses neurite outgrowth (Alabed et al., 2007). GSK3β phosphorylates L-CRMP4 at Thr622 and attenuates RhoA-binding and cancels the suppression (Alabed et al., 2010). Myelin-associated inhibitors (MAIs) bring their inhibitory action through the inactivation of GSK3β and dephosphorylation of L-CRMP4 Thr622. Other known interactions are summarized in Supplementary Table S3.
Neuronal Subcellular Localization of CRMPs
Each CRMP has a different subcellular localization in a developmental-stage dependent manner. In primary cultured cortical neurons, CRMP1 and CRMP2 are predominantly expressed in axons and, to a lesser extent, in the somatodendritic regions at 7 days in vitro (DIV 7; Makihara et al., 2016). By DIV14, while CRMP1 tends to localize to the presynaptic region, CRMP2 distributes to the axons and dendrites.
Alternative splicing also contributes to localization. L-CRMP2 localizes to the distal axons of hippocampal neurons (Balastik et al., 2015). Studies in mice in which CRMP3 has been knocked-out revealed that CRMP3 regulates the dendritic growth of hippocampal neurons (Quach et al., 2013). CRMP4 distributes along axons and dendrites of hippocampal neurons and facilitates their growth through its interaction with actin and tubulin (Khazaei et al., 2014; Cha et al., 2016). CRMP5 is involved in the dendritic growth of hippocampal neurons (Yamashita et al., 2011). The effect of CRMP5 on axon outgrowth is inconsistent i.e., it can both variably promote and inhibit it (Brot et al., 2014; Gong et al., 2016); however, the inhibitory effect may reflect Thr516 phosphorylation of CRMP5 by GSK3β (Brot et al., 2014).
The neuronal growth cone consists of an actin-rich peripheral region and a tubulin-rich central domain. The peripheral region is further divided into filopodia and lamellipodia. CRMP1 localizes to both the peripheral and central domains, while CRMP2 localizes to the central domain in chick DRG growth cones (Higurashi et al., 2012). Focal inactivation of CRMP2 by micro-CALI support the role of CRMP2 in microtubule extension and stabilization. CRMP4 localizes to both the peripheral and central regions of hippocampal growth cones (Khazaei et al., 2014). Peripheral and central CRMP4 participate in filopodial F-actin bundling and microtubule elongation, respectively. CRMP5 predominantly localizes to the peripheral region of hippocampal growth cones to promote filopodial formation (Hotta et al., 2005).
Nuclear-translocation of calpain-digested CRMPs has already been discussed above under the “Proteolysis” section.
The Relation of CRMPs and Axon Guidance Molecules
Sema3A, a prototypical inhibitory axonal guidance molecule, regulates axonal projection of various neurons in the peripheral and central nervous system (CNS; Nakamura et al., 2000). It also participates in the dendritic growth and synapse formation and maturation (Goshima et al., 2016). The role of the CRMPs in Sema3A-signaling has been studied both in vitro and in vivo. Sema3A binds the NRP1 and Plexin-A receptor complex, which activates at least three distinct downstream signaling pathways; the phosphorylation cascade, the small-G-protein cascade, and MICAL-mediated oxidation. CRMPs are involved in all of these signaling pathways.
Sema3A activates Cdk5 and GSK3β kinases, which sequentially phosphorylates CRMP2 Ser522 and Thr509 residues, respectively. Phosphorylation of CRMP2 at Tyr32 by Fyn or Fes has also been shown to be involved in Sema3A-signaling (Yamashita et al., 2012; Figure 2). Phosphorylation makes CRMP2 alter its binding partners from tubulin-dimer to other downstream molecules such as CaV2.2 (Brittain et al., 2012). CRMP1 also acts on molecules downstream of Sema3A by being phosphorylated at Ser522 by Cdk5 (Yamashita et al., 2012). CRMP2 binds and activates α2-chimaerin, a Rac-GTPase activating protein, to downregulate Rac. This action is involved in the Sema3A response in DRG neurons (Brown et al., 2004). CRMP2 binds and activates MICAL by releasing its autoinhibition domain (Schmidt et al., 2008).
Sema3a-knockout mice demonstrate that Sema3A restricts peripheral neuron projections while facilitating the formation of dendrites and synapses in CNS. Sema3a−/− mice showed an overshooting and defasciculation of trigeminal branches and DRG neurons (Taniguchi et al., 1997). Crmp2−/− mice show CRMP2 to be involved in the peripheral projection of the trigeminal ophthalmic branches (Ziak et al., 2020). However, as the irregular peripheral projection of DRG axons is absent in Crmp2−/− mice, CRMP2 may mediate additional guidance cues such as ephrin-A5 and Slit2 in this projection (Jayasena et al., 2005; Kubilus and Linsenmayer, 2010).
The involvement of CRMPs in Sema3A-signaling is more evident in CNS development. Synapse formation of cortical pyramidal neurons is reduced in Sema3a−/−, Crmp2−/−, and Crmp2−/− mutants compared to wild-type (Makihara et al., 2016). Sema3a+/–; Crmp1+/– and Sema3a+/–; Crmp2+/– double heterozygous mice exhibit a similar phenotype, indicating that both CRMP1 and CRMP2 act as downstream molecules of Sema3A-regulated synapse formation. However, as Crmp1+/–; Crmp2+/– double heterozygous showed normal phenotype, CRMP1 and CRMP2 may mediate different intracellular signaling pathways.
Considering the obvious role of CRMPs in synapse maturation, it is noteworthy that the “lithium-response (LiR) pathway” governs the phosphorylation state of CRMP2, hence yielding insight into the pathogenesis of bipolar disorder (BPD; Tobe et al., 2017). Proteomic and phospho-proteomic analysis of human-induced pluripotent stem cells (hiPSCs) and their neuronal derivatives showed that the “set-point” for the ratio of inactive phosphorylated CRMP2 to active non-phosphorylated CRMP2 is elevated uniquely in LiR BPD patients, but not in patients with other psychiatric (including lithium-nonresponsive BPD) or neurological disorders. Lithium (and other CRMP2 pathway modulators) lowers pCRMP2, increasing dendritic spine area and density. Actual human BPD brains show similarly elevated CRMP2 ratios and diminished spine densities; lithium therapy normalizes the ratios and spines. Behaviorally, transgenic mice that reproduce lithium’s postulated site-of-action in dephosphorylating CRMP2 emulate lithium responsiveness in BPD.
The CRMPs also play roles as downstream target genes of signaling of other axon guidance molecules, morphogens, and cytokines in neuronal development and function. BMP-Smad1 signal suppresses CRMP2 gene expression (Sun et al., 2010). TGF-β signaling regulates neuronal morphogenesis through the suppression of CRMP2 expression during brain development (Nakashima et al., 2018). Nicotine administration causes gene up-regulation of CRMP2 in adult mice during nicotine-induced hippocampal long-term-potentiation (Kadoyama et al., 2015).
Sema3F binds Neuropilin-2 (NRP2)/Plexin-A complex and activates intracellular signaling like Sema3A. Sema3f and Nrp2 knockout mice showed axon guidance defects including disorganized anterior commissure and pruning defect of hippocampal mossy fiber axons in the infrapyramidal bundle (Chen et al., 2000; Sahay et al., 2003). Crmp2−/− mice exhibit reduction of the corpus callosum, hypoplastic anterior commissure, and defective pruning of CA3 infrapyramidal bundle and corticospinal visual axons (Ziak et al., 2020). As Crmp2−/− mice show normal pruning of the hippocampo-septal bundle, of which projection is regulated by Sema3A, CRMP2 is involved in Sema3F-regulated axon guidance rather than Sema3A-guidance.
Reelin regulates neuronal cell migration including neocortex six-layer formation and hippocampal lamination (Yamamoto et al., 2009). CRMP1 is involved in Reelin signaling (Yamashita et al., 2006). Radial migration of cortical neurons is delayed in Crmp2−/− brain. Dab1, an adaptor protein of Reelin signaling, is co-localized with CRMP1 in migrating neurons. Homozygous Dab1 yotari mutant mice, Dab1 yot/yot, exhibit disrupted hippocampal lamination. A similar phenotype is observed in Crmp2−/−; Dab1+/yot mice but not in Crmp2−/− or Dab1+/yot mutants. This genetic augmentation suggests that CRMP1 is involved in the Reelin-regulated neuronal cell migration.
Nogo, an inhibitory signal for neurite outgrowth after spinal cord injury (SCI), exerts the action through NgR1 and its associated transmembrane proteins. It has been shown that CRMP2 is involved in the downstream of Nogo-signaling by the Rho-kinase phosphorylation at Thr555 (Mimura et al., 2006; Petratos et al., 2012). As Nogo downstream signaling, L-CRMP4 (CRMP4b) binds to RhoA and interferes with neurite outgrowth (Alabed et al., 2007). NgR1 forms a receptor complex with Plexin-A2, which associates with CRMP2 and CRMP4 in Nogo dependent manner (Sekine et al., 2019).
Repulsive guidance molecule-a (RGMa) exerts its inhibition through the phosphorylation of CRMP2 by Rho-kinase and GSK3β (Wang et al., 2013). Chondroitin sulfate proteoglycan (CSPG) is a major inhibitor of axonal regeneration after CNS trauma. Knockout mice studies revealed that CRMP4 mediates the action of CSPG and/or other inhibitory molecules related to SCI (Nagai et al., 2015, 2016). The functional recovery of motor and sensory neurons from SCI is accelerated in Crmp4−/− mice. Sema4D binds Plexin-B1 and downregulates R-Ras and PI3K-Akt signaling. This brings the activation of GSK3β and CRMP2 phosphorylation at Thr514 (Ito et al., 2006). Plexin-A also inactivates R-Ras by the same mechanism in Sema3A-signaling. In vivo Sema4D-CRMP2 relation has yet to be examined.
CRMPs in CNS Regeneration and Degeneration
After nerve injury, peripheral nervous system (PNS) axons form growth cones and regenerate (Ertürk et al., 2007). However, axons in the CNS fail to form tips and instead become dystrophic retraction bulbs (Ertürk et al., 2007; Bradke et al., 2012). Growth cones contain organized microtubules that form tight bundles along with axonal axis, whereas retraction bulbs have disassembled microtubules. The importance of the microtubule state for growth cone structure is supported by the fact that the application of the microtubule-destabilizing agent nocodazole transforms the growth cone into a retraction bulb-like structure in vitro, resulting in axonal growth arrest (Ertürk et al., 2007).
Phosphorylation of CRMP2 at its C-terminal domain by serine/threonine (Ser/Thr) kinases causes growth cone collapse via microtubule destabilization, while inhibition of C-terminal phosphorylation stabilizes the microtubules (Yamashita and Goshima, 2012; Nagai et al., 2017). CRMP2 phosphorylation is also induced during Wallerian degeneration after optic nerve injury. During Wallerian degradation, zinc/RING finger protein 1 (ZNRF1)-dependent protein kinase B (Akt) degradation increases GSK3β activity and results in an increase in CRMP2 phosphorylation at Thr514 (Wakatsuki et al., 2011). The introduction of the CRMP2-Thr514Ala (CRMP2T514A) virus suppresses the Wallerian degradation of the optic nerve in vivo (Wakatsuki et al., 2011). To delineate the in vivo role of CRMP2 phosphorylation at Ser522, the CRMP2S522A/S522A (CRMP2KI/KI) mouse was generated (Yamashita et al., 2012). Due to the phosphorylation of Ser522 by Cdk5 functions, as priming phosphorylation followed GSK3β phosphorylation of Thr509/Thr514/Ser518, 4 sites of phosphorylation in the C-terminal domain of CRMP2 are eliminated in the CRMP2KI/KI mice (Yamashita et al., 2012). Wallerian degeneration following optic nerve injury was found to be suppressed in CRMP2KI/KI mice (Kinoshita et al., 2019). Axonal damage of the optic nerve induces retinal ganglion cell death in the wild-type mice (Duan et al., 2015). However, suppression of retinal ganglion cell death is observed in CRMP2KI/KI mice (Kondo et al., 2019). Moreover, axonal regeneration was promoted in CRMP2KI/KI mice. CRMP2KI/KI mice showed higher expression of the regenerative axonal marker growth-associated protein 43 (GAP43). Tracing of the axon by injecting an anterograde tracer into the eye showed elongated optic nerves in CRMP2KI/KI mice, while labeled axons were limited in wild-type mice after optic nerve injury. These results are in agreement with the observation that optic nerve regeneration occurs after the intravitreal administration of the CRMP2T514A virus (Leibinger et al., 2017). Overexpression of wild-type CRMP2 via a viral infection promoted regeneration of the hypoglossal nerve in the adult rats (Suzuki et al., 2003). In an SCI model, microtubule-stabilizing agents have been found to promote functional recovery and serotonergic axon regeneration (Hellal et al., 2011). CRMP2KI/KI mice also showed better functional, motor and sensory recovery, and serotonergic axon regeneration (Nagai et al., 2016). CRMP2KI/KI DRG neurons showed an enhanced neurotrophic response to brain-derived neurotrophic factor and a hindered inhibitory response against CSPG. These alternations, which were in response to external factors, may also be linked to the recovery in CRMP2KI/KI mice after SCI.
Recently, the mutant superoxide dismutase (SOD)1G93A mouse model of amyotrophic lateral sclerosis (ALS) was crossed with the CRMP2KI/KI mice to assess the genetic inhibition of CRMP2 phosphorylation. Compared to baseline, CRMP2KI/KI mice x SOD1G93A mice developed a slower degeneration of axons and neuromuscular junctions and a delayed progression of motor symptoms (Numata-Uematsu et al., 2019). The myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis (EAE) mouse model of multiple sclerosis (MS) showed an increase in CRMP2 phosphorylation at Thr555 in a Nogo-dependent manner (Petratos et al., 2012). The anti-Nogo-A antibody prevented the development of EAE and increased CRMP2 phosphorylation at Thr555. A recent study further demonstrated that abrogation of the NgR1/pCRMP2 signaling cascade maintains Kinesin-1-dependent anterograde axonal transport to limit the inflammation-mediated axonopathy and demyelination of the EAE model (Lee et al., 2019).
Methyl-phenyl-tetrahydropyridine (MPTP) readily penetrates the blood-brain barrier and enters the brain where it is converted into 1-methyl-4-phenylpyridinium (MPP+) by MAO-B in astrocytes. MPP+ is transported by the dopamine (DA) transporter into DA nerve terminals, and it destroys dopaminergic neurons, thereby causing the symptoms similar to those of Parkinson’s disease (PD). MPTP administration in non-human primates and aged rodents are often used to model PD. The elevation of CRMP2 phosphorylation at Thr514 through Akt/GSK3β was reported in an MPP+-PD model in dopaminergic neurons in vitro (Fang et al., 2015). Elevation of CRMP2 phosphorylation at Ser522 was observed in dopaminergic neurons in the substantia nigra compacta (SNc) in vivo (Togashi et al., 2019). Production of p25 and elevation of Cdk5 activity have been reported in an MPTP-induced PD mouse model (Smith et al., 2003; Cheung and Ip, 2012). Therefore, increased phosphorylation of CRMP2 at Ser522 is consistent with these studies. CRMP2KI/KI mice showed increased axonal viability in the nigro-striatal pathway in an MPTP-induced PD model (Togashi et al., 2019).
Another member of the CRMPs, CRMP4, is reported to be involved in the signal pathway of myelin-associated inhibitors (MAIs; Alabed et al., 2007, 2010; Nagai et al., 2015). MAIs activate ras homolog family member A (RhoA), which interacts with L-CRMP4 to inhibit axonal growth in vitro. CRMP4 also interacts with CSPG through the NgR/GSK3β pathway. After SCI, CRMP4−/− mice showed motor and sensory axonal growth (Nagai et al., 2015, 2016). Furthermore, the deletion of CRMP4 prevents DA neuronal loss in SNc and increases axonal viability in the DA neurons in the striatum (Tonouchi et al., 2016).
The Role of CRMPs in Inflammatory Cells and Glia
In SCIs, scar formation occurs at the lesion site where astrocytes are recruited by the pro-inflammatory cytokines secreted from the activated microglia/macrophages. The compaction and seclusion of infiltrating inflammatory cells in the lesion center occur during the sub-acute phase of SCI. It is generally accepted that glial scars inhibit axonal growth, by physically interacting with the distal tip of axons (Filous et al., 2014) or by secreting extracellular matrix molecules such as CSPGs (Tan et al., 2011; Burnside and Bradbury, 2014; Cregg et al., 2014) and inhibitory axon guidance molecules such as Sema3A (Kaneko et al., 2006).
A reduction in scar formation after SCI in CRMP2KI/KI mice has been reported (Nagai et al., 2015). In rat SCI, activation of GSK3β in reactive astrocytes is involved in the infiltration of inflammatory cells and scar formation by Wnt signaling-mediated β1-integrin expression (Renault-Mihara et al., 2011). Compaction of scar formation was observed in the GSK3β inhibitor treatment of SCI. Therefore, one possible explanation for the reduced scar formation in CRMP2KI/KI after SCI is that it is mediated by the inhibition of CRMP2 phosphorylation.
After SCI, CRMP4 expression is reported in activated microglia/macrophages (Nagai et al., 2015). In cultured BV-2 microglial cells, microglia express CRMP4 after lipopolysaccharide stimulation (Manivannan et al., 2013). CRMP4 is involved in the association of F-actin, cytokine release, migration, and phagocytosis in BV-2 cells. Injection of Zymosan, macrophage-activating agent, to the spinal cord of Crmp4−/− mice, showed reduced microglial activation (Nagai et al., 2015). In Crmp4−/− mice with SCIs, reduction of scar formation promoted axonal growth and significant locomotor recovery was observed.
Inflammatory responses have been implicated in other causes of neuronal degeneration, including PD. Disease progression has been linked to the secretion of inflammatory cytokines that engage neighboring cells, including astrocytes, which, in turn, induce autocrine and paracrine responses that amplify the inflammation, leading to further neurodegeneration (Niranjan, 2014). Reduced inflammatory response and suppressed DA neuron death after MPTP injection have been observed in CRMP4−/− mice (Tonouchi et al., 2016). Since cell death of DA neurons by MPTP injection was suppressed, the reduced inflammatory response may be a secondary consequence of the limited release of factors from dying neurons. MS is a chronic inflammatory, demyelinating, and neurodegenerative disorder of the CNS. In an EAE mouse model of MS, the importance of CRMP2 Ser522 phosphorylation was demonstrated using CRMP2KI/KI mice (Moutal et al., 2019). CRMP2 is also expressed in the immune system and plays a critical role in T lymphocyte polarization and migration (Vincent et al., 2005). C-X-C motif chemokine 12 (CXCL12)/SDF1 treatment activates T lymphocyte migration by switching the dephosphorylation of the GSK3β site (Thr509/Thr514) of CRMP2 (Varrin-Doyer et al., 2009). CXCL12/SDF1 also induces tyrosine phosphorylation at Tyr479 via Yes kinase, and the introduction of CRMP2-Tyr479Phe expression suppresses the migration of Jurkat cells, indicating the importance of tyrosine phosphorylation of CRMP2 (Varrin-Doyer et al., 2009).
CRMP2-Interacting Drugs
Several small molecules that have been shown to bind CRMP2 are lacosamide (Wilson et al., 2012, 2014), lanthionine ketimine (LK; Hensley et al., 2010a, 2013; Hensley and Harris-White, 2015), edonerpic maleate (Abe et al., 2018) and naringenin (Ghofrani et al., 2015; Lawal et al., 2018). These molecules are considered candidates for application in the treatment of some of the CRMP2-related pathological conditions discussed above.
(R)-Lacosamide inhibits CRMP2-mediated neurite outgrowth in cultured cortical neurons. (R)-lacosamide reduces CRMP2-mediated tubulin polymerization in vitro (Wilson et al., 2012). It seems to prevent posttraumatic axon sprouting in vivo. An in vitro study showed that (S)-lacosamide, a stereoisomer of the clinically used antiepileptic drug (R)-lacosamide, impairs the ability of CRMP2 to enhance tubulin polymerization in vitro without altering tubulin-binding (Wilson et al., 2014).
Lanthionine ketamine-ethyl ester (LKE) is a synthetic cell-penetrating ester derivative of LK, an endogenous sulfur amino acid metabolite in the mammalian brain (Hensley et al., 2010a). LK and LKE bind directly to CRMP2 and change the binding affinity of CRMP2 to its binding partners such as tubulin dimers and neurofilaments. LKE administration reduces CRMP2-tubulin affinity while enhancing CRMP2-neurofilament binding. The neurite growth-promoting action of LKE has been reported in NSC-34 mouse motor neuron-like cells and primary chick DRG neurons (Hensley et al., 2010a). LKE has also been shown to have a neuroprotective effect on these cells from oxidative stress insults. When applied to the SOD1G93A transgenic mouse model of ALS, LKE was reported to delay progressive neurodegeneration (Hensley et al., 2010b). LKE treatment also substantially diminished cognitive decline and brain amyloid-β (Aβ) peptide deposition and phospho-tau accumulation in the 3 × Tg-AD mouse model of AD, reducing the density of Iba1-positive microglia (Hensley et al., 2013). In this study, LKE normalized CRMP2 phosphorylation at Thr514 and suppressed neuroinflammation in the brains of these mice (Hensley et al., 2013). LKE has also been tested in a mouse model of SCI and was reported to benefit the recovery of motor function and reduce post-traumatic neuroinflammation (Kotaka et al., 2017).
Edonerpic maleate has been shown to bind CRMP2, and facilitate experience-driven synaptic glutamate α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic-acid (AMPA) receptor delivery and accelerate motor function recovery after motor cortex cryoinjury in mice in a training-dependent manner through cortical reorganization (Abe et al., 2018). Edonerpic maleate decreased the amount of phosphorylated form of CRMP2, and activated actin-depolymerizing factor (ADF)/cofilin, thereby leading to the trafficking of AMPA receptors into the spine surface under plasticity-inducing conditions. The drug failed to augment recovery in CRMP2-deficient mice, suggesting a CRMP2-dependent action (Abe et al., 2018).
Naringenin has demonstrated the ability to bind and decrease CRMP2 phosphorylation (Yang et al., 2016; Lawal et al., 2018). This action of changing CRMP2’s phosphorylated status and hence the binding of cytoskeletal elements may account for the ability of naringenin to improve AD pathology and cognitive deficits in mouse models of AD. For example, naringenin has been shown to significantly improve the performance of Aβ-injected rats in passive avoidance and radial arm maze tasks (Ghofrani et al., 2015). In a 5xFAD mouse model of AD, naringenin ameliorated memory deficits and decreased amyloid plaques and phosphorylated tau (p-tau; Yang et al., 2016). Some of the beneficial effects of naringenin in rodent models of AD may also be related to its anti-inflammatory actions (Park et al., 2012; Wu et al., 2016).
Conclusion
The CRMPs family of proteins appear to coordinately be involved in several coordinated biological events including axon guidance, target recognition, synapse maturation, and dendritic branching. The CRMPs possess the ability to interact with various kinds of molecules, thereby being involved in these many processes. Although the CRMPs themselves are regulated by versatile enzymes having a wide substrate spectrum, successful chemical modulators or therapeutic agents may be developed to act on such protein-protein interaction interfaces.
Author Contributions
FN, TO, and YG contributed to the writing and editing of this review.
Funding
FN is funded by a Grant-in-Aid for Scientific Research(C); (nos. 2450044, 16K07062). TO is funded by a Grant-in-Aid for Scientific Research(C); (no. 26430043). YG is funded by a Grants-in-Aid for Scientific Research in a priority Area (no. 17082006), Targeted Proteins Research Program (no. 0761890004), and by Global COE Program, Innovative Integration between Medicine and Engineering Based on Information Communication Technology (no. 1542140002), and Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems (no. 42890001) from the Ministry of Education, Science, Sports, and Culture.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We sincerely thank Drs. Yukio Sasaki, Naoya Yamashita, Kohtaro Takei, Evan Snyder, Kenneth Hensley, Ritsuko Ohtani-Kaneko, Hiroshi Kiyonari, Go Shioi, Pappachan Kolattukudy and Jerome Honnorat for collaboration. We also thank Dr. EY Snyder for his critical reading of our manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2020.00188/full#supplementary-material.
References
Abe, H., Jitsuki, S., Nakajima, W., Murata, Y., Jitsuki-Takahashi, A., Katsuno, Y., et al. (2018). CRMP2-binding compound, edonerpic maleate, accelerates motor function recovery from brain damage. Science 360, 50–57. doi: 10.1126/science.aao2300
Alabed, Y. Z., Pool, M., Ong Tone, S., and Fournier, A. E. (2007). Identification of CRMP4 as a convergent regulator of axon outgrowth inhibition. J. Neurosci. 27, 1702–1711. doi: 10.1523/jneurosci.5055-06.2007
Alabed, Y. Z., Pool, M., Ong Tone, S., Sutherland, C., and Fournier, A. E. (2010). GSK3 β regulates myelin-dependent axon outgrowth inhibition through CRMP4. J. Neurosci. 30, 5635–5643. doi: 10.1523/jneurosci.6154-09.2010
Arimura, N., Inagaki, N., Chihara, K., Menager, C., Nakamura, N., Amano, M., et al. (2000). Phosphorylation of collapsin response mediator protein-2 by Rho-kinase. Evidence for two separate signaling pathways for growth cone collapse. J. Biol. Chem. 275, 23973–23980. doi: 10.1074/jbc.m001032200
Arimura, N., Kimura, T., Nakamuta, S., Taya, S., Funahashi, Y., Hattori, A., et al. (2009). Anterograde transport of TrkB in axons is mediated by direct interaction with Slp1 and Rab27. Dev. Cell 16, 675–686. doi: 10.1016/j.devcel.2009.03.005
Arimura, N., Menager, C., Kawano, Y., Yoshimura, T., Kawabata, S., Hattori, A., et al. (2005). Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol. Cell. Biol. 25, 9973–9984. doi: 10.1128/mcb.25.22.9973-9984.2005
Aylsworth, A., Jiang, S. X., Desbois, A., and Hou, S. T. (2009). Characterization of the role of full-length CRMP3 and its calpain-cleaved product in inhibiting microtubule polymerization and neurite outgrowth. Exp. Cell Res. 315, 2856–2868. doi: 10.1016/j.yexcr.2009.06.014
Balastik, M., Zhou, X. Z., Alberich-Jorda, M., Weissova, R., Ziak, J., Pazyra-Murphy, M. F., et al. (2015). Prolyl isomerase pin1 regulates axon guidance by stabilizing CRMP2A selectively in distal axons. Cell Rep. 13, 812–828. doi: 10.1016/j.celrep.2015.09.026
Boudreau, A. C., Ferrario, C. R., Glucksman, M. J., and Wolf, M. E. (2009). Signaling pathway adaptations and novel protein kinase A substrates related to behavioral sensitization to cocaine. J. Neurochem. 110, 363–377. doi: 10.1111/j.1471-4159.2009.06140.x
Bradke, F., Fawcett, J. W., and Spira, M. E. (2012). Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat. Rev. Neurosci. 13, 183–193. doi: 10.1038/nrn3176
Bretin, S., Rogemond, V., Marin, P., Maus, M., Torrens, Y., Honnorat, J., et al. (2006). Calpain product of WT-CRMP2 reduces the amount of surface NR2B NMDA receptor subunit. J. Neurochem. 98, 1252–1265. doi: 10.1111/j.1471-4159.2006.03969.x
Brittain, J. M., Duarte, D. B., Wilson, S. M., Zhu, W., Ballard, C., Johnson, P. L., et al. (2011). Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nat. Med. 17, 822–829. doi: 10.1038/nm.2345
Brittain, J. M., Piekarz, A. D., Wang, Y., Kondo, T., Cummins, T. R., and Khanna, R. (2009). An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J. Biol. Chem. 284, 31375–31390. doi: 10.1074/jbc.m109.009951
Brittain, J. M., Wang, Y., Eruvwetere, O., and Khanna, R. (2012). Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2.2. FEBS Lett. 586, 3813–3818. doi: 10.1016/j.febslet.2012.09.022
Brot, S., Smaoune, H., Youssef-Issa, M., Malleval, C., Benetollo, C., Besancon, R., et al. (2014). Collapsin response-mediator protein 5 (CRMP5) phosphorylation at threonine 516 regulates neurite outgrowth inhibition. Eur. J. Neurosci. 40, 3010–3020. doi: 10.1111/ejn.12674
Brown, M., Jacobs, T., Eickholt, B., Ferrari, G., Teo, M., Monfries, C., et al. (2004). α2-chimaerin, cyclin-dependent Kinase 5/p35 and its target collapsin response mediator protein-2 are essential components in semaphorin 3A-induced growth-cone collapse. J. Neurosci. 24, 8994–9004. doi: 10.1523/jneurosci.3184-04.2004
Brustovetsky, T., Pellman, J. J., Yang, X. F., Khanna, R., and Brustovetsky, N. (2014). Collapsin response mediator protein 2 (CRMP2) interacts with N-methyl-D-aspartate (NMDA) receptor and Na+/Ca2+ exchanger and regulates their functional activity. J. Biol. Chem. 289, 7470–7482. doi: 10.1074/jbc.m113.518472
Buel, G. R., Rush, J., and Ballif, B. A. (2010). Fyn promotes phosphorylation of collapsin response mediator protein 1 at tyrosine 504, a novel, isoform-specific regulatory site. J. Cell. Biochem. 111, 20–28. doi: 10.1002/jcb.22659
Burnside, E. R., and Bradbury, E. J. (2014). Manipulating the extracellular matrix and its role in brain and spinal cord plasticity and repair. Neuropathol. Appl. Neurobiol. 40, 26–59. doi: 10.1111/nan.12114
Cha, C., Zhang, J., Ji, Z., Tan, M., Li, S., Wu, F., et al. (2016). CRMP4 regulates dendritic growth and maturation via the interaction with actin cytoskeleton in cultured hippocampal neurons. Brain Res. Bull. 124, 286–294. doi: 10.1016/j.brainresbull.2016.06.008
Chen, H., Bagri, A., Zupicich, J. A., Zou, Y., Stoeckli, E., Pleasure, S. J., et al. (2000). Neuropilin-2 regulates the development of selective cranial and sensory nerves and hippocampal mossy fiber projections. Neuron 25, 43–56. doi: 10.1016/s0896-6273(00)80870-3
Cheung, Z. H., and Ip, N. Y. (2012). Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 22, 169–175. doi: 10.1016/j.tcb.2011.11.003
Cole, A. R., Causeret, F., Yadirgi, G., Hastie, C. J., Mclauchlan, H., Mcmanus, E. J., et al. (2006). Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. J. Biol. Chem. 281, 16591–16598. doi: 10.1074/jbc.m513344200
Cregg, J. M., Depaul, M. A., Filous, A. R., Lang, B. T., Tran, A., and Silver, J. (2014). Functional regeneration beyond the glial scar. Exp. Neurol. 253, 197–207. doi: 10.1016/j.expneurol.2013.12.024
Deo, R. C., Schmidt, E. F., Elhabazi, A., Togashi, H., Burley, S. K., and Strittmatter, S. M. (2004). Structural bases for CRMP function in plexin-dependent semaphorin3A signaling. EMBO J. 23, 9–22. doi: 10.1038/sj.emboj.7600021
Duan, X., Qiao, M., Bei, F., Kim, I. J., He, Z., and Sanes, J. R. (2015). Subtype-specific regeneration of retinal ganglion cells following axotomy: effects of osteopontin and mTOR signaling. Neuron 85, 1244–1256. doi: 10.1016/j.neuron.2015.02.017
Dustrude, E. T., Moutal, A., Yang, X., Wang, Y., Khanna, M., and Khanna, R. (2016). Hierarchical CRMP2 posttranslational modifications control NaV1.7 function. Proc. Natl. Acad. Sci. U S A 113, E8443–E8452. doi: 10.1073/pnas.1610531113
Dustrude, E. T., Wilson, S. M., Ju, W., Xiao, Y., and Khanna, R. (2013). CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. J. Biol. Chem. 288, 24316–24331. doi: 10.1074/jbc.m113.474924
Ertürk, A., Hellal, F., Enes, J., and Bradke, F. (2007). Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J. Neurosci. 27, 9169–9180. doi: 10.1523/JNEUROSCI.0612-07.2007
Fang, W., Gao, G., Zhao, H., Xia, Y., Guo, X., Li, N., et al. (2015). Role of the Akt/GSK-3β/CRMP-2 pathway in axon degeneration of dopaminergic neurons resulting from MPP+ toxicity. Brain Res. 1602, 9–19. doi: 10.1016/j.brainres.2014.08.030
Filous, A. R., Tran, A., Howell, C. J., Busch, S. A., Evans, T. A., Stallcup, W. B., et al. (2014). Entrapment via synaptic-like connections between NG2 proteoglycan+ cells and dystrophic axons in the lesion plays a role in regeneration failure after spinal cord injury. J. Neurosci. 34, 16369–16384. doi: 10.1523/jneurosci.1309-14.2014
Fukata, Y., Itoh, T. J., Kimura, T., Menager, C., Nishimura, T., Shiromizu, T., et al. (2002). CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 4, 583–591. doi: 10.1038/ncb825
Ghofrani, S., Joghataei, M. T., Mohseni, S., Baluchnejadmojarad, T., Bagheri, M., Khamse, S., et al. (2015). Naringenin improves learning and memory in an Alzheimer’s disease rat model: insights into the underlying mechanisms. Eur. J. Pharmacol. 764, 195–201. doi: 10.1016/j.ejphar.2015.07.001
Gojkovic, Z., Rislund, L., Andersen, B., Sandrini, M. P., Cook, P. F., Schnackerz, K. D., et al. (2003). Dihydropyrimidine amidohydrolases and dihydroorotases share the same origin and several enzymatic properties. Nucleic Acids Res. 31, 1683–1692. doi: 10.1093/nar/gkg258
Gong, X., Tan, M., Gao, Y., Chen, K., and Guo, G. (2016). CRMP5 interacts with actin to regulate neurite outgrowth. Mol. Med. Rep. 13, 1179–1185. doi: 10.3892/mmr.2015.4662
Goshima, Y., Nakamura, F., Strittmatter, P., and Strittmatter, S. M. (1995). Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature 376, 509–514. doi: 10.1038/376509a0
Goshima, Y., Yamashita, N., Nakamura, F., and Sasaki, Y. (2016). Regulation of dendritic development by semaphorin 3A through novel intracellular remote signaling. Cell Adh. Migr. 10, 627–640. doi: 10.1080/19336918.2016.1210758
Gu, Y., and Ihara, Y. (2000). Evidence that collapsin response mediator protein-2 is involved in the dynamics of microtubules. J. Biol. Chem. 275, 17917–17920. doi: 10.1074/jbc.c000179200
Hellal, F., Hurtado, A., Ruschel, J., Flynn, K. C., Laskowski, C. J., Umlauf, M., et al. (2011). Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 331, 928–931. doi: 10.1126/science.1201148
Hensley, K., Christov, A., Kamat, S., Zhang, X. C., Jackson, K. W., Snow, S., et al. (2010a). Proteomic identification of binding partners for the brain metabolite lanthionine ketimine (LK) and documentation of LK effects on microglia and motoneuron cell cultures. J. Neurosci. 30, 2979–2988. doi: 10.1523/JNEUROSCI.5247-09.2010
Hensley, K., Venkova, K., and Christov, A. (2010b). Emerging biological importance of central nervous system lanthionines. Molecules 15, 5581–5594. doi: 10.3390/molecules15085581
Hensley, K., Gabbita, S. P., Venkova, K., Hristov, A., Johnson, M. F., Eslami, P., et al. (2013). A derivative of the brain metabolite lanthionine ketimine improves cognition and diminishes pathology in the 3 x Tg-AD mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol. 72, 955–969. doi: 10.1097/nen.0b013e3182a74372
Hensley, K., and Harris-White, M. E. (2015). Redox regulation of autophagy in healthy brain and neurodegeneration. Neurobiol. Dis. 84, 50–59. doi: 10.1016/j.nbd.2015.03.002
Hensley, K., and Kursula, P. (2016). Collapsin response mediator protein-2 (CRMP2) is a plausible etiological factor and potential therapeutic target in Alzheimer’s disease: comparison and contrast with microtubule-associated protein tau. J. Alzheimers Dis. 53, 1–14. doi: 10.3233/jad-160076
Higurashi, M., Iketani, M., Takei, K., Yamashita, N., Aoki, R., Kawahara, N., et al. (2012). Localized role of CRMP1 and CRMP2 in neurite outgrowth and growth cone steering. Dev. Neurobiol. 72, 1528–1540. doi: 10.1002/dneu.22017
Hotta, A., Inatome, R., Yuasa-Kawada, J., Qin, Q., Yamamura, H., and Yanagi, S. (2005). Critical role of collapsin response mediator protein-associated molecule CRAM for filopodia and growth cone development in neurons. Mol. Biol. Cell 16, 32–39. doi: 10.1091/mbc.e04-08-0679
Hou, S. T., Jiang, S. X., Aylsworth, A., Cooke, M., and Zhou, L. (2013). Collapsin response mediator protein 3 deacetylates histone H4 to mediate nuclear condensation and neuronal death. Sci. Rep. 3:1350. doi: 10.1038/srep01350
Hou, S. T., Jiang, S. X., Aylsworth, A., Ferguson, G., Slinn, J., Hu, H., et al. (2009). CaMKII phosphorylates collapsin response mediator protein 2 and modulates axonal damage during glutamate excitotoxicity. J. Neurochem. 111, 870–881. doi: 10.1111/j.1471-4159.2009.06375.x
Hou, S. T., Jiang, S. X., Desbois, A., Huang, D., Kelly, J., Tessier, L., et al. (2006). Calpain-cleaved collapsin response mediator protein-3 induces neuronal death after glutamate toxicity and cerebral ischemia. J. Neurosci. 26, 2241–2249. doi: 10.1523/JNEUROSCI.4485-05.2006
Ito, Y., Oinuma, I., Katoh, H., Kaibuchi, K., and Negishi, M. (2006). Sema4D/plexin-B1 activates GSK-3β through R-Ras GAP activity, inducing growth cone collapse. EMBO Rep. 7, 704–709. doi: 10.1038/sj.embor.7400737
Jayasena, C. S., Flood, W. D., and Koblar, S. A. (2005). High EphA3 expressing ophthalmic trigeminal sensory axons are sensitive to ephrin-A5-Fc: implications for lobe specific axon guidance. Neuroscience 135, 97–109. doi: 10.1016/j.neuroscience.2005.05.052
Jiang, S. X., Kappler, J., Zurakowski, B., Desbois, A., Aylsworth, A., and Hou, S. T. (2007). Calpain cleavage of collapsin response mediator proteins in ischemic mouse brain. Eur. J. Neurosci. 26, 801–809. doi: 10.1111/j.1460-9568.2007.05715.x
Ju, W., Li, Q., Wilson, S. M., Brittain, J. M., Meroueh, L., and Khanna, R. (2013). SUMOylation alters CRMP2 regulation of calcium influx in sensory neurons. Channels 7, 153–159. doi: 10.4161/chan.24224
Kadoyama, K., Matsuura, K., Nakamura-Hirota, T., Takano, M., Otani, M., and Matsuyama, S. (2015). Changes in the expression of collapsin response mediator protein-2 during synaptic plasticity in the mouse hippocampus. J. Neurosci. Res. 93, 1684–1692. doi: 10.1002/jnr.23626
Kaneko, S., Iwanami, A., Nakamura, M., Kishino, A., Kikuchi, K., Shibata, S., et al. (2006). A selective Sema3A inhibitor enhances regenerative responses and functional recovery of the injured spinal cord. Nat. Med. 12, 1380–1389. doi: 10.1038/nm1505
Khazaei, M. R., Girouard, M. P., Alchini, R., Ong Tone, S., Shimada, T., Bechstedt, S., et al. (2014). Collapsin response mediator protein 4 regulates growth cone dynamics through the actin and microtubule cytoskeleton. J. Biol. Chem. 289, 30133–30143. doi: 10.1074/jbc.m114.570440
Kimura, T., Watanabe, H., Iwamatsu, A., and Kaibuchi, K. (2005). Tubulin and CRMP-2 complex is transported via Kinesin-1. J. Neurochem. 93, 1371–1382. doi: 10.1111/j.1471-4159.2005.03063.x
Kinoshita, Y., Kondo, S., Takahashi, K., Nagai, J., Wakatsuki, S., Araki, T., et al. (2019). Genetic inhibition of CRMP2 phosphorylation delays Wallerian degeneration after optic nerve injury. Biochem. Biophys. Res. Commun. 514, 1037–1039. doi: 10.1016/j.bbrc.2019.05.060
Kondo, S., Takahashi, K., Kinoshita, Y., Nagai, J., Wakatsuki, S., Araki, T., et al. (2019). Genetic inhibition of CRMP2 phosphorylation at serine 522 promotes axonal regeneration after optic nerve injury. Sci. Rep. 9:7188. doi: 10.1038/s41598-019-43658-w
Kotaka, K., Nagai, J., Hensley, K., and Ohshima, T. (2017). Lanthionine ketimine ester promotes locomotor recovery after spinal cord injury by reducing neuroinflammation and promoting axon growth. Biochem. Biophys. Res. Commun. 483, 759–764. doi: 10.1016/j.bbrc.2016.12.069
Kowara, R., Chen, Q., Milliken, M., and Chakravarthy, B. (2005). Calpain-mediated truncation of dihydropyrimidinase-like 3 protein (DPYSL3) in response to NMDA and H2O2 toxicity. J. Neurochem. 95, 466–474. doi: 10.1111/j.1471-4159.2005.03383.x
Kubilus, J. K., and Linsenmayer, T. F. (2010). Developmental corneal innervation: interactions between nerves and specialized apical corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 51, 782–789. doi: 10.1167/iovs.09-3942
Lawal, M. F., Olotu, F. A., Agoni, C., and Soliman, M. E. (2018). Exploring the C-terminal tail dynamics: structural and molecular perspectives into the therapeutic activities of novel CRMP-2 inhibitors, naringenin and naringenin-7-O-glucuronide, in the treatment of Alzheimer’s disease. Chem. Biodivers 15:e1800437. doi: 10.1002/cbdv.201800437
Lee, J. Y., Kim, M. J., Thomas, S., Oorschot, V., Ramm, G., Aui, P. M., et al. (2019). Limiting neuronal nogo receptor 1 signaling during experimental autoimmune encephalomyelitis preserves axonal transport and abrogates inflammatory demyelination. J. Neurosci. 39, 5562–5580. doi: 10.1523/JNEUROSCI.1760-18.2019
Leibinger, M., Andreadaki, A., Golla, R., Levin, E., Hilla, A. M., Diekmann, H., et al. (2017). Boosting CNS axon regeneration by harnessing antagonistic effects of GSK3 activity. Proc. Natl. Acad. Sci. U S A 114, E5454–E5463. doi: 10.1073/pnas.1621225114
Leung, T., Ng, Y., Cheong, A., Ng, C. H., Tan, I., Hall, C., et al. (2002). p80 ROKα binding protein is a novel splice variant of CRMP-1 which associates with CRMP-2 and modulates RhoA-induced neuronal morphology. FEBS Lett. 532, 445–449. doi: 10.1016/s0014-5793(02)03736-5
Lin, P. C., Chan, P. M., Hall, C., and Manser, E. (2011). Collapsin response mediator proteins (CRMPs) are a new class of microtubule-associated protein (MAP) that selectively interacts with assembled microtubules via a taxol-sensitive binding interaction. J. Biol. Chem. 286, 41466–41478. doi: 10.1074/jbc.m111.283580
Makihara, H., Nakai, S., Ohkubo, W., Yamashita, N., Nakamura, F., Kiyonari, H., et al. (2016). CRMP1 and CRMP2 have synergistic but distinct roles in dendritic development. Genes Cells 21, 994–1005. doi: 10.1111/gtc.12399
Manivannan, J., Tay, S. S., Ling, E. A., and Dheen, S. T. (2013). Dihydropyrimidinase-like 3 regulates the inflammatory response of activated microglia. Neuroscience 253, 40–54. doi: 10.1016/j.neuroscience.2013.08.023
Mimura, F., Yamagishi, S., Arimura, N., Fujitani, M., Kubo, T., Kaibuchi, K., et al. (2006). Myelin-associated glycoprotein inhibits microtubule assembly by a Rho-kinase-dependent mechanism. J. Biol. Chem. 281, 15970–15979. doi: 10.1074/jbc.m510934200
Morinaka, A., Yamada, M., Itofusa, R., Funato, Y., Yoshimura, Y., Nakamura, F., et al. (2011). Thioredoxin mediates oxidation-dependent phosphorylation of CRMP2 and growth cone collapse. Sci. Signal. 4:ra26. doi: 10.1126/scisignal.2001127
Moutal, A., Kalinin, S., Kowal, K., Marangoni, N., Dupree, J., Lin, S. X., et al. (2019). Neuronal conditional knockout of collapsin response mediator protein 2 ameliorates disease severity in a mouse model of multiple sclerosis. ASN Neuro 11:1759091419892090. doi: 10.1177/1759091419892090
Nagai, J., Baba, R., and Ohshima, T. (2017). CRMPs function in neurons and glial cells: potential therapeutic targets for neurodegenerative diseases and CNS injury. Mol. Neurobiol. 54, 4243–4256. doi: 10.1007/s12035-016-0005-1
Nagai, J., Kitamura, Y., Owada, K., Yamashita, N., Takei, K., Goshima, Y., et al. (2015). Crmp4 deletion promotes recovery from spinal cord injury by neuroprotection and limited scar formation. Sci. Rep. 5:8269. doi: 10.1038/srep08269
Nagai, J., Takaya, R., Piao, W., Goshima, Y., and Ohshima, T. (2016). Deletion of Crmp4 attenuates CSPG-induced inhibition of axonal growth and induces nociceptive recovery after spinal cord injury. Mol. Cell. Neurosci. 74, 42–48. doi: 10.1016/j.mcn.2016.03.006
Nakamura, F., Kalb, R. G., and Strittmatter, S. M. (2000). Molecular basis of semaphorin-mediated axon guidance. J. Neurobiol. 44, 219–229. doi: 10.1002/1097-4695(200008)44:2<219::aid-neu11>3.0.co;2-w
Nakamura, F., Kumeta, K., Hida, T., Isono, T., Nakayama, Y., Kuramata-Matsuoka, E., et al. (2014). Amino- and carboxyl-terminal domains of Filamin-A interact with CRMP1 to mediate Sema3A signalling. Nat. Commun. 5:5325. doi: 10.1038/ncomms6325
Nakashima, H., Tsujimura, K., Irie, K., Ishizu, M., Pan, M., Kameda, T., et al. (2018). Canonical TGF-β signaling negatively regulates neuronal morphogenesis through TGIF/smad complex-mediated CRMP2 suppression. J. Neurosci. 38, 4791–4810. doi: 10.1523/JNEUROSCI.2423-17.2018
Niranjan, R. (2014). The role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson’s disease: focus on astrocytes. Mol. Neurobiol. 49, 28–38. doi: 10.1007/s12035-013-8483-x
Nishimura, T., Fukata, Y., Kato, K., Yamaguchi, T., Matsuura, Y., Kamiguchi, H., et al. (2003). CRMP-2 regulates polarized Numb-mediated endocytosis for axon growth. Nat. Cell Biol. 5, 819–826. doi: 10.1038/ncb1039
Niwa, S., Nakamura, F., Tomabechi, Y., Aoki, M., Shigematsu, H., Matsumoto, T., et al. (2017). Structural basis for CRMP2-induced axonal microtubule formation. Sci. Rep. 7:10681. doi: 10.1038/s41598-017-11031-4
Numata-Uematsu, Y., Wakatsuki, S., Nagano, S., Shibata, M., Sakai, K., Ichinohe, N., et al. (2019). Inhibition of collapsin response mediator protein-2 phosphorylation ameliorates motor phenotype of ALS model mice expressing SOD1G93A. Neurosci. Res. 139, 63–68. doi: 10.1016/j.neures.2018.08.016
Pan, S. H., Chao, Y. C., Hung, P. F., Chen, H. Y., Yang, S. C., Chang, Y. L., et al. (2011). The ability of LCRMP-1 to promote cancer invasion by enhancing filopodia formation is antagonized by CRMP-1. J. Clin. Invest. 121, 3189–3205. doi: 10.1172/jci42975
Park, H. Y., Kim, G. Y., and Choi, Y. H. (2012). Naringenin attenuates the release of pro-inflammatory mediators from lipopolysaccharide-stimulated BV2 microglia by inactivating nuclear factor-kappaB and inhibiting mitogen-activated protein kinases. Int. J. Mol. Med. 30, 204–210. doi: 10.3892/ijmm.2012.979
Petratos, S., Ozturk, E., Azari, M. F., Kenny, R., Lee, J. Y., Magee, K. A., et al. (2012). Limiting multiple sclerosis related axonopathy by blocking Nogo receptor and CRMP-2 phosphorylation. Brain 135, 1794–1818. doi: 10.1093/brain/aws100
Ponnusamy, R., Lebedev, A. A., Pahlow, S., and Lohkamp, B. (2014). Crystal structure of human CRMP-4: correction of intensities for lattice-translocation disorder. Acta Crystallogr. D Biol. Crystallogr. 70, 1680–1694. doi: 10.1107/S1399004714006634
Ponnusamy, R., and Lohkamp, B. (2013). Insights into the oligomerization of CRMPs: crystal structure of human collapsin response mediator protein 5. J. Neurochem. 125, 855–868. doi: 10.1111/jnc.12188
Quach, T. T., Honnorat, J., Kolattukudy, P. E., Khanna, R., and Duchemin, A. M. (2015). CRMPs: critical molecules for neurite morphogenesis and neuropsychiatric diseases. Mol. Psychiatry 20, 1037–1045. doi: 10.1038/mp.2015.77
Quach, T. T., Wilson, S. M., Rogemond, V., Chounlamountri, N., Kolattukudy, P. E., Martinez, S., et al. (2013). Mapping CRMP3 domains involved in dendrite morphogenesis and voltage-gated calcium channel regulation. J. Cell Sci. 126, 4262–4273. doi: 10.1242/jcs.131409
Renault-Mihara, F., Katoh, H., Ikegami, T., Iwanami, A., Mukaino, M., Yasuda, A., et al. (2011). Beneficial compaction of spinal cord lesion by migrating astrocytes through glycogen synthase kinase-3 inhibition. EMBO Mol. Med. 3, 682–696. doi: 10.1002/emmm.201100179
Rogemond, V., Auger, C., Giraudon, P., Becchi, M., Auvergnon, N., Belin, M. F., et al. (2008). Processing and nuclear localization of CRMP2 during brain development induce neurite outgrowth inhibition. J. Biol. Chem. 283, 14751–14761. doi: 10.1074/jbc.m708480200
Rosslenbroich, V., Dai, L., Baader, S. L., Noegel, A. A., Gieselmann, V., and Kappler, J. (2005). Collapsin response mediator protein-4 regulates F-actin bundling. Exp. Cell Res. 310, 434–444. doi: 10.1016/j.yexcr.2005.08.005
Sahay, A., Molliver, M. E., Ginty, D. D., and Kolodkin, A. L. (2003). Semaphorin 3F is critical for development of limbic system circuitry and is required in neurons for selective CNS axon guidance events. J. Neurosci. 23, 6671–6680. doi: 10.1523/JNEUROSCI.23-17-06671.2003
Schmidt, E. F., Shim, S. O., and Strittmatter, S. M. (2008). Release of MICAL autoinhibition by semaphorin-plexin signaling promotes interaction with collapsin response mediator protein. J. Neurosci. 28, 2287–2297. doi: 10.1523/JNEUROSCI.5646-07.2008
Schmidt, E. F., and Strittmatter, S. M. (2007). The CRMP family of proteins and their role in Sema3A signaling. Adv. Exp. Med. Biol. 600, 1–11. doi: 10.1007/978-0-387-70956-7_1
Sekine, Y., Algarate, P. T., Cafferty, W. B. J., and Strittmatter, S. M. (2019). Plexina2 and CRMP2 signaling complex is activated by Nogo-A-liganded Ngr1 to restrict corticospinal axon sprouting after trauma. J. Neurosci. 39, 3204–3216. doi: 10.1523/JNEUROSCI.2996-18.2019
Smith, P. D., Crocker, S. J., Jackson-Lewis, V., Jordan-Sciutto, K. L., Hayley, S., Mount, M. P., et al. (2003). Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U S A 100, 13650–13655. doi: 10.1073/pnas.2232515100
Stenmark, P., Ogg, D., Flodin, S., Flores, A., Kotenyova, T., Nyman, T., et al. (2007). The structure of human collapsin response mediator protein 2, a regulator of axonal growth. J. Neurochem. 101, 906–917. doi: 10.1111/j.1471-4159.2006.04401.x
Sumi, T., Imasaki, T., Aoki, M., Sakai, N., Nitta, E., Shirouzu, M., et al. (2018). Structural insights into the altering function of CRMP2 by phosphorylation. Cell Struct. Funct. 43, 15–23. doi: 10.1247/csf.17025
Sun, Y., Fei, T., Yang, T., Zhang, F., Chen, Y. G., Li, H., et al. (2010). The suppression of CRMP2 expression by bone morphogenetic protein (BMP)-SMAD gradient signaling controls multiple stages of neuronal development. J. Biol. Chem. 285, 39039–39050. doi: 10.1074/jbc.m110.168351
Suzuki, Y., Nakagomi, S., Namikawa, K., Kiryu-Seo, S., Inagaki, N., Kaibuchi, K., et al. (2003). Collapsin response mediator protein-2 accelerates axon regeneration of nerve-injured motor neurons of rat. J. Neurochem. 86, 1042–1050. doi: 10.1046/j.1471-4159.2003.01920.x
Tan, C. L., Kwok, J. C., Patani, R., Ffrench-Constant, C., Chandran, S., and Fawcett, J. W. (2011). Integrin activation promotes axon growth on inhibitory chondroitin sulfate proteoglycans by enhancing integrin signaling. J. Neurosci. 31, 6289–6295. doi: 10.1523/JNEUROSCI.0008-11.2011
Taniguchi, M., Yuasa, S., Fujisawa, H., Naruse, I., Saga, S., Mishina, M., et al. (1997). Disruption of semaphorin III/D gene causes severe abnormality in peripheral nerve projection. Neuron 19, 519–530. doi: 10.1016/s0896-6273(00)80368-2
Terman, J. R., Mao, T., Pasterkamp, R. J., Yu, H. H., and Kolodkin, A. L. (2002). MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell 109, 887–900. doi: 10.1016/s0092-8674(02)00794-8
Tobe, B. T. D., Crain, A. M., Winquist, A. M., Calabrese, B., Makihara, H., Zhao, W. N., et al. (2017). Probing the lithium-response pathway in hiPSCs implicates the phosphoregulatory set-point for a cytoskeletal modulator in bipolar pathogenesis. Proc. Natl. Acad. Sci. U S A 114, E4462–E4471. doi: 10.1073/pnas.1700111114
Togashi, K., Hasegawa, M., Nagai, J., Tonouchi, A., Masukawa, D., Hensley, K., et al. (2019). Genetic suppression of collapsin response mediator protein 2 phosphorylation improves outcome in methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s model mice. Genes Cells 24, 31–40. doi: 10.1111/gtc.12651
Tonouchi, A., Nagai, J., Togashi, K., Goshima, Y., and Ohshima, T. (2016). Loss of collapsin response mediator protein 4 suppresses dopaminergic neuron death in an 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse model of Parkinson’s disease. J. Neurochem. 137, 795–805. doi: 10.1111/jnc.13617
Tsutiya, A., Nakano, Y., Hansen-Kiss, E., Kelly, B., Nishihara, M., Goshima, Y., et al. (2017). Human CRMP4 mutation and disrupted Crmp4 expression in mice are associated with ASD characteristics and sexual dimorphism. Sci. Rep. 7:16812. doi: 10.1038/s41598-017-16782-8
Uchida, Y., Ohshima, T., Yamashita, N., Ogawara, M., Sasaki, Y., Nakamura, F., et al. (2009). Semaphorin3A signaling mediated by Fyn-dependent tyrosine phosphorylation of collapsin response mediator protein 2 at tyrosine 32. J. Biol. Chem. 284, 27393–27401. doi: 10.1074/jbc.m109.000240
Varrin-Doyer, M., Vincent, P., Cavagna, S., Auvergnon, N., Noraz, N., Rogemond, V., et al. (2009). Phosphorylation of collapsin response mediator protein 2 on Tyr-479 regulates CXCL12-induced T lymphocyte migration. J. Biol. Chem. 284, 13265–13276. doi: 10.1074/jbc.M807664200
Vincent, P., Collette, Y., Marignier, R., Vuaillat, C., Rogemond, V., Davoust, N., et al. (2005). A role for the neuronal protein collapsin response mediator protein 2 in T lymphocyte polarization and migration. J. Immunol. 175, 7650–7660. doi: 10.4049/jimmunol.175.11.7650
Wakatsuki, S., Saitoh, F., and Araki, T. (2011). ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat. Cell Biol. 13, 1415–1423. doi: 10.1038/ncb2373
Wang, L. H., and Strittmatter, S. M. (1996). A family of rat CRMP genes is differentially expressed in the nervous system. J. Neurosci. 16, 6197–6207. doi: 10.1523/JNEUROSCI.16-19-06197.1996
Wang, T., Wu, X., Yin, C., Klebe, D., Zhang, J. H., and Qin, X. (2013). CRMP-2 is involved in axon growth inhibition induced by RGMa in vitro and in vivo. Mol. Neurobiol. 47, 903–913. doi: 10.1007/s12035-012-8385-3
Wilson, S. M., Moutal, A., Melemedjian, O. K., Wang, Y., Ju, W., Francois-Moutal, L., et al. (2014). The functionalized amino acid (S)-Lacosamide subverts CRMP2-mediated tubulin polymerization to prevent constitutive and activity-dependent increase in neurite outgrowth. Front. Cell. Neurosci. 8:196. doi: 10.3389/fncel.2014.00196
Wilson, S. M., Xiong, W., Wang, Y., Ping, X., Head, J. D., Brittain, J. M., et al. (2012). Prevention of posttraumatic axon sprouting by blocking collapsin response mediator protein 2-mediated neurite outgrowth and tubulin polymerization. Neuroscience 210, 451–466. doi: 10.1016/j.neuroscience.2012.02.038
Wu, L. H., Lin, C., Lin, H. Y., Liu, Y. S., Wu, C. Y., Tsai, C. F., et al. (2016). Naringenin suppresses neuroinflammatory responses through inducing suppressor of cytokine signaling 3 expression. Mol. Neurobiol. 53, 1080–1091. doi: 10.1007/s12035-014-9042-9
Yamamoto, T., Setsu, T., Okuyama-Yamamoto, A., and Terashima, T. (2009). Histological study in the brain of the reelin/Dab1-compound mutant mouse. Anat. Sci. Int. 84, 200–209. doi: 10.1007/s12565-008-0009-7
Yamane, M., Yamashita, N., Hida, T., Kamiya, Y., Nakamura, F., Kolattukudy, P., et al. (2017). A functional coupling between CRMP1 and Nav1.7 for retrograde propagation of Semaphorin3A signaling. J. Cell Sci. 130, 1393–1403. doi: 10.1242/jcs.199737
Yamashita, N., and Goshima, Y. (2012). Collapsin response mediator proteins regulate neuronal development and plasticity by switching their phosphorylation status. Mol. Neurobiol. 45, 234–246. doi: 10.1007/s12035-012-8242-4
Yamashita, N., Mosinger, B., Roy, A., Miyazaki, M., Ugajin, K., Nakamura, F., et al. (2011). CRMP5 (collapsin response mediator protein 5) regulates dendritic development and synaptic plasticity in the cerebellar Purkinje cells. J. Neurosci. 31, 1773–1779. doi: 10.1523/JNEUROSCI.5337-10.2011
Yamashita, N., Ohshima, T., Nakamura, F., Kolattukudy, P., Honnorat, J., Mikoshiba, K., et al. (2012). Phosphorylation of CRMP2 (collapsin response mediator protein 2) is involved in proper dendritic field organization. J. Neurosci. 32, 1360–1365. doi: 10.1523/JNEUROSCI.5563-11.2012
Yamashita, N., Uchida, Y., Ohshima, T., Hirai, S., Nakamura, F., Taniguchi, M., et al. (2006). Collapsin response mediator protein 1 mediates reelin signaling in cortical neuronal migration. J. Neurosci. 26, 13357–13362. doi: 10.1523/JNEUROSCI.4276-06.2006
Yang, X., Zhang, X., Li, Y., Han, S., Howells, D. W., Li, S., et al. (2016). Conventional protein kinase Cβ-mediated phosphorylation inhibits collapsin response-mediated protein 2 proteolysis and alleviates ischemic injury in cultured cortical neurons and ischemic stroke-induced mice. J. Neurochem. 137, 446–459. doi: 10.1111/jnc.13538
Yoshimura, T., Kawano, Y., Arimura, N., Kawabata, S., Kikuchi, A., and Kaibuchi, K. (2005). GSK-3β regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120, 137–149. doi: 10.1016/j.cell.2004.11.012
Yu-Kemp, H. C., Kemp, J. P. Jr., and Brieher, W. M. (2017). CRMP-1 enhances EVL-mediated actin elongation to build lamellipodia and the actin cortex. J. Cell Biol. 216, 2463–2479. doi: 10.1083/jcb.201606084
Zhang, J., Zhao, B., Zhu, X., Li, J., Wu, F., Li, S., et al. (2018). Phosphorylation and SUMOylation of CRMP2 regulate the formation and maturation of dendritic spines. Brain Res. Bull. 139, 21–30. doi: 10.1016/j.brainresbull.2018.02.004
Zheng, Y., Sethi, R., Mangala, L. S., Taylor, C., Goldsmith, J., Wang, M., et al. (2018). Tuning microtubule dynamics to enhance cancer therapy by modulating FER-mediated CRMP2 phosphorylation. Nat. Commun. 9:476. doi: 10.1038/s41467-017-02811-7
Zhu, L. Q., Zheng, H. Y., Peng, C. X., Liu, D., Li, H. L., Wang, Q., et al. (2010). Protein phosphatase 2A facilitates axonogenesis by dephosphorylating CRMP2. J. Neurosci. 30, 3839–3848. doi: 10.1523/JNEUROSCI.5174-09.2010
Keywords: CRMP, neuronal development, regeneration, structural biology, posttranslational modifications, phosphorylation, neurological disorders, drug target
Citation: Nakamura F, Ohshima T and Goshima Y (2020) Collapsin Response Mediator Proteins: Their Biological Functions and Pathophysiology in Neuronal Development and Regeneration. Front. Cell. Neurosci. 14:188. doi: 10.3389/fncel.2020.00188
Received: 01 April 2020; Accepted: 29 May 2020;
Published: 23 June 2020.
Edited by:
Juan Pablo Henríquez, University of Concepcion, ChileReviewed by:
Anne Marie Duchemin, The Ohio State University, United StatesLisette Leyton, University of Chile, Chile
Copyright © 2020 Nakamura, Ohshima and Goshima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshio Goshima, Z29zaGltYUBtZWQueW9rb2hhbWEtY3UuYWMuanA=
† These authors have contributed equally to this work