
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci., 14 January 2020
Sec. Cellular Neuropathology
Volume 13 - 2019 | https://doi.org/10.3389/fncel.2019.00587
This article is part of the Research TopicEpilepsy and Neurodevelopmental DiseasesView all 11 articles
 Karen M. J. van Loo*
Karen M. J. van Loo* Albert J. Becker
Albert J. BeckerEpilepsy is a common neurological disorder characterized by recurrent uncontrolled seizures and has an idiopathic “genetic” etiology or a symptomatic “acquired” component. Genetic studies have revealed that many epilepsy susceptibility genes encode ion channels, including voltage-gated sodium, potassium and calcium channels. The high prevalence of ion channels in epilepsy pathogenesis led to the causative concept of “ion channelopathies,” which can be elicited by specific mutations in the coding or promoter regions of genes in genetic epilepsies. Intriguingly, expression changes of the same ion channel genes by augmentation of specific transcription factors (TFs) early after an insult can underlie acquired epilepsies. In this study, we review how the transcriptional regulation of ion channels in both genetic and acquired epilepsies can be controlled, and compare these epilepsy “ion channelopathies” with other neurodevelopmental disorders.
Epilepsy is a severe chronic brain disorder characterized by recurrent seizure activity due to aberrant neuronal network activity (Fisher et al., 2014; Fisher, 2015). Despite many years of research, the underlying mechanisms that orchestrate seizure activity are still not fully understood. This is also reflected in the fact that treatment strategies for epilepsy with antiepileptic drugs (AEDs) are insufficient in about one-third of epilepsy patients (Kwan and Sander, 2004). This relatively high level of pharmacoresistance, together with the often severe side effects of AEDs, asks for a better understanding of its etiology and pathogenesis (Löscher et al., 2013).
Nowadays, it is generally accepted that both genetic as well as environmental factors, such as head trauma, brain tumors, brain infection, stroke, autoimmune diseases, status epilepticus (SE) and hippocampal sclerosis (Engel, 1996; Bien and Elger, 2007; Bien et al., 2007; Liu et al., 2016; Pitkänen et al., 2016; Vezzani et al., 2016) can play a role in the etiopathogenesis of epilepsy. Epilepsies with such a causal injury to the central nervous system (CNS) are called acquired or symptomatic epilepsies, whereas those lacking a clear predisposing cause, are called idiopathic or genetic epilepsies (Shorvon, 2011).
In the last decades, enormous progress has been made in the discovery of epilepsy genes, resulting in a current list of approximately 1,000 epilepsy-associated genes (reviewed by Wang et al., 2017). Since many of the genes annotated on this list are ion channels, the theory was born that epilepsy is a disease of “ion channelopathies” (Wallace et al., 1998; Reid et al., 2009). Ion channels are pore-forming membrane proteins involved in maintaining ion homeostasis and the generation and propagation of neuronal action potentials. A disturbance in the neuronal ionic flow might result in hyperexcitability, which can form the basis for seizure activity (Raimondo et al., 2015). In general, ion channels can be divided into two main groups, depending on their mode of activation (Brenowitz et al., 2017). Voltage-gated ion channels are activated by changes in membrane potential and ligand-gated ion channels are opened in response to specific ligands binding to the extracellular domain of the ion channel (Alexander et al., 2015a,b).
In this study, we focus on the transcriptional mechanisms involved in channelopathy-induced epilepsy. We review how the expression of ion channel genes can be affected and compare these mechanisms between genetic and acquired epilepsies. In addition, we also summarize how these transcriptional mechanisms can play a role in the etiopathogenesis of other neurodevelopmental disorders.
For decades, scientists try to unravel the molecular background of epilepsies. In 1995, the first epilepsy-associated ion channel was identified; a mutation in a strongly conserved amino acid residue in the acetylcholine receptor alpha 4 subunit (CHRNA4) correlated with autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE; Steinlein et al., 1995). After this first discovery, many other ion channels were reported to be linked to epilepsy, including genes belonging to the voltage-gated sodium, potassium, calcium and hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. Besides the voltage-gated ion channels, also several ligand-gated ion channel genes were identified as epilepsy-associated genes, including ionotropic glutamate receptors, GABAA receptors and nicotinic acetylcholine receptors (Table 1; reviewed by Lerche et al., 2013; Wang et al., 2017; Wei et al., 2017; Oyrer et al., 2018).
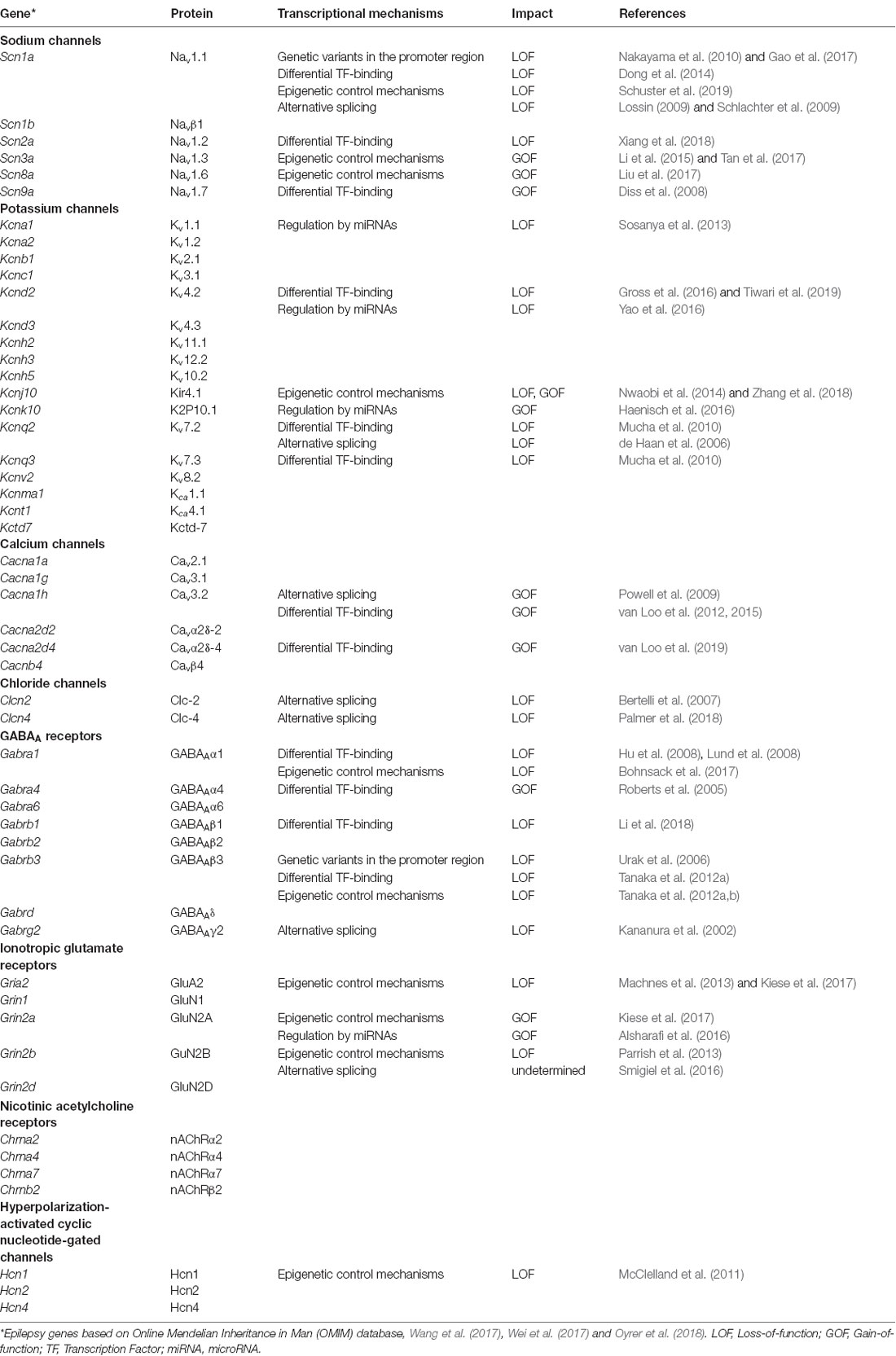
Table 1. Transcriptional channelopathies implicated in epilepsies.
Currently, it is thought that genetic epilepsy can be the result of: (i) rare variants with high penetrance (also known as monogenic or “common-disease-rare-variant model”) or of (ii) common variants with low penetrance (also known as polygenic or “common-disease-common-variants model”; Reich and Lander, 2001; Gibson, 2012; Saint Pierre and Génin, 2014). Such rare variants (or mutations) can nowadays be identified by deep sequencing approaches (e.g., exome sequencing or whole-genome sequencing; Dunn et al., 2018), whereas for the identification of common variants (also known as single nucleotide polymorphisms, SNPs), genome-wide association studies are indispensable in large cohorts of patients and controls (International League Against Epilepsy Consortium on Complex Epilepsies, 2018). However, common variants are often difficult to link unequivocally to disease, since these variants contribute only minimally and might also require an additional environmental factor for a pathological outcome.
In epilepsy, both rare as well as common variants have been identified in ion channel genes. Mutations in the sodium channel SCN1A, probably the most studied and best-documented epilepsy gene, can cause a spectrum of epilepsy syndromes including Dravet syndrome and genetic (generalized) epilepsy with febrile Seizures Plus (GEFS+; Brunklaus and Zuberi, 2014), whereas a common variant within an intron of the same gene (rs7587026), was found to associate with mesial temporal lobe epilepsy (mTLE; Kasperaviciute et al., 2013).
Mostly, genetic channelopathies are the result of variants within the coding region of the gene. Both missense mutations (mutations causing an amino acid change), as well as nonsense mutations (mutations causing a premature stop codon), can underlie epilepsy pathogenesis by inducing a loss-of-function (LOF) or a gain-of-function (GOF) channelopathy. In addition, also deletions and duplications of (part of) the gene can strongly affect normal channel function (Borlot et al., 2017; Monlong et al., 2018). Since the focus of this review is on the transcriptional regulation of ion channels, listing all genetic variants within the coding regions of ion channel genes is beyond the scope of this article (for reviews, see Deng et al., 2014; Wei et al., 2017; Zhang et al., 2019).
Genetic epilepsies can also be the result of a genetic variant positioned in the promoter region, a splice site, or a regulatory region of the gene. How can variants outside the coding region induce a channelopathy? For SCN1A for example, a microdeletion in the 5’-promoter region was found in patients with Dravet syndrome (Nakayama et al., 2010), and another heterozygous mutation in the promoter region (h1u-1962 T >G) was identified in a patient with partial epilepsy and febrile seizures (Gao et al., 2017). Functional analysis of this SCN1A h1u-1962 T >G variant revealed a reduction of SCN1A promoter activity by 42.1% compared to the wild-type variant (Gao et al., 2017), explaining the relatively mild phenotypical impairment caused by this non-coding variant when compared with effects caused by SCN1A coding variants that can result in null expression.
A genetic variant can also be located at a splice site, resulting in alternative splicing of the ion channel gene. This process allows a single gene to produce alternative ion channels with different functional characteristics. In particular, for SCN1A many alternative splicing mutations have been identified in epilepsy pathogenesis (Lossin, 2009; Thompson et al., 2011; Carvill et al., 2018; Table 1).
If a genetic variant is located within the binding site of an activating or repressing transcription factor (TF) or a repressor, it may result in altered regulation of the transcriptional machinery. For example, four different haplotypes, consisting of 13 SNPs located in the 5’ region of the GABRB3 gene were found to segregate with childhood absence epilepsy (CAE; Urak et al., 2006). The GABRB3 gene encodes the β3 subunit of the GABAA receptor which mediates phasic (synaptic) and tonic (perisynaptic) inhibition (Farrant and Nusser, 2005; Hirose, 2014). Functional analysis of these haplotypes revealed a reduced transcriptional activity of the GABRB3-haplotype-2 promoter (overrepresented in CAE) compared to the GABRB3-haplotype-1 promoter (overrepresented in controls). The difference in expression could be explained by reduced binding of the TF N-Oct3 to the GABRB3-haplotype-2 promoter, resulting in decreased expression of the GABRB3 gene (Urak et al., 2006). The reduced β3 levels observed in CAE patients might cause a loss of inhibitory properties of the receptor, eventually causing seizure activity.
Acquired epilepsies are epilepsies, which are on the consequence of an environmental factor. These epilepsies can be: (i) completely dependent on environmental factors; or (ii) can be caused by an interaction of environmental factor(s) with a predisposition genome. In the latter case, the presence of common susceptibility variants (e.g., SNPs or CNVs) can lower the threshold of the environmental factor for inducing an epileptic outcome. Most of our current knowledge of acquired epilepsy pathogenesis comes from the use of animal models, in which insults causing TLE can be mimicked in rodents using approaches like traumatic brain injury, kindling, or by applying one of the chemo-convulsants pilocarpine or kainic acid to induce SE (reviewed by Jefferys et al., 2016; Lévesque et al., 2016; Becker, 2018; Nirwan et al., 2018). Numerous studies using animal models for TLE have provided valuable information on epilepsy pathogenesis, resulting in a list of several channels involved in acquired epilepsies, including but not limited to HCN channels (Chen et al., 2001; Shah et al., 2004; Marcelin et al., 2009; Jung et al., 2011; Arnold et al., 2019), the A-type potassium channel Kv4.2 (Bernard et al., 2004; Monaghan et al., 2008), Kir2 channels (Young et al., 2009), small-conductance (SK) calcium-activated potassium channels (Oliveira et al., 2010), big potassium channels (BK-channels; Pacheco Otalora et al., 2008; Shruti et al., 2008), persistent sodium channels (Agrawal et al., 2003; Chen et al., 2011), the T-type calcium channel CaV3.2 (Su et al., 2002; Becker et al., 2008) and the calcium channel subunit α2δ4 (van Loo et al., 2019).
Currently, one of the main questions in epilepsy research is how the expression of ion channel genes in acquired epilepsies can be regulated. The transcriptional regulation of ion channels in acquired epilepsy can occur for example via differential expression of transcriptional activators or repressors. After a brain insult, a transient increase of activity-regulated TFs is evident (e.g., Egr-4, Fos, Jun and Arc), which can as a consequence dysregulate the transcriptional machinery of many genes, including ion channel genes (Herdegen et al., 1993; Beer et al., 1998; Herdegen and Leah, 1998; Honkaniemi and Sharp, 1999). To date, several transcriptional mechanisms have already been identified in the context of channelopathies and epilepsy pathogenesis (Table 1).
For CaV3.2, we recently performed an in-depth promoter analysis, examining the molecular mechanisms involved in the transcriptional augmentation of this channel early after pilocarpine-induced SE (Becker et al., 2008). Here, we observed a highly-sophisticated mechanism of transcriptional regulation: the increase of CaV3.2 expression was found to be mediated by metal-regulatory transcription factor 1 (MTF1) upon a rise in intracellular zinc ([Zn2+]i); denoted as the Zn2+-MTF1-CaV3.2 cascade (van Loo et al., 2015). A rise in [Zn2+]i, often seen after a transient insult to the brain (Assaf and Chung, 1984; Zhao et al., 2014), can activate MTF1, which then binds to metal-responsive elements within the CaV3.2 promoter region. Consequently, this results in increased CaV3.2 expression, a larger T-type current and increased burst-firing behavior (van Loo et al., 2015). In this way, the Zn2+-MTF1-CaV3.2 cascade can enhance hippocampal network excitability, resulting in seizure activity. Besides the Zn2+-MTF1-CaV3.2 cascade, also other TFs were found to control CaV3.2 expression, including Egr1 and RE1-silencing transcription factor (REST; van Loo et al., 2012). Such a multifactorial regulation by several TFs, thought to be a general phenomenon of ion channel regulation, severely complicates pharmacological intervention.
The transcription of ion channels in acquired epilepsies can also be regulated at the epigenetic level: both DNA methylation at cytosine residues, as well as changes in histone modifications (e.g., acetylation or methylation) can strongly affect the transcriptional machinery (reviewed by Hauser et al., 2018). Methylation of DNA generally occurs at cytosines within the 5’-cytosine-guanine-3’ context (CpG). Gene promoters often contain large clusters of CpGs (referred to as CpG islands), which are mostly hypomethylated and are linked to transcriptional activation. An increase in DNA methylation may cause reduced transcriptional activity due to physical inhibition of TF binding to their cognate DNA binding motif, or by binding of repressor proteins known as methyl-CpG binding domain proteins (MBDs) to the methylated DNA. In the latter case, MBDs can recruit histone deacetylases (HDAC1 and HDAC2) to the methylated DNA, resulting in the silencing of the corresponding gene (Clouaire and Stancheva, 2008). To date, several epilepsy-channelopathies have been described to be caused by an epigenetic control mechanism (Table 1).
The transcriptional machinery of ion channels in acquired epilepsies can also be influenced by small non-coding RNAs, such as microRNAs (miRNAs). miRNAs are 22 nucleotides noncoding RNAs that can regulate gene expression by associating with the RNA-induced silencing complex (RISC). The RISC complex then uses the miRNA as a template for recognizing the complementary mRNA of the ion channel gene (Ranganathan and Sivasankar, 2014). The main function of miRNAs appears to be the regulation at the post-transcriptional level: either by hindering protein translation or by enhancing mRNA degradation. Nowadays, it is also debated that miRNAs can have a nuclear function by modulating gene expression directly at the transcriptional level (reviewed by Catalanotto et al., 2016). Numerous miRNAs have been identified in relation to epilepsy pathogenesis (reviewed by Reschke and Henshall, 2015; Henshall et al., 2016; Shao and Chen, 2017; Tiwari et al., 2018), and also several ion channels appear to be under control of miRNAs, including Kv1.1, Kv4.2, Kcnk10 and Grin2a (Sosanya et al., 2013; Alsharafi et al., 2016; Gross et al., 2016; Haenisch et al., 2016; Tiwari et al., 2019; Table 1).
To date, it is generally accepted that ion channelopathies are not unique for epilepsy pathogenesis, but have gained considerable attention for the pathogenesis of several neurodevelopmental disorders, including pathology aspects of autism spectrum disorders (ASDs), schizophrenia, bipolar disorder, major depressive disorder and migraine (reviewed by Imbrici et al., 2013; Schmunk and Gargus, 2013; Albury et al., 2017). Seizures have been noted as a comorbidity feature of neurodevelopmental disorders (Hyde and Weinberger, 1997; Canitano, 2007; Mula et al., 2008; Liao et al., 2018; Salpekar and Mula, 2018; Strasser et al., 2018), which overall may point to the emergence of a functionally impaired neuronal network. For many neurodevelopmental disorders, several genetic variations (both rare mutations as well as common variants) in the coding regions of ion channel genes have been identified and reviewed elsewhere (Imbrici et al., 2013; Schmunk and Gargus, 2013; Daghsni et al., 2018; Weiss and Zamponi, 2019). Interestingly, also the mechanisms described above to be involved in the transcriptional regulation of ion channels in epilepsy pathogenesis, have been observed in the regulation of ion channels in other neurodevelopmental diseases. For example, transcriptional regulation by presence of genetic variants within the promoter region was observed for Grin2a and Grin2b and resulted in schizophrenia pathogenesis (Miyatake et al., 2002; Itokawa et al., 2003; Liu et al., 2015); alternative splicing was documented for Gabrb2, Grin2b and Gabra3 and resulted in mental retardation and ASD (Zhao et al., 2009; Endele et al., 2010; O’Roak et al., 2012; Piton et al., 2013); differential expression of TFs was observed for Scn10a, Kcnq1, Cacna1c, and Grin1 in schizophrenia and other psychiatric disorders (Rannals et al., 2016; Billingsley et al., 2018; Page et al., 2018); epigenetic control mechanisms were described for Gabrb2, Gabrb3, Gria, and Chrna7 in ASD, schizophrenia and Rett syndrome (Samaco et al., 2005; Yasui et al., 2011; Kordi-Tamandani et al., 2013; Zong et al., 2017) and regulation by miRNAs was observed for Cacna1c, Cacnb1, Grin2b and NMDAR in schizophrenia and ASD (Kocerha et al., 2009; Guan et al., 2014; Cammaerts et al., 2015; Zhang et al., 2015; Kichukova et al., 2017).
Many of these “transcriptional channelopathies” apparently are rather specific, since they are mostly associated with only one individual neurodevelopmental disorder. However, a few examples exist, in which a comparable transcriptional regulatory mechanism has been observed for channelopathies in epilepsy and other neurodevelopmental disorders, hinting at an explanation for the comorbidity seen between the different disorders. One such example is the GABRB3 gene, an important neurodevelopmental gene and besides epilepsy also associated with Angelman syndrome, Rett syndrome and ASD (Tanaka et al., 2012b). Differential expression of GABRB3 in CAE can be caused by genetic variants within the promoter region (Urak et al., 2006). Interestingly, one of these variants is also associated with schizophrenia and heroin dependence (Chen et al., 2014b; Liu et al., 2019), whereas other genetic variants within the same regulatory region are correlated with ASD (Chen et al., 2014a).
Another example of comparable transcriptional control mechanisms in epilepsy and neurodevelopmental disorders was observed for the NMDAR gene Grin2b. The expression of Grin2b was significantly decreased in the kainic acid-induced SE model and correlated with increased DNA methylation levels at specific CpGs located within the Grin2b locus. Additionally, interfering with the DNA methylation levels prior to SE using a DNA-methyltransferase inhibitor, prevented the Grin2b DNA methylation increase after SE and resulted in augmented Grin2b mRNA and protein expression (Parrish et al., 2013).
Such a glutamatergic hypofunction caused by an epigenetic control mechanism in Grin2b in the epilepsy model can also contribute to the pathophysiology of other neurodevelopmental disorders (Coyle et al., 2002; Lau and Zukin, 2007). Recently, it was reported that in a mouse model for schizophrenia, Grin2b expression was also under control of epigenetic control mechanisms. Here, the reduction in Grin2b expression was caused by an increase in H3K27me3 and REST at the Grin2b promoter (Gulchina et al., 2017). We may thus assume that different neurodevelopmental disorders are associated with a channelopathy with similar underlying transcriptional mechanisms.
Although we see comparable transcriptional mechanisms, no large overlap exists between the individual regulatory players in epilepsy pathogenesis and other neurodevelopmental disorders. Of course, this can also be explained by the fact that most specific mechanisms simply have not been analyzed in all neurodevelopmental disorders, or not in an analogous manner, making a direct comparison at the moment impossible. Further studies will reveal whether the altered diseases-associated expression of more proteins is based on a (partly) general underlying transcriptional phenomenon, possibly explaining the comorbidity between epilepsy and other neurodevelopmental disorders.
In this study, we reviewed the mechanisms involved in the transcriptional regulation of channelopathies in genetic and acquired epilepsies. Although a large amount of data exists, our understanding of transcriptional mechanisms governing ion channel expression is far from complete and requires further detailed investigation, not only for epilepsy pathogenesis, but also for other neurodevelopmental disorders. A better understanding of the underlying mechanisms might result in the development of novel drugs and may provide opportunities for better-individualized treatment strategies.
Both authors contributed to the writing and editing of the manuscript.
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 1089: KL, AB), FOR 2715 (AB), BMBF (EraNet DeCipher to AB), the European Union’s Seventh Framework Programme (FP7/2007–2013) under grant agreement n°602102 (EPITARGET; AB), Else Kröner-Fresenius-Foundation ‘Promotionskolleg NeuroImmunology’ (AB) as well as the BONFOR program of the University of Bonn Medical Center.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past co-authorship with one of the authors AB.
Agrawal, N., Alonso, A., and Ragsdale, D. S. (2003). Increased persistent sodium currents in rat entorhinal cortex layer V neurons in a post-status epilepticus model of temporal lobe epilepsy. Epilepsia 44, 1601–1604. doi: 10.1111/j.0013-9580.2003.23103.x
Albury, C. L., Stuart, S., Haupt, L. M., and Griffiths, L. R. (2017). Ion channelopathies and migraine pathogenesis. Mol. Genet. Genomics 292, 729–739. doi: 10.1007/s00438-017-1317-1
Alexander, S. P., Catterall, W. A., Kelly, E., Marrion, N., Peters, J. A., Benson, H. E., et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: voltage-gated ion channels. Br. J. Pharmacol. 172, 5904–5941. doi: 10.1111/bph.13349
Alexander, S. P., Peters, J. A., Kelly, E., Marrion, N., Benson, H. E., Faccenda, E., et al. (2015b). The concise guide to PHARMACOLOGY 2015/16: ligand-gated ion channels. Br. J. Pharmacol. 172, 5870–5903. doi: 10.1111/bph.13350
Alsharafi, W. A., Xiao, B., and Li, J. (2016). MicroRNA-139–5p negatively regulates NR2A-containing NMDA receptor in the rat pilocarpine model and patients with temporal lobe epilepsy. Epilepsia 57, 1931–1940. doi: 10.1111/epi.13568
Arnold, E. C., McMurray, C., Gray, R., and Johnston, D. (2019). Epilepsy-induced reduction in hcn channel expression contributes to an increased excitability in dorsal, but not ventral, hippocampal CA1 neurons. eNeuro 6:ENEURO.0036-19.2019. doi: 10.1523/eneuro.0036-19.2019
Assaf, S. Y., and Chung, S. H. (1984). Release of endogenous Zn2+ from brain tissue during activity. Nature 308, 734–736. doi: 10.1038/308734a0
Becker, A. J. (2018). Review: animal models of acquired epilepsy: insights into mechanisms of human epileptogenesis. Neuropathol. Appl. Neurobiol. 44, 112–129. doi: 10.1111/nan.12451
Becker, A. J., Pitsch, J., Sochivko, D., Opitz, T., Staniek, M., Chen, C. C., et al. (2008). Transcriptional upregulation of Cav3.2 mediates epileptogenesis in the pilocarpine model of epilepsy. J. Neurosci. 28, 13341–13353. doi: 10.1523/JNEUROSCI.1421-08.2008
Beer, J., Mielke, K., Zipp, M., Zimmermann, M., and Herdegen, T. (1998). Expression of c-jun, junB, c-fos, fra-1 and fra-2 mRNA in the rat brain following seizure activity and axotomy. Brain Res. 794, 255–266. doi: 10.1016/s0006-8993(98)00233-9
Bernard, C., Anderson, A., Becker, A., Poolos, N. P., Beck, H., and Johnston, D. (2004). Acquired dendritic channelopathy in temporal lobe epilepsy. Science 305, 532–535. doi: 10.1126/science.1097065
Bertelli, M., Cecchin, S., Lapucci, C., de Gemmis, P., Danieli, D., d’Amore, E. S., et al. (2007). Quantification of chloride channel 2 (CLCN2) gene isoforms in normal versus lesion- and epilepsy-associated brain tissue. Biochim. Biophys. Acta 1772, 15–20. doi: 10.1016/j.bbadis.2006.10.015
Bien, C. G., and Elger, C. E. (2007). Limbic encephalitis: a cause of temporal lobe epilepsy with onset in adult life. Epilepsy Behav. 10, 529–538. doi: 10.1016/j.yebeh.2007.03.011
Bien, C. G., Urbach, H., Schramm, J., Soeder, B. M., Becker, A. J., Voltz, R., et al. (2007). Limbic encephalitis as a precipitating event in adult-onset temporal lobe epilepsy. Neurology 69, 1236–1244. doi: 10.1212/01.wnl.0000276946.08412.ef
Billingsley, K. J., Manca, M., Gianfrancesco, O., Collier, D. A., Sharp, H., Bubb, V. J., et al. (2018). Regulatory characterisation of the schizophrenia-associated CACNA1C proximal promoter and the potential role for the transcription factor EZH2 in schizophrenia aetiology. Schizophr. Res. 199, 168–175. doi: 10.1016/j.schres.2018.02.036
Bohnsack, J. P., Patel, V. K., and Morrow, A. L. (2017). Ethanol exposure regulates gabra1 expression via histone deacetylation at the promoter in cultured cortical neurons. J. Pharmacol. Exp. Ther. 363, 1–11. doi: 10.1124/jpet.117.242446
Borlot, F., Regan, B. M., Bassett, A. S., Stavropoulos, D. J., and Andrade, D. M. (2017). Prevalence of pathogenic copy number variation in adults with pediatric-onset epilepsy and intellectual disability. JAMA Neurol. 74, 1301–1311. doi: 10.1001/jamaneurol.2017.1775
Brenowitz, S., Duguid, I., and Kammermeier, P. J. (2017). Ion channels: history, diversity, and impact. Cold Spring Harb. Protoc. 2017:pdb.top092288. doi: 10.1101/pdb.top092288
Brunklaus, A., and Zuberi, S. M. (2014). Dravet syndrome—from epileptic encephalopathy to channelopathy. Epilepsia 55, 979–984. doi: 10.1111/epi.12652
Cammaerts, S., Strazisar, M., Smets, B., Weckhuysen, S., Nordin, A., De Jonghe, P., et al. (2015). Schizophrenia-associated MIR204 regulates noncoding RNAs and affects neurotransmitter and ion channel gene sets. PLoS One 10:e0144428. doi: 10.1371/journal.pone.0144428
Canitano, R. (2007). Epilepsy in autism spectrum disorders. Eur. Child Adolesc. Psychiatry 16, 61–66. doi: 10.1007/s00787-006-0563-2
Carvill, G. L., Engel, K. L., Ramamurthy, A., Cochran, J. N., Roovers, J., Stamberger, H., et al. (2018). Aberrant inclusion of a poison exon causes dravet syndrome and related SCN1A-associated genetic epilepsies. Am. J. Hum. Genet. 103, 1022–1029. doi: 10.1016/j.ajhg.2018.10.023
Catalanotto, C., Cogoni, C., and Zardo, G. (2016). MicroRNA in control of gene expression: an overview of nuclear functions. Int. J. Mol. Sci. 17:E1712. doi: 10.3390/ijms17101712
Chen, K., Aradi, I., Thon, N., Eghbal-Ahmadi, M., Baram, T. Z., and Soltesz, I. (2001). Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat. Med. 7, 331–337. doi: 10.1038/85480
Chen, C. H., Huang, C. C., Cheng, M. C., Chiu, Y. N., Tsai, W. C., Wu, Y. Y., et al. (2014a). Genetic analysis of GABRB3 as a candidate gene of autism spectrum disorders. Mol. Autism 5:36. doi: 10.1186/2040-2392-5-36
Chen, C. H., Huang, C. C., and Liao, D. L. (2014b). Association analysis of GABRB3 promoter variants with heroin dependence. PLoS One 9:e102227. doi: 10.1371/journal.pone.0102227
Chen, S., Su, H., Yue, C., Remy, S., Royeck, M., Sochivko, D., et al. (2011). An increase in persistent sodium current contributes to intrinsic neuronal bursting after status epilepticus. J. Neurophysiol. 105, 117–129. doi: 10.1152/jn.00184.2010
Clouaire, T., and Stancheva, I. (2008). Methyl-CpG binding proteins: specialized transcriptional repressors or structural components of chromatin? Epigenomes 65, 1509–1522. doi: 10.1007/s00018-008-7324-y
Coyle, J. T., Tsai, G., and Goff, D. C. (2002). Ionotropic glutamate receptors as therapeutic targets in schizophrenia. Curr. Drug Targets CNS Neurol. Disord. 1, 183–189. doi: 10.2174/1568007024606212
Daghsni, M., Rima, M., Fajloun, Z., Ronjat, M., Brusés, J. L., M’Rad, R., et al. (2018). Autism throughout genetics: perusal of the implication of ion channels. Brain Behav. 8:e00978. doi: 10.1002/brb3.978
de Haan, G. J., Pinto, D., Carton, D., Bader, A., Witte, J., Peters, E., et al. (2006). A novel splicing mutation in KCNQ2 in a multigenerational family with BFNC followed for 25 years. Epilepsia 47, 851–859. doi: 10.1111/j.1528-1167.2006.00552.x
Deng, H., Xiu, X., and Song, Z. (2014). The molecular biology of genetic-based epilepsies. Mol. Neurobiol. 49, 352–367. doi: 10.1007/s12035-013-8523-6
Diss, J. K., Calissano, M., Gascoyne, D., Djamgoz, M. B., and Latchman, D. S. (2008). Identification and characterization of the promoter region of the Nav1.7 voltage-gated sodium channel gene (SCN9A). Mol. Cell. Neurosci. 37, 537–547. doi: 10.1016/j.mcn.2007.12.002
Dong, Z. F., Tang, L. J., Deng, G. F., Zeng, T., Liu, S. J., Wan, R. P., et al. (2014). Transcription of the human sodium channel SCN1A gene is repressed by a scaffolding protein RACK1. Mol. Neurobiol. 50, 438–448. doi: 10.1007/s12035-014-8633-9
Dunn, P., Albury, C. L., Maksemous, N., Benton, M. C., Sutherland, H. G., Smith, R. A., et al. (2018). Next generation sequencing methods for diagnosis of epilepsy syndromes. Front. Genet. 9:20. doi: 10.3389/fgene.2018.00020
Endele, S., Rosenberger, G., Geider, K., Popp, B., Tamer, C., Stefanova, I., et al. (2010). Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat. Genet. 42, 1021–1026. doi: 10.1038/ng.677
Engel, J. Jr. (1996). Introduction to temporal lobe epilepsy. Epilepsy Res. 26, 141–150. doi: 10.1016/s0920-1211(96)00043-5
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat. Rev. Neurosci. 6, 215–229. doi: 10.1038/nrn1625
Fisher, R. S. (2015). Redefining epilepsy. Curr. Opin. Neurol. 28, 130–135. doi: 10.1097/WCO.0000000000000174
Fisher, R. S., Acevedo, C., Arzimanoglou, A., Bogacz, A., Cross, J. H., Elger, C. E., et al. (2014). ILAE official report: a practical clinical definition of epilepsy. Epilepsia 55, 475–482. doi: 10.1111/epi.12550
Gao, Q. W., Hua, L. D., Wang, J., Fan, C. X., Deng, W. Y., Li, B., et al. (2017). A point mutation in SCN1A 5’ genomic region decreases the promoter activity and is associated with mild epilepsy and seizure aggravation induced by antiepileptic drug. Mol. Neurobiol. 54, 2428–2434. doi: 10.1007/s12035-016-9800-y
Gibson, G. (2012). Rare and common variants: twenty arguments. Nat. Rev. Genet. 13, 135–145. doi: 10.1038/nrg3118
Gross, C., Yao, X., Engel, T., Tiwari, D., Xing, L., Rowley, S., et al. (2016). MicroRNA-mediated downregulation of the potassium channel Kv4.2 contributes to seizure onset. Cell Rep. 17, 37–45. doi: 10.1016/j.celrep.2016.08.074
Guan, F., Zhang, B., Yan, T., Li, L., Liu, F., Li, T., et al. (2014). MIR137 gene and target gene CACNA1C of miR-137 contribute to schizophrenia susceptibility in Han Chinese. Schizophr. Res. 152, 97–104. doi: 10.1016/j.schres.2013.11.004
Gulchina, Y., Xu, S. J., Snyder, M. A., Elefant, F., and Gao, W. J. (2017). Epigenetic mechanisms underlying NMDA receptor hypofunction in the prefrontal cortex of juvenile animals in the MAM model for schizophrenia. J. Neurochem. 143, 320–333. doi: 10.1111/jnc.14101
Haenisch, S., von Ruden, E. L., Wahmkow, H., Rettenbeck, M. L., Michler, C., Russmann, V., et al. (2016). miRNA-187–3p-mediated regulation of the KCNK10/TREK-2 potassium channel in a rat epilepsy model. ACS Chem. Neurosci. 7, 1585–1594. doi: 10.1021/acschemneuro.6b00222
Hauser, R. M., Henshall, D. C., and Lubin, F. D. (2018). The epigenetics of epilepsy and its progression. Neuroscientist 24, 186–200. doi: 10.1177/1073858417705840
Henshall, D. C., Hamer, H. M., Pasterkamp, R. J., Goldstein, D. B., Kjems, J., Prehn, J. H. M., et al. (2016). MicroRNAs in epilepsy: pathophysiology and clinical utility. Lancet Neurol. 15, 1368–1376. doi: 10.1016/S1474-4422(16)30246-0
Herdegen, T., and Leah, J. D. (1998). Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res. Rev. 28, 370–490. doi: 10.1016/s0165-0173(98)00018-6
Herdegen, T., Sandkuhler, J., Gass, P., Kiessling, M., Bravo, R., and Zimmermann, M. (1993). JUN, FOS, KROX and CREB transcription factor proteins in the rat cortex: basal expression and induction by spreading depression and epileptic seizures. J. Comp. Neurol. 333, 271–288. doi: 10.1002/cne.903330212
Hirose, S. (2014). Mutant GABAA receptor subunits in genetic (idiopathic) epilepsy. Prog. Brain Res. 213, 55–85. doi: 10.1016/b978-0-444-63326-2.00003-x
Honkaniemi, J., and Sharp, F. R. (1999). Prolonged expression of zinc finger immediate-early gene mRNAs and decreased protein synthesis following kainic acid induced seizures. Eur. J. Neurosci. 11, 10–17. doi: 10.1046/j.1460-9568.1999.00401.x
Hu, Y., Lund, I. V., Gravielle, M. C., Farb, D. H., Brooks-Kayal, A. R., and Russek, S. J. (2008). Surface expression of GABAA receptors is transcriptionally controlled by the interplay of cAMP-response element-binding protein and its binding partner inducible cAMP early repressor. J. Biol. Chem. 283, 9328–9340. doi: 10.1074/jbc.m705110200
Hyde, T. M., and Weinberger, D. R. (1997). Seizures and schizophrenia. Schizophr. Bull. 23, 611–622. doi: 10.1093/schbul/23.4.611
Imbrici, P., Camerino, D. C., and Tricarico, D. (2013). Major channels involved in neuropsychiatric disorders and therapeutic perspectives. Front. Genet. 4:76. doi: 10.3389/fgene.2013.00076
International League Against Epilepsy Consortium on Complex Epilepsies. (2018). Genome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nat. Commun. 9:5269. doi: 10.1038/s41467-018-07524-z
Itokawa, M., Yamada, K., Yoshitsugu, K., Toyota, T., Suga, T., Ohba, H., et al. (2003). A microsatellite repeat in the promoter of the N-methyl-D-aspartate receptor 2A subunit (GRIN2A) gene suppresses transcriptional activity and correlates with chronic outcome in schizophrenia. Pharmacogenetics 13, 271–278. doi: 10.1097/00008571-200305000-00006
Jefferys, J., Steinhäuser, C., and Bedner, P. (2016). Chemically-induced TLE models: topical application. J. Neurosci. Methods 260, 53–61. doi: 10.1016/j.jneumeth.2015.04.011
Jung, S., Warner, L. N., Pitsch, J., Becker, A. J., and Poolos, N. P. (2011). Rapid loss of dendritic HCN channel expression in hippocampal pyramidal neurons following status epilepticus. J. Neurosci. 31, 14291–14295. doi: 10.1523/jneurosci.1148-11.2011
Kananura, C., Haug, K., Sander, T., Runge, U., Gu, W., Hallmann, K., et al. (2002). A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch. Neurol. 59, 1137–1141. doi: 10.1001/archneur.59.7.1137
Kasperaviciute, D., Catarino, C. B., Matarin, M., Leu, C., Novy, J., Tostevin, A., et al. (2013). Epilepsy, hippocampal sclerosis and febrile seizures linked by common genetic variation around SCN1A. Brain 136, 3140–3150. doi: 10.1093/brain/awt233
Kichukova, T. M., Popov, N. T., Ivanov, I. S., and Vachev, T. I. (2017). Profiling of circulating serum MicroRNAs in children with autism spectrum disorder using stem-loop qRT-PCR assay. Folia Med. 59, 43–52. doi: 10.1515/folmed-2017-0009
Kiese, K., Jablonski, J., Hackenbracht, J., Wrosch, J. K., Groemer, T. W., Kornhuber, J., et al. (2017). Epigenetic control of epilepsy target genes contributes to a cellular memory of epileptogenesis in cultured rat hippocampal neurons. Acta Neuropathol. Commun. 5:79. doi: 10.1186/s40478-017-0485-x
Kocerha, J., Faghihi, M. A., Lopez-Toledano, M. A., Huang, J., Ramsey, A. J., Caron, M. G., et al. (2009). MicroRNA-219 modulates NMDA receptor-mediated neurobehavioral dysfunction. Proc. Natl. Acad. Sci. U S A 106, 3507–3512. doi: 10.1073/pnas.0805854106
Kordi-Tamandani, D. M., Dahmardeh, N., and Torkamanzehi, A. (2013). Evaluation of hypermethylation and expression pattern of GMR2, GMR5, GMR8 and GRIA3 in patients with schizophrenia. Gene 515, 163–166. doi: 10.1016/j.gene.2012.10.075
Kwan, P., and Sander, J. W. (2004). The natural history of epilepsy: an epidemiological view. J. Neurol. Neurosurg. Psychiatry 75, 1376–1381. doi: 10.1136/jnnp.2004.045690
Lau, C. G., and Zukin, R. S. (2007). NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 8, 413–426. doi: 10.1038/nrn2153
Lerche, H., Shah, M., Beck, H., Noebels, J., Johnston, D., and Vincent, A. (2013). Ion channels in genetic and acquired forms of epilepsy. J. Physiol. 591, 753–764. doi: 10.1113/jphysiol.2012.240606
Lévesque, M., Avoli, M., and Bernard, C. (2016). Animal models of temporal lobe epilepsy following systemic chemoconvulsant administration. J. Neurosci. Methods 260, 45–52. doi: 10.1016/j.jneumeth.2015.03.009
Li, Z., Cogswell, M., Hixson, K., Brooks-Kayal, A. R., and Russek, S. J. (2018). Nuclear respiratory factor 1 (NRF-1) controls the activity dependent transcription of the GABA-A receptor β 1 subunit gene in neurons. Front. Mol. Neurosci. 11:285. doi: 10.3389/fnmol.2018.00285
Li, H. J., Wan, R. P., Tang, L. J., Liu, S. J., Zhao, Q. H., Gao, M. M., et al. (2015). Alteration of Scn3a expression is mediated via CpG methylation and MBD2 in mouse hippocampus during postnatal development and seizure condition. Biochim. Biophys. Acta 1849, 1–9. doi: 10.1016/j.bbagrm.2014.11.004
Liao, J., Tian, X., Wang, H., and Xiao, Z. (2018). Epilepsy and migraine-Are they comorbidity? Genes Dis. 5, 112–118. doi: 10.1016/j.gendis.2018.04.007
Liu, R., Dang, W., Du, Y., Zhou, Q., Liu, Z., and Jiao, K. (2015). Correlation of functional GRIN2A gene promoter polymorphisms with schizophrenia and serum D-serine levels. Gene 568, 25–30. doi: 10.1016/j.gene.2015.05.011
Liu, Y., Ding, M., Liu, Y. P., Zhang, X. C., Xing, J. X., Xuan, J. F., et al. (2019). Functional analysis of haplotypes and promoter activity at the 5′ region of the human GABRB3 gene and associations with schizophrenia. Mol. Genet. Genomic Med. 7:e652. doi: 10.1002/mgg3.652
Liu, Y., Lai, S., Ma, W., Ke, W., Zhang, C., Liu, S., et al. (2017). CDYL suppresses epileptogenesis in mice through repression of axonal Nav1.6 sodium channel expression. Nat. Commun. 8:355. doi: 10.1038/s41467-017-00368-z
Liu, S., Yu, W., and Lu, Y. (2016). The causes of new-onset epilepsy and seizures in the elderly. Neuropsychiatr. Dis. Treat. 12, 1425–1434. doi: 10.2147/ndt.s107905
Löscher, W., Klitgaard, H., Twyman, R. E., and Schmidt, D. (2013). New avenues for anti-epileptic drug discovery and development. Nat. Rev. Drug Discov. 12, 757–776. doi: 10.1038/nrd4126
Lossin, C. (2009). A catalog of SCN1A variants. Brain Dev. 31, 114–130. doi: 10.1016/j.braindev.2008.07.011
Lund, I. V., Hu, Y., Raol, Y. H., Benham, R. S., Faris, R., Russek, S. J., et al. (2008). BDNF selectively regulates GABAA receptor transcription by activation of the JAK/STAT pathway. Sci. Signal. 1:ra9. doi: 10.1126/scisignal.1162396
Machnes, Z. M., Huang, T. C., Chang, P. K., Gill, R., Reist, N., Dezsi, G., et al. (2013). DNA methylation mediates persistent epileptiform activity in vitro and in vivo. PLoS One 8:e76299. doi: 10.1371/journal.pone.0076299
Marcelin, B., Chauviere, L., Becker, A., Migliore, M., Esclapez, M., and Bernard, C. (2009). h channel-dependent deficit of theta oscillation resonance and phase shift in temporal lobe epilepsy. Neurobiol. Dis. 33, 436–447. doi: 10.1016/j.nbd.2008.11.019
McClelland, S., Flynn, C., Dube, C., Richichi, C., Zha, Q., Ghestem, A., et al. (2011). Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy. Ann. Neurol. 70, 454–464. doi: 10.1002/ana.22479
Miyatake, R., Furukawa, A., and Suwaki, H. (2002). Identification of a novel variant of the human NR2B gene promoter region and its possible association with schizophrenia. Mol. Psychiatry 7, 1101–1106. doi: 10.1038/sj.mp.4001152
Monaghan, M. M., Menegola, M., Vacher, H., Rhodes, K. J., and Trimmer, J. S. (2008). Altered expression and localization of hippocampal A-type potassium channel subunits in the pilocarpine-induced model of temporal lobe epilepsy. Neuroscience 156, 550–562. doi: 10.1016/j.neuroscience.2008.07.057
Monlong, J., Girard, S. L., Meloche, C., Cadieux-Dion, M., Andrade, D. M., Lafreniere, R. G., et al. (2018). Global characterization of copy number variants in epilepsy patients from whole genome sequencing. PLoS Genet. 14:e1007285. doi: 10.1371/journal.pgen.1007285
Mucha, M., Ooi, L., Linley, J. E., Mordaka, P., Dalle, C., Robertson, B., et al. (2010). Transcriptional control of KCNQ channel genes and the regulation of neuronal excitability. J. Neurosci. 30, 13235–13245. doi: 10.1523/jneurosci.1981-10.2010
Mula, M., Schmitz, B., Jauch, R., Cavanna, A., Cantello, R., Monaco, F., et al. (2008). On the prevalence of bipolar disorder in epilepsy. Epilepsy Behav. 13, 658–661. doi: 10.1016/j.yebeh.2008.08.002
Nakayama, T., Ogiwara, I., Ito, K., Kaneda, M., Mazaki, E., Osaka, H., et al. (2010). Deletions of SCN1A 5′ genomic region with promoter activity in Dravet syndrome. Hum. Mutat. 31, 820–829. doi: 10.1002/humu.21275
Nirwan, N., Vyas, P., and Vohora, D. (2018). Animal models of status epilepticus and temporal lobe epilepsy: a narrative review. Rev. Neurosci. 29, 757–770. doi: 10.1515/revneuro-2017-0086
Nwaobi, S. E., Lin, E., Peramsetty, S. R., and Olsen, M. L. (2014). DNA methylation functions as a critical regulator of Kir4.1 expression during CNS development. Glia 62, 411–427. doi: 10.1002/glia.22613
Oliveira, M. S., Skinner, F., Arshadmansab, M. F., Garcia, I., Mello, C. F., Knaus, H. G., et al. (2010). Altered expression and function of small-conductance (SK) Ca2+-activated K+ channels in pilocarpine-treated epileptic rats. Brain Res. 1348, 187–199. doi: 10.1016/j.brainres.2010.05.095
O’Roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. doi: 10.1126/science.1227764
Oyrer, J., Maljevic, S., Scheffer, I. E., Berkovic, S. F., Petrou, S., and Reid, C. A. (2018). Ion channels in genetic epilepsy: from genes and mechanisms to disease-targeted therapies. Pharmacol. Rev. 70, 142–173. doi: 10.1124/pr.117.014456
Pacheco Otalora, L. F., Hernandez, E. F., Arshadmansab, M. F., Francisco, S., Willis, M., Ermolinsky, B., et al. (2008). Down-regulation of BK channel expression in the pilocarpine model of temporal lobe epilepsy. Brain Res. 1200, 116–131. doi: 10.1016/j.brainres.2008.01.017
Page, S. C., Hamersky, G. R., Gallo, R. A., Rannals, M. D., Calcaterra, N. E., Campbell, M. N., et al. (2018). The schizophrenia- and autism-associated gene, transcription factor 4 regulates the columnar distribution of layer 2/3 prefrontal pyramidal neurons in an activity-dependent manner. Mol. Psychiatry 23, 304–315. doi: 10.1038/mp.2017.37
Palmer, E. E., Stuhlmann, T., Weinert, S., Haan, E., Van Esch, H., Holvoet, M., et al. (2018). De novo and inherited mutations in the X-linked gene CLCN4 are associated with syndromic intellectual disability and behavior and seizure disorders in males and females. Mol. Psychiatry 23, 222–230. doi: 10.1038/mp.2016.135
Parrish, R. R., Albertson, A. J., Buckingham, S. C., Hablitz, J. J., Mascia, K. L., Davis Haselden, W., et al. (2013). Status epilepticus triggers early and late alterations in brain-derived neurotrophic factor and NMDA glutamate receptor Grin2b DNA methylation levels in the hippocampus. Neuroscience 248, 602–619. doi: 10.1016/j.neuroscience.2013.06.029
Pitkänen, A., Roivainen, R., and Lukasiuk, K. (2016). Development of epilepsy after ischaemic stroke. Lancet Neurol. 15, 185–197. doi: 10.1016/s1474-4422(15)00248-3
Piton, A., Jouan, L., Rochefort, D., Dobrzeniecka, S., Lachapelle, K., Dion, P. A., et al. (2013). Analysis of the effects of rare variants on splicing identifies alterations in GABAA receptor genes in autism spectrum disorder individuals. Eur. J. Hum. Genet. 21, 749–756. doi: 10.1038/ejhg.2012.243
Powell, K. L., Cain, S. M., Ng, C., Sirdesai, S., David, L. S., Kyi, M., et al. (2009). A Cav3.2 T-type calcium channel point mutation has splice-variant-specific effects on function and segregates with seizure expression in a polygenic rat model of absence epilepsy. J. Neurosci. 29, 371–380. doi: 10.1523/JNEUROSCI.5295-08.2009
Raimondo, J. V., Burman, R. J., Katz, A. A., and Akerman, C. J. (2015). Ion dynamics during seizures. Front. Cell Neurosci. 9:419. doi: 10.3389/fncel.2015.00419
Ranganathan, K., and Sivasankar, V. (2014). MicroRNAs - biology and clinical applications. J. Oral Maxillofac. Pathol. 18, 229–234. doi: 10.4103/0973-029x.140762
Rannals, M. D., Hamersky, G. R., Page, S. C., Campbell, M. N., Briley, A., Gallo, R. A., et al. (2016). Psychiatric risk gene transcription factor 4 regulates intrinsic excitability of prefrontal neurons via repression of SCN10a and KCNQ1. Neuron 90, 43–55. doi: 10.1016/j.neuron.2016.02.021
Reich, D. E., and Lander, E. S. (2001). On the allelic spectrum of human disease. Trends Genet. 17, 502–510. doi: 10.1016/s0168-9525(01)02410-6
Reid, C. A., Berkovic, S. F., and Petrou, S. (2009). Mechanisms of human inherited epilepsies. Prog. Neurobiol. 87, 41–57. doi: 10.1016/j.pneurobio.2008.09.016
Reschke, C. R., and Henshall, D. C. (2015). microRNA and epilepsy. Adv. Exp. Med. Biol. 888, 41–70. doi: 10.1007/978-3-319-22671-2_4
Roberts, D. S., Raol, Y. H., Bandyopadhyay, S., Lund, I. V., Budreck, E. C., Passini, M. A., et al. (2005). Egr3 stimulation of GABRA4 promoter activity as a mechanism for seizure-induced up-regulation of GABAA receptor α4 subunit expression. Proc. Natl. Acad. Sci. U S A 102, 11894–11899. doi: 10.1073/pnas.0501434102
Saint Pierre, A., and Génin, E. (2014). How important are rare variants in common disease? Brief. Funct. Genomics 13, 353–361. doi: 10.1093/bfgp/elu025
Salpekar, J. A., and Mula, M. (2018). Common psychiatric comorbidities in epilepsy: how big of a problem is it? Epilepsy Behav. 98, 293–297. doi: 10.1016/j.yebeh.2018.07.023
Samaco, R. C., Hogart, A., and LaSalle, J. M. (2005). Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 14, 483–492. doi: 10.1093/hmg/ddi045
Schlachter, K., Gruber-Sedlmayr, U., Stogmann, E., Lausecker, M., Hotzy, C., Balzar, J., et al. (2009). A splice site variant in the sodium channel gene SCN1A confers risk of febrile seizures. Neurology 72, 974–978. doi: 10.1212/01.wnl.0000344401.02915.00
Schmunk, G., and Gargus, J. J. (2013). Channelopathy pathogenesis in autism spectrum disorders. Front. Genet. 4:222. doi: 10.3389/fgene.2013.00222
Schuster, J., Laan, L., Klar, J., Jin, Z., Huss, M., Korol, S., et al. (2019). Transcriptomes of Dravet syndrome iPSC derived GABAergic cells reveal dysregulated pathways for chromatin remodeling and neurodevelopment. Neurobiol. Dis. 132:104583. doi: 10.1016/j.nbd.2019.104583
Shah, M. M., Anderson, A. E., Leung, V., Lin, X., and Johnston, D. (2004). Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron 44, 495–508. doi: 10.1016/j.neuron.2004.10.011
Shao, Y., and Chen, Y. (2017). Pathophysiology and clinical utility of non-coding RNAs in epilepsy. Front. Mol. Neurosci. 10:249. doi: 10.3389/fnmol.2017.00249
Shorvon, S. D. (2011). The etiologic classification of epilepsy. Epilepsia 52, 1052–1057. doi: 10.1111/j.1528-1167.2011.03041.x
Shruti, S., Clem, R. L., and Barth, A. L. (2008). A seizure-induced gain-of-function in BK channels is associated with elevated firing activity in neocortical pyramidal neurons. Neurobiol. Dis. 30, 323–330. doi: 10.1016/j.nbd.2008.02.002
Smigiel, R., Kostrzewa, G., Kosinska, J., Pollak, A., Stawinski, P., Szmida, E., et al. (2016). Further evidence for GRIN2B mutation as the cause of severe epileptic encephalopathy. Am. J. Med. Genet. A 170, 3265–3270. doi: 10.1002/ajmg.a.37887
Sosanya, N. M., Huang, P. P., Cacheaux, L. P., Chen, C. J., Nguyen, K., Perrone-Bizzozero, N. I., et al. (2013). Degradation of high affinity HuD targets releases Kv1.1 mRNA from miR-129 repression by mTORC1. J. Cell Biol. 202, 53–69. doi: 10.1083/jcb.201212089
Steinlein, O. K., Mulley, J. C., Propping, P., Wallace, R. H., Phillips, H. A., Sutherland, G. R., et al. (1995). A missense mutation in the neuronal nicotinic acetylcholine receptor α 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 11, 201–203. doi: 10.1038/ng1095-201
Strasser, L., Downes, M., Kung, J., Cross, J. H., and De Haan, M. (2018). Prevalence and risk factors for autism spectrum disorder in epilepsy: a systematic review and meta-analysis. Dev. Med. Child Neurol. 60, 19–29. doi: 10.1111/dmcn.13598
Su, H., Sochivko, D., Becker, A., Chen, J., Jiang, Y., Yaari, Y., et al. (2002). Upregulation of a T-type Ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus. J. Neurosci. 22, 3645–3655. doi: 10.1523/jneurosci.22-09-03645.2002
Tan, N. N., Tang, H. L., Lin, G. W., Chen, Y. H., Lu, P., Li, H. J., et al. (2017). Epigenetic downregulation of Scn3a expression by valproate: a possible role in its anticonvulsant activity. Mol. Neurobiol. 54, 2831–2842. doi: 10.1007/s12035-016-9871-9
Tanaka, M., Bailey, J. N., Bai, D., Ishikawa-Brush, Y., Delgado-Escueta, A. V., and Olsen, R. W. (2012a). Effects on promoter activity of common SNPs in 5′ region of GABRB3 exon 1A. Epilepsia 53, 1450–1456. doi: 10.1111/j.1528-1167.2012.03572.x
Tanaka, M., DeLorey, T. M., Delgado-Escueta, A., and Olsen, R. W. (2012b). “GABRB3, Epilepsy and Neurodevelopment,” in Jasper’s Basic Mechanisms of the Epilepsies, eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, and A. V. Delgado-Escueta (Bethesda, MD: Oxford University Press), 887–899.
Thompson, C. H., Kahlig, K. M., and George, A. L. Jr. (2011). SCN1A splice variants exhibit divergent sensitivity to commonly used antiepileptic drugs. Epilepsia 52, 1000–1009. doi: 10.1111/j.1528-1167.2011.03040.x
Tiwari, D., Brager, D. H., Rymer, J. K., Bunk, A. T., White, A. R., Elsayed, N. A., et al. (2019). MicroRNA inhibition upregulates hippocampal A-type potassium current and reduces seizure frequency in a mouse model of epilepsy. Neurobiol. Dis. 130:104508. doi: 10.1016/j.nbd.2019.104508
Tiwari, D., Peariso, K., and Gross, C. (2018). MicroRNA-induced silencing in epilepsy: opportunities and challenges for clinical application. Dev. Dyn. 247, 94–110. doi: 10.1002/dvdy.24582
Urak, L., Feucht, M., Fathi, N., Hornik, K., and Fuchs, K. (2006). A GABRB3 promoter haplotype associated with childhood absence epilepsy impairs transcriptional activity. Hum. Mol. Genet. 15, 2533–2541. doi: 10.1093/hmg/ddl174
van Loo, K. M. J., Rummel, C. K., Pitsch, J., Muller, J. A., Bikbaev, A. F., Martinez-Chavez, E., et al. (2019). Calcium channel subunit α2δ4 is regulated by early growth response 1 and facilitates epileptogenesis. J. Neurosci. 39, 3175–3187. doi: 10.1523/jneurosci.1731-18.2019
van Loo, K. M., Schaub, C., Pernhorst, K., Yaari, Y., Beck, H., Schoch, S., et al. (2012). Transcriptional regulation of T-type calcium channel CaV3.2: bi-directionality by early growth response 1 (Egr1) and repressor element 1 (RE-1) protein-silencing transcription factor (REST). J. Biol. Chem. 287, 15489–15501. doi: 10.1074/jbc.m111.310763
van Loo, K. M., Schaub, C., Pitsch, J., Kulbida, R., Opitz, T., Ekstein, D., et al. (2015). Zinc regulates a key transcriptional pathway for epileptogenesis via metal-regulatory transcription factor 1. Nat. Commun. 6:8688. doi: 10.1038/ncomms9688
Vezzani, A., Fujinami, R. S., White, H. S., Preux, P. M., Blumcke, I., Sander, J. W., et al. (2016). Infections, inflammation and epilepsy. Acta Neuropathol. 131, 211–234. doi: 10.1007/s00401-015-1481-5
Wallace, R. H., Wang, D. W., Singh, R., Scheffer, I. E., George, A. L. Jr., Phillips, H. A., et al. (1998). Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel β1 subunit gene SCN1B. Nat. Genet. 19, 366–370. doi: 10.1038/1252
Wang, J., Lin, Z. J., Liu, L., Xu, H. Q., Shi, Y. W., Yi, Y. H., et al. (2017). Epilepsy-associated genes. Seizure 44, 11–20. doi: 10.1016/j.seizure.2016.11.030
Wei, F., Yan, L. M., Su, T., He, N., Lin, Z. J., Wang, J., et al. (2017). Ion channel genes and epilepsy: functional alteration, pathogenic potential and mechanism of epilepsy. Neurosci. Bull. 33, 455–477. doi: 10.1007/s12264-017-0134-1
Weiss, N., and Zamponi, G. W. (2019). Genetic T-type calcium channelopathies. J. Med Genet. 57, 1–10. doi: 10.1136/jmedgenet-2019-106163
Xiang, J., Wen, F., Zhang, L., and Zhou, Y. (2018). FOXD3 inhibits SCN2A gene transcription in intractable epilepsy cell models. Exp. Neurol. 302, 14–21. doi: 10.1016/j.expneurol.2017.12.012
Yao, J. J., Zhao, Q. R., Liu, D. D., Chow, C. W., and Mei, Y. A. (2016). Neuritin up-regulates Kv4.2 α-subunit of potassium channel expression and affects neuronal excitability by regulating the calcium-calcineurin-NFATc4 signaling pathway. J. Biol. Chem. 291, 17369–17381. doi: 10.1074/jbc.m115.708883
Yasui, D. H., Scoles, H. A., Horike, S., Meguro-Horike, M., Dunaway, K. W., Schroeder, D. I., et al. (2011). 15q11.2–13.3 chromatin analysis reveals epigenetic regulation of CHRNA7 with deficiencies in Rett and autism brain. Hum. Mol. Genet. 20, 4311–4323. doi: 10.1093/hmg/ddr357
Young, C. C., Stegen, M., Bernard, R., Muller, M., Bischofberger, J., Veh, R. W., et al. (2009). Upregulation of inward rectifier K+ (Kir2) channels in dentate gyrus granule cells in temporal lobe epilepsy. J. Physiol. 587, 4213–4233. doi: 10.1113/jphysiol.2009.170746
Zhang, Y., Fan, M., Wang, Q., He, G., Fu, Y., Li, H., et al. (2015). Polymorphisms in MicroRNA Genes and genes involving in NMDAR signaling and schizophrenia: a case-control study in chinese han population. Sci. Rep. 5:12984. doi: 10.1038/srep12984
Zhang, S. P., Zhang, M., Tao, H., Luo, Y., He, T., Wang, C. H., et al. (2018). Dimethylation of Histone 3 Lysine 9 is sensitive to the epileptic activity and affects the transcriptional regulation of the potassium channel Kcnj10 gene in epileptic rats. Mol. Med. Rep. 17, 1368–1374. doi: 10.3892/mmr.2017.7942
Zhang, S., Zhu, Y., Cheng, J., and Tao, J. (2019). Ion Channels in Epilepsy: Blasting Fuse for Neuronal Hyperexcitability. IntechOpen. 1–12. doi: 10.5772/intechopen.83698
Zhao, Y., Pan, R., Li, S., Luo, Y., Yan, F., Yin, J., et al. (2014). Chelating intracellularly accumulated zinc decreased ischemic brain injury through reducing neuronal apoptotic death. Stroke 45, 1139–1147. doi: 10.1161/strokeaha.113.004296
Zhao, C., Xu, Z., Wang, F., Chen, J., Ng, S. K., Wong, P. W., et al. (2009). Alternative-splicing in the exon-10 region of GABAA receptor β2. subunit gene: relationships between novel isoforms and psychotic disorders. PLoS One 4:e6977. doi: 10.1371/journal.pone.0006977
Keywords: genetic and acquired epilepsies, ion channels, channelopathies, transcriptional regulation, neurodevelopmental disorders
Citation: van Loo KMJ and Becker AJ (2020) Transcriptional Regulation of Channelopathies in Genetic and Acquired Epilepsies. Front. Cell. Neurosci. 13:587. doi: 10.3389/fncel.2019.00587
Received: 12 September 2019; Accepted: 23 December 2019;
Published: 14 January 2020.
Edited by:
Eleonora Aronica, University Medical Center Amsterdam, NetherlandsReviewed by:
Hee Jung Chung, University of Illinois at Urbana-Champaign, United StatesCopyright © 2020 van Loo and Becker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karen M. J. van Loo, a2FyZW4udmFuX2xvb0B1a2IudW5pLWJvbm4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.