 Feihui Zhou1
Feihui Zhou1 Rong Liu2Pengcheng Han3Xingkui Zhang1Zhigao Li1Shen Zhang1Chang Liu1Yang Xia1Zhiwei Tang1*
Rong Liu2Pengcheng Han3Xingkui Zhang1Zhigao Li1Shen Zhang1Chang Liu1Yang Xia1Zhiwei Tang1*- 1Department of Neurosurgery, The First Affiliated Hospital of Kunming Medical University, Kunming, China
- 2Key Laboratory of Animal Models and Human Disease Mechanisms of Chinese Academy of Sciences, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China
- 3Department of Pathology and Laboratory Medicine, Medical University of South Carolina, Charleston, SC, United States
Objective: To investigate the effect and the underlying mechanism of Pertussis toxin (PTX) on microglia in the setting of cerebral ischemia.
Methods: We tested the effect of PTX 400 ng/days on middle cerebral artery occlusion stroke model by evaluating the neurologic function, infarct size, microglial distribution, and activation. In parallel, we also tested the effect of PTX on primary cultured microglia by evaluating microglial proliferation, activation, cytokine release, and CX3CR1 expression.
Results: PTX reduced the poststroke infarct size, improved the neurologic function as evaluated by Longa score, and reduced microglial aggregation and activation in the infarcted area. Further, PTX significantly decreased lipopolysaccharide-stimulated microglial proliferation, the release of interleukin 1β (IL-1β) and tumor necrosis factor α (TNF-α), and the expression of CX3CR1.
Interpretation: PTX treatment in stroke reduced microglial accumulation and activation in the infarct zone, resulting in a better functional outcome. The benefits of PTX treatment may be attributed to the reduced production of proinflammatory cytokine such as IL-1β and TNF-α and reduced expression of chemokine CX3CR1.
Introduction
Stroke is the second leading cause of mortality worldwide and has overtaken heart disease and become the primary cause of death in east Asian population (Moskowitz et al., 2010; Liu et al., 2011). Ischemic stroke constitutes 85% of all stroke incidences; the increasing morbidity and mortality associated with stroke pose a huge socioeconomic burden to aging society worldwide (Di Carlo, 2009). It remains a huge challenge to establish an effective therapy, but incessant efforts to search for better therapeutic approach are clinically demanding and highly valuable.
Inflammatory reactions inevitably follow a stroke event due to the hypoxia-related cell damage and tissue necrosis. The natural pathologic course starts from aseptic inflammation involving a wide spectrum of inflammatory factors. Microglial activation is a crucial step in the pathogenesis of aseptic inflammation (Xiong et al., 2016; Zhang et al., 2017).
Microglia are perhaps the most important intrinsic cerebral immune cells, constituting approximately 5% to 10% of total number of brain cells (Chan et al., 2007; Li and Barres, 2018). Microglia are the first group of immune cells that reacts to stroke event; they are activated by multiple cytokines and plasma proteins (Zhang et al., 2017). The activated microglia are key immune cells in central nervous system, as they maintain microenvironmental homeostasis by repairing the damage (Davalos et al., 2005; Kim and de Vellis, 2005). Microglia aggregate toward necrotic tissue and their peripheral regions. The accumulated microglia at early stage are M2 type, phagocytizing and removing cell debris, necrotic tissue, and toxic metabolites. After approximately 24 h, M2 microglia are superseded by M1 subtypes, which release additional proinflammatory cytokines and exacerbate cell damage (Perego et al., 2011; Wang et al., 2011, 2013). In this context, microglial activation in necrotic tissue might be a detrimental factor to accelerate neuronal death by spilling over inflammatory cytokines to by-standing neurons. Therefore, it would be beneficial to inhibit microglial activation after stroke onset. Suggested by several recent studies, however, microglial functions are heterogeneous, and their role in stroke is not uniform. Instead, microglia may play diverse functions, depending on the time and location during stroke pathogenesis (Hu et al., 2012; Xiong et al., 2016). The complexity and importance of microglia in stroke remain elusive and deserve in-depth study.
Pertussis toxin (PTX) is an exotoxin produced by Bordetella pertussis. PTX is used to prepare mouse model of experimental autoimmune encephalomyelitis (EAE; Tang et al., 2013). We previously found that low dose of PTX (50 ng/ml) may reduce neuronal calcium influx, thus minimizing neuronal damage and protecting cell viability in stroke (Tang et al., 2015). Although PTX, at least at a restricted dose, offers a direct protective effect on neuron, their potential pharmacologic mechanism in stroke protection may include additional key steps. We also found previously that PTX paradoxically attenuates inflammation in EAE model if we administer a relative low dose (Tang et al., 2013). Therefore, we hypothesize that inhibiting inflammatory components may contribute to the protective effect of PTX in experimental stroke. Specifically, we hypothesize that PTX modulates microglial function by inhibiting their activation and proliferation, thus ameliorating poststroke brain damage.
Materials and Methods
Transient Middle Cerebral Artery Occlusion Animal Model
All procedures are approved by the Institutional Animal Care and Use Committee of the first hospital of Kunming Medical University. All studies were conducted in accordance with the US Public Health Service’s Policy on Humane Care and Use of Laboratory Animals. The procedure for transient middle cerebral artery occlusion (tMCAO) model was described previously (Chen et al., 2005; Liesz et al., 2009; Wang et al., 2018). Briefly, 20–25 g C57BL/6 male adult mice were anesthetized with intraperitoneal injection of 1 mg ketamine and 0.5 mg xylazine dissolved in 1 ml normal saline, and the injection dose was 0.01 ml/g. Then, the animals were stabilized on the operating table with neck fully exposed, shaved and disinfected with 75% alcohol, and placed under the operating microscope. The skin is cut along the neck midline with blunt dissection of subcutaneous tissue. After exposure of the right internal carotid, external carotid, and common carotid arteries, a small incision was made in the common carotid artery, and a 6–0 surgical nylon monofilament with rounded tip entered the internal carotid artery approximately 10 mm until stopped by slight resistance. The filament was left in place for 60 min and then withdrawn for reperfusion. Throughout the procedure, body temperature was maintained at 37°C ± 0.5°C. Postoperatively, the animals were placed in an incubator with constant ambient temperature. Sham-operated control mice received the same surgical procedure without inserting a filament. They were separately housed and injected with 1 ml normal saline twice a day.
Randomization and Treatment
After establishing tMCAO stroke model, we randomized mice into a therapy group (n = 12) and a placebo control group (n = 12). Sham group also was established at the same time including 12 mice. The therapy group was treated with PTX 400 ng/days dissolved in 1 ml normal saline, and the control group and sham group were given 1 ml normal saline intraperitoneally, which lasted for 3 days.
Neurological Deficit Assessments (Longa Score)
Neurological deficit assessments were performed by investigators who were blinded to the experimental groups, as described previously (Tang et al., 2015; Yin et al., 2015; Xie et al., 2018). Briefly, rating scale was used as follows: score 0 was defined as the complete absence of neurological deficit; score 1 was defined as that front paws incompletely extended and mild neurological deficit; score 2 was defined as lateral turning while walking and moderate neurological deficits; score 3 was defined as lateral jumping of animal body and severe neurological deficits; score 4 was defined as lack of “conscious” response to noxious stimuli.
Microglial Culture
The method was previously described in detail (Lian et al., 2016). Briefly, the mice born within 24 h were sacrificed by decapitation under sterilized condition. The brain tissue was placed in a 35-mm Petri dish containing an anatomical fluid and placed on ice. Tissues were dissected under a dissecting microscope; the cortex and hippocampus were preserved, and the meninges were carefully removed. The brain tissue was smashed, collected in a centrifuge tube, and placed vertically. Then the tissue was mixed gently with 0.25% trypsin and incubated in a 37°C for 10 min. After gentle trituration, the cell suspension was filtered with 70-μm cell strainer, and the supernatant was collected after centrifugation 1,600× g for 5 min. Cell counting was performed using a hemocytometer. The cell suspension was placed into a polylysine-coated Petri dish at approximately 5 × 104/ml. The Petri dishes were placed in a 37°C incubator, and the media was changed at 24 h after inoculation and then changed 50% of the volume every 72 h until used for lipopolysaccharide (LPS; 400 ng/ml) stimulation or other experiments (PTX, 50 ng/ml).
2,3,5-Triphenyltetrazolium Chloride Staining
As previously described (Wang et al., 2018), the animals were quickly anesthetized, and the brains were removed rapidly and frozen at −20°C for 5 min. Coronal slices were cut anterior to posterior at 2-mm interval, and sections were immersed in 2% triphenyltetrazolium chloride (TTC; Sigma–Aldrich, St. Louis, MO, USA) at 37°C for 20 min. The presence of infarction was determined by the area that stained negative with TTC and was quantitated with ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Immunohistochemistry for in vivo and in vitro Microglia
Seventy-two hours after MCAO, terminally anesthetized mice were perfused intracardially with saline followed by 4% paraformaldehyde. The fixed brains were embedded in paraffin and cut into serial 5-μm-thick coronal slices. Immunohistochemistry was performed with antibodies against IBA-1 (Fujifilm-Wako, Tokyo, Japan) to identify microglia, anti-CX3CR1 antibody (Invitrogen, Carlsbad, CA, USA) to identify Chemokine receptor, and anti-Ki67 antibody (#9027; Cell Signaling, Danvers, MA, USA) to identify proliferative cells. The secondary antibodies we used were goat anti-mouse immunoglobulin G (IgG; cat. #401215; Millipore, Burlington, MA, USA) and goat anti-rabbit IgG (cat. #AP307P, Lot #2899737; Millipore, Burlington, MA, USA).
For cultured microglia, the cells were fixed in 4% paraformaldehyde, treated with 0.3% Triton X-100 for 20 min, washed twice with phosphate-buffered saline, and stained with antibodies against IBA-1 to identify microglia and CX3CR1 to identify chemokine receptor. The nuclear counter stain was performed using 0.5 μg/ml DAPI. Results are presented as IBA-1+ or CX3CR1+ cells per images of 20× magnification field images.
Microglial Proliferation Assay
We used two independent assays to assess microglial cell numbers after LPS stimulation and compared them with PTX-treated cells. MTT assay was performed using Vybrant MTT Cell Proliferation Assay Kit (V13154; Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer instruction. Briefly, cultured primary cells were divided into various treatment group, control group, and blank group (see “Results” section). Five percent MTT solution is added and incubated at 37 degrees for 2 h. After removing medium and adding dimethyl sulfoxide with gentle shaking, we measured the absorbance at 570 nm.
Sulforhodamine B (SRB) assay was performed using CYTOSCANTM SRB Assay kit (cat. 786213; G-Biosciences, St. Louis, MO, USA) according to the manufacturer instruction. Briefly, microglia were plated in 48-well plates at densities varying from 1 × 104 to 4 × 104 cells/well. One day after plating, the cells were treated with vehicle control, LPS, LPS + PTX, and PTX separately at indicated dosage for another 48 h. The cells were then fixed with 10% trichloroacetic acid followed by staining with 0.4% (wt/vol) SRB for 30 min at room temperature, and extra dye was washed with 1% acetic acid. Finally, 10 mM Tris base was added to dissolve the dye, and the optical densities at 530 nm were measured using a spectrophotometric plate reader (Bio-Tek, USA). Data were expressed as percentage of viable cells compared to the control culture.
Western Blot Analysis
The brain tissue of the affected side of MCAO mice was crushed in liquid nitrogen, and the tissue was placed on lysate (RIPA + 1% protein inhibiting enzyme) to be lysed on ice. After centrifugation, the supernatant was used for Western blot analysis. Anti–IBA-1 (1:500), anti-CX3CR1 (1:500), and anti–β-actin (1:1,000) were used as primary antibodies. Protein detection and quantification were performed according to the manufacturer’s protocol.
Enzyme-Linked Immunosorbent Assay
Microglial cultured media were harvested to measure interleukin 1β (IL-1β) and tumor necrosis factor α (TNF-α) using Quantikine enzyme-linked immunosorbent assay (ELISA) kit (cat. number MTA00B for TNF-α, cat. MLB00C for IL-1β; R&D System, Minneapolis, MN, USA). Briefly, specimen was measured in parallel with a series of standard diluted control sample, which was used to establish standard curve. The optical density of each well was measured using a microplate reader set to 450 nm. Wavelength correction was performed at 540 and 570 nm.
Statistical Analysis
The results are presented as the means ± SEM. Statistical differences between two groups were evaluated by the two-tailed unpaired Student’s t-test. Multiple comparisons were performed with two-way analysis of variance (ANOVA) accompanied by Bonferroni post hoc test. P < 0.05 was considered significant.
Results
PTX Reduced Poststroke Infarct Volume and Functional Deficit
Stroke animals in PTX treatment group received daily intra-abdominal injection of PTX (400 ng per day, 1 ml) for three consecutive days. Stroke animals in the control group received daily intra-abdominal injection of 1 ml saline. The animals were sacrificed 72 h poststroke, and the brain slices were stained with TTC to evaluate infarct volume. PTX therapy significantly reduced infarct volume of therapy group (average infarct percentage = 34% ± 8%) compared to that of the saline-treated group (52% ± 12%, P < 0.05, Student’s t-test), suggesting PTX therapy effectively reduces cell death associated with MCAO-induced ischemia (Figures 1A,B). Further, Longa scores were assessed at 24, 48, and72 h poststroke. At 24 and 48 h poststroke, the average Longa score in the therapy group was not different from that of the saline-treated group. At 72 h poststroke, however, the average score was 1.6 in the therapy group, significantly reduced compared to 2.3 in the control group (P < 0.05), suggesting that 3 days’ treatment with PTX prevented functional deficit (Figure 1C). The lack of therapeutic effect at early stage is perhaps due to insufficient time for the neurological dysfunction to be fully developed. These data are consistent with our previous study, supporting the hypothesis that PTX therapy reduces necrosis and functional deficit (Tang et al., 2015).
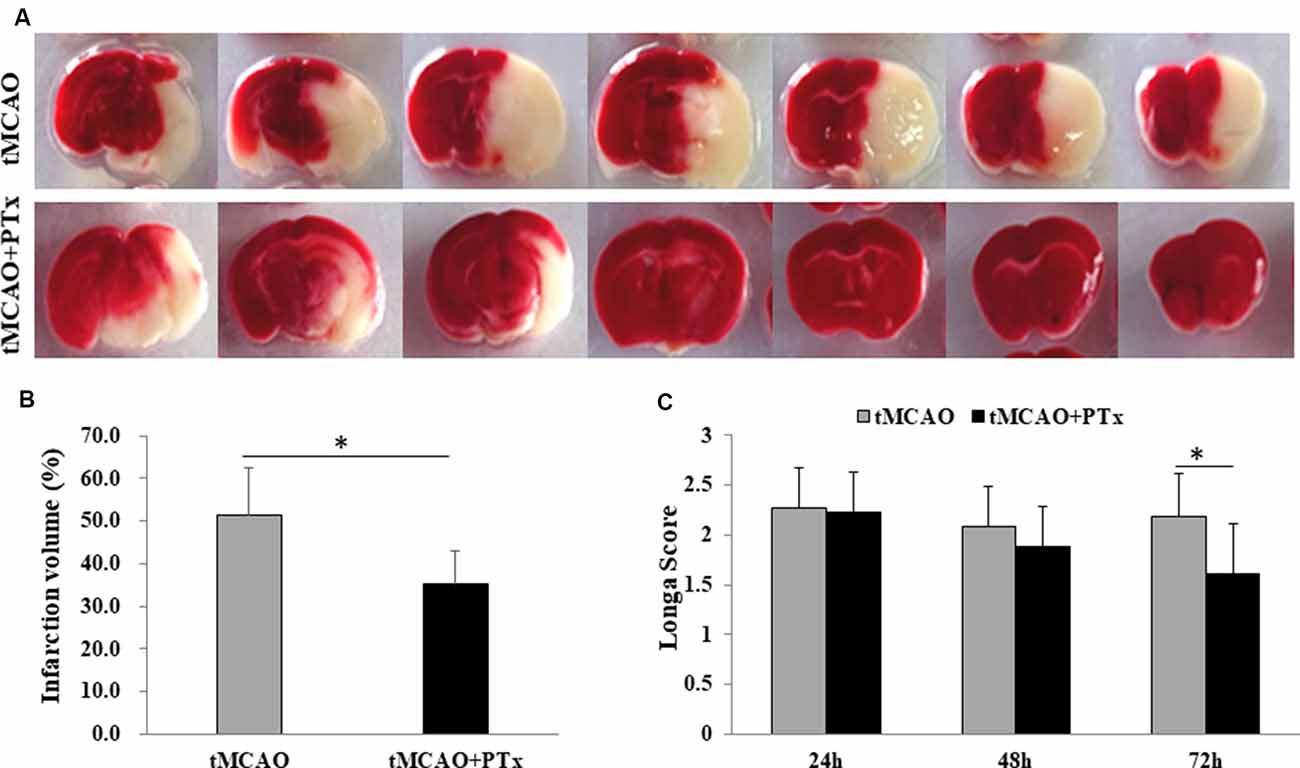
Figure 1. Pertussis toxin (PTX) reduced poststroke infarct volume and functional deficit. (A) Representative images of triphenyltetrazolium chloride (TTC)-stained brain slice in stroke mice with or without PTX treatment. The infarction area (non-stained region) was quantified in each slice and cumulated as a percentage of total volume. Note PTX-treated animals showed relative less area of infarction. (B) Statistical comparison of infarction volume between stroke mice with or without PTX treatment. (C) Neurological deficit was assessed using Longa score (*P < 0.05).
PTX Reduced Microglial Quantity After MCAO-Induced Ischemia
We evaluate microglial aggregation in MCAO ischemic brain area by counting IBA-1–positive cells. At 72 h poststroke, massive IBA-1–positive cells accumulated in necrotic regions, and their morphology transformed from resting state into enlarged, amoeba-like cell body. The quantity of IBA-1–positive microglia was reduced in the PTX-treated group, and they presented as branch-like morphology, a shape in between the activated and resting states (Figure 2A). The IBA-1–positive cell count was 25 ± 4 per 200× microscopic field in non-stroke (sham surgery) mice brain. The average count was 183 ± 46 at 72 h poststroke in saline-treated stroke mice, but was as low as 63 ± 22 in PTX-treated stroke mice (P < 0.05, one-way ANOVA with post hoc Bonferroni test; Figure 2B). Consistent with cell count, the IBA1 expression level was significantly reduced by 21% in PTX-treated mice, as compared to the saline-treated MCAO group (P < 0.05, one-way ANOVA with post hoc Bonferroni test; Figures 2C,D).
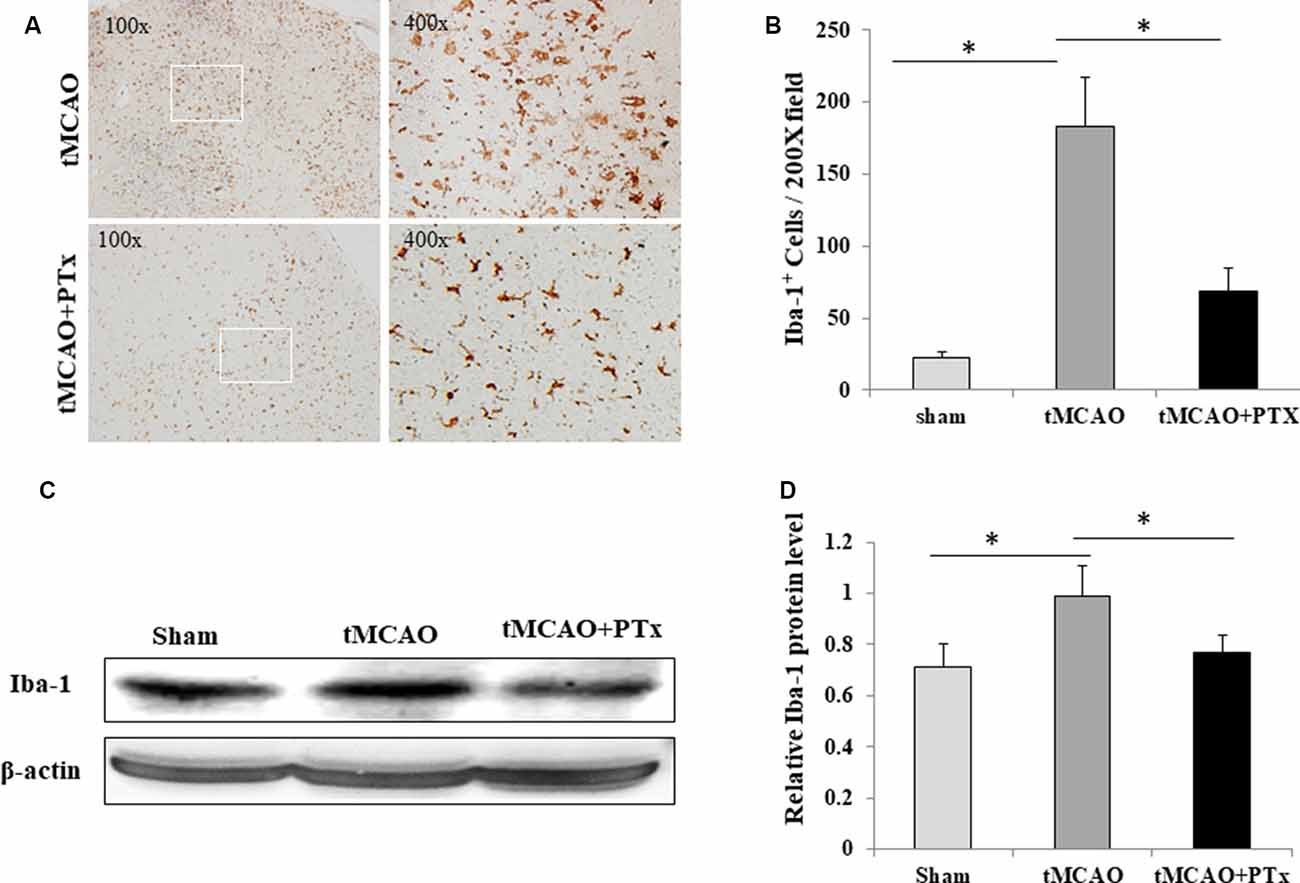
Figure 2. PTX reduced microglial quantity after MCAO-induced ischemia. (A) Representative images of IBA-1–stained microglia in brain sections obtained from transient middle cerebral artery occlusion (tMCAO) mice. (B) Statistical comparison of IBA-1+ cell counts between MCAO mice treated with or without PTX. (C,D) The ratio of IBA-1/β-actin immunoblot density was compared between tMCAO mice with or without PTX treatment (*P < 0.05).
PTX Reduced Microglial Proliferation in vivo and in vitro
The source of microglia in stroke may come from blood-borne macrophages that enter central nervous system through damaged blood–brain barrier. Alternatively, resident microglia in brain may rapidly proliferate. As IBA-1 is a specific marker for microglia, it is likely that PTX reduced microglial cell number by inhibiting their proliferation. To evaluate intrinsic microglial proliferative capability, first we count the Ki67-positive cells. At 72 h poststroke, massive Ki67-positive cells was observed in necrotic regions, and PTX treatment could reduce Ki67-positive cells (Figure 3C). The Ki67-positive cell count was 3 ± 2 per 200× microscopic field in nonstroke (sham surgery) mice brain. The average count was 42 ± 9 at 72 h poststroke in saline-treated stroke mice, but was as low as 12 ± 5 in PTX-treated stroke mice (P < 0.05, one-way ANOVA with post hoc Bonferroni test; Figure 3D). Then, we stimulated primary cultured microglia with LPS, mimicking the pathophysiologic process in vivo (Lund et al., 2006). Lipopolysaccharide significantly increased microglial proliferation resulting in a larger population of surviving cells that is 20% higher than non-stimulated microglia. PTX inhibited LPS-stimulated microglial proliferation by approximately 30% (Figure 3A). Consistently, SRB assay revealed a similar suppression of LPS-stimulated microglia (Figure 3B).
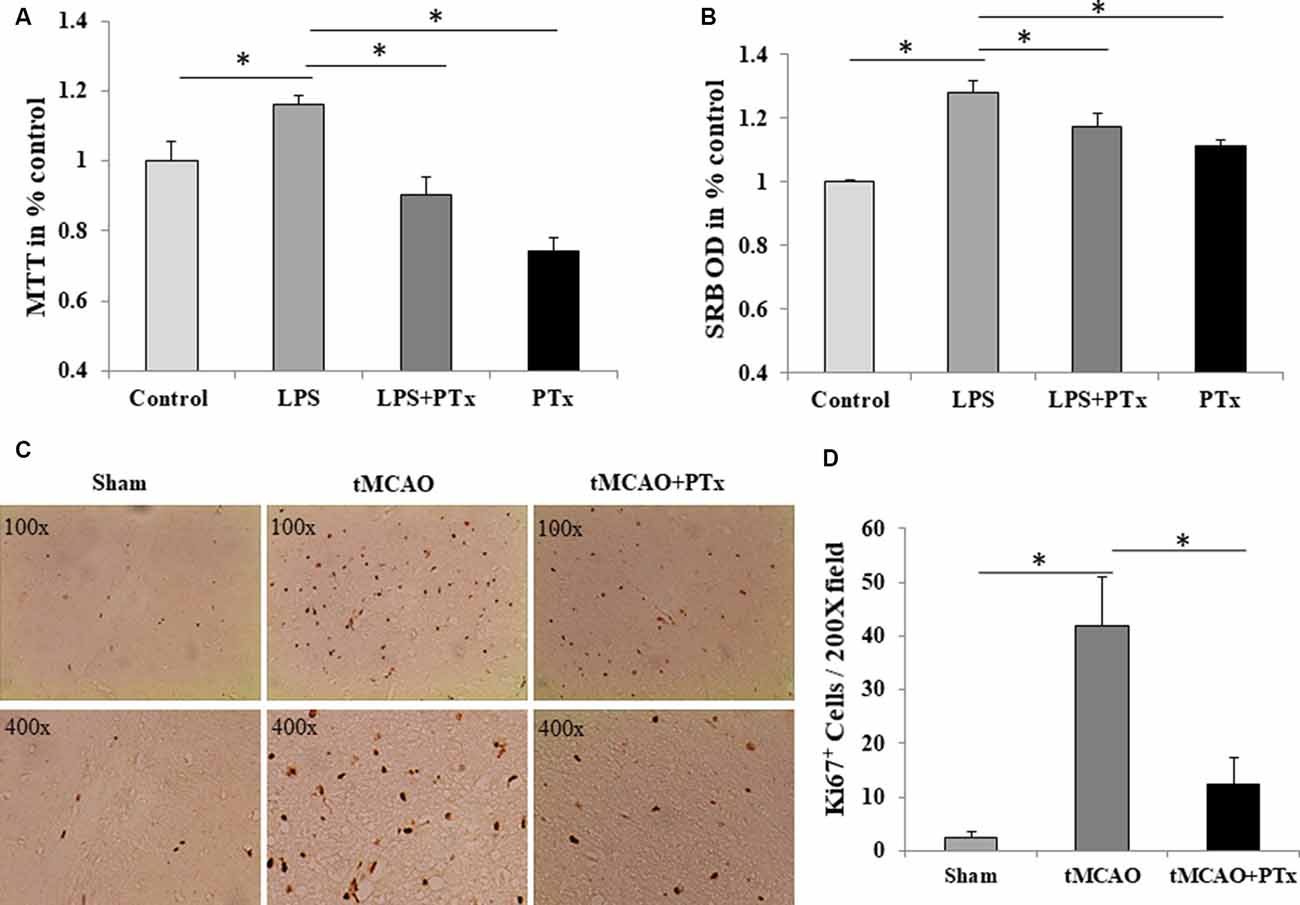
Figure 3. PTX reduced microglial proliferation in vivo and in vitro. (A) PTX treatment (50 ng/ml) reduced MTT quantity compared to lipopolysaccharide (LPS) stimulated microglia. (B) PTX treatment (50 ng/ml) reduced Sulforhodamine B (SRB) quantity compared to LPS stimulated microglia (*P < 0.05). (C) Representative images of Ki67+-stained cells in brain sections obtained from tMCAO mice. (D) Statistical comparison of Ki67+ cell counts between MCAO mice treated with or without PTX (*P < 0.05).
PTX Reduced Microglial Cytokine Release in Response to LPS Stimulation
We evaluate microglial cytokine release by measuring representative inflammatory factors, IL-1β and TNF-α, using ELISA assay. Cultured microglia were added with 50 ng/ml PTX (treatment group), LPS only, LPS + PTX, or with equal volume culture media (control group). The cultured cells and media were harvested at 24 h after LPS stimulation. When the cultured cells were treated with PTX only without LPS stimulation, the microglia showed a similar morphology as control, suggesting that PTX, at least at this concentration of 50 ng/ml, is not toxic to microglia. Lipopolysaccharide-stimulated microglia morphed into a distinct large amoeboid cell, whereas microglia treated with PTX demonstrated morphology similar to resting microglia with long spread cell body and scant branches (Figure 4A). We harvest the media to measure IL-1β and TNF-α in the conditioned media. In LPS-stimulated media, IL-1β was increased from 0.128 ± 0.003 to 0.165 ± 0.001 pg/ml, and TNF-α was increased from 0.128 ± 0.001 to 0.75 ± 0.003 pg/ml. In the PTX treatment group, IL-1β was reduced from 0.165 ± 0.001 to 0.091 ± 0.0004 pg/ml, and TNF-α was reduced from 0.75 ± 0.003 to 0.46 ± 0.005 pg/ml (Figures 4B,C; P < 0.05, one-way ANOVA with post hoc Bonferroni test).
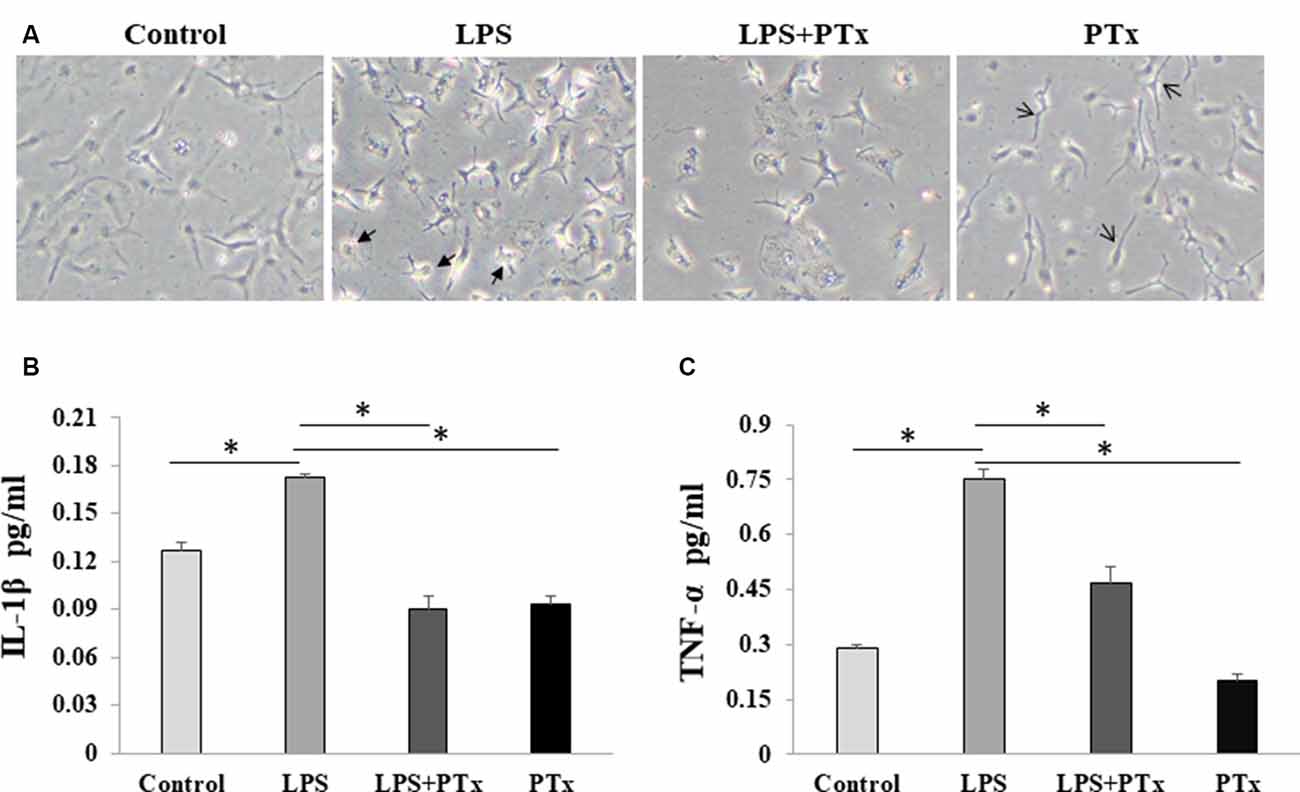
Figure 4. PTX reduced microglial cytokine release in response to LPS stimulation. (A) Representative images of cultured microglia at the time of harvest at 24 h. Two kinds of arrows indicate the distinct large amoeboid (thick arrow symbol) and long spread body cells (thin arrow symbol) and long spread body cells. (B) ELISA quantification of IL-1β in conditioned media. (C) ELISA quantification of TNF-α in conditioned media (*P < 0.05).
PTX Reduced Expression of CX3CCR1 by Microglia
In primary cultured microglia without LPS stimulation, CX3CR1 expression is low with the average fluorescent intensity of 0.02 ± 0.001. Lipopolysaccharide-stimulated microglia demonstrated significant increase in CX3CR1 fluorescent intensity up to 0.043 ± 0.003. PTX treatment of LPS-stimulated microglia reduced CX3CR1 fluorescent intensity down to 0.025 ± 0.003. PTX treatment alone without LPS stimulation was similar to control (Figures 5A,B). Consistent with cultured microglia in vitro, CX3CR1 expression in MCAO stroke mouse was increased 30% as compared to non-stroke sham-surgical mice. PTX–treated stroke mice decreased the CX3CR1 expression back to a level similar to sham-surgical mice (Figures 5C,D).
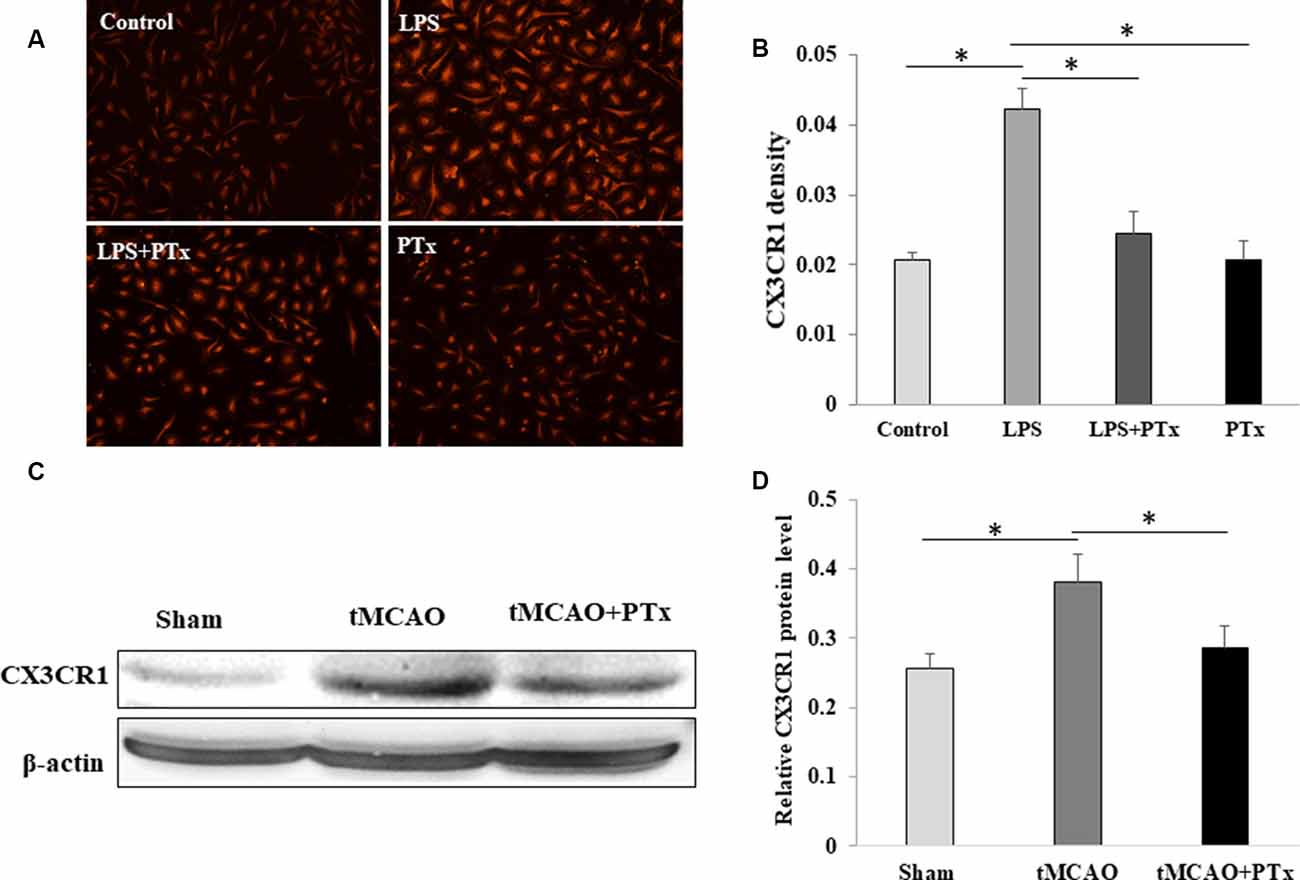
Figure 5. PTX reduced expression of CX3CCR1 by microglia. (A,B) Representative images (A) and statistical comparison (B) of fluorescent density of CX3CR1-binding antibody in cultured microglia. Note PTX decreased CX3CR1 fluorescent density. (C,D) The density ratio of CX3CR1/β-actin was decreased in PTX treated stroke mice (*P < 0.05).
Discussion
The primary goal for modern stroke therapy is to salvage tissue in ischemic penumbra aiming to minimize long-term functional deficit. Since the proposal of penumbra concept in 1970s, it is still the primary therapeutic goal as of today (Campbell et al., 2014; Davis and Donnan, 2014; Baron, 2018). While early reperfusion by thrombolysis or thrombectomy is a crucial step, reducing the cell vulnerability is equally essential to minimize reperfusion injury. The infarct tissue inevitably induces inflammation by activating microglia, the resident scavenging cells in the central nervous system. While microglia play an important role in phagocytosing the necrotic tissue, they release multiple proinflammatory factors to recruit lymphocytes and neutrophils (Neumann et al., 2018). The large amount of proinflammatory cytokines may damage the neighboring neurons in the penumbra. Therefore, fine-tuning the microglial reaction after stroke is a potential therapeutic target in tandem with neuroprotection, and recent studies provide encouraging progress (Bok et al., 2017).
In this study, we showed that PTX decreased infarct volume in MCAO stroke model at 72 h poststroke, a timeline consistent with cerebral perfusion kinetics(Tang et al., 2015). In this our study, we focused on the acute stage of ischemic stroke; thus, we investigated the effects of PTX on stroke at 24 and 72 h after treatment. While the PTX did not reduce the infarct volume and improve the neurological function at 24 h after stroke, and the IBA-1+ cells increased only mildly, we did not observe that PTX can inhibit microglia activation at this time point, whereas at 72 h, PTX inhibited microglia activation and decreased infarct volume dramatically. In different disease, the time point that PTX inhibits inflammatory responses is different (Andreasen and Carbonetti, 2008; Connelly et al., 2012). PTX also protected neuronal apoptosis; the underlying mechanisms include amelioration of calcium overload (Tang et al., 2015). However, given the potent effect of PTX, a direct neuroprotection is unlikely the only mechanism. PTX may have even broader effects on multiple players in the context of ischemic stroke. Therefore, we tested the effects of PTX on microglial activation in this study.
Microglial proliferation was observed in our tMCAO model as indicated by increased IBA-1+ and Ki67+ cells. PTX effectively reduced the number of microglia; the reason should include both limiting the recruitment of microglia and inhibiting the proliferation of microglia. Several articles have reported that PTX could inhibit microglia migration (Yin et al., 2010). In our study, we focus on the inhibition of microglia proliferation by PTX. We found cell proliferation increased remarkably 72 h after stroke, whereas PTX can inhibit this proliferation. This in vivo effect was modeled in vitro by stimulating cultured microglia with LPS but suppressed by PTX. The cultured microglia were assessed by two independent methods: MTT and SRB assays. Sulforhodamine B measures the level of constitutive protein corresponding to the amount of live cell because dead cells were washed away prior to the assay. MTT assay is predicated not only on cell number but also on mitochondrial quantity. Therefore, the suppressive effect of PTX appears larger in MTT assay than that in SRB assay. Nevertheless, both assays confirmed the inhibitory effects of PTX on LPS-stimulated microglial proliferation.
PTX could change microglia morphology. The authors showed that PTX induced a dose-dependent morphological transformation. At 125 pg/ml, PTX almost does not change microglial morphology. Typical change into small and rounded macrophages happened at 125 ng/ml; at 12.5 mg/ml, many cells showed lamellar growth surfaces and short filopodial processes indicative of amoeboid morphology (Kalla et al., 2003). In our study, we use 50 ng/ml of PTX to treat the culture microglia, which did not change the cells into small and rounded macrophages; the microglia showed a similar morphology as control, suggesting that PTX, at least at this concentration of 50 ng/ml, is not toxic to microglia, and it can inhibit the LPS effect to microglia. In another article (Ahmad et al., 2019), at 2.75 ng/ml, PTX can inhibit maturation of human monocyte precursors into the more phagocytic macrophage cells. It is similar to our data. All these indicated that low concentration of PTX is not toxic to microglia, and it can inhibit microglia change to macrophages.
Upon microglial activation, multiple cytokines are released from microglia. Among these, IL-1β is the immediate early response cytokine (Kim et al., 2006), which further orchestrates underlying inflammatory response (Kaushik et al., 2013). TNF-α is a signal linking inflammation and excitotoxicity (Olmos and Lladó, 2014). Activated microglia secrete TNF-α, which further stimulates additional microglia via autocrine mechanism (Kuno et al., 2005). Meanwhile, TNF-α has a direct effect on glutamate receptor inducing unabated calcium influx resulting in excitotoxicity (Han and Whelan, 2010). Therefore, we chose to analyze microglia-secreted IL-1β and TNF-α as the most relevant and representative cytokines in our study, because IL-1β is an early marker for microglial activation, and TNF-α is an effector cytokine for neuronal damage. Our data showed that PTX reduced LPS-stimulated IL-1β and TNF-α secretion. Although LPS and their targeted TLR are not known to be associated with G-protein signaling, secondary signaling (e.g., purinergic receptor signal) may be subject to G-protein–related modulation (Netea et al., 2009), hence a potential step that can be blocked by PTX.
The chemokine fractalkine (CX3CL1)/CX3C chemokine receptor 1 (CX3CR1) system is a critical signaling system modulating the communication between neurons and microglia (Harrison et al., 1998; Mizuno et al., 2003). CX3C chemokine receptor 1 is expressed predominantly in astrocytes and microglia (Maciejewski-Lenoir et al., 1999), but moderately in neurons as well (Meucci et al., 2000; Wang et al., 2018). Our previous work demonstrated that CX3CR1-mediated microglia activation was associated with neuronal apoptosis in the penumbra 72 h after transient cerebral artery occlusion in mice, and CX3CR1 deficiency suppresses the neurotoxic effect of microglia (Tang et al., 2014). Therefore, CX3CR1 upregulation is a necessary event in microglia-mediated neurotoxicity. Here, we show the suppressive effects of PTX on microglial CX3CR1 expression in vitro and in vivo, suggesting that CX3CL1–CX3CR1 is a likely target of PTX. In fact, CX3CR1 belongs to a family of G-protein–coupled receptor functionally interacting with Gαi, a target of PTX (Poniatowski et al., 2017). Further study is needed to elucidate the mechanism why blocking a downstream signaling protein provides a negative feedback to upstream receptor and reduced the expression level of CX3CR1.
The mechanism that PTX inhibits microglia remains elusive. Nevertheless, it has been noted that PTX suppresses the function of pulmonary alveolar macrophage (Carbonetti et al., 2007), perhaps as a strategy to fend host innate immune system and establish B. pertussis infection. PTX targeted several G-protein signaling pathways in macrophage (Lattin et al., 2007; Carbonetti, 2010). Given that microglia and macrophage are derived from common embryonic cell lineage and share common molecular mechanisms in their functions (Li and Barres, 2018), it is not surprising to observe that PTX inhibits microglial proliferation and inflammatory response. Indeed, microglial inhibition by PTX was also observed in EAE; the total number of IBA-1–positive cells was reduced by 69.6% with PTX treatment (Yin et al., 2014).
The suppression of microglial activation by PTX is in tandem with a direct neuroprotective effect of PTX. We previously demonstrated that PTX significantly reduced calcium influx via glutamate receptor and voltage-gated calcium channel (Tang et al., 2015). Further, our recent data suggest that neuronal CX3CR1 upregulation may be directly related to glutamate receptor–mediated calcium influx (Tang et al., 2014). Therefore, it is possible that PTX blocks CX3CR1 signaling in neurons, hence indirectly reduced calcium overload. One potential possibility is that reduced cell death decreases microglial activation; hence, the suppressive effect of PTX on microglia may be attributed to an indirect outcome subsequent to better neuronal protection. While this indirect mechanism certainly exists, PTX likely exerts direct effect on microglia; as shown in this study, primary cultured microglia in vitro were equally suppressed by PTX as well as in vivo. Therefore, PTX provides a neuroprotective effect through multiple nonexclusive mechanisms involving both neurons and microglia.
Conclusion
We demonstrated here that PTX ameliorated postischemic microglial proliferation, activation, cytokine release, and the expression of proinflammatory signal molecules. The suppression of microglia in alignment with direct neuroprotection suggests that PTX has a multifaceted therapeutic function, a unique advantage in treating ischemic stroke.
Data Availability Statement
All datasets generated for this study are included in the article.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of the first hospital of Kunming Medical University.
Author Contributions
FZ is the first author, she is in charge of most of the experimental works and writing. ZT is the corresponding author, he is in charge of project funding application, project design and writing. RL is in charge of experimental guidance and project design. PH is in charge of article modification. XZ and ZL are in charge of making animal model. SZ and CL are in charge of immunohistochemistry and TTC. YX is in charge of ELISA.
Funding
This work was supported by the National Natural Science Foundation of China (81360204 and 81760220 to ZT), Yunnan Applied Basic Research Projects (2017FE467-135, 2017FB111, 2017NS057 and 2016NS058 to ZT), and The Yunnan province research innovation team for the effect of neurodevelopmental regulatory factors in neural repair and clinical application (Kunming Medical University, 2018HC084).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahmad, J. N., Holubova, J., Benada, O., Kofronova, O., Stehlik, L., Vasakova, M., et al. (2019). Bordetella adenylate cyclase toxin inhibits monocyte-to-macrophage transition and dedifferentiates human alveolar macrophages into monocyte-like cells. mBio 10:e01743-19. doi: 10.1128/mbio.01743-19
Andreasen, C., and Carbonetti, N. H. (2008). Pertussis toxin inhibits early chemokine production to delay neutrophil recruitment in response to Bordetella pertussis respiratory tract infection in mice. Infect. Immun. 76, 5139–5148. doi: 10.1128/iai.00895-08
Baron, J. C. (2018). Protecting the ischaemic penumbra as an adjunct to thrombectomy for acute stroke. Nat. Rev. Neurol. 14, 325–337. doi: 10.1038/s41582-018-0002-2
Bok, S., Kim, Y. E., Woo, Y., Kim, S., Kang, S. J., Lee, Y., et al. (2017). Hypoxia-inducible factor-1α regulates microglial functions affecting neuronal survival in the acute phase of ischemic stroke in mice. Oncotarget 67, 111508–111521. doi: 10.18632/oncotarget.22851
Campbell, B. C., Donnan, G. A., and Davis, S. M. (2014). Vessel occlusion, penumbra, and reperfusion—translating theory to practice. Front. Neurol. 5:194. doi: 10.3389/fneur.2014.00194
Carbonetti, N. H. (2010). Pertussis toxin and adenylate cyclase toxin: key virulence factors of Bordetella pertussis and cell biology tools. Future Microbiol. 5, 455–469. doi: 10.2217/fmb.09.133
Carbonetti, N. H., Artamonova, G. V., Van Rooijen, N., and Ayala, V. I. (2007). Pertussis toxin targets airway macrophages to promote Bordetella pertussis infection of the respiratory tract. Infect. Immun. 75, 1713–1720. doi: 10.1128/iai.01578-06
Chan, W. Y., Kohsaka, S., and Rezaie, P. (2007). The origin and cell lineage of microglia: new concepts. Brain Res. Rev. 53, 344–354. doi: 10.1016/j.brainresrev.2006.11.002
Chen, J., Zacharek, A., Zhang, C., Jiang, H., Li, Y., Roberts, C., et al. (2005). Endothelial nitric oxide synthase regulates brain-derived neurotrophic factor expression and neurogenesis after stroke in mice. J. Neurosci. 9, 2366–2375. doi: 10.1523/JNEUROSCI.5071-04.2005
Connelly, C. E., Sun, Y., and Carbonetti, N. H. (2012). Pertussis toxin exacerbates and prolongs airway inflammatory responses during Bordetella pertussis infection. Infect. Immun. 80, 4317–4332. doi: 10.1128/iai.00808-12
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 6, 752–758. doi: 10.1038/nn1472
Davis, S., and Donnan, G. A. (2014). Time is Penumbra: imaging, selection and outcom. The Johann jacob wepfer award 2014. Cerebrovasc. Dis. 38, 59–72. doi: 10.1159/000365503
Di Carlo, A. (2009). Human and economic burden of stroke. Age Ageing 38, 4–5. doi: 10.1093/ageing/afn282
Han, P., and Whelan, P. J. (2010). Tumor necrosis factor α enhances glutamatergic transmission onto spinal motoneurons. J. Neurotrauma 27, 287–292. doi: 10.1089/neu.2009.1016
Harrison, J. K., Jiang, Y., Chen, S., Xia, Y., Maciejewski, D., McNamara, R. K., et al. (1998). Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. U S A 18, 10896–10901. doi: 10.1073/pnas.95.18.10896
Hu, X., Li, P., Guo, Y., Wang, H., Leak, R. K., Chen, S., et al. (2012). Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 11, 3063–3070. doi: 10.1161/strokeaha.112.659656
Kalla, R., Bohatschek, M., Kloss, C. U., Krol, J., Von Maltzan, X., and Raivich, G. (2003). Loss of microglial ramification in microglia-astrocyte cocultures: involvement of adenylate cyclase, calcium, phosphatase, and Gi-protein systems. Glia 41, 50–63. doi: 10.1002/glia.10176
Kaushik, D. K., Thounaojam, M. C., Kumawat, K. L., Gupta, M., and Basu, A. (2013). Interleukin-1β orchestrates underlying inflammatory responses in microglia via Kruppel-like factor 4. J. Neurochem. 127, 233–244. doi: 10.1111/jnc.12382
Kim, S. U., and de Vellis, J. (2005). Microglia in health and disease. J. Neurosci. Res. 81, 302–313. doi: 10.1002/jnr.20562
Kim, Y. J., Hwang, S. Y., Oh, E. S., Oh, S., and Han, I. O. (2006). IL-1β, an immediate early protein secreted by activated microglia, induces iNOS/NO in C6 astrocytoma cells through p38 MAPK and NF-κB pathways. J. Neurosci. Res. 84, 1037–1046. doi: 10.1002/jnr.21011
Kuno, R., Wang, J., Kawanokuchi, J., Takeuchi, H., Mizuno, T., and Suzumura, A. (2005). Autocrine activation of microglia by tumor necrosis factor-α. J. Neuroimmunol. 162, 89–96. doi: 10.1016/j.jneuroim.2005.01.015
Lattin, J., Zidar, D. A., Schroder, K., Kellie, S., Hume, D. A., and Sweet, M. J. (2007). G-protein-coupled receptor expression, function, and signaling in macrophages. J. Leukoc. Biol. 82, 16–32. doi: 10.1189/jlb.0107051
Li, Q., and Barres, B. A. (2018). Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225–242. doi: 10.1038/nri.2017.125
Lian, H., Roy, E., and Zheng, H. (2016). Protocol for primary microglial culture preparation. Bio Protoc. 6:e1988. doi: 10.21769/BioProtoc.1988
Liesz, A., Suri-Payer, E., Veltkamp, C., Doerr, H., Sommer, C., Rivest, S., et al. (2009). Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 2, 192–199. doi: 10.1038/nm.1927
Liu, L., Wang, D., Wong, K. S., and Wang, Y. (2011). Stroke and stroke care in China: huge burden, significant workload and a national priority. Stroke 42, 3651–3654. doi: 10.1161/strokeaha.111.635755
Lund, S., Christensen, K. V., Hedtjarn, M., Mortensen, A. L., Hagberg, H., Falsig, J., et al. (2006). The dynamics of the LPS triggered inflammatory response of murine microglia under different culture and in vivo conditions. J. Neuroimmunol. 180, 71–87. doi: 10.1016/j.jneuroim.2006.07.007
Maciejewski-Lenoir, D., Chen, S., Feng, L., Maki, R., and Bacon, K. B. (1999). Characterization of fractalkine in rat brain cells: migratory and activation signals for CX3CR-1-expressing microglia. J. Immunol. 163, 1628–1635.
Meucci, O., Fatatis, A., Simen, A. A., and Miller, R. J. (2000). Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc. Natl. Acad. Sci. U S A 97, 8075–8080. doi: 10.1073/pnas.090017497
Mizuno, T., Kawanokuchi, J., Numata, K., and Suzumura, A. (2003). Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 979, 65–70. doi: 10.1016/s0006-8993(03)02867-1
Moskowitz, M. A., Lo, E. H., and Iadecola, C. (2010). The science of stroke: mechanisms in search of treatments. Neuron 67, 181–198. doi: 10.1016/j.neuron.2010.08.019
Netea, M. G., Nold-Petry, C. A., Nold, M. F., Joosten, L. A., Opitz, B., van der Meer, J. H., et al. (2009). Differential requirement for the activation of the inflammasome for processing and release of IL-1β in monocytes and macrophages. Blood 10, 2324–2335. doi: 10.1182/blood-2008-03-146720
Neumann, J., Henneberg, S., von Kenne, S., Nolte, N., Muller, A. J., Schraven, B., et al. (2018). Beware the intruder: real time observation of infiltrated neutrophils and neutrophil-Microglia interaction during stroke in vivo. PLoS One 13:e0193970. doi: 10.1371/journal.pone.0193970
Olmos, G., and Lladó, J. (2014). Tumor necrosis factor α: a link between neuroinflammation and excitotoxicity. Mediators Inflamm. 2014:861231. doi: 10.1155/2014/861231
Perego, C., Fumagalli, S., and De Simoni, M. G. (2011). Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflammation 8:174. doi: 10.1186/1742-2094-8-174
Poniatowski, L. A., Wojdasiewicz, P., Krawczyk, M., Szukiewicz, D., Gasik, R., Kubaszewski, L., et al. (2017). Analysis of the role of CX3CL1 (Fractalkine) and its receptor CX3CR1 in traumatic brain and spinal cord injury: insight into recent advances in actions of neurochemokine agents. Mol. Neurobiol. 3, 2167–2188. doi: 10.1007/s12035-016-9787-4
Tang, Z., Gan, Y., Liu, Q., Yin, J. X., Liu, Q., Shi, J., et al. (2014). CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. J. Neuroinflammation 11:26. doi: 10.1186/1742-2094-11-26
Tang, Z., Li, S., Han, P., Yin, J., Gan, Y., Liu, Q., et al. (2015). Pertussis toxin reduces calcium influx to protect ischemic stroke in a middle cerebral artery occlusion model. J. Neurochem. 5, 998–1006. doi: 10.1111/jnc.13359
Tang, Z., Yin, J. X., Han, P., Gan, Y., Coons, S. W., Wang, C., et al. (2013). Pertussis toxin attenuates experimental autoimmune encephalomyelitis by upregulating neuronal vascular endothelial growth factor. Neuroreport 9, 469–475. doi: 10.1097/wnr.0b013e3283619fc8
Wang, J., Gan, Y., Han, P., Yin, J., Liu, Q., Ghanian, S., et al. (2018). Ischemia-induced neuronal cell death is mediated by chemokine receptor CX3CR1. Sci. Rep. 1:556. doi: 10.1038/s41598-017-18774-0
Wang, Y. C., Lin, S., and Yang, Q. W. (2011). Toll-like receptors in cerebral ischemic inflammatory injury. J. Neuroinflammation 8:134. doi: 10.1186/1742-2094-8-134
Wang, G., Zhang, J., Hu, X., Zhang, L., Mao, L., Jiang, X., et al. (2013). Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J. Cereb. Blood Flow Metab. 12, 1864–1874. doi: 10.1038/jcbfm.2013.146
Xie, C. J., Gu, A. P., Cai, J., Wu, Y., and Chen, R. C. (2018). Curcumin protects neural cells against ischemic injury in N2a cells and mouse brain with ischemic stroke. Brain Behav. 8:e00921. doi: 10.1002/brb3.921
Xiong, X. Y., Liu, L., and Yang, Q. W. (2016). Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 142, 23–44. doi: 10.1016/j.pneurobio.2016.05.001
Yin, J., Han, P., Tang, Z., Liu, Q., and Shi, J. (2015). Sirtuin 3 mediates neuroprotection of ketones against ischemic stroke. J. Cereb. Blood Flow Metab. 35, 1783–1789. doi: 10.1038/jcbfm.2015.123
Yin, J. X., Tang, Z., Gan, Y., Li, L., Shi, F., Coons, S., et al. (2014). Pertussis toxin modulates microglia and T cell profile to protect experimental autoimmune encephalomyelitis. Neuropharmacology 81, 1–5. doi: 10.1016/j.neuropharm.2014.01.027
Yin, J. X., Tu, J. L., Lin, H. J., Shi, F. D., Liu, R. L., Zhao, C. B., et al. (2010). Centrally administered pertussis toxin inhibits microglia migration to the spinal cord and prevents dissemination of disease in an EAE mouse model. PLoS One 5:e12400. doi: 10.1371/journal.pone.0012400
Keywords: pertussis toxin, stroke, microglia, neuroprotection, neuroinflammation
Citation: Zhou F, Liu R, Han P, Zhang X, Li Z, Zhang S, Liu C, Xia Y and Tang Z (2020) Pertussis Toxin Ameliorates Microglial Activation Associated With Ischemic Stroke. Front. Cell. Neurosci. 14:152. doi: 10.3389/fncel.2020.00152
Received: 26 January 2020; Accepted: 08 May 2020;
Published: 26 June 2020.
Edited by:
Veronica Ines Brito, Vall d’Hebron University Hospital, SpainReviewed by:
Karen Scanlon, University of Maryland School of Medicine, United StatesCiaran Skerry, University of Maryland, Baltimore, United States
Copyright © 2020 Zhou, Liu, Han, Zhang, Li, Zhang, Liu, Xia and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiwei Tang, dGFuZ3poaXdlaTc3NTVAaG90bWFpbC5jb20=