
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 11 February 2020
Sec. Cellular Neuropathology
Volume 14 - 2020 | https://doi.org/10.3389/fncel.2020.00016
This article is part of the Research Topic The Role of Cellular Senescence in Health and Disease View all 16 articles
 Carmen Martínez-Cué*
Carmen Martínez-Cué* Noemí Rueda
Noemí RuedaCellular senescence is a homeostatic biological process characterized by a permanent state of cell cycle arrest that can contribute to the decline of the regenerative potential and function of tissues. The increased presence of senescent cells in different neurodegenerative diseases suggests the contribution of senescence in the pathophysiology of these disorders. Although several factors can induce senescence, DNA damage, oxidative stress, neuroinflammation, and altered proteostasis have been shown to play a role in its onset. Oxidative stress contributes to accelerated aging and cognitive dysfunction stages affecting neurogenesis, neuronal differentiation, connectivity, and survival. During later life stages, it is implicated in the progression of cognitive decline, synapse loss, and neuronal degeneration. Also, neuroinflammation exacerbates oxidative stress, synaptic dysfunction, and neuronal death through the harmful effects of pro-inflammatory cytokines on cell proliferation and maturation. Both oxidative stress and neuroinflammation can induce DNA damage and alterations in DNA repair that, in turn, can exacerbate them. Another important feature associated with senescence is altered proteostasis. Because of the disruption in the function and balance of the proteome, senescence can modify the proper synthesis, folding, quality control, and degradation rate of proteins producing, in some diseases, misfolded proteins or aggregation of abnormal proteins. There is an extensive body of literature that associates cellular senescence with several neurodegenerative disorders including Alzheimer’s disease (AD), Down syndrome (DS), and Parkinson’s disease (PD). This review summarizes the evidence of the shared neuropathological events in these neurodegenerative diseases and the implication of cellular senescence in their onset or aggravation. Understanding the role that cellular senescence plays in them could help to develop new therapeutic strategies.
Cellular senescence, a homeostatic process that reduces proliferation and helps to prevent the propagation of damaged cells (Vicencio et al., 2008; Coppé et al., 2010; Faragher et al., 2017; Yanagi et al., 2017), has been proposed to be a type of cell differentiation (Stein et al., 1991). Among its most relevant biological functions are the counteraction of uncontrolled cell proliferation, which avoids the formation of tumors, and the facilitation of the elimination of cells that are damaged or that are no longer necessary (Kültz, 2005). Cellular senescence has an essential physiological role during development (Muñoz-Espín and Serrano, 2014; Barbouti et al., 2019).
According to its duration and characteristics, senescence has been categorized into acute or chronic senescence (van Deursen, 2014). Acute senescence is implicated in normal biological processes during embryonic development or tissue repair. While, chronic senescence, induced by prolonged exposure to stress (Salama et al., 2014; van Deursen, 2014; Childs et al., 2015; Velarde and Menon, 2016; Watanabe et al., 2017; Ogrodnik et al., 2019), produces cellular and tissue alterations. Although both types of senescent cells display similar characteristics in vitro and in vivo, they are triggered by different stimuli and have differential consequences on the tissue.
Chronic cellular senescence contributes to functional alterations associated with aging and neurodegenerative diseases (Howcroft et al., 2013); the number of cells with this phenotype increases with healthy aging (Herbig et al., 2004; Lawless et al., 2010).
Different categories of chronic senescence have been identified, including replicative senescence, stress-induced premature senescence (SIPS), and mitochondrial dysfunction-associated senescence, among others (Kuilman et al., 2010).
Replicative senescence is the process in which human fibroblasts cultured in vitro end up displaying the hallmarks of senescence (a rise in protein p21 levels cell cycle arrest, SA-β-Gal activation, and morphological alterations; Romanov et al., 2012; Sanders et al., 2013). Replicative senescence is currently considered a model of aging (Chen et al., 2007).
Different types of cells of the central nervous system (CNS) can become senescent, including astrocytes (Pertusa et al., 2007; Mansour et al., 2008; Salminen et al., 2011), microglia (Evans et al., 2003; Flanary and Streit, 2004; Flanary et al., 2007; Bitto et al., 2010), oligodendrocytes (Al-Mashhadi et al., 2015), neurons (Sedelnikova et al., 2004; Jurk et al., 2012), and Neural Stem Cells (NSCs; Ferrón et al., 2004; He et al., 2013; Li et al., 2016). Senescence of these cell types have been implicated in the etiopathology of several neurodegenerative diseases (Streit et al., 2004, 2009; Conde and Streit, 2006; Baker et al., 2011; Bhat et al., 2012; He et al., 2013; Nasrabady et al., 2018; Ohashi et al., 2018), including Alzheimer’s disease (AD), Parkinson Disease (PD), frontotemporal dementia, amyotrophic lateral sclerosis, and multiple sclerosis (Yurov et al., 2014; Biron-Shental et al., 2015). This review summarizes the main findings on the role of cellular senescence in the neurodegenerative diseases AD, Down syndrome (DS), and PD.
Senescent cells are characterized by:
(i) Permanent cell cycle arrest, due to the blockade to the entrance to the S phase of the cycle (Stein et al., 1999; Krenning et al., 2014).
(ii) Senescence-associated secretory phenotype (SASP), which consists of the synthesis and release of proinflammatory chemokines, cytokines, growth factors and metalloproteinases (Coppé et al., 2010; Acosta et al., 2013; Chen et al., 2015) responsible of neuroinflammatory processes (Coppé et al., 2010; Freund et al., 2010; Özcan et al., 2016).
During aging and in neurodegenerative diseases, microglial cells display altered morphology characteristic of senescence (Flanary and Streit, 2004; Streit et al., 2004; Flanary et al., 2007). However, it has to be taken into account that increased release of pro-inflammatory cytokines due to microglia or astroglia activation is not necessarily related to cellular senescence. During healthy aging, the brain suffers mild chronic inflammation (Yankner et al., 2008). There is evidence that this state is caused by the dysregulation of microglial activation (Sheng et al., 1998; Frank et al., 2006; von Bernhardi, 2007; Mosher and Wyss-Coray, 2014; von Bernhardi et al., 2015a), which enhances the release of numerous proinflammatory cytokines, including IL1α, IL-2, IL-6, IL-8, IL-12, IL-15, IL-17, IL-18, IL-22, IL-23, IL6, IFN-γ and TNFα (Sheng et al., 1998; Njie et al., 2012; von Bernhardi et al., 2015b; Minciullo et al., 2016; Ventura et al., 2017; Rea et al., 2018). Inflammation induces a higher release of proinflammatory cytokines in old than in young cells (Combrinck et al., 2002; Cunningham et al., 2005; Sierra et al., 2007; Henry et al., 2009).
In neurodegenerative diseases, neuronal damage also dysregulates microglia activation and induces an increase in the release of pro-inflammatory mediators (von Bernhardi, 2007; López-Otín et al., 2013).
(iii) Altered mitochondrial function and morphology, which is one of the most important triggers of senescence, mainly through the induction of oxidative stress, which will alter cellular signaling and SASP (Takahashi et al., 2005; Passos et al., 2007, 2010; Correia-Melo and Passos, 2015).
High levels of oxidative stress have been demonstrated to induce cellular senescence during aging (De Haan et al., 1996), promoting neuronal DNA damage (Chow and Herrup, 2015), a deregulated DDR (Sedelnikova et al., 2004), an alteration in the entry and progression of the cell cycle and changes in cell morphology (Monti et al., 1992), premature replicative senescence and an accelerated rate of telomere attrition per cellular replication (von Zglinicki et al., 2000; Serra et al., 2003).
Mitochondria is a major site of production as well as a target of reactive oxygen species (ROS), and its endogenous antioxidant pathways are essential to maintain physiological redox signaling (Brand, 2014). Furthermore, mitochondrial ATP production is necessary for neural activity. But aging deteriorates mitochondrial integrity and function leading to reduced ATP production, to enhanced ROS formation, mutations in mitochondrial DNA (Richter et al., 1988; Shigenaga et al., 1994), facilitating neurodegeneration (Dröge and Schipper, 2007).
Finally, mitochondrial ROS can also induce telomere attrition, and dysfunction (Passos et al., 2007) and mitochondrial dysfunction can also facilitate senescence by inducing alterations in the cell metabolism (Ziegler et al., 2015; Liguori et al., 2018).
(iv) Changes in cellular metabolism, partially due to the altered mitochondrial function, producing an increase in lysosomal senescence-associated-β-galactosidase (Lee et al., 2006; Weichhart, 2018), accumulation of lipofuscin in the cytoplasm (Georgakopoulou et al., 2013; Höhn and Grune, 2013), and a reduction in fatty acid synthesis (Wu et al., 2017). Due to the high energy demand, and increased oxidative stress the cells can accumulate high levels of DNA damage. This damage increases during normal aging, when the DNA repair capacity is reduced (Maynard et al., 2015).
(v) DNA damage that alters chromatin structure and activates the DNA damage response (DDR) (Nakamura et al., 2008; Rodier et al., 2011).
During replicative senescence, telomeres, DNA–proteins that protect chromosome ends from degradation, and inappropriate recombinations or fusions shorten with every cell division (Blackburn, 1991; Zakian, 1995; Chan and Blackburn, 2004; Hockemeyer et al., 2005). Telomere shortening reduces the protection of the ends of the chromosomes and leaves them exposed to DNA damage that can be similar to DNA double-strand break (d’Adda di Fagagna et al., 2003; Takai et al., 2003; Herbig et al., 2004; Rodier et al., 2009, 2011) that triggers the DDR. During senescence, the increase in DDR leaves these cells unable to perform DNA repair (Galbiati et al., 2017). Under these circumstances, the cell stops dividing (Harley et al., 1990; Harley, 1991; de Lange, 2002; d’Adda di Fagagna et al., 2003) leading to the cell cycle arrest characteristic of senescence.
Telomere shortening has been proposed to be a major mechanism in aging, and age-related pathology and a marker of cellular senescence (Greider and Blackburn, 1987; Olovnikov, 1996; Bernatdotte et al., 2016). The role of telomere attrition in aging and its correlation with senescence has been demonstrated in studies in primates, mice and humans (Blasco et al., 1997; Lee et al., 1998; Herbig et al., 2006; Hewitt et al., 2012; Kong et al., 2013; López-Otín et al., 2013; Birch et al., 2015).
Besides cellular replication, several stimuli can affect telomeres. There is evidence that mitochondrial dysfunction induces telomere damage, and that telomere damage can produce mitochondrial alterations (Zheng et al., 2019). Besides, oxidative stress accelerates telomere damage producing senescence (von Zglinicki et al., 2000; von Zglinicki, 2002; Saretzki et al., 2003; Serra et al., 2003), and the intracellular oxidative stress of a cell correlates with telomere attrition and with its replicative potential (Richter and von Zglinicki, 2007).
Although, as mentioned above, many studies have shown a relation between telomere attrition, senescence and aging (Harley et al., 1990; Allsopp et al., 1992; Hao et al., 2005; Heidinger et al., 2012; Kaul et al., 2012; Reichert et al., 2013), telomere length does not always correlate with senescence (Karlseder et al., 2002; Stewart et al., 2003).
Several reports failed to associate telomere length and mortality risk in elder humans (Bischoff et al., 2006; Li et al., 2015). Also, some studies have demonstrated that telomere dysfunction is not dependent on its length. In mice, and baboons longer telomeres with DNA-damage have been associated with aging (Fumagalli et al., 2012; Hewitt et al., 2012; Jurk et al., 2014).
Finally, as previously mentioned, telomere dysfunction is not only related to senescence in healthy aging but it is also found in patients with dementia (Kota et al., 2015), AD (Cai et al., 2013), and PD (Maeda et al., 2012).
The continuous activation of DDR produces senescence in neurons (Fielder et al., 2017). DNA damage induces senescence, enhanced oxidative stress and an associated increase in β-galactosidase activity in neurons of old mice (Jurk et al., 2012). Because the efficiency of DNA repair decreases with age, and more complex, and less efficient DNA repair mechanisms are used, the increased DNA damage can lead to neurodegeneration (Vaidya et al., 2014).
(vi) Epigenetic modifications. Epigenomic changes in senescent cells include an imbalance in repressive and active histone marks, heterochromatic alterations including formation of senescence-associated heterochromatic foci (SAHF), histone variants, altered nucleosomal composition, alterations in DNA methylation pattern, alterations in nuclear lamina-chromatin interactions and on 3D genome organization that contributes to cellular dysfunction (Yang and Sen, 2018; Wagner, 2019). Micro-RNAs, also participate in the regulation of cellular senescence (Komseli et al., 2018); resistance to apoptotic death, controlled by the p53 and p21 stress response pathway (Tang et al., 2006).
Epigenetic modification such as histone modifications and specific DNA methylation changes, including alterations in the activity of methylation enzymes (Vanyushin et al., 1973), occur during aging and are associated with the neuropathology and the progression of various neurodegenerative diseases (Berson et al., 2018; Prasad and Jho, 2019; Wagner, 2019). During aging, these changes in DNA methylation and histone modifications alter chromatin architecture (Tsurumi and Li, 2012). Aged tissues also display alterations in the expression of some micro RNAs (Cencioni et al., 2013).
(vii) Morphological changes. Senescent cells display changes in the organization of nuclear lamina that modify nuclear morphology and gene expression (Freund et al., 2012). Due to cytoskeletal rearrangements, senescent cells display an increased size, a flat and irregular shape and changes in cell membrane composition (Ohno-Iwashita et al., 2010; Druelle et al., 2016; Cormenier et al., 2018).
(viii) Altered proteostasis. Senescence cells display increased unfolded protein response (UPR) associated with endoplasmic reticulum (ER) stress, which participates in the increase in ER size and the changes in the shape and size of these cells (Ohno-Iwashita et al., 2010; Cormenier et al., 2018).
Proteostasis refers to the balance and correct function of the proteome and requires proper synthesis, folding, quality control, and degradation rate of proteins. In eukaryotic cells, it depends on the accurate regulation of the proteasome, on the lysosomal system and autophagy, an intracellular degradation system of damaged proteins (López-Otín et al., 2013). Senescent cells lose protein homeostasis due to nucleolar dysfunction, autophagy and lysosomal anomalies and alterations in UPR indicative of ER stress. Correct proteostasis reduces the secretion of inflammatory cytokines (Tanaka and Matsuda, 2014), while its alteration (including protein misfolding aggregation and deposition) are a hallmark of many neurodegenerative diseases (see below for PD and AD).
In summary, some of the factors that trigger senescence in pathological conditions are oxidative SIPS (Hernandez-Segura et al., 2018), mitochondrial dysfunction (Wiley et al., 2017), DNA-damage (Dörr et al., 2013), telomere attrition or dysfunction (Hayflick and Moorhead, 1961), aberrant gene activation (Gorgoulis and Halazonetis, 2010), epigenetic modifications (Yang and Sen, 2018; Wagner, 2019), and impaired autophagy (Kang et al., 2011), although other stressors can also induce senescence (Figure 1).
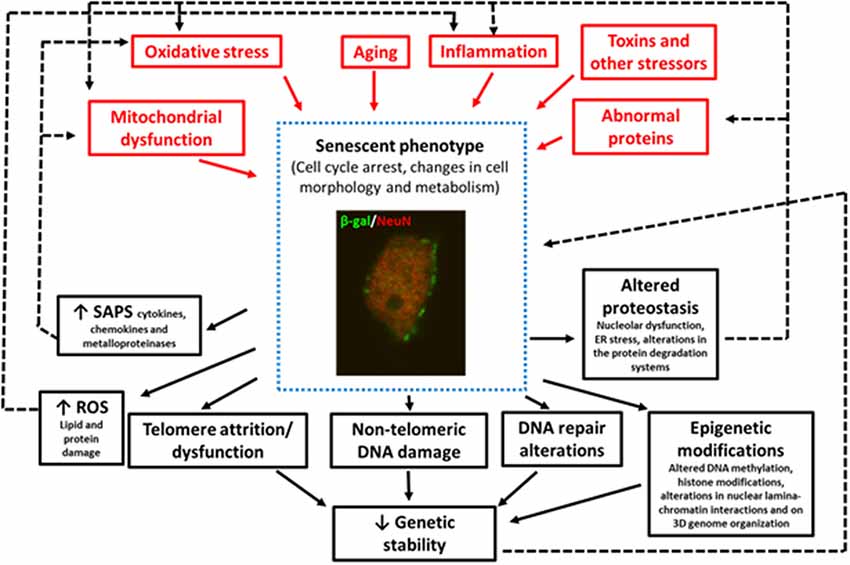
Figure 1. Image of a granular neuron (NeuN+) of the hippocampus of an aged wildtype mouse showing senescent phenotypes SA-β-Gal expression, and changes in cell morphology. This figure summarizes the triggering stimuli (in red) and the consequences (in black) of the increase in the number of cells with senescent phenotypes in neurodegenerative diseases. The black dotted lines represent the positive feedback mechanism that aggravates aging and neurodegeneration.
Finally, as previously mentioned, there is evidence that cellular senescence contributes to the pathogenesis of different neurodegenerative diseases through various mechanisms (Figure 1) including:
(i) The promotion of chronic inflammation: (Coppé et al., 2010). Thus, neuroinflammation is not only a trigger but also a result of senescence that can perpetuate the damage to the cell or neighboring cells (Nelson et al., 2012; Acosta et al., 2013; Ribezzo et al., 2016).
(ii) The promotion of oxidative stress and mitochondrial dysfunction. There is a positive feedback mechanism between senescence, oxidative stress, and mitochondrial dysfunction (Pole et al., 2016; Figure 1).
(iii) The reduction of the regenerative capacities of the nervous system: adult neurogenesis has been demonstrated in rodents (Kriegstein and Alvarez-Buylla, 2009; Ming and Song, 2011) and humans (Cipriani et al., 2018). Importantly, it has been recently demonstrated that neurogenesis is reduced in aged individuals and AD patients (Moreno-Jiménez et al., 2019). Due to the cell cycle arrest associated with cellular senescence, the reduced regenerative capacities of the brain (i.e., neurogenesis), would facilitate neurodegeneration.
(iv) Loss of function: finally, cell-cycle arrest and other alterations associated with cellular senescence alter the function of neurons and different CNS cell types (Purcell et al., 2014).
Aging increases the number of senescent cells (Rodier and Campisi, 2011) and the loss of neurons (De Stefano et al., 2016), compromising brain function and triggering or aggravating a neurodegenerative disease. Because senescent cells cannot maintain tissue function or repair its damage, if this state is maintained chronically, cellular aging and degenerative diseases would be aggravated.
In this review, we summarize the data that demonstrate the role of cellular senescence in the neurodegenerative diseases AD, DS, and PD (Table 1). Despite the differences in pathology among these three diseases, they are characterized by senescence and progressive loss of neurons leading to functional alterations (Nussbaum and Ellis, 2003).
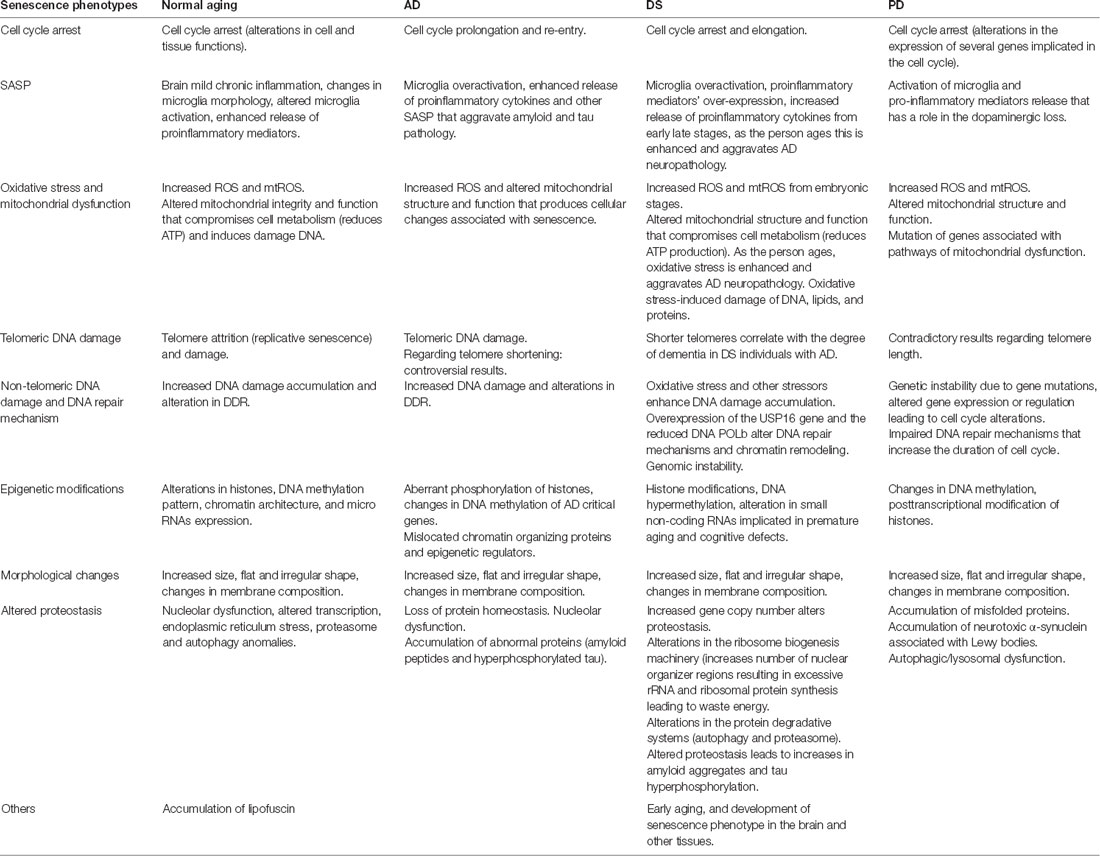
Table 1. Summary of senescent phenotypes in normal aging, Alzheimer’s disease (AD), Down syndrome (DS), Parkinson Disease (PD).
AD is a chronic neurodegenerative disease that accounts for 60–70% of cases of dementia. The main pathological hallmarks of AD are dementia and cognitive impairment, amyloid plaques, neurofibrillary tangles (NFTs) of hyperphosphorylated tau proteins, and loss of neurons and synapses (Selkoe and Hardy, 2016). However, there is increasing evidence that some pathological events that appear years earlier than the former have a prominent role in the development of amyloid plaques and NFTs. These events include increased oxidative stress, neuroinflammation, and cellular senescence due to DNA damage and altered proteostasis. Numerous reports have demonstrated their role in the increase in Aβ burden, tau hyperphosphorylation, neuronal death and an accelerated cognitive decline (Hardy and Higgins, 1992; Gitter et al., 1995; Chong, 1997; Weldon et al., 1998; Eikelenboom et al., 2006; Hardy, 2006; Sipos et al., 2007; Wilcock, 2012).
Cellular senescence has been demonstrated to play an important role in the onset and aggravation of AD (Bhat et al., 2012; Boccardi et al., 2015). Increased senescence is found in different cell types of AD brains, including astrocytes, microglia, and neurons as demonstrated by their enhanced SA-β-gal expression (He et al., 2013), p53 expression, a mediator of cellular senescence (Arendt et al., 1996; McShea et al., 1997; Luth et al., 2000; Yates et al., 2015), an increase in the release of SASP components (Erusalimsky, 2009), DNA damage (Myung et al., 2008), telomere attrition or damage (Flanary and Streit, 2004), and senescence-like morphological changes (Streit et al., 2004).
Increased levels of senescent cells with higher SA-β-gal and p53 levels are also found and in plasma samples from AD patients and mouse models of AD (de la Monte et al., 1997; Tiribuzi et al., 2011; Magini et al., 2015; Caldeira et al., 2017).
Several studies have demonstrated an association between senescence and AD neurodegeneration. In cultured neurons, the expression of several senescent-associated genes was upregulated after the exposure to Aβ (Wei et al., 2016). These results were also confirmed in vivo, using the 5× FAD mouse model of AD that displays progressive Aβ deposition. After 7 months of age, this model shows the upregulation of some essential senescent—related genes in the hippocampus (Wei et al., 2016). Besides, a murine model that overexpresses human tau and develop NFT deposition also shows increased expression of several senescent—associated genes in the hippocampus and cortex (Bussian et al., 2018).
Besides, the administration of Aβ oligomers to oligodendrocyte progenitor cells can also induce senescence (Zhang et al., 2019) and in murine neural stem cells, Aβ42 peptides increase the number of SA-β-Gal positive cells (He et al., 2013). The removal of senescent cells in mice reduces Aβ accumulation and enhances their cognitive abilities (Zhang et al., 2019). Besides, hyperphosphorylation of tau can induce senescence in glial cells (Musi et al., 2018).
The fact that many of the early alterations found in AD (i.e., neuroinflammation, oxidative stress, DNA damage and changes in DNA repair and altered proteostasis) trigger or are associated with cellular senescence, has led to the suggestion that this process has a crucial role in the etiopathology of AD.
The brains of AD patients and of mouse models of AD have higher levels of inflammation due to microglia activation that produces pro-inflammatory cytokines and other SASP mediators (Streit et al., 2004; Hickman et al., 2008; Rawji et al., 2016; Olivieri et al., 2018). Neuroinflammation in the brain of AD patients is influenced by many SASPs mediators (Bauer et al., 1991; Huell et al., 1995; Kiecolt-Glaser et al., 2003) that have a prominent role in the onset of senescence (Flanary and Streit, 2004; Flanary et al., 2007). IL-6, a proinflammatory cytokine, is upregulated in the aged brain and AD (Bauer et al., 1991; Huell et al., 1995; Kiecolt-Glaser et al., 2003) and its overexpression has been shown to induce neurodegeneration (Campbell et al., 1993). The brains and lymphocytes of AD patients and AD mouse models also present increased activity of the SASP regulator p38MAPK (Sun et al., 2003). This enhanced p38MAPK activity upregulates the levels of the pro-inflammatory cytokines IL-6, IL-1, TGF-β and TNF-α levels in AD brains (Bauer et al., 1991; Huell et al., 1995; Freund et al., 2012; Lai et al., 2017; Rea et al., 2018), CSF and serum (Wood et al., 1993; Cacabelos et al., 1994; Blum-Degen et al., 1995; Luterman et al., 2000; Swardfager et al., 2010; Tarkowski et al., 2003; Gezen-Ak et al., 2013; Dursun et al., 2015). AD brains also present increased levels of other SASPs mediators: the metalloproteinases MMP-1, MMP-3, and MMP-10 (Leake et al., 2000; Yoshiyama et al., 2000; Bjerke et al., 2011; Horstmann et al., 2018). The fact that SASP meditators have a prominent role in the onset of senescence (Flanary and Streit, 2004; Flanary et al., 2007), provide support for the relationship between neuroinflammation and senescence in AD neuropathology.
Neuroinflammation aggravates AD (Guerreiro et al., 2013) mainly because it increases APP and Aβ expression in this disease. Pro-inflammatory cytokines induce the formation of Aβ oligomers, the phosphorylation of tau, and ROS production (Sastre et al., 2003; Blurton-Jones and Laferla, 2006; Steele et al., 2007). In turn, Aβ peptides and APP activate glial cells (Dickson et al., 1993; Barger and Harmon, 1997), produce an enhanced release of pro-inflammatory mediators such as IL-1 and IFNγ in the brains of AD patients (Ho et al., 2005; Meager, 2004, 2005). Under these circumstances, in AD, microglia internalize less Aβ (Floden and Combs, 2011; Njie et al., 2012) and are less able to process it (Nixon et al., 2001; Hickman et al., 2008; Mawuenyega et al., 2010). The higher release of pro-inflammatory cytokines reduces the ability of the cells to remove Aβ and facilitates its accumulation. Thus, there is a positive feedback between cytokine release, Aβ, and APP expression, phosphorylated tau and neurodegeneration (Wilcock and Griffin, 2013).
Oxidative stress is another crucial mechanism that contributes to accelerating aging and cognitive dysfunction in AD and during aging (López-Otín et al., 2013). AD patients and mouse models of AD, present increased oxidative stress and mitochondrial dysfunction, similar to that observed in senescence. These events are present in the early stages of AD and precede the major pathologic hallmarks, such as senile plaques and NFTs (Yates et al., 2015; Ott et al., 2018).
In AD mitochondrial function and structure are impaired (Cadonic et al., 2016; Tai et al., 2017), which has a critical role in the progression of SIPS (Gao et al., 2017). Besides, the enhanced production of ROS decreases ATP synthesis. Because DNA repair is a mechanism that requires a high amount of energy, and DNA is heavily damaged in cells exposed to oxidative stress (Monti et al., 1992), these cells would need a high amount of energy to perform DNA repair. Thus, mitochondrial dysfunction reduces the ability of the cell to repair these alterations.
As mentioned in the “Introduction” section, during aging, high levels of oxidative stress induce in cells a state of senescence that displays its main pathological characteristics, including DNA damage, altered DDR, alterations in the cell cycle and cell morphology and telomere damage (Monti et al., 1992; De Haan et al., 1996; von Zglinicki et al., 2000; Serra et al., 2003; Chow and Herrup, 2015; Kawanishi and Oikawa, 2004; Jennings et al., 2000; Liu et al., 2003). Thus, a link between oxidative stress and cellular senescence has been postulated in AD.
Evidence for the relation between oxidative stress and cellular senescence in AD comes from studies in mouse models of AD in which the animals were subjected to chronic oxidative stress (SIPS) and found cellular changes identical to those found in other types of senescence: i.e., an increase in SA-β-gal expression, cell cycle arrest and alterations in cellular morphology (Toussaint et al., 2000; Ma et al., 2014). Besides, fibroblasts from AD patients produced more ROS, a slowing in the growth rate, an increase in the expression of p53 and p21 and senescence-like phenotype (Toussaint et al., 2000; Naderi et al., 2006).
As mentioned above, DNA damage and alterations in DNA repair are two of the main characteristics of cellular senescence (Sedelnikova et al., 2004) and are associated with aging (Brosh and Bohr, 2007). In AD, enhanced DNA damage and reduced DDR has also been demonstrated, and these alterations seem to accelerate the progression of the disease (Lovell et al., 1999).
Telomere attrition and damage have been proposed to be a potential contributor in the pathogenesis of several neurological disorders including AD (Eitan et al., 2014; Boccardi et al., 2015; Forero et al., 2016). However, there are contradictory reports on the role of telomeres alterations on AD neuropathology.
Some studies have shown that telomere shortening or alteration might be implicated in AD pathology (Aβ burden, hyperphosphorylation of tau and dementia) (Grodstein et al., 2008; Jenkins et al., 2008; Guan et al., 2012).
In monocytes of AD patients, shorter telomeres were found (Hochstrasser et al., 2012). Shorter telomere length was also found in patients with AD than in those with mild cognitive impairment than in healthy controls (Scarabino et al., 2017). Moreover, a meta-analysis of 13 studies concluded that there is evidence that AD patients are more likely to present telomere attrition (Forero et al., 2016).
However, other studies did not find changes in the length of the telomeres of patients with AD (Hinterberger et al., 2017). The telomere length of tissue obtained from the cerebellum of AD patients did not differ from age-matched controls (Lukens et al., 2009). In studies that evaluated telomere length in patients with different types of dementia (Zekry et al., 2010) or with dementia of AD types, no differences were found between affected patients and controls (Takata et al., 2012; Hinterberger et al., 2017). Also, in a mouse model of AD, telomere shortening reduces Aβ burden and improves cognition (Rolyan et al., 2011), although the animals still showed enhanced DNA damage and neurodegeneration.
Therefore, more studies are necessary to clarify the role of telomere attrition in AD neuropathology.
Additionally, an association between reduced lifespan, elevated levels of DNA damage, and a prolongation of the cell cycle duration have been demonstrated (Weirich-Schwaiger et al., 1994). In AD there is also evidence that there are significant alterations of cell cycle re-entry of post-mitotic neurons (Vincent et al., 1996; Nagy, 2005; Herrup and Yang, 2007).
During senescence, the accumulated DNA errors alter cellular functions such as transcription and DNA repair (Hackett et al., 1981; Cleaver et al., 1983; Mayne, 1984; Friedberg, 1985; Klocker et al., 1985). These alterations are more evident during aging (Staiano-Coico et al., 1983; Nette et al., 1984; Dutkowski et al., 1985; Mayer et al., 1989; Roth et al., 1989). Moreover, in AD, an enhanced DDR is found in the hippocampus and lymphocytes, as demonstrated by the presence of elevated levels of the phosphorylated histone γH2AX (H2A histone family member X) (Silva et al., 2014; Siddiqui et al., 2018).
In neurodegenerative diseases, epigenetic modifications, such as alteration in DNA methylation occur early in the disease process, affect to particular genes and correlate with misfolded proteins in specific brain regions (Armstrong et al., 2019; Prasad and Jho, 2019).
AD, PD, and DS among other neurodegenerative diseases have common aberrant DNA methylation profile that mainly affects the expression of critical genes involved in various signaling pathways that are implicated in several pathological hallmarks such as presence of Aβ plaques, NFT or α-synuclein inclusions that are present in some of them (Sanchez-Mut et al., 2016; Armstrong et al., 2019; Prasad and Jho, 2019).
Among the epigenetic alterations found in AD, DNA methylation and histone modifications show evident deregulation. It has been demonstrated that in AD brains changes in DNA methylation affect to a few common gene loci which play an essential role in the formation of Aβ plaques (Lord and Cruchaga, 2014; Watson et al., 2016; Qazi et al., 2018; Armstrong et al., 2019; Esposito and Sherr, 2019; Prasad and Jho, 2019). Among them, of particular interest are the differences in methylation in different regions of the APP gene promoters in the brains of humans with AD and these changes have been associated with neuropathological markers of this disorder (Bradley-Whitman and Lovell, 2013). Besides, in AD brains histone modifications have been linked to the reduction in transcription of genes that are implicated in neuronal physiology and increased the transcription of genes that are normally silenced (Berson et al., 2018). The phosphorylated histone H3 presents an altered localization of the cytoplasm, while the levels of the acetylated histone H4 are decreased in AD patients (Kwon et al., 2016).
Also, chromatin organizing proteins and epigenetic regulators are mislocated in AD, affecting chromatin architecture (Winick-Ng and Rylett, 2018). Finally, in AD, other epigenetic modifications such as alterations in chromatin remodelers, phosphorylation of histones have been found (Esposito and Sherr, 2019), and might be associated with senescence.
Altered proteostasis is a phenotypic hallmark of senescent cells. The integrity of the nucleolar ribosome biogenesis machinery and the ER, as well as the correct function of the degradation machinery (ubiquitin-proteasome and lysosomal pathways), are essential for maintaining proteostasis (Labbadia and Morimoto, 2015). The perturbation of proteostasis leads to misfolded protein accumulation and proteotoxic stress (Hetz and Mollereau, 2014; Hipp et al., 2014), which also contributes to neurodegeneration. Senescent cells lose protein homeostasis due to nucleolar dysfunction, autophagy and lysosomal anomalies and alterations in UPR indicative of ER stress. The majority of neurodegenerative disorders, present impairments of degradative compartments such as lysosomes and autophagosomes.
Protein dysfunction has been associated with numerous human neurodegenerative diseases such as AD, PD, amyotrophic lateral sclerosis, spinocerebellar ataxia, and Huntington’s disease (Yurov et al., 2014). In these diseases, high levels of different toxic protein aggregates due to a loss of protein homeostasis have been found. In particular, in AD, the formation of Aβ plaques (Ow and Dunstan, 2014), which is directly associated with the expression of SASP- associated factors (Bhat et al., 2012), and of NFT, composed by hyperphosphorylated tau protein, increase with the acquisition of a senescent phenotype and at the same time both induce senescence (Zare-Shahabadi et al., 2015; Chung et al., 2018; Mendelsohn and Larrick, 2018).
These alterations have been demonstrated to contribute to the etiology and progression of AD (Ihara et al., 2012; Nixon, 2013; Hetz and Mollereau, 2014). In this disorder, the most common form of protein aggregation, in Aβ plaques (Ow and Dunstan, 2014), which is directly associated with the expression of SASP- associated factors (Bhat et al., 2012), is one of the primary triggers and consequences of cellular senescence. The other abnormal protein found in AD brains is hyperphosphorylated tau in the form of NFTs, which have been reported to induce senescence (Mendelsohn and Larrick, 2018).
DS, or trisomy of chromosome 21, is the primary cause of cognitive disability of genetic origin (Shin et al., 2009). These cognitive alterations are due to defects in growth and differentiation of the CNS that appear during early prenatal stages (Haydar and Reeves, 2012; Lott, 2012). DS is characterized by premature aging and early appearance (around the fourth decade of life) of neuropathology identical to the one found in sporadic AD, including amyloid plaques, NFTs, neurodegeneration, and synapse loss (Teipel and Hampel, 2006; Sabbagh et al., 2011; Cenini et al., 2012; Lott, 2012; Wilcock and Griffin, 2013; Casanova et al., 1985; McGeer et al., 1985). Both, premature aging and AD pathology are thought to be the leading causes of the earlier mortality of this population (Schupf and Sergievsky, 2002; Zigman and Lott, 2007; Prasher et al., 2008; Esbensen, 2010).
The DS brain also present neuroinflammatory changes typical of AD, such as microglial activation and increased release of pro-inflammatory cytokines (Griffin et al., 1989; Park et al., 2005; Griffin, 2006; Wilcock and Griffin, 2013). There is compelling evidence that, in DS, the increased release of cytokines due to microglia activation, enhances the production of ROS, aggravates synaptic dysfunction, and neurodegeneration, and reduces neurogenesis (Town et al., 2005; Fuster-Matanzo et al., 2013; Llorens-Martín et al., 2014; Lyman et al., 2014; Rosi et al., 2012). The aggravation produced by neuroinflammation on neurodegeneration and the reduced neurogenesis (see Rueda et al., 2012), render DS individuals more susceptible to neuropathological events that can accelerate the onset of dementia (Teipel and Hampel, 2006).
Enhanced microglial activation also produces an increased expression of proinflammatory cytokines in different areas of the brain of a mouse model of DS (Hunter et al., 2004; Lockrow et al., 2011; Roberson et al., 2012; Rueda et al., 2018). Reducing neuroinflammation by administering an antibody against the pro-inflammatory cytokine IL17A to these mice, reduces cellular senescence in the hippocampus and cortex of these animals, improves their cognitive abilities and reduces some of the signs of neurodegeneration (Rueda et al., 2018). These results suggest a direct link between cellular senescence, neuroinflammation and neurodegeneration in DS.
Another crucial mechanism that contributes to accelerating aging and cognitive dysfunction in DS is oxidative stress. In this syndrome, it is present from embryonic stages producing a reduction in neuronal proliferation, differentiation, connectivity, and survival (Monti et al., 1992; Busciglio and Yankner, 1995; Perluigi et al., 2011). As the person with DS ages, the levels of oxidative stress increases, contributing to the progression of cognitive and neuronal degeneration (Busciglio et al., 2007; Lockrow et al., 2009; Shichiri et al., 2011; Perluigi and Butterfield, 2012; Rueda et al., 2012; Parisotto et al., 2016). As previously mentioned, oxidative stress is an essential factor that causes cellular senescence (Monti et al., 1992; He et al., 2013; Rodríguez-Sureda et al., 2015).
Furthermore, mitochondrial dysfunction is also present in DS cells during embryonic life (Perluigi et al., 2011), leading to profound alterations in energy metabolism due to the reduced synthesis of ATP synthesis (Valenti et al., 2010). ATP is also reduced in vitro in DS fibroblasts, which could contribute to premature cell aging (Rodríguez-Sureda et al., 2015).
In DS, early induction of senescence by aberrant intracellular oxidant activity and antioxidant defense has been demonstrated in fibroblasts, skin tissue, lymphocytes (De Haan et al., 1996; Kalanj-Bognar et al., 2002; Cristofalo et al., 2004; Kimura et al., 2005; Biron-Shental et al., 2015; Rodríguez-Sureda et al., 2015) and in the amniotic fluid of women carrying DS fetuses (Perluigi et al., 2011; Amiel et al., 2013). Thus, senescence is present in DS from prenatal stages and can be responsible for different altered phenotypes. But, enhanced senescence is a prominent phenomenon during all the life stages of a DS individual and is aggravated with aging and the appearance of AD neuropathology.
In agreement with these results, it has been demonstrated that the hippocampus of a mouse model of DS shows higher amounts of oxidative damage (protein and lipid oxidative damage), Aβ expression and tau phosphorylation, as well as increased density of senescent cells in different hippocampal regions (i.e., DG, CA1, CA3, and hilus; Corrales et al., 2013; Parisotto et al., 2016; García-Cerro et al., 2017; Rueda et al., 2018). Other authors have also found the early presence of SA-β-gal activity in both cultured fibroblasts and skin tissue from DS mice (Contestabile et al., 2009b). Finally, chronic administration of melatonin, a potent antioxidant, to this mouse model of DS reduces hippocampal senescence and oxidative damage, improves the cognitive abilities of these mice, enhances neurogenesis and reduces their neurodegenerative phenotype (Corrales et al., 2013, 2014; Parisotto et al., 2016). These results provide support for the association between oxidative stress and senescence in DS.
Hematopoietic stem cells and satellite cells of skeletal myofibers in the Ts65Dn mouse model of DS accumulate DNA damage and prematurely develop a senescent phenotype (Adorno et al., 2013; Wang et al., 2016; Pawlikowski et al., 2018). Furthermore, in this murine model, the ubiquitin-specific-peptidase 16 (Usp16) gene is triplicated, leading to the overexpression of the Usp16 enzyme that controls the ubiquitination state of the histone 2A (Ub-H2A) and contributes to regulate the DDR chromatin remodeling and cell cycle progression (Joo et al., 2007). Thus, in these mice, both the overexpression of the Usp16 gene, which may affect the DNA damage/repair signaling pathways and the high levels of brain oxidative stress and neuroinflammation may produce and excessive accumulation of DNA damage leading to genomic instability and neurodegeneration or premature cellular senescence.
In DS, there is also evidence of prenatal accumulation of DNA damage and of the reduction of the ability to repair DNA (Agarwal et al., 1970; Raji et al., 1998; Pogribna et al., 2001; Cabelof et al., 2009; Nižetić and Groet, 2012; Di Domenico et al., 2015). Besides, in response to DNA damage, DNA polymerase b (POLb), whose function is to participate in base excision repair (BER) is induced; however, in DS fibroblasts its expression is reduced, thereby reducing the ability to repair DNA damage. This reduction is sufficient to induce senescence (Ahmed et al., 2018; Cabelof et al., 2002, 2003; Cabelof, 2007).
In DS, both alterations in the DNA damage/repair signaling pathways, and the high levels of brain oxidative stress may produce and excessive accumulation of DNA damage leading to genomic instability and neurodegeneration or premature cellular senescence. Hence, this mechanism might play an essential role in the onset and aggravation of the AD-associated cognitive decline.
DNA repair mechanisms eliminate damages and elongate cell cycle duration to give the repair enzymes time to work. However, alterations in DNA repair mechanisms producing accumulated DNA damage can delay the cell cycle until mitotic activity ceases, leading to premature senescence and sometimes cell death. In DS, enhanced DNA damage and impairment in DNA repair impairs mitosis and delays the cell cycle leading to senescence. There is evidence of delayed cell cycle in DS individuals and mouse models of this syndrome (Contestabile et al., 2007, 2009a).
As mentioned above, telomere attrition is one of the best-characterized senescence-triggering mechanisms. Telomeres are shorter in DS amniocytes, and placentas of fetuses with DS present an increased percentage of trophoblasts with senescence phenotypes (Sukenik-Halevy et al., 2011; Amiel et al., 2013; Biron-Shental et al., 2015). For this reason, it has been suggested that it could be an early biomarker of premature senescence (Vaziri et al., 1993). Also, a significantly higher loss of telomere sequences was observed in vitro and in vivo in lymphocytes from DS individuals (0–45 years old) (Vaziri et al., 1993; de Arruda Cardoso Smith et al., 2004). Jenkins et al. (2008) also demonstrated that shorter telomere length correlates with the degree of dementia and AD-associated neurodegeneration in DS individuals. These results suggest that senescence can play an essential role in the aging of the immune system in DS (Vaziri et al., 1993), leading to the altered microglia function and neuroinflammation. Thus, in DS, this type of DNA damage can induce senescence from prenatal stages through the entire lifespan of the individual.
Epigenetic alterations such as DNA and histone modifications are also found in patients with DS. Among them, DNA hypermethylation, histone modifications, histone core variants and changes in small non-coding RNAs are implicated in several phenotypic characteristics of DS such as memory impairment, premature aging and neurodevelopmental defects (Mentis, 2016).
Although some of these alterations are associated with premature aging in DS, the exact implication of epigenetic modifications in cellular senescence found in this syndrome needs to be elucidated.
Abnormalities in chromosome number disrupt proteostasis (Oromendia et al., 2012; Oromendia and Amon, 2014; Stingele et al., 2012). Gene copy number changes due to aneuploidy can affect the number of proteins expressed, their functions and the protein quality-control and repair machinery of the cell (Sheltzer et al., 2011; Oromendia et al., 2012; Thorburn et al., 2013; Yurov et al., 2014).
Thus, in DS, the extra chromosome can alter proteostasis (Lockstone et al., 2007). Trisomy may also affect the 3D nuclear architecture of the genome resulting in changes in gene interactions and gene expression that may impact proteostasis regulation (Vermunt et al., 2019).
Regarding protein synthesis, only a few studies have analyzed the ribosome biogenesis machinery in DS. Demirtas (2009), using a classic silver staining procedure to study the arrangement of the nucleolar organizer regions (NORs), found that cells from DS infants presented an increased number of AgNOR dots. Because ribosome biogenesis is a highly energy-consuming process, these authors suggest that this response to trisomy 21 might result in unnecessary rRNA and ribosomal protein synthesis, which leads to a waste in energy. Also, reduced expression of specific transcription, splicing, and translation factors has been found in cortices of fetuses with DS (Freidl et al., 2001), suggesting a deranged protein synthesis in them.
Moreover, in DS, both intracellular degradative systems, UPS and autophagy, seem to be affected. Chronic exposure to oxidative stress, as occurs in the DS brain, also causes protein oxidation of members of the proteostasis network, resulting in accumulation of unfolded/damaged protein aggregates and dysfunction of intracellular degradative system, such as autophagy and the ubiquitin-proteasome systems (UPS), contributing to neurodegeneration (Di Domenico et al., 2013; Hetz and Mollereau, 2014). In the hippocampus of Ts65Dn mice altered autophagy, due to reduction of autophagosome formation, has been found (Tramutola et al., 2016). In DS human fibroblasts and in the brain of two segmental trisomy models of DS, the Dp16 and Dp17 mice, increased ubiquitination, and disrupted proteasome activity has been reported (Aivazidis et al., 2017). Finally, in the cerebellum of the Ts65Dn mouse, a dysfunction of the UPS, due to reduced proteasome activity and a parallel increase of ubiquitinated proteins, has been implicated in the degeneration of Purkinje cells (Necchi et al., 2011). Consequently, ubiquitinated or polyubiquitinated proteins are not degraded at a normal rate and are stored in the nuclei of cerebellar neurons of this model (Necchi et al., 2011).
Besides, disruption in the proteostasis network could contribute to the accumulation of protein aggregates, such as amyloid deposits and NFTs (Di Domenico et al., 2013). Alterations of the proteostasis network are present in individuals with DS years before the age-related cognitive decline, and AD-associated dementia is detected.
In summary, DS is characterized by early senescence that continues throughout the lifespan of the individual and is very likely aggravated by the appearance of AD neuropathology.
PD is a progressive neurodegenerative disorder characterized by the loss of dopaminergic neurons in the substantia nigra, the presence of cytoplasmic protein aggregates, known as Lewy bodies, that contain a variety of proteins, including ubiquitin and α-synuclein (Nussbaum and Ellis, 2003; Thomas and Beal, 2011). These pathological features result in an impairment of motor control, including difficulty to initiate movements, loss of balance, rigidity, and tremor, and cognitive deterioration (Poewe et al., 2017). The most important risk factors for the development of PD are aging, a genetic predisposition and exposure to toxins (Chinta et al., 2013).
Similarly to AD and DS, PD neuropathology is aggravated by neuroinflammation mitochondrial dysfunction, α-synuclein accumulation, oxidative stress, and cellular senescence (Poewe et al., 2017).
There is widespread evidence that cellular senescence plays an essential role in the pathogenesis of PD. Patients with PD present enhanced levels of SA-β-gal in their CSF (van Dijk et al., 2013) and in their brain tissue (Chinta et al., 2018). Higher numbers of senescent astrocytes are also present in the substantia nigra of patients with PD (Chinta et al., 2018). Besides, paraquat, a herbicide that has been proposed to be implicated in the appearance of some types of PD, induces senescence in human astrocytes (Chinta et al., 2018).
Similarly to AD and DS, inflammation is prominent in PD (Chinta et al., 2018). Activation of microglia is thought to be one of the main determinants of dopaminergic loss in the substantia nigra (McGeer et al., 1988), hippocampus, cingulate and temporal cortex (Imamura et al., 2003). α-synuclein aggregation activates microglial cells in PD (Zhang et al., 2005). The result of microglia activation is in the increased levels of the pro-inflammatory mediators IL-1, IL-6, TNFγ and TNF-α in the CSF, serum, and dopaminergic regions of the striatum from patients with PD (Mogi et al., 1994a,b; Blum-Degen et al., 1995; McCoy et al., 2006; Mount et al., 2007; Brodacki et al., 2008; Scalzo et al., 2010; Lindqvist et al., 2012; Dursun et al., 2015).
The high presence of senescent cells and aged astrocytes in PDs brains suggests that senescence-induced neuroinflammation might be an important mechanism for PD neurodegeneration and a potential therapeutic target for this disease.
Oxidative stress plays an essential role in the etiology and progression of PD (Trist et al., 2019). Even in the early stages of the disease, before a significant loss of dopaminergic neurons, PD patients present elevated oxidative stress (Ferrer et al., 2011). Thus, oxidative stress could be partially responsible for neurodegeneration, in addition to been enhanced as a result of the neuronal loss. As demonstrated in AD and DS, it seems that positive feedback is produced between both insults that probably aggravates the disease.
Mitochondrial dysfunction also plays a vital role in PD etiopathology. In familial PD, different mutations in the genes associated with pathways of mitochondrial dysfunction have been demonstrated, and some of these pathways are also altered in sporadic PD (Park et al., 2018). The production of ROS during ATP synthesis is higher in dopaminergic neurons of the substantia nigra. Due to their size and complexity, they need more elevated amounts of ATP to maintain resting membrane potential, propagate action potentials, and enable synaptic transmission (Pissadaki and Bolam, 2013).
These alterations in mitochondrial function and oxidative stress are likely to be a result of senescence.
DNA damage and repair have been proposed as important actors altering the function of the dopaminergic system in PD (Sepe et al., 2016). Telomere shortening plays a causal role in cellular aging (Hastie et al., 1990), and it has been proposed as a potential contributor in the pathogenesis of PD (Anitha et al., 2019).
The studies of telomere length in PD patients had provided contradictory results. Some of them provide support for shorter telomere length in different cell types or tissues (Tomac and Hoffer, 2001; Guan et al., 2008; Koliada et al., 2014) while others do not find evidence of this alteration in PD (Guan et al., 2008; Wang et al., 2008; Eerola et al., 2010; Watfa et al., 2011; Maeda et al., 2012; Degerman et al., 2014; Eitan et al., 2014; Forero et al., 2016). A meta-analysis of all these studies concluded that PD patients do not present changes in telomere length (Forero et al., 2016). Thus, new studies are necessary to clarify the putative role of replicative senescence and telomere shortening in different brain regions and cell types in PD.
However, it has been demonstrated that PD is characterized by genetic instability due to gene mutations, altered gene expression, or regulation. The expression of some genes implicated in the cell cycle, such as p16INK4a (Chinta et al., 2018) and pRb (Ihara et al., 2012), are upregulated in PD brains. This enhanced genetic expression is likely to produce alterations in the cell cycle in this disorder.
Furthermore, alterations in epigenetic modulation seem to play an important role in the etiopathology of PD. The brains of these patients present changes in DNA methylation, post-translational modifications of histones and of microRNAs that regulate pathways involved in the pathophysiology of PD (Armstrong et al., 2019; Prasad and Jho, 2019; Renani et al., 2019).
However, the role of the alterations of DNA methylation in the pathological hallmarks of this disease, including cellular senescence, is still unclear.
Another characteristic that PD shares with other neurodegenerative diseases is altered proteostasis. In PD, there is an accumulation of misfolded proteins in the brain (Martinez et al., 2019). All patients with PD present insoluble α-synuclein fibrils, which damage neurons, and are associated with intracellular Lewy bodies (Melki, 2015). Alterations in the proteostasis network seem to play a prominent role in the protein aggregation found in PD, mainly due to changes in the function of the ER, the organelle involved in protein folding and quality control. ER stress produces the UPR to try to restore proteostasis.
In this disease, the increase in senescent cells is thought to be directly associated with the enhanced aggregation of α-synuclein (Chinta et al., 2018).
It has been proposed that mitochondrial dysfunction and oxidative stress could be partially responsible for this α- synuclein accumulation (Dias et al., 2013; Rocha et al., 2018).
Finally, the autophagic/lysosomal dysfunction also plays an essential role in the pathogenesis of the disease, since different genetic mutations that produce defects in these pathways are found in PD patients (Rivero-Ríos et al., 2016; Pitcairn et al., 2018).
In summary, cellular senescence is also a pathological event of crucial importance in PD, and shares with other neurodegenerative diseases many of the factors that trigger it and aggravate the neuropathology (i.e., oxidative stress, mitochondrial dysfunction, altered proteostasis, among others).
Senescent cells accumulate with aging and in neurodegenerative pathologies, and there is extensive evidence that they might be implicated in the etiopathology of these diseases. Here, we reviewed the data that demonstrate that cellular senescence is a common pathological feature of PD, AD, and DS. These three diseases also share some of the mechanisms that promote senescence and that allow this permanent state of cell arrest to induce or aggravate neurodegeneration. Furthermore, extensive senescence has also been found in other neurodegenerative diseases such as Huntington Disease, Multiple Sclerosis, and Rett syndrome. Thus, senescence seems to be a common mechanism for neurodegeneration that needs to be further investigated.
At present, there are no efficient neuroprotective treatments that can prevent or delay the progression of the disease for AD in individuals with or without DS or for PD. Thus, new neuroprotective therapeutic approaches are needed.
Because of the central role of cellular senescence in the etiopathology of different neurodegenerative diseases, therapies that reduce senescence could be a promising approach to prevent the loss of cells and the alteration of their function. These therapies have been more extensively investigated in the case of AD (He et al., 2013; Kirkland and Tchkonia, 2017).
Several therapeutic strategies, known as senotherapies, to selective eliminate tissue-specific senescent cells, have been developed (Childs et al., 2017; Kritsilis et al., 2018; Kim and Kim, 2019). However, almost none have been tested in models of different neurodegenerative diseases, except AD. Clearance of senescent cells extended lifespan in normal mice (Baker et al., 2016) and in a progeroid mouse model (Baker et al., 2011). Finally, a recent work has demonstrated that pharmacological elimination of senescent astrocytes and microglia cells using a senolytic compound (ABT263) attenuates tau phosphorylation, modulates tau aggregation, prevents the upregulation of senescence-associated genes in the cortex and hippocampus and improves short-term memory in a transgenic mouse model that overexpresses human tau (Kirkland and Tchkonia, 2017; Bussian et al., 2018). Thus, senotherapeutic agents may serve as powerful tools to prevent neurodegenerative diseases.
Importantly, in many of these disorders, senescence is an early event that appears years before other signs of neurodegeneration and in the case of DS, even at embryonic stages, providing a broad therapeutic window to prevent or delay some of the pathological processes associated.
Finally, many of the mechanisms implicated in the onset of senescence and that also appear years earlier than neurodegeneration (i.e., oxidative stress, mitochondrial damage, neuroinflammation, DNA damage, altered proteostasis), are also a result of senescence, therefore aggravating the disease (Figure 1). Thus, therapies targeting these earlier events could exert a double benefit preventing the onset of the neurodegenerative disease or delaying its progress.
CM-C and NR wrote and edited the manuscript.
This study was supported by Fundación Tatiana Pérez de Guzmán el Bueno, IDIVAL, the grant reference (NVAL 19/23) and the Spanish Ministry of Economy and Competitiveness (Ministerio de Economía y Competitividad; PSI-2016-76194-R, AEI/FEDER, EU).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acosta, J. C., Banito, A., Wuestefeld, T., Georgilis, A., Janich, P., Morton, J. P., et al. (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990. doi: 10.1038/ncb2784
Adorno, M., Sikandar, S., Mitra, S. S., Kuo, A., Nicolis Di Robilant, B., Haro-Acosta, V., et al. (2013). Usp16 contributes to somatic stem-cell defects in Down’s syndrome. Nature 501, 380–384. doi: 10.1038/nature12530
Agarwal, S. S., Blumberg, B. S., Gerstley, B. J., London, W. T., Sutnick, A. I., and Loeb, L. A. (1970). DNA polymerase activity as an index of lymphocyte stimulation: studies in Down’s syndrome. J. Clin. Invest. 49, 161–169. doi: 10.1172/jci106215
Ahmed, A. A., Smoczer, C., Pace, B., Patterson, D., and Cress Cabelof, D. (2018). Loss of DNA polymerase β induces cellular senescence. Environ. Mol. Mutagen. 59, 603–612. doi: 10.1002/em.22206
Aivazidis, S., Coughlan, C. M., Rauniyar, A. K., Jiang, H., Liggett, L. A., Maclean, K. N., et al. (2017). The burden of trisomy 21 disrupts the proteostasis network in Down syndrome. PLoS One 12:e0176307. doi: 10.1371/journal.pone.0176307
Allsopp, R. C., Vaziri, H., Pattersont, C., Goldsteint, S., Younglai, E. V., Futcher, A. B., et al. (1992). Telomere length predicts replicative capacity of human fibroblasts. Proc. Natl. Acad. Sci. U S A 89, 10114–10118. doi: 10.1073/pnas.89.21.10114
Al-Mashhadi, S., Simpson, J. E., Heath, P. R., Dickman, M., Forster, G., Matthews, F. E., et al. (2015). Oxidative glial cell damage associated with white matter lesions in the aging human brain. Brain Pathol. 25, 565–574. doi: 10.1111/bpa.12216
Amiel, A., Fejgin, M. D., Liberman, M., Sharon, Y., Kidron, D., and Biron-Shental, T. (2013). Senescence in amniocytes and placentas from trisomy 21 pregnancies. J. Matern. Fetal Neonatal Med. 26, 1086–1089. doi: 10.3109/14767058.2013.768982
Anitha, A., Thanseem, I., Vasu, M. M., Viswambharan, V., and Poovathinal, S. A. (2019). Telomeres in neurological disorders. Adv. Clin. Chem. 90, 81–132. doi: 10.1016/bs.acc.2019.01.003
Arendt, T., Rödel, L., Gärtner, U., and Holzer, M. (1996). Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer’s disease. Neuroreport 7, 3047–3049. doi: 10.1097/00001756-199611250-00050
Armstrong, M. J., Jin, Y., Allen, E. G., and Jin, P. (2019). Diverse and dynamic DNA modifications in brain and diseases. Hum. Mol. Genet. 28, R241–R253. doi: 10.1093/hmg/ddz179
Baker, D. J., Childs, B. G., Durik, M., Wijers, M. E., Siebe, C. J., Zhong, J., et al. (2016). Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189. doi: 10.1038/nature16932
Baker, D. J., Wijshake, T., Tchkonia, T., LeBrasseur, N. K., Childs, B. G., van de Sluis, B., et al. (2011). Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236. doi: 10.1038/nature10600
Barbouti, A., Evangelou, K., Pateras, I. S., Papoudou-Bai, A., Patereli, A., Stefanaki, K., et al. (2019). In situ evidence of cellular senescence in Thymic Epithelial Cells (TECs) during human thymic involution. Mech. Ageing Dev. 177, 88–90. doi: 10.1016/j.mad.2018.02.005
Barger, S. W., and Harmon, A. D. (1997). Microglial activation by alzhelmer amyloid precursor protein and modulation by apolipoprotein E. Nature 388, 878–881. doi: 10.1038/42257
Bauer, J., Strauss, S., Schreiter-Gasser, U., Ganter, U., Schlegel, P., Witt, I., et al. (1991). Interleukin-6 and α-2-macroglobulin indicate an acute-phase state in Alzheimer’s disease cortices. FEBS Lett. 285, 111–114. doi: 10.1016/0014-5793(91)80737-n
Bernatdotte, A., Mikhelson, V. M. K., and Spivak, I. M. (2016). Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 8, 1–9. doi: 10.18632/aging.100871
Berson, A., Nativio, R., Berger, S. L., and Bonini, N. M. (2018). Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 41, 587–598. doi: 10.1016/j.tins.2018.05.005
Bhat, R., Crowe, E. P., Bitto, A., Moh, M., Katsetos, C. D., Garcia, F. U., et al. (2012). Astrocyte senescence as a component of Alzheimer’s disease. PLoS One 7:e45069. doi: 10.1371/journal.pone.0045069
Birch, J., Anderson, R. K., Correia-Melo, C., Jurk, D., Hewitt, G., Marques, F. M., et al. (2015). DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L1124–L1137. doi: 10.1152/ajplung.00293.2015
Biron-Shental, T., Liberman, M., Sharvit, M., Sukenik-Halevy, R., and Amiel, A. (2015). Amniocytes from aneuploidy embryos have enhanced random aneuploidy and signs of senescence—can these findings be related to medical problems? Gene 562, 232–235. doi: 10.1016/j.gene.2015.02.075
Bischoff, C., Petersen, H., Graakjaer, J., Andersen-Ranberg, K., Vaupel, J., Bohr, V., et al. (2006). No association between telomere length and survival among the elderly and oldest old. Epidemiology 17, 190–194. doi: 10.1097/01.ede.0000199436.55248.10
Bitto, A., Sell, C., Crowe, E., Lorenzini, A., Malaguti, M., Hrelia, S., et al. (2010). Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 316, 2961–2968. doi: 10.1016/j.yexcr.2010.06.021
Bjerke, M., Zetterberg, H., Edman, Å., Blennow, K., Wallin, A., and Andreasson, U. (2011). Cerebrospinal fluid matrix metalloproteinases and tissue inhibitor of metalloproteinases in combination with subcortical and cortical biomarkers in vascular dementia and Alzheimer’s disease. J. Alzheimers Dis. 27, 665–676. doi: 10.3233/jad-2011-110566
Blackburn, E. H. (1991). Structure and function of telomeres. Nature 350, 569–573. doi: 10.1038/350569a0
Blasco, M. A., Lee, H. W., Hande, M. P., Samper, E., Lansdorp, P. M., DePinho, R. A., et al. (1997). Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91, 25–34. doi: 10.1016/s0092-8674(01)80006-4
Blum-Degen, D., Müller, T., Kuhn, W., Gerlach, M., Przuntek, H., and Riederer, P. (1995). Interleukin-1 β and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 202, 17–20. doi: 10.1016/0304-3940(95)12192-7
Blurton-Jones, M., and Laferla, F. M. (2006). Pathways by which Aβ facilitates tau pathology. Curr. Alzheimer Res. 3, 437–448. doi: 10.2174/156720506779025242
Boccardi, V., Pelini, L., Ercolani, S., Ruggiero, C., and Mecocci, P. (2015). From cellular senescence to Alzheimer’s disease: the role of telomere shortening. Ageing Res. Rev. 22, 1–8. doi: 10.1016/j.arr.2015.04.003
Bradley-Whitman, M. A., and Lovell, M. A. (2013). Epigenetic changes in the progression of Alzheimer’s disease. Mech. Ageing Dev. 134, 486–495. doi: 10.1016/j.mad.2013.08.005
Brand, M. D. (2014). The role of mitochondria in longevity and healthspan. Longev. Healthspan 3:7. doi: 10.1186/2046-2395-3-7
Brodacki, B., Staszewski, J., Toczyłowska, B., Kozłowska, E., Drela, N., Chalimoniuk, M., et al. (2008). Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNF, and INF concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 441, 158–162. doi: 10.1016/j.neulet.2008.06.040
Brosh, R. M., and Bohr, V. A. (2007). Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Res. 35, 7527–7544. doi: 10.1093/nar/gkm1008
Busciglio, J., Pelsman, A., Helguera, P., Ashur-Fabian, O., Pinhasov, A., Brenneman, D. E., et al. (2007). NAP and ADNF-9 protect normal and Down’s syndrome cortical neurons from oxidative damage and apoptosis. Curr. Pharm. Des. 13, 1091–1098. doi: 10.2174/138161207780618957
Busciglio, J., and Yankner, B. A. (1995). Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature 378, 776–779. doi: 10.1038/378776a0
Bussian, T. J., Aziz, A., Meyer, C. F., Swenson, B. L., van Deursen, J. M., and Baker, D. J. (2018). Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582. doi: 10.1038/s41586-018-0543-y
Cabelof, D. C. (2007). Aging and base excision repair: in need of a comprehensive approach. DNA Repair 6, 1399–1402. doi: 10.1016/j.dnarep.2007.06.006
Cabelof, D. C., Guo, Z., Raffoul, J. J., Sobol, R. W., Wilson, S. H., Richardson, A., et al. (2003). Base excision repair deficiency caused by polymerase β haploinsufficiency: accelerated DNA damage and increased mutational response to carcinogens. Cancer Res. 63, 5799–5807.
Cabelof, D. C., Patel, H. V., Chen, Q., van Remmen, H., Matherly, L. H., Ge, Y., et al. (2009). Mutational spectrum at GATA1 provides insights into mutagenesis and leukemogenesis in Down syndrome. Blood 114, 2753–2763. doi: 10.1182/blood-2008-11-190330
Cabelof, D. C., Raffoul, J. J., Yanamadala, S., Ganir, C., Guo, Z., and Heydari, A. R. (2002). Attenuation of DNA polymerase β-dependent base excision repair and increased DMS-induced mutagenicity in aged mice. Mutat. Res. 500, 135–145. doi: 10.1016/s0027-5107(02)00003-9
Cacabelos, R., Alvarez, X. A., Fernández-Novoa, L., Franco, A., Mangues, R., Pellicer, A., et al. (1994). Brain interleukin-1β in Alzheimer’s disease and vascular dementia. Methods Find. Exp. Clin. Pharmacol. 16, 141–151.
Cadonic, C., Sabbir, M. G., and Albensi, B. C. (2016). Mechanisms of mitochondrial dysfunction in Alzheimer’s disease. Mol. Neurobiol. 53, 6078–6090. doi: 10.1007/s12035-015-9515-5
Cai, Z., Yan, L.-J., and Ratka, A. (2013). Telomere shortening and Alzheimer’s disease. Neuromolecular. Med. 15, 25–48. doi: 10.1007/s12017-012-8207-9
Caldeira, C., Cunha, C., Vaz, A. R., Falcão, A. S., Barateiro, A., Seixas, E., et al. (2017). Key aging-associated alterations in primary microglia response to β-amyloid stimulation. Front. Aging Neurosci. 9:277. doi: 10.3389/fnagi.2017.00277
Campbell, I. L., Abraham, C. R., Masliah, E., Kemper, P., Inglis, J. D., Oldstone, M. B., et al. (1993). Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. U S A 90, 10061–10065. doi: 10.1073/pnas.90.21.10061
Casanova, M. F., Walker, L. C., Whitehouse, P. J., and Price, D. L. (1985). Abnormalities of the nucleus basalis in Down’s syndrome. Ann. Neurol. 18, 310–313. doi: 10.1002/ana.410180306
Cencioni, C., Spallotta, F., Martelli, F., Valente, S., Mai, A., Zeiher, A., et al. (2013). Oxidative stress and epigenetic regulation in ageing and age-related diseases. Int. J. Mol. Sci. 14, 17643–17663. doi: 10.3390/ijms140917643
Cenini, G., Dowling, A. L., Beckett, T. L., Barone, E., Mancuso, C., Murphy, M. P., et al. (2012). Association between frontal cortex oxidative damage and β-amyloid as a function of age in Down syndrome. Biochim. Biophys. Acta 1822, 130–138. doi: 10.1016/j.bbadis.2011.10.001
Chan, S. R. W. L., and Blackburn, E. H. (2004). Telomeres and telomerase. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 109–122. doi: 10.1098/rstb.2003.1370
Chen, Y., McMillan-Ward, E., Kong, J., Israels, S. J., and Gibson, S. B. (2007). Mitochondrial electron–transport–chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J. Cell Sci. 120, 4155–4166. doi: 10.1242/jcs.011163
Chen, H., Ruiz, P. D., McKimpson, W. M., Novikov, L., Kitsis, R. N., and Gamble, M. J. (2015). MacroH2A1 and ATM play opposing roles in paracrine senescence and the senescence-associated secretory phenotype. Mol. Cell 59, 719–731. doi: 10.1016/j.molcel.2015.07.011
Childs, B. G., Durik, M., Baker, D. J., and van Deursen, J. M. (2015). Cellular senescence in aging and age–related disease: from mechanisms to therapy. Nat. Med. 21, 1424–1435. doi: 10.1038/nm.4000
Childs, B. G., Gluscevic, M., Baker, D. J., Laberge, R. M., Marquess, D., Dananberg, J., et al. (2017). Senescent cells: an emerging target for diseases of ageing. Nat. Rev. Drug Discov. 16, 718–735. doi: 10.1038/nrd.2017.116
Chinta, S. J., Lieu, C. A., Demaria, M., Laberge, R. M., Campisi, J., and Andersen, J. K. (2013). Environmental stress, ageing and glial cell senescence: a novel mechanistic link to Parkinson’s disease? J. Intern. Med. 273, 429–436. doi: 10.1111/joim.12029
Chinta, S. J., Woods, G., Demaria, M., Rane, A., Zou, Y., Mcquade, A., et al. (2018). Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 22, 930–940. doi: 10.1016/j.celrep.2017.12.092
Chong, Y. (1997). Effect of a carboxy-terminal fragment of the Alzheimer’s amyloid precursor protein on expression of proinflammatory cytokines in rat glial cells. Life Sci. 61, 2323–2333. doi: 10.1016/s0024-3205(97)00936-3
Chow, H. M., and Herrup, K. (2015). Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 16, 672–684. doi: 10.1038/nrn4020
Chung, K. M., Hernández, N., Sproul, A., and Yu, W. H. (2018). Alzheimer’s disease and the autophagic-lysosomal system. Neurosci. Lett. 697, 49–58. doi: 10.1016/j.neulet.2018.05.017
Cipriani, S., Ferrer, I., Aronica, E., Kovacs, G. G., Verney, C., Nardelli, J., et al. (2018). Hippocampal radial glial subtypes and their neurogenic potential in human fetuses and healthy and Alzheimer’s disease adults. Cereb. Cortex 28, 2458–2478. doi: 10.1093/cercor/bhy096
Cleaver, J. E., Kaufmann, W. K., Kapp, L. N., and Park, S. D. (1983). Replicon size and excision repair as factors in the inhibition and recovery of DNA synthesis from ultraviolet damage, Biochim. Biochim. Biophys. Acta 739, 207–215. doi: 10.1016/0167-4781(83)90031-3
Combrinck, M., Perry, V., and Cunningham, C. (2002). Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience 112, 7–11. doi: 10.1016/s0306-4522(02)00030-1
Conde, J. R., and Streit, W. J. (2006). Microglia in the aging brain. J. Neuropathol. Exp. Neurol. 65, 199–203. doi: 10.1097/01.jnen.0000202887.22082.63
Contestabile, A., Fila, T., Bartesaghi, R., and Ciani, E. (2009a). Cell cycle elongation impairs proliferation of cerebellar granule cell precursors in the Ts65Dn mouse, an animal model for Down syndrome. Brain Pathol. 19, 224–237. doi: 10.1111/j.1750-3639.2008.00168.x
Contestabile, A., Fila, T., Cappellini, A., Bartesaghi, R., and Ciani, E. (2009b). Widespread impairment of cell proliferation in the neonate Ts65Dn mouse, a model for Down syndrome. Cell Prolif. 42, 171–181. doi: 10.1111/j.1365-2184.2009.00587.x
Contestabile, A., Fila, T., Ceccarelli, C., Bonasoni, P., Bonapace, L., Santini, D., et al. (2007). Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with Down syndrome and in Ts65Dn mice. Hippocampus 17, 665–678. doi: 10.1002/hipo.20308
Coppé, J.-P., Desprez, P.-Y., Krtolica, A., and Campisi, J. (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118. doi: 10.1146/annurev-pathol-121808-102144
Cormenier, J., Martin, N., Deslé, J., Salazar-Cardozo, C., Pourtier, A., Abbadie, C., et al. (2018). The ATF6 arm of the Unfolded Protein Response mediates replicative senescence in human fibroblasts through a COX2/prostaglandin E2 intracrine pathway. Mech. Ageing Dev. 170, 82–91. doi: 10.1016/j.mad.2017.08.003
Corrales, A., Martínez, P., García, S., Vidal, V., García, E., Flórez, J., et al. (2013). Long-term oral administration of melatonin improves spatial learning and memory and protects against cholinergic degeneration in middle-aged Ts65Dn mice, a model of Down syndrome. J. Pineal Res. 54, 346–358. doi: 10.1111/jpi.12037
Corrales, A., Vidal, R., García, S., Vidal, V., Martínez, P., García, E., et al. (2014). Chronic melatonin treatment rescues electrophysiological and neuromorphological deficits in a mouse model of Down syndrome. J. Pineal Res. 56, 51–61. doi: 10.1111/jpi.12097
Correia-Melo, C., and Passos, J. F. (2015). Mitochondria: are they causal players in cellular senescence? Biochim. Biophys. Acta 1847, 1373–1379. doi: 10.1016/j.bbabio.2015.05.017
Cristofalo, V. J., Lorenzini, A., Allen, R. G., Torres, C., and Tresini, M. (2004). Replicative senescence: a critical review. Mech. Ageing Dev. 125, 827–848. doi: 10.1016/j.mad.2004.07.010
Cunningham, C., Wilcockson, D., Campion, S., Lunnon, K., and Perry, V. (2005). Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J. Neurosci. 25, 9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005
d’Adda di Fagagna, F., Reaper, P. M., Clay-Farrace, L., Fiegler, H., Carr, P., Von Zglinicki, T., et al. (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198. doi: 10.1038/nature02118
de Arruda Cardoso Smith, M., Borsatto-Galera, B., Feller, R. I., Gonçalves, A., Oyama, R. S., Segato, R., et al. (2004). Telomeres on chromosome 21 and aging in lymphocytes and gingival fibroblasts from individuals with Down syndrome. J. Oral Sci. 46, 171–177. doi: 10.2334/josnusd.46.171
De Haan, J. B., Cristiano, F., Iannello, R., Bladier, C., Kelner, M. J., and Kola, I. (1996). Elevation in the ratio of Cu/Zn-superoxide dismutase to glutathione peroxidase activity induces features of cellular senescence and this effect is mediated by hydrogen peroxide. Hum. Mol. Genet. 5, 283–292. doi: 10.1093/hmg/5.2.283
de la Monte, S. M., Sohn, Y. K., and Wands, J. R. (1997). Correlates of p53- and Fas (CD95)-mediated apoptosis in Alzheimer’s disease. J. Neurol. Sci. 152, 73–83. doi: 10.1016/s0022-510x(97)00131-7
de Lange, T. (2002). Protection of mammalian telomeres. Oncogene 21, 532–540. doi: 10.1038/sj.onc.1205080
De Stefano, N., Stromillo, M. L., Giorgio, A., Bartolozzi, M. L., Battaglini, M., Baldini, M., et al. (2016). Establishing pathological cut-offs of brain atrophy rates in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 87, 93–99. doi: 10.1136/jnnp-2014-309903
Degerman, S., Domellof, M., Landfors, M., Linder, J., Lundin, M., Haraldsson, S., et al. (2014). Long leukocyte telomere length at diagnosis is a risk factor for dementia progression in idiopathic parkinsonism. PLoS One 9:e113387. doi: 10.1371/journal.pone.0113387
Demirtas, H. (2009). AgNOR status in Down’s syndrome infants and a plausible phenotype formation hypothesis. Micron 40, 511–518. doi: 10.1016/j.micron.2009.02.014
Di Domenico, F., Coccia, R., Cocciolo, A., Murphy, M. P., Cenini, G., Head, E., et al. (2013). Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: redox proteomics analysis of human brain. Biochim. Biophys. Acta 1832, 1249–1259. doi: 10.1016/j.bbadis.2013.04.013
Dias, V., Junn, E., and Mouradian, M. M. (2013). The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 3, 461–491. doi: 10.3233/JPD-130230
Dickson, D. W., Lee, S. C., Mattiace, L. A., Yen, S. H., and Brosnan, C. (1993). Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia 7, 75–83. doi: 10.1002/glia.440070113
Di Domenico, F., Pupo, G., Mancuso, C., Barone, E., Paolini, F., Arena, A., et al. (2015). Bach1 overexpression in Down syndrome correlates with the alteration of the HO-1/BVR-A system: insights for transition to Alzheimer disease. J. Alzheimers Dis. 44, 1107–1120. doi: 10.3233/Jad-141254
Dörr, J. R., Yu, Y., Milanovic, M., Beuster, G., Zasada, C., Däbritz, J. H. M., et al. (2013). Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501, 421–425. doi: 10.1038/nature12437
Dröge, W., and Schipper, H. (2007). Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell 6, 361–370. doi: 10.1111/j.1474-9726.2007.00294.x
Druelle, C., Drullion, C., Deslé, J., Martin, N., Saas, L., Cormenier, J., et al. (2016). ATF6α regulates morphological changes associated with senescence in human fibroblasts. Oncotarget 7, 67699–67715. doi: 10.18632/oncotarget.11505
Dursun, E., Gezen-Ak, D., Hană gası, H., Bilgiç, B., Lohmann, E., Ertan, S., et al. (2015). The interleukin 1α, interleukin 1β, interleukin 6 and α-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J. Neuroimmunol. 283, 50–57. doi: 10.1016/j.jneuroim.2015.04.014
Dutkowski, R. T., Lesh, R., Staiano-Coico, L., Thaler, H., Darlington, G. J., and Weksler, M. E. (1985). Increased chromosomal instability in lymphocytes from elderly humans. Mutat. Res. 149, 505–512. doi: 10.1016/0027-5107(85)90169-1
Eerola, J., Kananen, L., Manninen, K., Hellstrom, O., Tienari, P. J., and Hovatta, I. (2010). No evidence for shorter leukocyte telomere length in Parkinson’s disease patients. J. Gerontol. A Biol. Sci. Med. Sci. 65A, 1181–1184. doi: 10.1093/gerona/glq125
Eikelenboom, P., Veerhuis, R., Scheper, W., Rozemuller, A. J., van Gool, W. A., and Hoozemans, J. J. (2006). The significance of neuroinflammation in understanding Alzheimer’s disease. J. Neural Transm. 113, 1685–1695. doi: 10.1007/s00702-006-0575-6
Eitan, E., Hutchison, E. R., and Mattson, M. P. (2014). Telomere shortening in neurological disorders: an abundance of unanswered questions. Trends Neurosci. 37, 256–263. doi: 10.1016/j.tins.2014.02.010
Erusalimsky, J. D. (2009). Vascular endothelial senescence: from mechanisms to pathophysiology. J. Appl. Physiol. 106, 326–332. doi: 10.1152/japplphysiol.91353.2008
Esbensen, A. J. (2010). Health conditions associated with aging and end of life of adults with Down syndrome. Int. Rev. Res. Ment. Retard. 39, 107–126. doi: 10.1016/s0074-7750(10)39004-5
Esposito, M., and Sherr, G. L. (2019). Epigenetic modifications in Alzheimer’s neuropathology and therapeutics. Front. Neurosci. 13:476. doi: 10.3389/fnins.2019.00476
Evans, R. J., Wyllie, F. S., Wynford-Thomas, D., Kipling, D., and Jones, C. J. (2003). A P53-dependent, telomere-independent proliferative life span barrier in human astrocytes consistent with the molecular genetics of glioma development. Cancer Res. 63, 4854–4861.
Faragher, R. G. A., McArdle, A., Willows, A., and Ostler, E. L. (2017). Senescence in the aging process. F1000Res 6:1219. doi: 10.12688/f1000research.10903.1
Ferrer, I., Martinez, A., Blanco, R., Dalfó, E., and Carmona, M. (2011). Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: preclinical Parkinson disease. J. Neural Transm. 118, 821–839. doi: 10.1007/s00702-010-0482-8
Ferrón, S., Mira, H., Franco, S., Cano-Jaimez, M., Bellmunt, E., Ramírez, C., et al. (2004). Telomere shortening and chromosomal instability abrogates proliferation of adult but not embryonic neural stem cells. Development 131, 4059–4070. doi: 10.1242/dev.01215
Fielder, E., von Zglinicki, T., and Jurk, D. (2017). The DNA damage response in neurons: die by apoptosis or survive in a senescence-like state? J. Alzheimers Dis. 60, S107–S131. doi: 10.3233/JAD-161221
Flanary, B. E., Sammons, N. W., Nguyen, C., Walker, D., and Streit, W. J. (2007). Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 10, 61–74. doi: 10.1089/rej.2006.9096
Flanary, B. E., and Streit, W. J. (2004). Progressive telomere shortening occurs in cultured rat microglia, but not astrocytes. Glia 45, 75–88. doi: 10.1002/glia.10301
Floden, A. M., and Combs, C. K. (2011). Microglia demonstrate age-dependent interaction with amyloid-β fibrils. J. Alzheimers Dis. 25, 279–293. doi: 10.3233/JAD-2011-101014
Forero, D. A., González-Giraldo, Y., López-Quintero, C., Castro-Vega, L. J., Barreto, G. E., Perry, G., et al. (2016). Meta-analysis of telomere length in Alzheimer’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 71, 1069–1073. doi: 10.1093/gerona/glw053
Frank, M., Barrientos, R., Biedenkapp, J., Rudy, J., Watkins, L., and Maier, S. (2006). mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol. Aging 27, 717–722. doi: 10.1016/j.neurobiolaging.2005.03.013
Freidl, M., Gulesserian, T., Lubec, G., Fountoulakis, M., and Lubec, B. (2001). Deterioration of the transcriptional, splicing and elongation machinery in brain of fetal Down syndrome. J. Neural Transm. 61, 47–57. doi: 10.1007/978-3-7091-6262-0_4
Freund, A., Laberge, R.-M., Demaria, M., and Campisi, J. (2012). Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 23, 2066–2075. doi: 10.1091/mbc.E11-10-0884
Freund, A., Orjalo, A. V., Desprez, P.-Y., and Campisi, J. (2010). Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 16, 238–246. doi: 10.1016/j.molmed.2010.03.003
Fumagalli, M., Rossiello, F., Clerici, M., Barozzi, S., Cittaro, D., Kaplunov, J. M., et al. (2012). Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365. doi: 10.1038/ncb2466
Fuster-Matanzo, A., Llorens-Martín, M., Hernández, F., and Avila, J. (2013). Role of neuroinflammation in adult neurogenesis and Alzheimer disease: therapeutic approaches. Mediators Inflamm. 2013:260925. doi: 10.1155/2013/260925
Galbiati, A., Beauséjour, C., and d’Adda di Fagagna, F. (2017). A novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging Cell 16, 422–427. doi: 10.1111/acel.12573
Gao, J., Wang, L., Liu, J., Xie, F., Su, B., and Wang, X. (2017). Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants 6:25. doi: 10.3390/antiox6020025
García-Cerro, S., Rueda, N., Vidal, V., Lantigua, S., and Martínez-Cué, C. (2017). Normalizing the gene dosage of Dyrk1A in a mouse model of Down syndrome rescues several Alzheimer’s disease phenotypes. Neurobiol. Dis. 106, 76–88. doi: 10.1016/j.nbd.2017.06.010
Georgakopoulou, E. A., Tsimaratou, K., Evangelou, K., Fernandez-Marcos, P. J., Zoumpourlis, V., Trougakos, I. P., et al. (2013). Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 5, 37–50. doi: 10.18632/aging.100527
Gezen-Ak, D., Dursun, E., Hanăgası, H., Bilgiç, B., Lohman, E., Araz, Ö. S., et al. (2013). BDNF, TNFα, HSP90, CFH, and IL-10 serum levels in patients with early or late onset Alzheimer’s disease or mild cognitive impairment. J. Alzheimers Dis. 37, 185–195. doi: 10.3233/JAD-130497
Gitter, B. D., Cox, L. M., Rydel, R. E., and May, P. C. (1995). Amyloid β peptide potentiates cytokine secretion by interleukin-1β-activated human astrocytoma cells. Proc. Natl. Acad. Sci. U S A 92, 10738–10741. doi: 10.1073/pnas.92.23.10738
Gorgoulis, V. G., and Halazonetis, T. D. (2010). Oncogene-induced senescence: the bright and dark side of the response. Curr. Opin. Cell Biol 22, 816–827. doi: 10.1016/j.ceb.2010.07.013
Greider, C. W., and Blackburn, E. H. (1987). The telomere terminal transferase of tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 51, 887–898. doi: 10.1016/0092-8674(87)90576-9
Griffin, W. S. (2006). Inflammation and neurodegenerative diseases. Am. J. Clin. Nutr. 83, 470–474. doi: 10.1093/ajcn/83.2.470S
Griffin, W. S., Stanley, L. C., Ling, C., White, L., MacLeod, V., Perrot, L. J., et al. (1989). Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. U S A 86, 7611–7615. doi: 10.1073/pnas.86.19.7611
Grodstein, F., van Oijen, M., Irizarry, M. C., Rosas, H. D., Hyman, B. T., Growdon, J. H., et al. (2008). Shorter telomeres may mark early risk of dementia: preliminary analysis of 62 participants from the nurses’ health study. PLoS One 3:e1590. doi: 10.1371/journal.pone.0001590
Guan, J. Z., Guan, W. P., Maeda, T., and Makino, N. (2012). Effect of vitamin E administration on the elevated oxygen stress and the telomeric and subtelomeric status in Alzheimer’s disease. Gerontology 58, 62–69. doi: 10.1159/000327821
Guan, J. Z., Maeda, T., Sugano, M., Oyama, J., Higuchi, Y., Suzuki, T., et al. (2008). A percentage analysis of the telomere length in Parkinson’s disease patients. J. Gerontol. A Biol. Sci. Med. Sci. 63, 467–473. doi: 10.1093/gerona/63.5.467
Guerreiro, R., Brás, J., and Hardy, J. (2013). Snapshot: genetics of Alzheimer’s disease. Cell 155, 968–968.e1. doi: 10.1016/j.cell.2013.10.037
Hackett, P. B., Varmus, H. E., and Bishop, J. M. (1981). Repair of lesions which cause premature termination of transcription in chicken embryo cells irradiated with ultraviolet light. Virology 112, 752–756. doi: 10.1016/0042-6822(81)90320-2
Hao, L.-Y., Armanios, M., Strong, M., Karim, B., Feldser, D. M., Huso, D., et al. (2005). Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell 123, 1121–1131. doi: 10.1016/j.cell.2005.11.020
Hardy, J. (2006). Has the amyloid cascade hypothesis for Alzheimer’s disease been proved? Curr. Alzheimer Res. 3, 71–73. doi: 10.2174/156720506775697098
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Harley, C. B. (1991). Telomere loss: mitotic clock or genetic time bomb? Mutat. Res. 256, 271–282. doi: 10.1016/0921-8734(91)90018-7
Harley, C. B., Futcher, A. B., and Greider, C. W. (1990). Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460. doi: 10.1038/345458a0
Hastie, N. D., Dempster, M., Dunlop, M. G., Thompson, A. M., Green, D. K., and Allshire, R. C. (1990). Telomere reduction in human colorectal carcinoma and with ageing. Nature 346, 866–868. doi: 10.1038/346866a0
Haydar, T. F., and Reeves, R. H. (2012). Trisomy 21 and early brain development. Trends Neurosci. 35, 81–91. doi: 10.1016/j.tins.2011.11.001
Hayflick, L., and Moorhead, P. S. (1961). The serial cultivation of human diploid strains. Exp. Cell Res. 25, 585–621. doi: 10.1016/0014-4827(61)90192-6
He, N., Jin, W.-L., Lok, K.-H., Wang, Y., Yin, M., and Wang, Z.-J. (2013). Amyloid-β(1–42) oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis. 4:e924. doi: 10.1038/cddis.2013.437
Heidinger, B. J., Blount, J. D., Boner, W., Griffiths, K., Metcalfe, N. B., and Monaghan, P. (2012). Telomere length in early life predicts lifespan. Proc. Natl. Acad. Sci. U S A 109, 1743–1748. doi: 10.1073/pnas.1113306109
Henry, C., Huang, Y., Wynne, A., and Godbout, J. (2009). Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behav. Immun. 23, 309–317. doi: 10.1016/j.bbi.2008.09.002
Herbig, U., Ferreira, M., Condel, L., Carey, D., and Sedivy, J. M. (2006). Cellular senescence in aging primates. Science 311:1257. doi: 10.1126/science.1122446
Herbig, U., Jobling, W. A., Chen, B. P. C., Chen, D. J., and Sedivy, J. M. (2004). Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a. Mol. Cell 14, 501–513. doi: 10.1016/s1097-2765(04)00256-4
Hernandez-Segura, A., Nehme, J., and Demaria, M. (2018). Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453. doi: 10.1016/j.tcb.2018.02.001
Herrup, K., and Yang, Y. (2007). Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat. Rev. Neurosci. 8, 368–378. doi: 10.1038/nrn2124
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. doi: 10.1038/nrn3689
Hewitt, G., Jurk, D., Marques, F. D., Correia-Melo, C., Hardy, T., Gackowska, A., et al. (2012). Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 3:708. doi: 10.1038/ncomms1708
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360. doi: 10.1523/JNEUROSCI.0616-08
Hinterberger, M., Fischer, P., Huber, K., Krugluger, W., and Zehetmayer, S. (2017). Leukocyte telomere length is linked to vascular risk factors not to Alzheimer’s disease in the VITA study. J. Neural Transm. 124, 809–819. doi: 10.1007/s00702-017-1721-z
Hipp, M. S., Park, S. H., and Hartl, F. U. (2014). Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 24, 506–514. doi: 10.1016/j.tcb.2014.05.003
Ho, G. J., Drego, R., Hakimian, E., and Masliah, E. (2005). Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Curr. Drug Targets. Inflamm. Allergy 4, 247–256. doi: 10.2174/1568010053586237
Hochstrasser, T., Marksteiner, J., and Humpel, C. (2012). Telomere length is age-dependent and reduced in monocytes of Alzheimer patients. Exp. Gerontol. 47, 160–163. doi: 10.1016/j.exger.2011.11.012
Hockemeyer, D., Sfeir, A. J., Shay, J. W., Wright, W. E., and de Lange, T. (2005). POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J. 24, 2667–2678. doi: 10.1038/sj.emboj.7600733
Höhn, A., and Grune, T. (2013). Lipofuscin: formation, effects and role of macroautophagy. Redox Biol. 1, 140–144. doi: 10.1016/j.redox.2013.01.006
Horstmann, S., Budig, L., Gardner, H., Koziol, J., Deuschle, M., Schilling, C., et al. (2018). Matrix metalloproteinases in peripheral blood and cerebrospinal fluid in patients with Alzheimer’s disease. Int. Psychogeriatr. C Int. Psychogeriatr. Assoc. 226, 966–972. doi: 10.1017/S1041610210000827
Howcroft, T. K., Campisi, J., Louis, G. B., Smith, M. T., Wise, B., Wyss-Coray, T., et al. (2013). The role of inflammation in age-related disease. Aging 5, 84–93. doi: 10.18632/aging.100531
Huell, M., Strauss, S., Volk, B., Berger, M., and Bauer, J. (1995). Interleukin-6 is present in early stages of plaque formation and is restricted to the brains of Alzheimer’s disease patients. Acta Neuropathol. 89, 544–551. doi: 10.1007/bf00571510
Hunter, C. L., Bachman, D., and Granholm, A. C. (2004). Minocycline prevents cholinergic loss in a mouse model of Down’s syndrome. Ann. Neurol. 56, 675–688. doi: 10.1002/ana.20250
Ihara, Y., Morishima-Kawashima, M., and Nixon, R. (2012). The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006361. doi: 10.1101/cshperspect.a006361
Imamura, K., Hishikawa, N., Sawada, M., Nagatsu, T., Yoshida, M., and Hashizume, Y. (2003). Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 106, 518–526. doi: 10.1007/s00401-003-0766-2
Jenkins, E. C., Ye, L., Gu, H., Ni, S. A., Duncan, C. J., Velinov, M., et al. (2008). Increased “absence” of telomeres may indicate Alzheimer’s disease/dementia status in older individuals with Down syndrome. Neurosci. Lett. 440, 340–343. doi: 10.1016/j.neulet.2008.05.098
Jennings, B. J., Ozanne, S. E., and Hales, C. N. (2000). Nutrition, oxidative damage, telomere shortening and cellular senescence: individual or connected agents of aging? Mol. Genet. Metab. 71, 32–42. doi: 10.1006/mgme.2000.3077
Joo, H.-Y., Zhai, L., Yang, C., Nie, S., Erdjument-Bromage, H., Tempst, P., et al. (2007). Regulation of cell cycle progression and gene expression by H2A deubiquitination. Nature 449, 1068–1072. doi: 10.1038/nature06256
Jurk, D., Wang, C., Miwa, S., Maddick, M., Korolchuk, V., Tsolou, A., et al. (2012). Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004. doi: 10.1111/j.1474-9726.2012.00870.x
Jurk, D., Wilson, C., Passos, J. F., Oakley, F., Correia-Melo, C., Greaves, L., et al. (2014). Chronic inflammation induces telomeredysfunction and accelerates ageing in mice. Nat. Commun. 2:4172. doi: 10.1038/ncomms5172
Kalanj-Bognar, S., Rundek, T., Furac, I., Demarin, V., and Cosović, C. (2002). Leukocyte lysosomal enzymes in Alzheimer’s disease and Down’s syndrome. J. Gerontol. A Biol. Sci. Med. Sci. 57, B16–B21. doi: 10.1093/gerona/57.1.b16
Kang, H. T., Lee, K. B., Kim, S. Y., Choi, H. R., and Park, S. C. (2011). Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS One 6:e23367. doi: 10.1371/journal.pone.0023367
Karlseder, J., Smogorzewska, F. A., and de Lange, T. (2002). Senescence induced by altered telomere state, not telomere loss. Science 295, 2446–2449. doi: 10.1126/science.1069523
Kaul, Z., Cesare, A. J., Huschtscha, L. I., Neumann, A. A., and Reddel, R. R. (2012). Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep. 13, 52–59. doi: 10.1038/embor.2011.227
Kawanishi, S., and Oikawa, S. (2004). Mechanism of telomere shortening by oxidative stress. Ann. N Y Acad. Sci. 1019, 278–284. doi: 10.1196/annals.1297.047
Kiecolt-Glaser, J. K., Preacher, K. J., MacCallum, R. C., Atkinson, C., Malarkey, W. B., and Glaser, R. (2003). Chronic stress and age-related increases in the proinflammatory cytokine IL-6. Proc. Natl. Acad. Sci. U S A 100, 9090–9095. doi: 10.1073/pnas.1531903100
Kim, E. C., and Kim, J. R. (2019). Senotherapeutics: emerging strategy for healthy aging and age-related disease. BMB Rep. 52, 47–55. doi: 10.5483/bmbrep.2019.52.1.293
Kimura, M., Cao, X., Skurnick, J., Cody, M., Soteropoulos, P., and Aviv, A. (2005). Proliferation dynamics in cultured skin fibroblasts from Down syndrome subjects. Free Radic. Biol. Med. 39, 374–380. doi: 10.1016/j.freeradbiomed.2005.03.023
Kirkland, J. L., and Tchkonia, T. (2017). Cellular senescence: a translational perspective. EBioMedicine 21, 21–28. doi: 10.1016/j.ebiom.2017.04.013
Klocker, H., Schneider, R., Burtscher, H. J., Auer, B., Hirsch-Kauffmann, M., and Schweiger, M. (1985). Transient expression of a plasmid gene, a tool to study DNA repair in human cells: defect of DNA repair in Cockayne syndrome; one thymine cyclobutane dimer is sufficient to block transcription. Eur. J. Cell Biol. 39, 346–351.
Koliada, A. K., Vaiserman, A. M., Krasnenkov, D. S., and Karaban’, I. N. (2014). The study of telomere length in patients with Parkinson’s disease. Zh. Nevrol. Psikhiatr. Im. S S Korsakova 114, 58–61.
Komseli, E.-S., Pateras, I. S., Krejsgaard, T., Stawiski, K., Rizou, S. V., Polyzos, A., et al. (2018). A prototypical non-malignant epithelial model to study genome dynamics and concurrently monitor micro-RNAs and proteins in situ during oncogene-induced senescence. BMC Genomics 19:37. doi: 10.1186/s12864-017-4375-1
Kong, C. M., Lee, X. W., and Wang, X. (2013). Telomere shortening in human diseases. FEBS J. 280, 3180–3193. doi: 10.1111/febs.12326
Kota, L. N., Bharath, S., Purushottam, M., Moily, N. S., Sivakumar, P. T., Varghese, M., et al. (2015). Reduced telomere length in neurodegenerative disorders may suggest shared biology. J. Neuropsychiatry Clin. Neurosci. 27, e92–e96. doi: 10.1176/appi.neuropsych.13100240
Krenning, L., Feringa, F. M., Shaltiel, I. A., Van den Berg, J., and Medema, R. H. (2014). Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol. Cell 55, 59–72. doi: 10.1016/j.molcel.2014.05.007
Kriegstein, A., and Alvarez-Buylla, A. (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149–184. doi: 10.1146/annurev.neuro.051508.135600
Kritsilis, M. V., Rizou, S. V., Koutsoudaki, P. N., Evangelou, K., Gorgoulis, V. G., and Papadopoulos, D. (2018). Ageing, cellular senescence and neurodegenerative disease. Int. J. Mol. Sci. 19:E2937. doi: 10.3390/ijms19102937
Kuilman, T., Michaloglou, C., Mooi, W. J., and Peeper, D. S. (2010). The essence of senescence. Genes Dev. 24, 2463–2479. doi: 10.1101/gad.1971610
Kültz, D. (2005). Molecular and evolutionary basis of the cellular stress response. Annu. Rev. Physiol. 67, 225–257. doi: 10.1146/annurev.physiol.67.040403.103635
Kwon, M. J., Kim, S., Han, M. H., and Lee, S. B. (2016). Epigenetic changes in neurodegenerative diseases. Mol. Cells 39, 783–789. doi: 10.14348/molcells.2016.0233
Labbadia, J., and Morimoto, R. I. (2015). The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464. doi: 10.1146/annurev-biochem-060614-033955
Lai, K. S. P., Liu, C. S., Rau, A., Lanctôt, K. L., Köhler, C. A., Pakosh, M., et al. (2017). Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 88, 876–882. doi: 10.1136/jnnp-2017-316201
Lawless, C., Wang, C., Jurk, D., Merz, A., Zglinicki, T. V., and Passos, J. F. (2010). Quantitative assessment of markers for cell senescence. Exp. Gerontol. 45, 772–778. doi: 10.1016/j.exger.2010.01.018
Leake, A., Morris, C., and Whateley, J. (2000). Brain matrix metalloproteinase 1 levels are elevated in Alzheimer’s disease. Neurosci. Lett. 291, 201–203. doi: 10.1016/s0304-3940(00)01418-x
Lee, H. W., Blasco, M. A., Gottlieb, G. J., Horner, J. W. II., Greider, C. W., and DePinho, R. A. (1998). Essential role of mouse telomerase in highly proliferative organs. Nature 392, 569–574. doi: 10.1038/33345
Lee, B. Y., Han, J. A., Im, J. S., Morrone, A., Johung, K., Goodwin, E. C., et al. (2006). Senescence-associated β-galactosidase is lysosomal-galactosidase. Aging Cell 5, 187–195. doi: 10.1111/j.1474-9726.2006.00199.x
Li, H., Hedmer, M., Wojdacz, T., Hossain, M. B., Lindh, C. H., Tinnerberg, H. Ô., et al. (2015). Oxidative stress, telomere shortening, and DNA methylation in relation to low-to-moderate occupational exposure to welding fumes. Environ. Mol. Mutagen. 56, 684–693. doi: 10.1002/em.21958
Li, W.-Q., Wang, Z., Liu, S., Hu, Y., Yin, M., and Lu, Y. (2016). N-stearoyl-L-tyrosine inhibits the senescence of neural stem/progenitor cells induced by Aβ 1–42 via the CB2 receptor. Stem Cells Int. 2016:7419389. doi: 10.1155/2016/7419389
Liguori, I., Russo, G., Curcio, F., Bulli, G., Aran, L., Della-Morte, D., et al. (2018). Oxidative stress, aging, and diseases. Clin. Interv. Aging 13, 757–772. doi: 10.2147/CIA.S158513
Lindqvist, D., Kaufman, E., Brundin, L., Hall, S., Surova, Y., and Hansson, O. (2012). Non-motor symptoms in patients with Parkinson’s disease—correlations with inflammatory cytokines in serum. PLoS One 7:e47387. doi: 10.1371/journal.pone.0047387
Liu, L., Trimarchi, J. R., Navarro, P., Blasco, M. A., and Keefe, D. L. (2003). Oxidative stress contributes to arsenic-induced telomere attrition, chromosome instability, and apoptosis. J. Biol. Chem. 278, 31998–32004. doi: 10.1074/jbc.m303553200
Llorens-Martín, M., Jurado-Arjona, J., Fuster-Matanzo, A., Hernández, F., Rábano, A., and Ávila, J. (2014). Peripherally triggered and GSK-3β driven brain inflammation differentially skew adult hippocampal neurogenesis, behavioral pattern separation and microglial activation in response to ibuprofen. Transl. Psychiatry 4:e463. doi: 10.1038/tp.2014.92
Lockrow, J., Boger, H., Bimonte-Nelson, H., and Granholm, A. C. (2011). Effects of long-term memantine on memory and neuropathology in Ts65Dn mice, a model for Down syndrome. Behav. Brain Res. 221, 610–622. doi: 10.1016/j.bbr.2010.03.036
Lockrow, J., Prakasam, A., Huang, P., Bimonte-Nelson, H., Sambamurti, K., and Granholm, A. C. (2009). Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Exp. Neurol. 216, 278–289. doi: 10.1016/j.expneurol.2008.11.021
Lockstone, H. E., Harris, L. W., Khaitovich, P., Weickert, C. S., Webster, M. J., and Bahn, S. (2007). Gene expression profiling in the adult Down syndrome brain. Genomics 90, 647–660. doi: 10.1016/j.ygeno.2007.08.005
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Lord, J., and Cruchaga, C. (2014). The epigenetic landscape of Alzheimer’s disease. Nat. Neurosci. 17, 1138–1140. doi: 10.1038/nn.3792
Lott, I. T. (2012). Neurological phenotypes for Down syndrome across the life span. Prog. Brain Res. 197, 101–121. doi: 10.1016/b978-0-444-54299-1.00006-6
Lovell, M. A., Gabbita, S. P., and Markesbery, W. R. (1999). Increased DNA oxidation and decreased levels of repair products in Alzheimer’s disease ventricular CSF. J. Neurochem. 72, 771–776. doi: 10.1046/j.1471-4159.1999.0720771.x
Lukens, J. N., Van Deerlin, V., Clark, C. M., Xie, S. X., and Johnson, F. B. (2009). Comparisons of telomere lengths in peripheral blood and cerebellum in Alzheimer’s disease. Alzheimers Dement. 5, 463–469. doi: 10.1016/j.jalz.2009.05.666
Luterman, J. D., Haroutunian, V., Yemul, S., Ho, L., Purohit, D., Aisen, P. S., et al. (2000). Cytokine gene expression as a function of the clinical progression of Alzheimer disease dementia. Arch. Neurol. 57, 1153–1160. doi: 10.1001/archneur.57.8.1153
Luth, I. H. J., Holzer, M., Gertz, H.-J., and Arendt, T. (2000). Aberrant expression of nNOS in pyramidal neurons in Alzheimer’s disease is highly co-localized with p21 ras and p16. Brain Res. 852, 45–55. doi: 10.1016/s0006-8993(99)02178-2
Lyman, M., Lloyd, D. G., Ji, X., Vizcaychipi, M. P., and Ma, D. (2014). Neuroinflammation: the role and consequences. Neurosci. Res. 79, 1–12. doi: 10.1016/j.neures.2013.10.004
Ma, D., Stokes, K., Mahngar, K., Domazet-Damjanov, D., Sikorska, M., and Pandey, S. (2014). Inhibition of stress induced premature senescence in presenilin-1 mutated cells with water soluble Coenzyme Q10. Mitochondrion 17, 106–115. doi: 10.1016/j.mito.2014.07.004
Maeda, T., Guan, J. Z., Koyanagi, M., Higuchi, Y., and Makino, N. (2012). Aging-associated alteration of telomere length and subtelomeric status in female patients with Parkinson’s disease. J. Neurogenet. 26, 245–251. doi: 10.3109/01677063.2011.651665
Magini, A., Polchi, A., Tozzi, A., Tancini, B., Tantucci, M., Urbanelli, L., et al. (2015). Abnormal cortical lysosomal β-hexosaminidase and β-galactosidase activity at post-synaptic sites during Alzheimer’s disease progression. Int. J. Biochem. Cell Biol. 58, 62–70. doi: 10.1016/j.biocel.2014.11.001
Mansour, H., Chamberlain, C. G., Weible, M. W. II., Hughes, S., Chu, Y., and Chan-Ling, T. (2008). Aging-related changes in astrocytes in the rat retina: imbalance between cell proliferation and cell death reduces astrocyte availability. Aging Cell 7, 526–540. doi: 10.1111/j.1474-9726.2008.00402.x
Martinez, A., Lopez, N., Gonzalez, C., and Hetz, C. (2019). Targeting of the unfolded protein response (UPR) as therapy for Parkinson’s disease. Biol. Cell 111, 161–168. doi: 10.1111/boc.201800068
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330:1774. doi: 10.1126/science.1197623
Mayer, P. J., Lange, C. S., Bradley, M. O., and Nichols, W. W. (1989). Age-dependent decline in rejoining of X-ray-induced DNA double-strand breaks in normal human lymphocytes. Mutation Res. 219, 95–100. doi: 10.1016/0921-8734(89)90019-2
Maynard, S., Fang, E. F., Scheibye-Knudsen, M., Croteau, D. L., and Bohr, V. A. (2015). DNA damage, DNA repair, aging, and neurodegeneration. Cold Spring Harb. Perspect. Med. 5:a025130. doi: 10.1101/cshperspect.a025130
Mayne, L. V. (1984). Inhibitors of DNA synthesis (aphidicolin and araC/HU) prevent the recovery of RNA synthesis after UV-irradiation. Mutat. Res. 131, 187–191. doi: 10.1016/0167-8817(84)90023-3
McCoy, M., Martinez, T., Ruhn, K., Szymkowski, D., Smith, C., Botterman, B., et al. (2006). Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J. Neurosci. 26, 9365–9375. doi: 10.1523/JNEUROSCI.1504-06.2006
McGeer, P., Itagaki, S., Boyes, B., and McGeer, E. (1988). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291. doi: 10.1212/wnl.38.8.1285
McGeer, E. G., Norman, M., Boyes, B., O’Kusky, J., Suzuki, J., and McGeer, P. L. (1985). Acetylcholine and aromatic amine systems in postmortem brain of an infant with Down’s syndrome. Exp. Neurol. 87, 557–570. doi: 10.1016/0014-4886(85)90184-0
McShea, A., Harris, P. L., Webster, K. R., Wahl, A. F., and Smith, M. A. (1997). Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am. J. Pathol. 150, 1933–1939.
Meager, A. (2004). “Cytokines: interleukins,” in Encyclopedia of Molecular Cell Biology and Molecular Medecine, ed. R. Meyers (Weinheim: Wilhey-VCH), 115–151.
Meager, A. (2005). “Viral inhibitors and immune response mediators: the interferons,” in Encyclopedia of Molecular Cell Biology and Molecular Medecine, ed. R. Meyers (Weinheim: Wilhey-VCH), 387–421.
Melki, R. (2015). Role of different α-synuclein strains in synucleinopathies, similarities with other neurodegenerative diseases. J. Parkinsons Dis. 5, 217–227. doi: 10.3233/jpd-150543
Mendelsohn, A. R., and Larrick, J. W. (2018). Cellular senescence as the key intermediate in tau-mediated neurodegeneration. Rejuvenation Res. 21, 572–579. doi: 10.1089/rej.2018.2155
Mentis, A. F. (2016). Epigenomic engineering for Down syndrome. Neurosci. Biobehav. Rev. 71, 323–327. doi: 10.1016/j.neubiorev.2016.09.012
Minciullo, P. L., Catalano, A., Mandraffino, G., Casciaro, M., Crucitti, A., Maltese, G., et al. (2016). Inflammaging and anti-iInflammaging: the role of cytokines in extreme longevity. Arch. Immunol. Ther. Exp. 64, 111–126. doi: 10.1007/s00005-015-0377-3
Ming, G., and Song, H. (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702. doi: 10.1016/j.neuron.2011.05.001
Mogi, M., Harada, M., Kondo, T., Riederer, P., Inagaki, H., Minami, M., et al. (1994a). Interleukin-1, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci. Lett. 180, 147–150. doi: 10.1016/0304-3940(94)90508-8
Mogi, M., Harada, M., Riederer, P., Narabayashi, H., Fujita, K., and Nagatsu, T. (1994b). Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 165, 208–210. doi: 10.1016/0304-3940(94)90746-3
Monti, D., Grassilli, E., Troiano, L., Cossarizza, A., Salvioli, S., Barbieri, D., et al. (1992). Senescence, immortalization, and apoptosis. An intriguing relationship. Ann. N Y Acad. Sci. 673, 70–82. doi: 10.1111/j.1749-6632.1992.tb27438.x
Moreno-Jiménez, E. P., Flor-García, M., Terreros-Roncal, J., Rábano, A., Cafini, F., Pallas-Bazarra, N., et al. (2019). Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 25, 554–560. doi: 10.1038/s41591-019-0375-9
Mosher, K. I., and Wyss-Coray, T. (2014). Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 88, 594–604. doi: 10.1016/j.bcp.2014.01.008
Mount, M., Lira, A., Grimes, D., Smith, P., Faucher, S., Slack, R., et al. (2007). Involvement of interferon-γ in microglial-mediated loss of dopaminergic neurons. J. Neurosci. 27, 3328–3337. doi: 10.1523/JNEUROSCI.5321-06.2007
Muñoz-Espín, D., and Serrano, M. (2014). Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496. doi: 10.1038/nrm3823
Musi, N., Valentine, J. M., Sickora, K. R., Baeuerle, E., Thompson, C. S., Shen, Q., et al. (2018). Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 17:e12840. doi: 10.1111/acel.12840
Myung, N.-H., Zhu, X., Kruman, I. I., Castellani, R. J., Petersen, R. B., Siedlak, S. L., et al. (2008). Evidence of DNA damage in Alzheimer disease: phosphorylation of histone H2AX in astrocytes. Age 30, 209–215. doi: 10.1007/s11357-008-9050-7
Naderi, J., Lopez, C., and Pandey, S. (2006). Chronically increased oxidative stress in fibroblasts from Alzheimer’s disease patients causes early senescence and renders resistance to apoptosis by oxidative stress. Mech. Ageing Dev. 127, 25–35. doi: 10.1016/j.mad.2005.08.006
Nagy, Z. (2005). The last neuronal division: a unifying hypothesis for the pathogenesis of Alzheimer’s disease. J. Cell. Mol. Med. 9, 531–541. doi: 10.1111/j.1582-4934.2005.tb00485.x
Nakamura, A. J., Chiang, Y. J., Hathcock, K. S., Horikawa, I., Sedelnikova, O. A., Hodes, R. J., et al. (2008). Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenetics Chromatin 1:6. doi: 10.1186/1756-8935-1-6
Nasrabady, S. E., Rizvi, B., Goldman, J. E., and Brickman, A. M. (2018). White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 6:22. doi: 10.1186/s40478-018-0515-3
Necchi, D., Lomoio, S., and Scherini, E. (2011). Dysfunction of the ubiquitin-proteasome system in the cerebellum of aging Ts65Dn mice. Exp. Neurol. 232, 114–118. doi: 10.1016/j.expneurol.2011.08.009
Nelson, G., Wordsworth, J., Wang, C., Jurk, D., Lawless, C., Martin-Ruiz, C., et al. (2012). A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11, 345–349. doi: 10.1111/j.1474-9726.2012.00795.x
Nette, E. G., Xi, Y.-P., Sun, Y.-K., Andrews, A. D., and King, D. W. (1984). A correlation between aging and DNA repair in human epidermal cells. Mech. Ageing Dev. 24, 283–292. doi: 10.1016/0047-6374(84)90114-3
Nixon, R. A., Mathews, P. M., and Cataldo, A. M. (2001). The neuronal endosomal-lysosomal system in Alzheimer’s disease. J. Alzheimers Dis. 3, 97–107. doi: 10.3233/jad-2001-3114
Nixon, R. A. (2013). The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997. doi: 10.1038/nm.3232
Nižetić, D., and Groet, J. (2012). Tumorigenesis in Down’s syndrome: big lessons from a small chromosome. Nat. Rev. Cancer 12, 721–732. doi: 10.1038/nrc3355
Njie, E., Boelen, E., Stassen, F., Steinbusch, H., Borchelt, D., and Streit, W. (2012). Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 33, 195.e1–195.e12. doi: 10.1016/j.neurobiolaging.2010.05.008
Nussbaum, R. L., and Ellis, C. E. (2003). Alzheimer’s disease and Parkinson’s disease. N. Engl. J. Med. 348, 1356–1364. doi: 10.1056/NEJM2003ra020003
Ogrodnik, M., Salmonowicz, H., and Gladyshev, V. N. (2019). Integrating cellular enescence with the concept of damage accumulation in aging: relevance for clearance of senescent cells. Aging Cell 2019:e2841. doi: 10.1111/acel.12841
Ohashi, M., Korsakova, E., Allen, D., Lee, P., Fu, K., Vargas, B. S., et al. (2018). Loss of MECP2 leads to activation of P53 and neuronal senescence. Stem Cell Reports 10, 1453–1463. doi: 10.1016/j.stemcr.2018.04.001
Ohno-Iwashita, Y., Shimada, Y., Hayashi, M., and Inomata, M. (2010). Plasma membrane microdomains in aging and disease. Geriatr. Gerontol. Int. 10, S41–S52. doi: 10.1111/j.1447-0594.2010.00600.x
Olivieri, F., Prattichizzo, F., Grillari, J., and Balistreri, C. R. (2018). Cellular senescence and inflammaging in age-related diseases. Med. Inflamm. 2018:9076485. doi: 10.1155/2018/9076485
Olovnikov, A. (1996). Telomeres, telomerase, and aging: origin of the theory. Exp. Gerontol. 31, 443–448. doi: 10.1016/0531-5565(96)00005-8
Oromendia, A. B., and Amon, A. (2014). Aneuploidy: implications for protein homeostasis and disease. Dis. Model. Mech. 7, 15–20. doi: 10.1242/dmm.013391
Oromendia, A. B., Dodgson, S. E., and Amon, A. (2012). Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 26, 2696–2708. doi: 10.1101/gad.207407.112
Ott, B. R., Jones, R. N., Daiello, L. A., de la Monte, S. M., Stopa, E. G., Johanson, C. E., et al. (2018). Blood-cerebrospinal fluid barrier gradients in mild cognitive impairment and Alzheimer’s disease: relationship to inflammatory cytokines and chemokines. Front. Aging Neurosci. 10:245. doi: 10.3389/fnagi.2018.00245
Ow, S. Y., and Dunstan, D. E. (2014). A brief overview of amyloids and Alzheimer’s disease. Protein Sci. 23, 1315–1331. doi: 10.1002/pro.2524
Özcan, S., Alessio, N., Acar, M. B., Mert, E., Omerli, F., Peluso, G., et al. (2016). Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging 8, 1316–1329. doi: 10.18632/aging.100971
Parisotto, E. B., Vidal, V., García-Cerro, S., Lantigua, S., Wilhelm Filho, D., Sanchez-Barceló, E. J., et al. (2016). Chronic melatonin administration reduced oxidative damage and cellular senescence in the hippocampus of a mouse model of down syndrome. Neurochem. Res. 41, 2904–2913. doi: 10.1007/s11064-016-2008-8
Park, J.-S., Davis, R. L., and Sue, C. M. (2018). Mitochondrial dysfunction in Parkinson’s disease: new mechanistic insights and therapeutic perspectives. Curr. Neurol. Neurosci. Rep. 18:21. doi: 10.1007/s11910-018-0829-3
Park, J. I., Strock, C. J., Ball, D. W., and Nelkin, B. D. (2005). Interleukin-1β can mediate growth arrest and differentiation via the leukemia inhibitory factor/JAK/ STAT pathway in medullary thyroid carcinoma cells. Cytokine 29, 125–134. doi: 10.1016/j.cyto.2004.10.005
Passos, J. F., Nelson, G., Wang, C., Richter, T., Simillion, C., Proctor, C. J., et al. (2010). Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6:347. doi: 10.1038/msb.2010.5
Passos, J. F., Saretzki, G., and von Zglinicki, T. (2007). DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res. 35, 7505–7513. doi: 10.1093/nar/gkm893
Pawlikowski, B., Betta, N. D., Elston, T., Williams, D. A., and Olwin, B. B. (2018). Muscle stem cell dysfunction impairs muscle regeneration in a mouse model of Down syndrome. Sci. Rep. 8:4309. doi: 10.1038/s41598-018-22342-5
Perluigi, M., and Butterfield, D. A. (2012). Oxidative stress and down syndrome: a route toward alzheimer-like dementia. Curr. Gerontol. Geriatr. Res. 2012:724904. doi: 10.1155/2012/724904
Perluigi, M., di Domenico, F., Fiorini, A., Cocciolo, A., Giorgi, A., Foppoli, C., et al. (2011). Oxidative stress occurs early in Down syndrome pregnancy: a redox proteomics analysis of amniotic fluid. Proteomics Clin. Appl. 5, 167–178. doi: 10.1002/prca.201000121
Pertusa, M., García-Matas, S., Rodríguez-Farré, E., Sanfeliu, C., and Cristòfol, R. (2007). Astrocytes aged in vitro show a decreased neuroprotective capacity. J. Neurochem. 101, 794–805. doi: 10.1111/j.1471-4159.2006.04369.x
Pissadaki, E. K., and Bolam, J. P. (2013). The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 7:13. doi: 10.3389/fncom.2013.00013
Pitcairn, C., Wani, W. Y., and Mazzulli, J. R. (2018). Dysregulation of the autophagic-lysosomal pathway in Gaucher and Parkinson’s disease. Neurobiol. Dis. 122, 72–82. doi: 10.1016/j.nbd.2018.03.008
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primers 3:17013. doi: 10.1038/nrdp.2017.13
Pogribna, M., Melnyk, S., Pogribny, I., Chango, A., Yi, P., and James, S. J. (2001). Homocysteine metabolism in children with Down syndrome: in vitro modulation. Am. J. Hum. Genet. 69, 88–95. doi: 10.1086/321262
Pole, A., Dimri, M., and Dimri, G. P. (2016). Oxidative stress, cellular senescence and ageing. AIMS Mol. Sci. 3, 300–324. doi: 10.3934/molsci.2016.3.300
Prasad, R., and Jho, E. H. (2019). A concise review of human brain methylome during aging and neurodegenerative diseases. BMB Rep. 52, 577–588. doi: 10.5483/bmbrep.2019.52.10.215
Prasher, V. P., Sajith, S. G., Rees, S. D., Patel, A., Tewari, S., Schupf, N., et al. (2008). Significant effect of APOE epsilon 4 genotype on the risk of dementia in Alzheimer’s disease and mortality in persons with Down syndrome. Int. J. Geriatr. Psychiatry 23, 1134–1140. doi: 10.1002/gps.2039
Purcell, M., Kruger, A., and Tainsky, M. A. (2014). Gene expression profiling of replicative and induced senescence. Cell Cycle 13, 3927–3937. doi: 10.4161/15384101.2014.973327
Qazi, T. J., Quan, Z., Mir, A., and Qing, H. (2018). Epigenetics in Alzheimer’s disease: perspective of DNA methylation. Mol. Neurobiol. 55, 1026–1044. doi: 10.1007/s12035-016-0357-6
Raji, N. S., Surekha, A., and Rao, K. S. (1998). Improved DNA-repair parameters in PHA-stimulated peripheral blood lymphocytes of human subjects with low body mass index. Mech. Ageing Dev. 104, 133–148. doi: 10.1016/s0047-6374(98)00062-1
Rawji, K. S., Mishra, M. K., Michaels, N. J., Rivest, S., Stys, P. K., and Yong, V. W. (2016). Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain 139, 653–661. doi: 10.1093/brain/awv395
Rea, I. M., Gibson, D. S., McGilligan, V., McNerlan, S. E., Alexander, H. D., and Ross, O. A. (2018). Age and age-related diseases: role of inflammation triggers and cytokines. Front. Immunol. 9:586. doi: 10.3389/fimmu.2018.00586
Reichert, S., Criscuolo, F., Verinaud, E., Zahn, S., and Massemin, S. (2013). Telomere length correlations among somatic tissues in adult zebra finches. PLoS One 8:e81496. doi: 10.1371/journal.pone.0081496
Renani, P. G., Taheri, F., Rostami, D., Farahani, N., Abdolkarimi, H., Abdollahi, E., et al. (2019). Involvement of aberrant regulation of epigenetic mechanisms in the pathogenesis of Parkinson’s disease and epigenetic-based therapies. J. Cell. Physiol. 234, 19307–19319. doi: 10.1002/jcp.28622
Ribezzo, F., Shiloh, Y., and Schumacher, B. (2016). Systemic DNA damage responses in aging and diseases. Semin. Cancer Biol. 37–38, 26–35. doi: 10.1016/j.semcancer.2015.12.005
Richter, C., Park, J., and Ames, B. (1988). Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. U S A 85, 6465–6467. doi: 10.1073/pnas.85.17.6465
Richter, T., and von Zglinicki, T. (2007). A continuous correlation between oxidative stress and telomere shortening in fibroblasts. Exp. Gerontol. 42, 1039–1042. doi: 10.1016/j.exger.2007.08.005
Rivero-Ríos, P., Madero-Pérez, J., Fernández, B., and Hilfiker, S. (2016). Targeting the autophagy/lysosomal degradation pathway in Parkinson’s disease. Curr. Neuropharmacol. 14, 238–249. doi: 10.2174/1570159x13666151030103027
Roberson, R., Kuddo, T., Horowitz, K., Caballero, M., and Spong, C. Y. (2012). Cytokine and chemokine alterations in Down syndrome. Am. J. Perinatol. 29, 705–708. doi: 10.1055/s-0032-1314892
Rocha, E. M., De Miranda, B., and Sanders, L. H. (2018). α-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 109, 249–257. doi: 10.1016/j.nbd.2017.04.004
Rodier, F., and Campisi, J. (2011). Four faces of cellular senescence. J. Cell Biol. 192, 547–556. doi: 10.1083/jcb.201009094
Rodier, F., Coppé, J. P., Patil, C. K., Hoeijmakers, W. A., Muñoz, D. P., Raza, S. R., et al. (2009). Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979. doi: 10.1038/ncb1909
Rodier, F., Muñoz, D. P., Teachenor, R., Chu, V., Le, O., Bhaumik, D., et al. (2011). DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J. Cell Sci. 124, 68–81. doi: 10.1242/jcs.071340
Rodríguez-Sureda, V., Vilches, A., Sánchez, O., Audí, L., and Domínguez, C. (2015). Intracellular oxidant activity, antioxidant enzyme defense system, and cell senescence in fibroblasts with trisomy 21. Oxid. Med. Cell. Longev. 2015:509241. doi: 10.1155/2015/509241
Rolyan, H., Scheffold, A., Heinrich, A., Begus-Nahrmann, Y., Langkopf, B. H., Holter, S. M., et al. (2011). Telomere shortening reduces Alzheimer’s disease amyloid pathology in mice. Brain 134, 2044–2056. doi: 10.1093/brain/awr133
Romanov, V. S., Pospelov, V. A., and Pospelova, T. V. (2012). Cyclin dependent kinase inhibitor p21Waf1: contemporary view on its role in senescence and oncogenesis. Biochemistry 77, 575–584. doi: 10.1134/s000629791206003x
Rosi, S., Belarbi, K., Ferguson, R. A., Fishman, K., Fishman, K., Obenaus, A., et al. (2012). Trauma induced alterations in cognition and Arc expression are reduced by previous exposure to 56Fe irradiation. Hippocampus 22, 544–554. doi: 10.1002/hipo.20920
Roth, M., Emmons, L. R., Häner, M., Müller, H. J., and Boyle, J. M. (1989). Age-related decrease in an early step of DNA-repair of normal human lymphocytes exposed to ultraviolet-irradiation. Exp. Cell Res. 180, 171–177. doi: 10.1016/0014-4827(89)90221-8
Rueda, N., Flórez, J., and Martínez-Cué, C. (2012). Mouse models of Down syndrome as a tool to unravel the causes of mental disabilities. Neural Plast. 2012:584071. doi: 10.1155/2012/584071
Rueda, N., Vidal, V., García-Cerro, S., Narcís, J. O., Llorens-Martín, M., Corrales, A., et al. (2018). Anti-IL17 treatment ameliorates Down syndrome phenotypes in mice. Brain Behav. Immun. 73, 235–251. doi: 10.1016/j.bbi.2018.05.008
Sabbagh, M. N., Fleisher, A., Chen, K., Rogers, J., Berk, C., Reiman, E., et al. (2011). Positron emission tomography and neuropathologic estimates of fibrillar amyloid-β in a patient with Down syndrome and Alzheimer disease. Arch. Neurol. 68, 1461–1466. doi: 10.1001/archneurol.2011.535
Salama, R., Sadaie, M., Hoare, M., and Narita, M. (2014). Cellular senescence and its effector programs. Genes Dev. 28, 99–114. doi: 10.1101/gad.235184.113
Salminen, A., Ojala, J., Kaarniranta, K., Haapasalo, A., Hiltunen, M., and Soininen, H. (2011). Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 34, 3–11. doi: 10.1111/j.1460-9568.2011.07738.x
Sanchez-Mut, J. V., Heyn, H., Vidal, E., Moran, S., Sayols, S., Delgado-Morales, R., et al. (2016). Human DNA methylomes of neurodegenerative diseasesshow common epigenomic patterns. Transl. Psychiatry 6:e718. doi: 10.1038/tp.2015.214
Sanders, Y. Y., Liu, H., Zhang, X., Hecker, L., Bernard, K., Desai, L., et al. (2013). Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Biol. 1, 8–16. doi: 10.1016/j.redox.2012.11.004
Saretzki, G., Murphy, M. P., and von Zglinicki, T. (2003). MitoQ counteracts telomere shortening and elongates lifespan of fibroblasts under mild oxidative stress. Aging Cell 2, 141–143. doi: 10.1046/j.1474-9728.2003.00040.x
Sastre, M., Dewachter, I., Landreth, G. E., Willson, T. M., Klockgether, T., van Leuven, F., et al. (2003). Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-γ agonists modulate immunostimulated processing of amyloid precursor protein through regulation of β-secretase. J. Neurosci. 23, 9796–97804. doi: 10.1523/JNEUROSCI.23-30-09796.2003
Scalzo, P., Kümmer, A., Cardoso, F., and Teixeira, A. L. (2010). Serum levels of interleukin-6 are elevated in patients with Parkinson’s disease and correlate with physical performance. Neurosci. Lett. 468, 56–58. doi: 10.1016/j.neulet.2009.10.062
Scarabino, D., Broggio, E., Gambina, G., and Corbo, R. M. (2017). Leukocyte telomere length in mild cognitive impairment and Alzheimer’s disease patients. Exp. Gerontol. 98, 143–147. doi: 10.1016/j.exger.2017.08.025
Schupf, N., and Sergievsky, G. H. (2002). Genetic and host factors for dementia in Down’s syndrome. Br. J. Psychiatry 180, 405–410. doi: 10.1192/bjp.180.5.405
Sedelnikova, O. A., Horikawa, I., Zimonjic, D. B., Popescu, N. C., Bonner, W. M., and Barrett, J. C. (2004). Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 6, 168–170. doi: 10.1038/ncb1095
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Sepe, S., Milanese, C., Gabriels, S., Derks, K. W., Payan-Gomez, C., van IJcken, W. F., et al. (2016). Inefficient DNA repair is an aging related modifier of Parkinson’s disease. Cell Rep. 15, 1866–1875. doi: 10.1016/j.celrep.2016.04.071
Serra, V., von Zglinicki, T., Lorenz, M., and Saretzki, G. (2003). Extracellular superoxide dismutase is a major antioxidant in human fibroblasts and slows telomere shortening. J. Biol. Chem. 278, 6824–6830. doi: 10.1074/jbc.m207939200
Sheltzer, J. M., Blank, H. M., Pfau, S. J., Tange, Y., George, B. M., Humpton, T. J., et al. (2011). Aneuploidy drives genomic instability in yeast. Science 333, 1026–1030. doi: 10.1126/science.1206412
Sheng, J., Mrak, R., and Griffin, W. (1998). Enlarged and phagocytic, but not primed, interleukin-1 α-immunoreactive microglia increase with age in normal human brain. Acta Neuropathol. 95, 229–234. doi: 10.1007/s004010050792
Shichiri, M., Yoshida, Y., Ishida, N., Hagihara, Y., Iwahashi, H., Tamai, H., et al. (2011). α-tocopherol suppresses lipid peroxidation and behavioral and cognitive impairments in the Ts65Dn mouse model of Down syndrome. Free Radic. Biol. Med. 50, 1801–1811. doi: 10.1016/j.freeradbiomed.2011.03.023
Shigenaga, M., Hagen, T., and Ames, B. (1994). Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. U S A 91, 10771–10778. doi: 10.1073/pnas.91.23.10771
Shin, M., Besser, L. M., Kucik, J. E., Lu, C., Siffel, C., and Correa, A. (2009). Prevalence of Down syndrome among children and adolescents in 10 regions of the United States. Pediatrics 124, 1565–1571. doi: 10.1542/peds.2009-074
Siddiqui, M. S., Francois, M., Hecker, J., Faunt, J., Fenech, M. F., and Leifert, W. R. (2018). H2AX is increased in peripheral blood lymphocytes of Alzheimer’s disease patients in the South Australian Neurodegeneration, Nutrition and DNA Damage (SAND) study of aging. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 829–830, 6–18. doi: 10.1016/j.mrgentox.2018.03.001
Sierra, A., Gottfried-Blackmore, A., McEwen, B., and Bulloch, K. (2007). Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55, 412–424. doi: 10.1002/glia.20468
Silva, A. R. T., Santos, A. C. F., Farfel, J. M., Grinberg, L. T., Ferretti, R. E. L., Campos, A. H. J. F. M., et al. (2014). Repair of oxidative DNA damage, cell-cycle regulation and neuronal death may influence the clinical manifestation of Alzheimer’s disease. PLoS One 9:e99897. doi: 10.1371/journal.pone.0099897
Sipos, E., Kurunczi, A., Kasza, A., Horváth, J., Felszeghy, K., Laroche, S., et al. (2007). β-amyloid pathology in the entorhinal cortex of rats induces memory deficits: implications for Alzheimer’s disease. Neuroscience 147, 28–36. doi: 10.1016/j.neuroscience.2007.04.011
Staiano-Coico, L., Darzynkiewicz, Z., Hefton, J. M., Dutkowski, R., Darlington, G. J., and Weksler, M. E. (1983). Increased sensitivity of lymphocytes from people over 65 to cell cycle arrest and chromosomal damage. Science 219, 1335–1337. doi: 10.1126/science.6828861
Steele, M., Stuchbury, G., and Münch, G. (2007). The molecular basis of the prevention of Alzheimer’s disease through healthy nutrition. Exp. Gerontol. 42, 28–36. doi: 10.1016/j.exger.2006.06.002
Stein, G. H., Drullinger, L. F., Robetorye, R. S., Pereira-Smith, O. M., and Smith, J. R. (1991). Senescent cells fail to express cdc2, cycA, and cycB inresponse to mitogen stimulation. Proc. Natl. Acad. Sci. U S A 88, 11012–11016. doi: 10.1073/pnas.88.24.11012
Stein, G. H., Drullinger, L. F., Soulard, A., and Dulić, V. (1999). Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol. 19, 2109–2117. doi: 10.1128/mcb.19.3.2109
Stewart, S. A., Ben-Porath, I., Carey, V. J., O’Connor, B. F., Hahn, W. C., and Weinberg, R. A. (2003). Erosion of the telomeric single-strand overhang at replicative senescence. Nat. Genet. 33, 492–496. doi: 10.1038/ng1127
Stingele, S., Stoehr, G., Peplowska, K., Cox, J., Mann, M., and Storchova, Z. (2012). Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol. 8:608. doi: 10.1038/msb.2012.40
Streit, W. J., Braak, H., Xue, Q.-S., and Bechmann, I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 118, 475–485. doi: 10.1007/s00401-009-0556-6
Streit, W. J., Sammons, N. W., Kuhns, A. J., and Sparks, D. L. (2004). Dystrophic microglia in the aging human brain. Glia 45, 208–212. doi: 10.1002/glia.10319
Sukenik-Halevy, R., Biron-Shental, T., Sharony, R., Fejgin, M. D., and Amiel, A. (2011). Telomeres in trisomy 21 amniocytes. Cytogenet. Genome Res. 135, 12–18. doi: 10.1159/000329714
Sun, A., Liu, M., Nguyen, X. V., and Bing, G. (2003). P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 183, 394–405. doi: 10.1016/s0014-4886(03)00180-8
Swardfager, W., Lanctôt, K., Rothenburg, L., Wong, A., Cappell, J., and Herrmann, N. (2010). A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 68, 930–941. doi: 10.1016/j.biopsych.2010.06.012
Tai, H., Wang, Z., Gong, H., Han, X., Zhou, J., Wang, X., et al. (2017). Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 13, 99–113. doi: 10.1080/15548627.2016.1247143
Takahashi, Y., Karbowski, M., Yamaguchi, H., Kazi, A., Wu, J., Sebti, S. M., et al. (2005). Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol. Cell. Biol. 25, 9369–9382. doi: 10.1128/mcb.25.21.9369-9382.2005
Takai, H., Smogorzewska, A., and de Lange, T. (2003). DNA damage foci at dysfunctional telomeres. Curr. Biol. 13, 1549–1556. doi: 10.1016/s0960-9822(03)00542-6
Takata, Y., Kikukawa, M., Hanyu, H., Koyama, S., Shimizu, S., Umahara, T., et al. (2012). Association between ApoE phenotypes and telomere erosion in Alzheimer’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 67, 330–335. doi: 10.1093/gerona/glr185
Tanaka, K., and Matsuda, N. (2014). Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta 1843, 197–204. doi: 10.1016/j.bbamcr.2013.03.012
Tang, Y., Luo, J., Zhang, W., and Gu, W. (2006). Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 24, 827–839. doi: 10.1016/j.molcel.2006.11.021
Tarkowski, E., Liljeroth, A.-M., Minthon, L., Tarkowski, A., Wallin, A., and Blennow, K. (2003). Cerebral pattern of pro-and anti-inflammatory cytokines in dementias. Brain Res. Bull. 61, 255–260. doi: 10.1016/s0361-9230(03)00088-1
Teipel, S. J., and Hampel, H. (2006). Neuroanatomy of Down syndrome in vivo: a model of preclinical Alzheimer’s disease. Behav. Genet. 36, 405–415. doi: 10.1007/s10519-006-9047-x
Thomas, B., and Beal, M. F. (2011). Molecular insights into Parkinson’s disease. F1000 Med. Rep. 3:7. doi: 10.3410/M3-7
Thorburn, R. R., Gonzalez, C., Brar, G. A., Christen, S., Carlile, T. M., Ingolia, N. T., et al. (2013). Aneuploid yeast strains exhibit defects in cell growth and passage through START. Mol. Biol. Cell 24, 1274–1289. doi: 10.1091/mbc.e12-07-0520
Tiribuzi, R., Orlacchio, A. A., Crispoltoni, L., Maiotti, M., Zampolini, M., De Angelis, M., et al. (2011). Lysosomal β-galactosidase and β-hexosaminidase activities correlate with clinical stages of dementia associated with Alzheimer’s disease and type 2 diabetes mellitus. J. Alzheimers Dis. 24, 785–797. doi: 10.3233/jad-2011-100525
Tomac, A. C., and Hoffer, B. J. (2001). Assignment of the mouse Park2 (PARKIN), the homologue to a new human Parkinson candidate gene, to the telomeric region of mouse 17A3.2–3.3, by in situ hybridization. Cytogenet. Cell Genet. 95, 120–121. doi: 10.1159/000057032
Toussaint, O., Medrano, E. E., and von Zglinicki, T. (2000). Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 35, 927–945. doi: 10.1016/s0531-5565(00)00180-7
Town, T., Nikolic, V., and Tan, J. (2005). The microglial “activation” continuum: from innate to adaptive responses. J. Neuroinflammation 2:24. doi: 10.1186/1742-2094-2-24
Tramutola, A., Pupo, G., Di Domenico, F., Barone, E., Arena, A., Lanzillotta, C., et al. (2016). Activation of p53 in Down syndrome and in the Ts65Dn mouse brain is associated with a proapoptotic phenotype. J. Alzheimers Dis. 52, 359–371. doi: 10.3233/jad-151105
Trist, B. G., Hare, D. J., and Double, K. L. (2019). Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 20:e13031. doi: 10.1111/acel.13031
Tsurumi, A., and Li, W. (2012). Global heterochromatin loss: a unifying theory of aging? Epigenetics 7, 680–688. doi: 10.4161/epi.20540
Vaidya, A., Mao, Z., Tian, X., Spencer, B., Seluanov, A., and Gorbunova, V. (2014). Knock-in reporter mice demonstrate that DNA repair by non-homologous end joining declines with age. PLoS Genet. 10:e1004511. doi: 10.1371/journal.pgen.1004511
Valenti, D., Tullo, A., Caratozzolo, M. F., Merafina, R. S., Scartezzini, P., Marra, E., et al. (2010). Impairment of F1F0-ATPase, adenine nucleotide translocator and adenylate kinase causes mitochondrial energy deficit in human skin fibroblasts with chromosome 21 trisomy. Biochem. J. 431, 299–310. doi: 10.1042/bj20100581
van Deursen, J. M. (2014). The role of senescent cells in aging. Nature 509, 439–446. doi: 10.1038/nature13193
van Dijk, K. D., Persichetti, E., Chiasserini, D., Eusebi, P., Beccari, T., Calabresi, P., et al. (2013). Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov. Disord. 28, 747–754. doi: 10.1002/mds.25495
Vanyushin, B., Nemirovsky, L., Klimenko, V., Vasiliev, V., and Belozersky, A. (1973). The 5-methylcytosine in DNA of rats. Tissue and age specificity and the changes induced byhydrocortisone and other agents. Gerontologia 19, 138–152. doi: 10.1159/000211967
Vaziri, H., Schächter, F., Uchida, I., Wei, L., Zhu, X., Effros, R., et al. (1993). Loss of telomeric DNA during aging of normal and trisomy 21 human lymphocytes. Am. J. Hum. Genet. 52, 661–667.
Velarde, M. C., and Menon, R. (2016). Positive and negative effects of cellular senescence during female reproductive aging and pregnancy. J. Endocrinol. 230, R59–R76. doi: 10.1530/joe-16-0018
Ventura, M. T., Casciaro, M., Gangemi, S., and Buquicchio, R. (2017). Immunosenescence in aging: between immune cells depletion and cytokines up-regulation. Clin. Mol. Allergy 15:21. doi: 10.1186/s12948-017-0077-0
Vermunt, M. W., Zhang, D., and Blobel, G. A. (2019). The interdependence of gene-regulatory elements and the 3D genome. J. Cell Biol. 218, 12–26. doi: 10.1083/jcb.201809040
Vicencio, J. M., Galluzzi, L., Tajeddine, N., Ortiz, C., Criollo, A., Tasdemir, E., et al. (2008). Senescence, apoptosis or autophagy? When a damaged cell must decide its path—a mini-review. Gerontology 54, 92–99. doi: 10.1159/000129697
Vincent, I., Rosado, M., and Davies, P. (1996). Mitotic mechanisms in Alzheimer’s disease? J. Cell Biol. 132, 413–425. doi: 10.1083/jcb.132.3.413
von Bernhardi, R. (2007). Glial cell dysregulation: a new perspective on Alzheimer disease. Neurotox. Res. 12, 215–232. doi: 10.1007/bf03033906
von Bernhardi, R., Cornejo, F., Parada, G., and Eugenín, J. (2015a). Role of TGFβ signaling in the pathogenesis of Alzheimer’s disease. Front. Cell Neurosci. 9:426. doi: 10.3389/fncel.2015.00426
von Bernhardi, R., Eugenin-von Bernhardi, L., and Eugenin, J. (2015b). Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 7:124. doi: 10.3389/fnagi.2015.00124
von Zglinicki, T. (2002). Oxidative stress shortens telomeres. Trends Biochem. Sci. 27, 339–344. doi: 10.1016/s0968-0004(02)02110-2
von Zglinicki, T., Pilger, R., and Sitte, N. (2000). Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic. Biol. Med. 28, 64–74. doi: 10.1016/s0891-5849(99)00207-5
Wagner, W. (2019). The link between epigenetic clocks for aging and senescence. Front. Genet. 10:303. doi: 10.3389/fgene.2019.00303
Wang, Y., Chang, J., Shao, L., Feng, W., Luo, Y., Chow, M., et al. (2016). Hematopoietic stem cells from Ts65Dn mice are deficient in the repair of DNA double-strand Breaks. Radiat. Res. 185, 630–637. doi: 10.1667/rr14407.1
Wang, H., Chen, H., Gao, X., McGrath, M., Deer, D., De Vivo, I., et al. (2008). Telomere length and risk of Parkinson’s disease. Mov. Disord. 23, 302–305. doi: 10.1002/mds.21867
Watanabe, S., Kawamoto, S., Ohtani, N., and Hara, E. (2017). Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 108, 563–569. doi: 10.1111/cas.13184
Watfa, G., Dragonas, C., Brosche, T., Dittrich, R., Sieber, C. C., Alecu, C., et al. (2011). Study of telomere length and different markers of oxidative stress in patients with Parkinson’s disease. J. Nutr. Health Aging 15, 277–281. doi: 10.1007/s12603-010-0275-7
Watson, C. T., Roussos, P., Garg, P., Ho, D. J., Azam, N., Katsel, P. L., et al. (2016). Genome-wide12 DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med. 8:5. doi: 10.1186/s13073-015-0258-8
Wei, Z., Chen, X. C., Song, Y., Pan, X. D., Dai, X. M., Zhang, J., et al. (2016). Amyloid β protein aggravates neuronal senescence and cognitive deficits in 5XFAD mouse model of Alzheimer’s disease. Chin. Med. J. 129, 1835–1844. doi: 10.4103/0366-6999.186646
Weichhart, T. (2018). mTOR as regulator of lifespan, aging, and cellular senescence: a mini-review. Gerontology 64, 127–134. doi: 10.1159/000484629
Weirich-Schwaiger, H., Weirich, H. G., Gruber, B., Schweiger, M., and Hirsch-Kauffmann, M. (1994). Correlation between senescence and DNA repair in cells from young and old individuals and in premature aging syndromes. Mutat. Res. 316, 37–48. doi: 10.1016/0921-8734(94)90006-x
Weldon, D. T., Rogers, S. D., Ghilardi, J. R., Finke, M. P., Cleary, J. P., O’Hare, E., et al. (1998). Fibrillar β-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase and loss of a select population of neurons in the rat CNS in vivo. J. Neurosci. 18, 2161–2173. doi: 10.1523/JNEUROSCI.18-06-02161.1998
Wilcock, D. M. (2012). Neuroinflammation in the aging down syndrome brain; Lessons from Alzheimer’s disease. Curr. Gerontol. Geriatr. Res. 2012:170276. doi: 10.1155/2012/170276
Wilcock, D. M., and Griffin, W. S. (2013). Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflammation 10:84. doi: 10.1186/1742-2094-10-84
Wiley, C. D., Flynn, J. M., Morrissey, C., Lebofsky, R., Shuga, J., Dong, X., et al. (2017). Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 16, 1043–1050. doi: 10.1111/acel.12632
Winick-Ng, W., and Rylett, R. J. (2018). Into the fourth dimension: dysregulation of genome architecture in aging and Alzheimer’s disease. Front. Mol. Neurosci. 11:60. doi: 10.3389/fnmol.2018.00060
Wood, J. A., Wood, P. L., Ryan, R., Graff-Radford, N. R., Pilapil, C., Robitaille, Y., et al. (1993). Cytokine indices in Alzheimer’s temporal cortex: no changes in mature IL-1 β or IL-1RA but increases in the associated acute phase proteins IL-6, α2-macroglobulin and C-reactive protein. Brain Res. 629, 245–252. doi: 10.1016/0006-8993(93)91327-o
Wu, M., Ye, H., Shao, C., Zheng, X., Li, Q., Wang, L., et al. (2017). Metabolomics-proteomics combined approach identifies differential metabolism-associated molecular events between senescence and apoptosis. J. Proteome Res. 16, 2250–2261. doi: 10.1021/acs.jproteome.7b00111
Yanagi, S., Tsubouchi, H., Miura, A., Matsuo, A., Matsumoto, N., and Nakazato, M. (2017). The impacts of cellular senescence in elderly pneumonia and in age-related lung diseases that increase the risk of respiratory infections. Int. J. Mol. Sci. 18:E503. doi: 10.3390/ijms18030503
Yang, N., and Sen, P. (2018). The senescent cell epigenome. Aging 10, 3590–3609. doi: 10.18632/aging.101617
Yankner, B., Lu, T., and Loerch, P. (2008). The aging brain. Annu. Rev. Pathol. 3, 41–66. doi: 10.1146/annurev.pathmechdis.2.010506.092044
Yates, S. C., Zafar, A., Rabai, E. M., Foxall, J. B., Nagy, S., Morrison, K. E., et al. (2015). The effects of two polymorphisms on p21cip1 function and their association with Alzheimer’s disease in a population of european descent. PLoS One 10:e0114050. doi: 10.1371/journal.pone.0114050
Yoshiyama, Y., Asahina, M., and Hattori, T. (2000). Selective distribution of matrix metalloproteinase-3 (MMP-3) in Alzheimer’s disease brain. Acta Neuropathol. 99, 91–95. doi: 10.1007/pl00007428
Yurov, Y. B., Vorsanova, S. G., Liehr, T., Kolotii, A. D., and Iourov, I. Y. (2014). X chromosome aneuploidy in the Alzheimer’s disease brain. Mol. Cytogenet. 7:20. doi: 10.1186/1755-8166-7-20
Zakian, V. A. (1995). Telomeres: beginning to understand the end. Science 270, 1601–1607. doi: 10.1126/science.270.5242.1601
Zare-Shahabadi, A., Masliah, E., Johnson, G. V. W., and Rezaei, N. (2015). Autophagy in Alzheimer’s disease. Rev. Neurosci. 26, 385–395. doi: 10.1515/revneuro-2014-0076
Zekry, D., Herrmann, F. R., Irminger-Finger, I., Graf, C., Genet, C., Vitale, A. M., et al. (2010). Telomere length and ApoE polymorphism in mild cognitive impairment, degenerative and vascular dementia. J. Neurol. Sci. 299, 108–111. doi: 10.1016/j.jns.2010.07.019
Zhang, P., Kishimoto, Y., Grammatikakis, I., Gottimukkala, K., Cutler, R. G., Zhang, S., et al. (2019). Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 22, 719–728. doi: 10.1038/s41593-019-0372-9
Zhang, W., Wang, T., Pei, Z., Miller, D., Wu, X., Block, M., et al. (2005). Aggregated α-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 19, 533–542. doi: 10.1096/fj.04-2751com
Zheng, Q., Huang, J., and Wang, G. (2019). Mitochondria, telomeres and telomerase subunits. Front. Cell Dev. Biol. 7:274. doi: 10.3389/fcell.2019.00274
Ziegler, D. V., Wiley, C. D., and Velarde, M. C. (2015). Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell 14, 1–7. doi: 10.1111/acel.12287
Keywords: senescence, Alzheimer’s disease, Down syndrome, Parkinsion’s disease, neurodegenaration
Citation: Martínez-Cué C and Rueda N (2020) Cellular Senescence in Neurodegenerative Diseases. Front. Cell. Neurosci. 14:16. doi: 10.3389/fncel.2020.00016
Received: 15 November 2019; Accepted: 21 January 2020;
Published: 11 February 2020.
Edited by:
Vassilis G. Gorgoulis, National and Kapodistrian University of Athens, GreeceReviewed by:
Claudio Torres, Drexel University, United StatesCopyright © 2020 Martínez-Cué and Rueda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmen Martínez-Cué, bWFydGluZWNAdW5pY2FuLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.