
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci., 30 June 2020
Sec. Cellular Neuropathology
Volume 14 - 2020 | https://doi.org/10.3389/fncel.2020.00178
This article is part of the Research TopicThe Role of Cellular Senescence in Health and DiseaseView all 16 articles
 Dimitrios Papadopoulos1*
Dimitrios Papadopoulos1* Roberta Magliozzi2Dimos D. Mitsikostas3
Roberta Magliozzi2Dimos D. Mitsikostas3 Vassilis G. Gorgoulis1
Vassilis G. Gorgoulis1 Richard S. Nicholas4,5*
Richard S. Nicholas4,5*Aging is one of the most important risk factors for the development of several neurodegenerative diseases including progressive multiple sclerosis (MS). Cellular senescence (CS) is a key biological process underlying aging. Several stressors associated with aging and MS pathology, such as oxidative stress, mitochondrial dysfunction, cytokines and replicative exhaustion are known triggers of cellular senescence. Senescent cells exhibit stereotypical metabolic and functional changes, which include cell-cycle arrest and acquiring a pro-inflammatory phenotype secreting cytokines, growth factors, metalloproteinases and reactive oxygen species. They accumulate with aging and can convert neighboring cells to senescence in a paracrine manner. In MS, accelerated cellular senescence may drive disease progression by promoting chronic non-remitting inflammation, loss or altered immune, glial and neuronal function, failure of remyelination, impaired blood-brain barrier integrity and ultimately neurodegeneration. Here we discuss the evidence linking cellular senescence to the pathogenesis of MS and the putative role of senolytic and senomorphic agents as neuroprotective therapies in tackling disease progression.
Multiple sclerosis (MS) is a chronic, immune mediated disease of unknown etiology characterized by inflammatory demyelination, astrogliosis, neuronal and axonal loss involving the brain and spinal cord. The majority of MS patients follow an initial course with relapses and remissions (RR-MS) followed by a phase of progressive accumulation of disability termed secondary progressive (SP-MS). Ten to 15% of patients with primary progressive MS (PP-MS) exhibit gradual worsening from the start and typically PP-MS presents at an older age than RR-MS (Compston and Coles, 2008). The pathogenesis of the progressive disease courses (P-MS) is poorly understood. However, epidemiological evidence indicates that age is the strongest predictor for the transition from the relapsing phase, which is considered primarily inflammatory to the secondary progressive phase of the disease, which is mainly neurodegenerative (Trapp and Nave, 2008; Scalfari et al., 2011).
Although our understanding of the biological basis of aging remains incomplete, the prevailing hypothesis postulates that it is driven by the accumulation of irreparable molecular and cellular damage leading to an increased risk of functional decline, disease and ultimately death. Aging exhibits a great diversity of phenotypes and a loose connection between biological and chronological age, probably due to the stochastic nature of molecular damage and the complexity of the interaction between genetic and environmental factors (Kirkwood et al., 2005; Kirkwood and Melov, 2011). Aging is the most important risk factor for the development of neurodegenerative disease (Hou et al., 2019). Cellular senescence (CS) has been recognized as a key biological process underling normal aging (López-Otín et al., 2013; Gorgoulis et al., 2019) and evidence suggest that the accumulation of senescent cells with time may contribute to the pathogenesis of age-related and neurodegenerative disease (Kritsilis et al., 2018). Here, we review the data that support a role for cellular senescence in the pathogenesis of MS.
Several cell stressors have been identified as triggers of CS, which among others include oxidative stress, mitochondrial dysfunction, replicative stress, cytokines, irradiation, genotoxic agents presented in detail in Gorgoulis et al. (2019). Oxidative stress, mitochondrial dysfunction and cytokines, such as TGF-beta are key features of MS pathology (Gilgun-Sherki et al., 2004; Mahad et al., 2009; Haider et al., 2011; Elkjaer et al., 2019). Most of these triggering factors are associated with DNA damage and activate the signal transduction system of DNA damage response (DDR) (Nakamura et al., 2008). If the damage is irreparable DDR may elicit CS (Rodier and Campisi, 2011).
The senescent phenotype is typically associated with several metabolic and functional changes including stable cell cycle arrest, the expression of a senescence-associated secretory phenotype (SASP) and the accumulation of dysfunctional mitochondria (Coppé et al., 2010; Munoz-Espin and Serrano, 2014; Correia-Melo et al., 2016). SASP consists of pro-inflammatory cytokines, growth factors, cytotoxic mediators, metalloproteinases and reactive oxygen species (ROS). These are capable of affecting neighboring cells and converting them to senescence in a paracrine manner (Kuilman and Peeper, 2009; Acosta et al., 2013). Other changes characteristic of the senescent state are described in greater detail elsewhere (Munoz-Espin and Serrano, 2014; Gorgoulis et al., 2019). The senescence-associated changes reported specifically for CNS cells are summarized in Table 1.
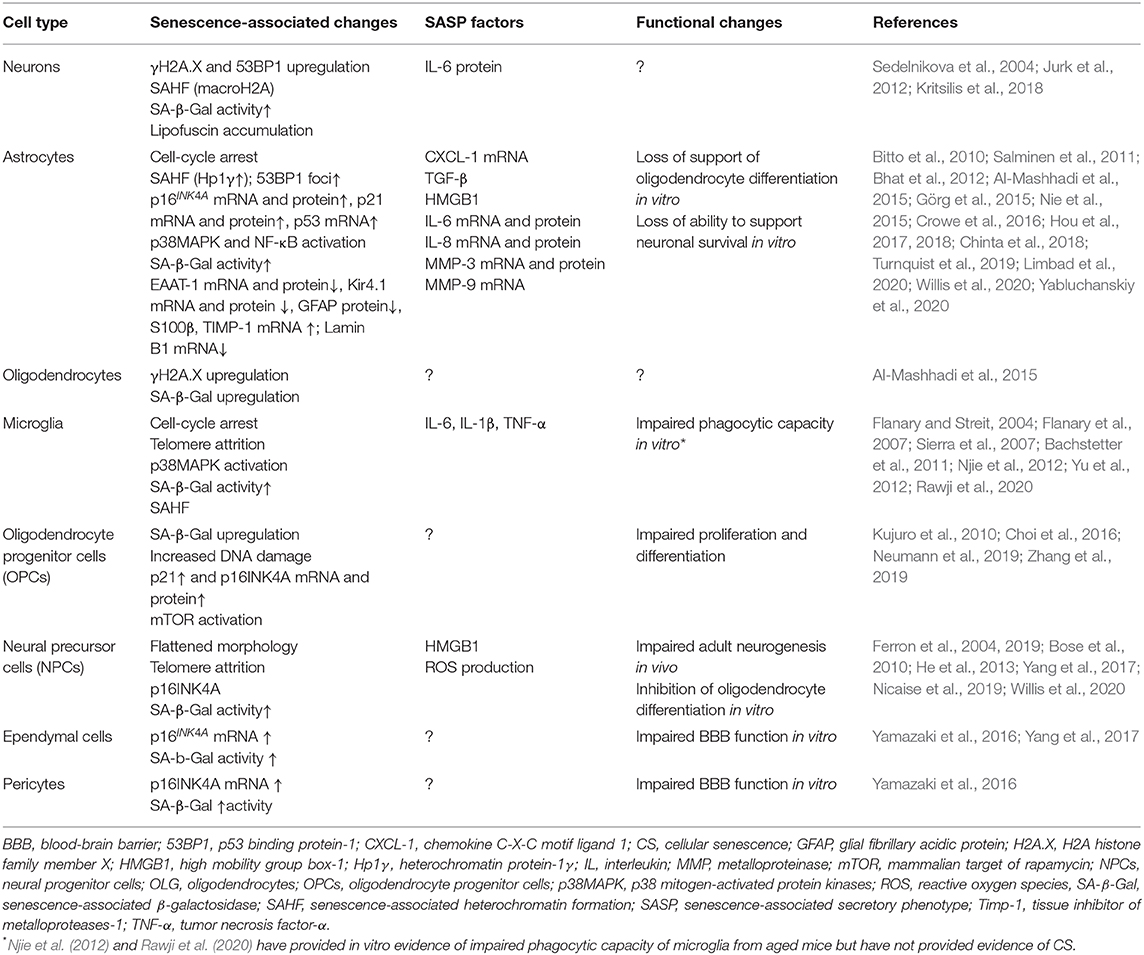
Table 1. Observed hallmarks, features, and functional changes associated with CS in CNS cells.
Although CS is a homeostatic response aiming to prevent the proliferation and neoplastic conversion of damaged cells (Munoz-Espin and Serrano, 2014) it also has a role in development (Rajagopalan and Long, 2012; Barbouti et al., 2019). Damaged senescent cells remain viable and metabolically active, they accumulate with aging and evidence suggests that their build-up may promote neurodegeneration (Rodier and Campisi, 2011). The detrimental effects of CS on the brain are due to the pro-inflammatory milieu formed by senescent cells that act as sources of inflammatory mediators (Coppé et al., 2010). CS-associated cell-cycle arrest may exhaust the regenerative capacities of adult progenitors, such as oligodendrocyte progenitor cells (OPCs) responsible for myelin repair. In addition, CS along with replication arrest is associated with extensive changes in gene expression, which indicate severe loss or alteration of physiological cell function (Purcell et al., 2014). Finally, endothelial cell senescence may compromise blood-brain barrier (BBB) integrity (Yamazaki et al., 2016), which is essential for preserving brain tissue homeostasis (Berthiaume et al., 2018).
Both innate and adaptive components of the immune response are known to play key roles in the immunopathogenesis of MS (Weissert, 2013; Hemmer et al., 2015). Microglial cells, the resident representative of the innate immune response in the CNS (Ransohoff and Brown, 2012) are known to become senescent under specific circumstances. Cultured microglial cells can become senescent in response to chronic inflammatory stimulation by lipopolysaccharide treatment (Yu et al., 2012). Rat and human microglial cells from AD patients have been shown to undergo replicative senescence due to telomere shortening (Flanary and Streit, 2004; Flanary et al., 2007). With aging, microglial cells exhibit a dystrophic phenotype associated with functional changes, which seem to be distinct from the typical microglial reaction (Streit et al., 2004, 2009; Conde and Streit, 2006). Aged microglia exhibit decreased migratory and phagocytic capacity and secrete constitutively greater amounts of interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) in culture (Njie et al., 2012; Rawji et al., 2020). The constitutive secretion of proinflammatory cytokines by microglia from aged mice is consistent with the SASP of senescent cells. Myelin clearance is a prerequisite for remyelination to occur (Kotter et al., 2001, 2006; Cantuti-Castelvetri et al., 2018) and impaired phagocytotic capacity of aged microglia and macrophages could hinder myelin repair in older patients. Nevertheless, the presence of senescent microglia and macrophages in MS and its models has not been shown.
With regard to the adaptive component of the immune response in MS, there is evidence of premature immunosenescence with T cell changes resembling those seen in the elderly (Thewissen et al., 2005). An expansion of CD4(+) CD28(–) T cells and a reduction of T-cell receptor excision circles (TREC) has been observed in the peripheral blood of MS patients (Thewissen et al., 2005). Peripheral blood CD4(+) CD28(-) T cells in patients and healthy controls exhibit an effector-memory T cell phenotype with cytotoxic properties, as they secrete cytotoxic granules in response to polyclonal stimuli and MS-related autoantigens. These CD4(+)CD28(-) T cells are attracted by increased levels of fractalkine and IL-15 and accumulate in demyelinated lesions (Broux et al., 2012, 2015). Similarly, a population of IgD(-)−CD27(-)CD11c (+)(CD21low) B cells, which have been associated with aging was demonstrated in higher proportions in the CSF and peripheral blood of MS patients than age-matched healthy controls. These IgD(-)−CD27(-)CD11c(+)(CD21low) B cells produced proinflammatory cytokines upon ex vivo stimulation and showed MHC class-II expression and costimulatory molecule expression capable to induce proinflammatory T cell responses. Their presence indicates that premature senescence of B cells may promote inflammation and thereby contribute to disease progression in MS (Claes et al., 2016).
Senescent neurons and glia accumulating in the MS brain and secreting SASP-related inflammatory mediators may represent an alternative source of inflammation independent of the immune cells that bring about innate and adaptive immune responses. We have previously provided evidence of senescent glial cells and neurons showing lipofuscin+ senescent glial cells in acute and chronic actively demyelinated white matter lesions from SP-MS cases using GL13 histochemistry (Kritsilis et al., 2018). All resident CNS cell types that under some circumstances acquire a senescent phenotype may become sources of parenchymal inflammation. The documented ability of senescent cells to convert neighboring cells to senescence via paracrine action of SASP mediators is consistent with the progressive nature of disability seen in P-MS (Acosta et al., 2013; Chen et al., 2015). Current therapeutic strategies fail to tackle disability progression in P-MS despite being efficacious in preventing MS relapses and new lesion formation thought to be mediated by adaptive immune responses (Pardo and Jones, 2017). This is consistent with the concept of resident glia and neurons secreting SASP-related factors that maintain a low-burning yet persistent and self-enhancing inflammatory environment not affected by immunomodulators and immunosuppressants. The glial cell types, which are prone to senescence in the MS lesions remain to be specified.
Endogenous myelin repair (remyelination) of axons surviving inflammatory demyelination is known to occur in MS (Patani et al., 2007). Remyelination contributes to restoration of impulse conduction along axons traversing demyelinating plaques and exerts a neuroprotective effect on remyelinated axons preventing axonal degeneration associated with chronic demyelination (Kornek et al., 2000; Franklin and Goldman, 2015; Mei et al., 2016). Adult oligodendrocyte progenitor cells (OPCs) comprising 5–10% of all CNS cells are primarily responsible for carrying out myelin repair following demyelinating events (Reynolds et al., 2002; Tripathi et al., 2010). The efficiency of myelin repair is known to gradually decline with time and it is shown to be least extensive in the SP-MS phase of the disease (Bramow et al., 2010). This suggests that age-related remyelination failure may contribute to disability progression seen at the advanced stages of MS (Kuhlmann et al., 2008; Goldschmidt et al., 2009). Aging has been shown to reduce the capacity for remyelination in several animal models of demyelination (Shen et al., 2008; Hampton et al., 2012; Cantuti-Castelvetri et al., 2018) in which remyelination inefficacy is associated with impaired recruitment of OPCs into demyelinated lesions and slower differentiation into myelinating oligodendrocytes (Sim et al., 2002). OPCs from aged rats show features of CS with increased levels of DNA damage, mitochondrial dysfunction and p38MAPK mRNA upregulation (Neumann et al., 2019). In addition, recent experimental evidence indicates that murine astrocytes aged in culture develop a pro-inflammatory senescence-like phenotype and lose their ability to support oligodendrocyte differentiation (Willis et al., 2020).
However, OPC senescence may not only be associated with aging. In experimental autoimmune encephalomyelitis (EAE) model of MS in young mice, OPCs exhibited cell-cycle arrest linked to an upregulation of sirtuin 1 (SIRT1) transcription, suggesting that failure of OPC proliferation may be due to CS (Prozorovski et al., 2019). Furthermore, in vitro exposure of cultured murine OPCs to Aβ oligomers triggered CS and inhibited myelin sheet formation indicating that toxic factors can elicit senescence in OPCs (Horiuchi et al., 2012; Zhang et al., 2019). In the APP/PS1 model of Alzheimer's disease OPCs expressing upregulated p16, p21, and senescence-associated-β-Galactosidase (SA-β-Gal) markers of CS have been identified in association with Aβ plaques and treatments aiming to remove senescent OPCs (senolytics) attenuated neuroinflammation and cognitive deficits, indicating that OPC SASP promotes neuroinflammation and functional impairment (Zhang et al., 2019).
Other progenitor cells including neural progenitor cells (NPCs) and mesenchymal stem cells (MSCs) also have a role in promoting remyelination and tissue repair (Nicaise et al., 2017; Rivera et al., 2019). It is documented that the adult CNS harbors multipotent neural progenitor cells (NPCs) that can produce neurons, astrocytes, and oligodendrocytes (Weiss et al., 1996; Johansson et al., 1999). They are thought to primarily reside in the subventricular zone (SVZ) and the subgranular zone (SGZ) of the dentate gyrus (Kriegstein and Alvarez-Buylla, 2009; Ming and Song, 2011). Most NPCs in the adult brain exist in a quiescent state (Ding et al., 2020) unless CNS injury or specific stimuli elicit their proliferation, migration and differentiation (neurogenesis) (Mothe and Tator, 2005). Evidence supports the functional significance of NPCs as hippocampal neurogenesis is critical for cognition (Suh et al., 2009; Christian et al., 2014) and its disruption is associated with cognitive impairment (Aimone et al., 2014). In addition, studies have demonstrated that adult NPCs from the subventricular zone (SVZ) and the spinal cord contribute to the generation of new oligodendrocytes and myelin repair in models of demyelination (Nait-Oumesmar et al., 1999; Danilov et al., 2006; Menn et al., 2006; Xing et al., 2014; Maeda et al., 2019). Thus, NPCs could provide an alternative source of myelinating oligodendrocytes and probably also a source of neurons in demyelinated MS lesions (Chang et al., 2008).
Aging is associated with progressive reduction in adult neurogenesis (Lugert et al., 2010; Cipriani et al., 2018), which is ascribed to a diminution of the pool of stem cells capable of activation and division (Lugert et al., 2010) and it is associated with functional impairment (Hollands et al., 2017). Accumulating evidence suggests that these NPCs are also prone to senescence. Cultured NPCs exhibit characteristics of senescence, such as enlarged and flattened morphology, increased levels of SA-β-Gal and p16 and decreased level of phospho-Retinoblastoma (pRb) upon long term incubation with Aβ oligomers (Ferron et al., 2004; He et al., 2013; Li et al., 2016). Cell cycle arrest of adult progenitor cells in the context of CS or inhibitory paracrine stimuli by neighboring senescent cells may impair progenitor proliferation, reduce the regenerative capacities of the CNS and render it susceptible to neurodegeneration. This notion is supported by in vivo evidence from the BUBR1 KO progeroid mouse model in which adult neurogenesis was impaired in the SGZ and SVZ in an age-dependent manner (Yang et al., 2017). In MS, SOX2+ NPCs from demyelinated white matter lesions of autopsy material and NPCs from induced pluripotent stem cell lines from P-MS patients were found to express markers of CS. These senescent progenitor cells exhibited impaired capacity to support oligodendrocyte maturation in vitro, compared to NPCs from age-matched controls. Proteomic and transcriptomic analysis of the P-MS NPC secretome identified high-mobility group box-1 (HMGB1) as a senescence-associated inhibitor of oligodendrocyte differentiation, which induces expression of epigenetic regulators. HMGB1 was found to be expressed by progenitor cells in MS white matter lesions (Nicaise et al., 2019). Failure of spontaneous remyelination in MS may be at least partly due to conversion of OPCs and other neural progenitor cells to a state of CS induced by MS-specific triggers including oxidative stress, chronic inflammation, mitochondrial dysfunction and aging.
Neuroaxonal loss is the pathological correlate of irreversible disability (Trapp et al., 1998; Papadopoulos et al., 2006). Although axonal loss can be an early feature of MS pathology associated with inflammatory lesion formation, in P-MS new focal inflammatory demyelinating plaques are rare (Trapp and Nave, 2008). Neuroaxonal loss in P-MS is driven by neurodegenerative mechanisms, which are poorly understood (Lassmann et al., 2007, 2012; Trapp and Nave, 2008).
Recently, telomere length of white blood cells (WBCs) used as a measure of WBC biological age was found decreased in P-MS patients compared to aged-matched controls (Habib et al., 2020). Moreover, shorter telomere lengths correlated with greater brain atrophy and higher levels of disability (Krysko et al., 2019), suggesting that biological aging contributes to neuroaxonal loss and disability progression in MS. Total brain atrophy, as assessed by MRI, reflects primarily gray matter atrophy due to neurodegeneration (Filippi et al., 2012) and correlates with long-term disability in MS (Fisniku et al., 2008; Filippi et al., 2013). Combined longitudinal MRI-based brain morphometry and brain age estimation using machine learning, revealed accelerated progressive brain aging in MS patients compared to healthy controls, which was related to brain atrophy and increased white matter lesion load (Høgestøl et al., 2019; Cole et al., 2020). Healthy aging is associated with brain cell loss, which may account up to 0.4% of brain volume per year (De Stefano et al., 2016). Both apoptotic and senescent cells are cleared by the immune system in a highly regulated manner and may contribute to age-related brain volume loss (Hoenicke and Zender, 2012; Ovadya et al., 2018).
Although post-mitotic cells do not fit the strict definition of CS, there is evidence of neurons developing a senescence-like phenotype. Neurons of aged mice have been shown to accumulate hallmarks of cellular senescence including double-strand DNA breaks, heterochromatinization, upregulation of SA-β- Gal, p38MAPK activation and production of SASP-related mediators including ROS and IL-6 (Sedelnikova et al., 2004; Jurk et al., 2012). The demonstration of neuronal granular cytoplasmic lipofuscin deposits in subpial demyelinated cortical lesions and normal appearing cortex from SP-MS cases using GL13 histochemistry supports the notion that human neurons may also acquire a senescence-like phenotype in MS (Kritsilis et al., 2018).
Although the functional state of senescent cells has not been fully elucidated, CS is accompanied by changes in gene expression and phenotypic changes, which constitute serious restrictions in the functionality of cells (Purcell et al., 2014). The number of senescent cells increases with age (Rodier and Campisi, 2011). When the number of dysfunctional senescent cells exceeds a certain threshold in a brain with reduced reserves due to age and MS-related cell loss, brain tissue function is likely to become compromised (Oost et al., 2018).
Neuronal survival strongly depends on the functional integrity of glial cells. Cultured astrocytes from aging rats have been found to upregulate the activity of SA-β-Gal, a marker of CS while they showed a reduced ability to maintain survival of co-cultured neurons, thus associating astrocyte senescence with neurodegeneration (Pertusa et al., 2007). Senescent astrocytes expressing P16INK4A and secreting metalloproteinase-1 (MMP-1) have been found in post-mortem tissues of Alzheimer's disease patients (Bhat et al., 2012). Primary human astrocytes made senescent by X-irradiation were found to downregulate genes encoding glutamate and potassium transporters leading to neuronal death in co-culture assays. These findings indicate that excitotoxicity, a recognized mechanism of neurodegeneration in MS (Werner et al., 2001) may result from impaired homeostatic capacities of senescent astrocytes (Limbad et al., 2020). Notably, dysregulated splicing of several genes from human senescent astrocytes has been demonstrated and an association between peripheral blood GFAPa, TAU3 and p14ARF isoform levels and cognitive decline has been demonstrated, indicating a link between astrocyte senescence and disability (Lye et al., 2019). Although no evidence of astrocyte senescence has been shown in association with MS pathology, their key role in neuron-glial crosstalk, regulation of neuronal metabolic and ion homeostasis and modulation of synaptic transmission via glutamate suggest that age-related astrocytic senescence may promote neuronal dysfunction and degeneration, contributing to MS progression.
Age-related accumulation of senescent endothelial cells is linked to impaired tight junction structure and compromised blood-brain barrier (BBB) function (Farrall and Wardlaw, 2009; Yamazaki et al., 2016; Castellazzi et al., 2020). Several lines of evidence from human studies and experimental animal models support a key role for fibrinogen in neuroinflammation (Davalos and Akassoglou, 2012). Blood-derived fibrinogen has been shown in vivo to interact with microglia via the CD11b/CD18 integrin receptor leading to perivascular microglial activation and axonal loss (Davalos et al., 2012). Fibrinogen has been found at the edge of chronic active lesions, which exhibit ongoing inflammatory demyelination, but not in inactive lesions, suggesting that fibrinogen may play a role in sustained inflammation even in the chronic setting. Endothelial senescence leading to a constantly leaky BBB may permit fibrinogen to diffuse into the brain parenchyma and drive axonal damage and loss mediated by persistent microglia activation as well as inhibition of remyelination (Petersen et al., 2018). The sustained nature of age-related BBB leakiness is consistent with the putative role of CS in neuroaxonal loss-mediated disability progression in MS.
Currently, there is an unmet need for neuroprotective treatments that can effectively prevent disability progression in MS. A growing body of evidence implicates CS in the pathogenesis of neurodegeneration in a number of settings (Martínez-Cué and Rueda, 2020), rendering CS a promising target for neuroprotection. Anti-senescent or senotherapeutic approaches may involve the selective death of senescent cells (senolysis) to reduce the load of senescent cells and their detrimental effects on tissues. Alternatively, senotherapy may be based on the modulation of the senescent cell phenotype (senomorphism) in order to block the damaging effects of the SASP or other senescence-associated mediators (Kirkland et al., 2017). Senotherapy in MS would aim at preventing senescence-associated chronic inflammation, loss of cell function and neuroaxonal loss and promoting remyelination. Compounds with senolytic or senomorphic actions have been studied in vitro and in vivo with promising results (Myrianthopoulos et al., 2019; Thoppil and Riabowol, 2020). Evidence from genetically modified mice support the neuroprotective potential of senolytic manipulation. Lifelong elimination of p16INK4A cells in BubR1 progeroid mice by activation of an INK-ATTAC transgene substantially delayed age-related disease, whereas late life elimination of p16INK4A cells attenuated these age-related pathologies (Baker et al., 2011). In addition, a senolytic compound (ABT263) was shown to attenuate tau phosphorylation and aggregation and to improve memory deficits in the PS19 transgenic model of tau-dependent neurodegeneration, by removing senescent glial cells (Bussian et al., 2018).
Fasting and metformin treatment could reverse the senescent state of rat OPCs and improve remyelination capacity (Neumann et al., 2019). Furthermore, rapamycin treatment modified the senescent state of progressive MS patient-derived NPCs produced from induced pluripotent stem cells and improved their capacity to promote OPC differentiation in vitro, providing evidence that senomorphic treatment can promote remyelination in MS (Nicaise et al., 2019). Simvastatin has shown efficacy in delaying brain atrophy and disability progression in MS trials (Chataway et al., 2014). This neuroprotective effect may be mediated via its senomorphic actions, which include downregulation of p38MAPK activation, SASP markers, TNFa, and GM-CSF as shown in other settings (Liu et al., 2015; Ayad et al., 2018). Many promising compounds with senolytic or senomorphic activity, such as metformin or simvastatin used with different indications could be repurposed and used as neuroprotectants combined with currently available immunomodulators. Immune-mediated physiological clearance of senescent cells could potentially be therapeutically enhanced by medications or vaccines aimed at priming the immune response to remove specific senescent populations (Burton and Stolzing, 2018; Song et al., 2020). Reprogramming of senescent cells may be another approach (Tamanini et al., 2018; Mahmoudi et al., 2019).
On the other hand, treatments aiming at the disease processes that precede and accelerate CS, such as inflammation, oxidative stress and mitochondrial dysfunction at the earliest stages of the disease may delay CS and hinder CS-related neurodegeneration. Furthermore, senescence-inducing practices and medications including exposure to ionizing radiation and DNA-damaging chemotherapeutics should be avoided. Interestingly, approved MS treatments, such as corticosteroids, beta-interferons and mitoxantrone should be re-evaluated for their long-term effects given that they have been shown to promote CS (Moiseeva et al., 2006; Ikeda et al., 2010; Poulsen et al., 2014).
Aging is an important risk factor for the development of several neurodegenerative diseases including P-MS, where the neurodegenerative component dominates. A primary causative role of CS in MS is highly unlikely given the great diversity which characterizes aging-related neurodegenerative pathologies that have CS as a common feature. However, CS may be a shared mechanism, which substantially contributes to the pathogenesis and impact of neurodegenerative diseases and thereby may determine disease susceptibility, age at disease presentation and rate of progression. In MS, senescence may be responsible for chronic non-remitting inflammation, which is not amenable to immunomodulation, lost or altered glial and neuronal function, failure of remyelination, impaired BBB integrity and neurodegeneration (Figure 1).
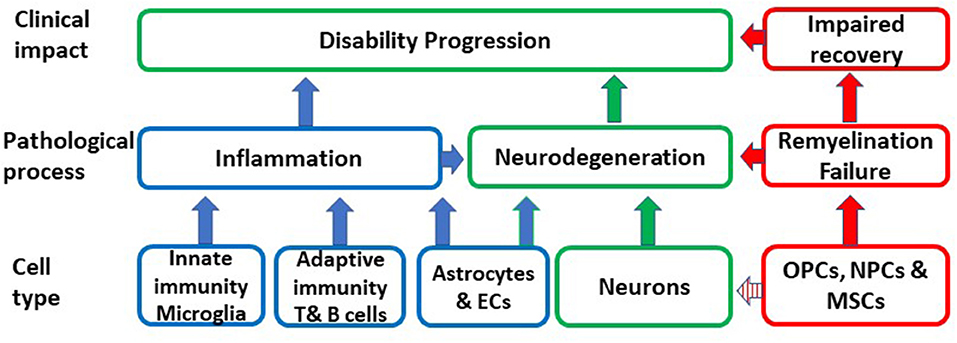
Figure 1. Schematic representation of the putative impact of the conversion of different cell types to senescence on inflammation, remyelination, neurodegeneration and ultimately on disability progression. OPCs, oligodendrocyte progenitors; MSCs, mesenchymal stem cells; NPCs, neural progenitor cells; EC, endothelial cells.
Nevertheless, current evidence for a role of CS in disability progression in MS is intriguing but limited and indirect. Shedding light on CS and its role in neurodegeneration is essential to safely exploit it therapeutically. To facilitate these efforts a thorough histopathological investigation of post-mortem MS tissue at various disease stages and levels of disability would inform us of the extent, timing, particular cell types converted to senescence and all features of pathology associated with the accumulation of senescent cells. A more concise understanding of the biology of CS of neural cells, its triggers and mediators is required. Advanced human 3D and organoid culture techniques (Marton et al., 2019; Yoon et al., 2019) could help identify the specific factors that induce CS and the contribution of each cell type to tissue injury. Transgenic animal models of demyelination could provide mechanistic evidence to disentangle the detrimental effects of senescent cell types and their mediators and become platforms on which to test senotherapeutic agents. The complex physiological and pathophysiological roles of CS, along with the cell-type specific variability in senescence triggers and phenotypes, necessitates a cautious approach to avoid pitfalls when dealing with such a multifaceted biological process. If senescent cells are proven to drive neurodegeneration senotherapy may become the groundbreaking neuroprotective strategy to prevent and potentially reverse progressive disability in MS.
Literature search by DP, RM, DM, and RN. The first draft of the manuscript was written by DP and RN. RM, DM, and VG critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Publication fees were covered by the Imperial College fund for open access (OA) article processing charges (APCs).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acosta, J. C., Banito, A., Wuestefeld, T., Georgilis, A., Janich, P., Morton, J. P., et al. (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990. doi: 10.1038/ncb2784
Aimone, J. B., Li, Y., Lee, S. W., Clemenson, G. D., Deng, W., and Gage, F. H. (2014). Regulation and function of adult neurogenesis: from genes to cognition. Physiol. Rev. 94, 991–1026. doi: 10.1152/physrev.00004.2014
Al-Mashhadi, S., Simpson, J. E., Heath, P. R., Dickman, M., Forster, G., Matthews, F. E., et al. (2015). Oxidative glial cell damage associated with white matter lesions in the aging human brain. Brain Pathol. 25, 565–574. doi: 10.1111/bpa.12216
Ayad, M. T., Taylor, B. D., and Menon, R. (2018). Regulation of p38 mitogen-activated kinase-mediated fetal membrane senescence by statins. Am. J. Reprod. Immunol. 80:e12999. doi: 10.1111/aji.12999
Bachstetter, A. D., Xing, B., de Almeida, L., Dimayuga, E. R., Watterson, D. M., and Van Eldik, L. J. (2011). Microglial p38α MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Aβ). J. Neuroinflammation 11:175. doi: 10.1186/1742-2094-8-79
Baker, D. J., Wijshake, T., Tchkonia, T., LeBrasseur, N. K., Childs, B. G., van de Sluis, B., et al. (2011). Clearance of p16Ink4a-positive senescent cells delays ageing- associated disorders. Nature 479, 232–236. doi: 10.1038/nature10600
Barbouti, A., Evangelou, K., Pateras, I. S., Papoudou-Bai, A., Patereli, A., Stefanaki, K., et al. (2019). In situ evidence of cellular senescence in thymic epithelial cells (TECs) during human thymic involution. Mech. Ageing Dev. 177, 88–90. doi: 10.1016/j.mad.2018.02.005
Berthiaume, A. A., Hartmann, D. A., Majesky, M. W., Bhat, N. R., and Shih, A. Y. (2018). Pericyte structural remodeling in cerebrovascular health and homeostasis. Front. Aging Neurosci. 10:210. doi: 10.3389/fnagi.2018.00210
Bhat, R., Crowe, E. P., Bitto, A., Moh, M., Katsetos, C. D., Garcia, F. U., et al. (2012). Astrocyte senescence as a component of Alzheimer's disease. PLoS ONE 7:e0045069. doi: 10.1371/journal.pone.0045069
Bitto, A., Sell, C., Crowe, E., Lorenzini, A., Malaguti, M., Hrelia, S., et al. (2010). Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 316, 2961–2968. doi: 10.1016/j.yexcr.2010.06.021
Bose, R., Moors, M., Tofighi, R., Cascante, A., Hermanson, O., and Ceccatelli, S. (2010). Glucocorticoids induce long-lasting effects in neural stem cells resulting in senescence-related alterations. Cell Death Dis. 1:e92. doi: 10.1038/cddis.2010.60
Bramow, S., Frischer, J. M., Lassmann, H., Koch-Henriksen, N., Lucchinetti, C. F., Sørensen, P. S., et al. (2010). Demyelination versus remyelination in progressive multiple sclerosis. Brain 133, 2983–2998. doi: 10.1093/brain/awq250
Broux, B., Mizee, M. R., Vanheusden, M., van der Pol, S., van Horssen, J., Van Wijmeersch, B., et al. (2015). IL-15 amplifies the pathogenic properties of CD4+CD28− T cells in multiple sclerosis. J. Immunol. 194, 2099–2109. doi: 10.4049/jimmunol.1401547
Broux, B., Pannemans, K., Zhang, X., Markovic-Plese, S., Broekmans, T., Eijnde, B. O., et al. (2012). CX(3)CR1 drives cytotoxic CD4(+)CD28(-) T cells into the brain of multiple sclerosis patients. J. Autoimmun. 38, 10–19. doi: 10.1016/j.jaut.2011.11.006
Burton, D. G. A., and Stolzing, A. (2018). Cellular senescence: immunosurveillance and future immunotherapy. Ageing Res. Rev. 43, 17–25. doi: 10.1016/j.arr.2018.02.001
Bussian, T. J., Aziz, A., Meyer, C. F., Swenson, B. L., van Deursen, J. M., and Baker, D. J. (2018). Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582. doi: 10.1038/s41586-018-0543-y
Cantuti-Castelvetri, L., Fitzner, D., Bosch-Queralt, M., Weil, M. T., Su, M., Sen, P., et al. (2018). Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 359, 684–688. doi: 10.1126/science.aan4183
Castellazzi, M., Morotti, A., Tamborino, C., Alessi, F., Pilotto, S., Baldi, E., et al. (2020). Increased age and male sex are independently associated with higher frequency of blood-cerebrospinal fluid barrier dysfunction using the albumin quotient. Fluids Barriers CNS 17:14. doi: 10.1186/s12987-020-0173-2
Chang, A., Smith, M. C., Yin, X., Fox, R. J., Staugaitis, S. M., and Trapp, B. D. (2008). Neurogenesis in the chronic lesions of multiple sclerosis. Brain 131, 2366–2375. doi: 10.1093/brain/awn157
Chataway, J., Schuerer, N., Alsanousi, A., Chan, D., MacManus, D., Hunter, K., et al. (2014). Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet 383, 2213–2221. doi: 10.1016/S0140-6736(13)62242-4
Chen, H., Ruiz, P. D., McKimpson, W. M., Novikov, L., Kitsis, R. N., and Gamble, M. J. (2015). MacroH2A1 and ATM play opposing roles in paracrine senescence and the senescence-associated secretory phenotype. Mol. Cell 59, 719–731. doi: 10.1016/j.molcel.2015.07.011
Chinta, S. J., Woods, G., Demaria, M., Rane, A., Zou, Y., Mcquade, A., et al. (2018). Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson's disease. Cell Rep. 22, 930–940. doi: 10.1016/j.celrep.2017.12.092
Choi, C.-I., Yoo, K. H., Hussaini, S. M. Q., Jeon, B. T., Welby, J., Gan, H., et al. (2016). The progeroid gene BubR1 regulates axon myelination and motor function. Aging (Albany NY) 8, 2667–2688. doi: 10.18632/aging.101032
Christian, K. M., Song, H., and Ming, G. L. (2014). Functions and dysfunctions of adult hippocampal neurogenesis. Annu. Rev. Neurosci. 37, 243–262. doi: 10.1146/annurev-neuro-071013-014134
Cipriani, S., Ferrer, I., Aronica, E., Kovacs, G. G., Verney, C., Nardelli, J., et al. (2018). Hippocampal radial glial subtypes and their neurogenic potential in human fetuses and healthy and Alzheimer's disease adults. Cereb. Cortex 28, 2458–2478. doi: 10.1093/cercor/bhy096
Claes, N., Fraussen, J., Vanheusden, M., Hellings, N., Stinissen, P., Van Wijmeersch, B., et al. (2016). Age-associated B cells with proinflammatory characteristics are expanded in a proportion of multiple sclerosis patients. J. Immunol. 197, 4576–4583. doi: 10.4049/jimmunol.1502448
Cole, J. H., Raffel, J., Friede, T., Eshaghi, A., Brownlee, W., Chard, D., et al. (2020). Accelerated brain ageing and disability in multiple sclerosis. Ann Neurol. doi: 10.1101/584888. [Epub ahead of print].
Compston, A., and Coles, A. (2008). Multiple sclerosis. Lancet 372, 1502–1517. doi: 10.1016/S0140-6736(08)61620-7
Conde, J. R., and Streit, W. J. (2006). Microglia in the aging brain. J. Neuropathol. Exp. Neurol. 65, 199–203. doi: 10.1097/01.jnen.0000202887.22082.63
Coppé, J.-P., Desprez, P.-Y., Krtolica, A., and Campisi, J. (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 5, 99–118. doi: 10.1146/annurev-pathol-121808-102144
Correia-Melo, C., Marques, F. D., Anderson, R., Hewitt, G., Hewitt, R., Cole, J., et al. (2016). Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 35, 724–742. doi: 10.15252/embj.201592862
Crowe, E. P., Tuzer, F., Gregory, B. D., Donahue, G., Gosai, S. J., Cohen, J., et al. (2016). Changes in the transcriptome of human astrocytes accompanying oxidative stress-induced senescence. Front. Aging Neurosci. 8:208. doi: 10.3389/fnagi.2016.00208
Danilov, A. I., Covacu, R., Moe, M. C., Langmoen, I. A., Johansson, C. B., Olsson, T., et al. (2006). Neurogenesis in the adult spinal cord in an experimental model of multiple sclerosis. Eur. J. Neurosci. 23, 394–400. doi: 10.1111/j.1460-9568.2005.04563.x
Davalos, D., and Akassoglou, K. (2012). Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 34, 43–62 doi: 10.1007/s00281-011-0290-8
Davalos, D., Ryu, J. K., Merlini, M., Baeten, K. M., Le Moan, N., Petersen, M. A., et al. (2012). Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 3:1227. doi: 10.1038/ncomms2230
De Stefano, N., Stromillo, M. L., Giorgio, A., Bartolozzi, M. L., Battaglini, M., Baldini, M., et al. (2016). Establishing pathological cut-offs of brain atrophy rates in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 87, 93–99. doi: 10.1136/jnnp-2014-309903
Ding, W. Y., Huang, J., and Wang, H. (2020). Waking up quiescent neural stem cells: molecular mechanisms and implications in neurodevelopmental disorders. PLoS Genet. 16:e1008653. doi: 10.1371/journal.pgen.1008653
Elkjaer, M. L., Frisch, T., Reynolds, R., Kacprowski, T., Burton, M., Kruse, T. A., et al. (2019). Molecular signature of different lesion types in the brain white matter of patients with progressive multiple sclerosis. Acta Neuropathol. Commun. 7:205. doi: 10.1186/s40478-019-0855-7
Farrall, A. J., and Wardlaw, J. M. (2009). Blood-brain barrier: ageing and microvascular disease–systematic review and meta-analysis. Neurobiol. Aging. 30, 337–352 doi: 10.1016/j.neurobiolaging.2007.07.015
Ferron, S., Mira, H., Franco, S., Cano-Jaimez, M., Bellmunt, E., Ramírez, C., et al. (2004). Telomere shortening and chromosomal instability abrogates proliferation of adult but not embryonic neural stem cells. Development. 131, 4059–4070. doi: 10.1242/dev.01215
Ferron, S. R., Marques-Torrejon, M. A., Mira, H., Flores, I., Taylor, K., Blasco, M. A., et al. (2019). Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J. Neurosci. 29, 14394–14407. doi: 10.1523/JNEUROSCI.3836-09.2009
Filippi, M., Preziosa, P., Copetti, M., Riccitelli, G., Horsfield, M. A., Martinelli, V., et al. (2013). Gray matter damage predicts the accumulation of disability 13 years later in MS. Neurology 81, 1759–1767. doi: 10.1212/01.wnl.0000435551.90824.d0
Filippi, M., Rocca, M. A., Barkhof, F., Brück, W., Chen, J. T., Comi, G., et al. (2012). Association between pathological and MRI findings in multiple sclerosis. Lancet Neurol. 11, 349–360. doi: 10.1016/S1474-4422(12)70003-0
Fisniku, L. K., Chard, D. T., Jackson, J. S., Anderson, V. M., Altmann, D. R., Miszkiel, K. A., et al. (2008). Gray matter atrophy is related to long-term disability in multiple sclerosis: GM atrophy and disability in MS. Ann. Neurol. 64, 247–254. doi: 10.1002/ana.21423
Flanary, B. E., Sammons, N. W., Nguyen, C., Walker, D., and Streit, W. J. (2007). Evidence that aging and amyloid promote microglial cell senescence. Rejuv. Res. 10, 61–74. doi: 10.1089/rej.2006.9096
Flanary, B. E., and Streit, W. J. (2004). Progressive telomere shortening occurs in cultured rat microglia, but not astrocytes. Glia 45, 75–88. doi: 10.1002/glia.10301
Franklin, R. J. M., and Goldman, S. A. (2015). Glia disease and repair- Remyelination. Cold Spring Harb. Perspect. Biol. 7, 1–28. doi: 10.1101/cshperspect.a020594
Gilgun-Sherki, Y., Melamed, E., and Offen, D. (2004). The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J. Neurol. 251, 261–268. doi: 10.1007/s00415-004-0348-9
Goldschmidt, T., Antel, J., König, F., Brück, W., and Kuhlmann, T. (2009). Remyelination capacity of the MS brain decreases with disease chronicity. Neurology 72, 1914–1921. doi: 10.1212/WNL.0b013e3181a8260a
Görg, B., Karababa, A., Shafigullina, A., Bidmon, H. J., and Häussinger, D. (2015). Ammonia-induced senescence in cultured rat astrocytes and in human cerebral cortex in hepatic encephalopathy. Glia 63, 37–50. doi: 10.1002/glia.22731
Gorgoulis, V., Adams, P. D., Alimonti, A., Bennett, D. C., Bischof, O., Bishop, C., et al. (2019). Cellular senescence: defining a path forward. Cell 179, 813–827. doi: 10.1016/j.cell.2019.10.005
Habib, R., Ocklenburg, S., Hoffjan, S., Haghikia, A., Epplen, J. T., and Arning, L. (2020). Association between shorter leukocyte telomeres and multiple sclerosis. J. Neuroimmunol. 341:577187. doi: 10.1016/j.jneuroim.2020.577187
Haider, L., Fischer, M. T., Frischer, J. M., Bauer, J., Höftberger, R., Botond, G., et al. (2011). Oxidative damage in multiple sclerosis lesions. Brain. 134, 1914–1924. doi: 10.1093/brain/awr128
Hampton, D. W., Innes, N., Merkler, D., Zhao, C., Franklin, R. J. M., and Chandran, S. (2012). Focal immune-mediated white matter demyelination reveals an age associated increase in axonal vulnerability and decreased remyelination efficiency. Am. J. Pathol. 180, 1897–1905. doi: 10.1016/j.ajpath.2012.01.018
He, N., Jin, W.-L., Lok, K.-H., Wang, Y., Yin, M., and Wang, Z.-J. (2013). Amyloid-_1–42 oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis. 4:e924. doi: 10.1038/cddis.2013.437
Hemmer, B., Kerschensteiner, M., and Korn, T. (2015). Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 14, 406–419. doi: 10.1016/S1474-4422(14)70305-9
Hoenicke, L., and Zender, L. (2012). Immune surveillance of senescent cells–biological significance in cancer- and non-cancer pathologies. Carcinogenesis 33, 1123–1126. doi: 10.1093/carcin/bgs124
Høgestøl, E. A., Kaufmann, T., Nygaard, G. O., Beyer, M. K., Sowa, P., Nordvik, J. E., et al. (2019). Cross-sectional and longitudinal MRI brain scans reveal accelerated brain aging in multiple sclerosis. Front. Neurol. 10:450. doi: 10.3389/fneur.2019.00450
Hollands, C., Tobin, M. K., Hsu, M., Musaraca, K., Yu, T.-S., Mishra, R., et al. (2017). Depletion of adult neurogenesis exacerbates cognitive deficits in Alzheimer's disease by compromising hippocampal inhibition. Mol. Neurodegener. 12:64. doi: 10.1186/s13024-017-0207-7
Horiuchi, M., Maezawa, I., Itoh, A., Wakayama, K., Jin, L. W., Itoh, T., et al. (2012). Amyloid β1-42 oligomer inhibits myelin sheet formation in vitro. Neurobiol. Aging 33, 499–509. doi: 10.1016/j.neurobiolaging.2010.05.007
Hou, J., Cui, C., Kim, S., Sung, C., and Choi, C. (2018). Ginsenoside F1 suppresses astrocytic senescence-associated secretory phenotype. Chem. Biol. Interact. 283, 75–83. doi: 10.1016/j.cbi.2018.02.002
Hou, J., Kim, S., Sung, C., and Choi, C. (2017). Ginsenoside Rg3 prevents oxidative stress-induced astrocytic senescence and ameliorates senescence paracrine effects on glioblastoma. Molecules. 22:1516. doi: 10.3390/molecules22091516
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. doi: 10.1038/s41582-019-0244-7
Ikeda, M., Kurose, A., Takatori, E., Sugiyama, T., Traganos, F., Darzynkiewicz, Z., et al. (2010). DNA damage detected with gammaH2AX in endometrioid adenocarcinoma cell lines. Int. J. Oncol. 36, 1081–1088. doi: 10.3892/ijo_00000589
Johansson, C. B., Svensson, M., Wallstedt, L., Janson, A. M., and Frisen, J. (1999). Neural stem cells in the adult human brain. Exp. Cell Res. 253, 733–736. doi: 10.1006/excr.1999.4678
Jurk, D., Wang, C., Miwa, S., Maddick, M., Korolchuk, V., Tsolou, A., et al. (2012). Post-mitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004. doi: 10.1111/j.1474-9726.2012.00870.x
Kirkland, J. L., Tchkonia, T., Zhu, Y., Niedernhofer, L. J., and Robbins, P. D. (2017). The clinical potential of senolytic drugs. J. Am. Geriatr. Soc. 65, 2297–2301. doi: 10.1111/jgs.14969
Kirkwood, T. B. L., Feder, M., Finch, C. E., Franceschi, C., Globerson, A., Klingenberg, C. P., et al. (2005). What accounts for the wide variation in life span of genetically identical organisms reared in a constant environment? Mech. Ageing Dev. 126, 439–443. doi: 10.1016/j.mad.2004.09.008
Kirkwood, T. B. L., and Melov, S. (2011). On the programmed/non-programmed nature of ageing within the life history. Curr. Biol. 21, R701–R707. doi: 10.1016/j.cub.2011.07.020
Kornek, B., Storch, M. K., Weissert, R., Wallstroem, E., Stefferl, A., Olsson, T., et al. (2000). Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 157, 267–276. doi: 10.1016/S0002-9440(10)64537-3
Kotter, M. R, Li, W. W., Zhao, C., and Franklin, R.J. (2006). Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J. Neurosci. 26, 328–332. doi: 10.1523/JNEUROSCI.2615-05.2006
Kotter, M. R., Setzu, A., Sim, F. J., Van Rooijen, N., and Franklin, R. J. (2001). Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia 35, 204–212. doi: 10.1002/glia.1085
Kriegstein, A., and Alvarez-Buylla, A. (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149–184. doi: 10.1146/annurev.neuro.051508.135600
Kritsilis, M., Rizou, S. V., Koutsoudaki, P. N., Evangelou, K., Gorgoulis, V. G., and Papadopoulos, D. (2018). Ageing, cellular senescence and neurodegenerative disease. Int. J. Mol. Sci. 19:2937. doi: 10.3390/ijms19102937
Krysko, K. M., Henry, R. G., Cree, B. A. C., Lin, J., Caillier, S., Santaniello, A., et al. (2019). Telomere length is associated with disability progression in multiple sclerosis. Ann. Neurol. 86, 671–682. doi: 10.1002/ana.25592
Kuhlmann, T., Miron, V., Cuo, Q., Wegner, C., Antel, J., and Brück, W. (2008). Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 131, 1749–1758. doi: 10.1093/brain/awn096
Kuilman, T., and Peeper, D. S. (2009). Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 9, 81–94. doi: 10.1038/nrc2560
Kujuro, Y., Suzuki, N., and Kondo, T. (2010). Esophageal cancer-related gene 4 is a secreted inducer of cell senescence expressed by aged CNS precursor cells. Proc. Natl. Acad. Sci. U.S.A. 107, 8259–8264. doi: 10.1073/pnas.0911446107
Lassmann, H., Brück, W., and Lucchinetti, C. F. (2007). The immunopathology of multiple sclerosis: an overview. Brain Pathol. 17, 210–218. doi: 10.1111/j.1750-3639.2007.00064.x
Lassmann, H., van Horssen, J., and Mahad, D. (2012). Progressive multiple sclerosis: pathology and pathogenesis. Nat. Rev. Neurol. 8, 647–656. doi: 10.1038/nrneurol.2012.168
Li, W.-Q., Wang, Z., Liu, S., Hu, Y., Yin, M., and Lu, Y. (2016). N-stearoyl-L-tyrosine inhibits the senescence of neural stem/progenitor cells induced by A1-42 via the CB2 receptor. Stem Cells Int. 2016:7419389. doi: 10.1155/2016/7419389
Limbad, C., Oron, T. R., Alimirah, F., Davalos, A. R., Tracy, T. E., Gan, L., et al. (2020). Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS ONE 15:e0227887. doi: 10.1371/journal.pone.0227887
Liu, S., Uppal, H., Demaria, M., Desprez, P. Y., Campisi, J., and Kapahi, P. (2015). Simvastatin suppresses breast cancer cell proliferation induced by senescent cells. Sci. Rep. 5:17895. doi: 10.1038/srep17895
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Lugert, S., Basak, O., Knuckles, P., Haussler, U., Fabel, K., Götz, M., et al. (2010). Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell. Stem Cell. 6, 445–456. doi: 10.1016/j.stem.2010.03.017
Lye, J. J., Latorre, E., Lee, B. P., Bandinelli, S., Holley, J. E., Gutowski, N. J., et al. (2019). Astrocyte senescence may drive alterations in GFAPα, CDKN2A p14ARF, and TAU3 transcript expression and contribute to cognitive decline. Geroscience 41, 561–573. doi: 10.1007/s11357-019-00100-3
Maeda, Y., Nakagomi, N., Nakano-Doi, A., Ishikawa, H., Tatsumi, Y., Bando, Y., et al. (2019). Potential of adult endogenous neural stem/progenitor cells in the spinal cord to contribute to remyelination in experimental autoimmune encephalomyelitis. Cells 8:E1025. doi: 10.3390/cells8091025
Mahad, D. J., Ziabreva, I., Campbell, G., Lax, N., White, K., Hanson, P. S., et al. (2009). Mitochondrial changes within axons in multiple sclerosis. Brain 132, 1161–1174. doi: 10.1093/brain/awp046
Mahmoudi, S., Xu, L., and Brunet, A. (2019). Turning back time with emerging rejuvenation strategies. Nat. Cell Biol. 21, 32–43. doi: 10.1038/s41556-018-0206-0
Martínez-Cué, C., and Rueda, N. (2020). cellular senescence in neurodegenerative diseases. Front. Cell. Neurosci. 14:16. doi: 10.3389/fncel.2020.00016
Marton, R. M., Miura, Y., Sloan, S. A., Li, Q., Revah, O., Levy, R. J., et al. (2019). Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 22, 484–491. doi: 10.1038/s41593-018-0316-9
Mei, F., Lehmann-Horn, K., Shen, Y. A. A., Rankin, K. A., Stebbins, K. J., Lorrain, D. S., et al. (2016). Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. Elife 5:e13. doi: 10.7554/eLife.18246.013
Menn, B., Garcia-Verdugo, J. M., Yaschine, C., Gonzalez-Perez, O., Rowitch, D., and Alvarez-Buylla, A. (2006). Origin of oligodendrocytes in the subventricular zone of the adult brain. J. Neurosci. 26, 7907–7918. doi: 10.1523/JNEUROSCI.1299-06.2006
Ming, G., and Song, H. (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702. doi: 10.1016/j.neuron.2011.05.001
Moiseeva, O., Mallette, F. A., Mukhopadhyay, U. K., Moores, A., and Ferbeyre, G. (2006). DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol. Biol. Cell. 17, 1583–1592. doi: 10.1091/mbc.e05-09-0858
Mothe, A. J., and Tator, C. H. (2005). Proliferation, migration, and differentiation of endogenous ependymal region stem/progenitor cells following minimal spinal cord injury in the adult rat. Neuroscience 131, 177–187. doi: 10.1016/j.neuroscience.2004.10.011
Munoz-Espin, D., and Serrano, M. (2014). Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell. Biol. 15, 482–496. doi: 10.1038/nrm3823
Myrianthopoulos, V., Evangelou, K., Vasileiou, P. V. S., Cooks, T., Vassilakopoulos, T. P., Pangalis, G. A., et al. (2019). Senescence and senotherapeutics: a new field in cancer therapy. Pharmacol. Ther. 193, 31–49. doi: 10.1016/j.pharmthera.2018.08.006
Nait-Oumesmar, B., Decker, L., Lachapelle, F., Avellana-Adalid, V., Bachelin, C., and Baron-Van Evercooren, A. (1999). Progenitor cells of the adult mouse subventricular zone proliferate, migrate and differentiate into oligodendrocytes after demyelination. Eur. J. Neurosci. 11, 4357–4366. doi: 10.1046/j.1460-9568.1999.00873.x
Nakamura, A. J., Chiang, Y. J., Hathcock, K. S., Horikawa, I., Sedelnikova, O. A., Hodes, R. J., et al. (2008). Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenet. Chromatin 1:6. doi: 10.1186/1756-8935-1-6
Neumann, B., Baror, R., Zhao, C., Segel, M., Dietmann, S., Rawji, K. S., et al. (2019). Metformin restores CNS remyelination capacity by rejuvenating aged stem cells. Cell. Stem Cell. 25, 473–485.e8. doi: 10.1016/j.stem.2019.08.015
Nicaise, A. M., Banda, E., Guzzo, R. M., Russomanno, K., Castro-Borrero, W., Willis, C. M., et al. (2017). iPS-derived neural progenitor cells from PPMS patients reveal defect in myelin injury response. Exp. Neurol. 288, 114–121. doi: 10.1016/j.expneurol.2016.11.012
Nicaise, A. M., Wagstaff, L. J., Willis, C. M., Paisie, C., Chandok, H., Robson, P., et al. (2019). Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 116, 9030–9039. doi: 10.1073/pnas.1818348116
Nie, X., Liang, L., Xi, H., Jiang, S., Jiang, J., Tang, C., et al. (2015). 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces premature senescence of astrocytes via WNT/-catenin signaling and ROS production. J. Appl. Toxicol. 35, 851–860. doi: 10.1002/jat.3084
Njie, E. G., Boelen, E., Stassen, F. R., Steinbusch, H. W., Borchelt, D. R., and Streit, W. J. (2012). Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 33, 195.e1–195.e12. doi: 10.1016/j.neurobiolaging.2010.05.008
Oost, W., Talma, N., Meilof, J. F., and Laman, J. D. (2018). Targeting senescence to delay progression of multiple sclerosis. J. Mol. Med. (Berl). 96, 1153–1166. doi: 10.1007/s00109-018-1686-x
Ovadya, Y., Landsberger, T., Leins, H., Vadai, E., Gal, H., Biran, A., et al. (2018). Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 9:5435. doi: 10.1038/s41467-018-07825-3
Papadopoulos, D., Pham-Dinh, D., and Reynolds, R. (2006). Axon loss is responsible for chronic neurological deficit following inflammatory demyelination in the rat. Exp. Neurol. 197, 373–385. doi: 10.1016/j.expneurol.2005.10.033
Pardo, G., and Jones, D. E. (2017). The sequence of disease-modifying therapies in relapsing multiple sclerosis: safety and immunologic considerations. J. Neurol. 264, 2351–2374. doi: 10.1007/s00415-017-8594-9
Patani, R., Balaratnam, M., Vora, A., and Reynolds, R. (2007). Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol. Appl. Neurobiol. 33, 277–287. doi: 10.1111/j.1365-2990.2007.00805.x
Pertusa, M., García-Matas, S., Rodríguez-Farré, E., Sanfeliu, C., and Cristòfol, R. (2007). Astrocytes aged in vitro show a decreased neuroprotective capacity. J. Neurochem. 101, 794–805. doi: 10.1111/j.1471-4159.2006.04369.x
Petersen, M. A., Ryu, J. K., and Akassoglou, K. (2018). Fibrinogen in neurological diseases: mechanisms, imaging and therapeutics. Nat. Rev. Neurosci. 19, 283–301. doi: 10.1038/nrn.2018.13
Poulsen, R. C., Watts, A. C., Murphy, R. J., Snelling, S. J., Carr, A. J., and Hulley, P. A. (2014). Glucocorticoids induce senescence in primary human tenocytes by inhibition of sirtuin 1 and activation of the p53/p21 pathway: in vivo and in vitro evidence. Ann. Rheum. Dis. 73, 1405–1413. doi: 10.1136/annrheumdis-2012-203146
Prozorovski, T., Ingwersen, J., Lukas, D., Göttle, P., Koop, B., Graf, J., et al. (2019). Regulation of sirtuin expression in autoimmune neuroinflammation: induction of SIRT1 in oligodendrocyte progenitor cells. Neurosci. Lett. 704, 116–125. doi: 10.1016/j.neulet.2019.04.007
Purcell, M., Kruger, A., and Tainsky, M. A. (2014). Gene expression profiling of replicative and induced senescence. Cell Cycle 13, 3927–3937. doi: 10.4161/15384101.2014.973327
Rajagopalan, S., and Long, E. O. (2012). Cellular senescence induced by CD158d reprograms natural killer cells to promote vascular remodeling. Proc. Natl. Acad. Sci. U.S.A. 109, 20596–20601. doi: 10.1073/pnas.1208248109
Ransohoff, R. M., and Brown, M. A. (2012). Innate immunity in the central nervous system. J. Clin. Invest. 122, 1164–1171. doi: 10.1172/JCI58644
Rawji, K. S., Young, A. M. H., Ghosh, T., Michaels, N. J., Mirzaei, R., Kappen, J., et al. (2020). Niacin-mediated rejuvenation of macrophage/microglia enhances remyelination of the aging central nervous system. Acta Neuropathol. 139, 893–909. doi: 10.1007/s00401-020-02129-7
Reynolds, R., Dawson, M., Papadopoulos, D., Polito, A., Di Bello, I. C., Pham-Dinh, D., et al. (2002). The response of NG2-expressing oligodendrocyte progenitors to demyelination in MOG-EAE and MS. J. Neurocytol. 31, 523–536. doi: 10.1023/A:1025747832215
Rivera, F. J., de la Fuente, A. G., Zhao, C., Silva, M. E., Gonzalez, G. A., Wodnar, R., et al. (2019). Aging restricts the ability of mesenchymal stem cells to promote the generation of oligodendrocytes during remyelination. Glia 67, 1510–1525. doi: 10.1002/glia.23624
Rodier, F., and Campisi, J. (2011). Four faces of cellular senescence. J. Cell Biol. 192, 547–556. doi: 10.1083/jcb.201009094
Salminen, A., Ojala, J., Kaarniranta, K., Haapasalo, A., Hiltunen, M., and Soininen, H. (2011). Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 34, 3–11. doi: 10.1111/j.1460-9568.2011.07738.x
Scalfari, A., Neuhaus, A., Daumer, M., Ebers, G. C., and Muraro, P. A. (2011). Age and disability accumulation in multiple sclerosis. Neurology 77, 1246–1252. doi: 10.1212/WNL.0b013e318230a17d
Sedelnikova, O. A., Horikawa, I., Zimonjic, D. B., Popescu, N. C., Bonner, W. M., and Barrett, J. C. (2004). Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 6, 168–170. doi: 10.1038/ncb1095
Shen, S., Sandoval, J., Swiss, V. A., Li, J., Dupree, J., Franklin, R. J. M., et al. (2008). Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat. Neurosci. 11, 1024–1034. doi: 10.1038/nn.2172
Sierra, A., Gottfried-Blackmore, A. C., McEwen, B. S., and Bulloch, K. (2007). Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55, 412–424. doi: 10.1002/glia.20468
Sim, F. J., Zhao, C., Penderis, J., and Franklin, R. J. (2002). The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J. Neurosci. 22, 2451–2459. doi: 10.1523/JNEUROSCI.22-07-02451.2002
Song, P., An, J., and Zou, M-H. (2020). Immune clearance of senescent cells to combat ageing and chronic diseases. Cells 9:671. doi: 10.3390/cells9030671
Streit, W. J., Braak, H., Xue, Q.-S., Bechmann, I., Qing-Shan, A., Ae, X., et al. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 118, 475–485. doi: 10.1007/s00401-009-0556-6
Streit, W. J., Sammons, N. W., Kuhns, A. J., and Sparks, D. L. (2004). Dystrophic microglia in the aging human brain. Glia 45, 208–212. doi: 10.1002/glia.10319
Suh, H., Deng, W., and Gage, F. H. (2009). Signaling in adult neurogenesis. Annu. Rev. Cell Dev. Biol. 25, 253–275. doi: 10.1146/annurev.cellbio.042308.113256
Tamanini, S., Comi, G. P., and Corti, S. (2018). In vivo transient and partial cell reprogramming to pluripotency as a therapeutic tool for neurodegenerative diseases. Mol. Neurobiol. 55, 6850–6862. doi: 10.1007/s12035-018-0888-0
Thewissen, M., Linsen, L., Somers, V., Geusens, P., Raus, J., and Stinissen, P. (2005). Premature immunosenescence in rheumatoid arthritis and multiple sclerosis patients. Ann. N. Y. Acad. Sci. 1051, 255–262. doi: 10.1196/annals.1361.066
Thoppil, H., and Riabowol, K. (2020). Senolytics: a translational bridge between cellular senescence and organismal aging. Front. Cell. Dev. Biol. 7:367. doi: 10.3389/fcell.2019.00367
Trapp, B. D., and Nave, K. A. (2008). Multiple sclerosis: an immune or neurodegenerative disorder? Annu. Rev. Neurosci. 31, 247–269. doi: 10.1146/annurev.neuro.30.051606.094313
Trapp, B. D., Peterson, J., Ransohoff, R. M., Rudick, R., Mörk, S., and Bö, L. (1998). Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 338, 278–285. doi: 10.1056/NEJM199801293380502
Tripathi, R. B., Rivers, L. E., Young, K. M., Jamen, F., and Richardson, W. D. (2010). NG2 glia generate new oligodendrocytes but few astrocytes in a murine experimental autoimmune encephalomyelitis model of demyelinating disease. J. Neurosci. 30, 16383–16390. doi: 10.1523/JNEUROSCI.3411-10.2010
Turnquist, C., Beck, J. A., Horikawa, I., Obiorah, I. E., Von Muhlinen, N., Vojtesek, B., et al. (2019). Radiation-induced astrocyte senescence is rescued by Δ133p53. Neuro Oncol. 21, 474–485. doi: 10.1093/neuonc/noz001
Weiss, S., Dunne, C., Hewson, J., Wohl, C., Wheatley, M., Peterson, A. C., et al. (1996). Multipotent CNS stem cells are present in the adult mammalian spinal cord and ventricular neuroaxis. J. Neurosci. 16, 7599–7609. doi: 10.1523/JNEUROSCI.16-23-07599.1996
Weissert, R. (2013). The immune pathogenesis of multiple sclerosis. J. Neuroimmune Pharmacol. 8, 857–866. doi: 10.1007/s11481-013-9467-3
Werner, P., Pitt, D., and Raine, C. S. (2001). Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann. Neurol. 50, 169–180. doi: 10.1002/ana.1077
Willis, C. M., Nicaise, A. M., Bongarzone, E. R., Givogri, M., Reiter, C. R., Heintz, O., et al. (2020). Astrocyte support for oligodendrocyte differentiation can be conveyed via extracellular vesicles but diminishes with age. Sci. Rep. 10:828. doi: 10.1038/s41598-020-57663-x
Xing, Y. L., Röth, P. T., Stratton, J. A., Chuang, B. H., Danne, J., Ellis, S. L., et al. (2014). Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. J. Neurosci. 34, 14128–14146. doi: 10.1523/JNEUROSCI.3491-13.2014
Yabluchanskiy, A., Tarantini, S., Balasubramanian, P., Kiss, T., Csipo, T., Fülöp, G. A., et al. (2020). Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation–induced impairment of neurovascular coupling responses protecting cognitive function in mice. Geroscience. 42, 409–428. doi: 10.1007/s11357-020-00154-8
Yamazaki, Y., Baker, D. J., Tachibana, M., Liu, C. C., van Deursen, J. M., Brott, T. G., et al. (2016). Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke 47, 1068–1077. doi: 10.1161/STROKEAHA.115.010835
Yang, Z., Jun, H., Choi, C.-I., Yoo, K. H., Cho, C. H., Hussaini, S. M. Q., et al. (2017). Age-related decline in BubR1 impairs adult hippocampal neurogenesis. Aging Cell, 16, 598–601. doi: 10.1111/acel.12594
Yoon, S. J., Elahi, L. S., Paşca, A. M., Marton, R. M., Gordon, A., Revah, O., et al. (2019). Reliability of human cortical organoid generation. Nat. Methods 16, 75–78. doi: 10.1038/s41592-018-0255-0
Yu, H. M., Zhao, Y. M., Luo, X. G., Feng, Y., Ren, Y., Shang, H., et al. (2012). Repeated lipopolysaccharide stimulation induces cellular senescence in BV2 cells. Neuroimmunomodulation 19, 131–136. doi: 10.1159/000330254
Zhang, P., Kishimoto, Y., Grammatikakis, I., Gottimukkala, K., Cutler, R. G., Zhang, S., et al. (2019). Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer's disease model. Nat. Neurosci. 22, 719–728. doi: 10.1038/s41593-019-0372-9
Keywords: multiple sclerosis, cellular senescence, inflammation, remyelination, neurodegeneration, neuroprotection, senolytics
Citation: Papadopoulos D, Magliozzi R, Mitsikostas DD, Gorgoulis VG and Nicholas RS (2020) Aging, Cellular Senescence, and Progressive Multiple Sclerosis. Front. Cell. Neurosci. 14:178. doi: 10.3389/fncel.2020.00178
Received: 30 March 2020; Accepted: 22 May 2020;
Published: 30 June 2020.
Edited by:
Kempuraj Duraisamy, University of Missouri, United StatesReviewed by:
Cory Willis, University of Cambridge, United KingdomCopyright © 2020 Papadopoulos, Magliozzi, Mitsikostas, Gorgoulis and Nicholas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dimitrios Papadopoulos, ZC5wYXBhZG9wb3Vsb3NAb3RlbmV0Lmdy; Richard S. Nicholas, ci5uaWNob2xhc0BpbXBlcmlhbC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.